

Summary
Four types of primary cells—dermal fibroblasts, dermal microvascular endothelial cells, epidermal keratinocytes, and epidermal melanocytes—can be isolated simultaneously from a single human skin sample, without the use of xenogeneic murine feeder cells. This protocol describes the procedures for isolation of these cells from adult full-thickness skin obtained from surgical discard tissue. The cells isolated using this protocol contain stem cell populations and are competent to form functional skin tissue in three-dimensional reconstructed skin models.
For complete details on the use and execution of this profile, please refer to Supp et al. (2002), Boyce et al. (2015), Boyce et al. (2017a), Boyce et al. (2017b), and Supp et al. (2019).
Subject areas: Cell Biology, Cell culture, Cell isolation, Stem Cells, Tissue Engineering
Graphical abstract
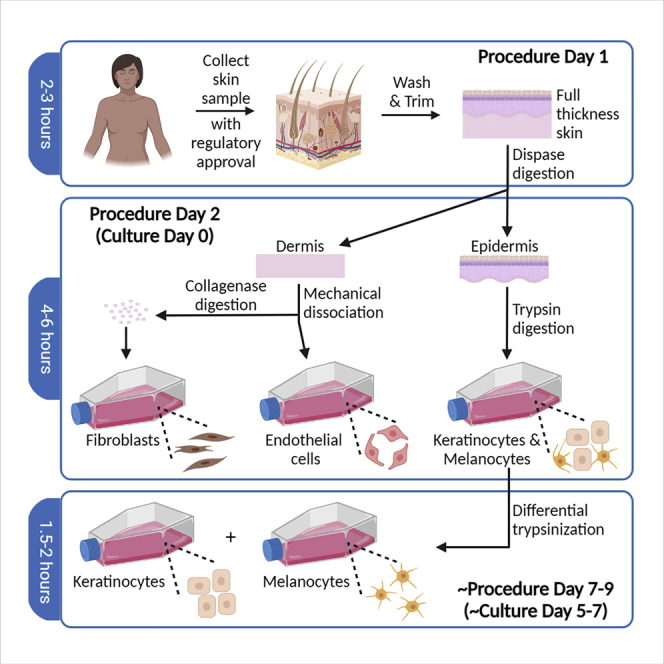
Highlights
-
•
Procedures are described for isolation of up to four cell types from human skin
-
•
A feeder cell layer for culture of primary human keratinocytes is not required
-
•
Specific media formulations and supplements are described for each cell type
-
•
Isolated cells are competent to form skin equivalents with stratified epidermal layers
Four types of primary cells—dermal fibroblasts, dermal microvascular endothelial cells, epidermal keratinocytes, and epidermal melanocytes—can be isolated simultaneously from a single human skin sample, without the use of xenogeneic murine feeder cells. This protocol describes the procedures for isolation of these cells from adult full-thickness skin obtained from surgical discard tissue. The cells isolated using this protocol contain stem cell populations and are competent to form functional skin tissue in three-dimensional reconstructed skin models.
Before you begin
Tissue sources: The procedures described here are for isolation and culture of primary cells from full-thickness human skin samples, including the establishment of primary epidermal keratinocyte cultures without the use of murine 3T3 feeder cells. This protocol can also be used to isolate cells from split-thickness (e.g., dermatomed) human skin, with modifications as indicated below. For isolation of cells from normal human skin, surgical discard tissue is often used. A common source for cell isolation is neonatal foreskin because this tissue is readily available and the cells are often highly proliferative due to the young age of the donor. However, note that skin structure varies at different anatomical sites, and foreskin is a very specialized type of skin that may not be suitable for all investigative purposes. The procedures described here have been used for isolation of cells from full thickness skin excised during breast reductions, abdominoplasties, panniculectomies, facelifts, limb and digit amputations, and circumcision (foreskin), in addition to excised keloids and hypertrophic scars, and skin from patients with epidermolysis bullosa (Hahn et al., 2013; Supp et al., 2002, 2019, 2020). It has also been used to isolate cells from split-thickness skin samples (Boyce et al., 2017b).
These procedures have been optimized for human skin and primary human cells, and may not be suitable for isolation of cells from other species. Human primary cells isolated using these procedures have been utilized for preparation of engineered skin substitutes, demonstrating that the cells retain their capacity to differentiate into stratified skin-like tissue (Supp et al., 2002, Supp et al., 2019, 2020). Further, long-term engraftment of engineered skin substitutes prepared with these primary human skin cells, in both animal models and human patients in clinical trials, demonstrates the presence of sufficient stem cell populations to support re-establishment of the epidermal stem cell niche in vivo (Boyce et al., 2002, 2006, 2017b).
Terminology: Primary cells are cultured directly from tissue and are distinct from “cell lines.” The term “cell line” generally refers to cells that have been adapted to culture and are often immortalized, and therefore may contain genetic or functional changes compared to their tissue of origin. (Hayflick, 1965; Pan et al., 2009). Primary cells can undergo a finite number of population doublings in vitro and are often referred to as “cell strains” to distinguish these primary cells from “cell lines” (Hayflick, 1965). We refer to primary human skin cells isolated from different individuals as different “donor cell strains.”
Record keeping: If you will be culturing cells from multiple donors over time, we recommend that you develop a database or other system to record and track cell inventory, with unique identifiers used for each donor cell strain and cell type. For most purposes, this database will contain de-identified information only (without Protected Health Information, or PHI, which can be used to link the cells to the donor) so that it can be accessed by most laboratory personnel. Information linking PHI to the donor cell strain, if necessary and available, should be maintained in a secure file location according to your institution’s regulations governing human subjects research. Some demographic information that may be useful for investigational purposes, including age (not date of birth, which may be regarded as PHI), race/ethnicity, sex, and body site, should be included in the database, as these variables can affect the results of your research. Consult with your institution’s Institutional Review Board (IRB) or other regulatory office to ensure that this information can be recorded for de-identified samples, because local regulations may vary.
Regulatory considerations: For the procedures described and illustrated here, approval was obtained from the University of Cincinnati IRB. The University of Cincinnati IRB determined that collection of deidentified surgical discard tissue does not constitute Human Subjects Research and is therefore exempt from the requirement to obtain informed consent from patients. Consult your institution’s IRB for regulatory guidance regarding collection of human skin from surgical discard tissue. Some institutions permit collection of de-identified tissue following protocol review, without patient consent, but this may depend on the specific content of surgical consent forms signed by patients undergoing elective surgical procedures. Many institutions require an approved IRB protocol and informed consent of patients for collection of tissue, even surgical discard tissue, used for research. Make sure that you have IRB approval, and an IRB approved protocol if required, before collecting any tissue samples from human patients. Establishing a collegial relationship with surgeons at your institution will facilitate access to tissue samples from patients.
Additional regulatory approval at your institution may be required for handling of human skin and human skin-derived primary cells in the laboratory. Because human tissues that have not been previously screened may pose a risk due to the potential for carrying transmissible pathogens, they must be handled using Universal Precautions and Biosafety Level II (BSL2) procedures. Consult your Institutional Biosafety Committee (IBC), or similar regulatory body at your institution, to determine whether a specific IBC protocol is required for use of human-derived tissues and cells.
Reagent and media preparation
General considerations
-
1.Dedicated bottles should be used for media storage.
-
a.Check glass media storage bottles frequently and discard any bottles with cracks or chips, especially around mouth and neck of bottle.
-
b.Reusable glass media storage bottles can be washed in a commercial-style dishwasher, but for best results we recommend rinsing bottles (3×) with Milli-Q water after dishwashing; let dry before autoclaving bottles to sterilize. Rinsing is important to remove chemical and other contaminants that can affect cultures, as cultured cells are very sensitive to even low levels of toxins that may be present in the media (Nims and Price, 2017).
-
a.
-
2.Ensure the quality of all reagents used for preparing media, and exercise caution while using and storing media to preserve quality.
-
a.Use fresh Milli-Q purified water (typically 18.2 MΩ at 25°C), or water of similar quality, for all components unless otherwise specified.
-
b.For best results, store all components as advised by manufacturer(s) and discard by the expiration date.
-
c.Most media formulations contain some ingredients that are light-sensitive. Bottles of media should be stored in a cold room with the light off, or in a refrigerator with a solid (not glass) door.
-
d.When working with media in a biosafety cabinet (i.e., tissue culture hood), try to work with the light off as much as possible to minimize exposure of media to light.
-
e.Warm the media in a covered water bath at 37°C for the minimum amount of time required, and do not leave bottles out on a lab bench in the light for extended periods of time.
-
f.For growth factors and other supplements added to media, we recommend preparing stock solutions and storing aliquots of these stocks frozen at -20°C to avoid multiple freeze-thaw cycles that can diminish potency.
-
g.Once supplemented (e.g., after addition of growth factors and antibiotic/antimycotic solution), the media should be used within one week for optimal performance. Although the media may be usable beyond one week, the efficacy of certain components can drop over time, particularly at 37°C, decreasing overall performance. It is better to supplement smaller volumes of media than to use for extended periods. If you do need to use over a week after supplementation, be aware that cellular growth and experimental endpoints may be affected.
-
a.
-
3.Biosafety:
-
a.Human skin samples are generally handled using Biosafety Level II (BSL2) precautions, but you should check with your institution’s Biosafety Committee to ensure compliance with all applicable regulations and safety standards.
-
b.Use personal protective equipment (PPE) and work carefully to protect both staff and cultures.
-
c.All plasticware and other materials that contact human skin or primary cells must be disposed of as biohazardous waste. Check with your institution to determine appropriate disposal measures.
-
a.
-
4.Sterile technique:
-
a.All procedures should be performed in a biological safety cabinet (i.e., tissue culture hood) using aseptic technique.
-
b.The tissue culture hood should be thoroughly decontaminated before and after use. If your hood is equipped with an ultraviolet (UV) light for decontamination purposes, turn this on and wait for at least 20 min (or as recommended by the manufacturer) prior to use.
 CRITICAL: Personnel should not be working in the hood, or in the immediate vicinity, while the UV light is on, because personal injury (especially to eyes and skin) may result from UV exposure. Do not place any reagents, tissues, or cells in the hood, or near the hood, while the UV light is on.
CRITICAL: Personnel should not be working in the hood, or in the immediate vicinity, while the UV light is on, because personal injury (especially to eyes and skin) may result from UV exposure. Do not place any reagents, tissues, or cells in the hood, or near the hood, while the UV light is on. -
c.The UV light should not be used as the sole method for decontamination. We recommend wiping all surfaces thoroughly with 70% alcohol prior to use, and for decontamination after use. Either 70% isopropanol or 70% ethanol can be used for this purpose. Additionally, wipe all bottles—especially media bottles that have been warmed in a 37°C water bath—with 70% alcohol before placing in hood.
-
d.Do not use bleach to clean stainless steel surfaces because it is corrosive and may damage surfaces. If required to use bleach on stainless steel, make sure to thoroughly rinse all contacted surfaces after use.
-
e.Instruments used for this procedure (e.g., forceps, scissors, scalpel) must be sterilized immediately before use and repeatedly throughout the procedure. A glass bead sterilizer can be used for this purpose, but make sure to wipe instruments clean before placing in the pre-heated sterilizer. Heat instruments for 10–15 s to sterilize; if left in glass beads for longer periods, the handle may become hot and can cause burns. Allow to cool before use. A sterile tissue culture dish or lid can be used for short-term storage of sterilized instruments (see Figure 2A). Alternatively, stainless steel instruments can be sterilized by briefly rinsing in 95% ethanol, followed by flaming over a Bunsen burner and cooling prior to use, if permitted by institutional regulations. However, most modern biosafety cabinets are not equipped for use of a Bunsen burner, and sterilization by ethanol and flame presents a serious risk of fire that should be avoided if another method for sterilization (such as a glass bead sterilizer) is available.
-
f.To reduce risk of contaminating cultures, use individually wrapped sterile disposable plugged pipets and change pipets frequently. Use caution to avoid touching any surfaces—including insides of bottles or sterile flasks or dishes—and never place a pipet back into a bottle of media if it has touched any surface. Avoid overfilling pipets and discard if cotton plug becomes wet with liquid due to overfilling.
-
g.When working with culture media, avoid drips of media around the threads at the necks of bottles and in bottle caps because these can facilitate contamination of the bottles’ contents.
-
h.To control microbial growth in the water bath used for warming media and thawing reagents, add 1 mL of Clear Bath (see key resources table) to each gallon of deionized water added to water bath. Alternatively, a dry bath can be used.
-
i.To control microbial growth in water pan used in incubator to provide saturated humidity, use deionized water supplemented with 5% Lysol (see key resources table).
-
j.Wear disposable gloves for all procedures.
-
k.Clean lab coats are recommended for preparation of media and reagents and are essential during work with sterile cells and materials in the tissue culture hood.
-
a.
Figure 2.

Illustration of steps involved in establishing primary cell cultures from human skin
(A) Dishes arranged in tissue culture hood for decontamination and rinsing of skin tissue, showing dishes containing 5% Dettol (whitish solution) and HBS rinses (pinkish solution).
(B) Cutting skin into strips measuring ∼2 cm × 0.25 cm after trimming and decontaminating, and prior to Dispase digestion.
(C) Separation of epidermis from dermis following overnight (∼16 h) Dispase digestion.
(D) Strips of dermis (left) and epidermis (right) after Dispase digestion and separation.
(E) Extrusion of HDMEC from dermal strips.
(F) Mincing of dermal strips prior to collagenase digestion.
(G) Suspension of minced dermis in collagenase.
(H) T150 flask containing dermal fibroblasts and dermal “pieces” following collagenase digestion, rinsing, and inoculation.
(I) Filtration of digested epidermal tissue through cell strainer following trypsin treatment.
HEPES-Buffered Saline (HBS): Combine the following components in ∼9.7 L of Milli-Q water in a large (20 L) carboy and stir well to solubilize. Adjust pH to 7.40, then bring volume to 10 L and filter sterilize using a sterile 0.2 μm Millipak filter and a peristaltic pump. Dispense into sterile polypropylene media storage bottles. Store at 4°C for up to 6 months.
