

Summary
Most tumor cells reactivate telomerase to ensure unlimited proliferation, whereas the expression of human telomerase reverse transcriptase (hTERT) is tightly regulated and rate-limiting for telomerase activity maintenance. Several general transcription factors (TFs) have been found in regulating hTERT transcription; however, a systematic study is lacking. Here we performed an inducible CRISPR/Cas9 KO screen using an hTERT core promoter-driven reporter. We identified numerous positive regulators including an E3 ligase DTX2. In telomerase-positive cancer cells, DTX2 depletion downregulated hTERT transcription and telomerase activity, contributing to progressive telomere shortening, growth arrest, and increased apoptosis. Utilizing BioID, we characterized multiple TFs as DTX2 proximal proteins, among which NFIC functioned corporately with DTX2 in promoting hTERT transcription. Further analysis demonstrated that DTX2 mediated K63-linked ubiquitination of NFIC, which facilitated NFIC binding to the hTERT promoter and enhanced hTERT expression. These findings highlight a new hTERT regulatory pathway that may be exploited for potential cancer therapeutics.
Subject areas: biological sciences, human physiology, molecular biology, cancer
Graphical abstract
Highlights
-
•
An inducible CRISPR/Cas9 screen identifies regulators for hTERT transcription
-
•
DTX2 deficiency leads to telomere shortening and cell growth arrest
-
•
DTX2 mediates ubiquitination on NFIC, stabilizing NFIC binding on hTERT promoter
-
•
DTX2-NFIC functions corporately to promote hTERT transcription and tumorigenesis
Biological sciences; Human Physiology; Molecular biology; Cancer
Introduction
Telomeres are crucial to maintaining the natural ends of linear chromosomes (Blackburn et al., 2015). Telomere DNA undergoes progressive shortening with each cell division (Harley et al., 1990; Victorelli and Passos, 2017). Critically short telomeres can trigger growth arrest and cell death (Hayashi et al., 2015; Saretzki et al., 1999), and cells that escape from such crisis may eventually lead to tumorigenesis (Maciejowski and de Lange, 2017; Wright et al., 1989). Human telomeres consist of TTAGGG tandem repeats that are elongated by the telomerase complex, with the reverse transcriptase hTERT and RNA component hTR/hTERC as its core components (Blackburn and Collins, 2011; Wang et al., 2019). Telomerase activity appears undetectable in most normal human somatic cells but present in ∼90% of cancer and immortalized cells (Shay, 2013), underscoring the importance of upregulated telomerase activity in cancer development. Understanding the pathways that regulate telomerase expression and activity promises to provide valuable insight into cancer development and treatment.
Unlike hTERC, which is highly and ubiquitously expressed even in tissues with no detectable telomerase activity, hTERT expression appears suppressed in nearly all normal somatic cells (Shay, 2016; Zhang et al., 2016) and rate-limiting for maintaining cellular telomerase activity, given its positive correlation with telomerase activity (Ramlee et al., 2016; Weinrich et al., 1997). hTERT expression can be controlled at the transcriptional, posttranscriptional (e.g., alternative splicing), and posttranslational (e.g., phosphorylation) level. Transcriptionally, hTERT suppression in normal cells and its reactivation in cancer cells may occur through distinct but interconnected mechanisms, including regulation through transcription factors, mutations in the hTERT promoter, and epigenetic modifications of the hTERT locus (e.g., DNA methylation and histone acetylation). Low hTERT expression in human somatic cells largely attributes to the condensed heterochromatin that surrounds the hTERT gene (Wang et al., 2009; Zhang et al., 2016). However, in telomerase-positive tumor cells, the chromatin region near the hTERT transcription start site (TSS) becomes relatively open, as evidenced by the presence of DNaseI-hypersensitive sites (Wang and Zhu, 2003; Zhu et al., 2010).
Multifunctional transcription factors such as c-MYC, NF-κB, and STAT3 can upregulate hTERT transcription in cancer cells (Ramlee et al., 2016). Transcription factor binding appears to concentrate within 400 bps upstream of hTERT TSS, which contains E-box and GC repeats and recruits factors such as Sp1/3, AP-2, USFs, and Myc family proteins (Ramlee et al., 2016). In addition, oncoproteins including HER2, Ras, and Raf are also capable of stimulating hTERT transcription via ETS transcription factors (Goueli and Janknecht, 2004). To date, there remains a dearth of information regarding specific positive regulators of hTERT transcription, and systematic studies are needed to elucidate the regulatory network that enables hTERT reactivation in cancer cells.
Here, we performed an inducible CRISPR/Cas9 knockout (KO) screen to identify positive regulators of hTERT expression, utilizing two sgRNA libraries respectively targeting nuclear genes and genes of unknown function. Our screen revealed multiple candidates that could upregulate hTERT expression including the E3 ubiquitin ligase DTX2. DTX2 KO in telomerase-positive cancer cells significantly reduced hTERT mRNA level as well as telomerase activity, resulting in progressive telomere shortening and cell growth arrest. A large-scale affinity purification screen using proximity-dependent biotin identification (BioID) further identified nuclear factor I C (NFIC) as a DTX2-associating protein. We demonstrated that DTX2 could interact with NFIC and mediate K63-linked ubiquitination of NFIC to facilitate its binding to the hTERT core promoter, which in turn enhanced hTERT transcription. Our work presents a more comprehensive list of candidate positive regulators of hTERT expression and highlights DTX2-mediated ubiquitination of transcription factors as a novel regulatory pathway for hTERT transcription and telomere maintenance.
Results
An inducible CRISPR/Cas9 screen identifies positive regulators for hTERT transcription
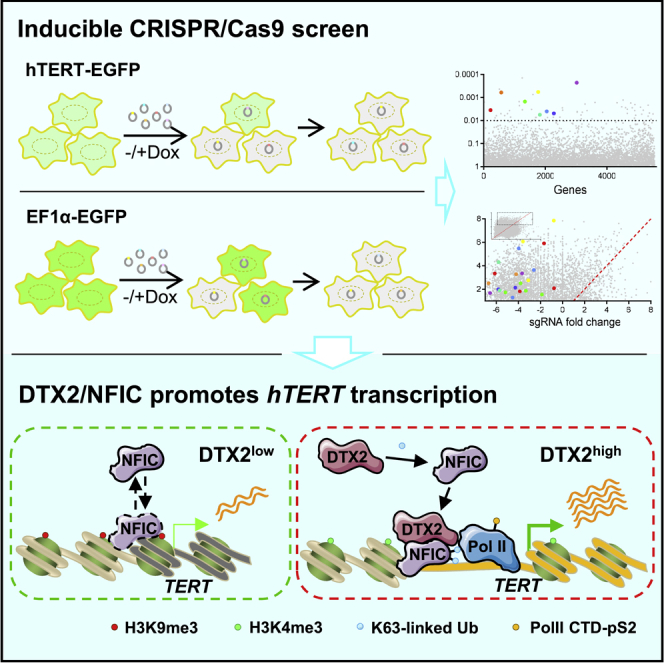
To systematically search for hTERT transcriptional regulators, we stably expressed EGFP driven by the hTERT core promoter (Higashi et al., 2014) (hTERT-EGFP) in the telomerase-positive HeLa line that also expressed doxycycline (Dox)-inducible Cas9 (HeLa-iCas9) (Kim et al., 2017) (Figure 1A). HeLa-iCas9 cells expressing EF1α promoter-driven EGFP were also generated (EF1α-EGFP) to help exclude general factors regulating transcription and protein stability. EGFP+ cells were enriched by fluorescence-activated cell sorting (FACS) and then transduced with two lentiviral sgRNA libraries respectively targeting 3,733 nuclear genes and 1,808 genes of unknown function (10 sgRNAs/gene) (Wang et al., 2014). Cells cultured without Dox (i.e., basal groups) served as the baseline for all subsequent analysis. Upon Dox-induced Cas9 expression, cells with specific gene KO and loss of EGFP signal were enriched by FACS as sorted groups. Cells from basal groups and sorted groups were then processed for next-generation sequencing (NGS) to calculate sgRNA abundance.
Figure 1.

An inducible CRISPR/Cas9 screen identifies positive regulators of hTERT transcription
(A) HeLa cells stably expressing inducible Cas9 (iCas9) and EGFP driven by the hTERT core promoter (hTERT-EGFP) or EF1α promoter (EF1α-EGFP) were sorted for EGFP+ cells, which were then infected with lentiviral sgRNA libraries (MOI of 0.3) and selected with blasticidin. Selected cells cultured without doxycycline (Dox) served as basal groups. Dox-induced cells (1 μg/mL, 7 days) went through two rounds of FACS sorting to enrich for EGFP− cells (sorted group). All groups were subsequently analyzed by next-generation sequencing (NGS) to determine individual sgRNA abundance.
(B) MAGeCK analysis was performed (see STAR Methods) to identify top candidate genes (p < 0.01) in regulating hTERT transcription.
(C) The enrichment of specific sgRNAs for genes whose KO led to decreased hTERT transcription is shown in upper left corner, where each dot represents one sgRNA. Fold changes of sgRNA reads (sorted over basal groups) were transformed with Log2 and plotted using Graphpad Prism7. The rectangular area is magnified below. The dotted red line indicates where the fold change of sgRNA reads is the same for both hTERT-EGFP and EF1α-EGFP samples.
(D and E) CRISPR KO cell pools were generated using HEK293T iCas9 (D) or HeLa iCas9 (E) cells for the genes indicated. Cells were cultured with −/+Dox (1 μg/mL) for 48 h before RT-qPCR analysis. Relative hTERT mRNA levels were quantified and plotted. Data were shown as mean ± SD (N = 3). p values were calculated using Student’s t test. ∗p< 0.05; ∗∗p< 0.01; ∗∗∗p< 0.001. See also Figure S1, Tables S1, and S2.
NGS analysis revealed considerable shifts and decreased clustering in sgRNA distributions following KO induction (Figure S1A). Known hTERT transcription factors AP-2, USF1/2, NF-κB, and STAT3 were targeted by our sgRNA libraries, but sgRNA recovery was very low for these genes in both sorted groups, likely reflecting their importance for general transcription and cell survival. Overall, 257 potential positive regulators of hTERT transcription were identified (Figure S1B), including epigenetic regulators involved in histone acetylation (KAT2A and KAT6A) and H3K4 methylation (MLL4). MAGeCK analysis (Li et al., 2014) revealed several genes significantly enriched in the hTERT-EGFP group (with multiple high-ranking sgRNAs), which have never been implicated in hTERT transcriptional regulation (Figures 1B, 1C and S1C, and Tables S1, S2). Using HEK293T-iCas9 and HeLa-iCas9 KO cell pools (Figure S1D), we verified that depletion of ASXL2, DTX2, FGF1, or FOXD3 led to decreased hTERT mRNA levels (Figures 1D and 1E) coupled with reduced telomerase activities (Figures S1E and S1F), supporting a role of these genes in promoting hTERT transcription. In particular, loss of DTX2 gave rise to a prominent effect. Given its high expression in multiple cancers (according to TCGA and GTEx (Tang et al., 2017) (Figure S1G), we hypothesized that DTX2 might facilitate tumor progression through upregulating hTERT transcription.
