

Abstract
Nucleotide metabolism fuels normal DNA replication and is also primarily targeted by the DNA replication checkpoint when replication stalls. To reveal a comprehensive interconnection between genome maintenance and metabolism, we analyzed the metabolomic changes upon replication stress in the budding yeast S. cerevisiae. We found that upon treatment of cells with hydroxyurea, glucose is rapidly diverted to the oxidative pentose phosphate pathway (PPP). This effect is mediated by the AMP‐dependent kinase, SNF1, which phosphorylates the transcription factor Mig1, thereby relieving repression of the gene encoding the rate‐limiting enzyme of the PPP. Surprisingly, NADPH produced by the PPP is required for efficient recruitment of replication protein A (RPA) to single‐stranded DNA, providing the signal for the activation of the Mec1/ATR–Rad53/CHK1 checkpoint signaling kinase cascade. Thus, SNF1, best known as a central energy controller, determines a fast mode of replication checkpoint activation through a redox mechanism. These findings establish that SNF1 provides a hub with direct links to cellular metabolism, redox, and surveillance of DNA replication in eukaryotes.
Keywords: carbon metabolism, cell cycle checkpoints, DNA replication stress, genome stability, reductive/oxidative (redox)
Subject Categories: DNA Replication, Recombination & Repair; Metabolism
Genotoxic effects may underlie the long‐lasting proliferative arrest triggered by anti‐cancer drugs that cause G1 phase pausing.
Introduction
Genomic DNA is constantly exposed to numerous exogenous and endogenous agents that cause the slowing or stalling of replication forks and/or DNA synthesis (known as replication stress) (Zeman & Cimprich, 2014; Berti et al, 2020). Because the genome is exceptionally vulnerable during its replication in S phase of the cell cycle, all eukaryotic cells have evolved a highly conserved DNA replication checkpoint, also known as the S‐phase checkpoint, which surveys replication forks for potential threats and prevents fork collapse and damage accumulation until replication stress is relieved (Ciccia & Elledge, 2010; Hustedt et al, 2013; Maréchal & Zou, 2013; Awasthi et al, 2015; Blackford & Jackson, 2017; Giannattasio & Branzei, 2017; Lanz et al, 2019). Given its essential contribution to maintaining genome integrity, the DNA replication checkpoint currently is the target for many novel cancer therapeutics (Pearl et al, 2015).
The DNA replication checkpoint comprises a highly conserved protein kinase cascade mediated by Mec1 and Rad53 in budding yeast and by their orthologs ATR and CHK1 in humans (Pardo et al, 2016; Yazinski & Zou, 2016; Saldivar et al, 2017). At the head of the cascade in yeast, Mec1, a phosphoinositide 3‐kinase‐related protein kinase, and its partner protein Ddc2, recognize long stretches of single‐stranded (ss)DNA that is exposed during replication stress and bind replication protein A (RPA) (Choi et al, 2010; Yazinski & Zou, 2016; Deshpande et al, 2017). RPA, the main eukaryotic ssDNA‐binding factor, is a heterotrimer of Rfa1, Rfa2, and Rfa3 (Wold, 1997). Upon binding to RPA‐coated ssDNA, Mec1 is activated and phosphorylates many substrates, including histone H2A and mediator of replication checkpoint 1 (Mrc1). Mrc1 cooperates with Mec1 to phosphorylate Rad53 and activates its kinase activity. Hyperphosphorylated Rad53 then triggers a series of downstream effects, such as stalled fork stabilization, cell cycle arrest, inhibition of late replication origin firing, induction of DNA repair gene expression, and upregulation of the activity of ribonucleotide reductase (RNR), which is the rate‐limiting enzyme for the biogenesis of deoxyribonucleoside triphosphates (dNTPs) (Huang et al, 1998; Zhao et al, 2001; Pardo et al, 2016; Lanz et al, 2019). Overexpression of RNR genes or deletion of genes encoding RNR inhibitor proteins rescues the lethality of MEC1 and RAD53 deletion (Desany et al, 1998; Zhao et al, 1998), indicating the fundamental importance of dNTP biosynthesis in the DNA replication checkpoint.
In unperturbed conditions, metabolism fuels the DNA replication machines with energy (ATP) and building blocks (dNTPs) through nucleotide biogenesis as well (Zhu & Thompson, 2019). To investigate whether there is any link other than nucleotide biogenesis between metabolism and genome maintenance, we used untargeted metabolomics to analyze the changes that occur when yeast cells are subjected to replication stress by treatment with hydroxyurea (HU), an inhibitor of RNR that is commonly used to slow or stall replication forks. We report here that this replication stress rapidly diverts glucose from glycolysis to the pentose phosphate pathway (PPP), which generates NADPH and the nucleotide precursor ribose‐5‐phosphate (R5P). This enhanced flux through the PPP is achieved by the AMP‐activated protein kinase (AMPK) SNF1, an important regulator of energy metabolism. We show that SNF1 acts upstream of Mec1 and is required to timely activate the DNA replication checkpoint. Moreover, NADPH generated by the PPP provides the reducing environment necessary for RPA to bind to ssDNA at stalled replication forks. These findings suggest an important role for redox state in the DNA replication checkpoint activation and reveal an unanticipated early function of SNF1 in response to replication stress upstream of the activation of Mec1.
Results
Replication stress induces the PPP through Snf1 and Mig1
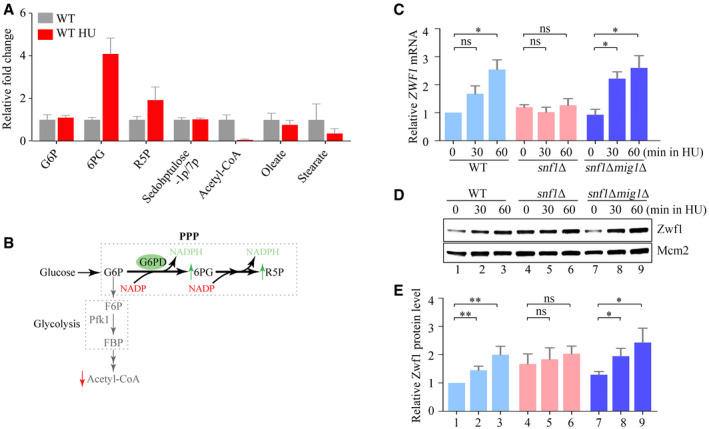
To systematically investigate the effect of replication stress on the metabolism, we analyzed the metabolome of Saccharomyces cerevisiae 60 min after the addition of 200 mM HU to exponentially growing cells. More than 9,200 mass spectral features were detected in negative ion mode (Appendix Fig S1A and B, Dataset EV1) and 16,100 in positive ion mode (Appendix Fig S1C and D, Dataset EV2). One of the metabolites that increased most upon HU treatment was 6‐phosphogluconate, an intermediate metabolite of the PPP; certain other metabolites of this pathway, including the nucleotide precursor R5P, were also elevated (Fig 1A). Acetyl‐CoA, by contrast, decreased substantially and glucose‐6‐phosphate remained constant (Fig 1A). These data indicate rapid diversion of glucose from glycolysis to the PPP when cells encounter replication stress (Fig 1B).
Figure 1. Replication stress induces the pentose phosphate pathway through Snf1 and Mig1.
-
AThe relative fold change of carbon metabolites with or without HU treatment for 60 min. Error bars represent standard deviations (SD) from six independent experiments.
-
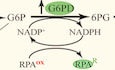
BThe diagram of carbon metabolism rewiring focused on glycolysis and PPP (pentose phosphate pathway).
-
CThe relative ZWF1 mRNA levels were measured by qPCR. Cells were grown into an exponential phase and treated by 200 mM HU for 0, 30, and 60 min. mRNA were prepared just as described in Methods and Materials. The relative mRNA levels of ZWF1 to ACTIN1 were determined by qPCR analyses. The value of the untreated WT sample was normalized to 1. Error bars represent standard deviations (SD) from at least three biological repeats. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05).