Components for preparation of HBS, 10 L of 1× solution
| Component | Final concentration, mM | Amount to add for 10 L |
|---|---|---|
| HEPES | 30.00 | 71.49 g |
| Phenol red (3.3 mM stock solution)∗ | 3.30E-3 | 10 mL |
| Dextrose | 10.00 | 18.02 g |
| Potassium chloride | 3.00 | 2.24 g |
| Sodium chloride | 131.71 | 76.97 g |
| Sodium phosphate heptahydrate | 1.00 | 2.68 g |
∗To prepare 3.3 mM phenol red stock solution, dissolve 0.62 g phenol red in Milli-Q water, bring to 500 mL final volume, filter sterilize and store at room temperature (20°C–22°C) for up to one year.
0.2 M Phosphate Buffer: Combine 23.00 g Sodium Phosphate Dibasic Anhydrous (Na2HPO4) and 5.24 g Sodium Phosphate Monobasic Monohydrate (NaH2PO4) with Milli-Q water and bring to a final volume of 1 L. Filter sterilize using a sterile 0.2 μm filter and store at room temperature (20°C–22°C).
Keratinocyte Growth Medium (KGM) and Keratinocyte Growth Medium-Low Calcium (KGM-LC): If you expect that your lab will be culturing large amounts of keratinocytes on an ongoing basis, you may want to consider preparing the required media in-house. Although this is more labor-intensive than purchasing commercially prepared solutions, it can be more cost-effective if you are using very large amounts (>50 L every 2–3 months), and in our experience can produce superior results. The culture medium for primary human keratinocytes used by our laboratory, modified MCDB-153, is prepared in-house and is based on formulations initially described over two decades ago by Boyce (Boyce, 1999), Boyce and Ham (Boyce and Ham, 1985), and others (Ham and McKeehan, 1979; Peehl and Ham, 1980; Pittelkow and Scott, 1986).
For laboratories that will not be using very large volumes of media, there are multiple commercially available options that are convenient and effective. Our recommended commercial medium for culture of primary human keratinocytes is Gibco® Medium 154CF, which we have tested in the procedures described here. Medium 154CF is provided with a separate 0.2 M calcium chloride stock solution, allowing the calcium chloride concentration to be varied according to specific experimental needs. Calcium is a major regulator of keratinocyte differentiation, with higher levels promoting keratinocyte differentiation in vivo and in vitro. In skin in vivo, a gradient of calcium ions--with low levels in the basal layer and high levels in the outer, terminally differentiated layer—regulates epidermal stratification (Bikle et al., 2012). In the presence of murine 3T3 feeder cells, 0.2 mM CaCl2 is sufficiently low to favor keratinocyte proliferation vs. differentiation. However, in the absence of a feeder cell layer, we have found that reducing the CaCl2 level in P0 keratinocyte cultures to 0.06 mM CaCl2 results in a more highly proliferative population, even if cells are transitioned to 0.2 mM CaCl2 after passage to passage 1 (P1). For standard (“regular”) KGM used on culture day 0 and for keratinocytes at P1 and beyond, calcium chloride should be added to a final concentration of 0.2 mM. For KGM-LC, used for culture of P0 keratinocytes (culture day 1 through end of P0 culture), calcium chloride should be added to a final concentration of 0.06 mM. Note that there are other sources of calcium in the non-supplemented medium, contributing ∼0.5–0.8 μM to the final overall calcium concentration.
Note: Whether preparing KGM with “regular” calcium chloride (0.2 mM) or KGM-LC (0.06 mM), the same supplements are used, as listed in the table below. Alternatively, a mixture of supplements can be purchased for use: Gibco™ Human Keratinocyte Growth Supplement (HKGS; see key resources table). Although the components of the commercial supplement vary slightly from the individual components listed here, comparable results can be obtained using the commercial supplement mixture. Note, however, that laboratories routinely culturing keratinocytes may realize a cost savings by purchasing the components individually. We recommend preparing stock solutions of the following supplements at the concentrations shown in the table below, using sterile solutions and following manufacturers’ instructions for reconstitution of solid reagents. These can be stored in sterile, disposable polypropylene tubes in aliquots (0.5, 1.0, or 5 mL) at −20°C, which can be thawed in a 37°C water bath to supplement 0.5 L of Medium-154CF at a time for preparation of KGM. Once supplemented, KGM should be used within one week for best results and most consistent performance. Store KGM at 4°C in the dark.
Supplements for 500 mL Medium 154CF for keratinocyte growth medium (KGM) and keratinocyte growth medium-low calcium (KGM-LC)
| Supplement | Stock concentration and solvent | Final concentration | Volume/500 mL medium |
|---|---|---|---|
| Calcium chloride for KGM | 0.2 M∗ in sterile H2O | 0.20 mM | 0.5 mL |
| Calcium chloride for KGM-LC | 0.2 M∗ in sterile H2O | 0.06 mM | 0.15 mL |
| Epidermal growth factor (EGF) | 1 μg/mL in sterile HBS | 1 ng/mL | 0.5 mL |
| Hydrocortisone | 0.5 mg/mL in 100% ethanol | 0.5 μg/mL | 0.5 mL |
| Human insulin | 5 mg/mL in sterile 10 mM HCl | 5 μg/mL | 0.5 mL |
| Bovine pituitary extract (BPE)∗∗ | 100% | 0.2% | 1 mL |
| Penicillin/Streptomycin/Fungizone (PSF) | 100× | 1× | 5 mL |
∗The calcium chloride 0.2M stock solution is provided with purchase of Medium 154CF, or can be made in-house.
∗∗We recommend centrifuging BPE to remove particulates prior to freezing aliquots to use in supplementing media. While not a required step, this will significantly reduce potential for debris that causes clouding of media (which could otherwise be confused with microbial contamination). Centrifuge for 20 min at 16,000 rpm (∼20,000 x g), 4°C, then sterile filter through a 0.45 μm filter, then finally a 0.2 μm filter, prior to freezing aliquots at -80°C.
Melanocyte Growth Medium (MGM): MGM is prepared using the same base medium used for keratinocytes (Medium 154CF with “regular” calcium chloride concentration, 0.2 mM). The supplements for culture of melanocytes are shown in the table, below. As with keratinocyte supplements, we recommend preparing stock solutions, following manufacturers’ recommendations for reconstitution of solid reagents, and storing aliquots at -20°C. These can be thawed to supplement 0.5 L of melanocyte medium at a time. MGM should be stored at 4°C in the dark and used within one week of supplementation for best and most consistent performance.
Supplements for 500 mL Melanocyte Growth Medium (MGM)
| Supplement | Stock concentration and solvent | Final concentration | Volume/500 mL medium |
|---|---|---|---|
| Bovine pituitary extract (BPE)∗∗ | 100% | 0.4% | 2 mL |
| Hydrocortisone | 0.5 mg/mL in 100% ethanol | 0.5 μg/mL | 0.5 mL |
| Human insulin | 5 mg/mL in sterile 10 mM HCl | 5 μg/mL | 0.5 mL |
| Triiodothyronine | 20 nM in sterile HBS | 20 pM | 0.5 mL |
| α-Melanocyte stimulating hormone (αMSH) | 1 μM in sterile HBS | 1 nM | 0.5 mL |
| Endothelin 1 | 0.5 μM in sterile HBS | 0.5 nM | 0.5 mL |
| Basic fibroblast growth factor (bFGF) | 1 μg/mL in 0.1% Bovine Serum Albumin (BSA) in Tris buffer pH 7.6 | 1 ng/mL | 0.5 mL |
| Penicillin/Streptomycin/Fungizone (PSF) | 100× | 1× | 5 mL |
∗∗See previous table for recommended centrifugation of BPE.
Fibroblast Growth Medium (FGM): For culture of fibroblasts, the base medium consists of Dulbecco’s Modified Eagle’s Medium (DMEM), low glucose with pyruvate. Although this media can usually be purchased in liquid form, it is simple and more cost-effective to prepare from powder. Dissolve DMEM powder in the appropriate amount of Milli-Q water, leaving some room for addition of sodium bicarbonate and adjusting pH. For each liter of DMEM, add 1.85 g of sodium bicarbonate and mix well. Adjust the pH to 7.15 and bring up to final volume with Milli-Q water. Filter sterilize through a sterile 0.2 μm Millipak filter using a peristaltic pump into sterile polypropylene or glass media storage bottles. Store at 4°C, protected from light, and use within 6 months.
Note: Supplements for use with DMEM to prepare FGM are listed in the table below. The combination of supplements used in FGM was developed to promote rapid in vitro proliferation of fibroblasts while still supporting their ability to contribute to morphogenesis of engineered skin substitutes upon 3-dimensional organotypic culture (Boyce et al., 2017b). Previous studies demonstrated that fibroblasts cultured in FGM display higher proliferation rates and lower expression of collagen type I genes than fibroblasts cultured in DMEM + 10% FBS, and fibroblasts cultured in FGM can produce and remodel extracellular matrix and promote epidermal stratification in organotypic culture (Boyce et al., 2017b; McFarland et al., 2011). We recommend preparing stock solutions of these supplements at the concentrations shown in the table below, following manufacturers’ instructions for reconstitution of solid reagents. These stock solutions can be stored in aliquots (1, 10, or 40 mL) at -20°C, and can be thawed as needed to supplement 1 L of DMEM at a time. Once supplemented, FGM should be used stored at 4°C in the dark and used within one week for best results.
Supplements for 1 L DMEM for fibroblast growth medium (FGM)
| Supplement | Stock concentration and solvent | Final concentration | Volume/1 liter medium |
|---|---|---|---|
| Epidermal growth factor (EGF) | 10 μg/mL in sterile HBS | 10 ng/mL | 1 mL |
| Hydrocortisone | 0.5 mg/mL in 100% ethanol | 0.5 μg/mL | 1 mL |
| Human insulin | 5 mg/mL in 10mM HCl | 5 μg/mL | 1 mL |
| Ascorbic acid 2-phosphate (AA2P) | 100 mM in sterile HBS | 0.1 mM | 1 mL |
| Fetal bovine serum (FBS) | 100% | 4.0% | 40 mL |
| Penicillin/Streptomycin/Fungizone (PSF) | 100× | 1× | 10 mL |
Endothelial Cell Growth Medium (ECGM): Medium 131 (commercially available; see key resources table) is our recommended base medium for human dermal microvascular endothelial cell (HDMEC) culture. It can be purchased as a sterile liquid, but it is also available (and more cost-effective) when purchased in powdered form (with L-glutamine). Prepare in 10 L batches following the manufacturer’s instructions and supplement with 1.18 g/L sodium bicarbonate. Adjust the pH to 7.3 and bring up to final volume with Milli-Q water. Filter sterilize into sterile polypropylene or glass media storage bottles, and store at 4°C protected from light for up to 3 months.
Note: To supplement Medium 131 for preparation of ECGM for culture of HDMECs, a cocktail containing all required supplements, called Microvascular Growth Supplement (MVGS), can be purchased from a commercial vendor (see key resources table). However, for laboratories that will be regularly culturing HDMECs, preparing and using individual supplement stock solutions is more cost-effective. The components of MVGS are listed in the table below, with recommended stock solution concentrations and required amounts for supplementation of 500 mL of Medium 131 to prepare ECGM. These can be prepared individually and stored as frozen stocks. Once supplemented, store ECGM at 4°C in the dark and use within one week for best results.
