

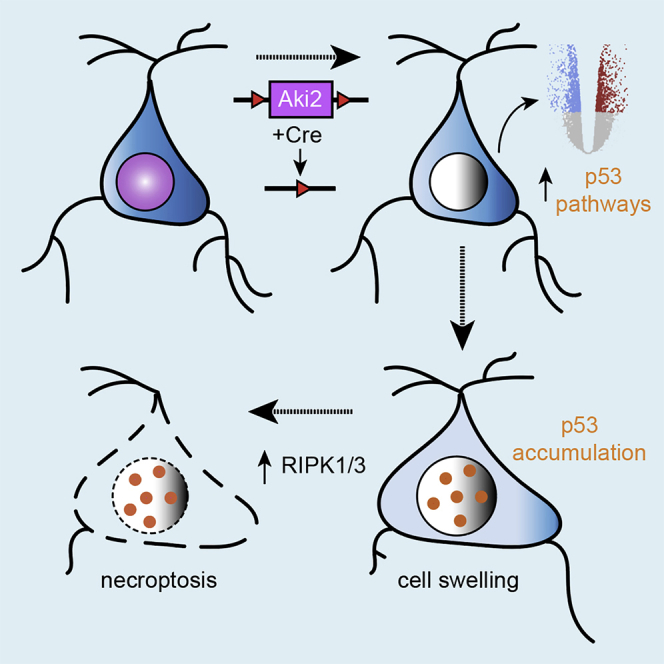
Summary
Proper gene regulation is critical for both neuronal development and maintenance as the brain matures. We previously demonstrated that Akirin2, an essential nuclear protein that interacts with transcription factors and chromatin remodeling complexes, is required for the embryonic formation of the cerebral cortex. Here we show that Akirin2 plays a mechanistically distinct role in maintaining healthy neurons during cortical maturation. Restricting Akirin2 loss to excitatory cortical neurons resulted in progressive neurodegeneration via necroptosis and severe cortical atrophy with age. Comparing transcriptomes from Akirin2-null postnatal neurons and cortical progenitors revealed that targets of the tumor suppressor p53, a regulator of both proliferation and cell death encoded by Trp53, were consistently upregulated. Reduction of Trp53 rescued neurodegeneration in Akirin2-null neurons. These data: (1) implicate Akirin2 as a critical neuronal maintenance protein, (2) identify p53 pathways as mediators of Akirin2 functions, and (3) suggest Akirin2 dysfunction may be relevant to neurodegenerative diseases.
Subject areas: biological sciences, molecular biology, neuroscience, cell biology
Graphical abstract
Highlights
-
•
Akirin2 is a nuclear protein involved in proliferation but maintained in postmitotic neurons
-
•
Postnatal Akirin2 knockout in cortical neurons leads to slow neurodegeneration
-
•
RNASeq identifies the dysregulation of p53-related genes in knockout neurons
-
•
Akirin2-null neurons die by necroptosis that depends on p53 upregulation
Biological sciences; Molecular biology; Neuroscience; Cell biology
Introduction
During development, neural progenitor cells become mitotically quiescent and terminally differentiate into a multitude of specific neuronal subtypes. Maintaining subtype identity and postmitotic status requires continuous expression of the proper gene patterns throughout a neuron’s long postmitotic life (Deneris and Hobert., 2014; Cholewa-Waclaw et al., 2016; Gallegos et al., 2018). This includes both activating cell-type-specific genes as well as suppressing nonneuronal and cell cycle-related genes. Dysregulation of gene networks can lead to defects in innervation (Lin et al., 1998; Arber et al., 2000; Kania and Jessell., 2003; Chen et al., 2013), dendritic and axonal deterioration (Kadkhodaei et al., 2013; Lipinski et al., 2020), loss of cell-type identity (Liu et al., 2010; Bovetti et al., 2013; Montana et al., 2013), neuronal network dysfunction (Chen et al., 2013; Kadkhodaei et al., 2013; Lipinski et al., 2020) and cell death (Ninkovic et al., 2010; von Schimmelmann et al., 2016). Thus, it is not surprising that disrupted gene regulation is associated with memory impairment (Barrett et al., 2011; Vogel-Ciernia et al., 2013), cognitive dysfunction, and neurodegenerative diseases (De Jager et al., 2014; Sanchez-Mut et al., 2016; Watson et al., 2016; Berson et al., 2018; Li et al., 2019). In fact, many studies of neurodegenerative diseases find dysregulation of gene expression in postmitotic neurons before symptom onset, implicating gene dysregulation as the earliest neuronal insult in these diseases (Yang et al., 2003; De Jager et al., 2014). Cell cycle genes, in particular, are ectopically expressed in neurodegenerative diseases, and postmitotic neurons gain phenotypes suggestive of re-entry into the cell cycle (Busser et al., 1998; Yang et al., 2003; McShea et al., 2007; Lee et al., 2009; Wang et al., 2009; Li et al., 2019). As postmitotic neurons are physically incapable of dividing and completing the cell cycle, this re-entry typically leads to cell death (Park et al., 2007). Proper regulation of gene expression patterns is critical for preventing such aberrant cell cycle re-entry and for postmitotic neuron maintenance and survival.
Akirin2 (Aki2) is a highly conserved nuclear protein that regulates gene expression by interacting with transcription factors (Nowak et al., 2012; Bonnay et al., 2014; Tartey et al., 2014) and members of chromatin remodeling complexes [reviewed by (Bosch et al., 2020)]. Invertebrates such as C. elegans and Drosophila harbor a single Akirin gene, while mammals have two homologs. Goto et al. (2008) generated both Akirin1 (Aki1) and Akirin2 (Aki2) null mice, whereas the former were viable and fertile with no outward defects, constitutive Aki2 knockouts exhibited early embryonic lethality, identifying Aki2 as the critical mammalian homolog (Goto et al., 2008). Utilizing an Emx1-Cre driver to delete a conditional Aki2 mutant allele in forebrain neural progenitor cells, we previously found an essential role for Aki2 in cerebral cortex development. Without Aki2, neural progenitor cells exited the cell cycle prematurely and underwent apoptosis, resulting in few cortical neurons after embryonic day (E) 13. These mice were severely microcephalic and nearly all died shortly after birth (Bosch et al., 2016). This work identified previously unknown roles for Akirins in embryonic brain development that is consistent with prior and subsequent studies. Together, current studies on Aki2 indicate that it regulates proliferation and differentiation while inhibiting apoptosis in a variety of tissues and cell types (Komiya et al., 2008; Krossa et al., 2015; Tartey et al., 2015; Liu et al., 2017; Bosch et al., 2018, 2019; Leng et al., 2019).
Here, we show that Aki2 expression continues throughout cortical development, albeit at lower levels in postnatal neurons. As postmitotic neurons have progressed beyond the stages of proliferation and differentiation, we hypothesized that residual Aki2 in the maturing postnatal brain serves distinct roles in postmitotic neurons. To test this hypothesis, we focused on subpopulations of maturing excitatory neurons to avoid neonatal lethality by utilizing a CaMKIIα-Cre driver (Tsien et al., 1996) that disrupted the Aki2 gene in postmitotic excitatory neurons of the cortex beginning around the third postnatal week. In contrast to embryonic cortical phenotypes in which massive apoptosis occurred within 24 h of Cre expression (Bosch et al., 2016), we found that postnatal Aki2 mutants are not immediately affected. Rather, we find that a slow process of neuronal death by necroptosis occurs over several months. Transcriptomic analysis revealed many genes dysregulated in the absence of neuronal Aki2 including a substantial subset that is downstream of the tumor suppressor, p53. A role for p53 downstream of Aki2 is supported by a significant upregulation of p53 protein in knockout cortices, a similar dysregulation of p53 target genes in Aki2-null embryonic telencephalon transcriptomes, and a rescue of postnatal neurodegeneration when Aki2 mutants are heterozygous for, or lack, p53. Together, these data identify a new role for Aki2 in postmitotic neuron maintenance, implicate p53 as a new functional partner underlying the pleiotropic roles played by the Akirin family of proteins, and suggest that Aki2 and its downstream partners may be relevant targets in neurodegenerative disorders.
Results
Neurons of the postnatal cerebral cortex express Akirin2
Gene expression databases suggest that postmitotic neurons express Aki2 transcript (Zhang et al., 2014; Mancarci et al., 2017; Sugino et al., 2019). Anti-Aki2 immunofluorescence (IF) (Figure 1A) and in situ hybridization with an antisense Aki2 probe (Figure S1A) confirmed that cells in all cortical layers expressed Aki2 protein and transcript. To assess Aki2 expression dynamics during cortical development, we western blotted cortical lysates at multiple ages using an anti-Aki2 antibody revealing that Aki2 expression peaked at P0, tapered off during postnatal development, stabilized at P21, and remained detectable into adulthood (Figure 1B). In contrast, mRNA remained consistent throughout development (Figure 1C), which is consistent with prior work (Fertuzinhos et al., 2014; Bosch et al., 2019) and suggests that Aki2 protein is posttranscriptionally regulated. Close inspection of western blots at a variety of exposures revealed 3 distinct bands of ∼22, 25, and 27kDa, as well as two higher molecular weight bands of ∼33 and ∼35kDa. Knockout and knockdown validation of the primary antibody used here (Figure 1F; Bosch et al., 2018; Bosch et al., 2019), confirmed that all five bands are specific to Aki2. This suggests that the cortex expresses several posttranslationally modified forms and/or unannotated Aki2 splice variants.
Figure 1.

Generation of an Akirin2 cortical-restricted knockout mouse model
(A) IF demonstrates widespread Aki2 protein expression across all cortical layers (I–VI) in the adult.
(B) Cortical lysates western blotted with anti-Aki2 antibody show multiple-specific bands around the expected molecular weight of 22 kDa, as well as two larger bands. Protein expression peaks ∼ P0 and slowly declines to a stable level of ∼20% relative to P0 (3 samples/age, normalized to β−tubulin signal within the same lane).
(C) Consistent Aki2 transcript levels across cortical maturation suggest that Aki2 protein is posttranscriptionally regulated (qPCR of 3 samples/age, with GAPDH as a reference gene and plotted as percent of P0 level). One-way ANOVA with Dunnet’s multiple comparisons test comparing every age to P0.
(D) Schematic of alleles and breeding strategy.
(E) Shortly after Cre expression onset, tdTomato labels many cells in cortical layers II–IV (Cux1-demarcated) and VI (FoxP2-demarcated), as well as hippocampal (hipp) dentate gyrus and CA1. Around 2/3 of Cre-positive cells reside in layers II–IV and ∼1/3 are FoxP2-negative layer VI neurons; few reside in layer V.
(F) Immunoblots of control and mutant (cKO) cortical lysates with anti-Aki2 indicate a significant reduction of protein levels (and confirm the specificity of all band sizes observed) at P35, with some expression remaining from Cre (−) cells. Values from 5 to 6 samples/genotype normalized to GAPDH signal and compared using an unpaired t-test. (G) At P30 Anti-Cre and anti-Aki2 antibody staining on cortical sections confirm Aki2 loss in most Cre-expressing mutant cells but not control neurons. By P50, loss of Aki2 IF is apparent in layers with significant Cre expression (II–IV, VI). D, dorsal; V, ventral; M, medial; L, lateral; wm, white matter; Scale bar: 300 μm in A, 500 μm in E, 100 μm in
(G) Data are shown as mean ± SEM, ∗∗p< 0.01, ∗∗∗∗p< 0.0001. See also Figure S1.
