

Summary
Here, we present a step-by-step protocol for three-dimensional reconstruction of astrocyte morphology, applied to the central amygdala oxytocin receptor-expressing astrocytes. This includes RNAse-free perfusion, combination of RNAscope and immunohistochemistry, and confocal imaging. This protocol provides detailed information about tissue handling and a comprehensive description of the RNAScope technique to label rat and mouse oxytocin receptor mRNA. We also describe three-dimensional reconstruction that allows the assessment of more than 70 different cellular parameters, powerful for studying astrocyte morphology and astrocyte-astrocyte interactions.
For complete details on the use and execution of this protocol, please refer to Wahis et al. (2021) and Althammer et al. (2020).
Subject areas: Microscopy, Molecular Biology, Antibody, In Situ Hybridization, Neuroscience
Graphical abstract
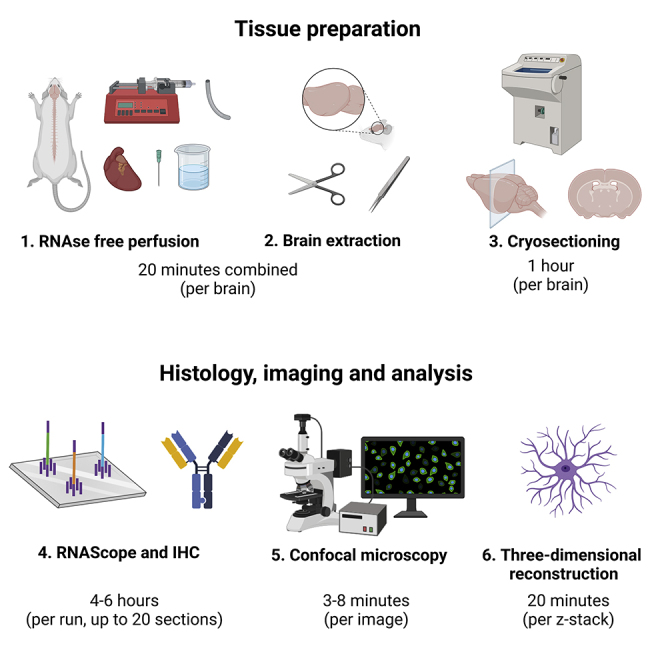
Highlights
-
•
RNAse-free perfusion and brain extraction from rat or mouse
-
•
Astrocyte-specific immunohistochemistry and confocal image acquisition
-
•
RNAScope in situ hybridization for oxytocin receptor mRNA
-
•
Three-dimensional reconstruction of oxytocin-receptor expressing astrocytes
Here we present a step-by-step protocol for three-dimensional reconstruction of astrocyte morphology, applied to the central amygdala oxytocin receptor-expressing astrocytes. This includes RNAse-free perfusion, combination of RNAscope and immunohistochemistry, and confocal imaging. This protocol provides detailed information about tissue handling and a comprehensive description of the RNAScope technique to label rat and mouse oxytocin receptor mRNA. We also describe three-dimensional reconstruction that allows the assessment of more than 70 different cellular parameters, powerful for studying astrocyte morphology and astrocyte-astrocyte interactions.
Before you begin
Astrocytes play a critical role in brain homeostasis by modulating neurotransmitter release, buffering extracellular K+, taking up active part in glucose metabolism, releasing various growth factors and playing a pivotal role for the formation of the blood-brain-barrier (Escartin et al., 2021; Sofroniew and Vinters, 2010). The shape, function and genetics of astrocytes can drastically differ depending on the species, sex, age, brain region and condition, with reactive astrocytes displaying severe morphological changes including somatic swelling, astrogliosis and retraction of astrocytic processes (Escartin et al., 2021). We recently described a subpopulation of astrocytes in the central amygdala of rats and mice that were positive for oxytocin receptor mRNA (Wahis et al., 2021). In these astrocytes, the prosocial neuropeptide oxytocin fine-tunes the intra-amygdala CeL→CeM neuronal pathway and promotes anxiolysis as well as the rodent equivalent of comfort. Intriguingly, we found that this subpopulation of oxytocin receptor-expressing astrocytes is morphologically distinct from other astrocytes, thus highlighting the importance of three-dimensional reconstruction for the assessment of morphometric parameters and astro-astrocytic contacts (Althammer et al., 2020; Wahis et al., 2021). While the general methodology and techniques described in this protocol can certainly be used for other types of astrocytes in a variety of brain regions, we would like to emphasize that this step-by-step protocol has been optimized to achieve reliable results for OTR-positive astrocytes in the CeL of rats and mice. In addition, we acknowledge that ethical guidelines and standards greatly vary between countries, states and institutions; thus, some of the described approaches for anesthesia and pain management might not be applicable to every lab and need to be verified before starting the experimental procedure to be in line with specific institute-approved guidelines.
Setting up perfusion pump (Part 1&2)
Timing: 10 min
-
1.
Always check that the perfusion pump is functional before starting to induce the anesthesia.
-
2.
Make sure that each tube is placed in the correct solution (PBS and PFA).
-
3.
Place a 18G needle on the tip of the main tube.
-
4.
Start by filling the PFA tube until the solution reaches the valve (main tube) and then switch to PBS solution.
-
5.
Once the main tube is filled with PBS, let the pump run for 5-10 seconds and check for any leaks.
-
6.
Check if there are any air bubbles inside of the tube and remove them by firmly flicking the respective parts of the tube.
-
7.
Once you removed all air bubbles, let the pump run for another 30 seconds.
-
8.
Proceed with induction of anesthesia.
Preparation and induction of anesthesia (Part 1&2)
-
9.
Before you can start with the perfusion, you need to induce deep anesthesia to avoid suffering of the animal during the procedure. It is important that you review the guidelines provided by the local authorities.
-
10.
Inject appropriate amount of anesthetic (ketamine/xylazine at 80/20 mg/kgbw or pentobarbital at 800mg/kgbw) in accordance with approved ethical guidelines or place animal in a box with sufficient amounts of Isoflurane (3-5mL per animal).
-
11.
Wait until breathing stops – this usually takes 3-7 minutes. If the animal is still breathing after 10 minutes, repeat Step 2.
-
12.
Test pedal reflex by firmly pinching the toes with a forceps.
-
13.
Proceed with the perfusion protocol.
Preparing the RNAScope setup (Part 4)
-
14.