-
D, EThe relative levels of endogenous Zwf1 protein with a GFP tag were analyzed by immunoblots in WT, snf1Δ, or snf1Δmig1Δ cells (D). Protein extracts were probed with an anti‐GFP antibody. Mcm2 was applied as a loading control. Three independent experiments were carried out and the relative fold changes were measured by Image J (E). Error bars represent standard deviations (SD) from three biological repeats. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05; **0.001 < P‐values <0.01).
Source data are available online for this figure.
In the first step of the PPP oxidative phase, glucose‐6‐phosphate is converted to 6‐phosphogluconolactone by the rate‐limiting enzyme glucose‐6‐phosphate dehydrogenase (G6PD) (Yang et al, 2018). This enzyme is encoded by the ZWF1 gene in yeast. To see whether the increased PPP flux might be explained by increased expression of the ZWF1 gene, we used quantitative PCR (qPCR) to measure ZWF1 mRNA levels relative to those of a control gene ACT1 60 min after HU addition and found a ~2.5‐fold increase (Fig 1C).
To determine the factors involved in ZWF1 induction, we used bioinformatics to identify potential binding sites for transcription factors in the ZWF1 gene and its regulatory regions (Appendix Fig S1E). Among them, a predicted binding site for the transcription factor Mig1 caught our attention because this factor is the main transcriptional repressor regulated by the yeast AMPK ortholog, SNF1 (Treitel et al, 1998; Hardie, 2014). To investigate whether SNF1 and Mig1 are involved in the metabolic reprogramming induced by HU, we measured ZWF1 mRNA levels in the absence of the genes that encode these factors. Deletion of SNF1, which encodes the kinase subunit Snf1, abolished the induction of ZWF1 mRNA by HU, and the induction was restored when MIG1 was also deleted (Fig 1C). Consistently, the protein level of Zwf1 was induced by HU in a Snf1–Mig1‐dependent manner as well (Fig 1D and E). These findings imply that upon replicative stress, Snf1 diverts glucose into the PPP by relieving the repression of ZWF1 transcription by Mig1.
SNF1 promotes Mec1–Rad53 checkpoint activation
To determine whether Snf1‐dependent metabolic reprogramming contributes to cell survival in the presence of replication stress, we compared the growth of cells in which the SNF1 gene was deleted (snf1Δ) with the growth of wild‐type (WT) cells in the presence of HU. Fivefold serial dilutions of snf1Δ and WT cells were spotted on plates with or without HU. Whereas 200 mM HU was sufficient to kill snf1Δ cells completely, WT cells grew normally at this concentration (Appendix Fig S2A). snf1Δ cells were more sensitive to HU than were ddc1Δ dpb11ΔC cells lacking the genes encoding two well‐established Mec1 activators (Appendix Fig S2A) (Navadgi‐Patil & Burgers, 2009). Meanwhile, snf1Δ cells were sensitive to DNA damage reagents like methyl methanesulfonate (MMS) and bleomycin as well (Appendix Fig S2A and B). These data indicate that Snf1 is crucial for survival when cells encounter replication stress or DNA damage.
SNF1 is a heterotrimeric kinase complex comprising the catalytic subunit Snf1, one of three beta‐subunits (Sip1, Sip2, or Gal83) and the stimulating subunit (Snf4) (Hedbacker & Carlson, 2008). Deleting the genes encoding Sip1, Sip2, or Gal83 conferred no HU sensitivity; only deleting all three genes resulted in HU sensitivity similar to that of snf1Δ (Appendix Fig S2C). Deletion of the gene encoding the Snf4 subunit, snf4Δ, resulted in a HU sensitivity similar to that of snf1Δ cells (Fig 2A and Appendix Fig S2D). This finding contradicted a previous study, which reported that snf4Δ cells were much less sensitive to HU than were snf1Δ cells (Dubacq et al, 2004). To confirm our finding, we deleted SNF4 in the BY4741 background used by Dubacq and colleagues. As in the W303 background, snf4Δ was highly sensitive to HU (Fig 2A and Appendix Fig S2D). Moreover, its sensitivity to HU and non‐fermentable sugar (sucrose) was complemented by re‐introducing SNF4 (Appendix Fig S2E). These data suggest that all three subunits of the SNF1 kinase complex are similarly required to manage replication stress.
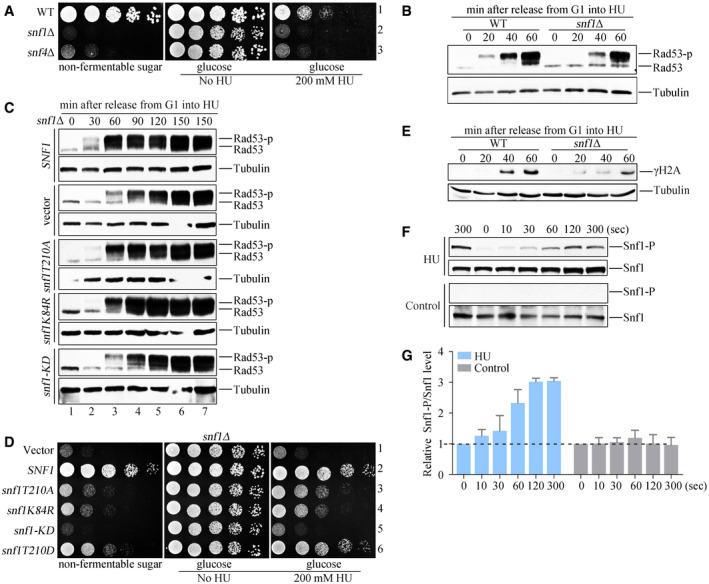
Figure 2. The kinase activity of SNF1 is critical for the timely activation of Mec1 and Rad53.
-
AThe snf1Δ and snf4Δ mutant cells are hypersensitive to HU. Fivefold serial dilutions of the overnight cultures were assayed on normal growth media (YPD) with or without 200 mM HU. A plate in which 2% glucose was replaced by 2% non‐fermentable carbon (galactose) was applied as a control. All plates were cultured at 30°C for 48 h before photography.
-
BRad53 hyperphosphorylation of WT and snf1Δ in response to HU. Cells were synchronized in G1 and released into the fresh medium containing 200 mM HU for the indicated time. Cell extracts were prepared for immunoblotting against anti‐Rad53 antibodies. Tubulin was probed as a loading control.
-
CRad53 phosphorylation in various snf1 alleles in response to HU. The plasmids expressing the indicated snf1 alleles were transformed into snf1Δ. Rad53 phosphorylation was detected as above.
-
DThe snf1 kinase‐defective mutants are sensitive to HU. Yeast spot assays were performed as in Fig 2A. The phospho‐mimetic snf1T210D (a constitutive active mutant) was applied as a control for non‐phosphorylatable snf1T210A.
-
EH2A S129 phosphorylation (namely γ‐H2A) of WT and snf1Δ in response to HU. γ‐H2A was detected by immunoblotting.
-
F, GHU induces a rapid Snf1 T210 phosphorylation. Exponential cells were treated by 200 mM HU for the indicated time. Protein extracts were prepared as described in the Methods and Materials. Three independent experiments were done. Quantitation of Snf1 T210 and Snf1 signals was performed by Image J. The relative ratio of Snf1 T210/Snf1 was shown in (G). Error bars represent standard deviations (SD) from three biological repeats.
Source data are available online for this figure.