It has been shown that telomerase inhibition in cells with short telomeres can cause cell growth arrest and even apoptosis (Zhang et al., 1999). Inhibition of hTERT could induce immediate cell growth arrest by blocking them in S phase, whereas apoptosis might be induced via extra-telomeric effects on activation of p53 and p21 (Celeghin et al., 2016; Luo et al., 2009). When we knocked down DTX2 in telomerase-positive cancer lines (e.g., HT-1080, Hs578T, and MDA-MB-231) with relatively short telomeres (Figure S1H), growth arrest and increased apoptosis were readily detectable (Figures 2A–2D and S2A–S2H). In comparison, the effect of DTX2 knockdown in cancer lines (e.g., HeLa and DLD1) with longer telomeres was modest, although still comparable to hTERT knockdown (Figures S2I–S2P). Therefore, DTX2 may be especially important for maintaining telomerase expression and in turn cell growth and viability in telomerase-positive cancer cells that harbor short telomeres.
Figure 2.

DTX2 positively regulates hTERT transcription and is necessary for cancer cell growth
(A–D) HT-1080 cells stably expressing DTX2 or hTERT shRNAs were generated. Cells infected with pLKO1 vector viruses served as controls. Relative DTX2 (A) and hTERT (B) mRNA levels were determined by RT-qPCR. Cell growth curve was examined by CCK8 assay (C). Data were shown as mean ± SD (N = 4). ∗∗∗p< 0.001. The Annexin V+/PI− or Annexin V+/PI+ cells were measured by flow cytometry and defined as early or late apoptotic cells, respectively (D).
(E–H) HeLa cells expressing Flag-HA-tagged DTX2 (FH-DTX2) or EGFP (FH-EGFP) were generated. Protein expression was examined by western blotting with an anti-Flag antibody (E). Relative hTERT mRNA levels (F) and telomerase activity (G) were determined by RT-qPCR and qTRAP, respectively. Data were shown as mean ± SD (N = 3). ∗∗∗p< 0.001. Telomere restriction fragment (TRF) analysis was performed to assess average telomere length at the indicated population doublings (PDs) with quantification using ImageQuant 5.2 (H).
(I–K) Two DTX2 knockout (KO) HeLa cell lines (#20 & #39) were generated. Empty px458 vector transfected cells served as controls. All cells were examined for relative hTERT mRNA level (I), telomerase activity (J), and average telomere length (K) as above. Data were shown as mean ± SD (N = 3). ∗∗p< 0.01; ∗∗∗p< 0.001.(L–O) FH-EGFP or FH-DTX2 were ectopically expressed in DTX2 KO line #20. Protein expression (L), relative hTERT mRNA level (M), telomerase activity (N), and average telomere length (O) were assessed as above. Data are shown as mean ± SD (N = 3). ∗∗p< 001; ∗∗∗p< 0.001. For all panels, p values were calculated using Student’s t test, and average telomere length change is given beneath TRF blots. See also Figures S2 and S3.
Increase of telomerase activity is found in the majority of human cancers (Roake and Artandi, 2020). Whether modulation of DTX2 affects tumorigenicity of cancer cells remains to be addressed. Cell growth in soft agar is a gold standard in vitro assessment of cell malignant transformation, which is indicative for anchorage-independent growth (Anderson et al., 2007; Thierbach and Steinberg, 2009). Similar to hTERT knockdown, DTX2 knockdown significantly hampered the colony forming ability of MDA-MB-231 (Figure S1Q) and HeLa (Figure S1R) cells. These data suggested that targeting DTX2 suppressed anchorage-independent growth of cancer cells.
DTX2 promotes hTERT transcription dependent of both RING and WWE domains
To further probe the activity of DTX2, Flag-HA (FH)-tagged DTX2 were expressed in HeLa and HT-1080 cells. Compared with the control group, DTX2 significantly upregulated hTERT mRNA level and telomerase activity (Figures 2E–2G and S3A–S3C). Moreover, cells with DTX2 expression also exhibited a faster rate of telomere lengthening (Figures 2H and S3D). Next, we examined two DTX2 KO clones (#20 and #39) and found markedly reduced hTERT mRNA level and telomerase activity, as well as accelerated telomere shortening (Figures 2I–2K and S3E). To exclude possible CRISPR/Cas9 off-target effects, FH- or SFB-tagged DTX2 were reintroduced in DTX2 KO line #20. Rescue expression of DTX2 was able to elevate hTERT mRNA level and telomerase activity and maintain telomere length (Figures 2L–2O and S3F–S3I). These data together support the notion that DTX2 positively regulates hTERT transcription in telomerase-positive cancer cells.
DTX2 has two N-terminal WWE motifs, a low complexity region (LC) and a C-terminal RING finger (Figure 3A) (Schultz et al., 2000). We compared three truncation mutants of DTX2 for their ability to rescue DTX2 KO cells. DTX2 mutants lacking regions containing the RING or WWE domains failed to rescue the decrease in hTERT mRNA level, telomerase activity, or telomere length in DTX2 KO cells (Figures 3B–3E). These data indicate that both RING and WWE domains are indispensable to DTX2 function.
Figure 3.

DTX2 promotes hTERT transcription dependent of both RING and WWE domains
(A) Schematic diagram of full-length and truncated DTX2 proteins. WWE, two tandem WWE motifs. LC, low complexity region. RING, region containing RING-H2 finger and C-terminal region.
(B–E) Full-length FH-DTX2 and truncation mutants were ectopically expressed in DTX2 KO line #20. Protein expression (B), relative hTERT mRNA level (C), telomerase activity (D), and average telomere length (E) were assessed. Data were shown as mean ± SD (N = 3). p values were calculated using Student’s t test. ∗p< 0.05; ∗∗p< 0.01; n.s., not significant. Average telomere length change is given beneath the TRF blot.
DTX2 can localize to the nucleus and bind to the hTERT core promoter
DTX2 has three potential nuclear localization signals (NLS) (Lin and Hu, 2013) and is predicted to localize in the nucleoplasm based on the Human Protein Atlas (Thul and Lindskog, 2018). Because none of the commercially available DTX2 antibodies worked satisfactorily in our assays (data not shown), we examined the subcellular localization of DTX2 in HeLa cells expressing Flag-HA-tagged full-length DTX2 or truncation mutants. For comparison, the central region deletion mutant (ΔCEN) was also included (Figure 4A). Immunofluorescence (IF) assay revealed that full-length DTX2 signals were more prominent in the nucleus (Figure 4B). Cell fractionation assay showed that full-length DTX2 signals were readily detectable in both cytosolic and nuclear fractions (Figure 4C). Interestingly, respective deletion of the WWE, RING, and LC domains had little effect on DTX2 localization, whereas the ΔCEN mutant showed drastically reduced nuclear DTX2 signals in both IF and fractionation assays (Figures 4B and 4C).
Figure 4.

DTX2 can localize to the nucleus and bind to the hTERT core promoter
(A) Schematic diagram of predicted nuclear localization signals (NLS) of DTX2 and the ΔCEN mutant.
(B and C) Full-length FH-DTX2 and its truncation mutants were expressed in HeLa cells. The localization of various DTX2 proteins was examined by immunostaining with an anti-HA antibody (scale bar: 10 μm) (B) and western blotting following cell fractionation using the indicated antibodies (C). FH-EGFP-expressing cells served as negative controls for fractionation.
(D and E) HeLa cells expressing full-length DTX2 or the ΔCEN mutant were assayed by RT-qPCR and qTRAP to determine the relative hTERT mRNA level (D) and telomerase activity (E). Data were shown as mean ± SD (N = 3). p values were calculated using Student’s t test. ∗p< 0.05; ∗∗p<0.01; n.s., not significant.
(F and G) DTX2 KO cells (#20) expressing full-length DTX2 or the ΔCEN mutant were assayed by RT-qPCR and qTRAP to determine the relative hTERT mRNA level (F) and telomerase activity (G). Data were shown as mean ± SD (N = 3). p values were calculated using Student’s t test. ∗p< 0.05; ∗∗∗p< 0.001; n.s., not significant.
(H) DTX2 KO line #20 expressing FH-EGFP or FH-DTX2 were fractionated for the chromatin association assay. Various fractions were subjected to immunoblot analysis with the indicated antibodies. WCE, whole cell extract. CE, cytosolic extract. Wash 1 & 2, nucleoplasmic fraction. Soluble & insoluble, chromatin fractions sensitive or resistant to micrococcal nuclease digestion.
(I) ChIP-qPCR analysis was done using cells from (H) and an anti-HA antibody. Primers span areas within the hTERT core promoter region (−113/+54 and −476/−242), upstream (∼3.7 kb) of hTERT TSS (−3,783/−3,646), or in the last exon (+41,480/+41,590). Enrichment of specific regions was normalized to input. Data were shown as mean ± SD (N = 3). p values were calculated using Student’s t test. ∗p< 0.05; ∗∗p< 0.01.
(J and K) ChIP-qPCR analysis was done using cells from (H) and anti-H3K4me3, anti-H3K9me3, and anti-H3K27me3 antibodies. Primers span areas within the hTERT core promoter region [−113/+54 (J) and −476/−242 (K)]. Enrichment of specific regions was normalized to input. Data were shown as mean ± SD (N = 2). p values were calculated using Student’s t test. ∗p< 0.05; ∗∗p< 0.01. (L) ChIP-qPCR analysis was done using cells from (H) and anti-Pol II-pS2 antibody. Primers span areas in the last exon of hTERT (+41,480/+41,590). Enrichment of specific regions was normalized to input. Data were shown as mean ± SD (N = 2). p values were calculated using Student’s t test. ∗p< 0.05.
