

Abstract
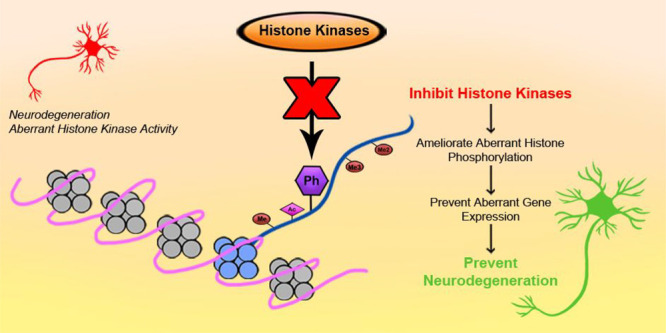
Breakthroughs in understanding the epigenetic mechanisms involved in neurodegenerative disease have highlighted “epidrugs” as a potential avenue for therapeutic development. Here, we expand on the future of epidrugs against neurodegeneration and discuss promising novel targets underexploited thus far: histone kinases.
1. Introduction
An aging population has led to a drastic increase in the prevalence of neurodegenerative diseases.1 Among these disorders, amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) affect different neuronal types in distinct locations of the nervous system causing motor and behavioral symptoms, respectively. Due to an overlap in disease mechanisms, these two disorders are now thought to lie on two ends of a disease continuum. The vast majority of ALS/FTD cases are sporadic, while a small proportion run in families.1 Several genes have been linked to ALS/FTD pathology, including superoxide dismutase 1 (SOD1), fused in sarcoma (FUS), TAR DNA-binding protein 43 (TDP-43), and chromosome 9 open reading frame 72 (C9orf72). A hallmark of both familial and sporadic ALS/FTD is the cytoplasmic mislocalization and aggregation of the SOD1, FUS, TDP-43, and c9orf72 proteins.1
As genetics alone fails to explain the etiology of neurodegenerative disease, recent work has explored epigenetic mechanisms in this context. Epigenetics may provide novel insight into the development of these diseases. Epigenetic channels include the post-translational modification (PTM) of histones. DNA wraps around the core histone proteins H2A, H2B, H3, and H4, which can undergo acetylation, phosphorylation, and mono-, di-, or even trimethylation among many other modifications on specific residues.2 These PTMs are “written” and “erased” by proteins that can add and remove them. For example, histone acetylation is installed by histone acetyltransferases (HAT) and is removed by histone deacetylases (HDAC).3 The presence or absence of certain histone PTMs results in changes in gene regulation either by directly impacting the binding of DNA to histones or by serving as a platform for the binding of “reader” proteins.2
New data exploring epigenetic dysregulation in neurodegenerative disease has highlighted histone PTMs as potential markers of disease progression and as novel drug targets. For instance, our own work revealed unique histone PTM landscapes connected to distinct ALS/FTD proteinopathies in yeast models.4 Furthermore, frontal cortices and cerebella of c9orf72ALS/FTD (c9ALS/FTD) patients bear increased trimethylation of lysine residues on histones H3 and H4.5 Moreover, mutant c9orf72 mice as well as cortices from patients with c9ALS/FTD display increased H3K4me3 and H3K27me3.6 Together, these findings call attention to the role of histone PTMs in neurodegeneration.