Activation of the DNA replication checkpoint is necessary for cells to survive the replication stress induced by HU. To investigate whether SNF1 protects cells from HU by activating this checkpoint, we used Rad53 phosphorylation as a readout for checkpoint activation (Fig 2B). WT or snf1Δ cells were synchronized in G1 phase by using α‐factor, then the α‐factor was removed and 200 mM HU was added. After various times up to 60 min, the cells were analyzed by immunoblotting with an antibody against Rad53. The phosphorylated forms of Rad53, which migrate more slowly than the unphosphorylated protein, appeared in snf1Δ cells only after 40 min, a delay of about 20 min when compared with the appearance of these forms in WT cells. The checkpoint activation defect was unlikely caused by the G1/S transition delay reported in snf1Δ because of two reasons: First, high concentrations of glucose (5%) can restore the cell cycle defect (Fig EV1A) (Pessina et al, 2010; Busnelli et al, 2013), but not timely Rad53 activation (Fig EV1B) and HU sensitivity (Fig EV1C) of snf1Δ. Second, as mentioned above, snf4Δ exhibits the same HU sensitivity and checkpoint defects (Figs 2A and EV1D and E) as snf1Δ, but with nearly WT cell cycle progression (Fig EV1A) and origin unwinding (as shown below in Fig 3F). These results suggest that Snf1 plays a critical role in efficient replication checkpoint activation in a cell‐cycle‐independent manner.
Figure EV1. SNF1 complex is required for timely checkpoint activation.
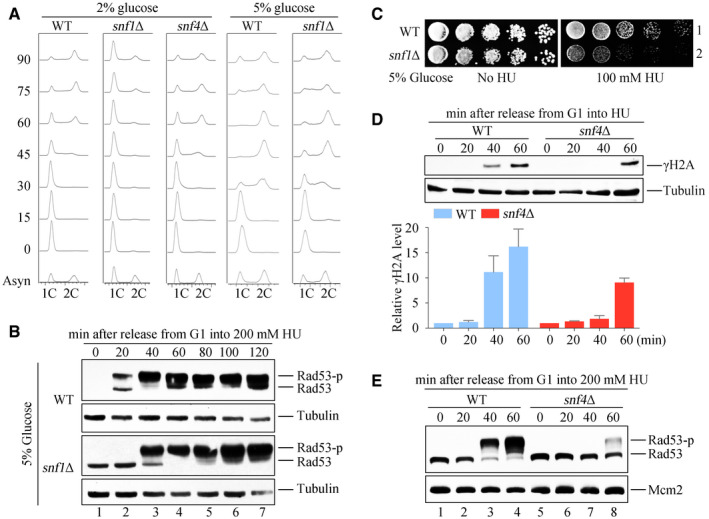
- DNA content was monitored by flow cytometry. Cells were grown in media containing 2 or 5% glucose and exponential cells were arrested into G1 phase by alpha‐factor before release into corresponding fresh media for the indicated time.
- Rad53 hyperphosphorylation of WT and snf1Δ in response to HU. Cells were synchronized in G1 and released into the fresh medium (5% glucose) containing 200 mM HU for the indicated time. Cell extracts were prepared for immunoblotting against anti‐Rad53 antibodies. Tubulin was probed as a loading control.
- 5% glucose cannot rescue the HU sensitivity of snf1Δ. WT and snf1Δ strains were grown and spotted on HU plates with 5% glucose. All plates were cultured at 30°C for 48 h before photography.
- H2A S129 phosphorylation (namely γ‐H2A) of WT and snf4Δ in response to HU. γ‐H2A was detected by immunoblotting (upper panel). Three independent experiments were performed and quantified by Image J (lower panel). Error bars represent standard deviations (SD) from three biological repeats.
- Rad53 phosphorylation of WT and snf4Δ in response to HU. Rad53 phosphorylation was detected as above. Mcm2 was probed as a loading control.
Source data are available online for this figure.
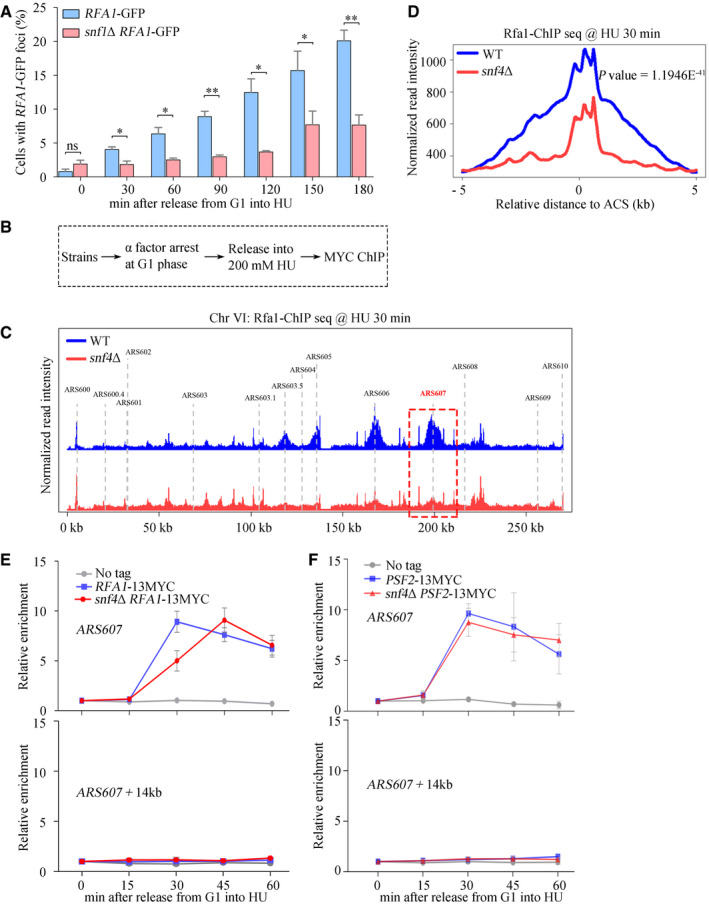
Figure 3. SNF1 affects RPA recruitment on ssDNA.
-
AA significant delay in RPA foci formation in the absence of Snf1. The percentage of cells with RPA foci was calculated and plotted against the incubation time in 200 mM HU. Error bars represent SD from three biological repeats. At least 200 cells were analyzed for each experiment of three independent experiments. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05; **0.001 < P‐values < 0.01).
-
BThe experimental scheme for the Rfa1–13MYC ChIP in the indicated yeast strains.
-
CSnap shot of Rfa1 ChIP‐Seq at Chromosome VI. Cells were synchronized in G1 and released into the fresh medium containing 200 mM HU for 30 min. The sequencing reads were mapped to the yeast reference genome. The red dashed box indicated the relative read intensity of ARS607.
-
DThe average Rfa1 ChIP‐seq read density from cells released into HU medium around ACS sites. ACS, ARS consensus sequence.
-
E, FSNF1 is required for efficient recruitment of Rfa1, but not Psf2, to HU‐stalled replication forks. Cells were grown and synchronized in G1 by α‐factor before release into the fresh medium supplemented with 200 mM HU for the indicated time. Cell extracts were prepared and subjected to MYC–ChIP of Rfa1–13MYC (E) or Psf2–13MYC (F). The amounts of DNA in the precipitates were quantified by qPCR. Error bars represent SD from three biological repeats.
The kinase activity of SNF1 requires its ATP‐binding motif (containing residue K84) and phosphorylation of the activation loop on residue T210 by its upstream kinases (Hedbacker & Carlson, 2008). To test whether the kinase activity of SNF1 is important for induction of the DNA replication checkpoint in response to HU, we constructed plasmids expressing kinase‐defective snf1mutants (T210A, K84R, and the double mutant T210AK84R hereafter referred as snf1‐KD) and introduced them into snf1Δ cells (Estruch et al, 1992). Upon treatment with HU, Rad53 hyperphosphorylation was delayed in the cells expressing the snf1‐T210AK84R double mutant (as in snf1Δ cells), whereas the cells expressing snf1 single mutants (snf1‐K84R or snf1‐T210A) had a relatively modest defect in Rad53 activation (Fig 2C). The strength of the defect in Rad53 kinase activation in the snf1Δ cells expressing the various snf1 mutants correlated with their HU sensitivity (Fig 2D). Additionally, when the stimulatory subunit Snf4 of SNF1 was deleted, Rad53 phosphorylation was dramatically compromised as well (Fig EV1D and E). Collectively, these data indicate that the kinase activity of SNF1 is required for efficient Rad53 activation.