Selective Akirin2 knockout in maturing cortical neurons
We previously found that Aki2 is essential for the normal proliferation, differentiation, and survival of cortical progenitor cells (Bosch et al., 2016). Given its maintained, albeit lower, expression in the postmitotic cortex, we hypothesized that Aki2 plays a distinct role in postmitotic neurons. To explore this, we knocked out Aki2 in postnatal excitatory forebrain neurons by crossing mice harboring a conditional Aki2 mutant allele (Goto et al., 2008) to the CaMKIIα-Cre line T29-1 (Tsien et al., 1996). We also included the Ai14 tdTomato Cre-reporter allele (Figure 1D). Using this reporter line, we found initial tdTomato expression spanning the somato-motor and somatosensory cortices in the rostral-caudal axis (Figure S1C) that begins in the third postnatal week (Figures 1E, S1B, and S1C, http://www.informatics.jax.org/recombinase/specificity?id=MGI:2177650&system=nervous+system). Although Cre was also active in hippocampal CA1, we limited our analysis to the cortex where 61.38 ± 1.21% (mean ± SEM) of tdTomato-positive cells were found in layers II–IV and 33.69% ± 1.43% in layer VI, with very few in layer V (Figure 1E). There are two neuronal subpopulations in layer VI: FoxP2-negative corticocortical projection neurons and FoxP2-positive corticothalamic projection neurons (Kast et al., 2019). Close examination of layer VI revealed that most FoxP2-positive neurons lacked the Cre reporter; instead, Cre expression was limited to FoxP2-negative layer VI neurons (Figure 1E). Although it was difficult to localize CaMK-Cre activity as development proceeded because tdTomato protein spread to the neuropil, obfuscating individual cell bodies (Figure S1D), an anti-Cre antibody showed an expression pattern at P30 similar to that of tdTomato (Figure 1G), which is also consistent with reported GFP expression driven by the CaMKIIα promoter (Wang et al., 2013) and the loss of Aki2 IF (Figure 1G). Western blot of cortical lysates revealed a ∼70% reduction of Aki2 protein (Figure 1F) in mutants; the remaining signal from Cre-negative cells was expected. Altogether, these results indicate that in the CaMK-Cre;Aki2fl/fl cortex, layers II–IV have the most Aki2-null neurons followed by FoxP2-negative layer VI neurons, while layer V and FoxP2-positive layer VI neurons are almost completely spared.
Akirin2 mutants exhibit gradual neurodegeneration and cortical atrophy
CaMK-Cre;Aki2fl/fl mutants were indistinguishable from controls until 8 weeks of age, at which time mutant animals stopped gaining weight; males weighed significantly less than controls by 12 weeks of age, while females weighed significantly less by 18 weeks of age (Figure 2A). Mutants behaved similarly to their control littermates until ∼ P60, at which time they were distinguishable by their hyperactivity, running in circles, and rearing up on their hindlimbs and scratching at the cage repetitively. At P150, the oldest age at which these mice were maintained, mutants typically exhibited kyphosis (Figure 2B) and were largely inactive. Analysis of whole brains revealed slow atrophy of the cortex while other Cre-negative structures (e.g., the cerebellum) remained unchanged (Figure 2C). Measuring cortical thickness found that the mutant cortex was significantly thinner by P35 and continued to atrophy with age (Figure 2D). The thinning cortex was obvious at P50 where Golgi-staining consistently labeled fewer neurons in the mutant cortex (Figure 2E). Consistent with neuronal loss, a sensitive silver stain for degenerating neurons identified many impregnated neurons in mutants but not controls at P28 (Figure 2F).
Figure 2.

Progressive cortical atrophy in the absence of neuronal Akirin2
(A) Comparison of control mouse (n = 6–17/age) and gender-matched mutant weights (n = 4–13/age) indicates that mutants stop gaining weight at ∼8 weeks.
(B) By 21 weeks CaMK-Cre;Aki2fl/fl (cKO) mutants are clearly smaller and exhibit kyphosis.
(C) Control (Ctl) and cKO brains were collected at the indicated time points. There is clear progressive atrophy of cKO cortex (outlined in blue) but not Cre-negative cerebellum (outlined in purple).
(D) Cortical thickness measurements on coronal cryosections (4 mice/genotype) indicate significant cortical atrophy in the mutant by P35. See Figures 3A and 2E for representative images.
(E) Golgi and (F) NeuroSilver staining reveal a thinner cortex (E; P50) with degenerating neurons (F; P28). Insets, magnified areas of main image. Scale bar: 100 μm. Data are shown as mean ± SEM, ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001, multiple t-tests with Holm–Sidak method corrections.
To determine the spatiotemporal extent of cell loss, we used NeuN IF to label and estimate the number of neurons in cortical sections (Figure 3A). The number of neurons did not significantly differ between control and mutant cortices at P35; the fact that the cortex is already thinner by this age thus suggests that neuropil loss may precede frank cell death. By P50, the mutant cortex had significantly fewer neurons with progressive loss at P90 and P150, by which point almost half of all neurons were lost (Figure 3B). Cell loss differed by cortical layer in a manner consistent with Cre expression (Figure 1E). Significant loss over time was observed in layers II–IV (Figure 3D), which express Cre highly, as well as in Cre-positive FoxP2-negative layer VI neurons (Figure S2). In contrast, in subpopulations with sparse Cre expression (Layer V and FoxP2-positive populations of layer VI) numbers did not significantly decrease (Figure 3D). This indicates that death is, at least initially, cell autonomous.
Figure 3.

Neuronal loss in the absence of Akirin2
(A and B) Quantification of neurons labeled by NeuN or (C,D) the number belonging to each immunostained population (n = 3–4 mice/genotype/age) show significant neuronal loss begins at P50 and progress with age (B). Cre-positive (mutant) neuronal populations are lost first (D, Cux1, Layer II–IV), while Cre-negative populations (D, Ctip2, FoxP2) remain normal. ∗∗p< 0.01, ∗∗∗p< 0.001, ∗∗∗∗p< 0.0001 by two-way ANOVA with Sidak’s multiple comparison tests comparing control to age-matched cKO. Data are shown as mean SEM. cp, caudoputamen; wm, white matter; dashed lines delineate cortical edges. Scale bar: 200 μm. See also Figure S2.
In response to neuronal injury, nearby astrocytes activate and upregulate glial fibrillary acidic protein (GFAP) (Pekny and Nilsson, 2005). Additionally, microglia increases their phagocytic consumption of cellular debris, increasing their lysosomes (Brown and Neher, 2014). We asked whether the activation of glial cells–all of which are Cre-negative and retain Aki2 expression (Zhang et al., 2014; Mancarci et al., 2017) in our model–occurred in the mutant cortex. We measured the mean fluorescence intensity of GFAP IF (Figures 4A and 4B) and the area immunostained with anti-CD68, a marker of lysosomes (Figures 4C and 4D). By P50, GFAP mean fluorescence intensity was nearly 5-fold higher (Figure 4B), while CD68 IF increased by over 10-fold by P50, in the mutant cortex (Figure 4D). Co-staining for CD68 and P2Y12, a microglial-specific receptor, confirmed that the lysosomes were microglial-specific and demonstrated that microglia in the mutant cortex took on the characteristic ameboid morphology of activated microglia (Figure 4C) (Stence et al., 2001). Interestingly, we found that initially only layers II–IV and VI (where some neurons lose Aki2 expression) contained activated glia while layer V (where Aki2 expression is unaltered) did not. Over time, however, glial activation spread to layer V at later ages.
Figure 4.

Glial activation accompanies cortical neuron loss in Akirin2 mutants.
Quantification of cryosections immunostained for GFAP (A) or CD68 (C) show remarkable increases in astrocyte (B) and microglia (D) activation at the ages indicated. Initial signal was most prevalent in the layers containing significant numbers of Aki2-null neurons (II–IV and VI). Colocalization of CD68 with the microglial marker P2Y12 confirms the activation of microglia (C). ∗∗∗∗p< 0.0001 by two-way ANOVA with Sidak’s multiple comparisons test comparing control to age-matched mutants. Data are shown as mean ± SEM n = , 3–4 mice/genotype/age. wm, white matter; dashed lines delineate cortical edges. Scale bar: 300 μm.
Cortical neurons undergo necroptosis in the absence of Akirin2
Because previous studies found that Aki2 suppresses apoptosis in several contexts (Krossa et al., 2015; Tartey et al., 2015; Bosch et al., 2016), we initially expected neuronal loss in CaMK-Cre;Aki2fl/fl cortex to be apoptotic in nature. Surprisingly, neither IF nor western blotting with an antibody for the apoptotic marker, cleaved caspase 3 (CC3), nor TUNEL staining for apoptotic nuclei, revealed any sign of excessive apoptosis in the mutant cortex at any age examined (Figures S3A–S3C). In apoptosis, cells typically shrink; in contrast, mutant cortical neurons appeared larger (Figure 5A). To confirm this, we measured NeuN area–which labels both the nucleus and the somatic cytoplasm (Lind et al., 2005) in a pattern that overlaps with Nissl stain (Figure S3D). We found that in layers II–IV of P50 mutant cortices, coincident with the first significant cell loss, neurons were significantly larger. As expected, cell size was not significantly different in largely Cre-negative layers V and VI, where significant neuronal loss does not occur at any age examined (Figure 5B).
Figure 5.

Cortical neurons undergo necroptosis in the absence of Akirin2
(A) NeuN IF indicates mutant neurons appear larger than control neurons at P50.
(B) Neuronal size was estimated using NeuN area (nucleus + somatic cytoplasm) on 10 neurons//layer (II–IV, V, VI)/cryosection and average size per mouse plotted as % control (n = 4 mice/genotype/age) at each age indicated. Two-way ANOVA with Sidak’s multiple comparisons tests comparing control to age-matched mutant.
(C) Cortical lysates (n = 8–10) were immunoprobed for necroptosis-regulating kinases RIPK1 and RIPK3 at P50; M = molecular weight marker.
(D) Levels of the indicated protein were normalized to β-tubulin within the same lane, graphed as % of control, and significance determined using the Holm–Sidak method for 2 unpaired t-tests.
(E–H) Mice were injected daily for 30 days with vehicle (DMSO) or necroptosis inhibitor, Nec-1s, from ages P21–P50. The ability of Nec1s to rescue neuronal death was assessed via quantification of layer II–IV neurons (E,F) at P50. Glial activation was assessed by measuring GFAP fluorescence intensity (G,H) at P50. Statistical significance was determined using two-way ANOVA with Sidak’s multiple comparison test comparing control and cKO of the same treatment. Daily Nec-1s treatment significantly rescued neuronal number and degree of glial activation in the Aki2 mutant cortex. n = 4–5 mice/genotype/treatment. Data are shown as mean ± SEM ∗p< 0.05; ∗∗p< 0.01; ∗∗∗p< 0.001; ∗∗∗∗p< 0.0001. Scale bar: 150 μm. See also Figure S3.
An increase in cell size can indicate either autophagy or necroptosis (Kroemer et al., 2009). To seek signs of autophagy, we examined the lipidation of the microtubule-associated protein 1 light chain 3 (LC3) from LC3-I to LC3-II, a key step in the formation and lengthening of autophagosomes (Kabeya et al., 2000). We found no increase in the faster-migrating LC3II band versus the LC3I band in CaMK-Cre;Aki2fl/fl cortical lysates (Figure S3E). In contrast, levels of necroptosis mediators, receptor-interacting protein kinases RIPK1 and RIPK3, were robustly increased in mutant cortical lysates (Figures 5C and 5D), suggesting that neurons undergo necroptosis in the absence of Aki2. To test this hypothesis further, we injected a cohort of mice daily with the RIPK1-specific inhibitor, Nec-1s, or vehicle from P21 (shortly after Cre expression begins) until P50 (the age at which we first detect significant neuronal loss). We then assessed cell numbers and glial activation to determine if Nec-1s ameliorated phenotypes in the mutant cortex. We found that mutants receiving Nec-1s had numbers of Layer II–IV neurons (Figure 5E,F) comparable to controls, while mutants receiving vehicle exhibited the expected deficits. Similarly, GFAP levels were significantly increased in vehicle-treated mutants compared to vehicle-treated controls, but not in Nec-1s-treated mutants compared to Nec1s-treated controls (Figure 5G,H). Together, we interpret these results as a rescue of the Aki2 mutant phenotype by Nec-1s, which bolsters the other evidence–lack of apoptosis, lack of evidence for autophagy, upregulation of RIPK1/3, and neuronal swelling, indicating that cortical Aki2-null neurons die by necroptosis.