Expression of the ΔCEN mutant also failed to upregulate hTERT mRNA level and telomerase activity in parental HeLa cells or rescue the decrease of hTERT mRNA level and telomerase activity in DTX2 KO cells (Figures 4D–4G), indicating the importance of nuclear targeting for DTX2 function. DTX2 might regulate telomerase expression through association with the chromatin; we therefore carried out chromatin association assays using the FH-DTX2-expressing DTX2 KO line (Figure 4H). As expected, FH-DTX2 was readily detectable in the cytosolic fraction (CE). A significant amount of DTX2 proteins could also be detected in the micrococcal nuclease-resistant insoluble chromatin fraction, suggesting likely association of DTX2 with heterochromatin regions. Because the hTERT locus is embedded in a region of condensed chromatin (Zhang et al., 2016), we next tested whether DTX2 could bind to the hTERT promoter region via ChIP-qPCR. With FH-DTX2 rescue expression, anti-HA ChIP signals were stronger than controls in the hTERT core promoter region (−476 to −242, −113 to +54), but similar to controls at regions farther away from hTERT TSS (Figure 4I), suggesting that DTX2 may directly regulate hTERT transcription through binding to the hTERT promoter.
To understand how DTX2 regulates hTERT transcription, we tested whether DTX2 could cause epigenetic alternations on the hTERT promoter. ChIP-qPCR analysis revealed that H3K4 tri-methylation level was significantly upregulated, whereas H3K9 tri-methylation level was decreased within hTERT core promoter region (−476 to −242, −113/+54, relative to hTERT TSS), suggesting the transcription activation of hTERT upon DTX2 expression (Figures 4J and 4K). Although H3K27 tri-methylation level did not show apparent changes in the promoter region, the binding of phosphorylated Pol II-S2 (indicative of transcription elongation) was upregulated at the 3′-end of hTERT gene locus (Figure 4L), suggesting that DTX2 promotes hTERT transcription by recruiting Pol II with productive elongating activity.
BioID-mediated proximity labeling identifies DTX2-associating partners
DTX2 lacks a DNA-binding domain; we therefore hypothesized that it might be targeted to the hTERT promoter through association with other DNA-binding proteins. To search for DTX2-recruiting factors, we performed BioID-mediated proximity labeling (Gingras et al., 2019; Kim et al., 2016) using HeLa DTX2 KO cells expressing BioID-DTX2 fusion proteins. DTX2 nearby proteins (<10 nm) were labeled with biotin, captured by streptavidin, and subjected to mass spectrometry analysis (Figure 5A). The proximal proteome of BioID-DTX2 exhibited great divergence from the control group (Figure S4A), with 180 proteins showing ≥3-fold higher relative abundance (Figure 5B, Table S3). Functional enrichment analysis (Pathan et al., 2015) revealed that ∼10% of the DTX2-interacting partners possessed transcription factor activity, with 43 candidates implicated in transcriptional regulation (e.g., CREB regulated transcription coactivators, NF-I family, etc.) (Figure 5C). Notably, the list also contains several players involved in Notch signaling (e.g., NOTCH1-3, CTBP1, NUMB, and NCOR2), consistent with findings that Drosophila Deltex and human DELTEX family members interact with Notch (Matsuno et al., 1998; Zweifel et al., 2005).
Figure 5.

BioID-mediated proximity-labeling identifies DTX2-associating partners
(A) BioID was fused to the N-terminus of full-length DTX2 and expressed in HeLa cells. In the presence of biotin, BioID labels nearby proteins on lysine (K) residues. Biotinylated proteins are then pulled down by streptavidin-coupled beads and analyzed by mass spectrometry (MS). Each group was done in triplicates.
(B) Proteins identified in the BioID-DTX2 group were compared with those for BioID alone in a volcano plot graphed using Perseus. Red dots indicate highly enriched proteins (FOT>3, refer to STAR Methods).
(C) Molecular function analysis was performed using the FUNRICH software to analyze the proteins enriched in the BioID-DTX2 group from (B).
(D and E) HeLa cells treated for 24 h with different concentrations of the Notch inhibitor DBZ were harvested to assess the relative mRNA levels of HES1, DTX2, and hTERT (D) and telomerase activities (E). Data were shown as mean ± SD (N = 3). p values were calculated using two-way ANOVA. ∗∗∗p< 0.001; n.s., not significant.
(F and G) HeLa cells overexpressing DTX2 or NF-I family proteins were assessed for relative hTERT mRNA levels (F) and telomerase activities (G). Data were shown as mean ± SD (N = 3).
(H and I) DTX2 KO cells overexpressing DTX2 or NF-I family proteins were assessed for relative hTERT mRNA levels (H) and telomerase activities (I). Data were shown as mean ± SD (N = 3). For panels (F–I), p values were calculated using Student’s t test. ∗p< 0.05; ∗∗p< 0.01; n.s., not significant. See also Figures S4, S5, and Table S3.
DTX2 promotes hTERT transcription independent of Notch signaling
Drosophila Deltex activates Notch signaling by initiating endocytosis of the Notch receptor (Hori et al., 2004), whereas human DTX1 suppresses Notch/HES1 signaling to block osteosarcoma invasiveness (Zhang et al., 2010). To probe whether DTX2 could also regulate Notch signaling, we constructed a luciferase reporter driven by the promoter of the canonical Notch target HES1 (HES1P-wt), which contains the RBP-J binding site and can be activated by the Notch intracellular domain (NICD) (Luo et al., 2017). As expected, NICD expression led to significantly higher luciferase activities in cells expressing HES1P-wt, but not HES1P-mut with the RBP-J binding site mutated (GTGGGAA to GTGAAAA) (Luo et al., 2017), confirming the specificity of the reporter for Notch activation (Figure S4B). Co-expressing full-length DTX2 with the HES1P-wt reporter, however, had little effect on the luciferase activity (Figures S4C and S4D). Furthermore, overexpression or depletion of DTX2 respectively augmented or suppressed hTERT expression but minimally impacted HES1 mRNA levels (Figures S4E and S4F). These data argue against DTX2 functioning as a regulator of canonical Notch signaling.
Next, we examined whether Notch signaling played a role in regulating hTERT transcription. When HeLa cells were treated with Dibenzazepine (DBZ), the γ-secretase inhibitor that can suppress Notch receptor release (Groth et al., 2010), HES1 mRNA level was drastically reduced, whereas DTX2 and hTERT mRNA levels as well as telomerase activities remained unchanged (Figures 5D and 5E); this is supported by studies in human endothelial cells that overexpression of NOTCH had no effect on hTERT transcription (Giunco et al., 2015). Taken together, our data indicate that DTX2 likely regulates hTERT transcription independent of canonical Notch signaling.
DTX2 interacts with NFIC to regulate hTERT transcription
All four nuclear factor I (NF-I) family members, which have been implicated in development and cancer but never in telomerase regulation (Chen et al., 2017), were identified as DTX2-associating proteins (Table S3). NFIA, NFIB, and NFIC were among the top candidates and could each co-IP with DTX2 (Figure S4G). However, only NFIC overexpression significantly upregulated hTERT mRNA level and telomerase activity in HeLa cells, and rescued the decrease in hTERT mRNA level and telomerase activity in DTX2 KO cells (Figures 5F–5I), pointing to NFIC as the likeliest family member to facilitate DTX2-mediated telomerase regulation.
NF-I proteins contain highly conserved DNA-binding and dimerization regions in the N-terminus that include the pre-N and MH1 domains, but their C-terminal CTF transcription modulation regions vary considerably (Chen et al., 2017; Marchler-Bauer et al., 2017). To test which domain is required for DTX2-NFIC interaction, we constructed FH-tagged NFIC truncation mutants with the pre-N (Δpre-N), MH1 (ΔMH1), or CTF (ΔCTF) region deleted (Figure S5A). Co-IP experiment showed that pre-N or MH1 domain deletion severely impaired interaction between NFIC and DTX2, indicating that both regions were required for efficient DTX2-NFIC interaction (Figure S5B). In reciprocal co-IP experiments using cells co-expressing full-length NFIC with full-length or truncated DTX2, DTX2-NFIC interaction was completely abolished in the absence of the DTX2 central region, but not RING or WWE domains (Figure S5C). In fact, the central region alone was sufficient to co-IP with NFIC (Figure S5D). Given the importance of the CEN domain to DTX2 nuclear targeting and function, these observations suggest that DTX2 nuclear targeting enabled its interaction with NFIC.
DTX2 increases NFIC association with the hTERT promoter to augment hTERT transcription
To further confirm the role of NFIC in DTX2-mediated telomerase regulation, we generated DTX2/NFIC double KO (DKO) HeLa cells (Figures S6A and S6B). Unlike in DTX2 KO cells, expressing DTX2 in DKO cells did not rescue hTERT mRNA level or telomerase activity (Figures 6A, 6B, and S6C). In contrast, expression of NFIC significantly upregulated the level of both hTERT mRNA and telomerase activity in DKO cells, indicating that NFIC likely functioned downstream of DTX2. When we examined DKO cells by ChIP-qPCR, we could no longer detect any enrichment of DTX2 signals at the hTERT core promoter region (Figure 6C), suggesting that NFIC was crucial to DTX2 association with the hTERT promoter.
Figure 6.

DTX2 promotes NFIC association with the hTERT promoter for hTERT transcriptional activation
(A and B) FH-tagged EGFP, DTX2, or NFIC was ectopically expressed in the DTX2/NFIC double knockout (DKO) line #3. Relative hTERT mRNA level (A) and telomerase activity (B) were assessed. Data were shown as mean ± SD (N = 3). ∗∗p< 0.01; n.s., not significant.
(C) Cells from (A) were also examined by ChIP-qPCR with an anti-HA antibody and primers for the indicated region (relative to hTERT TSS). Enrichment of the examined regions in ChIP samples was normalized to input. Data were shown as mean ± SD (N = 3). ∗∗p< 0.01; ∗∗∗p< 0.001; n.s., not significant.
(D) HeLa cells stably expressing FH-NFIC were analyzed in ChIP-qPCR with an anti-HA antibody and primers for the indicated region (relative to hTERT TSS). Results were normalized to input. Data were shown as mean ± SD (N = 3). ∗p< 0.05; ∗∗p< 0.01.
(E) In the electrophoretic mobility shift assay (EMSA), cell lysate from HEK293T cells transiently expressing FH-EGFP or FH-NFIC was incubated with biotin-labeled double-stranded DNA probes containing the flanking sequences surrounding regions −19, −295, or −389 upstream of hTERT TSS. An anti-HA antibody was used to super-shift the protein-DNA complex. Reaction mixtures were resolved on a nondenaturing PAGE gel and blotted with streptavidin. Red arrow indicates protein-DNA complex. Red star indicates super-shifted complexes.