2. Current Progress in Epidrugs for ALS/FTD: A Focus on Histone Acetylation
The use of epidrugs, compounds that target enzymes involved in epigenetic processes, has garnered increasing interest for their use as therapeutic options for neurodegenerative diseases. Particularly, HDAC inhibition has been subject of intense research. Sodium phenylbutyrate and valproic acid, two FDA-approved pan-HDAC inhibitors, underwent phase II clinical trials for ALS.7,8 Treatment of ALS patients with sodium phenylbutyrate resulted in a drastic change in histone acetylation and in decreased ALS symptoms and extended patients’ lifespan.7 Similarly, valproic acid regulated histone acetylation levels and offered neuroprotective effects against glutamate-induced excitotoxicity in spinal cord cultures.8 Additionally, HDAC6 inhibitors Tubastatin A and ACY-738 reverse axonal transport deficits and increase α-tubulin acetylation in iPSC-derived motor neurons from ALS patients.9 Lastly, Trichostatin A, a preclinical pan-HDAC inhibitor, can reduce growth suppression related to FUS overexpression in a yeast model and ameliorate motor neuron degeneration in an ALS mouse model.10,11
3. Dysregulation of Kinases in ALS/FTD
Serine, tyrosine, and threonine side chains are phosphorylated by protein kinases and dephosphorylated by phosphatases. Features of ALS/FTD pathology may be attributed to alterations in activity of certain kinases with nonhistone targets. For example, TDP-43 is hyperphosphorylated in the cytoplasm of degenerating neurons of ALS/FTD patients. Casein kinase 1 (CK1) is a family of kinases that are dysregulated in neurodegenerative diseases.12 CK1ε and CK1δ increase TDP-43 phosphorylation at S409/410 while promoting the formation of aggregates.13
The mitogen-activated protein kinase (MAPK) pathway has also been implicated in neurodegeneration.14 This pathway regulates cell proliferation, differentiation, survival, and death. Several ALS-associated cellular pathophysiological defects can be linked back to abnormal phosphorylation of MAPK members, particularly MAP4K (MAP kinase kinase kinase kinase), making it a new target for novel therapeutics.14 Extracellular stimuli allow for the phosphorylation of MAP4K HGK (hepatocyte progenitor kinase-like/germinal center kinase-like kinase), triggering a cascade that leads to the phosphorylation of JNK (c-Jun N-terminal kinase). JNK in turn phosphorylates c-Jun, which regulates nuclear transcription and induces apoptosis.15 Indeed, c9ALS/FTD patient post-mortem tissues as well as TDP-43 transgenic mouse models have shown dysregulated HGK and hyperactivated JNK due to altered ER stress due to protein misfolding.14
Lastly, Aurora B kinase regulates axonal mitochondrial transport. Aurora B has several targets, such as the KNL1/Mis12 complex/NDC80 complex network, a key player in kinetochore-microtubule attachments necessary for the movement of mitochondria.16,17 In SOD1 ALS patient motor neurons, axonal mitochondrial trafficking is dysregulated; mitochondrial motility defects were rescued by small-molecule inhibition of Aurora B kinase.18 Further, Aurora B kinase has been recently implicated in neurite initiation and dendritic branching, processes that are altered in neurodegeneration.18,19 Interestingly, Aurora B kinase and JNK can also phosphorylate Histone H3 on Serine 10.20,21 This raises the compelling possibility that Aurora B kinase and JNK hyperactivation could be linked to neurodegeneration at least partially through their activity as histone kinases.
4. Changes in Histone Phosphorylation in ALS/FTD
Like histone acetylation, histone phosphorylation is highly dynamic. Addition of phosphate groups adds negative charge to histone proteins that results in the formation of a more open chromatin structure.22 Histone phosphorylation is typically a marker of DNA damage repair as well as chromatin remodeling.23 H2AXS139ph (yH2AX) and H3S10ph are among the most amply studied histone phosphorylation sites.