Turn on the hybridization oven at least 45 minutes before you want to start the actual experiment. It should reach a stable temperature of 40°C.
-
15.
Prepare the fluorescently-conjugated dyes Opal/TSA Plus Cy3/5. Follow the manufacturer’s instructions (https://www.akoyabio.com/phenoptics/opal-kits-reagents/opal-reagent-packs/) on how to dilute the dyes (usually 75μl DMSO, 1:100 diluted in Millipore water).
-
16.
Turn on the water bath and set it to 37°C. Place mRNA probes and wash buffer in the water bath at least 30 minutes before you want to start the RNAScope procedure.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GFAP, goat polyclonal, 1:500 | Abcam | ab53554 |
| Glutamine synthetase, mouse monoclonal, 1:500 | Merck Millipore | MAB 302, clone GS-6 |
| Chemicals, peptides, and recombinant proteins | ||
| Formaldehyde 37% | Sigma-Aldrich | 8187081000 |
| InvitrogenTM Nuclease-free water | Fisher Scientific | AM9930 |
| Veterinary Euthasol Solution 100mL Pentobarbital Sodium Vial | Henry Schein | Cat#1311760 |
| Wash buffer | Acdbio | Comes with Multiplex V2 RNAScope kit |
| VECTASHIELD® Antifade Mounting Medium with DAPI | Vector Laboratories | H-1200-10 |
| Critical commercial assays | ||
| RNAscope Multiplex Fluorescent Reagent Kit v2 | Acdbio | Cat#323100 |
| Oxytocin receptor (OTR) probe (rat) | Acdbio | Cat#483671 |
| Opal dye 570 Reagent pack (Cy3) or TSA dye (Cy3/Cy5) | Akoya Biosciences | FP1488001KT Or NEL745001KT |
| Experimental models: Organisms/strains | ||
| Wistar rat, wildtype female, adult, 10–14 weeks | Envigo Harlan | RccHan®:WIST |
| C57BL/6 mice, wildtype female, adult, 10–14 weeks | Envigo Harlan | C57BL/6JOlaHsd |
| Software and algorithms | ||
| Imaris 9.51 (or higher) for Neuroscientists | Oxford Instruments, Bitplane | https://imaris.oxinst.com/microscopy-imaging-software-free-trial |
| Zen Black Software 2.1 | Zeiss | https://www.zeiss.de/mikroskopie/produkte/mikroskopsoftware/zen-lite/zen-lite-download.html |
| Other | ||
| SuperfrostTM Plus slides | Fisher scientific | Cat#10149870 |
| CorningTM netwellsTM inserts | Merck | CLS3477-48AE |
| Fine bone trimmer | Roboz | RS-8469 |
| Surgical scissors | Fine Science Tools | Cat#14001-18 |
| Bone scissor | Fine Science Tools. | Cat#16030-12 |
| Fine scissors | Fine Science Tools. | Cat#14058-11 |
| Specimen discs 30mm | Leica Biosystems | Cat#14037008587 |
| Two-stop peristaltic perfusion pump | Cole-Parmer | GZ-07522-20 |
| Short syringe needle, 16 or 18 G | BD | 305197 or 305195 |
| Scintillation vials | Merck – Sigma-Aldrich | V7005 |
| RNAseZapTM wipes | Thermo Fisher Scientific | AM9786 |
| 500mL beaker with handle and spout | Corning | 1015P-500 |
| Hybridization oven | Acdbio | Comes with Multiplex V2 RNAScope kit |
| Slide rack | Acdbio | Comes with Multiplex V2 RNAScope kit |
| Slide tray | Acdbio | Comes with Multiplex V2 RNAScope kit |
| PAP pen | Acdbio | Comes with Multiplex V2 RNAScope kit |
| Humidifying paper | Acdbio | Comes with Multiplex V2 RNAScope kit |
| Corning®Netwells® | Merck – Sigma-Aldrich | CLS3477 |
| 6-Well plates | Merck – Sigma-Aldrich | CC302/CLS3335 |
| Confocal Microscope | Zeiss | LSM780 |
Materials and equipment
Required tools for RNAse-free perfusion & brain extraction (Parts 1&2):
| Setup | Surgical instruments | Consumable |
|---|---|---|
|
|
|
0.01 M PBS (pH 7.2–7.6) – recipe for 1000 mL
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCL | 0.15 M | 8.5 g |
| Na2HPO4 | 0.01 M | 1.4 g |
| NaH2PO4 | 0.01 M | 0.2 g |
PBS is stable at room temperature for up to 1 year. Always make sure to check the solution prior to use, there should be no debris floating around in the bottle and the solution should be transparent.
Required tools for Cryosectioning (Part 3):
| Setup | Surgical instruments | Consumable |
|---|---|---|
|
|
|
All images presented in Part 5 (Confocal microscopy) were obtained using the Zeiss LSM 780 (upright) confocal microscope (Zen Black software, 2.1) using the following settings:
-
•
Frame size 1024 × 1024 pixels
-
•
Pinhole: 30–50AU (make sure to use 1 Airy unit to align individual channels and to be consistent with section thickness. All channels should have identical thickness (e.g., 0.9 μm) for separate specimen layer scanning during z-stack acquisition).
-
•
Lasers: 488 nm (at 1.3 mW, laser power), 561 nm (at 1.5 mW, laser power) and 633 nm (at 3.0 mW, laser power).
-
•
Gain (Master, analog): 665–740 V (Range 0–1200)
-
•
Digital Offset: 0 for 488, −10 to −150 for 561 and 633.
-
•
Scan Area: Zoom 0.7 (maximum field of view).
-
•
Averaging: Averaging 2 frames and displaying the mean
-
•
Bit Depth: 8-bit, bidirectional scanning.
-
•
Scanning speed: 6–7, Pixel Dwell 1.5–1.7 μs and scan time: 1.75–2.0 ms.
-
•
Z-stack scanning: 1 μm steps, scanning the entire slice (20–50 μm).
-
•
Objective: 40× oil (Zeiss immersion oil 518 F).
-
•
Selection of channels: Oxytocin receptor probe (Alexa 594, Jackson ImmunoReseach, red, 561 nm laser), GFAP (Alexa 643, Jackson ImmunoReseach, magenta, 633 nm laser) and glutamine synthetase (Alexa 488, Jackson ImmunoReseach green, 488 nm laser).