Akirin2 loss disrupts gene expression patterns in both the postnatal and embryonic cortex
Aki2 regulates gene expression patterns in many organisms in a cell type- and context-dependent manner (Bosch et al., 2020). We thus hypothesized that Aki2 regulates overlapping, but distinct, gene patterns throughout the generation and maturation of cortical neurons. To test this, we performed transcriptomic analyses on cortical tissues using two conditional Aki2 knockout models: (1) the CaMK-Cre;Aki2fl/fl model described here and (2) the Emx1-Cre;Aki2fl/fl model described in Bosch et al. (2016), in which the Aki2 allele is disrupted in cortical neural progenitors starting at E9.5. We did this to: (1) generate hypotheses about what biological processes are regulated by Aki2 in the developing and maturing brain, a topic that remains unexplored and (2) to compare and contrast the embryonic versus postnatal differentially expressed genes (DEGs) to derive common, as well as context-specific, Aki2-dependent genes.
We performed RNA sequencing (RNASeq) on cortical tissue from CaMK-Cre;Aki2fl/fl mice and controls at P35. This allowed substantial time for existing Aki2 protein turnover after Cre onset but was before frank neuronal loss. Transcriptomic analysis using a 0.05 FDR cut-off, identified 295 genes significantly upregulated and 156 genes significantly downregulated in this model (Figure 6A). Gene set enrichment analysis (GSEA) identified vesicle targeting as the top overrepresented gene ontology (GO) biological process as well as several neuronal-specific GO biological processes involving membrane potential and action potential, and several pathways involved in regulating the mitotic cell cycle (Figures 6B and 6D).
Figure 6.

Akirin2 loss disrupts gene expression patterns in the postnatal cortex
(A) A volcano plot shows differentially expressed genes (DEGs) in the CaMK-Cre;Aki2fl/fl cortex (n = 3) compared to the control cortex (n = 4) at P35 with an FDR cutoff of 0.05. Genes in the p53 pathway are highlighted with black circles.
(B) Gene ontology (GO) analysis bar plot showing biological processes associated with DEGs identified in (A).
(C) qPCR confirmation of upregulated cell-cycle and p53 target genes in P35 Aki2 mutant biological and technical replicates (n = 10–13 mice) compared to P35 controls (n = 8–10 mice). Note the confirmation of significant (but not complete) Aki2 downregulation as expected. Data are shown as +/- SEM ∗∗p<0.01, ∗∗∗p<0.001, ∗∗∗∗p<0.001.
(D) Gene-concept network showing the relationship between the GO terms highlighted in B and the individual genes comprising each group. FC, fold change; FDR, false discovery rate. See also Figure S4 and Tables S3–S7
To gather comparative data on embryonic cortical neural progenitors, we also performed RNASeq on dorsal telencephalic tissue from E10.5 Emx1-Cre;Aki2fl/fl mice and their controls. We chose this time point as one where we can detect Aki2 loss, but at which significant apoptotic cell death has not yet occurred (Bosch et al., 2016). Using a 0.05 FDR cut-off, 49 genes were significantly upregulated while 16 genes were significantly downregulated (Figure S4A). Interestingly, GSEA revealed that these DEGs are involved in the p53-mediated apoptotic signaling pathway and in negatively regulating the transforming growth factor-β (TGF-β) receptor signaling pathway, in which the Aki2 C. elegans ortholog, Akirin, participates (Bowman et al., 2020) (Figures S4B and S4D). We previously found that Aki2 is essential for dorsal forebrain development by controlling cell cycle progression and suppressing apoptosis in cortical neural progenitor cells (Bosch et al., 2016). While GSEA of DEGs with an FDR cutoff of 0.05 identified the GO term hindbrain development, the individual genes involved (Figure S4D) control neurogenesis or neuron development in many brain regions, not exclusive to the hindbrain (Hattori et al., 2007; McKenna et al., 2011; Qu et al., 2013; Fernando et al., 2014; Bian et al., 2015; Okamoto et al., 2015; Fazel Darbandi et al., 2018; Donovan et al., 2019). Furthermore, relaxing the FDR cut-off to 0.1 results in a gene list enriched in DEGs associated with forebrain development, cell fate specification, and cell fate commitment (Figure S4C), further supporting Aki2’s role in these processes (Bosch et al., 2016; Liu et al., 2017).
To identify potential Aki2-dependent genes across multiple cell types and stages, we compared DEGs across both transcriptomes. We identified five genes (Trp53inp1, Ccng1, Eda2r, Susd6, Pls3) that were upregulated and one gene (Espn) that was downregulated in both datasets (Table S3). The KEGG pathways enriched in both transcriptomes converged on the p53 signaling pathway (Figure S4F). In fact, five of the six DEGs found in both transcriptomes are p53 target genes. The p53 protein, encoded by the Trp53 gene, is a transcription factor that responds to cellular stress to alter the transcription of genes that regulate an array of biological processes including necroptosis (Wang et al., 2016), apoptosis, cell- cycle arrest, and proliferation (Miller et al., 2000; Murray-Zmijewski et al., 2008). Interestingly, using ingenuity pathway analysis (IPA) (Krämer et al., 2013) to predict upstream transcriptional regulators of the two transcriptomes, we found that p53 was the most highly significant predicted regulator for both transcriptomes (Tables S4 and S5). p53 regulates 56 DEGs in the neuronal transcriptome (Table S6 and black outlined data points in Figure 6A), and 23 DEGs in the neural progenitor transcriptome (Table S7 and black outlined dots in Figure S4A).
Furthermore, several p53-linked biological processes (proliferation, differentiation, and apoptosis in Emx1-Cre;Aki2fl/fl mice and neurodegeneration and necroptosis in CaMK-Cre;Aki2fl/fl mice) are dysregulated in our mutant mouse models. We subsequently performed qPCR validation experiments choosing, based on preliminary DAVID (Huang da et al., 2009b; Huang da et al., 2009a) and IPA analyses, several p53 target genes, and 3 Aki2-dependent genes common to both transcriptomes. Because Liu et al., (2017) report Aki2 binding to Geminin in Xenopus, we also validated the purported upregulation of a geminin family member, Gmnc in the CaMK-Cre;Aki2fl/fl transcriptome. These independent analyses confirmed significant upregulation of 13/16 genes analyzed in CaMK-Cre;Aki2fl/fl and Emx1-Cre;Aki2fl/fl biological and technical replicates (Figures 6C and S4E) with one more trending higher (p = 0.0622).
Akirin2 inhibits P53-mediated cell death
The CaMK-Cre;Aki2fl/fl transcriptome and qPCR confirmation experiments (Figure 7A) indicated that Trp53 transcript levels did not change significantly in the mutant, yet many p53 target genes were significantly upregulated. This is consistent with the fact that p53 is regulated primarily through posttranslational modifications (PTMs) not transcript expression (Oren 1999). Thus, we measured p53 protein levels in cortical lysates, finding that at P35, when cell death is not yet significant, there was not yet a significant increase in p53 levels (Figures S5A and S5B). At P50, however, when cell death is significant, there was an unequivocal increase of p53 protein in CaMK-Cre;Aki2fl/fl mutant cortices (Figure 7B). Furthermore, immunostaining of CaMK-Cre;Aki2fl/fl cortical sections revealed that only Aki2-null neurons exhibited the upregulation of p53 protein levels (Figure 7C), suggesting that Aki2 suppresses p53 in the adult cortex. Because p53 is a major regulator of cell viability (Morrison and Kinoshita, 2000; Ranjan and Iwakuma, 2016), we tested the hypothesis that p53 mediates the neurodegeneration we have documented in the CaMK-Cre;Aki2fl/fl cortex. We crossed CaMK-Cre;Aki2fl/fl mice to p53 knockout mice (Jacks et al., 1994) to generate p53 heterozygous and p53 knockout Aki2 mutants (CaMK-Cre;Aki2fl/fl;Trp53+/−; CaMK-Cre;Aki2fl/fl;Trp53−/−). We confirmed reduced p53 levels in heterozygous mice and absence of p53 in knockout mice (Figures 7D–7F). If high p53 levels cause cell death, we expected that reducing those levels (by removing one allele) or knocking it out completely would rescue death and glial activation in CaMK-Cre;Aki2fl/fl mice. Indeed, we found a significant rescue: CaMK-Cre;Aki2fl/fl;Trp53+/− mice had normal numbers of Layer II–IV neurons (Figures 7G and S5C) and reduced glial activation compared to CaMK-Cre+;Aki2fl/fl mice (Figures 7H–7I, S5D–E). Similar results were observed in Aki2/p53 double knockout mice, indicating that reduced p53 levels in heterozygous mice were sufficient to rescue the bulk of Aki2 knockout phenotypes; such haploinsufficiency has been observed previously for p53-mediated cell death (Sakhi et al., 1996; Hirata and Cadet, 1997; Aloyz et al., 1998; Bae et al., 2005; Ghosh et al., 2018). Together, these data indicate that the absence of Aki2 in postmitotic neurons leads to the posttranscriptional upregulation of p53 and consequent neurodegeneration that requires p53-mediated pathways.
Figure 7.

Neurodegeneration in Akirin2 mutant cortex is mediated by p53
(A) qPCR identifies no significant upregulation of p53 transcript in the Aki2 mutant cortex compared to control (n = 6 mice/genotype). Significance determined using unpaired t-test
(B and C) In contrast, western blots (B) and IF of cortical cryosections (C) using an anti-p53 antibody reveal a massive increase in protein levels restricted to Aki2-null neurons (dashed circles, layers II–IV) in P50 mutants.
(D–F) Western blots (D) quantified in E, and IF of cortical cryosections (F) confirm decreased p53 protein levels in cKO; p53+/− mice (n = 4–5 mice) and loss of p53 protein in cKO;p53-/- cortices. Significance determined using unpaired t-test.
(G–I) p53 heterozygosity and KO rescue signs of neurodegeneration in P50 Aki2 mutant cortex. Data for control and cKO; Trp53+/+ were replotted from Figure 3 with the addition of 5–6 newly analyzed littermates of cKO; Trp53+/− (n = 4) and cKO; Trp53−/− (n = 3) mice. Reduction in p53 levels completely rescues neuronal numbers in layers II–IV (G) and aberrant glial activation (H,I) at P50. Data are shown as mean ± SEM ∗∗∗∗p< 0.0001. Statistical significance was determined using one-way ANOVA with Dunnet’s multiple comparison test comparing all genotypes to control. Scale bar: 100 μm.
Discussion
The small nuclear protein, Aki2, is broadly expressed in tissues with its most consistently reported functions including the control of cell proliferation and differentiation (Bosch et al., 2020). The expression of Aki2 in non-proliferating neurons suggested additional postnatal roles. Here, we have selectively removed Aki2 from many postmitotic neurons of the cerebral cortex. Following Aki2 excision, neurons (and animals) undergo a progressive decline in health, beginning with positive silver staining and cortical thinning without significant cell loss, suggestive of initial neurite degradation and neuropil depletion. By P50, ∼30 days after Aki2 excision, mutants exhibit significant neuron loss and cortical atrophy that worsens with age. Cell death and glial activation begin in cortical layers with the greatest number of Aki2-null cells and glial activation progress to primarily Cre-negative layers, providing evidence that cell death first occurs cell-autonomously; as mutants age and circuits degrade, there may be collateral loss of some neurons that retain Aki2 expression. Interestingly, unlike the apoptosis reported in Aki2-null cortical progenitors (Bosch et al., 2016) we present extensive convergent evidence that Aki2-null postmitotic neurons die via necroptosis in a p53-dependent manner. Transcriptomic analyses support a role for p53, as both mutant progenitor and postmitotic neuron transcriptomes were enriched for p53 target genes. Our data implicate p53 pathway proteins as novel mediators of Aki2 function and suggest that the mutation or dysregulation of Aki2 and its partners could contribute to a variety of neurodegenerative disorders.