Histone phosphorylation is understudied in the context of neurodegenerative disease. Early evidence ties yH2AX and H3S10ph to ALS/FTD. In a FUS yeast model, H3S10ph levels are altered.4 In yeast, this mark is installed by Ipl1, the yeast homologue of Aurora B kinase, further suggesting a role for Aurora B kinase in the etiology of ALS. C9orf72 has been also linked to dysregulation of histone phosphorylation levels. Spinal cord samples from c9ALS/FTD patients demonstrated up-regulation of yH2AX, as well as the phosphorylated form of the kinase ATM, while yH2AX levels were increased in c9ALS/FTD neuronal cells.24 Furthermore, H3S10ph perturbations might also be involved in c9ALS/FTD. Stable RNA-DNA hybrids (R-loops) are associated with c9orf72 mutations.25 Increased H3S10ph is associated with R-loops, and thus it is likely that H3S10ph is enhanced at the c9orf72 locus.26
5. Potential for Success: Recent Advances in Targeting Protein Kinases in ALS/FTD
CK1ε and CK1δ inhibitors reduce TDP-43 phosphorylation levels and lead to reduced aggregation, making casein kinases a new drug candidate in ALS.13,27 Stressed motor neurons derived from SOD1 mice displayed upregulated MAP4K4, which in turn can lead to increased JNK phosphorylation. Suppression of MAP4K4 in these same mice as well as in SOD1 iPS-derived motor neurons improved cell survival and neurite outgrowth.14 Additionally, inhibition of HGK and other MAP4Ks by Prosetin and MAP4K inhibitor 29 in motor neurons derived from human SOD1 mutant iPSCs resulted in increased motor neuron survival.14 Indeed, Prosetin, an orally administrable, brain penetrant small molecule, is currently undergoing Phase I clinical trials for ALS.28
6. The Future of Epidrugs: Targeting Histone Kinases in ALS/FTD
Despite significant progress in the use of epidrugs in the treatment of neurodegenerative disease, targeting histone phosphorylation remains a mostly untapped area. Stemming from intense exploration of kinase inhibitors as cancer therapeutics, there are several commercially available histone kinase inhibitors. Considering histone kinase hyperactivation and histone phosphorylation disruption in ALS/FTD, we propose exploring the repurposing of these compounds as an promising alternative in neurodegenerative disease treatment.
Current evidence points to a role for yH2AX and H3S10ph in ALS/FTD. The formation of yH2AX is controlled by the enzymes ATM, ATR, and DNA-PKcs, depending on the specific stressors.29 Currently, a number of ATM, ATR, and DNAPKcs inhibitors have been identified and several are undergoing clinical trials as cancer therapeutics.30,31 However, several difficulties arise in targeting yH2AX. First, many inhibitors of ATM and ATR are not useful as drug candidates due to their low specificity. Furthermore, inhibition of either ATM or ATR allows for the other to phosphorylate H2AX. Because of this, a combined treatment may be optimal to inhibit yH2AX formation.
H3S10 is phosphorylated by Aurora B kinase.18 The Aurora family of kinases has been difficult to study as the inhibition of individual members (such as Aurora B) is difficult due to lack of selectivity among inhibitor candidates. In fact, inhibition of Aurora kinases usually targets Aurora A and Aurora C, which are not involved in the phosphorylation of histone tails. Recently, the phosphate-based prodrug Barasertib (AZD1152) and the small molecule Hesperadin have been found to be strong inhibitors of Aurora B kinase.18,32 Use of Barasertib and Hesperadin may yield positive results in the context of neurodegenerative disease progression. Indeed, Aurora B kinase inhibition leads to neuroprotection in SOD1 models;18 it will be interesting to determine if this effect occurs through epigenetic channels.
Additionally, H3S10ph can also be installed by JNK.20 JNK inhibitors SP600125, AS601245, and JNK-IN-8 have shown promising results in several in vitro and in vivo studies.33 However, much like the Aurora family of kinases, JNK has several isoforms that have been associated with unique roles in the development of diabetes, asthma, various cancers, and even Parkinson’s disease.34 In particular, patient iPSC-derived motor neurons revealed activation of JNK3 by MAP4K4 and no change in levels of JNK1 or JNK2.35 Because of this, there is a need for the development of inhibitors selective enough for specific JNK isoforms. Several molecules with high specificity for JNK3 are currently in development, such as Quinazoline, Triazolothione 1, and a number of Pyridopyrimidinone derivatives.36−38 Quinazoline and Triazolothione 1 in particular might be promising molecules in preventing neurodegeneration, as they have shown good brain penetration and pharmacokinetic properties.36,37 All in all, chemical modulation of histone kinase activity could restore altered histone phosphorylation patterns, resolve gene expression defects, and lead to improved cell outcomes and neuroprotection (Figure 1).
Figure 1.