-
•
Emission/bandpass filter selection: 488 nm laser: MBS488, Plate/None/Mirror, BP505-530 nm; 561 nm laser: MBS561, Plate/None/Mirror, BP560-625 nm; 633 nm laser: MBS633, LP655 nm.
Step-by-step method details
RNAse-free perfusion & brain extraction (parts 1&2)
These parts are needed to perfuse and extract the rat and mouse brain. A brain that is cleared from blood, well fixated and undamaged will allow you to achieve optimal results. It is crucial that you work in a RNAse-free environment, as any contamination can lead to RNA degradation, which will ultimately negatively impact your RNAScope assay and the visualization of OTR mRNA.
-
1.
Set up perfusion pump and attach perfusion tubing and perfusion needle. First, run about 100 mL of tap water or saline through the tubing to remove any residue that might have accumulated inside of the tubing during previous uses. Then place open end of perfusion tube in beaker filled with cold 4% PFA (on ice). The volume of solution should be scaled to weight of animal and usually 200–300 mL will be sufficient for one 400g rat and 50–100 mL for a 30g mouse. Open valve (Cole Parmer Masterflex peristaltic perfusion pump with 2-way stop) and adjust to a slow steady drip (30–50 mL/min), and then close valve.
-
2.
Set up surgery site with scissors, forceps and clamps. Use a cleaning agent (e.g., Invitrogen RNAse Zap wipes, 9048-46-8) for removing RNase from all tools and surfaces. Give an appropriate amount of anesthetic to the animal (Isoflurane, CO2, ketamine/xylazine or pentobarbital). Results and data presented here and in Wahis et al., (2021), were exclusively obtained using pentobarbital or isoflurane, thus we cannot rule out that other means of anesthesia might affect the final results. Make sure that your anesthetic method has been approved and is in line with your animal protocol. In our case, we used 0.5 mL pentobarbital (Euthasol, Virbac, ANADA #200-071, Fort Worth, TX, USA, Pentobarbital, 80mg/kgbw intraperitoneal, i.p). and waited until the animal displayed signs of deep anesthesia. Once the animal is under anesthesia, place it on the operating table (inside the biosafety hood) on its back.
-
3.
To confirm deep anesthesia, use both palpebral and paw reflex control to easily ensure the depth of anesthesia. Animal must be unresponsive before proceeding with the following steps.
CRITICAL: Appropriate regulations for experiments with rats must be followed. The procedure complies with the animal surgery guidelines, including using appropriate anesthesia and analgesia to minimize pain.
-
4.
You may use some tape to hold the appendages so that the animal is securely fixed. We prefer using a styrofoam lid and four 18G (Birmingham gauge system) needles to pin down the animal.
-
5.
Lift the skin and make a small incision with scissor (or scalpel) through abdomen to verify the exact position and to avoid unnecessary damage to internal organs. Next, cut along the entire length of the diaphragm to expose the chest cavity. With sharp scissors, cut through the connective tissue at the bottom of diaphragm to allow access to rib cage.
-
6.
With large scissors, blunt side down, cut through ribs just left of the rib cage midline. An overview of all tools needed for the perfusion and extraction can be found on Figure 1A.
-
7.
Make one center or two end horizontal cuts through the rib cage, and open up thoracic cavity. Clamp open to expose heart and provide drainage for blood and fluids. We recommend performing the whole procedure on a styrofoam lid that is placed into a large plastic or metal tray.
-
8.
While holding heart steady with forceps (it should still be beating), insert about 10–15 mm of a needle directly into protrusion of left ventricle and insert (for mice, use a smaller gauge needle 21 or 23G an insert for 5–10 mm).
-
9.
Make cut in the right atrium (only use the tips of small scissor, 1–2 mm cut) with sharp scissors, and make sure solution is flowing freely. Start the perfusion with 1× PBS at 4°C. If fluid is not flowing freely or is coming from animal’s nostrils or mouth, reposition the needle (Figure 1B).
-
10.
When blood has been cleared from body (usually after 1–2 min) change to 4% paraformaldehyde solution (200–300 mL for rats and 50–100 mL for mice (depending on animal’s weight).
-
11.
Perfusion is almost complete when formalin-induced muscular contractions and lightened color of the liver (incision in the abdomen to expose liver is needed), due to the replacement of blood by the perfusion solution, are observed. Additionally, you may carefully check for stiffness in the neck of the animal (rigor), which is a good indicator of a sufficient perfusion.
-
12.Extraction of the brain:
-
a.Stop the perfusion and excise the brain by cutting the optic tract and carefully removing the brain from the skull. First, decapitate the animal using a guillotine. Cut along the anterior–posterior axis to remove the fur and expose the skull. Make sure that the spinal cord is visible – if not, use a large scissor to remove the remaining flesh. Use a bone cutter to make two cuts (left and right of the spinal cord) all the way to the bony external auditory canals (ear holes). You should hear a loud crack when making the cut. Be careful not to damage the brain (damaging the cerebellum is acceptable).
-
b.Use the bone cutter to make one more vertical cut above the spinal cord. Now, use a sturdy plier/forceps to peel off little pieces of the skull until the brain is completely exposed. You might have to remove/cut the dura mater first, if you see that the brain gets squished when removing skull pieces. Next, use a sturdy plier to crack the olfactory bone. Flip the brain upside down and use a spatula to carefully expose the optic tract. If you can’t see the optic tract yet you might need to use the bone cutter to make two more cuts in the top left and right corners to detach the brain from the skull. Use a feather scissor to cut the optic tract and now the brain should slide out of the skull without much resistance. Place brain in glass vial containing the same 4% PFA solution and fix for 4 more hours at 4°C before proceeding to dehydration and embedding. To save time, the above-mentioned steps can be performed for multiple brains prior to postfixation.
-
a.
-
13.
After 4 h of postfixation, transfer brains into RNAse free 30% sucrose (PBS) and store at 4°C until brains have sunk to the bottom of a sealable glass vial (usually 20 mL, big enough for rat brain with plastic lid) (this usually takes between one and a half to two days, 36–48 h). This step mainly serves as a dehydration (preventing the formation of ice crystals) and cryoprotection of the tissue before freezing it.
-
14.