Upstream of Rad53 phosphorylation, histone H2A is one of the direct substrates of Mec1. Like Rad53 phosphorylation, there was a delay in the appearance of phosphorylated H2A (γ‐H2A) in the absence of either the kinase subunit Snf1 (Fig 2E) or the stimulatory subunit Snf4 of SNF1 (Fig EV1D).
The data described above suggest that SNF1 may function upstream of Mec1. To test this possibility, we examined whether the SNF1 kinase is activated earlier than Mec1 by measuring phosphorylation of the Snf1 subunit on T210 by immunoblotting. We noticed that α‐factor addition could activate the kinase activity of Snf1 (Appendix Fig S2F); therefore, we treated exponential cells with HU, and observed Snf1 phosphorylation within 10 s (Fig 2F and G). Consistent with previous studies (Alcasabas et al, 2001; Zegerman & Diffley, 2010), both γ‐H2A and Rad53 phosphorylation were not seen before 20 min (Fig 2B and E). These data indicate that SNF1 is activated rapidly in response to HU, well before activation of Mec1.
SNF1 is required for RPA recruitment to HU‐stalled replication forks
To verify that SNF1 acts before Mec1, we used another technique based on the fact that RPA‐coated ssDNA activates Mec1: we expressed the Rfa1 subunit of RPA as a fusion protein with a green fluorescent protein (Rfa1–GFP) and compared the formation of green fluorescent foci in WT and snf1 mutants in response to HU treatment. Ninety minutes after the addition of 200 mM HU, RPA foci accumulated in the nuclei of WT cells as seen by fluorescence microscopy (Appendix Fig S3A). By contrast, many fewer snf1Δ cells contained RPA foci (Appendix Fig S3A). Quantification of the number of cells containing nuclei with RPA foci over a time‐course up to 180 min after addition of HU showed that the foci accumulated more rapidly in WT cells than in snf1Δ cells (Fig 3A). These data indicate a crucial role for SNF1 in the formation of RPA foci.
To directly measure the binding of RPA to HU‐stalled replication forks, we expressed Rfa1 as a fusion protein with a 13MYC tag in WT and mutant cells. We arrested the cells in G1 phase, allowed them to enter S phase synchronously in the presence of 200 mM HU for 30 min, then used chromatin immunoprecipitation (ChIP) with an antibody against the 13MYC tag to isolate the DNA bound to Rfa1–13MYC (Fig 3B). Untagged Rfa1 was used as a control. Combining the Chromatin immunoprecipitation with next‐generation sequencing (ChIP‐seq), we found that Rfa1 peaked at nearly all early origins across the genome (Figs 3C and EV2A), in good agreement with previous studies (Li et al, 2018). In the absence of Snf4, the average enrichment of Rfa1 across these origins decreased significantly (Figs 3C and D, and EV2A). These results indicate a requirement of Snf4 for RPA binding to HU‐stalled forks throughout the genome.
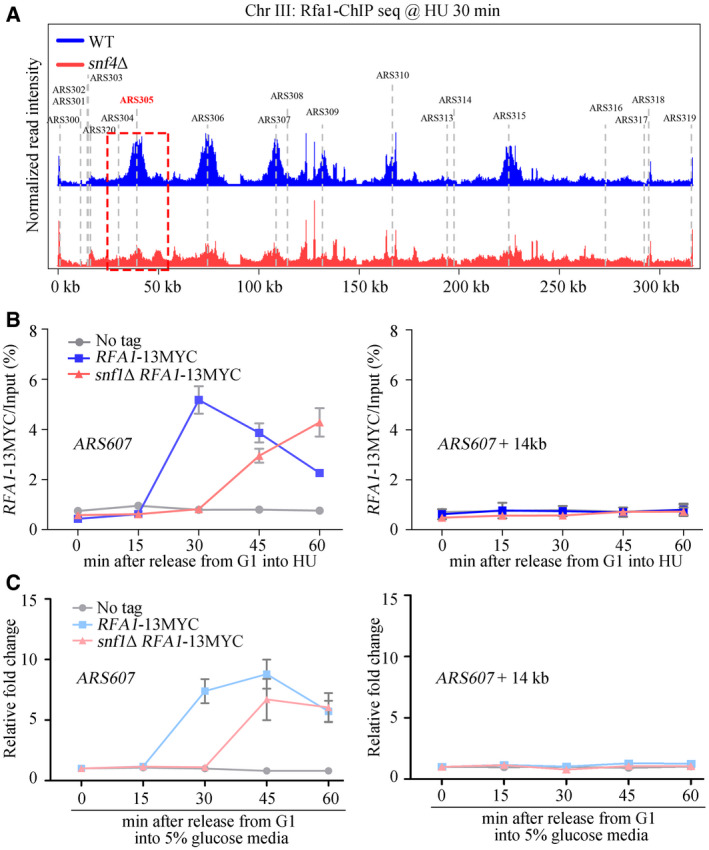
Figure EV2. SNF1 complex is required for RPA enrichment on early origins.
-
ASnap shot of Rfa1 ChIP‐Seq at Chromosome III. Cells were synchronized in G1 and released into the fresh medium containing 200 mM HU for 30 min. The sequencing reads were mapped to the yeast reference genome. The red dashed box indicated the relative read intensity of ARS305.
-
B, CSNF1 is required for efficient recruitment of Rfa1 to HU‐stalled replication forks, which is independent on cell cycle. Cells were grown and synchronized in G1 by α‐factor before release into the fresh medium containing 2% (B) or 5% (C) glucose supplemented with 200 mM HU for the indicated time. Cell extracts were prepared and subjected to MYC–ChIP of Rfa1–13MYC. The amounts of DNA in the precipitates were quantified by qPCR. Error bars represent SD from three biological repeats.
To corroborate the ChIP‐seq results, the amounts of DNA corresponding to early replication origins ARS607 and distal sites of replication (ARS607 + 14kb) were analyzed by qPCR. As expected, in WT cells, Rfa1–13MYC bound within 30 min to the early replication origins but did not bind to the distal sites, which remained unreplicated in the presence of a high concentration of HU (Liu et al, 2017) (Fig 3E). Deletion of SNF4 (Fig 3E) or SNF1 (Fig EV2B) caused a delay of at least 15 min in the peak of Rfa1–13MYC enrichment at HU‐stalled forks. Notably, the RPA‐loading delay persisted even in the presence of high concentration (5%) of glucose (Fig EV2C), suggesting that it is unlikely caused by the cell cycle defect in snf1Δ. This delay in the formation of RPA foci in snf mutants might reflect the involvement of SNF1 either in RPA recruitment or in ssDNA creation. To discriminate between these two possibilities, we analyzed whether loss of Snf4 affects binding of the replicative helicase, and therefore unwinding of the dsDNA. To do so, we expressed a subunit of the helicase, Psf2 as a fusion protein with a 13MYC tag (Psf2–13MYC) in WT and mutant cells and used ChIP‐qPCR to quantify its binding to early replication origins and distal sites, as before. The association of Psf2–13MYC with early origins was unaffected by the absence of Snf4 (Fig 3F), reflecting normal origin unwinding. Furthermore, ssDNA might be also created by nucleases like Mre11‐Rad50‐Xrs2 (MRX) behind the stalled forks. We performed ChIP‐qPCR of Xrs2, the subunit of MRX. In agreement with previous studies (Tittel‐Elmer et al, 2009; Seeber et al, 2016), Xrs2 was recruited to early origins 15–30 min after HU treatment (Appendix Fig S3B). Its binding pattern was not changed by SNF4 deletion either. Consistently, deleting either MRE11 or SAE2, encoding an endonuclease working together with MRX, exacerbated the HU sensitivity of snf1Δ or snf4Δ (Appendix Fig S3C, D, and E), indicating that SNF1 functions in parallel with MRX. Putting together, these data indicate that the delay in RPA binding to early replication origins in snf mutants is not due to delayed ssDNA creation. Thus, we conclude that SNF1 activates Mec1 by promoting RPA recruitment to ssDNA when there is replication stress.