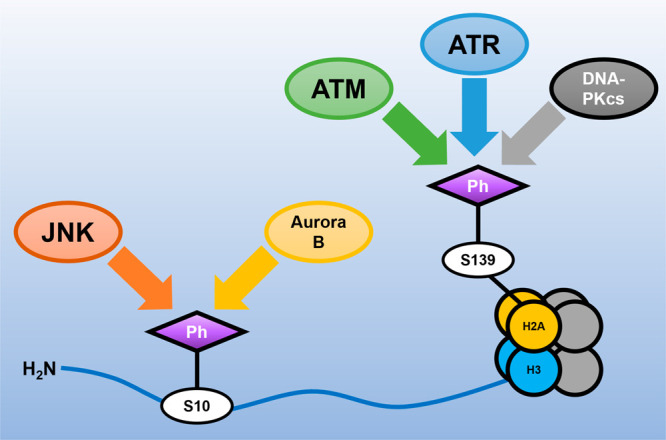
Histone kinases as emerging drug targets in ALS/FTD treatment. Schematic featuring histone kinases that are potentially dysregulated in ALS/FTD.
7. Conclusions
The use of HDAC inhibitors as potential treatments for ALS/FTD only scratches the surface of what other epidrugs can achieve. A more complete understanding of the epigenetic mechanisms of neurodegeneration is a key issue in the development of better medications with the potential to help thousands of patients. Another key issue is the design and synthesis of more selective kinase inhibitors. Despite these gaps, histone phosphorylation and kinases are an underexploited field within already accessible knowledge that would enable a novel approach to neurodegenerative disease therapy.
Acknowledgments
We thank Dr. Seth Bennett for critical review of the manuscript.
Author Contributions
The manuscript was written through contributions of both authors. Both authors have approved the final version of the manuscript.
Brooklyn College and CUNY supported M.P.T. The Graduate Center and Brooklyn College, CUNY supported S.N.C.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science special issue “Epigenetics 2022”.
References
- Gowland A.; et al. Predicting the future of ALS: the impact of demographic change and potential new treatments on the prevalence of ALS in the United Kingdom, 2020–2116. Amyotroph Lateral Scler Frontotemporal Degener 2019, 20, 264–274. 10.1080/21678421.2019.1587629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahid Z.; Simpson B.; Miao K. H.; Singh G.. Genetics, Histone Code. In StatPearls: StatPearls Publishing, 2021. [PubMed] [Google Scholar]
- Ruiz-Garcia A. B.; Sendra R.; Pamblanco M.; Tordera V. Gcn5p is involved in the acetylation of histone H3 in nucleosomes. FEBS Lett. 1997, 403, 186–190. 10.1016/S0014-5793(97)00049-5. [DOI] [PubMed] [Google Scholar]
- Chen K.; et al. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 838–848. 10.1021/acschemneuro.7b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzil V. V.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol 2013, 126, 895–905. 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. J. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019, 363, eaav2606. 10.1126/science.aav2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudkowicz M. E.; et al. Phase 2 study of sodium phenylbutyrate in ALS. Amyotroph Lateral Scler 2009, 10, 99–106. 10.1080/17482960802320487. [DOI] [PubMed] [Google Scholar]
- Piepers S.; et al. Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann. Neurol. 2009, 66, 227–234. 10.1002/ana.21620. [DOI] [PubMed] [Google Scholar]
- Guo W.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. 10.1038/s41467-017-00911-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett S. A.; et al. Trichostatin A Relieves Growth Suppression and Restores Histone Acetylation at Specific Sites in a FUS ALS/FTD Yeast Model. Biochemistry 2021, 60, 3671–3675. 10.1021/acs.biochem.1c00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo Y. E.; Ko C. P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. 10.1016/j.expneurol.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Cozza G.; Pinna L. A. Casein kinases as potential therapeutic targets. Expert Opin Ther Targets 2016, 20, 319–340. 10.1517/14728222.2016.1091883. [DOI] [PubMed] [Google Scholar]
- Brown D. G.; Shorter J.; Wobst H. J. Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia. Bioorg. Med. Chem. Lett. 2020, 30, 126942. 10.1016/j.bmcl.2019.126942. [DOI] [PubMed] [Google Scholar]
- Sahana T. G.; Zhang K. Mitogen-Activated Protein Kinase Pathway in Amyotrophic Lateral Sclerosis. Biomedicines 2021, 9, 969. 10.3390/biomedicines9080969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z.; et al. A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 1999, 274, 2118–2125. 10.1074/jbc.274.4.2118. [DOI] [PubMed] [Google Scholar]