(F–H) Dox-inducible DTX2-SFB (iDTX2-SFB) were reintroduced into DTX2 KO cells with FH-EGFP or FH-NFIC expression. Cells were cultured with −/+Dox (1 μg/mL) for 48 h before western blot (F), ChIP-qPCR analysis of indicated regions (relative to hTERT TSS) (G), and RT-qPCR for relative hTERT mRNA level (H). Data were shown as mean ± SD (N = 3). ∗∗p< 0.01; ∗∗∗p< 0.001. For all panels, p values were calculated using Student’s t test.
(I) DTX2/NFIC DKO cells stably expressing FH-tagged EGFP or NFIC were reintroduced with EGFP-DTX2 and examined by western blot using the indicated antibodies.
(J) ChIP-qPCR analysis was done using cells from (I) and anti-H3K4me3, anti-H3K9me3, and anti-H3K27me3 antibodies. Primers span areas within the hTERT core promoter region (−113/+54). Enrichment of specific regions was normalized to input. Data were shown as mean ± SD (N = 2). p values were calculated using Student’s t test. ∗∗p< 0.01; ∗∗∗p< 0.001.
(K) ChIP-qPCR analysis was done using cells from (I) and anti-Pol II-pS2 antibody. Primers span areas in the last exon of hTERT (+41,480/+41,590). Enrichment of specific regions was normalized to input. Data were shown as mean ± SD (N = 2). p values were calculated using Student’s t test. ∗∗p< 0.01. See also Figure S6.
NFI family members share identical DNA-binding motifs containing the same palindromic sequence and can bind DNA with half sites (albeit at lower affinity) (Gronostajski, 2000; Zenker et al., 2019). SELEX and ChIP-seq data from the JASPAR database (Fornes et al., 2020) indicate four potential DNA-binding motifs for NFIC, with T/CTGGC being the half-site consensus sequence (Figure S6D). Notably, three CTGGC motifs are located within the hTERT core promoter region (−389, −295, and −19 relative to ATG), which overlap with DTX2-associating regions (−476 to −242, −113 to +54) (Figure 4I). When we probed NFIC binding to the hTERT promoter by ChIP-qPCR using HeLa cells expressing FH-NFIC, we found enriched NFIC signals in the same regions bound by DTX2 (Figure 6D). Moreover, FH-NFIC bound to DNA probes containing CTGGC motifs in vitro (Figure 6E). Of the three potential binding sites, DNA probes with flanking sequences of both −19 and −389 (relative to ATG) could be bound by FH-NFIC but not FH-EGFP. More importantly, anti-HA antibodies together with DNA-bound FH-NFIC formed super-shift complexes, indicating specific and direct interaction between NFIC and the DNA probes.
To determine how DTX2 functions through NFIC, we reintroduced tetracycline-induced DTX2-SFB (iDTX2-SFB) into DTX2 KO cells and measured intensity of NFIC binding to the hTERT promoter by ChIP-qPCR. Upon Dox addition, NFIC-ChIP signals were significantly increased on the hTERT promoter (Figures 6F and 6G). Although NFIC expression alone upregulated hTERT expression in DTX2 KO cells, rescued DTX2 expression further elevated hTERT mRNA level (Figure 6H), suggesting that DTX2 might augment hTERT transcription by facilitating NFIC binding to the hTERT promoter.
To further examine whether the binding of DTX2-NFIC complex on hTERT promoter could lead to epigenetic changes, we performed ChIP-qPCR analysis in DTX2/NFIC DKO cells with re-expression of DTX2 (Figure 6I). As mentioned earlier, re-expression of DTX2 in DKO cells could not rescue hTERT transcription, whereas re-expression of NFIC could rescue this phenotype (Figure 6A). Consistently, we observed significant increase of H3K4me3 and decrease of H3K9me3 within the hTERT promoter region (Figure 6J), as well as accumulation of phosphorylated Pol II-S2 at the 3′-end of hTERT gene in the NFIC re-expression group (Figure 6K). Taken together, these data supported that DTX2-NFIC axis corporately regulates epigenetic changes in promoting hTERT transcription.
DTX2 mediates K63-linked ubiquitination of NFIC to enhance hTERT transcription
Although mammalian DTX family proteins can function as E3 ligases (Cui et al., 2012; Luo et al., 2017; Takeyama et al., 2003), very little is known about their physiological targets. To determine whether DTX2 could regulate NFIC through ubiquitination, we first carried out ubiquitination assays using HEK293T cells co-expressing HA-tagged ubiquitin (HA-Ub) with GST-NFIC and DTX2-SFB. Increased ubiquitin signals could be detected on GST-NFIC upon DTX2 expression (Figure 7A, left), indicating DTX2-dependent ubiquitination of NFIC in these cells. Two of the most common linkage types, K48- and K63-linked ubiquitination, are often associated with proteasome degradation and nonproteolytic functions, respectively (Wang et al., 2012). To understand the linkage types of DTX2-dependent ubiquitination, HA-tagged ubiquitin mutants containing only K48 or K63 were examined. With HA-Ub-K48, NFIC ubiquitination level appeared comparable between DTX2- and EGFP-expressing cells (Figure 7A, middle), suggesting involvement of other linkages not affecting NFIC stability. This is consistent with findings in iDTX2-SFB cells (Figure 6F), where FH-NFIC protein level remained unchanged upon dox-induced DTX2 expression. When HA-Ub-K63 was co-transfected with NFIC and DTX2, markedly increased NFIC ubiquitination was detected (Figure 7A, right), pointing to K63 as the primary linkage type.
Figure 7.

DTX2 mediates K63-linked ubiquitination of NFIC to enhance hTERT transcription
(A) GST-NFIC was transiently co-expressed in HEK293T cells with DTX2-SFB and HA-tagged Ub or Ub K48/K63 mutants as indicated. EGFP-SFB was used as a control. Cells were harvested for GST pull-down (PD) and western blotting as shown.
(B) GST-NFIC was transiently co-expressed in HEK293T cells with SFB-tagged DTX2 or the I/M∗ mutant as well as HA-tagged Ub or Ub K63 mutant as indicated. I/M∗, DTX2 with I414R and I457R mutations. EGFP-SFB was used as a control. Cells were harvested for GST pull-down (PD) and western blotting as shown.
(C) HA-Ub was transiently co-expressed in HEK293T cells with DTX2-SFB and GST-tagged WT or K/R point mutants of NFIC. EGFP-SFB was used as a control. Cells were harvested for GST pull-down (PD) and western blotting as shown.
(D–F) DTX2/NFIC DKO cells ectopically expressing FH-tagged WT or K/R point mutants of NFIC were examined for protein expression with an anti-Flag antibody (D), relative hTERT mRNA level by qPCR (E), and telomerase activity by qTRAP (F). Data were shown as mean ± SD (N = 3). ∗p< 0.05; ∗∗p< 0.01; n.s., not significant.
(G) ChIP-qPCR analysis was performed with an anti-HA antibody and primers for the indicated DNA regions (relative to hTERT TSS) using cells from (D). Data were shown as mean ± SD (N = 3). ∗∗p< 0.01; n.s., not significant. For panels E–G, p values were calculated using Student’s t test.
(H) In telomerase-positive cancer cells with low DTX2 expression, NFIC binds to the consensus half sites on the hTERT promoter with low affinity. Increased DTX2 expression promotes its interaction with NFIC and K63-linked ubiquitination at K65 and K99 sites, which helps stabilize the binding of the DTX2-NFIC complex to the hTERT core promoter and enhance hTERT transcription. (H) In telomerase-positive cancer cells with low DTX2 expression, NFIC binds to the consensus half sites on the hTERT promoter with low affinity. The hTERT gene remains transcription repressive with hypermethylation on H3K9. Increased DTX2 expression promotes its interaction with NFIC and K63-linked ubiquitination at K65 and K99 sites, which helps stabilize the binding of the DTX2-NFIC complex to the hTERT core promoter. The enrichment of DTX2-NFIC further upregulates H3K4me3 level and helps recruit phosphorylated Pol II-S2, therefore enhancing hTERT transcription. See also Figure S7.
Previous studies have identified key residues within the mouse Dtx2/Deltex2 RING domain (I411/M454), whose mutations blocked Dtx2 E3 ligase activity (Luo et al., 2017). When the orthologous residues were mutated in the RING domain (I/M∗, I414R/M457R) of DTX2, NFIC ubiquitination was noticeably reduced with either HA-Ub or HA-Ub-K63 (Figure 7B), indicating that the E3 ligase activity of DTX2 was essential for K63-linked ubiquitination of NFIC. In addition, re-expression of DTX2 I/M∗ could not rescue hTERT transcription in DTX2 KO cells (Figure S7A), suggesting the E3 ligase activity is required for hTERT transcription regulation. To map the NFIC domain required for ubiquitination, we co-transfected DTX2-SFB and HA-Ub into HEK293T cells with GST-tagged NFIC and its truncation mutants. Although ubiquitination of the Δpre-N and ΔCTF mutants remained efficient, ubiquitination of the ΔMH1 mutant was markedly impaired (Figure S7B), suggesting that the MH1 domain was critical to DTX2-mediated NFIC ubiquitination.
Based on in silico search using PhosphoSitePlus (Hornbeck et al., 2015) and UbiSite (Akimov et al., 2018), K to R point mutants of the five putative ubiquitin-conjugation sites on NFIC were generated. Consistently, two mutants within the MH1 domain, K65R and K99R, showed drastically reduced NFIC ubiquitination (Figure 7C), indicating these two residues as primary DTX2 ubiquitination sites. We then asked whether ubiquitination of K65/K99 was important for NFIC function by expressing single and double K-R mutants in the DKO cells (Figure 7D). Unlike wild-type (WT) NFIC, all three mutants failed to enhance hTERT mRNA level or telomerase activity and were unable to bind the hTERT promoter (Figures 7E–7G), underlining the importance of K63-linked ubiquitination of NFIC K65/K99 and the DTX2-NFIC axis to hTERT transcriptional regulation.
To examine whether DTX2-NFIC axis contributes to tumorigenesis, we performed the soft agar assays using DTX2/NFIC DKO cells with re-expression of DTX2, WT or mutant NFIC to determine their contribution to tumorigenesis. As shown in Figure S7C, HeLa cells with depletion of both DTX2 and NFIC barely formed colonies in soft agar. Although re-expression of DTX2 did not promote colony formation, re-expression of NFIC was sufficient to increase colony number in DKO cells. This is consistent with our previous data that NFIC functioned downstream of DTX2 to enhance hTERT transcription. Compared with WT NFIC, re-expression of K65R/K99R mutant led to fewer colonies, suggesting the DTX2-mediated ubiquitination of NFIC promotes tumorigenesis.