Neurodegeneration in CaMK-Cre;Aki2fl/fl mice: possible mechanisms
GSEA of the CaMK-Cre;Aki2fl/fl transcriptome reveals the dysregulation of genes controlling the cell cycle. This is striking because neurons terminally differentiate and must remain mitotically quiescent. Cyclin-dependent kinases (Cdks) together with cyclins regulate progression through cell cycle stages (Boward et al., 2016). Numerous studies reveal aberrant expression of cyclins, Cdks, and Cdk inhibitors in multiple neurodegenerative diseases. Together with the induction of signal transduction pathways and cytoskeletal changes reminiscent of developing neurons, this suggests that neurons in neurodegenerative tissues aberrantly re-enter the cell cycle (Zhu et al., 2007). This is believed to induce neuronal death (Park et al., 2007) as there is a lack of evidence for successful division in postmitotic neurons (Zhu et al., 2007). The CaMK-Cre;Aki2fl/fl mutant transcriptome is enriched in genes that regulate the G1/S mitotic transition, including several cyclins and Cdks.
Of particular interest to this study, the retinoblastoma tumor suppressor protein (pRb) suppresses the G1/S phase transition. In quiescent neurons, hypophosphorylated pRb represses members of the E2F protein family to block cell cycle progression (Lees et al., 1993). In response to a mitogenic stimulant, D-type cyclins are synthesized and bind to Cdk4 or Cdk6. The cyclin D-Cdk4/6 complex phosphorylates and represses pRb, leading to E2F activation (Giacinti and Giordano, 2006; Narasimha et al., 2014). Increased E2F-1 can result in G1/S cycle transition (Qin et al., 1994) or cell death (Hsieh et al., 1997; MacManus et al., 2003; Polager et al., 2008). Interestingly, expression of both E2f1 and Ccnd1 (encoding cyclin D1) is upregulated in CaMK-Cre;Aki2fl/fl cortices. It is thus possible that Aki2 represses cyclinD1; in Aki2 mutants, cyclinD1 may transactivate E2F-1 and trigger cell cycle re-entry and death. This would be in contrast to Aki2’s role in proliferating cells, where it activates the expression of cell cycle genes. Previous studies suggest that Aki2 induces cyclin D1 and cyclin D2 expression and is critical for recruiting the chromatin remodeling complex containing Brg1 to the cyclin D2 promoter in proliferating B cells (Tartey et al., 2015). This further supports the hypothesis that Aki2 has cell-type-dependent functions specific to each developmental stage.
P53 as a mediator of Akirin2 mutant phenotypes
Because the inactivation of the tumor suppressor protein, p53, is found in almost half of all human cancers (Kandoth et al., 2013; Lawrence et al., 2014), there has been extensive focus on its ability to drive cell cycle arrest and induce apoptosis. Studies suggest that p53 hyperactivation can explain the neurodegeneration and necroptosis we show here in CaMK-Cre;Aki2fl/fl mutants. Increased p53 levels–such as those observed in Aki2-null cortical neurons–are also found in the CNS of patients with neurodegenerative diseases, as well as in mouse models of such disorders (Miller et al., 2000; Simon et al., 2017; van Alstyne et al., 2018). Additionally, neurons upregulate p53 in response to insults such as excitotoxicity (Morrison et al., 1996; Xiang et al., 1996), ischemia (Banasiak and Haddad, 1998; Watanabe et al., 1999), traumatic brain injury (Plesnila et al., 2007), and neurotoxicity (Hirata and Cadet, 1997; Morrison and Kinoshita, 2000). In mouse models, ablation or reduction of p53 prevented cell death and/or rescued behavioral deficits (Herzog et al., 1998; Simon et al., 2017 Morrison and Kinoshita, 2000; Bae et al., 2005; Mfossa et al., 2020).
Although p53 activity is largely associated with apoptosis, previous work has identified p53 involvement in autophagy (Maiuri et al., 2010) and regulated forms of necrosis including both necroptosis and ferroptosis (Tu et al., 2009; Vaseva et al., 2012; Jiang et al., 2015; Ranjan and Iwakuma, 2016; Wang et al., 2016). p53 contributes to RIPK1/RIPK3-mediated necroptosis by upregulating levels of a long noncoding RNA (lncRNA) called necrosis-related factor (NRF). NRF inhibits miR-873, which in turn inhibits RIPK1 and RIPK3, the essential necroptosis initiators. Thus, the upregulation of p53 leads to the upregulation of NRF, increasing the inhibition of miR-873 and disinhibiting RIPK1 and RIPK3 (Ranjan and Iwakuma, 2016; Wang et al., 2016). Admittedly, neither NRF nor miR-873 were significant DEGs in the CaMK-Cre;Aki2fl/fl transcriptome. This may be because RNASeq was performed at P35, before obvious neuronal loss or p53 upregulation, or because nonubiquitous Cre activity in this model leaves many neurons with normal Aki2 expression, making it more difficult for Aki2-dependent expression changes to reach significance. In any case, our results are altogether consistent with p53 activation leading to necroptosis in CaMK-Cre;Aki2fl/fl mutant cortex.
Significant enrichment in p53-target genes in the Emx1-Cre;Aki2fl/fl transcriptome further supports the hypothesis that p53 mediates Aki2 function. In the developing nervous system, p53 regulates neural stem cell proliferation and progenitor differentiation (Ferreira and Kosik, 1996; Armesilla-Diaz et al., 2009; Forsberg et al., 2013; Quintens et al., 2015; Mfossa et al., 2020), as does Aki2 (Bosch et al., 2016; Liu et al., 2017). Interestingly, a recent preprint reports that p53 hyperactivation via γ-irradiation at E11 disrupts the cell cycle, triggers apoptosis, and leads to premature neurogenesis and ectopic neurons in the subventricular zone within 24 h. This results in microcephaly in newborn pups that is partially rescued by Emx1-Cre-mediated Trp53 ablation in neural progenitor cells (Mfossa et al., 2020). Furthermore, adherens junctions lining the ventricular surface were disrupted and several adherens junction proteins were downregulated after p53 activation (Mfossa et al., 2020). These aberrations closely resemble those found in the Emx1-Cre;Aki2fl/fl telencephalon (Bosch et al., 2016). Emx1-Cre;Aki2fl/fl mice displayed more severe microcephaly and apoptosis than did the hyperactive p53 mouse model in Mfossa et al. (2020). This is likely due to differing duration and/or intensity of p53 activation, which determines whether p53 triggers apoptosis or cell-cycle arrest (Kracikova et al., 2013).
Evidence for the repression of p53 by Akirin2
Neither databases (Stark et al., 2006; Szklarczyk et al., 2019) nor our own attempted co-immunoprecipitations (data not shown) provide evidence to support a physical interaction between Aki2 and p53. While this does not eliminate the possibility that such an interaction exists, it is worth considering alternative mechanisms through which Aki2 might indirectly repress p53. First, Aki2 could repress p53 by interacting with or modifying direct p53 regulators or signaling cascades that affect p53 stabilization. Mouse double minute 2 (MDM2) is an E3 ubiquitin ligase that tags p53 for proteasomal degradation to maintain low p53 protein levels in quiescent cells (Haupt et al., 1997; Honda et al., 1997; Kubbutat et al., 1997; Hafner et al., 2019). Over 300 known PTMs control p53 stabilization by interfering with p53-MDM2 binding (Hafner et al., 2019). If Aki2 represses any of the many proteins that participate in signaling cascades leading to p53 PTMs, loss of Aki2 could result in p53 activation. One potential candidate is the transcription factor, E2f1. E2f1 leads to the stabilization of p53 likely through PTMs (Kowalik et al., 1998) and is significantly upregulated in the CaMK-Cre;Aki2fl/fl cortex. Similar to the CaMK-Cre;Aki2fl/fl model presented here, the Pellizzoni and Mentis labs similarly showed that the upregulation of p53 leads to a caspase-independent nonapoptotic form of neuronal death in a spinal muscular atrophy mouse model (Simon et al., 2017). Furthermore, upregulation of p53 in this model is mediated by aberrant splicing of the two major p53 inhibitors MDM2 and MDM4 (van Alstyne et al., 2018). Interestingly, one GO term implicated in the CaMK-Cre;Aki2fl/fl transcriptome is “mRNA splicing via spliceosome.” Thus, it is possible that without Aki2, spliceosome formation is disrupted resulting in alternative splice variants of MDM2 and MDM4 that are unable to properly suppress p53 induction; this could be investigated in future studies.
Second, Aki2 could suppress p53 via NF-κB signaling. Initial studies on mammalian Aki2 and the Drosophila ortholog, Akirin, provide several lines of evidence that Akirin proteins interact with NF-κB transcription factor family members to activate genes in the fly Imd and related mammalian immune pathways (Goto et al., 2008; Bonnay et al., 2014; Tartey et al., 2014). NF-κB has a complex, but primarily antagonistic, relationship with p53 (Ak and Levine, 2010). For example, p53 is a tumor suppressor gene, while NF-κB is an oncogene; activation of p53 increases neuronal death concomitant with reduced NF-κB activity, while the inhibition of p53 increases neuronal survival concomitant with increased NF-κB (Plesnila et al., 2007). Because activation of one opposes the other, these two transcription factors cannot be activated within the same cell (Ak and Levine, 2010). Thus, it is possible that Aki2 indirectly regulates p53 through currently undefined NF-κB signaling pathways.