Supplements for 500 mL Medium 131 for endothelial cell growth medium (ECGM)
| Supplement | Stock concentration and solvent | Final concentration | Volume/500 mL medium |
|---|---|---|---|
| Fetal bovine serum | 100% | 5% | 25.0 mL |
| Basic fibroblast growth factor (bFGF) | 3 μg/mL in 0.1% BSA in Tris buffer pH 7.6 | 3 ng/mL | 0.5 mL |
| Hydrocortisone | 0.5 mg/mL in 100% ethanol | 1μg/mL | 1.0 mL |
| Epidermal growth factor (EGF) | 1 μg/mL in sterile HBS | 1 ng/mL | 0.5 mL |
| Heparin | 10,000 U/mL in sterile HBS | 10 U/mL | 0.5 mL |
| Dibutyryl cyclic AMP | 80 mM in sterile HBS | 0.08 mM | 0.5 mL |
| Penicillin/Streptomycin/Fungizone (PSF) | 100× | 1× | 10 mL |
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD31 antibody (mouse monoclonal) | BD Pharmingen | Cat#555444; RRID: AB_395837 |
| Anti-Cytokeratin 14 antibody (rabbit monoclonal) | Novus Biologicals | Cat#NBP2-67585; RRID: AB_2809501 |
| Anti-TYRP1 antibody (mouse monoclonal) | BioLegend | Cat#SIG-38150; RRID: AB_10175227 |
| Anti-Vimentin antibody (rabbit polyclonal) | Abcepta | Cat#AP2739a; RRID: AB_2216120 |
| Donkey-anti-Mouse IgG (H+L), Alexa Fluor 594 | Fisher Scientific | Cat#A21203; RRID: AB_141633 |
| Donkey-anti-Rabbit IgG (H+L), Alexa Fluor 594 | Fisher Scientific | Cat#A21207; RRID: AB_141637 |
| Chemicals, peptides, and recombinant proteins | ||
| HEPES | Sigma-Aldrich | Cat#H4034 |
| Dextrose | Fisher Scientific | Cat#D16-500 |
| Potassium chloride | Fisher Scientific | Cat#P217 |
| Sodium chloride | Fisher Scientific | Cat#S271 |
| Sodium phosphate dibasic anhydrous | Fisher Scientific | Cat#S375 |
| Sodium phosphate monobasic monohydrate | Fisher Scientific | Cat#S468 |
| Calcium chloride dihydrate | Fisher Scientific | Cat#C79 |
| Sodium bicarbonate | Fisher Scientific | Cat#S233 |
| Gibco™ Medium 154CF, Kit | Fisher Scientific | Cat#M154CF500 |
| Gibco™ Human Keratinocyte Growth Supplement | Fisher Scientific | Cat#S0015 |
| DMEM, powder, low glucose, pyruvate | Fisher Scientific | Cat#31-600-091 |
| Gibco Fetal Bovine Serum, Certified | Fisher Scientific | Cat#16-000-044 |
| Medium 131 | Sigma-Aldrich | Cat#M8537-10L |
| Microvascular Growth Supplement (MVGS) | Fisher Scientific | Cat#S00525 |
| Attachment Factor | Fisher Scientific | Cat#S006100 |
| Epidermal growth factor (EGF), recombinant human | PeproTech | Cat#AF-100-15 |
| Hydrocortisone | Sigma-Aldrich | Cat#H4001 |
| Insulin, recombinant human | Sigma-Aldrich | Cat#I2643 |
| Bovine pituitary extract | Hammond Cell Tech | Cat#1078-NZ |
| Penicillin/Streptomycin/Fungizone (PSF); Gibco Antibiotic-Antimycotic (100×) | Fisher Scientific | Cat#15-240-096 |
| Triiodothyronine | Sigma-Aldrich | Cat#T5516 |
| α-Melanocyte stimulating hormone (αMSH) | Sigma-Aldrich | Cat#M4135 |
| Endothelin 1 | Sigma-Aldrich | Cat#E7764 |
| Basic fibroblast growth factor (bFGF) | PeproTech | Cat#100-18B |
| Ascorbic acid 2-phosphate (AA2P) | Sigma-Aldrich | Cat#A8960 |
| Collagen I-Cell Culture Surface Coating Kit | ScienCell Research Laboratories | Cat#8188 |
| Trypsin | Sigma-Aldrich | Cat#T8003 |
| Ethylenediaminetetraacetic acid (EDTA), anhydrous | Sigma-Aldrich | Cat#EDS |
| 0.5 M EDTA solution | Sigma-Aldrich | Cat#E7889 |
| Dettol Liquid First Aid Antiseptic (4.8% Chloroxylenol) | Reckitt Benckiser | Cat#PL 44/5011 |
| Dispase II | Fisher Scientific | Cat#04942078001 |
| Collagenase, Type I | Worthington Biochemical Corp. | Cat#LS004196 |
| Lysol Disinfectant Solution | Andwin Scientific | Cat#21899-598 |
| Dimethyl sulfoxide (DMSO) | Sigma Aldrich | Cat#D2650 |
| Clear Bath | Spectrum Chemical | Cat#105540 |
| Other | ||
| Vectashield Plus Mounting Medium with DAPI | Vector Laboratories | Cat#H-2000 |
| Weck-Prep Blades (carbon steel razor blades) | Fisher Scientific | Cat#50-949-469 |
| Sterile 0.22 μm Millipak Disposable Filter Units | Fisher Scientific | Cat#MPGL06GH2 |
| PYREX™ Reusable Media Storage Bottles | Fisher Scientific | Cat#06-414-1D |
| Corning Centrifuge Tube Top Filters, 0.22 μm | Fisher Scientific | Cat#09-761-34 |
| Corning Centrifuge Tube Top Filters, 0.45 μm | Fisher Scientific | Cat#09-761-35 |
| Falcon 70 μm Cell Strainer | Fisher Scientific | Cat#08-771-2 |
| Cryovials, 2 mL | Fisher Scientific | Cat#03-374-059 |
| 50 mL Conical tubes (polypropylene) | Fisher Scientific | Cat#06-443-21 |
| pH Meter | ||
| Magnetic stir plate(s) | ||
| Water purification system, Milli-Q, Synthesis A10 or similar | ||
| Autoclave | ||
| Nalgene Polypropylene media storage bottles (optional) | United States Plastic Corp. | Cat#73107 |
| Analytical balance and weigh boats | ||
| Peristaltic pump for high volume liquid filtration | ||
| Biosafety cabinet (tissue culture hood; Class II type A2) | ||
| Phase-contrast microscope | ||
| Tissue-culture treated flasks (T75, T150, and T225 sizes) | Corning | Cat#431082 (T225 size) |
| Serological pipets: sterile, individually wrapped, sizes ranging from 2 mL–50 mL | ||
| Aspirating pipets: sterile, individually wrapped plastic or sterilized glass Pasteur pipets | ||
| Forceps: fine tip and blunt tip | ||
| Hemostat/clamp | ||
| Curved scissors | ||
| Sterile scalpel blades and handle | ||
| Glass bead sterilizer; e.g., Braintree Scientific Germinator 500 | Fisher Scientific | Cat#NC9956482 |
| Aspirating flask with long tubing, for liquid waste | ||
| Refrigerated centrifuge for spinning 50 mL conical tubes | ||
| Water-jacketed CO2 incubator with saturated humidity (water pan containing deionized water with 5% Lysol) set to 37°C, 5% CO2 | ||
| Water bath at 37°C for thawing/warming media & reagents | ||
Step-by-step method details
Day 1 of procedure (culture day -1)
Timing: 2–3 h
In the following steps, full thickness skin tissue will be decontaminated and trimmed, and enzyme digestion for separation of epidermis from dermis will be initiated prior to overnight (∼16 h) incubation.
-
1.Preparations before starting:
-
a.Prepare vacuum aspirator by adding bleach (or other disinfectant); volume should be sufficient to decontaminate liquid waste if vessel is filled (∼10% final concentration).
-
b.Decontaminate tissue culture hood.
-
c.Preheat glass bead sterilizer.
-
d.Prepare or thaw Dispase: Prepare a solution at a concentration of 2.4 U/mL in HBS. Note that activity can vary from lot-to-lot but is usually >0.5 units/mg. Use a minimum of 1 mL Dispase/cm2 of tissue. Filter sterilize the solution; this can be made in large batches and stored in aliquots (35 mL each) frozen at -20°C. Thaw at 37°C and store on ice until used in procedure.
-
e.Dettol First Aid Antiseptic Liquid: A 5% solution in Milli-Q water should be prepared and filter sterilized before use. Note that Dettol is a brown liquid that will appear milky after dilution. This can be stored at room temperature (20°C–22°C) after filter sterilization.
-
f.Warm HBS in 37°C water bath prior to use.
-
g.Collect tissue and transport to laboratory:
-
i.The procedure described below is intended for ∼20 cm2 full thickness human skin. The volume of specific reagents can be scaled down or up, as needed, for different size pieces of skin.
-
ii.Pieces of skin up to ∼35 cm2 can be processed as described here, although pieces measuring 10–20 cm2 in area should provide a good yield of cells for most purposes. For larger pieces, we recommend subdividing tissue and performing multiple isolations in parallel, as pieces of skin >35 cm2 are too large for the enzyme volumes described below.
-
iii.Although we have used this protocol to isolate cells from biopsies as small as 4 mm2 (with adjustments to reagent volumes), we recommend practicing with larger tissue pieces before attempting cell isolation from very small biopsies.
-
iv.The procedure can be used on skin that has been dermatomed (split thickness skin) by modifying the Dispase and Collagenase digestion steps, as detailed below. Note that initial procedures for split-thickness skin can be accomplished in a single day versus two days for full thickness skin. For isolation of HDMECs, full thickness skin is recommended for best results.
-
v.Freshly excised human skin should be used as soon as possible for optimal results. If immediate use is not possible, skin can be stored in a buffer solution (e.g., saline or HBS, preferably chilled to 4°C) for up to 1–2 h with minimal loss of cell viability. For longer storage (up to 24 h) skin should first be rinsed to remove as much blood as possible and stored in KGM at 4°C. If overnight storage is required (∼16 h), we recommend trimming excess subcutaneous tissue prior to storage to improve dermal and epidermal tissue preservation and enhance cell viability.
-
i.
-
a.
-
2.Decontamination and trimming skin tissue:
-
a.Cut the skin to desired size, and remove any necrotic tissue, if necessary. Any tissue showing signs of degradation or loss of structural integrity should be removed. Rinse the skin sample 2× with 20 mL HBS in a 100 mm dish (or other suitable vessel) to remove any residual blood products (Figures 1A and 1B).
-
b.Remove as much subcutaneous fat as possible. To do this, place the skin sample in a dish with the fat side facing up (Figure 1B). Add a small amount of HBS to the dish to prevent the tissue from drying out and help maintain skin viability while trimming (Figure 1C). Grasp the fat with fine tip forceps, and cut fat away with curved scissors (Figure 1C). Discard the fat. When subcutaneous fat is removed, the dermis will appear whitish in color (Figure 1D). Note: this will not be necessary for split thickness (dermatomed) skin or neonatal foreskin. If curved scissors are not available, other types of scissors or a scalpel may be used, but use care to remove only subcutaneous tissue and not dermal tissue or yield of dermal cell populations, especially HDMEC, may be reduced.
-
c.If desired, collect biopsies for histology or other uses (e.g., RNA isolation).
-
d.To decontaminate the tissue, two brief (15–20 s) rinses in 5% Dettol liquid antiseptic (20 mL/rinse) will be performed. To facilitate the process and control the length of time in Dettol, set up 6 100 mm dishes in the hood: 2 containing 20 mL 5% Dettol, and 4 containing 20 mL HBS (Figure 2A). Wash the skin quickly (∼15 s) by placing in a dish with Dettol and manipulating the tissue using sterile forceps so that all surfaces are exposed to Dettol. Remove from Dettol and rinse the skin twice for about 30 s each in HBS by moving to fresh dishes, manipulating the skin at each rinse to ensure all surfaces are exposed to HBS. Repeat the Dettol and two HBS rinses, and place skin in a clean, sterile 100 mm dish with 10 mL HBS. Aspirate to remove HBS and Dettol from used dishes, and discard dishes.CRITICAL: Do not leave in Dettol solution longer than recommended or cell yield and viability may be reduced. Make sure to rinse thoroughly with HBS to remove all traces of Dettol before proceeding.
-
e.Measure and record area of skin sample.
-
a.
-
3.Dispase digestion:
-
a.Place skin in a dish with ∼10 mL HBS and cut the skin sample into strips measuring ∼2 cm × 0.25 cm (Figure 2B). To simplify these cuts, clamp a razor blade (Weck-prep blade) in the teeth of a hemostat, and place the blade edge on the skin, perpendicular to the surface. The tissue will curl less if it is placed dermis side down during cutting. Cut strips by running a sterile scalpel blade along the edge of the razor blade (Figure 2B).
-
b.Transfer the tissue strips to a 100 mm dish containing 35 mL Dispase, place into a zippered plastic bag (to prevent spillage), and incubate overnight (∼16 h) at 4°C. Although other types of sterile containers can be used for this step, it is important for all tissue pieces to be exposed to the enzyme. For example, if using a tube instead of a dish for this incubation, the tissue pieces may settle to a small area in the bottom of the tube, hindering access of the enzyme to the tissue. If a tube is used instead of a dish, laying it on its side may allow for more even distribution of the tissue pieces in the enzyme solution.
-
a.
Note: If skin is split thickness (e.g., dermatomed) or very thin (e.g., neonatal foreskin), perform Dispase digestion at 37°C for 30–120 min. Rather than floating the tissue strips in Dispase in the 100 mm dish, place two layers of sterile cotton gauze in the dish and saturate with Dispase. Lay the tissue strips on the gauze with dermis side down, and incubate at 37°C for 30 min. Split-thickness skin tends to curl on itself more than full-thickness skin; laying the thin tissue pieces on a saturated gauze pad will help prevent this curling and ensure access of the enzyme to the tissue. Check for separation of epidermis from dermis after 30 min, and if epidermis is still tightly adhered, continue incubation at 37°C, checking every 30 min up to 2 h. Once epidermis easily separates from dermis, immediately move to the next steps in the procedure.
Figure 1.

Trimming human skin in preparation for primary cell isolation
(A) Abdominal skin with subcutaneous fat, collected from elective panniculectomy.
(B) Appearance of skin after initial trimming from surgical discard tissue, showing subcutaneous fat layer.
(C) Technique for removing subcutaneous fat using curved scissors.
(D) Skin after removal of subcutaneous fat layer, showing whitish appearance of dermal tissue. Size after trimming is ∼20 cm2.
Day 2 of procedure (culture day 0)
During procedures performed on day 2, primary cultures of HDMEC, fibroblasts, and keratinocytes (with melanocytes) will be isolated from digested tissue samples. This is considered culture day 0 for all four cell types.
-
4.Preparations before starting:
-
a.Prepare aspirator by adding bleach (or other disinfectant); volume should be sufficient to decontaminate liquid waste if vessel is filled (∼10% final concentration).
-
b.Decontaminate tissue culture hood. Pre-heat glass bead sterilizer. Turn on centrifuge and pre-cool to 4°C.
-
c.Prepare or thaw Trypsin/EDTA solution: Prepare a solution containing 0.025% Trypsin + 0.02% EDTA in HBS and filter sterilize. Store in aliquots at -20 C until use. Thaw (≥1 mL per cm2 of skin) at 37°C for the minimum time required and store on ice until needed in the procedure. Warm to 37°C immediately before use.Note: Extended incubation of trypsin at 37°C can lead to loss of enzyme activity. We recommend storing on ice after thawing and warming to 37°C for the minimum amount of time required, just prior to use.
-
d.Prepare Collagenase: Prepare a solution containing 625 U/mL in 30 mL KGM + 5% BPE (30 mL is enough for one primary culture, in most cases). BPE is added to neutralize trypsin activity in the collagenase enzyme preparation (Boyce, 1999). There can be lot-to-lot variability in collagenase activity, although it is usually >125 U/mg dry weight. We recommend storing aliquots of pre-weighed collagenase in snap-cap or screw-cap glass vials in a desiccator at 4°C for convenience. To determine the amount of collagenase (in mg) for each aliquot, use the following formula:To prepare a working collagenase solution, combine 28.5 mL KGM + 1.5 mL BPE in a 50 mL tube, and add small amounts to glass vial with collagenase to dissolve. Bring volume to 30 mL and filter sterilize using 0.22 μm 50 mL tube-top filter (or other suitable filter) and store on ice until ready to use. The estimated volume needed is 1.5 mL collagenase/cm2 skin.