Once brains have sunk to the bottom of the glass vial, carefully remove them by either pouring the solution or using a spatula. Brains should be carefully dried using a kim wipe, wrapped in aluminum foil and stored at −80°C for further analysis. While other methods of storage in the −80°C freezer such as tubes can be used too, we recommend using aluminum foil to provide a protective outer layer for the brains. This step is very important, because you want to conserve RNA as much as possible. If left too long in the sucrose solution, RNA starts degrading. Therefore, as soon as brains have sunken, block and store brains in −80°C for later analysis. While purified RNA is usually stable for days up to weeks at 4°C, the preparation of sucrose solution that is completely free of RNAses cannot always be guaranteed and thus we recommend to not wait longer than 24 h before transferring the brains to the −80°C freezer after they have sunk to the bottom of the glass vial.
-
15.Blocking and storing brains at −80°C:
-
a.It is very important that all these steps are performed under RNAse free conditions and as fast as possible to avoid RNA degradation. Therefore, using an RNase removing agent like RNAse Zap, clean all tools, tabletops, brain blocks and utensils you need for blocking and storing brains.
-
b.With tweezers, remove brain from brain jar (one at a time) and put it into a brain matrix. If no brain matrix is available, use a flat surface. Separate forebrain from hindbrain using a razor blade (Figure 1C). This step can also be performed immediately prior to sectioning the brain with the Cryostat. Minimize the time that the brains are at room temperature to avoid thawing of the tissue.
-
c.Put both fore and hindbrains on a piece of foil paper and close it tightly. Store immediately into a −80°C freezer or dry ice.
-
a.
Figure 1.

Perfusion and extraction of the rat brain
(A) Overview of tools needed for the perfusion and extraction of rat brains. The name and identity of all tools can be found in the key resource table.
(B) Schematic depiction of intraventricular perfusion of the rat heart. Perfusion needle must be inserted into the left ventricle, while the right atrium needs to be incised to allow the blood to flow out.
(C) Prior to cryosectioning, brains need to be cut with a razorblade to separate the forebrain from the hindbrain/brainstem. If available, use a coronal brain matrix to separate the brain. The bottom end of the brain will be glued onto a specimen disk using Tissue-TekTM O.C.T.TM compound. This figure was created with BioRender.com.
Cryosectioning (Part 3)
This part is necessary to slice the brain into thin sections, which will be used to perform the in situ hybridization and immunohistochemistry. The optimal slice is thick enough to remain stable and to allow reconstruction of astrocyte morphology, but thin enough to allow thorough penetration by mRNA probes and antibodies for robust visualization of mRNA and proteins. We recommend a slice thickness of 20–25 μm, although slightly thinner (10–15 μm) or thicker (30 μm) brain slices might works as well.
-
16.
Section brain tissue in a cryostat at 20–25 μm thickness and immediately place cross-sections in RNase-free PBS using a brush (always leave the brush in the cryostat, otherwise the brain sections will immediately thaw and stick to it) using regular well plates.
-
17.
Transfer brain sections from PBS to a new well using a brush and incubate sections in H2O2 (10%) for 10 min at room temperature (RT; 20°C–25°C) with free-floating brain sections.
-
18.
Transfer sections to well nets and wash them 3 × 5 min in RNAse-free PBS.
-
19.
Mount sections in RNAse-free PBS (pH 7.4) onto SuperFrostTM glass slides by submerging the slides in PBS, carefully moving the sections to the right position on the slide using a brush and then pinning down the section with the brush while moving the slide out of PBS at the same time. We recommend to not mount more than 2 sections on one slide.
-
20.
Draw circle around the sections with the PAP pen (comes with the acdbio RNAScope kit) and let air dry for 30 min. The circle should be as small as possible, without touching the brain sections. You want to minimize material consumption, but make sure to leave a ≅ 5 mm gap between the hydrophobic barrier and the brain tissue.
-
21.
Dip slides in 100% EtOH (molecular grade).
-
22.
Let slides dry in the dark overnight (at least 4 h) either in a box or simply by covering them with a styrofoam lid or aluminum foil etc.
RNAScope and IHC (Part 4)
This part accomplishes the visualization of OTR mRNA and the astrocytic GFAP and GS proteins in mounted brain slices. Astrocyte markers will be used to identify and three-dimensionally reconstruct astrocyte surface and filaments. The RNAScope in situ hybridization for OTR mRNA allows you to discriminate between OTR-positive and OTR-negative astrocytes.
-
23.
Remove RNAScope kit from 4°C and leave at room temperature (20°C–25°C) until you need to use the reagents.
-
24.
Place C1 probes in 40°C water bath for 10 min.
-
25.
C1 (channel 1) probes are ready-made and do not need to be diluted (order the oxytocin receptor/OTR probe as C1).
-
26.
Place concentrated acdbio wash buffer (WB; 50×) in 40°C water bath for 10 min. Dilute wash buffer to 1× (2940 mL dH2O + 60 mL 50× WB). Put wet humidifying paper in a plastic slide box with a sealable lid for the hybridization oven and then pre-warm the slide box and oven to 40°C for at least 30 min.
-
27.
Incubate sections in Pretreat 3 (Protease III, comes with acdbio kit) for 20 min at RT. Note: Protease treatment might have to be adjusted individually using shorter/longer incubation times depending on the tissue quality. Ideally, protease pre-treatment results in a strong mRNA vis
-
28.
Decant Pretreat/Protease 3 solution, place slides in a slide rack/wash container filled with dH2O and then wash 2 × in dH2O by moving the slide rack up and down (1 min each).
-
29.
Remove slides one at a time from the wash rack and remove the wash solution by placing the corner of a kimwipe adjacent to the sections.
-
30.
Immediately add 1–3 drops of OTR probe to sections and incubate at 40°C for 2 h.
-
31.
Decant solution and immediately place in WB. Rinse 2 × for 2 min at RT, with periodic agitation of the slides (washing steps should be performed in the wash tray supplied by the manufacturer by carefully tilting the tray up and down a few times each minute. 200 mL of washing buffer are sufficient for each step. Alternatively, classical coplin jars can be used and slides are placed in the washing buffer and moved up and down 3–5 times over 2 min).
-
32.
Repeat step 29 Immediately add 1–3 drops of AMP1 (Amplification solution 1, comes with acdbio kit) to sections and incubate at 40°C for 30 min.
-
33.
Repeat step 31
-
34.
Repeat step 29 Immediately add 1–3 drops of AMP2 (Amplification solution 2, comes with acdbio kit) to sections and incubate at 40°C for 30 min.
-
35.
Repeat step 31
-
36.