Limitations of the study
In closing, we note the caveat that in the CaMK-Cre;Aki2fl/fl mutant model used here, only a subpopulation (albeit a large one) of neurons in the cortex lacks Aki2. Because of this, the power of RNASeq is likely reduced, as the cortical transcriptomes analyzed include many neurons retaining Aki2 expression. Thus, the number of DEGs and the degree of their differential expression detected here is surely an underestimate. Future studies investigating a homogeneous population of Aki2-null neurons would aid in identifying further Aki2-dependent genes and would be a prerequisite for utilizing ChIP-Seq or ATAC-Seq approaches to identify neuronal genes directly regulated by Aki2. Nevertheless, the work presented here raises intriguing new insights into Aki2 function in the maturing brain that are worth exploring further, including the role of p53 pathway members as functional partners and the potential involvement of Aki2 dysregulation in a variety of human neurodegenerative disorders.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Primary antibodies for immunofluorescence | See Table S1 | N/A |
| Primary antibodies for Western Blot | See Table S1 | N/a |
| Sheep polyclonal Anti-digoxigenin-AP Fab fragments | Roche | Cat# 11093274910 |
| Chemicals, peptides, and recombinant proteins | ||
| cOmplete, Mini Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| PhosSTOP | Roche | Cat# 04906845001 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34580 |
| SuperSignal West Femto maximum sensitivity substrate | Thermo Fisher Scientific | Cat# 34095 |
| TRIzol Reagent | Ambion | Cat# 15596018 |
| Fluro-Gel with Tris Buffer | Electron Microscopy Services | Cat# 17985-11 |
| Nec-1s (7-Cl-O-Nec1) | Abcam | Cat# ab221984 |
| RNAlater | Invitrogen | Cat# AM7024 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23227 |
| mirVana miRNA Isolation Kit | Ambion/Life Technologies | Cat# AM1560 |
| High Capacity RNA-to-cDNA kit | Applied Biosystems/Thermo Fisher | Cat# 4368814 |
| SYBR Green I Master Mix | Roche | Cat# 04707516001 |
| FD NeuroSilver Kit II | FD Neurotechnologies, Inc. | Cat# PK301A |
| FD Rapid GolgiStain Kit | FD Neurotechnologies, Inc. | Cat# PK401 |
| FragEL DNA Fragmentation Kit | Novagen | Cat# QIA39 |
| TruSeq Stranded mRNA Library Prep kit | Illumina | Cat# 20020595 |
| TURBO DNA-free Kit | Invitrogen | Cat# AM1907 |
| Deposited data | ||
| Raw and analyzed RNA-Seq data | This paper | GEO: GSE178844 |
| Experimental models: Organisms/strains | ||
| Mouse: CaMK-Cre: B6.Cg-Tg(Camk2a-cre)T29-1 Stl/J | The Jackson Laboratory | JAX stock #005359 |
| Mouse: tdTomato reporter: B6:Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX stock #007914 |
| Mouse: Trp53 knockout: B6.129S2-Trp53tm1Tyj/J | The Jackson Laboratory | JAX stock #002101 |
| Mouse: Emx1-Cre: B6.129S2-Emx1tm1(cre)krj/J | The Jackson Laboratory | JAX stock #005628 |
| Mouse: Aki2fl/fl | Goto et al., 2008 | N/A |
| Oligonucleotides | ||
| Primers | See Table S2 | N/A |
| Software and algorithms | ||
| Image Studio Ver5.2 | LI-COR | https://www.licor.com/bio/image-studio/ |
| AxioVision Rel4.8 | Zeiss | https://www.micro-shop.zeiss.com/en/us/system/axiovision+software/software+axiovision/axiovision+program/410130-0909-000 |
| ImageJ/FIJI v1.52p | Schindelin et al., 2012; Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Adobe Photoshop Version 21.2.1 | Adobe | https://www.adobe.com/ |
| GraphPad Prism Software 8.1.2 | Graphpad Software (CA,USA) | https://www.graphpad.com/scientific-software/prism/ |
| qbase + software | Biogazelle Vandesompele et al., 2002 |
www.qbaseplus.com |
| bcbio-nextgen pipeline version 1.1.4 | https://github.com/bcbio/bcbio-nextgen, | |
| R | R Core Team, 2019 | N/A |
| STAR | Dobin et al., 2013 | N/A |
| featureCounts | Liao et al., 2014 | N/A |
| EDASeq R package | Risso et al., 2011 | N/A |
| RUVSeq | Risso et al., 2014 | N/A |
| edgeR quasi-likelihood pipeline | Robinson et al., 2010; McCarthy et al., 2012; Chen et al., 2016 | N/A |
| clusterProfiler R package | Yu et al., 2012 | N/A |
| Ingenuity Pathway Analysis | Qiagen Redwood City, CA, USA; Krämer et al., 2013 | www.qiagen.com/ingenuity |
| Other | ||
| LC3 control cell extracts: Chloroquine treated and nontreated HeLA cell lysates | Cell Signaling Technology | Cat# 11972 |
| Caspase-3 control cell extracts: Cytochrome C treated/untreated Jurkat cell lysates | Cell Signaling Technology | Cat# 9663 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dr. Joshua Weiner (joshua-weiner@uiowa.edu).
Materials availability
This study did not generate new unique reagents
Experimental model and subject details
Mice
All experiments included both male and female animals and were conducted in accordance with the University of Iowa’s Institutional Animal Care and Use Committee and NIH guidelines. All mice were healthy, not involved in previous procedures, naive to treatment, and kept under standard housing and husbandry conditions with food and water provided ad libitum and 12-hour light-dark cycles. The age of the mice used is indicated in each figure/figure legend and/or the text.
Transgenic mice
Akirin2fl/fl conditional mutant mice were initially provided by Dr. Osamu Takeuchi, Kyoto University (Goto et al., 2008; Bosch et al., 2016) and have been maintained in the Weiner laboratory colony long-term. These were crossed to the CaMKIIα-Cre line T29-1 (Tsien et al., 1996)(JAX stock #005359) to excise the floxed Aki2 allele in many postnatal forebrain excitatory cortical neurons. These are referred to to as CaMK-Cre;Aki2fl/fl in the manuscript and sometimes as cKO (conditional knockout) in several figures. As there were no obvious abnormalities in CaMK-Cre;Aki2fl/+, Aki2fl/fl, or Aki2fl/+ mice, all were used as controls. Similarly Aki2fl/fl;Trp53+/+ and Aki2fl/fl;Trp53+/− did not differ, thus both were used as controls in relevant experiments. To assess Cre-mediated excision and monitor knockout cells, we also crossed these mice to the Ai14-tdTomato Cre-reporter line (JAX stock #007914). The early telencephalic-restricted mutant Emx1-Cre;Aki2fl/fl mice were detailed in Bosch et al. (2016). Trp53 constitutive knockout mice (Jacks et al., 1994) (JAX stock #002101), Ai14-tdTomato reporter mice, CaMKIIα-Cre line T29-1 (JAX stock #005359), and Emx1-Cre (JAX stock #005628) lines were obtained from The Jackson Laboratory (Bar Harbor, ME). All lines were maintained on a C57BL/6J background.
Nec1s experiments
When possible, all mice receiving treatment were from the same litter. In 3 of the 5 experiments only one CaMK-Cre;Aki2fl/fl mouse was available in the litter for treatment. For 1 of those experiments an age-matched CaMK-Cre;Aki2fl/fl from a different litter received vehicle treatment. For the other experiment, no CaMK-Cre;Aki2fl/fl receiving vehicle was included. Which mouse in the litter received vehicle or drug was randomly chosen at the time of first injection. To reduce the confound of order of treatment, the order was randomized based on the order of which mice were retrieved from the cage.
Method details
Western blots
To prevent contamination of Aki2 signal from the highly-expressing blood leukocytes, mice were transcardially perfused with 15–25 mL of 1x phosphate-buffered saline (PBS). The cortex was homogenized in 1 mL RIPA buffer (0.1% SDS, 0.25% sodium deoxycholate, 1% NP-40, 0.15M NaCl, 50mM Tris-HCl (pH 7.4), 5mM NaF) containing both protease and phosphatase inhibitors (Roche) using a Dounce homogenizer and Wheaton overhead stirrer. After incubating at 4°C for 45 mins, samples were centrifuged at 16,000 X g for 10 minutes at 4°C and precipitate was removed. Protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein were separated via SDS/PAGE using TGX precast gels (Bio-Rad), transferred to nitrocellulose membranes using a Trans-Blot Turbo System (Bio-Rad), and analyzed using standard quantitative western blot methods (Pillai-Kastoori et al., 2020). Chloroquine treated and nontreated HeLa cells (Cell Signaling Technology #11972) were used as positive and negative controls, respectively, for LC3 probe. Cytochrome c treated or untreated Jurkat cells (Cell Signaling Technology #9663) were used as positive and negative controls, respectively, for cleaved caspase 3 probe. Membranes were blocked in 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBST). Primary antibodies were diluted in 5% BSA in TBST and membranes incubated overnight at 4oC. HRP-conjugated secondary antibodies were diluted in 5% milk in TBST and incubated at RT for 1 hour. Signal was detected using SuperSignal West Pico or Femto Enhanced Chemiluminescent Substrates (Thermo Fisher Scientific) on a LI-COR Odyssey Fc Imaging system. For all quantified probes, at least 3 biological replicates (i.e., samples from 3 mice) and at least 2 technical replicates (i.e. same samples run on different gels) per genotype or condition were run on the same gel and measured from the same image. Signal was measured using Image Studio Ver5.2 (LI-COR). All targets were normalized to a housekeeping protein (β−tubulin or GAPDH) as an internal loading control. Relative protein levels were calculated as % of control = . The % of control was averaged for all replicates and graphed. In instances in which protein level was measured across time, the earliest time point was used as the control in the equation above to show relative changes as maturation proceeded.
RNA extraction and quantitative RT-PCR (qPCR)
To eliminate signal from the blood, mice older than P7 were transcardially perfused with 15–20 mL cold DEPC-treated PBS. Cortices were homogenized in 1–2 mL of TRIzol (Thermo Fisher Scientific). RNA was extracted following the manufacturer’s protocol and purified using the mirVana miRNA Isolation Kit (Ambion/Life Technologies).
RNA was reverse transcribed using the High Capacity RNA-to-cDNA kit and qPCR performed using a Roche LightCycler480 with SYBR Green I Master Mix (Roche) and the primers listed in Table S2. PCR cycling parameters for 30 cycles were: 95°C 1 min, 55°C 15s, 72°C 1 min.
Immunofluorescence
Mice were transcardially perfused with 15–20 mL of PBS followed by 15–20 mL of 4% paraformaldehyde (PFA). Brains were collected and post-fixed in 4% PFA for 2 hours – overnight at 4oC and cryoprotected in 30% sucrose. For tissues to be stained with anti-Cre antibody, 2% PFA was used for transcardial perfusion and tissue was collected directly into 30% sucrose without post-fixation. Cryoprotected tissue was quickly frozen in OCT compound (Sakura Finetek) and 18 μm coronal cryosections were cut on a Leica CM1850 cryostat. Six coronal sections per slide were collected directly onto ten slides starting at the emergence of the third ventricle and guided by reference to an atlas, ensuring that similar brain regions were compared for every mouse. Tissue was blocked for 1 hour at room temperature (RT) in PBS containing 2.5% BSA 0.5% Triton X-100 and incubated in primary antibody (Table S1) overnight at 4°C. Sections were washed in PBS and incubated in the relevant secondary antibody conjugated to Alexa-Fluor 488, 568, or 647nm (Molecular Probes/Invitrogen) for 1 hour at RT. After washing in PBS, sections were counter stained with DAPI (4′,6-diamidino-2-pheylindole) and mounted in Fluoro-Gel (Electron Microscopy Services #17985–11).
In situ hybridization
Digoxigenin-UTP (Roche)-labeled Aki2 sense and antisense riboprobes were generated per manufacturer’s protocols as in (Bosch et al., 2016). In situ hybridization was performed following previously published methods (Grove et al., 1998; Wang et al., 2002). Fresh-frozen tissue was cryosectioned at 20 μm and dried at 65°C for 10 mins followed by RT for 10 mins. Tissue was fixed in 4% PFA for 10 min at RT followed by washes in PBS and a 10 min incubation in fresh acetylation solution (0.1M triethanolamine, 0.65% HCl, 0.25% acetic anhydride). After washing in PBS, slides were incubated in hybridization solution (50% formamide, 5x SSC, 5x Denhart’s solution, 250μg/ml tRNA, 500μg/ml salmon sperm DNA, 50μg/ml heparin) at RT for 2 hours. Slides were incubated with riboprobes at 65°C overnight. The following day, slides were washed over 4 hours in 0.2x SSC at 65°C followed by washes in TBST at RT. Slides were blocked in blocking solution (Roche DIG wash and block buffer set) diluted in 1x maleic acid for 1h at RT and riboprobes were detected by incubating in anti-digoxigenin-AP antibody (1:2000 in 1% sheep serum, Roche) overnight at 4°C. After several washes in TBST over 15 mins, slides were incubated in alkaline buffer for 10 mins (100mM NaCl, 100mM Tris-HCl (pH 9.5), 50mM MgCl2, 1% Tween-20), and developed using nitro blue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) at RT until sufficient color developed.
Imaging
A Leica SPE TCS Confocal Microscope and Leica Application Suite software were used for confocal and epifluorescent imaging. All epifluorescent and confocal images were captured using the same settings for each antibody. In situ hybridization was imaged on a Zeiss Axiophot microscope equipped with an Axiocam Hrc and using AxioVision Rel4.8. Images of silver and Golgi staining were captured using a Leica DMIRB inverted microscope. Brightness and contrast were adjusted equally on images using ImageJ/FIJI (Schindelin et al., 2012; Schneider et al., 2012) or Adobe Photoshop.