-
e.Prepare FGM (for fibroblasts), “regular” calcium (200 μM) KGM (for keratinocytes), and ECGM (for endothelial cells). Warm to 37°C in water bath before use, protecting from light.Note: KGM-LC (“low calcium,” 0.06 mM CaCl2) is used for feeding primary keratinocytes prior to passage (P0), but KGM with “regular” calcium (0.2 mM) is used during the initial isolation procedure (culture day 0) and the first overnight incubation after inoculation of flasks. After passaging (P1 and beyond), KGM with “regular” calcium (0.2 mM) will be used.
-
f.Prepare collagen-coated flasks for keratinocytes: Coat T225 flasks (∼1 per ∼10 cm2 of skin) with collagen I using the Collagen I-Cell Culture Surface Coating Kit according to manufacturer’s instructions with minor modifications. Dilute the 100× Collagen I Solvent provided with the kit 1:100 with sterile Milli-Q water and store at 4°C. To coat one T225 flask (225 cm2), dilute collagen I solution (1 mg/mL) 1:10 in 1× Collagen I Solvent and add 11.25 mL per flask (5 μg/cm2). Incubate for a minimum of 1 h at 37°C. Aspirate the collagen I solution and rinse each flask 3 times with 10 mL HBS. To use immediately, add 45 mL KGM to flask and pre-equilibrate in incubator at 37°C for at least 30 min. If not used immediately, air dry the flask in the hood and store at 4°C.Note: For best results, equilibrate all flasks with media in a 37°C, 5% CO2 incubator for ≥30 min before inoculation of cells. We recommend this during the primary procedure and for all later passages.
-
g.Prepare T150 flasks for fibroblast inoculation (1 per ∼10 cm2 of skin). Add 10 mL FGM to each flask and equilibrate in incubator ≥30 min before use.
-
h.Prepare Attachment Factor-coated flask(s) for HDMECs according to manufacturer’s instructions. Note that Attachment Factor is a 0.1% gelatin solution; a sterile solution of 0.1% gelatin in water can be used instead of the pre-made product. Coat T75 flask(s) (1 per ∼20 cm2 of skin) with 3–4 mL Attachment Factor: rock vigorously to completely cover growth surface and incubate in sealed flask for 30 min at 37°C or at least 2 h at room temperature (20°C–22°C). Aspirate liquid (do not rinse) and store flask tightly capped in hood at room temperature (20°C–22°C) for up to 24 h. Before use, add 15 mL ECGM per flask and equilibrate in incubator for ≥30 min before inoculating cells.
-
i.Prepare 100 mL KGM + 10% FBS (trypsin/EDTA “stop” solution) and store on ice until ready to use.
-
a.
-
5.Separate epidermis from dermis:
-
a.Remove tissue strips from Dispase and place in 100 mm dish containing 10 mL HBS. Prepare two additional 100 mm dishes with 20 mL HBS each; label for “epidermis” and “dermis”.
-
b.Separate dermis from epidermis. To do this, grasp the dermis with fine tipped forceps, grasp the epidermis with blunt forceps, and peel off epidermis (troubleshooting 2; Figure 2C). Place epidermal strips in a fresh dish (labeled “epidermis”) with 20 mL HBS (Figures 2C and 2D).
-
c.Place dish with epidermal strips in a plastic zippered bag to avoid spillage, and store in refrigerator or cold room (4°C) until ready to process for keratinocyte isolation. The epidermal strips can be stored for several hours, while the dermal strips are processed for isolation of HDMEC and fibroblasts, but do not store overnight. Keratinocytes should be isolated the same day to preserve cell viability.
-
a.
-
6.Isolation of HDMEC from dermis:
-
a.Mechanical force will be used to extrude HDMEC from vessels in the dermis. To do this, you will compress the tissue down the length of each dermal strip by squeezing with forceps, using pressure to force the endothelial cells out from vessels (imagine flattening a tube of toothpaste from the bottom up to get out the last bits of toothpaste). Grasp each dermal strip at one end with forceps, and use blunt forceps to squeeze the dermal strip near that end. Then, run the blunt forceps down the length of the strip, squeezing gently, to extrude the endothelial cells (Figure 2E and Methods video S1). Repeat ten times for each strip, then transfer strip to fresh dish with 20 mL HBS (labeled “dermis”) for subsequent isolation of fibroblasts.Note: This step must be done carefully because too much force will result in contamination of the HDMEC with fibroblasts, but too little force will result in low yield of endothelial cells. If large amounts of fibrous material are present after the extrusion procedure, this may indicate too much force was used. If this is observed, use less pressure for remaining strips (troubleshooting 4).
-
b.After scraping and moving all dermal strips to a new dish with fresh HBS, transfer the extruded cell suspension to a sterile 50 mL conical centrifuge tube. Rinse the dish used for extruding HDMEC several times with small amounts of ECGM, and add to conical tube.
-
c.Centrifuge for 7 min at 1000 rpm (∼250×g), 4°C, without brake.Note: Although spinning without the brake may add a small amount of time to each centrifugation step, we strongly recommend this for primary cells. Using the brake at the end of the spin can disrupt or dislodge the pellet, making it more difficult to remove the supernatant without disturbing the cells, and may cause mechanical damage to fragile primary cells.
-
d.Carefully aspirate or pipet off the supernatant. The cell pellet may be difficult to see, and/or may be loose; use care not to aspirate the cell pellet. Resuspend cell pellet in 2 mL ECGM per T75 flask. Inoculate prepared T75 flask(s) (coated with Attachment Factor and equilibrated with medium at 37°C; Day 2, step 4h) with 2 mL of the cell suspension per flask and incubate overnight (∼16 h) at 37°C, 5% CO2.Note: For isolation of HDMEC from skin pieces smaller than 10 cm2, T25 flask(s) may be used instead of T75 flasks.
-
a.
-
7.Isolation of dermal fibroblasts:
-
a.Warm collagenase (prepared in Day 2, step 4d) to 37°C immediately before use.
-
b.Carefully remove HBS from dish containing dermal strips using a 25mL pipet and using forceps to keep dermal strips away from pipet. Mince dermis into small pieces (<0.5–1 mm3). To mince the tissue, use a razor blade held by a hemostat and a scalpel (Figure 2F). Hold the blade firmly on top of the mass of dermal tissue, then slice along the edge of the razor blade with the scalpel (Figure 2F). Repeat many times, re-slicing in multiple directions, until tissue is finely minced. Try for pieces that are small enough to fit through the opening of a 50 mL pipet.
-
c.Add 10–20 mL of warm Collagenase to dish, and collect Collagenase solution and minced dermal pieces using a 50 mL (large bore) serological pipet (Figure 2G). Transfer to 50 mL conical tube, recovering as much of the minced dermis from the dish as possible.
-
d.Agitate the tissue suspension by pipetting up and down using a 50 mL serological pipet for 1–2 min.
-
e.Incubate tube with minced tissue in Collagenase for 30–120 min, depending on thickness of tissue, in a 37°C water bath. Full thickness tissue may require 60–120 min, whereas thin or dermatomed tissue may only require 30 min incubation.CRITICAL: Do not incubate minced dermal tissue in Collagenase for longer than 120 min.
-
f.Agitate the tissue (as in step 7d, above) every 15–30 min until tissue is sufficiently digested, making sure to clean the outside of the tube with 70% alcohol when moving from the water bath to the hood to avoid introducing contaminants. The tissue fragments should start to decrease in size and increase in number, the solution should become cloudy, and wisps of collagen and digested tissue may be visible. You should be able to reduce the size of the pipet from 50 mL to 25 mL as the tissue digests (troubleshooting 3).Note: To preserve cell viability, perform the minimal incubation in Collagenase required to release cells. Longer digestions may result in reduced colony forming efficiency and lower cellular growth rates. Clumps will still be visible after Collagenase digestion, particularly if full thickness skin is used. It is not necessary to digest full thickness skin in Collagenase until all clumps have dissolved. The tissue clumps will also be plated, allowing recovery of fibroblasts that migrate out of tissue pieces and onto plastic (as in an explant culture) in addition to fibroblasts released by collagenase digestion (Figure 2H).
-
g.Centrifuge at 1000 rpm (∼250×g) for 7 min at 4°C.
-
h.Carefully remove supernatant with a 50 mL pipet. The pellet will be loose, so we recommended using a pipet instead of aspirating when removing liquid to avoid disrupting the pellet.
-
i.Resuspend pellet in 30 mL FGM, and repeat steps 7G and 7H.
-
j.Resuspend pellet in FGM, using 5 mL of medium for each T150 flask to be inoculated.
-
k.Add cells to prepared flasks (pre-equilibrated with 10 mL FGM), using care to pipet the dermal cell suspension down the growth surface of the flask. Do not add additional medium to flask. Using a lower than normal volume of medium for this step improves adhesion of cells and tissue pieces to the growth surface of the flask.Note: This procedure enables recovery of fibroblasts released from collagen by Collagenase treatment, as well as fibroblasts that migrate out of remaining tissue pieces during the first few days of incubation, resulting in greater yields than either explant cultures or Collagenase digestion alone. Additionally, this allows for populations of fibroblasts more representative of the tissue as a whole, whereas explant cultures favor more migratory fibroblasts.
-
l.Label the flasks with #1 (Figure 2H) and place in incubator overnight (∼16 h).
 Pause point: If you need a short break during the procedure (1–2 h), this is an appropriate point to pause the protocol. However, do not wait overnight to isolate keratinocytes from epidermis.
Pause point: If you need a short break during the procedure (1–2 h), this is an appropriate point to pause the protocol. However, do not wait overnight to isolate keratinocytes from epidermis.
-
a.
-
8.Isolation of epidermal keratinocytes: In the steps outlined below, the strips will be incubated in trypsin with agitation, followed by two rinses/agitations in HBS to maximize recovery of cells. Between each of these incubation/agitation steps, the strips will be filtered through a sterile cell strainer and flow-through collected to recover released cells.
-
a.For convenience, set up a series of tubes containing the required volumes of solutions needed for the next several steps of the protocol; see Figure 2I for an example.
-
b.Remove dish with epidermal strips from refrigerator (step 5c, above). Transfer epidermal strips using sterile (heated & cooled) forceps to a 50 mL conical tube containing 20 mL pre-warmed trypsin/EDTA solution.
-
c.Coat the inside of a 25 mL serological pipet with KGM +10% FBS by pipetting the 10% FBS solution up and down a few times (and then fully emptying the pipet) before agitating the epidermal strips. The goal of this step is to leave a very thin coating of 10% FBS inside the pipet, which will help reduce sticking of epidermal strips on the inner surface of the pipet without significantly neutralizing the trypsin. Agitate the epidermal strips in the warm trypsin/EDTA solution for ∼5 min by repeatedly pipetting up and down using the coated 25 mL pipet.
-
d.Add 20 mL KGM + 10% FBS to the epidermal strips in trypsin/EDTA to neutralize the enzyme.
-
e.Using the coated 25 mL pipet, slowly filter the cell suspension through a 70 μm cell strainer into a clean, sterile 50 mL conical tube (Figure 2I). Handling the cell strainer in a sterile manner is simplified by clamping it in a sterilized hemostat, as shown in Figure 2I. Use care to avoid overloading the cell strainer, which can only handle a small volume at a time. Tilting the cell strainer at a ∼45 degree angle above the conical tube will allow for easier transfer and flow-through of liquid. Transfer the strained cell suspension to ice until ready to centrifuge.Note: Do not sit the cell strainer in the top of the 50 mL conical tube, although it fits and appears to be designed to be used this way. In our experience, this usually results in the cell suspension leaking out from the sides and dripping down the outside of 50 mL tube.
-
f.Using sterile (heated & cooled) forceps, transfer the epidermal strips from the cell strainer to a fresh 50 mL tube containing 20 mL HBS. Repeat agitation with a fresh 25 mL pipet, pre-coated with 10% FBS, for ∼4–5 min to release additional keratinocytes. You may notice the epidermal strips appearing thinner as cells are released.
-
g.Repeat filtration of epidermal strips through a 70 μm cell strainer into a fresh tube containing 20 mL KGM + 10% FBS. Place this tube on ice until ready to centrifuge. (Note: use a fresh cell strainer here, to avoid clogging that may occur if the strainer is re-used multiple times)
-
h.As above in step 8f, transfer the epidermal strips into a second tube containing 20 mL HBS and repeat agitation with 25 mL pipet for 4–5 min.
-
i.Repeat filtration of solution with epidermal strips a final time using a 70 μm cell strainer into a tube containing 20 mL KGM + 10% FBS.
-
j.Centrifuge all epidermal cell suspensions for 7 min at 1000 rpm (∼250×g) at 4°C.
-
k.Carefully remove the supernatants from all tubes by aspirating or pipetting, taking care not to disturb pellets. Resuspend cell pellets in cold KGM (200 μM calcium chloride) and combine into a single tube or sterile bottle. Remove an aliquot for counting, and place remaining cell suspension on ice until ready to inoculate flasks. Dilute the cell suspension 1:10 and count using a hemacytometer (Figure 3A).Note: We recommend counting using a hemacytometer so that only viable, proliferative cells are counted. These can be easily distinguished based on appearance, as illustrated in Figure 3A. Although the proliferative keratinocytes will probably not all exhibit the same shape in the hemacytometer, their size should be fairly uniform. Do not count dark or very large cells; count only refractory, uniformly sized cells.
-
l.Calculate the cell concentration in the undiluted cell suspension, and inoculate into collagen-coated T225 flasks pre-equilibrated with 45 mL KGM at 37°C, 5% CO2. A good yield of keratinocytes is ∼1 x 106 cells/cm2 of starting skin area. Inoculate into flasks at 0.5–1.0 × 105/cm2 (∼11.25–22.5 × 106 per T225 flask). Place flasks in incubator overnight (∼16 h).