Taken together, our data support the model: in telomerase positive cancer cells with low DTX2 expression, NFIC binds to the hTERT promoter (consensus half sites) with reduced affinity. Thus, hTERT gene is embedded in the transcription repressive chromatin with high level of H3K9 tri-methylation. Heightened DTX2 expression increases DTX2-NFIC association and K63-linked ubiquitination of NFIC K65/K99, which promotes DTX2-NFIC binding to the hTERT promoter. The enrichment of DTX2-NFIC complex on hTERT promoter increases H3K4me3 and recruits Pol II with productive elongating activity, therefore enhancing hTERT transcription (Figure 7H).
Discussion
In this study, we carried out systematic and functional mapping of the hTERT transcriptional regulatory network through an inducible CRISPR/Cas9 KO screen. The inducible strategy increased the chance of identifying positive regulators whose inhibition may affect cell growth and established baseline cells to account for random drifts. The control EF1α promoter was chosen for its human origin, robust activity across different cell types, and resistance to chromatin changes (Norrman et al., 2010). The EF1α reporter cells helped exclude factors important for transcription and protein stability in general. In this screen, many uncharacterized regulators for hTERT transcription were identified, providing a comprehensive view of hTERT regulation network.
DTX2 was discovered as an important hTERT transcription regulator through mediating ubiquitination of transcription factor NFIC. The function of DTX2 was rarely known until a recent study revealed multiple DNA-damage-repair-related proteins as DTX2-interacting partners and ubiquitination substrates (Ahmed et al., 2020). Especially, one of the substrates is poly-ADP ribose polymerase 1 (PARP1), which participates in DSB repair at telomeres (Doksani and de Lange, 2016; Harvey et al., 2018). Therefore, DTX2 may help maintain telomere homeostasis through NFIC-mediated hTERT transcriptional activation and PARP1-dependent telomere maintenance.
Our findings of NFIC as an hTERT transcriptional regulator downstream of DTX2 further underline the nonredundancy of NF-I proteins, which have shown opposite activities in different model systems (Chen et al., 2017). Promoter context, tissue/cell types, and interacting partners help determine their activity potential and mode of action (Chen et al., 2017). For example, NF-I members vary in their specificities for the CoRE response element of the WAP gene, depending on the presence of other transcription factors (Mukhopadhyay et al., 2001). In our case, reintroducing DTX2 in DTX2 KO cells significantly enhanced NFIC binding to the hTERT promoter and further increased hTERT transcription, due to DTX2-mediated K63-linked ubiquitination of NFIC K65/K99. Our findings support the notion that K63-linked ubiquitination may be essential to NFIC transactivation of hTERT expression and integral to DTX2-mediated transcriptional regulation.
Both Deltex and NFI family proteins have been implicated in cancer (Chen et al., 2017). For instance, DTX2 belongs to an 18-gene panel of prognostic biomarkers whose expression is associated with poor outcomes in metastatic breast cancer (Cheng et al., 2017). Our data also indicate that DTX2 is required for anchorage-independent growth of various cancer cells (Figures. S2Q and S2R). Tumor cells often reactivate telomerase to maintain telomere length and achieve unlimited cell proliferation. Therefore, targeting telomerase represents an attractive strategy (Guterres and Villanueva, 2020). The DTX2-NFIC axis offers a new avenue of research for telomerase inhibition through blocking DTX2-NFIC interaction. Further studies should not only shed more light on the mechanisms that govern hTERT expression in normal versus cancer cells but also uncover alternative therapeutics for treating cancer.
Limitations of the study
Increased expression of DTX2 is shown in various cancer types. In this study, we observed that depletion of DTX2 suppressed cancer cell growth via downregulating hTERT transcription especially in telomerase-positive cancer with short telomeres. We revealed the mechanism how DTX2-NFIC axis regulates hTERT transcription, but we did not identify the upstream signaling. Further study is needed to understand how DTX2-NFIC axis is regulated during cancer progression.
STAR★Methods
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Flag mouse mAb | Abmart | Cat# M20008, RRID:AB_2713960 |
| β-actin rabbit mAb | ABclonal | Cat# AC004, RRID:AB_2737399 |
| GAPDH mouse mAb | Abmart | Cat# M20006, RRID:AB_2737054 |
| α-tubulin mouse mAb | Sigma | Cat# T5168, RRID:AB_477579 |
| NFIC rabbit pAb | Cell Signaling Technology | Cat# 11911, RRID:AB_2797765 |
| HA mouse mAb (IF, WB) | Sigma | Cat# H9658, RRID: AB_260092 |
| HA rabbit pAb (WB) | Sigma | Cat# H6908, RRID:AB_260070 |
| HA rabbit mAb (EMSA, ChIP) | Cell Signaling Technology | Cat# 3724, RRID:AB_1549585 |
| Lamin A/C mouse mAb | Sigma | Cat# SAB4200236, RRID:AB_10743057 |
| Histone H3 rabbit pAb | Abcam | Cat# ab1791, RRID:AB_302613 |
| Myc mouse mAb | Sigma | Cat# M4439, RRID:AB_439694 |
| Rabbit IgG | Millipore | Cat# PP64, RRID:AB_97852 |
| H3K4me3 rabbit mAb | Cell Signaling Technology | Cat#9751, RRID:AB_2616028 |
| H3K9me3 rabbit pAb | Abcam | Cat# ab8898, RRID:AB_306848 |
| H3K27me3 rabbit pAb | Active Motif | Cat# 39155, RRID:AB_2561020 |
| PolII CTD-pS2 rat mAb | Active Motif | Cat# 61083, RRID:AB_2687450 |
| Donkey Anti-Rabbit IgG (Alexa Fluor 488) | Thermo Fisher Scientific | Cat# A-21206, RRID:AB_2535792 |
| Donkey anti-Mouse IgG (Alexa Fluor 555) | Thermo Fisher Scientific | Cat# A-31570, RRID:AB_2536180 |
| IRDye 800CW Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 926-32211, RRID:AB_621843 |
| IRDye 680RD Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926-68070, RRID:AB_10956588 |
| Chemicals, peptides, and recombinant proteins | ||
| Dibenzazepine | Selleckchem | Cat# S2711 |
| MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] | Sigma | Cat# V900888 |
| Critical commercial assays | ||
| RNAiso Plus | TAKARA | Cat# 9109 |
| EZ-press RNA Purification Kit | EZBioscience | Cat# B0004D |
| PrimeScript™ RT reagent Kit with gDNA Eraser | TAKARA | Cat# RR047 |
| 2×RealStar Green Fast Mixture (qTRAP) | GenStar | Cat# A301 |
| ChamQ Universal SYBR qPCR Master Mix (qPCR) | Vazyme | Cat# Q711-02 |
| Cell Counting Kit-8 (CCK-8) | Bimake | B34304 |
| AxyPrep Blood Genomic DNA Miniprep Kit | Axygen | Cat# AP-MN-BL-GDNA |
| Dual-Luciferase® Reporter Assay System | Promega | Cat# E1910 |
| Chemiluminescent Nucleic Acid Detection Module | Thermo Fisher Scientific | Cat# 89880 |
| Streptavidin Magnetic Beads | Thermo Fisher Scientific | Cat# 88817 |
| Protein A/G Magnetic Beads | Bimake | Cat# B23201 |
| Anti-Myc magnetic beads | Bimake | B26301 |
| Glutathione Sepharose® 4B | Merck | GE17-0756-01 |
| Deposited data | ||
| TERT CRISPR/Cas9 screen data analysis | This paper | PRJNA694462 |
| DTX2 BioID MS data analysis | This paper | PXD023795 |
| Mendeley dataset: unprocessed images | This paper | https://10.17632/jty4n5rd6y.1 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | N/A |
| HeLa | ATCC | N/A |
| HT-1080 | ATCC | N/A |
| Hs578T | ATCC | N/A |
| MDA-MB-231 | ATCC | N/A |
| MCF7 | ATCC | N/A |
| DLD1 | ATCC | N/A |
| Oligonucleotides: see Table S4 | ||
| Software and algorithms | ||
| GraphPad Prism 7.00 | N/A | https://www.graphpad.com/ |
| DOG 2.0 | N/A | http://dog.biocuckoo.org/ |
| MAGeCK analysis | N/A | (Li et al., 2014) |
| ImageQuant 5.2 | N/A | https://imagequanttl.software.informer.com/ |
| Maxquant 1.6.2.10 | N/A | https://www.maxquant.org/ |
| FunRich 3.1.3 | N/A | http://www.funrich.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhou Songyang (songyanz@mail.sysu.edu.cn).
Materials availability
The knockout cell lines and plasmids generated in this study are available from the Lead Contact without restriction.
Experimental model and subject details
Cell lines and cell culture
Cell lines HEK293T (human embryonic kidney), HeLa (cervical cancer), HT-1080 (fibrosarcoma), DLD1 (colorectal adenocarcinoma), Hs578T and MDA-MB-231 (breast adenocarcinoma) were purchased from ATCC. DLD1 cells were cultured in RPMI1640 (Gibco, 11,875-093) supplemented with 10% FBS (Excell biotech, FSP500). Other cells were cultured in DMEM (Corning; 10-013-CVR) supplemented with 10% FBS. All cell lines were confirmed to be free of mycoplasma contamination.
Method details
CRISPR/Cas9-mediated inducible knockout screen
An EF1α promoter-driven EGFP expression lentiviral vector (EF1α-EGFP) served as screening control. The hTERT core promoter (the region spanning the hTERT translation start site (TSS) and 454 bp upstream of it) (Higashi et al., 2014) was amplified using genomic DNA from HeLa cells to replace the EF1α promoter upstream of EGFP (hTERT-EGFP). HeLa or HEK293T cells were first transduced with lentiviruses encoding doxycycline (Dox)-inducible Cas9 (iCas9) (Kim et al., 2017) and selected with G418 (250 μg/mL) for 10 days. A clonal derivate for each cell line isolated by limiting dilution cloning and confirmed to have moderate iCas9 expression upon induction was used for all subsequent experiments described in this study. The HeLa-iCas9 line was used for the KO screen and subsequently transduced with either hTERT-EGFP or EF1α-EGFP lentiviruses and selected with puromycin (2 μg/mL) for 3 days. GFP-positive cells that were further enriched by FACS were used as reporter cell lines.