NeuroSilver and Golgi staining
For NeuroSilver staining, animals were transcardially perfused as for immunofluorescence, but replacing 1x PBS with 0.1M phosphate buffer (pH 7.4). Sections were cut on a Leica CM1850 cryostat at 60 μm and incubated in 4% PFA for 7 days at 4°C. Tissue was stained using the FD NeuroSilver Kit II (FD Neurotechnologies, Inc. Cat #PK301A) according to manufacturer’s protocol. For Golgi staining, the two cerebral hemispheres were separated from the rest of the brain and incubated with the solutions of the FD Rapid GolgiStain Kit (FD NeuroTechnologies, Inc) according to manufacturer’s protocol. One hundred-μm cryostat sections of telencephalon were mounted on slides and examined directly.
TUNEL staining
Eighteen-μm cryostat sections from the cerebrum were fixed in 4% PFA directly on slides and stained using the FragEL DNA Fragmentation Kit (QIA33; Millipore Sigma) according to manufacturer’s protocol. A positive control was generated by treating cortical sections with 20 μg/mL proteinase K for 10 minutes at RT followed by 1μg/μL DNase I in 1xTBS/1mM MgSO4 for 20 minutes at RT.
Nec-1s administration
Nec-1s (7-Cl-O-Nec1; Abcam #ab221984) was dissolved in DMSO, diluted in sterile saline, and administered at 6.25mg/kg via intraperitoneal injections daily beginning at P21 and ending at P50. Three to four mice per genotype were injected with Nec-1s or the equivalent amount of DMSO vehicle.
Transcriptomic analyses
CaMK-Cre;Aki2fl/fl transcriptome
After transcardial perfusion with 15-20mL of cold DEPC-treated PBS, total RNA was extracted as above from the cortices of 3 Aki2w/w and 1 CaMK-Cre;Aki2w/w controls and 3 CaMK-Cre;Ak2fl/fl mutants at P35. mRNA-enriched libraries were prepared by the Carver Center for Genomics (University of Iowa) using the Illumina TruSeq Stranded mRNA Library Prep kit and quantified using Turner BioSystems TBS-380 fluorometer and Invitrogen Quant-It Picogreen. The Genome Technologies service at The Jackson Laboratory (Bar Harbor, ME) subsequently QC’ed the libraries on an Agilent Bioanalyzer and sequenced them on an Illumina NextSeq 500, with 75 bp single end reads. At least 49M reads per sample were generated.
RNA used for qPCR validation was treated with DNase using the TURBO DNA-free Kit (Thermo Fisher) according to manufacturer’s protocol. Concentration and quality were assessed using a Trinean DropSense-16 Spectrophotometer and Agilent Bioanalyzer. All samples used had an RIN of 8.6 or higher. Two μg of RNA were reverse transcribed using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems/Thermo Fisher). qPCR was performed as above. The primers used are listed in Table S2. All qPCR validating CaMK-Cre transcriptomics used ribosomal protein L32 (RPL32) and transferrin receptor (Tfrc) as reference genes and relative expression levels were quantified using qbase + software (Biogazelle, Zwijnaarde, Belgium www.qbaseplus.com), employing the method described by (Vandesompele et al., 2002). Statistical differences were determined using two-tailed unpaired t-tests on log-transformed CNRQ values.
Emx1-Cre;Aki2fl/fl transcriptome
The telencephalon was carefully dissected from E10.5 embryos in cold PBS and placed into RNAlater (Invitrogen) at 4oC for a maximum of 1 week. RNAlater was carefully removed and the RNA was extracted and purified as above. RNA samples were assessed for purity using a nanodrop and for RNA integrity using the Experion automated electrophoresis station (BioRad). All samples used in the RNAseq experiments had an RQI of >8. Library preparation was performed using the Illumina TruSeq Stranded mRNA library prep kit and RNA sequencing was performed at the Iowa Institute of Human Genetics (University of Iowa). Libraries were generated with unique barcodes, combined equally into one pool and sequenced over 2 lanes of the Illumina HiSeq4000, using 75bp paired end read lengths.
For qPCR validation studies, RNA (90-100ng) collected from E10.5 telencephalon was reverse transcribed using the High Capacity RNA-to-cDNA kit and qPCR performed as above using the primers listed in Table S2. The ddC(t) method was used to calculate fold change using β-Actin as a reference gene as described previously (Bosch et al., 2019).
Bioinformatics
RNASeq data were processed with the bcbio-nextgen pipeline (https://github.com/bcbio/bcbio-nextgen, version 1.1.4). The pipeline uses STAR (Dobin et al., 2013) to align reads to the mm10 genome build (GENCODE release M10, Ensembl 89 annotation) and quantifies expression at the gene level with featureCounts (Liao et al., 2014). All further differential expression analyses were performed using R (R Core Team, 2019). For gene level count data, the R package EDASeq (Risso et al., 2011) was used to adjust for GC content effects (full quantile normalization) and account for sequencing depth (upper quartile normalization). Latent sources of variation in expression levels were assessed and accounted for using RUVSeq (RUVr mode using all features) (Risso et al., 2014). Differential expression analysis was conducted using the edgeR quasi-likelihood pipeline (Robinson et al., 2010; McCarthy et al., 2012; Chen et al., 2016). Gene ontology and KEGG pathway analysis was performed using the R package clusterProfiler (Yu et al., 2012). Ingenuity Pathway Analysis (Qiagen Redwood City, CA, USA, www.qiagen.com/ingenuity) (Krämer et al., 2013) was used for upstream analysis with molecule type restricted to transcriptional regulator.
All RNASeq data have been deposited in the NCBI Gene Expression Omnibus database, accession number GSE178844 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178844).
Quantification and statistical analysis
Cell quantifications
For cell counts, 3–6 coronal sections per mouse were imaged at the convergence of the primary motor and somatosensory cortices and analyzed using FIJI. Only brain regions determined to be similar based on histological landmarks and with reference to an atlas were included for analysis. The experimenter performing quantifications was blinded to genotype/treatment.
Cux1-positive cells in layers II-IV and FoxP2-positive cells in layers VI were counted in one (10x) field of view per section using the FIJI cell counter plug-in. Ctip2 expression, cell size, and cell density were used to identify Layer V, in which cells were counted in the same manner.
For NeuN, GFAP, and CD68 measurements, two 10x fields of view vertically spanning the cortex from white matter to pia were montaged in Adobe Photoshop. The background was subtracted with a rolling ball radius of 12 pixels and NeuN-positive cells were counted using analyze particles plugin with size = 20.5 – infinity; circularity = 0.2–1.00 in FIJI. The resulting images were manually inspected for mis-identified or conjoined cells for which the numbers were properly adjusted. On the same images, threshold was set to a level that optimized signal while reducing background. The individual areas of 10 cells in each of layers II-III, IV, V, and VI were measured in each image using FIJI. The average cell area in each layer per section is reported from at least 3 mice per genotype per age.
Average fluorescence intensity of GFAP staining and the area of CD68 as a percentage of the entire cortical area was measured within a polygon covering the entire cortex including white matter but excluding subcortical structures. The length of a line drawn from the dorsal edge of the lateral ventricle (including the white matter) to the dorsal edge of the pia (including Layer I) was used to estimate cortical thickness.
Statistical analysis
All statistical tests were performed using GraphPad Prism Software with significance level p< 0.05. Details of statistical tests used can be found in the accompanying figured legend. Outlier data points, determined using the ROUT method with a Q = 1, were excluded. The experimental unit for all studies is a single animal. The exact number of animals in each group for each experiment is indicated in the figure legend.
Acknowledgments
We would like to thank Drs. Maria Noterman, Andrew Pieper, Michael Dailey, Daniel Summers, and Diane Slusarski for generously contributing antibodies, reagents, and equipment, the Carver Center for Genomics and Iowa Institute of Human Genetics for assistance with transcriptomics and library preparation, and Dr. Sarit Smolikove and members of the Weiner Laboratory for helpful discussions. The Jackson Laboratory Shared Scientific Services are supported in part by a Basic Cancer Center Core Grant from the National Cancer Institute (CA34196). This work was supported by a Major Project Grant from the University of Iowa Office of the Vice President for Research to J.A.W. and J.R.M., by an Accelerator Grant from the Iowa Neuroscience Institute to J.A.W., and by NIH R01 NS055272 to J.A.W.
Author contributions
S.L.P., P.J.B., J.R.M., and J.A.W. conceived the experiments. S.L.P., P.J.B., B.J.I., M.P., and P.B. performed the experiments and collected and analyzed the data. E.B., M.P., and P.B. performed bioinformatic analyses. S.L.P., P.J.B., and J.A.W. wrote the article. All authors reviewed and edited the article. J.R.M., J.J.M, and R.W.B. provided expertise and supervised bioinformatic analyses
Declaration of interests
The authors declare no competing interests
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103814.