Mig1 mediates the checkpoint function of SNF1
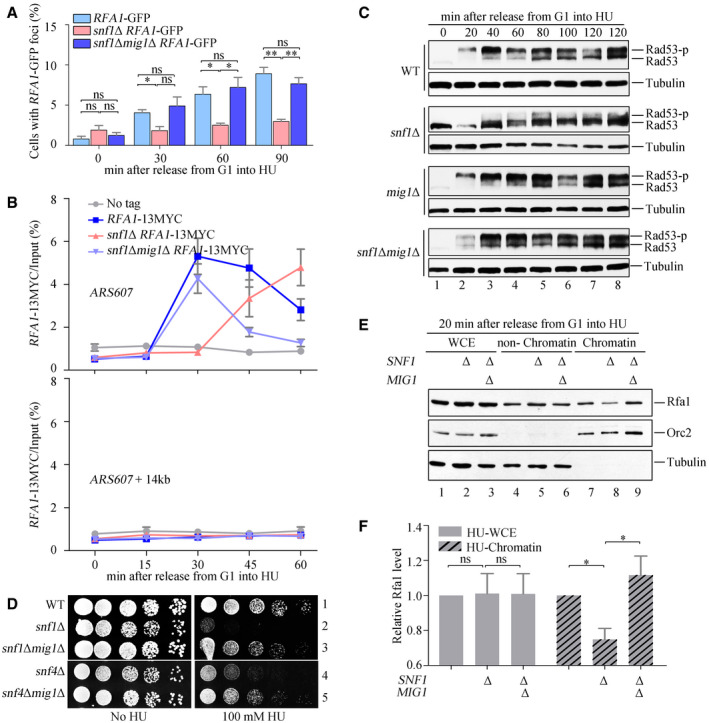
As already shown in Fig 1, SNF1 remodels glucose metabolism by antagonizing the transcription repressor Mig1. To investigate whether a similar mechanism might underlie the role of SNF1 in the DNA replication checkpoint, we studied the effect of deletion of the MIG1 gene on five different assays for the checkpoint in snf1Δ or snf4Δ cells treated with HU. Deletion of MIG1 rescued the checkpoint defects in snf1Δ or snf4Δ to nearly WT levels as determined by the formation of Rfa1–GFP foci (Fig 4A); Rfa1–13MYC binding to early replication origins (Fig 4B and Appendix Fig S4A); Rad53 phosphorylation (Figs 4C and EV3A); and growth sensitivity to HU (Fig 4D). We also assayed the association of endogenous Rfa1 with the chromatin and non‐chromatin fractions of snf1Δ and snf1Δmig1Δ cells by immunoblotting and found that Rfa1 association with the chromatin fraction in snf1Δ was restored to WT levels in the double mutant (Fig 4E and F). These immunoblots, furthermore, showed that Rfa1 protein levels in snf1Δ and snf1Δmig1Δ cells were similar to those seen in the WT. We conclude that SNF1 regulates RPA recruitment to chromatin by antagonizing Mig1.
Figure 4. mig1Δ rescues the checkpoint defects of snf1Δ.
-
A, BRfa1–GFP foci and Rfa1–13MYC ChIP assays were performed as in Fig 3. At least 200 cells were analyzed for each experiment of three independent experiments. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05; **0.001 < P‐values < 0.01) (A). Error bars represent SD from three biological repeats (B).
-
C, Dmig1Δ restores Rad53 phosphorylation and growth of the snf1 mutants in the presence of HU. The experiments were done as in Fig 2.
-
E, Fsnf1Δ shows less RPA on chromatin, which can be restored by mig1Δ. Cells were collected and fractionated into chromatin‐bound (Chromatin) and non‐chromatin‐bound fractions (non‐Chromatin) followed by immunoblotting. Tubulin and Orc2 were applied as controls for non‐chromatin and chromatin fractions, respectively. The signals were quantified by Image J and quantifications were shown in (F). Error bars represent SD from three biological repeats. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05).
Source data are available online for this figure.
Figure EV3. mig1Δ rescues the checkpoint defects of snf4Δ.
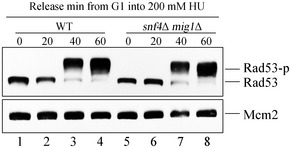
mig1Δ restores Rad53 phosphorylation of the snf4Δ in the presence of HU. The experiments were done as in Fig EV1B.
Source data are available online for this figure.
SNF1 phosphorylates Mig1 and promotes RPA recruitment by inhibiting its interaction with Ssn6
To study how SNF1 might antagonize Mig1 to promote RPA recruitment and thereby replication checkpoint activation, we constructed a series of mig1 truncation mutants and examined their ability to rescue the HU sensitivity of snf1Δ cells. Deletion of only the C‐terminal 24 amino acid residues (residues 481–504, referred to hereafter as Mig1C) rescued the HU sensitivity of snf1Δ cells as effectively as deleting the entire MIG1 gene (Appendix Fig S5A), indicating that Mig1C, negatively regulated by SNF1, is responsible for the HU sensitivity. This 24‐residue sequence contains two putative non‐canonical sites for phosphorylation by SNF1 (T489 and S495) (Fig 5A and Appendix Fig S5B).
Figure 5. Mig1 phosphorylation promotes RPA recruitment and HU resistance by attenuating its interaction with Ssn6.
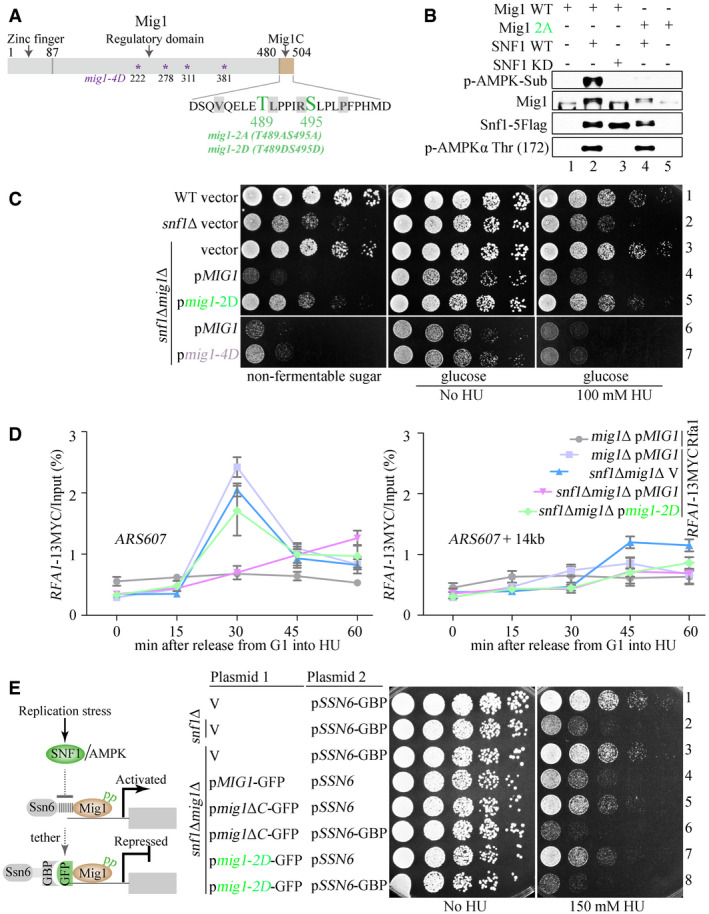
-
AT489 and S495, within Mig1C, are predicted to be the phosphorylation sites of SNF1 by global phosphorylation sites.