Two sgRNA libraries (Addgene, 51,047 & 51,043) were packaged as lentiviruses, which were transduced separately into the reporter cell lines at low MOI (∼0.3) with ≥100-fold coverage. Infected cells were selected with blasticidin (10 μg/mL) for 5 days. A portion of the cells were cultured without Dox and collected as the basal group. Cells that were cultured in Dox (1 μg/mL) for 7 days went through two rounds of FACS sorting for GFP-negative cells (sorted group).
High-throughput sequencing and data analysis
Genomic DNA was extracted from both basal and sorted groups for barcode amplification of the integrated sgRNAs for high-throughput sequencing (Novogene) (see Table S4 for barcodes). Sequencing data were first segregated into each sample group based on individual barcodes. Only sgRNAs that perfectly matched reference sequences from the sgRNA libraries were counted. Only sgRNAs that satisfied the following two conditions were considered as enriched, their fold change of reads in the hTERT-EGFP sorted group was ≥2-fold over its basal group, and their fold change of reads (sorted over basal) in hTERT-EGFP cells was more than their change in EF1α-EGFP cells. We also performed MAGeCK analysis (Li et al., 2014) using the sgRNA reads of sorted and basal groups from both hTERT-EGFP and EF1α-EGFP samples to identify significantly enriched genes. The corresponding genes with p< 0.05 were assigned as enriched genes. Genes with p< 0.01 and at least two sgRNAs significantly enriched were chosen as candidates. The high-throughput sequencing data have been deposited to the NCBI BioProject database, and is available at https://dataview.ncbi.nlm.nih.gov/object/PRJNA694462?reviewer=pi472nb79brvtoe17tbhcle6fq.
Generation of KO lines using dual-sgRNA strategies
For preliminary analysis, pooled KO cells were generated using two sgRNAs targeting non-overlapping regions for each candidate gene. The sgRNAs were cloned into the LentiGuide vector with the hygromycin or blasticidin selection marker (Kim et al., 2017). Lentiviruses encoding the sgRNAs were then packaged and transduced into iCas9-expressing HEK293T or HeLa cells. Gene knockout was induced by culturing the cells in Dox (1 μg/mL) for 48 h. For each targeted allele, this dual-sgRNA strategy may result in indels (at one or both sgRNA target sites) and/or deletions (precise or imprecise) of the intervening sequences between the two sgRNA target sites.
To generate clonal KO cells, two sgRNAs targeting different exons of the same gene were cloned into px458-based (Addgene, 134,449) vectors expressing Cas9-EGFP or Cas9-mScarlet and transfected into HeLa cells using Lipofectamine 3,000 (Thermo Fisher Scientific; L3000015). EGFP and mScarlet double-positive cells were sorted by FACS and seeded into 96-well plates 48 h after transfection for clonal isolation. For KO verification, the genomic region encompassing the intervening sequences between the two sgRNA targeting sites was amplified and ligated into pEASY-Blunt Zero vectors (Transgen, CB501) for Sanger sequencing.
RNA extraction and real-time qPCR
Total RNA was extracted using RNAiso Plus (TAKARA, 9109) or the EZ-press RNA Purification Kit (EZBioscience, B0004D) and reverse transcribed with the PrimeScript RT reagent Kit with gDNA Eraser (TAKARA, RR047). To quantify gene expression, real-time PCR was conducted using the ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711-03) on a LightCycler 480II real-time PCR system (Roche). The relative level of mRNA was quantified by 2−ΔΔCt normalized to GAPDH and control group.
Detection of relative telomerase activity (qTRAP)
The qTRAP (quantitative Telomere Repeat Amplification Protocol) assay was performed as previously described (Jiang et al., 2017). Briefly, diluted cell lysate (10-100 cells/μL) was used in 10 μL reaction mix containing 50 ng of TS and ACX primers, 1 mM EGTA, and the qPCR Supermix (GenStar, A301). Serially diluted telomerase-positive control samples were used to derive a standard curve based on which relative telomerase activities of test samples were calculated.
Analysis of average telomere length
Average telomere length was analyzed using the Telomere Restriction Fragment (TRF) assay (Liu, 2011). Briefly, cells were collected at different times points for genomic DNA extraction (Axygen, AP-MN-BL-GDNA) and restriction enzyme digestion (HinfI, RsaI, and MspI). Following agarose gel electrophoresis and gel drying, the gel was hybridized to γ-32P-labeled telomere DNA probes (TTAGGG)3 and quantified on the GE Typhoon FLA 9000 Gel Imaging Scanner. ImageQuant 5.2 was used to calculate average telomere length.
CCK-8 assay
Various cell lines were infected with lentiviruses encoding DTX2 or hTERT shRNAs (see Table S4 for shRNA sequences). At 72 h after infection, cells were seeded in 96-well plates (at 1,500 cells/well in 6 plates). Cell viability was determined daily using the Cell Counting Kit-8 (Bimake, B34304) and by measuring OD450 absorbance on a Biotek plate reader.
Cell apoptosis detection
Cells were collected one week after transduction with viruses encoding DTX2 or hTERT shRNAs, and then stained with Annexin V-FITC and PI following the instructions of the TransDetect Annexin V-FITC/PI Cell Apoptosis Detection Kit (TransGen, FA101). Fluorescence signals were detected using the Beckman CytoFLEX Flow Cytometer. Annexin V+/PI− and Annexin V+/PI+ populations were defined respectively as early and late apoptotic cells.
Soft agar assay
2×DMEM supplemented with 20% FBS and 2×antibiotics were mixed with 1.2% or 0.7% Noble agar solution (1:1 ratio) to generate 0.6% or 0.35% agar/medium mix, respectively. 0.5 mL of 0.6% agar/medium mix were added to each well of 24-well plate and solidified for 1 hour. 5,000 or 10,000 cells were resuspended in 0.5 mL of 0.35% agar/medium and seeded in each well. Incubate plates at 37°C in humidified incubator for 10 to 20 days. Feed cells two times per week with cell culture medium. Stain live colonies by adding 100 μL of 5 mg/mL MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] and incubate for 2 h. Images were captured with camera.
Immunostaining and confocal microscopy
Imaging analysis was carried out as described previously (Liu et al., 2018). Briefly, cells seeded on glass slides were fixed in 4% paraformaldehyde, permeabilized with 0.5% TritonX-100, and incubated with appropriate primary and secondary antibodies (see key resource table). The nucleus was stained with Hoechst 33,342 (1 μg/mL). Cells were visualized under a confocal laser scanning microscope (Leica TCS SP5).
Cell fractionation
Cell fractionation was carried out as previously described (Zhou et al., 2018). All steps were carried out at 4°C with protease inhibitors added to all solutions. Briefly, cells were incubated in hypotonic buffer (10 mM HEPES, pH 8.0; 1.5 mM MgCl2; 10 mM KCl) for 15 min and centrifuged at 2,000 g for 5 min. The pellet was incubated in cytosol lysis buffer (hypotonic buffer plus 0.5% NP-40) for 30 min and centrifuged at 13,000 g for 10 min. The supernatant was collected as the cytosol fraction. The pellet was washed in hypotonic buffer twice and lysed in nucleus lysis buffer (10 mM HEPES, pH 8.0; 1.5 mM MgCl2; 400 mM NaCl; 0.1 mM EDTA; 20% glycerol). The nuclear lysate was sonicated at 30% Ampl for 10 cycles of 2 s on and 8 s off, and centrifuged at 13,000 g for 15 min. The supernatant was collected as the nuclear fraction.
Chromatin association assay
This assay was performed as previously described (Macfarlan et al., 2005). Briefly, ∼107 cells were resuspended in NIB buffer (15 mM Tris-HCl, pH 7.4; 60 mM KCl; 15 mM MgCl2; 15 mM NaCl; 1 mM CaCl2) plus 0.3% NP-40 and centrifuged at 600 g for 3 min. The supernatant was collected as the cytosolic extraction. The nuclei were washed with NIB buffer plus 0.6% NP-40 and centrifuged again at 600 g for 3 min, with the supernatant designated as Wash 1. The nuclei were treated with micrococcal nuclease (Thermo, EN0181) in NIB buffer for 8 min at 37°C and then centrifuged at 12,000 g for 10 min. This supernatant was designated as Wash 2 and the pellet was centrifuged in 2 mM EDTA (pH 8.0) at 12,000 g for 10 min, with the supernatant and pellet retained as soluble and insoluble chromatin fractions respectively. The presence of nucleosomes in chromatin fractions was verified by DNA electrophoresis. All fractions were subjected to SDS-PAGE and immunoblot analysis with the appropriate antibodies (see key resource table).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as previously described (Liu et al., 2017). Briefly, ∼107 cells were fixed with 1% formaldehyde and quenched with 125 mM glycine for 15 min. Cells were pelleted and washed sequentially with Solution A (10 mM HEPES pH 7.9, 1 mM EDTA, 0.5 mM EGTA, 0.25% Triton X-100) and Solution B (10 mM HEPES pH 7.9, 1mM EDTA, 0.5 mM EGTA, 200 mM NaCl) before being lysed in Solution C (0.1% sodium deoxycholate, 150 mM NaCl, 50 mM HEPES pH 7.5, 0.1% SDS, 1% Triton X-100). The lysate was sonicated for 50 cycles of 30 s on and 30 s off (Bioruptor Pico) to ensure DNA fragmented to <1,000 bp. For each ChIP, 100 μL of clarified chromatin was incubated with indicated antibodies and then protein A/G magnetic beads (Bimake, B23202). Eluted chromatin DNA was ultimately purified using the QIAquick PCR purification kit (Qiagen, 28,106) for qPCR analysis (see Table S4 for primer sequences).
BioID-mediated proximity-labeling and MS analysis
BioID-mediated proximity labeling was carried out as described (Kim et al., 2016) with minor modifications. BioID was fused to the N-terminus of full-length DTX2 and cloned into a Flag-HA-tagging lentiviral vector for expression in HeLa DTX2 KO cells. About 4×107 cells were cultured in media containing 50 μM biotin for 16 h before being collected for lysis in 3 mL of RIPA buffer (10 mM Tris-Cl, pH 7.4; 140 mM NaCl; 0.1% SDS; 1 mM EDTA; 0.5 mM EGTA; 0.1% sodium deoxycholate; 1% Triton X-100; 1× protease and phosphatase inhibitors cocktail). The lysate was sonicated at 30% Ampl for 30 cycles of 2 s on and 8 s off (Sonics Vibra-Cell; VCX130PB) and centrifuged at 13,000 g for 10 min at 4°C. The supernatant was then incubated with Streptavidin magnetic beads (Thermo, 88,816) with rotation for 2 h at 4°C. The washed beads were then prepared for mass spectrometry (Wininnovate Bio Company) on Triple TOF 6600 (SCIEX, Concord, ON, Canada).