Supplemental information
Data and code availability
-
•
RNA-seq data have been deposited at the gene expression omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ak P., Levine A.J. p53 and NF-κB: different strategies for responding to stress lead to a functional antagonism. FASEB. J. 2010;24:3643–3652. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- Aloyz R.S., Bamji S.X., Pozniak C.D., Toma J.G., Atwal J., Kaplan D.R., Miller F.D. P53 is essential for developmental neuron death as regulated by the TrkA and p75 Neurotrophin receptors. J. Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S., Ladle D.R., Lin J.H., Frank E., Jessell T.M. ETS gene Er81 controls the formation of functional connections between group ia sensory afferents and motor neurons. Cell. 2000;101:485–498. doi: 10.1016/S0092-8674(00)80859-4. [DOI] [PubMed] [Google Scholar]
- Armesilla-Diaz A., Bragado P., del Valle I., Cuevas E., Lazaro I., Martin C., Cigudosa J.C., Silva A. p53 regulates the self-renewal and differentiation of neural precursors. Neuroscience. 2009;158:1378–1389. doi: 10.1016/j.neuroscience.2008.10.052. [DOI] [PubMed] [Google Scholar]
- Bae B.I., Xu H., Igarashi S., Fujimuro M., Agrawal N., Taya Y., Hayward S.D., Moran T.H., Montell C., Ross C.A., et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Banasiak K.J., Haddad G.G. Hypoxia-induced apoptosis: effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res. 1998;797:295–304. doi: 10.1016/S0006-8993(98)00286-8. [DOI] [PubMed] [Google Scholar]
- Barrett R.M., Malvaez M., Kramar E., Matheos D.P., Arrizon A., Cabrera S.M., Lynch G., Greene R.W., Wood M.A. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology. 2011;36:1545–1556. doi: 10.1038/npp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson A., Nativio R., Berger S.L., Bonini N.M. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 2018;41:587–598. doi: 10.1016/j.tins.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian W.J., Miao W.Y., He S.J., Wan Z.F., Luo Z.G., Yu X. A novel Wnt5a-Frizzled4 signaling pathway mediates activity-independent dendrite morphogenesis via the distal PDZ motif of Frizzled 4. Dev. Neurobiol. 2015;75:805–822. doi: 10.1002/dneu.22250. [DOI] [PubMed] [Google Scholar]
- Bonnay F., Nguyen X.H., Cohen-Berros E., Troxler L., Batsche E., Camonis J., Takeuchi O., Reichhart J.M., Matt N. Akirin specifies NF-kappaB selectivity of Drosophila innate immune response via chromatin remodeling. EMBO J. 2014;33:2349–2362. doi: 10.15252/embj.201488456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch P.J., Fuller L.C., Sleeth C.M., Weiner J.A. Akirin2 is essential for the formation of the cerebral cortex. Neural Dev. 2016;11:21. doi: 10.1186/s13064-016-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch P.J., Fuller L.C., Weiner J.A. An essential role for the nuclear protein Akirin2 in mouse limb interdigital tissue regression. Sci. Rep. 2018;8:12240. doi: 10.1038/s41598-018-30801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch P.J., Fuller L.C., Weiner J.A. A critical role for the nuclear protein Akirin2 in the formation of mammalian muscle in vivo. Genesis. 2019;57:e23286. doi: 10.1002/dvg.23286. [DOI] [PubMed] [Google Scholar]
- Bosch P.J., Peek S.L., Smolikove S., Weiner J.A. Akirin proteins in development and disease: critical roles and mechanisms of action. Cell Mol. Life Sci. 2020;77:4237–4254. doi: 10.1007/s00018-020-03531-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovetti S., Bonzano S., Garzotto D., Giannelli S.G., Iannielli A., Armentano M., Studer M., De Marchis S. COUP-TFI controls activity-dependent tyrosine hydroxylase expression in adult dopaminergic olfactory bulb interneurons. Development. 2013;140:4850–4859. doi: 10.1242/dev.089961. [DOI] [PubMed] [Google Scholar]
- Boward B., Wu T., Dalton S. Concise review: control of cell fate through cell cycle and pluripotency networks. Stem Cell. 2016;34:1427–1436. doi: 10.1002/stem.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman R., Balukoff N., Clemons A., Koury E., Ford T., Baxi K., Egydio de Carvalho C., Smolikove S. Akirin is required for muscle function and acts through the TGF-β Sma/Mab signaling pathway in caenorhabditis elegans development. G3 (Bethesda) 2020;10:387–400. doi: 10.1534/g3.119.400377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.C., Neher J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014;15:209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- Busser J., Geldmacher D.S., Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J. Neurosci. 1998;18:2801–2807. doi: 10.1523/jneurosci.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.R., Heck N., Lohof A.M., Rochefort C., Morel M.P., Wehrlé R., Doulazmi M., Marty S., Cannaya V., Avci H.X., et al. Mature Purkinje cells require the retinoic acid-related orphan receptor-α (RORα) to maintain climbing fiber mono-innervation and other adult characteristics. J. Neurosci. 2013;33:9546–9562. doi: 10.1523/jneurosci.2977-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Lun A.T., Smyth G.K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 2016;5:1438. doi: 10.12688/f1000research.8987.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewa-Waclaw J., Bird A., von Schimmelmann M., Schaefer A., Yu H., Song H., Madabhushi R., Tsai L.-H. The role of epigenetic mechanisms in the regulation of gene expression in the nervous system. J. Neurosci. 2016;36:11427–11434. doi: 10.1523/jneurosci.2492-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager P.L., Srivastava G., Lunnon K., Burgess J., Schalkwyk L.C., Yu L., Eaton M.L., Keenan B.T., Ernst J., McCabe C., et al. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014;17:1156–1163. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneris E.S., Hobert O. Maintenance of postmitotic neuronal cell identity. Nat. Neurosci. 2014;17:899–907. doi: 10.1038/nn.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan L.J., Spencer W.C., Kitt M.M., Eastman B.A., Lobur K.J., Jiao K., Silver J., Deneris E.S. Lmx1b is required at multiple stages to build expansive serotonergic axon architectures. Elife. 2019;8 doi: 10.7554/eLife.48788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazel Darbandi S., Robinson Schwartz S.E., Qi Q., Catta-Preta R., Pai E.L., Mandell J.D., Everitt A., Rubin A., Krasnoff R.A., Katzman S., et al. Neonatal Tbr1 dosage controls cortical layer 6 connectivity. Neuron. 2018;100:831–845.e837. doi: 10.1016/j.neuron.2018.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando C.V., Kele J., Bye C.R., Niclis J.C., Alsanie W., Blakely B.D., Stenman J., Turner B.J., Parish C.L. Diverse roles for Wnt7a in ventral midbrain neurogenesis and dopaminergic axon morphogenesis. Stem Cells Dev. 2014;23:1991–2003. doi: 10.1089/scd.2014.0166. [DOI] [PubMed] [Google Scholar]
- Ferreira A., Kosik K.S. Accelerated neuronal differentiation induced by p53 suppression. J. Cell Sci. 1996;109:1509–1516. doi: 10.1242/jcs.109.6.1509. [DOI] [PubMed] [Google Scholar]
- Fertuzinhos S., Li M., Kawasawa Y.I., Ivic V., Franjic D., Singh D., Crair M., Sestan N. Laminar and temporal expression dynamics of coding and noncoding RNAs in the mouse neocortex. Cell Rep. 2014;6:938–950. doi: 10.1016/j.celrep.2014.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg K., Wuttke A., Quadrato G., Chumakov P.M., Wizenmann A., Di Giovanni S. The tumor suppressor p53 fine-tunes reactive oxygen species levels and neurogenesis via PI3 kinase signaling. J. Neurosci. 2013;33:14318–14330. doi: 10.1523/jneurosci.1056-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos D.A., Chan U., Chen L.F., West A.E. Chromatin regulation of neuronal maturation and plasticity. Trends Neurosci. 2018;41:311–324. doi: 10.1016/j.tins.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., Wong S.K., Jiang Z., Liu B., Wang Y., Hao Q., Gorbunova V., Liu X., Zhou Z. Haploinsufficiency of Trp53 dramatically extends the lifespan of Sirt6-deficient mice. eLife. 2018;7:e32127. doi: 10.7554/eLife.32127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacinti C., Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- Goto A., Matsushita K., Gesellchen V., El Chamy L., Kuttenkeuler D., Takeuchi O., Hoffmann J.A., Akira S., Boutros M., Reichhart J.M. Akirins are highly conserved nuclear proteins required for NF-kappaB-dependent gene expression in drosophila and mice. Nat. Immunol. 2008;9:97–104. doi: 10.1038/ni1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove E.A., Tole S., Limon J., Yip L., Ragsdale C.W. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- Hafner A., Bulyk M.L., Jambhekar A., Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019;20:199–210. doi: 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- Hattori T., Baba K., Matsuzaki S., Honda A., Miyoshi K., Inoue K., Taniguchi M., Hashimoto H., Shintani N., Baba A., et al. A novel DISC1-interacting partner DISC1-Binding Zinc-finger protein: implication in the modulation of DISC1-dependent neurite outgrowth. Mol. Psychiatry. 2007;12:398–407. doi: 10.1038/sj.mp.4001945. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Herzog K.-H., Chong M.J., Kapsetaki M., Morgan J.I., McKinnon P.J. Requirement for atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280:1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- Hirata H., Cadet J.L. p53-knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J. Neurochem. 1997;69:780–790. doi: 10.1046/j.1471-4159.1997.69020780.x. [DOI] [PubMed] [Google Scholar]
- Honda R., Tanaka H., Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/S0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Hsieh J.K., Fredersdorf S., Kouzarides T., Martin K., Lu X. E2F1-induced apoptosis requires DNA binding but not transactivation and is inhibited by the retinoblastoma protein through direct interaction. Genes Dev. 1997;11:1840–1852. doi: 10.1101/gad.11.14.1840. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T., Weinberg R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jiang L., Kon N., Li T., Wang S.-J., Su T., Hibshoosh H., Baer R., Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo j. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadkhodaei B., Alvarsson A., Schintu N., Ramsköld D., Volakakis N., Joodmardi E., Yoshitake T., Kehr J., Decressac M., Björklund A., et al. Transcription factor Nurr1 maintains fiber integrity and nuclear-encoded mitochondrial gene expression in dopamine neurons. Proc. Natl. Acad. Sci. U S A. 2013;110:2360–2365. doi: 10.1073/pnas.1221077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania A., Jessell T.M. Topographic motor projections in the limb imposed by LIM homeodomain protein regulation of Ephrin-A:EphA interactions. Neuron. 2003;38:581–596. doi: 10.1016/S0896-6273(03)00292-7. [DOI] [PubMed] [Google Scholar]
- Kast R.J., Lanjewar A.L., Smith C.D., Levitt P. FOXP2 exhibits projection neuron class specific expression, but is not required for multiple aspects of cortical histogenesis. eLife. 2019;8:e42012. doi: 10.7554/eLife.42012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya Y., Kurabe N., Katagiri K., Ogawa M., Sugiyama A., Kawasaki Y., Tashiro F. A novel binding factor of 14-3-3beta functions as a transcriptional repressor and promotes anchorage-independent growth, tumorigenicity, and metastasis. J. Biol. Chem. 2008;283:18753–18764. doi: 10.1074/jbc.M802530200. [DOI] [PubMed] [Google Scholar]
- Kowalik T.F., DeGregori J., Leone G., Jakoi L., Nevins J.R. E2F1-specific induction of apoptosis and p53 accumulation, which is blocked by Mdm2. Cell Growth Differ. 1998;9:113–118. [PubMed] [Google Scholar]
- Kracikova M., Akiri G., George A., Sachidanandam R., Aaronson S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013;20:576–588. doi: 10.1038/cdd.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2013;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G., Galluzzi L., Vandenabeele P., Abrams J., Alnemri E.S., Baehrecke E.H., Blagosklonny M.V., El-Deiry W.S., Golstein P., Green D.R., et al. Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krossa S., Schmitt A.D., Hattermann K., Fritsch J., Scheidig A.J., Mehdorn H.M., Held-Feindt J. Down regulation of Akirin-2 increases chemosensitivity in human glioblastomas more efficiently than Twist-1. Oncotarget. 2015;6:21029–21045. doi: 10.18632/oncotarget.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat M.H.G., Jones S.N., Vousden K.H. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-g., Casadesus G., Zhu X., Castellani R.J., McShea A., Perry G., Petersen R.B., Bajic V., Smith M.A. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer's disease. Neurochem. Int. 2009;54:84–88. doi: 10.1016/j.neuint.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees J.A., Saito M., Vidal M., Valentine M., Look T., Harlow E., Dyson N., Helin K. The retinoblastoma protein binds to a family of E2F transcription factors. Mol. Cell Biol. 1993;13:7813–7825. doi: 10.1128/mcb.13.12.7813-7825.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng K., Xu Y., Kang P., Qin W., Cai H., Wang H., Ji D., Jiang X., Li J., Li Z., et al. Akirin2 is modulated by miR-490-3p and facilitates angiogenesis in cholangiocarcinoma through the IL-6/STAT3/VEGFA signaling pathway. Cell Death Dis. 2019;10:262. doi: 10.1038/s41419-019-1506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Marshall L., Oh G., Jakubowski J.L., Groot D., He Y., Wang T., Petronis A., Labrie V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer's disease pathology and cognitive symptoms. Nat. Commun. 2019;10:2246. doi: 10.1038/s41467-019-10101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lin J.H., Saito T., Anderson D.J., Lance-Jones C., Jessell T.M., Arber S. Functionally related motor neuron pool and muscle sensory afferent subtypes defined by coordinate ets gene expression. Cell. 1998;95:393–407. doi: 10.1016/S0092-8674(00)81770-5. [DOI] [PubMed] [Google Scholar]