-
BMig1 T489 and S495 are the main phosphorylation sites of SNF1 in response to HU. The SNF1 kinase complex was partially purified from HU‐treated Snf1‐5FLAG or snf1‐KD‐5FLAG yeast cells. Recombinant Mig1 or its mutant proteins (mig1‐2A, T489AS495A) were purified from E. coli. In vitro kinase assays were performed as described in Materials and Methods. Proteins were immunoblotted using the indicated antibodies. The anti‐AMPK substrate antibodies were used to probe the phosphorylation of the substrate Mig1. Less signals were observed when Mig1 was replaced by mig1‐2A. Phosphorylation of Snf1 Thr 172 indicates the activation status of the Snf1 kinase.
- C, D
-
EThe diagram of the strategy to reinforce the Mig1–Ssn6 interaction via a GFP and GBP pair (left panel). The persistent Mig1–Ssn6 association abolishes the phospho‐mimicking effect of mig1‐2D (right panel). Yeast spot assays were performed as in Fig 2A.
Source data are available online for this figure.
To directly test whether SNF1 phosphorylates Mig1 on T489 and S495, we performed in vitro kinase assays using purified proteins. WT SNF1 phosphorylated WT Mig1, as detected by an antibody specific for phospho‐(Ser/Thr) residues in substrates of AMPK, but did not phosphorylate the mig1‐2A mutant form in which both phosphorylation sites were mutated to Ala residues (Fig 5B). The kinase‐dead mutant of SNF1 (SNF1–KD) did not phosphorylate WT Mig1. These data indicate that SNF1 phosphorylates T489 and/or S495 in Mig1. During the regulation of sugar metabolism, by contrast, Mig1 is phosphorylated on residues S222, S278, S311, and S381 in its regulatory domain (Fig 5A) (Smith et al, 1999).
Phospho‐mimetic mutations of the two putative phosphorylation sites in Mig1 (mig1‐T489DS495D; mig1‐2D) were as effective as mig1Δ in rescuing the HU sensitivity of snf1Δ cells (Fig 5C, rows 3 and 5). By contrast, phospho‐mimetic mutations of the sites phosphorylated in response to carbon stress (mig1‐S222D, S278D, S311D, S381D, mig1‐4D) barely rescued the HU sensitivity of snf1Δ (Fig 5C, rows 6 and 7). Thus, SNF1 recognizes distinct sites in Mig1 depending on the type of stress the cells are exposed to. The mig1‐2D mutant was also as effective as mig1Δ at rescuing the defect of snf1Δ in recruiting Rfa1–13MYC to early replication origins (Fig 5D and Appendix Fig S5C), indicating that Mig1 T489 and S495 are functionally relevant phosphorylation sites of SNF1 in the DNA replication checkpoint.
Mig1 has been proposed to interact with its corepressor Ssn6 (Treitel & Carlson, 1995). To confirm a direct interaction between Mig1 and Ssn6, we used pull‐down assays with recombinant GST‐tagged Mig1 (GST‐Mig1) and showed that it can bind to 6His‐tagged Ssn6 (6His‐Ssn6), whereas a single phospho‐mimetic mutation in Mig1, S495D, was sufficient to weaken the interaction substantially (Appendix Fig S5D). We arrived at similar conclusions by using yeast two‐hybrid assays to study the interaction of Ssn6 with Mig1 and several mutant forms of the protein, including single and double phospho‐mimetic mutants (Appendix Fig S5E). Removing Mig1C or including a single S495D phospho‐mimetic mutation was sufficient to weaken the interaction (Appendix Fig S5D and E), indicating that phosphorylation of T489 and/or S495 may be sufficient to inhibit the interaction between Mig1 and Ssn6.
If Mig1 phosphorylation weakens its binding to Ssn6, the rescue effect of the phospho‐mimetic mutations might be abolished by reinforcing the interaction. To test this prediction, we expressed fusion proteins of mig1‐2D fused to GFP and Ssn6 fused to GFP‐binding protein (GBP). The interaction between GFP and GBP tethers mig1‐2D to Ssn6. This tethering was sufficient to abolish the ability of mig1‐2D to suppress the HU sensitivity of snf1Δ cells (Fig 5E). In the control in which Ssn6 was not tagged with GBP, mig1‐2D‐GFP retained its ability to suppress the HU sensitivity of snf1Δ cells robustly (Fig 5E). Similarly, tethering mig1ΔC–GFP to Ssn6–GBP abolished the suppression effect (Fig 5E). Together, these data support the notion that SNF1‐mediated phosphorylation of Mig1C results in dissociation of Mig1 from its corepressor Ssn6.
Zwf1 is the principal target of Mig1 in the DNA replication checkpoint
From the data in Fig 1, we concluded that the PPP induction in response to replicative stress requires SNF1 and Mig1, which relieve the transcriptional repression of ZWF1. To determine whether activation of the DNA replication checkpoint by SNF1 and Mig1 also requires ZWF1, we made triple‐mutant snf1Δmig1Δzwf1Δ cells. These cells grew normally in the absence of HU but, like snf1Δ, were highly sensitive to HU: when ZWF1, the target of Mig1, was deleted, the suppression of HU sensitivity seen in snf1Δmig1Δ double‐mutant cells was abolished (Fig 6A). Consistent with this, the number of cells with Rfa1–GFP foci induced by HU in the triple‐mutant cells was similar to that of snf1Δ cells (Fig 6B) as was the enrichment of Rfa1–13MYC at early replication origins (Fig 6C). Moreover, expression of Mig1 containing phospho‐mimetic mutations at the two phosphorylation sites (mig1‐2D) in snf1Δmig1Δ cells restored ZWF1 mRNA levels to those seen in WT cells (Fig 6D). Consistently, both mRNA and protein levels of Zwf1 were recovered by MIG1 deletion in the absence of another SNF1 subunit, Snf4 (Fig EV4A and B). These findings indicate that Zwf1 defines the primary target of the SNF1–Mig1 axis in response to replication stress.
Figure 6. Zwf1 is the principal target of Mig1 in replication stress response.
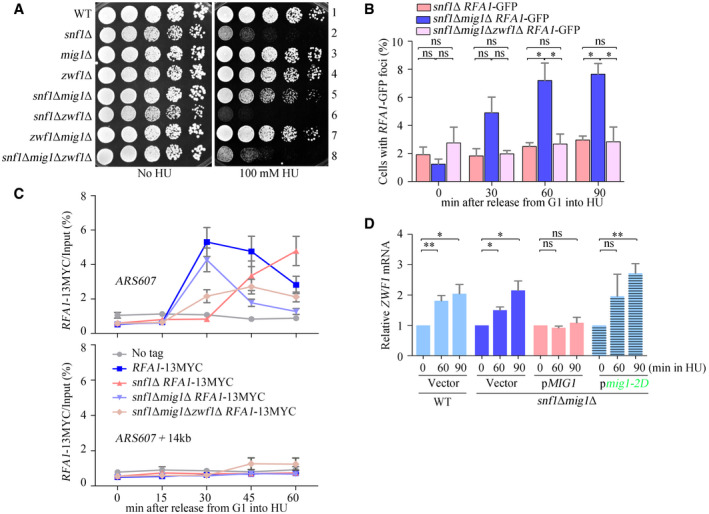
- ZWF1 is required for snf1Δmig1Δ growing in HU. Overnight cultures were spotted onto plates containing HU, and cultivated at 30°C for 48 h before photography. Yeast spot assays were performed as in Fig 2A.