Repeat step 29 Immediately add 1–3 drops of AMP3 (Amplification solution 3, comes with acdbio kit) to sections and incubate at 40°C for 15 min
-
37.
Repeat step 31.
-
38.
Repeat step 29 Immediately add 1–3 drops of HRP-C1 (horseradish peroxidase C1, comes with acdbio kit) to sections and incubate at 40°C for 15 min.
-
39.
Repeat step 31
-
40.
Prepare a 1:1000 dilution of Opal/TSA Plus Cy3/5 (or far red/ cyan colored solution) (Separate Kit from Akoya Bioscience/Perkin Elmer).
-
41.
Repeat step 29.
-
42.
Immediately add 1–3 drops of Opal/TSA Plus Cy3/5 (prediluted) to sections and incubate at 40°C for 30 min. Follow the manufacturer’s instructions (https://www.akoyabio.com/phenoptics/opal-kits-reagents/opal-reagent-packs/) on how to dilute the dyes (usually 75 μL DMSO, 1:100 diluted in Millipore water).
-
43.
Repeat step 31
-
44.
Repeat step 29 Immediately add 1–3 drops of HRP (horseradish peroxidase) blocker to sections and incubate at 40°C for 15 min.
-
45.
Repeat step 31
-
46.
Perform immunohistochemistry using GFAP and/or glutamine synthetase antibodies (we use 1:250 or 1:500 for both antibodies, Figure 2). No additional blocking step is required following the RNAScope procedure.
-
47.
Incubate sections with primary antibodies overnight (at least 12 h). In our hands, 2-day incubation with the primary antibodies did not significantly improve the signal intensity compared to overnight incubation (Figure 2D). Rinse 2× with dH2O for 2 min at RT.
-
48.
Incubate with secondary antibodies (Alexa488/594/643-conjugated, 1:250 or 1:500) for 4 h at RT. Rinse 2 × with dH2O for 2 min at RT.
-
49.
Carefully dry the sections using the edge of a kimwipe. Apply antifade mounting medium (roughly 60 μL, containing DAPI if needed). Make sure the mounting media has the same refractive index as the objective for improved image quality and dimension accuracy when obtaining images at high magnification.
-
50.
Coverslip the slide and remove any excess liquid from the edges of the slide using a kimwipe. You can gently tilt the slide to let the excess liquid flow down onto the kim wipe.
-
51.
Seal of the edges using regular clear nail polish to prevent the mounting media from escaping.
Figure 2.

Visualization of OTR-expressing astrocytes in the central amygdala
(A) Schematic depiction of the location of OTR-positive astrocytes in the central lateral amygdala.
(B) For the visualization of OTR-positive astrocytes after RNAScope and IHC, 25 μm thick z-stacks should be obtained at the confocal microscope.
(C) It is important to adjust the laser intensity prior to imaging using the negative control probe. All channels should be adjusted so that virtually no signal is visible for the negative RNA control probe to minimize the chances of false positive detection.
(D) Confocal image shows OTR-positive astrocytes labeled via GFAP, GS, DAPI and OTR mRNA probes. No drastic differences were observed when incubating 48 instead of 24 h with the primary antibodies. This figure was created with BioRender.com.
Confocal microscopy (Part 5)
While we are aware that different labs and institutes use entirely different confocal models and software, we believe it is still important to share the exact hardware, software and confocal settings used to obtain our data to provide a ballpark for individual adjustments and optimization. We strongly recommend collaborating with a microscopist as settings usually need to be adjusted for each experiment and can drastically impact the quality of the data obtained and, thus, ultimately affect the data analysis.
-
52.
Take your coverslipped sections to the confocal. Make sure that the nail polish completely dried before your star imaging (at least 24 h).
-
53.
Turn on the confocal at least 30 min before using it and make sure that laser power is stabilized (switch laser stabilizer from idle to on) before starting the imaging.
-
54.
Put some immersion oil on the slide and make sure to identify the correct brain region (i.e., central amygdala) using landmarks such as the optic tract and/or a rat brain atlas before starting the imaging process.
-
55.
Once the correct anatomical location has been verified using the eyepieces and fluorescent LEDs, switch to imaging mode.
-
56.
Adjust all three confocal channels (red, green and blue) according to the above-mentioned settings. We recommend not combining the channels so that each channel can be activated individually in the live mode.
-
57.
Once the channel settings are adjusted, use the GFAP signal (633 nm laser line, magenta) to set the top and bottom borders for your z-stack imaging (channels are acquired sequentially, not interwoven).
Note: z-stack imaging needs to be activated first by ticking the respective box at the top left corner of the software interface.
-
58.
Start imaging by clicking on ‘Start experiment’. The acquisition of one z-stack takes between 3 and 8 min.
-
59.
Files can be saved in the proprietary Zeiss format (czi) and directly imported into Imaris (see three-dimensional reconstruction below).
Note:Figure 1 provides a schematic depiction of the confocal microscopy process and example images showing OTR-positive and -negative astrocytes labeled via GFAP and GS.
Three-dimensional reconstruction (part 6)
This part is necessary for reconstructing astrocyte morphology, including surfaces and filaments. Based on the immunohistochemical staining for GFAP and GS, a three-dimensional model of individual astrocytes is created. This reconstruction allows you to assess more than 70 different cellular parameters and to compare complexity, domain size or cell volume of different astrocytes.
-
60.
Open z-stack file in Imaris by dragging and dropping the file into the Arena (Figure 3). Imaris supports most common image formats (czi, LIF etc.)
-
61.
Start by loading the file via double-clicking on the icon. A three-dimensional overview of immunohistochemical signals will appear and display all individually labeled channels. Individual channels (intensity and contrast) can be adjusted and turned off via Display Adjustment (ctrl+D).
-
62.
To create a new surface reconstruction, click on New Surface at the top left corner (blue drops symbol) of the Imaris menu. If you have previously saved settings, you can select them now via Favorite Creation Parameters (see below).
-
63.
Click on the blue arrowhead at the bottom left to continue. Select the channel you want to reconstruct. There are several things to consider when choosing Surfaces Detail and Local Contrast. If you start reconstructing astrocytes for the first time and don’t know what the ideal settings could be, we recommend using Surfaces Detail 0.3 μm and Background Subtraction (Local contrast) 1.2 μm. Note that these parameters are sensitive to the quality of your images. The better your immunohistochemical staining, the easier it is for the software to detect true signal and thus settings allowing a better voxel resolution (quality of three-dimensional pixels in Imaris) can be used. Choosing the proper settings for the semi-automated reconstruction pipeline is an iterative process. Once you find your ideal settings, you need to use the same settings for all your specimen within the same experiment to yield consistent results.