- Welburn J. P.; et al. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol. Cell 2010, 38, 383–392. 10.1016/j.molcel.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J.; et al. Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death Dis 2018, 9, 81. 10.1038/s41419-017-0145-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlevkov E.; et al. A High-Content Screen Identifies TPP1 and Aurora B as Regulators of Axonal Mitochondrial Transport. Cell Rep 2019, 28, 3224–3237.e5. 10.1016/j.celrep.2019.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazejewski S. M.; Bennison S. A.; Liu X.; Toyo-Oka K. High-throughput kinase inhibitor screening reveals roles for Aurora and Nuak kinases in neurite initiation and dendritic branching. Sci. Rep 2021, 11, 8156. 10.1038/s41598-021-87521-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V. K.; et al. A chromatin-modifying function of JNK during stem cell differentiation. Nat. Genet. 2012, 44, 94–100. 10.1038/ng.1036. [DOI] [PubMed] [Google Scholar]
- Chen C.; et al. Phosphorylation of histone H3 on Ser-10 by Aurora B is essential for chromosome condensation in porcine embryos during the first mitotic division. Histochem Cell Biol. 2017, 148, 73–83. 10.1007/s00418-017-1546-8. [DOI] [PubMed] [Google Scholar]
- Bannister A. J.; Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto D.; Avvakumov N.; Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. 10.4161/epi.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg M. A.; Konopka A.; Soo K. Y.; Ito D.; Atkin J. D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. 10.1093/hmg/ddx170. [DOI] [PubMed] [Google Scholar]
- Wang J.; Haeusler A. R.; Simko E. A. Emerging role of RNA*DNA hybrids in C9orf72-linked neurodegeneration. Cell Cycle 2015, 14, 526–532. 10.1080/15384101.2014.995490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano-Pozo M.; et al. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590. 10.1016/j.molcel.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Salado I. G.; et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. 10.1021/jm500065f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos P. H.; et al. Development of MAP4 Kinase Inhibitors as Motor Neuron-Protecting Agents. Cell Chem. Biol. 2019, 26, 1703–1715.e37. 10.1016/j.chembiol.2019.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M. H.; Oh D. Y. ATM in DNA repair in cancer. Pharmacol Ther 2019, 203, 107391. 10.1016/j.pharmthera.2019.07.002. [DOI] [PubMed] [Google Scholar]
- Huang R. X.; Zhou P. K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther 2020, 5, 60. 10.1038/s41392-020-0150-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok J. H. L.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. 10.1038/s41467-019-12836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S.; et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003, 161, 281–294. 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; et al. Selective inhibitors for JNK signalling: a potential targeted therapy in cancer. J. Enzyme Inhib Med. Chem. 2020, 35, 574–583. 10.1080/14756366.2020.1720013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005, 15, 36–42. 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- Wu C.; Watts M. E.; Rubin L. L. MAP4K4 Activation Mediates Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Cell Rep 2019, 26, 1143–1156.e5. 10.1016/j.celrep.2019.01.019. [DOI] [PubMed] [Google Scholar]
- He Y.; et al. Synthesis and SAR of novel quinazolines as potent and brain-penetrant c-jun N-terminal kinase (JNK) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1719–1723. 10.1016/j.bmcl.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitz R. J.; et al. Highly selective c-Jun N-terminal kinase (JNK) 3 inhibitors with in vitro CNS-like pharmacokinetic properties II. Central core replacement. Bioorg. Med. Chem. Lett. 2011, 21, 3726–3729. 10.1016/j.bmcl.2011.04.074. [DOI] [PubMed] [Google Scholar]
- Zheng K.; et al. Pyridopyrimidinone Derivatives as Potent and Selective c-Jun N-Terminal Kinase (JNK) Inhibitors. ACS Med. Chem. Lett. 2015, 6, 413–418. 10.1021/ml500474d. [DOI] [PMC free article] [PubMed] [Google Scholar]