-
a.
Figure 3.

Images of P0-P1 primary human keratinocytes in culture
(A) Image of P0 keratinocytes in hemacytometer, viewed through phase-contrast microscope. The 16-square area for counting is indicated by white arrows. Note that cell morphology is heterogeneous and only refractory (bright) cells of roughly uniform size should be counted. Dark looking cells and debris (black arrows) should not be counted.
(B) Human P0 keratinocytes at day 1, ∼24 h after inoculation into collagen-coated flask. Note that the cell population appears heterogeneous at this stage, with many cells appearing bright and raised from culture surface. Cells have irregular shapes, and some 2-cell colonies or dividing cells may be observed (white arrows).
(C) Human P0 keratinocytes at day 2 of culture. The cells exhibit diverse morphologies: rounded-up cells, which may be in the process of cell division (white arrow), can be seen, in addition to more flattened cells. This appearance is normal at P0 in KGM-LC. In KGM-LC, P0 keratinocytes may appear large and flat, and form loosely-associated colonies. Occasional elongated cells, most likely melanocytes, may be observed (black arrow).
(D) By culture day 6, P0 keratinocyte morphology appears more uniform, with numerous flattened, widely separated cells. A melanocyte is identified by its dendritic morphology (black arrow).
(E) P0 keratinocytes at day 7, immediately prior to melanocyte removal. Melanocytes are indicated by black arrows.
(F) Nearly confluent P0 keratinocytes at the time of harvesting for passage.
(G) P1 keratinocytes shown 48 h after inoculation.
(H) P1 keratinocytes 6 days after inoculation, at time of harvest (∼80%–85% confluent). Note tighter colonies and more uniform morphology after passage and culture in “regular” calcium (0.2 mM) KGM. Original image magnification: 10× (B–D); 4× (A, E–H).
This video briefly demonstrates the method used for mechanical extrusion of HDMEC from dermal strips after enzymatic separation of dermis from epidermis (as shown in Figure 2E). Following extrusion, the liquid containing HDMEC will be collected and centrifuged, and the cell pellet resuspended and plated as P0 HDMEC. The dermal strips will be placed in a separate vessel for subsequent isolation of fibroblasts
Day 3 of procedure (culture day 1)
-
9.HDMEC culture:
-
a.Rinse flasks 2–3 times with 5 mL HBS to remove debris, and replace with fresh ECGM, 15 mL per T75 flask. Incubate at 37°C, 5% CO2, for 48 h before changing media again.Note: Rinsing the flask the day after inoculation will help to remove any debris, such as collagen and bits of dermal tissue, which may contain non-adhered or loosely-adhered fibroblasts. This step will help reduce contamination of HDMEC by fibroblasts.
-
b.By days 1–2, small clusters of cells can be observed migrating from tissue pieces, and small colonies may be present (Figure 4A).
-
a.
-
10.Fibroblast culture:
-
a.Set up 2 T150 flasks for fibroblast pieces, and label as “#2” flasks. Add 10 mL FGM to each flask and equilibrate in incubator for ∼30 min.
-
b.Collect media and floating tissue pieces from #1 flasks and transfer to a sterile 50 mL conical tube. Replace with 25 mL fresh media per T150 flask and place #1 flasks back into the incubator.
-
c.Centrifuge collected tissue pieces at 1000 rpm (∼250×g) for 7 min at 4°C.
-
d.Carefully aspirate or pipet off supernatant (pellet will not be tight) and resuspend tissue pieces in 5 mL FGM. Inoculate half into each of the 2 T150 flasks prepared above.
-
a.
Note: On day 1, there may not be many fusiform cells visible in the #1 fibroblast flasks, but these should be clearly visible by days 2–3 in both #1 and #2 flasks (Figure 5A), including cells associated with small tissue pieces adhered to the plastic. Large clusters of cells should be easily visible by culture day 4 (Figures 5B and 5C).
-
11.Keratinocyte culture:
-
a.Supplement a bottle of KGM-LC (“low calcium,” 0.06 mM CaCl2).
-
b.Change P0 keratinocytes into KGM-LC one day after keratinocytes are initially inoculated: feed with 45 mL/T225 flask. Use this to feed keratinocytes until passaged to P1.
-
a.
Note: There may be few colonies visible on culture day 1, but by days 2–4, dividing cells and small colonies should be present (Figures 3B and 3C). It is important to note that the appearance of keratinocytes in KGM-LC on collagen-coated plastic differs dramatically from their appearance in regular KGM after passaging to P1 or P2. At P0, the cells may appear larger, darker, flatter, and more widely separated, compared to later passage cells grown in KGM without collagen-coated flasks.
Figure 4.

Images of P0-P1 primary human dermal microvascular endothelial cells (HDMECs) in culture
(A) Image of HDMEC P0 culture on day 1 after initiation. Note the appearance of cells associated with residual pieces of tissue. HDMECs are relatively compact at this stage and tightly associated with each other.
(B) Image of the same HDMEC P0 culture on day 4 after initiation. Note that the HDMECs now appear elongated but remain tightly associated in colonies. A contaminating fibroblast is observed (arrow).
(C) HDMEC at P0 culture day 6.
(D) HDMEC at P0 culture day 8, at time of harvest for passaging. White arrows indicate contaminating fibroblasts.
(E) P1 HDMEC, one day after passaging.
(F) P1 HDMEC at day 5. Contaminating fibroblasts are occasionally observed (white arrows in B, D, E, F). These tend to have more irregular shapes than HDMEC and are usually present as individual cells rather than colonies. Original image magnifications: 10× (A, B); 4× (C-F).
Figure 5.

Images of P0-P1 primary human fibroblasts in culture
(A) P0 fibroblasts in #1 flask at culture day 2, after removal of tissue “pieces.”
(B) P0 fibroblasts in #1 flasks at culture day 4.
(C) P0 fibroblasts in #2 flask on culture day 4, 3 days after removal from #1 flasks.
(D) P0 fibroblasts in #2 flasks on day 5, 4 days after removal from #1 flasks, at time of harvest for cryopreservation and/or passaging.
(E) P1 fibroblasts, 1 day after passage and inoculation.
(F) P1 fibroblasts, 5 days after inoculation, one day prior to planned harvest. Original image magnifications: 10× (A, B); 4× (C–F).
Day 4 of procedure (culture day 2) and beyond
-
12.HDMECs:
-
a.Change media every 48 h using 15 mL ECGM/T75 flask until cells approach confluence.
-
b.Passage the cells before confluence is reached; target density is 85%–90% confluence. If significant fibroblast contamination is present, harvest the cells at lower density.
-
a.
Note: HDMECs become more elongated by days 3–4 of P0 culture (Figure 4B). In contrast to contaminating fibroblasts, HDMECs generally form tight colonies with groups of cells aligned with each other (Figures 4B–4D).
Note: A low number of contaminating fibroblasts should be out-competed by HDMEC in ECGM. Practicing the technique used for extrusion of HDMEC from dermal strips will greatly improve the ratio of HDMEC to fibroblasts in your primary culture. However, the relative levels of fibroblast contamination vary from sample to sample. If necessary, HDMEC can be purified from contaminating fibroblasts by selecting CD31-expressing cells using fluorescence-activated flow sorting (FACS) or magnetic beads. For example, Bourland et al. published a procedure for immunomagnetic bead separation utilizing Dynabeads® CD31 that can be followed to eliminate contaminating fibroblasts from HDMEC cultures (Bourland et al., 2019).
-
13.Fibroblasts:
-
a.Change medium to fresh FGM in all #1 and #2 flasks, 25 mL per T150 flask. Continue to change using 25 mL FGM every 48 h until cells approach confluence (Figure 5D), then change 24 h prior harvesting and passaging or cryopreserving cells.
-
b.Do not wait until cells are confluent to passage fibroblasts; the cells are likely to be unevenly distributed across the growth surfaces of the flasks, with many very large colonies associated with residual tissue pieces. Additionally, the cell density is likely to differ in #1 vs. #2 flasks, depending on the efficiency of collagenase digestion. The cell viability and proliferative potential will be significantly reduced if you wait too long and cells progress well beyond confluence, which may result in cell cycle arrest due to contact inhibition (Chassot et al., 2008).
-
a.
-
14.Keratinocytes:
-
a.Continue to change medium every 48 h until cells approach confluence, then give one extra change the day before harvesting for passage to P1.
- b.
-
c.Passage the cells from P0 to P1 when they are ∼90%–95% confluent (Figure 3F).
-
a.
Note: P0 keratinocytes in KGM-LC will appear large and will not form tight colonies, and will appear to cover the growth surface of the flask (i.e., will appear confluent) at a lower cell density than later passage keratinocytes in KGM. At P0, it is acceptable to harvest keratinocytes when they appear nearly confluent, with relatively little empty space in the flask (Figure 3F). At P1 and beyond, do not allow cells to reach confluence prior to passage! Always try to passage before the entire growth surface is covered with cells (Figure 3H), as the cells will begin to terminally differentiate (and some cells in the culture will cease proliferating) when they reach or exceed confluence.
Isolation of epidermal melanocytes (on or around day 5–7 of procedure)
-
15.General considerations:
-
a.Melanocytes are removed from flasks by trypsin treatment much more easily than keratinocytes, allowing for separation by differential trypsin treatment. This differential trypsinization enables separation of melanocytes from keratinocytes; melanocytes will be collected and transferred to fresh flasks for culture in melanocyte-specific medium, while keratinocytes will remain in the original flasks to be further cultured in KGM-LC and subsequently passaged.
-
b.This separation procedure should be performed after keratinocyte colonies are visible, and melanocytes can be seen in these flasks (Figures 3D and 3E), but before keratinocytes begin to approach confluence. This usually occurs at 5–7 days after initial inoculation of P0 keratinocytes, but check flasks daily because different primary cultures can exhibit different growth characteristics. Separation will be much more difficult once keratinocytes become more densely packed in the flasks.
-
c.Use a 1:10 dilution of trypsin/EDTA solution when removing melanocytes from P0 keratinocytes, and at subsequent passages, to minimize contamination with keratinocytes.
-
d.Note that keratinocytes can be cultured without removal of melanocytes; the melanocytes will be mostly out-competed by keratinocytes after serial passaging, but may persist at low levels as “passengers” (i.e., contaminants) in the keratinocyte cultures. Cryopreservation of primary epidermal cultures will further diminish melanocyte numbers, because melanocyte viability is significantly diminished when frozen using parameters specific for keratinocytes; this is especially true for melanocytes with high melanin levels.
-
e.The removal of melanocytes from keratinocyte cultures is only necessary if you are interested in isolation of melanocytes, or if you are concerned about passenger melanocytes contaminating the keratinocyte cultures in downstream applications.
-
a.
-
16.Preparations before starting:
-
a.Warm HBS, MGM, FBS, and KGM-LC in a 37°C water bath.
-
b.Prepare 10 mL of a diluted trypsin/EDTA solution (1:10) for each T225 flask of keratinocytes. Mix 1 mL of 0.025% Trypsin + 0.02% EDTA with 9 mL HBS for each T225 flask. Store on ice until and warm to 37°C immediately before use.
-
c.Prepare 10 mL of a 10% FBS “stop” solution by mixing 1 mL of FBS with 9 mL MGM for each T225 flask.
-
d.Prepare FBS-coated flasks for primary melanocyte culture: Coat T75 flasks (1 per 1–3 T225 of keratinocytes) with 100% FBS (3 mL/T75), rock to cover entire growth surface, and incubate at 37°C for 30 min. Aspirate all the FBS to remove, and add 15 mL MGM to each T75 flask. Pre-equilibrate in incubator before inoculating melanocytes after removal from keratinocytes.
-
e.Turn on centrifuge and pre-chill to 4°C.
-
a.
-
17.Removal of melanocytes from keratinocyte cultures:
-
a.Aspirate medium from keratinocyte flasks (T225s).
-
b.Add 10 mL HBS to each flask, gently rock to rinse cell surface, and incubate at room temperature (20°C–22°C) for 3–4 min. Aspirate HBS and repeat wash step once.
-
c.Add 10 mL diluted, warmed trypsin/EDTA to each T225 flask. Rock very gently to ensure complete coverage of growth surface, and incubate 3–4 min at room temperature (20°C–22°C).
-
d.Quickly check under microscope; melanocytes should be releasing from flask, but keratinocytes should not. Do not wait until keratinocytes start rounding up and detaching from flasks, to avoid contamination of melanocytes with keratinocytes.Note: The phenotype of keratinocytes can vary from donor to donor, and some donor strains may adhere more tightly to flasks than others. It is very important to always check the cells under the microscope during this procedure to avoid removing keratinocytes with melanocytes.
-
e.Collect diluted trypsin/EDTA solution containing melanocytes, and add to a 50 mL conical tube containing 10% FBS.
-
f.Rinse each flask very gently with 10 mL HBS, and check under microscope. If keratinocytes still appear tightly adhered to flask, collect this HBS wash and add to tube(s) with collected melanocytes. If keratinocytes appear to be releasing from the flask, discard this wash solution to avoid contaminating melanocyte cultures with keratinocytes.
-
g.Add 45 mL KGM-LC to each T225 with keratinocytes and place back in incubator.
-
h.Centrifuge tubes with melanocytes at 1000 rpm (∼250×g) for 7 min at 4°C.
-
i.Resuspend melanocyte pellet in MGM; remove aliquot for counting. Inoculate melanocytes into pre-equilibrated FBS-coated flasks (prepared in step 15d, above) at ∼5 × 104/cm2 and place in incubator. The melanocytes are considered to be at passage P1 at this stage.