Mass spectrometry data were collected using the information-dependent analysis (IDA) mode. The LS-MS/MS raw data (in triplicates) were processed with the Maxquant software (Cox and Mann, 2008) for peptide search using the human proteome dataset (Swiss-Prot) (downloaded from UniProt on Oct 30, 2018). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository (Ma et al., 2019) with the dataset identifier PXD023795.
Contaminates as well as proteins that appeared in only one replicate were excluded. The iBAQ (Intensity Based Absolute Quantification) value of each protein from MS data was calculated using the Maxquant software and normalized to total iBAQs in each sample to generate the fraction of total (FOT) value (Zhou et al., 2017). Proteins whose FOT values in the BioID-DTX2 group were ≥3-fold over those in the control group were designated as proximal candidates.
Dual luciferase reporter assay
The genomic region from −178 to +65 relative to the transcription start site of human HES1 (core promoter) (Luo et al., 2017) was amplified using HEK293T genomic DNA and designated as HES1P wt. To block Notch activation, the RBP-J binding site GTGGGAA in the HES1 core promoter was mutated to GTGAAAA (Luo et al., 2017) and designated as HES1P mut. These sequences were respectively cloned into pGL4.10 (Promega, E6651) to drive firefly luciferase expression. An SV40 promoter-driven Renilla luciferase vector (pRL-SV40, Promega, E2231) served as an internal control. Expression plasmids and pRL-SV40 were co-transfected with pGL4.10-HES1P wt or pGL4.10-HES1P mut into HEK293T cells. The cells were then harvested 24 h after transfection for analysis using the Dual-Luciferase Reporter Assay System (Promega; E1910) following manufacturer’s instructions.
Co-immunoprecipitation (co-IP)
HEK293T cells were transiently transfected with expression plasmids using Polyethylenimine (Polyscience, 23,966). At 24 h after transfection, cells were lysed with NETN buffer and sonicated at 30% Ampl for 5 cycles of 2 s on and 8 s off (Sonics Vibra-Cell; VCX130PB). Clear lysate was incubated with indicated antibodies for 2-4 h and protein A/G magnetic beads for 1 hour. The beads were washed with NETN buffer for 6 times and the precipitates were analyzed by immunoblotting with appropriate antibodies (see key resource table).
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described (Hellman and Fried, 2007) with minor modifications. Briefly, cells were collected 24 h after transfection and lysed in Buffer A (20 mM HEPES, pH 7.9; 10 mM NaCl; 1 mM MgCl2; 0.15 mM spermine; 10% glycerol; 0.1 mM EDTA; 0.1 mM EGTA; 0.5 mM DTT). Annealed biotin-labeled dsDNA probes were added to lysate in Buffer W (20 mM HEPES, pH 7.9; 100 mM NaCl; 80 mM KCl; 10% glycerol; 1 mM MgCl2; 0.15 mM spermine; 0.1 mM EDTA; 0.1 mM EGTA; 0.5 mM DTT) for reactions to occur. Whenever appropriate, rabbit IgG or an anti-HA rabbit mAb (CST, 3,724) was also added to the reaction. The DNA-protein complex was separated on a 5% non-denaturing PAGE gel, blotted onto nylon membranes (GE Healthcare, RPN303B), and detected using the Chemiluminescent Nucleic Acid Detection Module (Thermo, 89,880).
Ubiquitination assay
Ubiquitination assays were carried out as described (Zheng et al., 2018). HEK293T cells were transiently transfected with expression plasmids for 24 h. Cells were lysed in NETN buffer plus 0.05% SDS. The lysate was sonicated at 30% Ampl for 5 cycles of 2 s on and 8 s off (Sonics Vibra-Cell; VCX130PB) and then centrifuged at 12,000 g for 10 min at 4°C. About 1% of lysate was saved as input, and the rest was precipitated with Glutathione Sepharose 4B (GE Healthcare, 17-0756-01). The precipitates were analyzed by immunoblotting with appropriate antibodies (see key resource table).
Quantification and statistical analysis
All quantitative experiments were performed with at least three independent biological repeats unless otherwise indicated. The results were analyzed and graphed using the GraphPad Prism 7 software. All data are presented as mean ± SD Unpaired two-tailed Student’s t test (two groups) or two-way ANOVA (multiple groups) was used to compare the mean of two or more samples, with p values and sample sizes indicated in the corresponding figure legends.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2018YFA0107003 to W.M.), National Natural Science Foundation of China (NSFC 31930058 to Z.S., 32170757 to W.M., 31871479 to W.M., 81871111 to F.L., 32071433 to F.L., 32070778 to S.W.), and Guangdong Basic and Applied Basic Research Foundation (2020B1515020044 to F.L., 2019A1515011552 to S.W.). The authors thank Dr. Dan Liu for her great efforts on manuscript revision, Mengjie Deng, Yingxia Wu, and Chuying Zhang for their excellent technical support, and members of the Songyang laboratory for their insightful discussions.
Author contributions
Z.S., W.M., and S.W. conceptualized the ideas, supervised the project, and edited the manuscript. F.L. and H.H. reviewed and edited the manuscript. Z.Z. designed and performed most of the experiments and wrote the manuscript. Y.L. designed and performed the CRISPR screen, and X.X. conducted the high-throughput sequencing data analysis. H.X. excluded the Notch signaling pathway in DTX2-mediated hTERT transcription regulation. S.W. helped with the BioID data analysis. Z.H. helped with the FACS and TRF experiment. S.L. helped with the ChIP-qPCR experiment. R.L. helped with the EMSA experiment. S.J. and J.C kindly provided ubiquitin expression plasmids and shared with the experiment procedure of ubiquitination assay. All authors discussed the results.
Declaration of interests
The authors declare no competing interests.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103813
Contributor Information
Su Wu, Email: wusu5@mail.sysu.edu.cn.
Wenbin Ma, Email: mawenbin@mail.sysu.edu.cn.
Zhou Songyang, Email: songyanz@mail.sysu.edu.cn.
Supplemental information
Data and code availability
-
•
The high-throughput sequencing data of CRISPR/Cas9 screen for hTERT transcription regulators have been uploaded to SRA database with accession PRJNA694462. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with accession PXD023795. All unprocessed images have been deposited at Mendeley Data (https://10.17632/jty4n5rd6y.1) and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ahmed S.F., Buetow L., Gabrielsen M., Lilla S., Chatrin C., Sibbet G.J., Zanivan S., Huang D.T. DELTEX2 C-terminal domain recognizes and recruits ADP-ribosylated proteins for ubiquitination. Sci Adv. 2020;6 doi: 10.1126/sciadv.abc0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimov V., Barrio-Hernandez I., Hansen S.V.F., Hallenborg P., Pedersen A.K., Bekker-Jensen D.B., Puglia M., Christensen S.D.K., Vanselow J.T., Nielsen M.M., et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018;25:631–640. doi: 10.1038/s41594-018-0084-y. [DOI] [PubMed] [Google Scholar]
- Anderson S.N., Towne D.L., Burns D.J., Warrior U. A high-throughput soft agar assay for identification of anticancer compound. J. Biomol. Screen. 2007;12:938–945. doi: 10.1177/1087057107306130. [DOI] [PubMed] [Google Scholar]
- Blackburn E.H., Collins K. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn E.H., Epel E.S., Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350:1193–1198. doi: 10.1126/science.aab3389. [DOI] [PubMed] [Google Scholar]
- Celeghin A., Giunco S., Freguja R., Zangrossi M., Nalio S., Dolcetti R., De Rossi A. Short-term inhibition of TERT induces telomere length-independent cell cycle arrest and apoptotic response in EBV-immortalized and transformed B cells. Cell Death Dis. 2016;7:e2562. doi: 10.1038/cddis.2016.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.S., Lim J.W.C., Richards L.J., Bunt J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett. 2017;410:124–138. doi: 10.1016/j.canlet.2017.09.015. [DOI] [PubMed] [Google Scholar]
- Cheng S.H., Huang T.T., Cheng Y.H., Tan T.B.K., Horng C.F., Wang Y.A., Brian N.S., Shih L.S., Yu B.L. Validation of the 18-gene classifier as a prognostic biomarker of distant metastasis in breast cancer. PLoS ONE. 2017;12:e0184372. doi: 10.1371/journal.pone.0184372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cui J., Li Y., Zhu L., Liu D., Songyang Z., Wang H.Y., Wang R.F. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat. Immunol. 2012;13:387–395. doi: 10.1038/ni.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y., de Lange T. Telomere-internal double-strand breaks are repaired by homologous recombination and PARP1/Lig3-dependent end-joining. Cell Rep. 2016;17:1646–1656. doi: 10.1016/j.celrep.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranasic D., et al. Jaspar 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.C., Abe K.T., Raught B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019;48:44–54. doi: 10.1016/j.cbpa.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Giunco S., Celeghin A., Gianesin K., Dolcetti R., Indraccolo S., De Rossi A. Cross talk between EBV and telomerase: the role of TERT and NOTCH2 in the switch of latent/lytic cycle of the virus. Cell Death Dis. 2015;6:e1774. doi: 10.1038/cddis.2015.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goueli B.S., Janknecht R. Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, Ras, or Raf. Mol. Cell Biol. 2004;24:25–35. doi: 10.1128/mcb.24.1.25-35.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronostajski R.M. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000;249:31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- Groth C., Alvord W.G., Quinones O.A., Fortini M.E. Pharmacological analysis of Drosophila melanogaster gamma-secretase with respect to differential proteolysis of Notch and APP. Mol. Pharmacol. 2010;77:567–574. doi: 10.1124/mol.109.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guterres A.N., Villanueva J. Targeting telomerase for cancer therapy. Oncogene. 2020;39:5811–5824. doi: 10.1038/s41388-020-01405-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley C.B., Futcher A.B., Greider C.W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Harvey A., Mielke N., Grimstead J.W., Jones R.E., Nguyen T., Mueller M., Baird D.M., Hendrickson E.A. PARP1 is required for preserving telomeric integrity but is dispensable for A-NHEJ. Oncotarget. 2018;9:34821–34837. doi: 10.18632/oncotarget.26201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M.T., Cesare A.J., Rivera T., Karlseder J. Cell death during crisis is mediated by mitotic telomere deprotection. Nature. 2015;522:492–496. doi: 10.1038/nature14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman L.M., Fried M.G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007;2:1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi K., Hazama S., Araki A., Yoshimura K., Iizuka N., Yoshino S., Noma T., Oka M. A novel cancer vaccine strategy with combined IL-18 and HSV-TK gene therapy driven by the hTERT promoter in a murine colorectal cancer model. Int. J. Oncol. 2014;45:1412–1420. doi: 10.3892/ijo.2014.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K., Fostier M., Ito M., Fuwa T.J., Go M.J., Okano H., Baron M., Matsuno K. Drosophila deltex mediates suppressor of Hairless-independent and late-endosomal activation of Notch signaling. Development. 2004;131:5527–5537. doi: 10.1242/dev.01448. [DOI] [PubMed] [Google Scholar]
- Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S., Tang M., Xin H., Huang J. In: Telomeres and Telomerase: Methods and Protocols. Songyang Z., editor. Springer New York; 2017. Assessing telomerase activities in mammalian cells using the quantitative PCR-based telomeric repeat amplification Protocol (qTRAP) pp. 95–101. [DOI] [PubMed] [Google Scholar]
- Kim D.I., Jensen S.C., Noble K.A., Kc B., Roux K.H., Motamedchaboki K., Roux K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell. 2016;27:1188–1196. doi: 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Li F., He Q., Deng T., Xu J., Jin F., Coarfa C., Putluri N., Liu D., Songyang Z. Systematic analysis of human telomeric dysfunction using inducible telosome/shelterin CRISPR/Cas9 knockout cells. Cell Discov. 2017;3:17034. doi: 10.1038/celldisc.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.R., Hu J. SeqNLS: nuclear localization signal prediction based on frequent pattern mining and linear motif scoring. PLoS ONE. 2013;8:e76864. doi: 10.1371/journal.pone.0076864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. Analysis of average telomere length in cultured human cells. Methods Mol. Biol. 2011;735:13–19. doi: 10.1007/978-1-61779-092-8_2. [DOI] [PubMed] [Google Scholar]
- Liu F., Feng X., Ma W. Analysis of telomere proteins by chromatin immunoprecipitation (ChIP) Methods Mol. Biol. 2017;1587:205–214. doi: 10.1007/978-1-4939-6892-3_19. [DOI] [PubMed] [Google Scholar]
- Liu Y., Liu F., Cao Y., Xu H., Wu Y., Wu S., Liu D., Zhao Y., Songyang Z., Ma W. Shwachman-diamond syndrome protein SBDS maintains human telomeres by regulating telomerase recruitment. Cell Rep. 2018;22:1849–1860. doi: 10.1016/j.celrep.2018.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D., de Morree A., Boutet S., Quach N., Natu V., Rustagi A., Rando T.A. Deltex2 represses MyoD expression and inhibits myogenic differentiation by acting as a negative regulator of Jmjd1c. Proc. Natl. Acad. Sci. U S A. 2017;114:E3071–E3080. doi: 10.1073/pnas.1613592114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Yi Y., Yao Z. Growth arrest in ovarian cancer cells by hTERT inhibition short-hairpin RNA targeting human telomerase reverse transcriptase induces immediate growth inhibition but not necessarily induces apoptosis in ovarian cancer cells. Cancer Invest. 2009;27:960–970. doi: 10.3109/07357900802491451. [DOI] [PubMed] [Google Scholar]
- Ma J., Chen T., Wu S., Yang C., Bai M., Shu K., Li K., Zhang G., Jin Z., He F., et al. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47:D1211–D1217. doi: 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan T., Kutney S., Altman B., Montross R., Yu J., Chakravarti D. Human THAP7 is a chromatin-associated, histone tail-binding protein that represses transcription via recruitment of HDAC3 and nuclear hormone receptor corepressor. J. Biol. Chem. 2005;280:7346–7358. doi: 10.1074/jbc.M411675200. [DOI] [PubMed] [Google Scholar]
- Maciejowski J., de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017;18:175–186. doi: 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A., Bo Y., Han L., He J., Lanczycki C.J., Lu S., Chitsaz F., Derbyshire M.K., Geer R.C., Gonzales N.R., et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45:D200–D203. doi: 10.1093/nar/gkw1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K., Eastman D., Mitsiades T., Quinn A.M., Carcanciu M.L., Ordentlich P., Kadesch T., Artavanis-Tsakonas S. Human deltex is a conserved regulator of Notch signalling. Nat. Genet. 1998;19:74–78. doi: 10.1038/ng0598-74. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S.S., Wyszomierski S.L., Gronostajski R.M., Rosen J.M. Differential interactions of specific nuclear factor I isoforms with the glucocorticoid receptor and STAT5 in the cooperative regulation of WAP gene transcription. Mol. Cell Biol. 2001;21:6859–6869. doi: 10.1128/MCB.21.20.6859-6869.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrman K., Fischer Y., Bonnamy B., Wolfhagen Sand F., Ravassard P., Semb H. Quantitative comparison of constitutive promoters in human ES cells. PLoS ONE. 2010;5:e12413. doi: 10.1371/journal.pone.0012413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathan M., Keerthikumar S., Ang C.S., Gangoda L., Quek C.Y., Williamson N.A., Mouradov D., Sieber O.M., Simpson R.J., Salim A., et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15:2597–2601. doi: 10.1002/pmic.201400515. [DOI] [PubMed] [Google Scholar]
- Ramlee M.K., Wang J., Toh W.X., Li S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes (Basel) 2016;7 doi: 10.3390/genes7080050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roake C.M., Artandi S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020;21:384–397. doi: 10.1038/s41580-020-0234-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saretzki G., Sitte N., Merkel U., Wurm R.E., von Zglinicki T. Telomere shortening triggers a p53-dependent cell cycle arrest via accumulation of G-rich single stranded DNA fragments. Oncogene. 1999;18:5148–5158. doi: 10.1038/sj.onc.1202898. [DOI] [PubMed] [Google Scholar]
- Schultz J., Copley R.R., Doerks T., Ponting C.P., Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay J.W. Are short telomeres predictive of advanced cancer? Cancer Discov. 2013;3:1096–1098. doi: 10.1158/2159-8290.CD-13-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay J.W. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016;6:584–593. doi: 10.1158/2159-8290.CD-16-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeyama K., Aguiar R.C., Gu L., He C., Freeman G.J., Kutok J.L., Aster J.C., Shipp M.A. The BAL-binding protein BBAP and related Deltex family members exhibit ubiquitin-protein isopeptide ligase activity. J. Biol. Chem. 2003;278:21930–21937. doi: 10.1074/jbc.M301157200. [DOI] [PubMed] [Google Scholar]
- Tang Z., Li C., Kang B., Gao G., Li C., Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierbach R., Steinberg P. Automated soft agar assay for the high-throughput screening of anticancer compounds. Anal Biochem. 2009;387:318–320. doi: 10.1016/j.ab.2009.01.029. [DOI] [PubMed] [Google Scholar]
- Thul P.J., Lindskog C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018;27:233–244. doi: 10.1002/pro.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victorelli S., Passos J.F. Telomeres and cell senescence - size matters not. EBioMedicine. 2017;21:14–20. doi: 10.1016/j.ebiom.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Gao Y., Li L., Jin G., Cai Z., Chao J.I., Lin H.K. K63-linked ubiquitination in kinase activation and cancer. Front Oncol. 2012;2:5. doi: 10.3389/fonc.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Zhao Y., Hu C., Zhu J. Differential repression of human and mouse TERT genes during cell differentiation. Nucleic Acids Res. 2009;37:2618–2629. doi: 10.1093/nar/gkp125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Zhu J. Evidence for a relief of repression mechanism for activation of the human telomerase reverse transcriptase promoter. J. Biol. Chem. 2003;278:18842–18850. doi: 10.1074/jbc.M209544200. [DOI] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Susac L., Feigon J. Structural biology of telomerase. Cold Spring Harb Perspect. Biol. 2019;11 doi: 10.1101/cshperspect.a032383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinrich S.L., Pruzan R., Ma L., Ouellette M., Tesmer V.M., Holt S.E., Bodnar A.G., Lichtsteiner S., Kim N.W., Trager J.B., et al. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 1997;17:498–502. doi: 10.1038/ng1297-498. [DOI] [PubMed] [Google Scholar]
- Wright W.E., Pereira-Smith O.M., Shay J.W. Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol. Cell Biol. 1989;9:3088–3092. doi: 10.1128/mcb.9.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenker M., Bunt J., Schanze I., Schanze D., Piper M., Priolo M., Gerkes E.H., Gronostajski R.M., Richards L.J., Vogt J., et al. Variants in nuclear factor I genes influence growth and development. Am. J. Med. Genet. C Semin. Med. Genet. 2019;181:611–626. doi: 10.1002/ajmg.c.31747. [DOI] [PubMed] [Google Scholar]
- Zhang F., Cheng D., Wang S., Zhu J. Human specific regulation of the telomerase reverse transcriptase gene. Genes (Basel) 2016;7 doi: 10.3390/genes7070030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Yang Y., Nolo R., Zweidler-McKay P.A., Hughes D.P. Regulation of NOTCH signaling by reciprocal inhibition of HES1 and Deltex 1 and its role in osteosarcoma invasiveness. Oncogene. 2010;29:2916–2926. doi: 10.1038/onc.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Mar V., Zhou W., Harrington L., Robinson M.O. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999;13:2388–2399. doi: 10.1101/gad.13.18.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Liu Q., Wu Y., Ma L., Zhang Z., Liu T., Jin S., She Y., Li Y.P., Cui J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018;37 doi: 10.15252/embj.201899347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Liu M., Xia X., Gong T., Feng J., Liu W., Liu Y., Zhen B., Wang Y., Ding C., Qin J. A mouse tissue transcription factor atlas. Nat Commun. 2017;8:15089. doi: 10.1038/ncomms15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Li M., Zhang L., Zhao H., Sahin O., Chen J., Zhao J.J., Songyang Z., Yu D. Oncogenic kinase-induced PKM2 tyrosine 105 phosphorylation converts Nononcogenic PKM2 to a tumor promoter and induces cancer stem-like cells. Cancer Res. 2018;78:2248–2261. doi: 10.1158/0008-5472.CAN-17-2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Zhao Y., Wang S. Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell. 2010;1:22–32. doi: 10.1007/s13238-010-0014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel M.E., Leahy D.J., Barrick D. Structure and Notch receptor binding of the tandem WWE domain of Deltex. Structure. 2005;13:1599–1611. doi: 10.1016/j.str.2005.07.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The high-throughput sequencing data of CRISPR/Cas9 screen for hTERT transcription regulators have been uploaded to SRA database with accession PRJNA694462. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with accession PXD023795. All unprocessed images have been deposited at Mendeley Data (https://10.17632/jty4n5rd6y.1) and are publicly available as of the date of publication.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.