Note: Before collecting large amounts of data for three-dimensional reconstruction, make sure that the slice preparation and quality of the immunohistochemical staining are sufficient for reconstruction via Imaris. Please see step 64 and troubleshooting section.
-
64.
Click on the blue forward button (Figure 4A, red box and arrow) at the bottom left to continue. Adjust the Threshold (Absolute Intensity) using your cursor and dragging the yellow line to the left or right (Figure 4A, blue box and arrow). It is paramount that the fluorescent channel that you want to reconstruct is visible during this process. Ideally, the Threshold should be adjusted so that you have close to 100% overlap between your immunohistochemical signal and the preview of the surface reconstruction (Figure 4A, bottom right panel).
-
65.
Click on either the blue or green arrow at the bottom right to finish the surface reconstruction (Figure 4A, red box). Carefully check if you are satisfied with the reconstruction by moving around and zooming in and out. Examples of sufficient and insufficient surface reconstructions are shown in Figure 4B. Click/unclick both the fluorescent channel and the surface reconstruction to check if there is sufficient overlap. If you need to refine, start over from step 63 If you are happy with the result, move to step 66.
-
66.
Before any usable data can be obtained, reconstructions need to be post-processed by filtering out small debris that will confound analysis. To do so, click on the gray Filter icon (Figure 5, red box and arrow). We recommend selecting the Volume filter (blue box and arrow), which you can find at the very bottom of the drop-down menu. Select a setting that filters out small debris/particles, which are clearly too small to be an individual astrocyte. We recommend co-staining the brain sections with DAPI and/or glutamine synthetase to help identify astrocyte somata.
-
67.
All particles that are to be removed will appear in yellow (Figure 5, bottom left panel). Click on the icon with the orange pencil, which is located directly left of the Filter icon (green box and arrow). By clicking Delete all selected particles will be removed from the current reconstruction. By clicking Mask All, you will create a new surface reconstruction, which only contains filtered, true astrocyte surfaces. You can repeat this process until you are satisfied with the final result. Each run will create a new template, so be sure to select the correct Channel when reconstructing astrocyte filaments (see step 71).
-
68.
The final step before collecting data on astrocyte surface parameters is to isolate astrocytes so that the software recognizes them as individual entities. To do so, click on each astrocyte individually. Selected astrocytes will appear in yellow (see Figure 6A). As astrocytes contact each other quite frequently, you will often see that 2 or more astrocytes will be selected at the same time, as their surface reconstructions will be connected to each other. To overcome this issue, you need to manually cut astrocyte processes at the contact points of two different cells. Use the Cut function, which you can find by clicking on the orange pencil icon. Cuts will always be done perpendicular to the 3D arena, so make sure that you bring your astrocytes in the correct position prior to cutting. Left click and a thin blue line will appear that tells you where the cut will be made (Figure 6A).
-
69.
At this point we also recommend manually deleting astrocytes that are too close to the x, y or z plane. Not removing incomplete astrocytes can confound the data set and lead to inconsistent data collection. Left click on the respective astrocytes and click delete on your keyboard or left click delete in the orange pencil tab. Fused astrocytes will now be separated (Figure 6B).
-
70.
Check a few pre-selected parameters such as surface area, cell volume or filament length on individual astrocytes simply by clicking on them (Figure 7A) after selecting Statistics. The final step is exporting all collected surface parameters as an Excel file (Figure 7A, red box and arrow, bottom right). Files can now be viewed in Excel using the respective parameter tabs (Figure 7B). Each row represents an individual cell (astrocyte) and each column provides information about the respective value and its units, categories of parameters, as well as the individual IDs for each astrocyte.
-
71.
Reconstruction of astrocyte filament is performed based on prior surface reconstruction. To do so, create a New Filaments tab (Figure 8, top left, green leaf, white box and arrow).
-
72.
It is very important to select the correct surface source channel, where all particles have been filtered out and fused astrocytes have been manually cut. Thus, you can either manually rename the respective channel or pick the masked source channel with the highest number (i.e., pick masked channel 4 over 3 etc.) The correct surface channel is usually the one listed at the bottom of the drop-down menu.
-
73.
Imaris provides automatically calculated starting and seed point values based on the previously reconstructed surface. These values usually work quite well and should be around 25–30 μm for starting points and 1–2 μm for seed points when reconstructing astrocyte filaments. Click the blue arrow to proceed to starting and seed point placement.
-
74.
Starting and seed point placement can be adjusted in analogy to the signal intensity thresholding (step 64). Adjust the respective lines so that you only have 1 starting point in the center of the astrocyte soma (Figure 8, green box and arrow). Seed points should overlap with bifurcations of astrocyte processes (Figures 9A and 9C). Occasionally, starting points need to be removed or placed manually, which can be done via shift left/right click.
-
75.
Click on the green arrow to finish filament reconstruction. Filament data can be exported in analogy to step 70.
Note: Choosing surface detail and local contrast is always a tradeoff between processing time, consistency, and false positives. Before starting to reconstruct and analyze astrocytes (or any other cell type via Imaris), define what cellular parameters are relevant for your study. Do you simply need a cell count? Do you need highly detailed reconstructions of filaments and surfaces? Do you need to be able to discriminate between two astrocytes in close proximity? Is it important to only include complete astrocytes in your analysis, which are not cut by x, y or z planes? Adjusting surfaces detail and local contrast can make a huge difference in terms of processing time, especially when you reconstruct the filaments based on the surface reconstructions. A less detailed astrocyte reconstruction could take 8 minutes per z-stack (100–200 MB), while a highly detailed reconstruction could easily take 15–20 min (500+MB). Assuming that you analyze tens or hundreds of z-stacks, the time that Imaris needs for simply computing surface and/or filaments increases rapidly. Thus, think ahead and make sure that your reconstructions only fulfill the minimum requirements for your respective study. In addition, several helpful tutorial videos can be found here:https://imaris.oxinst.com/tutorials.
Figure 3.

Opening confocal files in Imaris and creation of subfolders
(A) Confocal files can be imported in the Imaris arena by drag and dropping them into the software interface. Individual subfolders can be created by clicking New Folder at the top and will appear in the arena (red box).