-
j.24 h later, aspirate the medium from flasks containing the isolated melanocytes and replace with fresh MGM. Change medium to fresh MGM (15 mL/T75 flask) every 48 h until melanocytes are ready to harvest for passage.Note: If there are still melanocytes present in keratinocyte cultures after completion of this procedure, the differential trypsinization can be repeated on subsequent days. However, do not try to repeat on the same day or the subsequent yield of proliferative keratinocytes may be reduced due to damage from repeated trypsin exposure. Although a second removal of melanocytes will likely only produce small numbers of recovered cells, it will help to diminish or eliminate melanocytes from the keratinocyte cultures. Under conditions optimized for keratinocyte culture, melanocytes can still survive and even proliferate, albeit at low levels, which could otherwise interfere with some applications or experiments.Note: If there are keratinocytes present as contaminants in the melanocyte cultures, a second differential trypsinization procedure can be used to transfer melanocytes to fresh T75 flasks. Wait at least 2–3 days to do this to avoid damaging melanocytes.
-
a.
Harvesting P0 keratinocytes (and other cell types) for subculture or cryopreservation
-
18.General considerations:
-
a.The procedures for harvesting keratinocytes are described in detail here, with modifications presented below for fibroblasts, HDMEC, and melanocytes.
-
b.Tissue culture vessels & coatings:
-
i.Keratinocytes do not require any coating of flasks for culture at P1 and beyond; tissue culture-treated flasks without collagen coating are used, and medium containing 0.2 mM CaCl2 (“regular” calcium KGM) is used.
-
ii.For subcultures of HDMEC or melanocytes to P1 and beyond (described in steps 22–31, below), flasks coated with Attachment Factor or FBS, respectively, will still be used, as with P0 cultures. Fibroblasts do not require coated flasks at any stage of the procedure.
-
i.
-
c.Cell density prior to harvesting:
-
i.Ideally, cells should be subcultured prior to reaching confluence. Although this is important for all four primary cell types, it is most important for keratinocytes, which will begin to differentiate upon reaching confluence, resulting in decreased colony forming efficiency and substantially reduced proliferative potential.
-
ii.Note, however, that P0 keratinocytes form relatively loose colonies when cultured in KGM-LC, appear larger and flatter than at later passages, and will appear to reach confluence at a lower cell density per cm2 (Figure 3F) at P0 than cells in regular calcium (0.2 mM) KGM at P1 and beyond. Do not allow the density of P0 keratinocytes to exceed that shown in Figure 3F, if possible.
-
iii.It is better to harvest the cells sooner rather than later, as they will flatten and then start to differentiate and pile up if allowed to get overly dense before passage, resulting in lower overall proliferative capacity.
-
i.
-
a.
-
19.Preparations before starting:
-
a.Decontaminate tissue culture hood and prepare aspirating flask by adding a sufficient volume of bleach to decontaminate aspirated liquid (∼10% final concentration).
-
b.Prepare a dilute solution (685 μM) of EDTA by adding 68.5 μL of a 0.5M EDTA solution to 50 mL HBS. Sterile filter through a 0.22 μm tube top filter.
-
c.Warm HBS and KGM (“regular” calcium) in a 37°C water bath.
-
d.Pre-chill centrifuge to 4°C
-
e.Thaw 0.025% Trypsin + 0.02% EDTA (5 mL/T225) and store on ice; warm to 37°C just prior to use.
-
f.Prepare a “stop” solution consisting of 10% FBS in KGM (5 mL/T225) and store on ice.
-
a.
-
20.Harvest keratinocytes:
-
a.Remove flasks from incubator, and in tissue culture hood aspirate KGM from flasks.
-
b.Add 10 mL HBS per flask, and rock to rinse growth surface for 30 s–1 min. Aspirate to remove HBS.
-
c.Add 10 mL dilute EDTA solution to each flask and rock to even coat growth surface. Incubate for 5 min at room temperature (20°C–22°C). This will not remove keratinocytes, but will ease removal during the subsequent trypsin/EDTA treatment and will improve overall yield.
-
d.Aspirate to remove dilute EDTA solution, and add 5 mL warm trypsin/EDTA solution. Place in incubator at 37°C and incubate for 3–5 min.
-
e.Remove flasks from incubator and check under microscope. Cells should be rounded up but will probably still be attached to the flask. Gently tap flasks, and rock back and forth to loosen and remove cells. Check under microscope and continue tapping/rocking until most cells have released from surface. Work quickly and avoid leaving cells in trypsin/EDTA solution for more than 5–6 min total.Note: The strength of adhesion to the growth surface of the flask can vary widely among different donor cell strains, and may be affected by various pathological conditions; for example, keratinocytes isolated from keloids exhibit adhesion defects and are more easily removed by trypsin treatment than keratinocytes from normal skin (Hahn et al., 2013). If gentle tapping and rocking is insufficient to loosen keratinocytes from plastic, more vigorous rapping or banging of the flask can be tried, but use caution to avoid cracking the flask. We recommend hitting the flask harder, if necessary, to remove cells, rather than leaving them in trypsin for longer than 5–6 min.CRITICAL: Do not leave keratinocytes in trypsin for >6–7 min or viability and/or proliferative potential may be significantly reduced.
-
f.Collect cell suspension and add to tube(s) containing cold 10% FBS in KGM to neutralize the trypsin.
-
g.Add 10 mL HBS per flask to rinse; using a 10 mL pipet, spray the HBS rinse directly onto the growth surface several times to release any remaining cells. Collect this rinse and add it to the tube(s) with trypsinized cells in 10% FBS solution.
-
h.Centrifuge at 1000 rpm (∼250×g) for 7 min at 4°C.
-
i.Resuspend cells in KGM and remove a small amount to count using a hemacytometer. Keep keratinocytes on ice while counting and until ready to inoculate.
-
j.Note that collagen coating of flasks is not required for P1 cultures and beyond, and KGM with “regular” calcium (200 μM) should be used.
-
a.
-
21.Subculture:
-
a.Equilibrate flasks with “regular” calcium (200 μM) KGM for 30 min at 37°C, 5% CO2.
-
b.Inoculate keratinocytes (now P1) at a density of 2–10 × 103/cm2. The inoculation density will determine the number of days until the cells reach ∼80%–90% confluence. Table 1, below, shows estimated culture times for different densities, but expect there to be variability among different donor cell strains. In general, keratinocytes of a healthy, proliferative donor strain are expected to double in number approximately every 24 h. However, the growth rate will slow slightly at subsequent passages, and will drop off substantially after ∼P5 or P6. If cells are harvested at near-confluence (∼90%), expect to harvest ∼1 × 105 keratinocytes/cm2 (∼22.5 × 106 per T225 flask), although this will be lower for P0 keratinocytes, which appear larger and form looser colonies in KGM-LC than later passages in KGM.
-
a.
Table 1.
Recommended inoculation densities for different primary cell types
| Cell type | Density (cells/cm2) | Approximate time to reach ∼90% confluence |
|---|---|---|
| Keratinocytes | 2,000 | 6–7 days |
| 5,000 | 5–6 days | |
| 10,000 | 4–5 days | |
| Fibroblasts | 2,000 | 5–6 days |
| 5,000 | 4–5 days | |
| 10,000 | 3–4 days | |
| Melanocytesa | 5,000 | 6–7 days |
| 10,000 | 5–6 days | |
| Human Dermal Microvascular Endothelial Cells (HDMEC)a | 5,000 | 6–7 days |
| 10,000 | 5–6 days |
For melanocytes and HDMEC, we do not recommend inoculating into flasks at <5,000/cm2.
Modifications of harvest procedure for HDMEC and fibroblasts
-
22.
For HDMEC, coat flasks with Attachment Factor as described above (step 4h), then add ECGM to flask(s) and equilibrate in incubator. For fibroblasts, no coating is needed; equilibrate flask(s) with FGM in incubator.
-
23.
Harvest the cells at ∼90% confluence, when growth surface of flask is nearly covered. See Figures 4 and 5 for examples of P0 HDMEC (Figure 4D) and fibroblasts (Figure 5D) photographed just prior to harvest.
-
24.
Prepare tubes containing ECGM (for HDMEC) or FGM (for fibroblasts) for neutralization of trypsin during harvest. HDMEC and fibroblasts are cultured in media containing 4%–5% FBS, so it is not necessary to prepare a solution of media containing 10% FBS to neutralize trypsin. Instead, ensure that at least 2 mL of supplemented FGM or ECGM are used per 1 mL trypsin.
-
25.HDMEC and fibroblasts are more easily removed from tissue culture plastic than keratinocytes. Follow the procedures described above for keratinocytes (step 20), using 0.025% Trypsin + 0.02% EDTA to release cells from plastic, but use 3 mL for T150 flasks and 2 mL for T75 flasks, in addition to the following modifications.
-
a.Perform a brief HBS rinse (5–10 mL) prior to adding trypsin. The dilute EDTA rinse used for keratinocytes is not required for HDMEC and fibroblasts.
-
b.Treat with trypsin for ∼2–3 min to remove cells. This can usually be done at room temperature (20°C–22°C) rather than 37°C. Do not exceed 5 min.
-
c.Gentle tapping and rocking should be sufficient to dislodge HDMEC and fibroblasts after trypsin treatment.
-
d.Rinsing flasks with HBS after trypsinization is still recommended to increase recovery; add the rinse to the tube(s) with trypsinized cells.
-
a.
-
26.
Following harvesting cells for passaging to P1, inoculate into flasks as described above for keratinocytes (step 21b); note, however that different cell types have different doubling rates and thus will reach confluence at different times. Recommended inoculation densities are listed in Table 1, above. Do not inoculate HDMEC below <5,000/cm2, as this stresses the culture and may impede proliferation. Using the procedures and media formulations described here, fibroblasts should double in number every ∼16–20 h. HDMEC and melanocytes have slower growth rates, doubling every ∼2–3 days. For all primary cell types, there can be substantial donor-to-donor variability in growth characteristics, and doubling time will increase with increasing passage in culture.
Modifications of harvest procedure for melanocytes
-
27.
Harvest the cells at ∼90% confluence, when growth surface of flasks is nearly covered. See Figure 6D for example of melanocytes photographed just prior to harvest.
-
28.
Prepare 10% FBS in MGM for neutralization of trypsin during melanocyte harvest (≥1 mL per mL of trypsin).
-
29.
Prepare FBS-coated flask(s) as described above (step 16d), then add MGM to flask(s) and equilibrate in incubator. Use FBS-coated flasks for every passage of melanocytes.
-
30.Melanocytes are much more easily removed from tissue culture plastic than keratinocytes, and a 1:10 dilution of the trypsin/EDTA solution can be used for harvesting, as done when removing melanocytes from keratinocytes. Using a dilute solution at each passage will help eliminate any contaminating keratinocytes that may be present.
-
a.Perform a brief HBS rinse prior to adding trypsin; do not perform the initial rinse with EDTA, as done for keratinocytes.
-
b.Treat with trypsin at room temperature (20°C–22°C) for ∼2–3 min to loosen and remove cells. Do not exceed 5 min.
-
c.Gentle tapping and rocking should be sufficient to dislodge cells after trypsin treatment.
-
d.Rinsing flasks with HBS after trypsinization is recommended to increase recovery; add the rinse to the tube(s) with trypsinized cells.
-
a.
-
31.
After centrifugation, resuspension, and counting, inoculate into prepared FBS-coated, pre-equilibrated flasks at ≥5,000/cm2.
Figure 6.

Images of P1 primary human melanocytes in culture
(A) P1 melanocytes at culture day 2, 2 days after removal from P0 keratinocytes, photographed at 10× magnification.
(B) P1 melanocytes at culture day 5, photographed at 10× magnification.
(C) P1 melanocytes at culture day 5, photographed at 4× magnification. A small cluster of cells resembling keratinocytes is present (white arrow). Note that using a diluted trypsin solution when harvesting melanocytes, and trypsinizing for the minimum length of time required, will eliminate keratinocytes from successive passages.
(D) P1 melanocytes at day 8, at time of harvest for subculture. Some contaminating cells resembling keratinocytes are present (white arrows).
Cryopreservation of primary skin cells
-
32.General considerations:
-
a.Note that high melanin levels can reduce viability of melanocytes during cryopreservation and recovery if routine methods for freezing and thawing are followed. The procedures outlined here—particularly, slower freezing rate and high serum content of freezing medium—work well for melanocytes with high melanin contents (Boyce et al., 2017a).
-
b.Good record-keeping is particularly important if you will be storing large numbers of vials long-term for your research. We strongly recommend that you design and maintain an inventory of frozen cell stocks, noting the cell type, donor strain, passage number, cell density, and cryopreservation date on each vial.
-
a.
-
33.Technical considerations:
-
a.If you plan to freeze cells for future use, we recommend cryopreserving a portion of the cells at P0, and the rest after passage to P1. How many of each passage to cryopreserve will depend on your storage capacity and experimental needs.
-
b.As with harvesting cells for passage, we recommend harvesting cells for cryopreservation before reaching confluence, while cells are in log-phase growth.
-
c.We strongly recommend using a controlled rate cell freezer (e.g., Custom BioGenic Systems model 2100) for the most consistent and predictable results.
-
d.Frozen primary cells should be stored in liquid nitrogen or in a liquid nitrogen vapor phase freezer.
-
e.If storing in canes using a liquid nitrogen storage tank, we recommend storing only one cell type per cane to avoid mix-ups upon thawing.
-
f.Remember that when cells are recovered from cryopreservation, they will be one passage later than when they were frozen; the harvest for freezing counts as one passage. For example, if cells are frozen at P0, they should be considered P1 when thawed.
-
a.
-
34.Preparations before starting:
-
a.Prepare freezing media fresh before use by combining culture medium, FBS, and dimethyl sulfoxide (DMSO) in the amounts listed below in Table 2 for specific cell types. For keratinocytes, fibroblasts, and endothelial cells, medium is combined with FBS to yield ∼20% FBS in the freezing medium, with 10% DMSO. For melanocytes, 90% FBS + 10% DMSO is used. Filter sterilize using a 0.22 μm tube top filter, and store on ice until used.
-
b.Label sterile 2 mL cryovials with cell strain, cell type, cell number (density), and date.