- Rfa1–GFP foci were measured as in Fig 3. At least 200 cells were analyzed for each experiment of three independent experiments. Error bars represent SD from three biological repeats. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05).
- Rfa1–13MYC ChIP assays after HU treatment. Strains were synchronized in G1 phase by α‐factor prior to release into fresh media containing 200 mM HU for the indicated time points. Cell extracts were prepared as in Fig 3. Error bars represent SD from three biological repeats.
- Phosphorylation of Mig1 T489 S495 completely restores the ZWF1 mRNA levels in snf1Δ. The experiments were done as in Fig 1. Error bars represent SD from three biological repeats. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05; **0.001 < P‐values < 0.01).
Figure EV4. Zwf1 is the principal target of Mig1 in replication stress response.
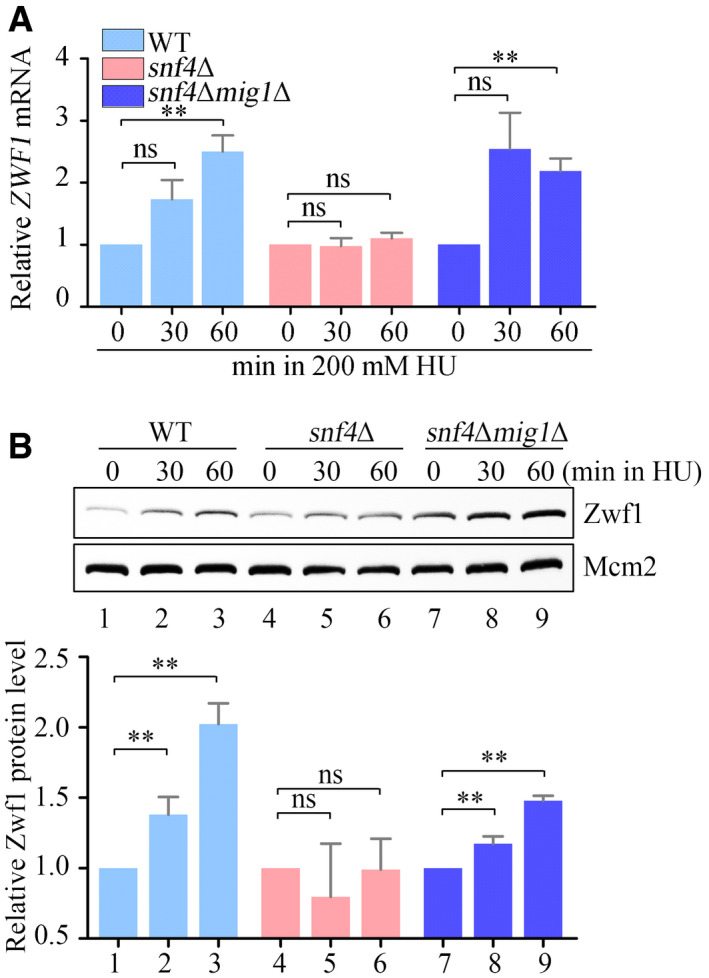
- The relative ZWF1 mRNA levels were measured by qPCR. Cells were grown into an exponential phase and treated by 200 mM HU for 0, 30, and 60 min. mRNA were prepared just as described in Materials and Methods. The relative mRNA levels of ZWF1 to ACTIN1 were determined by qPCR analyses. The value of the untreated WT sample was normalized to 1. Error bars represent standard deviations (SD) from at least three biological repeats. Statistical significance was evaluated based on Student’s t‐test (**0.001 < P‐values < 0.01).
- The relative levels of endogenous Zwf1 protein with a GFP tag were analyzed by immunoblots in WT, snf4Δ, or snf4Δmig1Δ cells (upper panel). Protein extracts were probed with an anti‐GFP antibody. Mcm2 was applied as a loading control. Three independent experiments were carried out and the relative fold changes were measured by Image J (lower panel). Statistical significance was evaluated based on Student’s t‐test (**0.001 < P‐values < 0.01).
Source data are available online for this figure.
On the other hand, we noted that cells in which only the ZWF1 gene is deleted were hardly sensitive to HU (Fig 6A, row 4), implying the existence of alternative targets of SNF1. To test this possibility, we carried out two experiments. First, in animals, the ortholog of SNF1, AMPK, directly phosphorylates and inhibits ACC1, the rate‐limiting enzyme in fatty acid synthesis (Jeon et al, 2012). When three conserved phosphorylation sites in yeast Acc1 (Chen et al, 2018) were mutated to Ala in the zwf1Δ background, the acc1‐3A zwf1Δ double mutant displayed an HU sensitivity similar to that of snf1Δ cells (Appendix Fig S6A). Second, AMPK, and its activator, the kinase LKB1, inhibit glycolysis (Faubert et al, 2014), which may also help to allocate glucose to other pathways including the PPP. Indeed, deleting PFK1, encoding the rate‐limiting enzyme in glycolysis, completely suppressed the HU sensitivity of snf1Δ (Appendix Fig S6B, row 4). Surprisingly, the pfk1Δ single mutant by itself was even more sensitive to HU than snf1Δ (Appendix Fig S6B, compare row 3 to 2). These data suggest that SNF1 might regulate carbon metabolism by Zwf1‐independent mechanisms to deal with replication stress as well.
NADPH produced by the PPP promotes the checkpoint activation
The main products of the oxidative PPP are R5P and NADPH, which cannot be produced by glycolysis. In Fig 1, we show a significant increase in R5P in response to HU. Unfortunately, our untargeted metabolomics could not identify NADPH. We then measured the cellular NADPH/NADP+ ratio using a sensitive enzymatic cycling assay (Wang et al, 2016). In WT cells treated for 60 min with HU, the NADPH/NADP+ ratio more than doubled; in snf1Δ cells, there was no such increase, but in snf1Δ mig1Δ cells a large increase in the NADPH/NADP+ ratio was seen (Fig 7A). These data confirm that the SNF1–Mig1–Zwf1 axis diverts glucose from glycolysis into the PPP upon replication stress.
Figure 7. α‐KG addition restores the defects of RPA recruitment and Rad53 activation in snf1Δ.
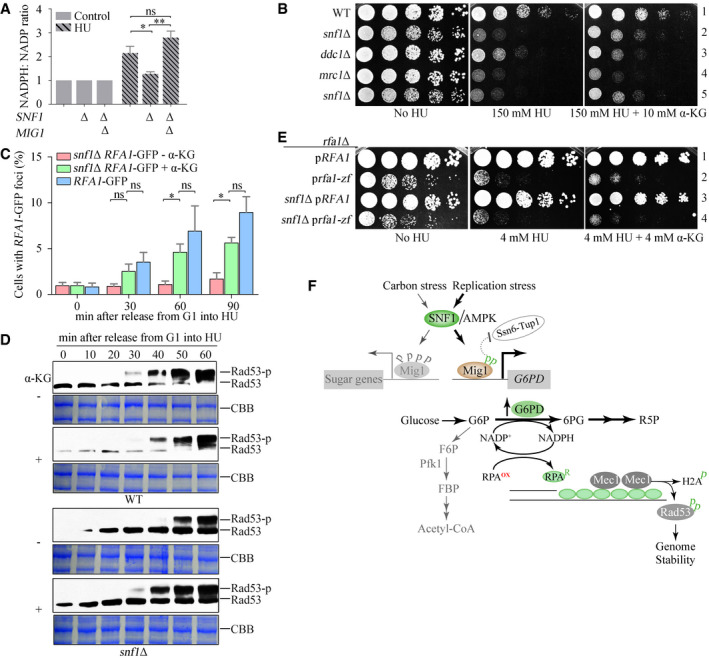
- The SNF1–Mig1 axis induces NADPH upon HU treatment. The relative ratios of cellular NADPH/NADP before or after HU treatment were measured as described in Materials and Methods. Error bars represent SD from three biological repeats. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05; **0.001 < P‐values < 0.01).