(B) Files can now be moved into the respective subfolders and opened by double-clicking on the icon (red box).
Figure 4.

Proper thresholding of signal intensity for astrocyte processes
(A) Each individual z-stack needs to be manually adjusted using the Threshold (Background Subtraction) function prior to reconstruction. Adjust the threshold using the left mouse click by dragging it to the correct position. Raw IHC (top left) and the three-dimensional surface preview should have near-perfect overlap (bottom right) before you continue. Examples of low (top right) and high (bottom left) thresholds are shown where a clear mismatch of raw fluorescence and surface reconstruction preview can be observed.
(B) Example images of insufficient (threshold too high) and sufficient (threshold optimal) surface reconstruction using.
Figure 5.

Filtering out debris and unspecific signal in the surface channel
After reconstruction, filter settings need to be adjusted to filter out small pieces of surface reconstruction that can confound the analysis from the unfiltered image (top left). Use Filter Type: Volume to set a threshold for the filter so that only complete, actual astrocytes remain (bottom left). Debris that are about to be removed appear in yellow, while the remaining surface reconstructions will appear in the original color. If the filter is set too coarse, actual astrocytes will appear yellow and would thus be removed (top right). Once the debris is removed, the remaining, filtered surface reconstructions are displayed in the original color (bottom right). Arrows and boxes show masking tab (green), filter tab (red) and filter type (blue).
Figure 6.

Separation of fused astrocytes by cutting through astrocyte processes
(A) Occasionally, Imaris accidentally recognizes two separate astrocytes as one entity (yellow astrocytes), when their filaments are very close together. If this happens, use the cut function (shift + left-click in the pencil mode) and a thin blue line (the line was artificially enhanced for better visibility in this image) will appear. This line indicates where the cut will be made (red arrows). The blue lines in the original image are shown in the enlarged inset on the right. (B) Once the cut has been made, astrocytes will be separated and clicking on them will only select one (see single astrocyte highlighted in yellow), but no longer both.
Figure 7.

Selection of individual astrocytes and parameter readout
(A) You can manually select reconstructed astrocytes by left-clicking on their surface (as indicated here in yellow). By clicking on the statistics icon (red box and arrow) and selecting different parameters from the drop-down menu the program provides information about various cellular parameters (i.e., surface area in μm2 in this example).
(B) All these parameters will appear in your Excel file after you export the data (red box) and can be looked up for each individual astrocyte, here shown for surface area in the current sheet.
Figure 8.

Starting point placement for filament reconstruction of astrocytes
Prior to filament reconstruction (start by clicking on the filament symbol, green leaf, white box and arrow), starting points need to be placed in the center of the astrocyte somata. While the software places those starting points automatically (simply by clicking the blue forward button, red box and arrow), they often need to be manually adjusted. Adjust the threshold by dragging the yellow line (blue box and arrow) to the left or right. Rotate the image and zoom in to make sure that each astrocyte has exactly one starting point (shown in cyan, see green boxes and arrows) in the center of their soma. You can manually add and remove starting points by hovering over the starting points and shift left click or delete/right click, respectively, if necessary.
Figure 9.

Seed point alignment for filament reconstruction
(A) In order to track filament length, number of filaments and number of bifurcations, Imaris places seed points (this is done automatically, by clicking on the blue forward button, see red box and arrow) along the astrocyte filaments based on IHC signal and surface reconstruction.
(B) Seed points appear mainly at the initial segment of the filament and increase in density in close proximity of a bifurcation. Usually, this feature does not need to be adjusted manually, but should be checked if obtained filament parameters seem inaccurate. As for the starting point placement, the threshold can be adjusted manually and should align nicely with the filament projections as depicted in the image.
(C) Seed points should align with the reconstructed filaments as shown in the overlay image.
Expected outcomes
Here we provide a detailed protocol for the visualization and three-dimensional reconstruction of OTR-expressing astrocytes in mice and rats. Immunohistochemical staining via GFAP and GS reliably labels astrocytic filaments and somata, respectively (Figure 2D). GS-positive, but GFAP negative cells most likely are oligodendrocytes and should not be included in subsequent analyses. OTR mRNA signal is mainly confined to astrocyte somata (Figure 2D), albeit we occasionally observed the presence of OTR mRNA in astrocytic processes. Three-dimensional reconstruction in Imaris is based on the immunohistochemical signal, depends on the overlap of immunosignal and predicted surface and provides a near-perfect model of astrocyte morphology (Figure 4B). For the central amygdala we reported that, on average, OTR-positive astrocytes are larger, more complex, have a bigger domain size and contact neighboring astrocytes more frequently than their OTR-negative counterparts. We rarely observed OTR-positive astrocytes with cell volume lower than 100 μm3 or more than 10,000 μm3 or surface areas lower than 500 μm2 or higher than 20,000 μm2. The potential presence of OTR-expressing astrocytes and, thus, their morphology, in other brain areas besides the amygdala is to the best of our knowledge currently unknown.
Quantification and statistical analysis
Imaris-generated data files are compatible with both Excel and Numbers and generated data sets can be easily imported to either software. For the statistical analysis common programs such as Graphpad Prism or SPSS can be used. In addition to the exported astrocyte parameters, Imaris also allows you to measure the signal intensity of astrocyte-confined mRNA transcripts. This can be achieved by manually creating spheres that precisely engulf the astrocytic soma and, thus, the mRNA signal. Use the Spot function (top left, orange symbol) and then clicking on the create tab (orange pencil). You can place the sphere via shift+left mouse click and manually adjust the size by rotating the mouse wheel or adjusting the diameter of the selected spot. You can now click on any spot and export data in analogy to step 70. The mRNA intensity can be found under the Mean Intensity tab in Excel.
A comprehensive selection of free Imaris and Fiji tutorial videos uploaded by IMB Microscopy can be found here: https://www.youtube.com/c/IMBMicroscopy/videos. We specifically recommend:
Introduction to Image Analysis: https://www.youtube.com/watch?v=qkgADgd7xu0, Introduction to Image Processing: https://www.youtube.com/watch?v=5tFz5z88JzY, Intermediate Fiji Workshop: https://www.youtube.com/watch?v=VnYQ3UAoKVE
3D quantification in Imaris:https://www.youtube.com/watch?v=8ldNUmWNbB0.