-
c.If using a controlled rate freezer, set up a blank vial (freezing media only) to include in freezing run. Turn on cell freezer and pre-chill before use.
-
a.
-
35.
Harvest cells as detailed above for each specific cell type, resuspend in growth medium, and store on ice while counting and performing calculations. Remove an aliquot of cells and count using a hemacytometer, then calculate the total number of cells harvested.
-
36.
Calculate the desired number of cells to freezer per vial, and the number of vials to be frozen. We recommend freezing 1–6 × 106 cells per vial. If 2 mL cryovials are used, assume a volume of 1.8 mL cells in freezing medium for each vial; this leaves sufficient space to allow for changes in volume and pressure due to changes in temperature during freezing and thawing.
-
37.
Pellet cells by centrifuging at 1000 rpm (∼250×g) for 7 min at 4°C.
-
38.
Aspirate medium and resuspend in the required volume of cold freezing medium to yield the correct number of cells per 1.8 mL.
Table 2.
Freezing media formulations for different primary cell types (to make 50 mL)
| Keratinocytes | Melanocytes | Fibroblasts | HDMEC | |
|---|---|---|---|---|
| Supplemented growth medium | 35.0 mL | – | 37.5 mL | 37.5 mL |
| FBS | 10.0 mL | 45.0 mL | 7.5 mL | 7.5 mL |
| DMSO | 5.0 mL | 5.0 mL | 5.0 mL | 5.0 mL |
Example: If you harvested 6.1 × 107 cells total, you can cryopreserve 10 vials at 6 × 106 each. Resuspend the cell pellet in 18.3 mL freezing medium, and aliquot 1.8 mL per vial.
-
39.
Aliquot cells into freezing vials on ice, then transfer to cell freezer for cryopreservation.
-
40.
For fibroblasts, keratinocytes, and HDMEC, a freezing rate of –10°C/min works well. However, for freezing melanocytes, a slower freezing rate of -1°C/min is recommended, particularly with melanocytes from darker skinned donors with high melanin content (Boyce et al., 2017a). If a controlled rate cell freezer is not available, other methods or equipment for cryopreservation (e.g., Mr. Frosty™) may be used; however, we cannot guarantee that other, less tightly-regulated methods to reduce temperature will work as well as cryopreservation using a controlled rate freezer.
-
41.
Quickly transfer vials of cells to liquid nitrogen for storage. Even brief increases in temperature can reduce cell viability over long-term storage, so it is important to work quickly to avoid allowing frozen vials to warm during transfer to storage vessels.
Caution: Utilize personal protective equipment (PPE) appropriate for use with liquid nitrogen, as the liquid is hazardous and can cause severe injury upon contact with skin. PPE for liquid nitrogen includes a face shield over safety goggles, or chemical splash goggles, and skin protection, including thermal insulated gloves, long sleeve lab coats, pants, and closed-toe shoes. For additional details, please consult the United States Department of Agriculture (USDA) website: https://www.ars.usda.gov/northeast-area/docs/safety-health-and-environmental-training/liquid-nitrogen-safety/.
Expected outcomes
Following the procedures outlined here, you should expect to obtain ∼1 × 106 keratinocytes/cm2 of skin, and these should reach ∼90% confluence in 6–8 days. Fibroblasts, endothelial cells, and melanocytes are not counted prior to inoculating primary (P0) flasks. However, fibroblasts should be ready to harvest for passage and/or cryopreservation in 5–6 days. Endothelial cells and melanocytes should be ready to passage in 6–8 days. Levels of tissue vascularization can vary greatly among different skin samples, which can affect the yield of endothelial cells isolated from the dermis. This is difficult to control, even when consistently using skin samples from the same body site.
Primary cells have finite lifespans in vitro. Following the procedures outlined here, including specific culture media formulations, you can expect highly proliferative cultures of keratinocytes, fibroblasts, and endothelial cells to at least P4 or P5; melanocytes may extend to P6 or P7. However, you should expect that doubling times may increase at later passages, with increasing proportions of senescent cells beyond ∼P5. Primary cells isolated using these procedures have been used for preparation of engineered skin substitutes (ESS), demonstrating the ability of cells to differentiate into functional skin tissue. For example, ESS containing autologous fibroblasts and keratinocytes were shown to provide long-term wound closure after treatment of large full-thickness burn wounds in clinical trials (Boyce et al., 2002, 2006, 2017b). Preclinical studies demonstrated that HDMEC isolated using this procedure were competent to form vascular analogs in ESS (Supp et al., 2002), and melanocytes not only produced and transferred melanin to keratinocytes in ESS, but were able to increase melanin production in response to UV irradiation and protect keratinocytes from UV-induced DNA damage (Boyce et al., 2020; Supp et al., 2020).
The four cell types isolated using this protocol—fibroblasts, keratinocytes, HDMEC, and melanocytes—have distinctive morphologies, and with experience it should be possible to easily distinguish these cell types from each other using light microscopy. Depending on the intended application, and for investigators new to primary culture of skin cells, it may be desirable to confirm the identities of these cell types. This can be done by immunofluorescence using antibodies for cell type-specific markers. Suggested markers for individual cell types include vimentin (VIM) for fibroblasts, keratin 14 (KRT14) for keratinocytes, platelet and endothelial cell adhesion molecule 1 (PECAM1; also known as CD31) for HDMEC, and tyrosinase related protein 1 (TYRP1; also known as Mel5) for melanocytes (Figure 7). Immunofluorescence can also be used to determine the homogeneity of cultures and/or identify contaminating cell types.
Figure 7.

Immunofluorescent staining of primary skin cells using cell type-specific markers
Immunofluorescence was used to validate the identify of individual primary cell types.
(A) Primary human dermal fibroblasts were labeled using a primary antibody against vimentin (1:200) followed by Alexa Fluor 594-labeled donkey anti-rabbit secondary antibody (1:400).
(B) Primary human keratinocytes were labeled using a primary antibody against keratin 14 (1:100) followed by Alexa Fluor 594-labeled donkey anti-rabbit secondary antibody (1:400).
(C) Primary human dermal microvascular endothelial cells (HDMEC) were labeled using a primary antibody against PECAM1/CD31 (1:100), followed by Alexa Fluor 594-labeled donkey anti-mouse secondary antibody (1:400).
(D) Primary human melanocytes were labeled with a primary antibody against tyrosinase-related protein 1 (1:100) followed by Alexa Fluor 594-labeled donkey anti-mouse secondary antibody (1:400). Cells were stained using standard immunofluorescence methods as described elsewhere (Hahn et al., 2021). For all cell types, specific staining is indicated by red fluorescence; nuclei were counterstained blue using mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI). Note that the cell morphologies may vary slightly from the phase-contrast images shown in Figures 3, 4, 5, and 6 because the cells were not cultured on tissue culture-treated plastic for staining. Fibroblasts and keratinocytes were cultured for immunofluorescence using chamber slides without coating; HDMEC and melanocytes were cultured using glass coverslips coated with 0.1% gelatin or FBS, respectively. The scale bar shown in A is the same for all panels.
There can be enormous person-to-person variability in skin characteristics. Likewise, there can be substantial variability in growth characteristics among primary cell cultures established from different donors. Thus, some variability is expected. Avoid introducing additional variability by using consistent technique and taking good notes, making sure to note any variations in procedures or unusual observations. In general, primary cultures established from skin of younger individuals will be more vigorous, with higher proliferative potential and possibly higher growth rates, than cultures from skin of older individuals. However, in our experience, there is not a specific cut-off age above which you can expect a significant change in yield or performance. Skin from teenaged patients will likely provide more proliferative cells than skin from a senior citizen, but there are numerous factors that can affect skin cell viability, in addition to age, making it difficult to predict performance in advance, particularly for de-identified samples where patients’ medical histories are unavailable.
Note that patient health status (e.g., diabetes, poor nutrition) can affect the characteristics of primary cultures, a fact that must be kept in mind when performing experiments with primary cells. In addition to age and health status, variability can result from differences in donor sex, ethnicity, skin pigmentation level, or body site for skin biopsy, among other factors. No single donor strain of any primary cell type can be considered representative of all “normal” cells without validation. Particularly when using primary skin cells to study skin disease, multiple donor cell strains for each group (i.e., pathological condition and normal controls) should be investigated to provide the most reliable data. A statistical power analysis is recommended, using estimates of variability or known data for individual donor strains, to determine group sizes and increase the likelihood of generating statistically significant data.
Body site tends to be an unappreciated source for variability among different primary skin cell cultures. In particular, some genes that are differentially expressed in different regions of the body, such as homeobox cluster (HOX) genes, retain their differential expression patterns when skin cells are cultured in vitro (Rinn et al., 2006, 2008). Further, differences in skin structure may lead to differences in phenotype or gene expression in vitro; for example, between cells isolated from foreskin, a specialized skin type, and trunk skin. This must be kept in mind when performing experiments with primary skin cells. Comparisons among different donor cell strains must be carefully controlled to avoid confounding factors due to differences in starting skin tissue sources.
This protocol can be used for isolation of just fibroblasts and/or keratinocytes if you do not need to isolate HDMEC and/or melanocytes. For isolation of fibroblasts from dermis without isolation of HDMEC, proceed directly from separating dermis from epidermis to mincing dermis and collagenase digestion. Few if any HDMEC will be present in your fibroblast cultures, and these will be rapidly out-competed by fibroblasts. If you do not need to isolate melanocytes, you can skip the differential trypsinization step and proceed as described above. Melanocytes will persist to varying degrees in the keratinocyte cultures (referred to as “passenger melanocytes”), which may affect downstream applications. However, these can be removed by differential trypsinization, without saving the cells if you do not need them. When cryopreserved at -10°C per minute, viability of melanocytes (particularly with high melanin levels) will be greatly reduced compared with keratinocytes, resulting in lower proportions of contaminating melanocytes following cryopreservation and recovery of keratinocytes.
Limitations
The protocol described here has been optimized for rapid proliferation and retention of differentiation capacity, as it was developed for tissue engineering of human skin and preparation of skin substitutes intended for treatment of traumatic injuries. These cells have limited in vitro lifespans, as explained above. We recommend performing experiments using cells at low passages (P1–P4) for best results. When comparing different donor cell strains, always compare cells at the same passage for the greatest consistency.
These procedures have not been tested using skin from other species, such as murine or porcine skin. Further, this protocol is intended for full-thickness human skin. If used for isolation of cells from split-thickness skin, it is important to modify the procedures as detailed above; for example, reducing the incubations for enzyme digestions. Yield of HDMEC may be reduced when isolated from split-thickness skin.
Although selective culture media is used for each cell type, the procedures described here do not guarantee pure populations of each cell type. Contaminating cells are often referred to as “passengers,” as they may be present and carried along non-specifically during in vitro culture. For example, as detailed above, HDMEC cultures are frequently contaminated with fibroblasts. If a heavy contamination is present, there is a risk of fibroblasts out-competing HDMECs and taking over the culture. Positive selection of HDMEC based on CD31 expression is one option to purify HDMECs (Bourland et al., 2019), but this is more difficult for keratinocytes, fibroblasts, and melanocytes, which lack suitable cell surface markers for easy positive selection. Differential trypsinization can be used to separate melanocytes or fibroblasts from keratinocytes, but low levels of contaminating cell populations may persist. To remove darkly pigmented passenger melanocytes from keratinocyte cultures, flow cytometry can be used to sort cells based on differences in scattering of light, as detailed by Swope et al. (Swope et al., 1997).
Troubleshooting
Problem 1
Low yield of isolated primary cells, and/or low cell viability. This may result from a number of causes, including poor storage of tissue prior to starting procedure, excessive time in Dettol during initial decontamination, or too much time in Dispase, collagenase, or trypsin.
Potential solution
Make sure that the skin has been handled properly before and during collection. Do not allow the skin to dry out, and keep cold if possible, to slow tissue degradation. Work quickly when trimming tissue so that the skin is not kept at room temperature (20°C–22°C) for too long, and does not dry out, before starting the procedure. Do not leave in Dettol rinses longer than 30 s, as Dettol can cause toxicity if cells are exposed for too long or it is not properly removed during rinses. Excessive incubation in Dispase, collagenase, and/or trypsin should be avoided. Make sure to use a sufficient volume of 10% FBS to neutralize trypsin following digestion of epidermal strips. When pipetting to resuspend cells after centrifugation, gently pipet up and down to avoid damaging cells. Never vortex primary cells.
Problem 2
Epidermis and dermis not separating easily following Dispase digestion (step 5).
Potential solution
If tissue strips are too big or too thick, the enzyme solution may not have adequate access to the dermal-epidermal basement membrane. Make sure that strips are no wider than 0.25 cm prior to digestion and that there is a sufficient volume of enzyme (1–2 mL/cm2) for the amount of tissue being digested. If dermis and epidermis are difficult to separate from each other following an overnight (∼16 h) incubation at 4°C in Dispase, try warming the solution to 37°C and incubating for 30 min to see if this facilitates separation.
Problem 3
Too many large dermal tissue pieces left after collagenase digestion and unable to transfer using 25 mL pipet (step 7).
Potential solution
While using a larger bore pipet may make transferring the solution easier, a better approach is to ensure that the dermal strips are finely minced prior to collagenase digestion. Do not extend the collagenase digestion beyond 2 h at 37°C as this can reduce fibroblast viability. Although this protocol uses collagenase to release fibroblasts from dermal tissue, we also recommend saving and plating the residual tissue “pieces” to increase your overall yield, allowing you to also recover fibroblasts that migrate from incompletely digested tissue. If you do not see fibroblasts adhering to the plastic flasks after 1–2 days, focus on the tissue pieces stuck to the flask; chances are good that you will see fibroblasts near these pieces and in the process of migrating out from the tissue. If not, a third round of collecting and plating those “pieces” can be done to try to recover as many fibroblasts as possible.
Problem 4
Low yield of HDMEC, or high contamination with dermal fibroblasts (step 6).