- Lind D., Franken S., Kappler J., Jankowski J., Schilling K. Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J. Neurosci. Res. 2005;79:295–302. doi: 10.1002/jnr.20354. [DOI] [PubMed] [Google Scholar]
- Lipinski M., Muñoz-Viana R., del Blanco B., Marquez-Galera A., Medrano-Relinque J., Caramés J.M., Szczepankiewicz A.A., Fernandez-Albert J., Navarrón C.M., Olivares R., et al. KAT3-dependent acetylation of cell type-specific genes maintains neuronal identity in the adult mouse brain. Nat. Commun. 2020;11:2588. doi: 10.1038/s41467-020-16246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Maejima T., Wyler S.C., Casadesus G., Herlitze S., Deneris E.S. Pet-1 is required across different stages of life to regulate serotonergic function. Nat. Neurosci. 2010;13:1190–1198. doi: 10.1038/nn.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Xia Y., Tang J., Ma L., Li C., Ma P., Mao B. Dual roles of Akirin2 protein during Xenopus neural development. J. Biol. Chem. 2017;292:5676–5684. doi: 10.1074/jbc.M117.777110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacManus J.P., Jian M., Preston E., Rasquinha I., Webster J., Zurakowski B. Absence of the transcription factor E2F1 attenuates brain injury and improves behavior after focal ischemia in mice. J. Cereb. Blood Flow Metab. 2003;23:1020–1028. doi: 10.1097/01.Wcb.0000084249.20114. [DOI] [PubMed] [Google Scholar]
- Maiuri M.C., Galluzzi L., Morselli E., Kepp O., Malik S.A., Kroemer G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Mancarci B.O., Toker L., Tripathy S.J., Li B., Rocco B., Sibille E., Pavlidis P. Cross-laboratory analysis of brain cell type transcriptomes with applications to interpretation of bulk tissue data. eNeuro. 2017;4 doi: 10.1523/eneuro.0212-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna W.L., Betancourt J., Larkin K.A., Abrams B., Guo C., Rubenstein J.L., Chen B. Tbr1 and Fezf2 regulate alternate corticofugal neuronal identities during neocortical development. J. Neurosci. 2011;31:549–564. doi: 10.1523/jneurosci.4131-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShea A., Lee H.G., Petersen R.B., Casadesus G., Vincent I., Linford N.J., Funk J.O., Shapiro R.A., Smith M.A. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim. Biophys. Acta. 2007;1772:467–472. doi: 10.1016/j.bbadis.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Mfossa A.-C.M., Verslegers M., Verreet T., Fida H.b., Mysara M., Van IJcken W.F.J., De Vos W.H., Moons L., Baatout S., Benotmane M.A., et al. p53 drives premature neuronal differentiation in response to radiation-induced DNA damage during early neurogenesis. bioRxiv. 2020 doi: 10.1101/2020.06.26.171132. [DOI] [Google Scholar]
- Miller F.D., Pozniak C.D., Walsh G.S. Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 2000;7:880–888. doi: 10.1038/sj.cdd.4400736. [DOI] [PubMed] [Google Scholar]
- Montana C.L., Kolesnikov A.V., Shen S.Q., Myers C.A., Kefalov V.J., Corbo J.C. Reprogramming of adult rod photoreceptors prevents retinal degeneration. Proc. Natl. Acad. Sci. U S A. 2013;110:1732–1737. doi: 10.1073/pnas.1214387110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison R.S., Wenzel H.J., Kinoshita Y., Robbins C.A., Donehower L.A., Schwartzkroin P.A. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J. Neurosci. 1996;16:1337–1345. doi: 10.1523/jneurosci.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison R.S., Kinoshita Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000;7:868–879. doi: 10.1038/sj.cdd.4400741. [DOI] [PubMed] [Google Scholar]
- Murray-Zmijewski F., Slee E.A., Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008;9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- Narasimha A.M., Kaulich M., Shapiro G.S., Choi Y.J., Sicinski P., Dowdy S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife. 2014;3:e02872. doi: 10.7554/eLife.02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkovic J., Pinto L., Petricca S., Lepier A., Sun J., Rieger M.A., Schroeder T., Cvekl A., Favor J., Götz M. The transcription factor Pax6 regulates survival of dopaminergic olfactory bulb neurons via crystallin αA. Neuron. 2010;68:682–694. doi: 10.1016/j.neuron.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak S.J., Aihara H., Gonzalez K., Nibu Y., Baylies M.K. Akirin links twist-regulated transcription with the Brahma chromatin remodeling complex during embryogenesis. PLoS Genet. 2012;8:e1002547. doi: 10.1371/journal.pgen.1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M., Iguchi T., Hattori T., Matsuzaki S., Koyama Y., Taniguchi M., Komada M., Xie M.J., Yagi H., Shimizu S., et al. DBZ regulates cortical cell positioning and neurite development by sustaining the anterograde transport of Lis1 and DISC1 through control of Ndel1 dual-phosphorylation. J. Neurosci. 2015;35:2942–2958. doi: 10.1523/jneurosci.5029-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren M. Regulation of the p53 tumor suppressor protein. J. Biol. Chem. 1999;274:36031–36034. doi: 10.1074/jbc.274.51.36031. [DOI] [PubMed] [Google Scholar]
- Park K.H.J., Hallows J.L., Chakrabarty P., Davies P., Vincent I. Conditional neuronal simian virus 40 T antigen expression induces alzheimer-like tau and amyloid pathology in mice. J. Neurosci. 2007;27:2969–2978. doi: 10.1523/jneurosci.0186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M., Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Pillai-Kastoori L., Schutz-Geschwender A.R., Harford J.A. A systematic approach to quantitative Western blot analysis. Anal. Biochem. 2020;593:113608. doi: 10.1016/j.ab.2020.113608. [DOI] [PubMed] [Google Scholar]
- Plesnila N., von Baumgarten L., Retiounskaia M., Engel D., Ardeshiri A., Zimmermann R., Hoffmann F., Landshamer S., Wagner E., Culmsee C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-κB transcriptional activity. Cell Death Differ. 2007;14:1529–1541. doi: 10.1038/sj.cdd.4402159. [DOI] [PubMed] [Google Scholar]
- Polager S., Ofir M., Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27:4860–4864. doi: 10.1038/onc.2008.117. [DOI] [PubMed] [Google Scholar]
- Qin X.Q., Livingston D.M., Kaelin W.G., Adams P.D. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. U S A. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Q., Sun G., Murai K., Ye P., Li W., Asuelime G., Cheung Y.T., Shi Y. Wnt7a regulates multiple steps of neurogenesis. Mol. Cell Biol. 2013;33:2551–2559. doi: 10.1128/mcb.00325-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintens R., Verreet T., Janssen A., Neefs M., Leysen L., Michaux A., Verslegers M., Samari N., Pani G., Verheyde J., et al. Identification of novel radiation-induced p53-dependent transcripts extensively regulated during mouse brain development. Biol. Open. 2015;4:331–344. doi: 10.1242/bio.20149969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2019. R: A Language and Environment for Statistical Computing. [Google Scholar]
- Ranjan A., Iwakuma T. Non-canonical cell death induced by p53. Int. J. Mol. Sci. 2016;17 doi: 10.3390/ijms17122068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D., Schwartz K., Sherlock G., Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinf. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D., Ngai J., Speed T.P., Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhi S., Gilmore W., Tran N.D., Schreiber S.S. p53-deficient mice are protected against adrenalectomy-induced apoptosis. Neuroreport. 1996;8:233–235. doi: 10.1097/00001756-199612200-00047. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mut J.V., Heyn H., Vidal E., Moran S., Sayols S., Delgado-Morales R., Schultz M.D., Ansoleaga B., Garcia-Esparcia P., Pons-Espinal M., et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl. Psychiatry. 2016;6:e718. doi: 10.1038/tp.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C.M., Dai Y., Van Alstyne M., Koutsioumpa C., Pagiazitis J.G., Chalif J.I., Wang X., Rabinowitz J.E., Henderson C.E., Pellizzoni L., et al. Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 2017;21:3767–3780. doi: 10.1016/j.celrep.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C., Breitkreutz B.J., Reguly T., Boucher L., Breitkreutz A., Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stence N., Waite M., Dailey M.E. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–266. doi: 10.1002/1098-1136(200103)33:3<256::AID-GLIA1024>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Sugino K., Clark E., Schulmann A., Shima Y., Wang L., Hunt D.L., Hooks B.M., Tränkner D., Chandrashekar J., Picard S., et al. Mapping the transcriptional diversity of genetically and anatomically defined cell populations in the mouse brain. eLife. 2019;8:e38619. doi: 10.7554/eLife.38619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gky1131. D607–d613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartey S., Matsushita K., Vandenbon A., Ori D., Imamura T., Mino T., Standley D.M., Hoffmann J.A., Reichhart J.M., Akira S., et al. Akirin2 is critical for inducing inflammatory genes by bridging IkappaB-zeta and the SWI/SNF complex. Embo j. 2014;33:2332–2348. doi: 10.15252/embj.201488447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartey S., Matsushita K., Imamura T., Wakabayashi A., Ori D., Mino T., Takeuchi O. Essential function for the nuclear protein Akirin2 in B Cell activation and humoral immune responses. J. Immunol. 2015;195:519–527. doi: 10.4049/jimmunol.1500373. [DOI] [PubMed] [Google Scholar]
- Tsien J.Z., Chen D.F., Gerber D., Tom C., Mercer E.H., Anderson D.J., Mayford M., Kandel E.R., Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Tu H.C., Ren D., Wang G.X., Chen D.Y., Westergard T.D., Kim H., Sasagawa S., Hsieh J.J., Cheng E.H. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc. Natl. Acad. Sci. U S A. 2009;106:1093–1098. doi: 10.1073/pnas.0808173106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Alstyne M., Simon C.M., Sardi S.P., Shihabuddin L.S., Mentis G.Z., Pellizzoni L. Dysregulation of Mdm2 and Mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev. 2018;32:1045–1059. doi: 10.1101/gad.316059.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. Research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva Angelina V., Marchenko Natalie D., Ji K., Tsirka Stella E., Holzmann S., Moll Ute M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A., Matheos D.P., Barrett R.M., Kramár E.A., Azzawi S., Chen Y., Magnan C.N., Zeller M., Sylvain A., Haettig J., et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat. Neurosci. 2013;16:552–561. doi: 10.1038/nn.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schimmelmann M., Feinberg P.A., Sullivan J.M., Ku S.M., Badimon A., Duff M.K., Wang Z., Lachmann A., Dewell S., Ma'ayan A., et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016;19:1321–1330. doi: 10.1038/nn.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Liu F., Liu C.Y., An T., Zhang J., Zhou L.Y., Wang M., Dong Y.H., Li N., Gao J.N., et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 2016;23:1394–1405. doi: 10.1038/cdd.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Bu B., Xie M., Zhang M., Yu Z., Tao D. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol. 2009;89:1–17. doi: 10.1016/j.pneurobio.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Wang X., Weiner J.A., Levi S., Craig A.M., Bradley A., Sanes J.R. Gamma protocadherins are required for survival of spinal interneurons. Neuron. 2002;36:843–854. doi: 10.1016/s0896-6273(02)01090-5. [DOI] [PubMed] [Google Scholar]
- Wang X., Zhang C., Szábo G., Sun Q.Q. Distribution of CaMKIIα expression in the brain in vivo, studied by CaMKIIα-GFP mice. Brain Res. 2013;1518:9–25. doi: 10.1016/j.brainres.2013.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H., Ohta S., Kumon Y., Sakaki S., Sakanaka M. Increase in p53 protein expression following cortical infarction in the spontaneously hypertensive rat. Brain Res. 1999;837:38–45. doi: 10.1016/s0006-8993(99)01652-2. [DOI] [PubMed] [Google Scholar]
- Watson C.T., Roussos P., Garg P., Ho D.J., Azam N., Katsel P.L., Haroutunian V., Sharp A.J. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016;8:5. doi: 10.1186/s13073-015-0258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H., Hochman D.W., Saya H., Fujiwara T., Schwartzkroin P.A., Morrison R.S. Evidence for p53-mediated modulation of neuronal viability. J. Neurosci. 1996;16:6753–6765. doi: 10.1523/jneurosci.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Mufson E.J., Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J. Neurosci. 2003;23:2557–2563. doi: 10.1523/jneurosci.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an r package for comparing biological themes among gene clusters. OMICS A. J. Integr. Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Chen K., Sloan S.A., Bennett M.L., Scholze A.R., O'Keeffe S., Phatnani H.P., Guarnieri P., Caneda C., Ruderisch N., et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014;34:11929–11947. doi: 10.1523/jneurosci.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Lee H.-g., Perry G., Smith M.A. Alzheimer disease, the two-hit hypothesis: an update. Biochim. Biophys. Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data have been deposited at the gene expression omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.