Limitations
Due to the reduced thickness (20–25 μm) that is needed for sufficient penetration of the RNAscope probe, each section inevitably contains less fluorescently-labeled astrocytes. This also means that several astrocytes in the field of view will be truncated in the z axis and will not be reconstructed in their entirety. Depending on the assessed condition, analyzed brain region or quality of the IHC staining and confocal images, severe fusion of astrocytes might occur. While it is technically possible to separate fused astrocytes in Imaris, it is very time-consuming and should only be performed if there is a small number of fused cells in each z-stack. Thus, it is always preferable to adjust staining, confocal imaging and reconstruction of surface in Imaris so that minimal fusion of astrocytes occurs and individual cells can be reconstructed and analyzed.
Troubleshooting
Problem 1
Absence of probe signal (steps 30–40).
Potential solution
There are a number of potential causes for absence of mRNA signal. We recommend a step-by-step approach to eliminate the most likely underlying candidates. (1) Make sure you ordered the correct probe (target, species) and applied sufficient amounts (50 μL per section). (2) Increase the duration of the H2O2 treatment. (3) Increase the duration of the protease pretreatment to allow for better tissue penetration. 4) Make sure you apply AMP 1–3 in the right order, right amounts and double-check whether you apply the correct HRP for the respective channels (C1–C3). (4) Always run a positive and negative control probe. This allows you to rule out problems with the dyes. If the problem persists, reach out to acdbio and ask for help with troubleshooting of your protocol. Depending on the individual case, additional pre-treatment such as various target retrieval steps might be necessary.
Problem 2
Absence of IHC signal (steps 46–48).
Potential solution
While we never experienced complete absence of the IHC signal, we noticed that a longer protease pre-treatment leads to a weaker IHC signal. Consider incubating with the primary antibody for a longer time (2 instead of 1 day) and increase the antibody concentration. If possible, reduce the time of the protease pre-treatment to eliminate less protein from the specimen. However, it is paramount that you make sure to still have a decent probe signal when adjusting.
Problem 3
Unspecific probe signal (steps 30–40).
Potential solution
The two most plausible reasons for this phenomenon are: (1) Cross-reactivity due to incorrect selection of probe sequence and (2) adhesion of fluorescent dyes to the specimen. Selection of the correct probe sequence is critical to avoid unspecific binding. Adhesion of fluorescent dyes can be prevented by thorough rinsing in between steps with periodic agitation of the slides.
Problem 4
Sections fall off the slides (steps 19–22).
Potential solution
Make sure to always use SuperFrostTM glass slides for mounting. Letting slices dry overnight (<12 h) is critical. If the problem persists, consider using a lower sodium PBS for mounting and make sure that the fixation of your tissue (perfusion and post-fixation) is sufficient.
Problem 5
Imaris performs surface reconstruction of astrocytes, but the final result is one giant blob and not individual astrocytes (step 64).
Potential solution
Adjust thresholding (Figure 1) and surface detail so that reconstruction processing is less coarse. Alternatively, revise your staining protocol to achieve a better signal-to-noise ratio (ratio of strength of immunohistochemical signal carrying information to that of unwanted interference/background; should not be lower than 2) and less background staining. Please see: https://www.olympus-lifescience.com/de/microscope-resource/primer/techniques/confocal/signaltonoise/
Problem 6
Imaris does not catch up to the IHC signal and reconstructs only a few or no astrocytes at all (step 64).
Potential solution
The most likely cause for this problem is a weak IHC signal. Try to adjust your laser settings at the confocal microscope so that you obtain ideal images with a sufficient signal-to-noise ratio (2–3 range), where astrocytic processes are clearly visible and distinct from the background. We recommend the consultation of a confocal microscopy expert to make sure that image acquisition yields optimal results. In addition, adjust thresholding (Figure 3) so that more IHC signal is converted into three-dimensional surface.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Javier Stern, jstern@gsu.edu.
Materials availability
This study did not generate new unique reagents.
Acknowledgements
This work was supported by DFG Postdoc Fellowship AL 2466/1-1 to F.A., National Heart, Lung, and Blood Institute Grant NIH HL090948 to J.E.S., National Institute of Neurological Disorders and Stroke Grant NIH NS094640 to J.E.S., and funding provided by the Center for Neuroinflammation and Cardiometabolic Diseases (CNCD) at Georgia State University, the German Research Foundation (DFG) grants GR 3619/8-1, GR 3619/13-1, GR 3619/15-1, GR 3619/16-1, Training Research Group (GRK) 2174 and SFB Consortium 1158-2 to V.G., the Centre National de la Recherche Scientifique contract UPR3212, the Université de Strasbourg contract UPR3212, ANR JCJC grant 19-CE16-0011-0 to A.C., and ANR-DFG grant GR 3619/701 to A.C. and V.G., National Heart, Lung, Blood Institute Grants NIH HL150750 and HL136595 to E.G.K. The graphical abstract and Figures 1 and 2 have been created with BioRender.com.
Author contributions
F.A. drafted the protocol and provided data and images. All other authors critically read and approved the manuscript.
Declaration of interests
The authors declare no conflict of interest.
Contributor Information
Ferdinand Althammer, Email: falthammer@gsu.edu, ferdinand.althammer@med.uni-heidelberg.de.
Alexandre Charlet, Email: acharlet@unistra.fr.
Javier E. Stern, Email: jstern@gsu.edu.
Data and code availability
This protocol did not include a particular code.
References
- Althammer F., Ferreira-Neto H.C., Rubaharan M., Roy R.K., Patel A.A., Murphy A., Cox D.N., Stern J.E. Three-dimensional morphometric analysis reveals time-dependent structural changes in microglia and astrocytes in the central amygdala and hypothalamic paraventricular nucleus of heart failure rats. J. Neuroinflammation. 2020;17:221. doi: 10.1186/s12974-020-01892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C., Galea E., Lakatos A., O'Callaghan J.P., Petzold G.C., Serrano-Pozo A., Steinhauser C., Volterra A., Carmignoto G., Agarwal A., et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021;24:312–325. doi: 10.1038/s41593-020-00783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew M.V., Vinters H.V. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahis J., Baudon A., Althammer F., Kerspern D., Goyon S., Hagiwara D., Lefevre A., Barteczko L., Boury-Jamot B., Bellanger B., et al. Astrocytes mediate the effect of oxytocin in the central amygdala on neuronal activity and affective states in rodents. Nat. Neurosci. 2021;24:529–541. doi: 10.1038/s41593-021-00800-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This protocol did not include a particular code.