Potential solution
Although HDMEC yield is dependent on tissue vascularity, there can be tremendous variability based on technique used during mechanical extrusion. Too much force will increase yield, but may also result in increased fibroblast contamination. It may take a bit of trial and error to learn the correct amount of force to exert on the dermal tissue strips to isolate only or mostly HDMEC. Rinsing the flasks on the day after inoculation to remove floating debris can help remove loosely adhered fibroblasts and floating cells associated with collagen and tissue debris. Purification of HDMEC based on positive selection for CD31, a cell surface protein specifically expressed on endothelial cells, can help overcome problems due to substantial fibroblast contamination (Bourland et al., 2019).
Problem 5
Melanocyte culture contaminated with dermal fibroblasts.
Potential solution
Keratinocyte contamination is easily monitored by visualizing under a microscope and using differential trypsinization to separate melanocytes from undesirable keratinocytes (detailed above). However, melanocyte cultures can become overrun with fibroblasts. Unfortunately, fibroblasts can proliferate in MGM, and may eventually start to out-compete melanocytes; this is more likely to be observed as the passage extends out to P4-P6, as the growth rate of melanocytes begins to drop. We suggest using immunohistochemistry or flow cytometry to stain a portion of the melanocytes with antibody to the melanocyte-specific marker TYRP1 to confirm melanocyte identity before utilizing in experiments where purity of the population is critical.
Problem 6
Culture medium appears cloudy and/or yellowish.
Potential solution
Clouding and/or yellowing of the culture medium likely indicates microbial contamination. Gross bacterial or fungal contamination is often evidenced by clouding of the media. Medium containing phenol red may change color upon contamination; for example, a yellowish color indicates acidification, which can occur with bacterial contamination (Nims and Price, 2017). When visually inspected using a phase-contrast microscope, bacteria may appear as small dark spots in the culture medium. These can sometimes be distinguished from cell debris because they may move slightly or appear to vibrate on their own when the medium is otherwise still. Fungal contaminants, such as yeast, may appear as small round cells or chains of cells, or may be observed as fluffy clumps floating at the surface of the media. If you are unsure whether contamination is present, you can test the media by taking a small aliquot of the suspect medium and adding it to a larger volume (∼50–100×) of medium without PSF and incubating for a few days at 37°C; if bacteria or fungi are present, the media will display increasing cloudiness over time during incubation. Another type of microbial contaminant, mycoplasma, can be difficult to detect as it is usually does not cause clouding of the medium, may be difficult to observe microscopically, and may be present at low levels without causing obvious cytotoxicity, although specific tests for mycoplasma are available (Nikfarjam and Farzaneh, 2012; Nims and Price, 2017). Once a culture displays signs of gross contamination, it is usually not possible to kill or remove all of the microbial contaminants or salvage the cultured cells. Adding increased amounts of PSF is not usually successful for eliminating gross contamination once present, and will likely damage the cells. For this reason, prevention of microbial contamination is key.
Pinpointing the source of contamination may be difficult but can aid in efforts to prevent future contamination. We prefer to discard any bottles of culture media that may be contaminated, or that have been opened while working with contaminated cells. However, if you do not wish to discard all suspect media, we recommend testing it for contamination by incubating cell-free medium at 37°C for several days to check for microbial growth. Once a contaminated culture is found, we highly recommend a thorough cleaning of the incubator to prevent the problem from spreading further.
Although bacterial contamination may be present in the tissue biopsy used for cell isolation, the decontamination procedure described here (step 2) is usually sufficient, and further treatment of tissue with Dettol will cause cytotoxicity. In our experience, bacterial contamination usually results from inadvertently touching surfaces that contact the culture or culture media, or touching pipets used for transferring media (this can be touching with hands or fingers, even if gloved, or contacting non-sterile surfaces in the hood). Bacteria or fungi may also be transferred from water baths used to warm media bottles. Use of a commercial sanitizer, such as Clear Bath (see key resources table), in the water bath can diminish this risk, as can thoroughly wiping bottles with alcohol (70% ethanol or isopropanol) prior to placing in the hood (Nikfarjam and Farzaneh, 2012). Fungal contamination is often due to airborne spores that may be present on hands or clothing. The risk of fungal contamination can be greatly diminished by wearing clean lab coats (ideally, with elastic or knit cuffs) and gloves for all tissue culture procedures. Lab coats should be washed often, especially after fungal contamination is observed. When working in a laminar flow biosafety cabinet, it is important to never pass your hands between the source and direction of air flow and an open media bottle or culture vessel. Also, using culture medium within a week of supplementation is recommended because the activity of the antibiotic/antimycotic solution, PSF, is diminished over time, particularly if the medium is kept for long periods at 37°C before use.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dorothy Supp (suppdm@ucmail.uc.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was funded by Shriners Hospitals for Children Medical Research Grants #85102-CIN-18, #71012-CIN-20, and #72005-CIN-21. The authors thank the surgeons who have graciously assisted with tissue collection, including Drs. W. John Kitzmiller, Elizabeth Dale, Ryan Gobble, Allison Lied, Petra Warner, Ann Schwentker, and Brian Pan, and the operating room staff of the University of Cincinnati Medical Center. We also acknowledge Dr. Steven Boyce for his contributions to the field. The graphical abstract was created with BioRender.com
Author contributions
Conceptualization, D.M.S. and H.M.P.; methodology, D.M.S., J.M.H., and H.M.P.; investigation, J.M.H., K.A.C., and K.L.M.; Writing—original draft, D.M.S. and J.M.H.; writing—review & editing, K.A.C., K.L.M., and H.M.P.; Supervision, D.M.S. and H.M.P.; funding acquisition, D.M.S. and H.M.P.
Declaration of interests
The authors have no competing interests to declare related to the content of this manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101172.
Contributor Information
Dorothy M. Supp, Email: suppdm@ucmail.uc.edu.
Jennifer M. Hahn, Email: hahnjf@ucmail.uc.edu.
Data and code availability
This study did not generate/analyze datasets or code.
References
- Bikle D.D., Xie Z., Tu C.L. Calcium regulation of keratinocyte differentiation. Expert Rev. Endocrinol. Metab. 2012;7:461–472. doi: 10.1586/eem.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourland J., Mayrand D., Tremblay N., Moulin V.J., Fradette J., Auger F.A. Isolation and culture of human dermal microvascular endothelial cells. Methods Mol. Biol. 2019;1993:79–90. doi: 10.1007/978-1-4939-9473-1_7. [DOI] [PubMed] [Google Scholar]
- Boyce S.T. Methods for the serum-free culture of keratinocytes and transplantation of collagen-gag-based skin substitutes. Methods Mol. Med. 1999;18:365–389. doi: 10.1385/0-89603-516-6:365. [DOI] [PubMed] [Google Scholar]
- Boyce S.T., Ham R.G. Cultivation, frozen storage, and clonal growth of normal human epidermal keratinocytes in serum-free media. J. Tiss. Cult. Meth. 1985;9:83–93. [Google Scholar]
- Boyce S.T., Kagan R.J., Greenhalgh D.G., Warner P., Yakuboff K.P., Palmieri T., Warden G.D. Cultured skin substitutes reduce requirements for harvesting of skin autograft for closure of excised, full-thickness burns. J. Trauma. 2006;60:821–829. doi: 10.1097/01.ta.0000196802.91829.cc. [DOI] [PubMed] [Google Scholar]
- Boyce S.T., Kagan R.J., Yakuboff K.P., Meyer N.A., Rieman M.T., Greenhalgh D.G., Warden G.D. Cultured skin substitutes reduce donor skin harvesting for closure of excised, full-thickness burns. Ann. Surg. 2002;235:269–279. doi: 10.1097/00000658-200202000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce S.T., Lloyd C.M., Kleiner M.C., Swope V.B., Abdel-Malek Z., Supp D.M. Restoration of cutaneous pigmentation by transplantation to mice of isogeneic human melanocytes in dermal-epidermal engineered skin substitutes. Pigment Cell Melanoma Res. 2017;30:531–540. doi: 10.1111/pcmr.12609. [DOI] [PubMed] [Google Scholar]
- Boyce S.T., Simpson P.S., Rieman M.T., Warner P.M., Yakuboff K.P., Bailey J.K., Nelson J.K., Fowler L.A., Kagan R.J. Randomized, paired-site comparison of autologous engineered skin substitutes and split-thickness skin graft for closure of extensive, full-thickness burns. J. Burn Care Res. 2017;38:61–70. doi: 10.1097/BCR.0000000000000401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce S.T., Supp D.M., Lloyd C.M. Exogenous keratinocyte growth factor is not required for pigmentation of skin substitutes with three isogeneic cell types. Tissue Eng. Part A. 2020;26:214–224. doi: 10.1089/ten.TEA.2019.0203. [DOI] [PubMed] [Google Scholar]
- Boyce S.T., Zimmerman R.L., Supp D.M. Tumorigenicity testing in athymic mice of cultured human melanocytes for transplantation in engineered skin substitutes. Cell Transpl. 2015;24:1423–1429. doi: 10.3727/096368914X683052. [DOI] [PubMed] [Google Scholar]
- Chassot A.A., Lossaint G., Turchi L., Meneguzzi G., Fisher D., Ponzio G., Dulic V. Confluence-induced cell cycle exit involves pre-mitotic CDK inhibition by p27(Kip1) and cyclin D1 downregulation. Cell Cycle. 2008;7:2038–2046. doi: 10.4161/cc.7.13.6233. [DOI] [PubMed] [Google Scholar]
- Hahn J.M., Glaser K., McFarland K.L., Aronow B.J., Boyce S.T., Supp D.M. Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen. 2013;21:530–544. doi: 10.1111/wrr.12060. [DOI] [PubMed] [Google Scholar]
- Hahn J.M., McFarland K.L., Combs K.A., Anness M.C., Supp D.M. Analysis of HOX gene expression and the effects of HOXA9 overexpression in fibroblasts derived from keloid lesions and normal skin. Wound Repair Regen. 2021;29:777–791. doi: 10.1111/wrr.12917. [DOI] [PubMed] [Google Scholar]
- Ham R.G., McKeehan W.L. Media and growth requirements. Methods Enzymol. 1979;58:44–93. doi: 10.1016/s0076-6879(79)58126-9. [DOI] [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- McFarland K.L., Glaser K., Hahn J.M., Boyce S.T., Supp D.M. Culture medium and cell density impact gene expression in normal skin and abnormal scar-derived fibroblasts. J. Burn Care Res. 2011;32:498–508. doi: 10.1097/BCR.0b013e3182223cb1. [DOI] [PubMed] [Google Scholar]
- Nikfarjam L., Farzaneh P. Prevention and detection of Mycoplasma contamination in cell culture. Cell J. 2012;13:203–212. [PMC free article] [PubMed] [Google Scholar]
- Nims R.W., Price P.J. Best practices for detecting and mitigating the risk of cell culture contaminants. In Vitro Cell. Dev. Biol. Anim. 2017;53:872–879. doi: 10.1007/s11626-017-0203-9. [DOI] [PubMed] [Google Scholar]
- Pan C., Kumar C., Bohl S., Klingmueller U., Mann M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell. Proteomics. 2009;8:443–450. doi: 10.1074/mcp.M800258-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peehl D.M., Ham R.G. Growth and differentiation of human keratinocytes without a feeder layer or conditioned medium. In Vitro. 1980;16:516–525. doi: 10.1007/BF02626465. [DOI] [PubMed] [Google Scholar]
- Pittelkow M.R., Scott R.E. New techniques for the in vitro culture of human skin keratinocytes and perspectives on their use for grafting of patients with extensive burns. Mayo Clin. Proc. 1986;61:771–777. doi: 10.1016/s0025-6196(12)64815-0. [DOI] [PubMed] [Google Scholar]
- Rinn J.L., Bondre C., Gladstone H.B., Brown P.O., Chang H.Y. Anatomic demarcation by positional variation in fibroblast gene expression programs. Plos. Genet. 2006;2:e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn J.L., Wang J.K., Allen N., Brugmann S.A., Mikels A.J., Liu H., Ridky T.W., Stadler H.S., Nusse R., Helms J.A., et al. A dermal HOX transcriptional program regulates site-specific epidermal fate. Genes Dev. 2008;22:303–307. doi: 10.1101/gad.1610508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supp D.M., Hahn J.M., Combs K.A., McFarland K.L., Schwentker A., Boissy R.E., Boyce S.T., Powell H.M., Lucky A.W. Collagen VII expression is required in both keratinocytes and fibroblasts for anchoring fibril formation in bilayer engineered skin substitutes. Cell Transpl. 2019;28:1242–1256. doi: 10.1177/0963689719857657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supp D.M., Hahn J.M., Lloyd C.M., Combs K.A., Swope V.B., Abdel-Malek Z., Boyce S.T. Light or dark pigmentation of engineered skin substitutes containing melanocytes protects against ultraviolet light-induced DNA damage in vivo. J. Burn Care Res. 2020;41:751–760. doi: 10.1093/jbcr/iraa029. [DOI] [PubMed] [Google Scholar]
- Supp D.M., Wilson-Landy K., Boyce S.T. Human dermal microvascular endothelial cells form vascular analogs in cultured skin substitutes after grafting to athymic mice. FASEB J. 2002;16:797–804. doi: 10.1096/fj.01-0868com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope V.B., Supp A.P., Cornelius J.R., Babcock G.F., Boyce S.T. Regulation of pigmentation in cultured skin substitutes by cytometric sorting of melanocytes and keratinocytes. J. Invest. Dermatol. 1997;109:289–295. doi: 10.1111/1523-1747.ep12335766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This video briefly demonstrates the method used for mechanical extrusion of HDMEC from dermal strips after enzymatic separation of dermis from epidermis (as shown in Figure 2E). Following extrusion, the liquid containing HDMEC will be collected and centrifuged, and the cell pellet resuspended and plated as P0 HDMEC. The dermal strips will be placed in a separate vessel for subsequent isolation of fibroblasts
Data Availability Statement
This study did not generate/analyze datasets or code.