- The effects of α‐KG on the HU sensitivity of snf1Δ and other checkpoint mutants. Yeast spot assays were performed as in Fig 2A.
- α‐KG restores the levels of Rfa1–GFP foci in snf1Δ. Rfa1–GFP foci were measured as in Fig 3. At least 200 cells were analyzed for each experiment. Error bars represent SD from three biological repeats. Statistical significance was evaluated based on Student’s t‐test (*0.01 < P‐values < 0.05).
- α‐KG restores Rad53 phosphorylation in snf1Δ. CBB indicates Coomassie bright blue (CBB) staining of the membrane as the loading control.
- The effects of α‐KG on the HU sensitivity of Rfa1 zinc finger mutants (C486S, C491S, C505S, C508S). Yeast spot assays were performed as in Fig 2A.
- The proposed working model. In brief, we uncover that sugar metabolism is rewired to protect genome stability by means of a redox mechanism. Mechanistically, an SNF1–Mig1–ZWF1–NADPH axis stimulates the ssDNA binding of RPA, thereby promoting a fast activation mode of Mec1–Rad53 kinase cascade to fight against replication threats.
Source data are available online for this figure.
To understand which metabolite(s) of the PPP might contribute to the checkpoint activation, we conducted a series of experiments. We first supplemented the growth medium of snf1∆ cells with R5P; this had no effect on their growth in HU (Appendix Fig S7A). R5P is a precursor for nucleotide and dNTP biogenesis, which previous studies have found of fundamental importance for the DNA replication checkpoint (Desany et al, 1998; Li et al, 2019b). To revisit this question, we overexpressed the four genes encoding RNR‐RNR1, RNR2, RNR3, and RNR4‐in snf1∆ cells and examined their resistance to HU. Whereas overexpression of RNR1 or RNR3 is known to suppress almost all checkpoint mutants, including mec1∆ and rad53∆ (Desany et al, 1998), overexpression of any one of the RNR genes had no effect on the growth of snf1∆ cells in HU (Appendix Fig S7B). These results, which are consistent with a previous study (Dubacq et al, 2004), indicate that the HU sensitivity and checkpoint defect of snf1∆ cells is not due to low levels of R5P and RNR, which are the precursor and rate‐limiting enzyme in dNTP biogenesis. Thus, the checkpoint function resulting from SNF1‐stimulated flux through the PPP is unlikely to depend on nucleotide and dNTP biogenesis, but might require NADPH.
If the role of SNF1, Mig1, and Zwf1 in the DNA replication checkpoint is primarily to produce NADPH, which is an important reducing agent for many biochemical reactions, we wondered whether other reductants might compensate for its role in the checkpoint response. To test this possibility, we supplemented the growth medium with 5 or 10 mM α‐ketoglutarate (α‐KG) and found that they improved the resistance of snf1∆ and snf4∆ cells to HU (Appendix Fig S7C). HU is thought to inhibit RNR by producing free radicals that quench the electron required for RNR catalysis (Davies et al, 2009; Singh & Xu, 2016). If so, one possible explanation for the ability of reductants to improve the resistance of snf1∆ and snf4∆ cells to HU is that they act as general antagonists of free radicals or other reactive oxygen species. We consider this possibility unlikely for two reasons: first, antioxidants that directly scavenge free radicals—Tiron and Tempol—did rescue H2O2 sensitivity but not HU sensitivity (Appendix Fig S7D) and second, α‐KG had no effect on the HU sensitivity of mutants defective in either a Mec1 activator (ddc1∆) or a checkpoint mediator (mrc1∆) (Fig 7B). These findings suggest that the rescue effect of α‐KG is specific to the mutants of the SNF1 complex.
Moreover, 10 mM α‐KG restored HU‐induced formation of RFA1–GFP foci (Fig 7C) and Rad53 phosphorylation (Fig 7D) in snf1∆ to nearly WT levels. By contrast, α‐KG caused a short delay in Rad53 phosphorylation in WT. These data strongly suggest that a proper reducing environment is important for RPA recruitment to stalled replication forks and for checkpoint activation.
The ssDNA‐binding activity of both human and yeast RPAs has been shown to be inhibited by H2O2 and restored by dithiothreitol in vitro (Park et al, 1999; Bochkareva et al, 2000; Wang et al, 2001). Four conserved Cys residues in the zinc finger (ZF) domain of the Rfa1 subunit of RPA are required for its redox regulation (Wang et al, 2001). To investigate the role of these residues, we mutated them to Ser. The resulting rfa1‐zf mutant (C486S, C491S, C505S, C508S) had a growth defect in normal conditions, which was exacerbated by the presence of HU, indicating that these Cys residues are crucial for the binding of RPA to ssDNA in both normal DNA replication and in response to replication stress. The rfa1‐zf mutant was epistatic with snf1∆ in terms of HU sensitivity (Fig 7E), suggesting that they function in the same pathway. Moreover, α‐KG failed to rescue the HU sensitivity of snf1∆ in the rfa1‐zf background (Fig 7E), consistent with the loss of redox‐sensing by this rfa1 allele. These data collectively suggest that RPA recruitment to ssDNA, the initial signal for the DNA replication checkpoint, is directly regulated by cellular redox state through SNF1‐dependent metabolic remodeling (Fig 7F).
Discussion
In this study, we set out to investigate the metabolomic changes that occur when yeast cells are subjected to replication stress. We show that upon HU treatment, SNF1 upregulates the gene encoding the rate‐limiting step of the oxidative PPP, thus diverting glucose from glycolysis to this pathway, which produces NADPH and R5P. The reducing environment created by NADPH production stimulates RPA binding to ssDNA at stalled replication forks, thereby promoting rapid activation of the Mec1–Rad53 kinase cascade. Thus, SNF1 is a crucial player in activating the DNA replication checkpoint, which maintains genome stability.
Cellular redox conditions appear to have distinct effects on the two branches of the DNA damage response: the DNA replication checkpoint controlled by Mec1 (or ATR in mammals) and the DNA damage checkpoint controlled by the related kinase Tel1 (or ATM in mammals). The oxidant H2O2 directly activates ATM by promoting disulfide bonding between two ATM molecules (Guo et al, 2010). By contrast, we show here that a reducing environment promotes Mec1 activation. Our finding is consistent with evidence that DNA replication is restricted to the reductive phase of the yeast metabolic cycle, i.e., glycolysis rather than respiration under nutrient‐limited conditions in continuous culture (Chen et al, 2007). The distinct redox requirements of these two related branches of the DNA damage response might reflect their division‐of‐labor and provide a potential means of transition between them. We are now investigating whether the mechanism identified in this study is conserved by AMPK and ATR in higher eukaryotes.
The reducing potential released by the PPP (i.e., NADPH) facilitates RPA recruitment to ssDNA but is probably not essential for it. When this pathway is inactive and the cellular NADPH/NADP+ ratio is relatively low, for example, RPA is still recruited to ssDNA and forms foci, albeit with a significant delay. This suggests that Mec1 might be activated by a slow mechanism in oxidizing conditions and a fast mechanism in reducing conditions. Such a tunable signaling system may allow cells to respond appropriately depending on the type, severity, and persistence of the stress.
When cells encounter replication stress, the SNF1‐mediated rapid response may have several physiological advantages. First, it redirects glucose to the oxidative PPP, which generates NADPH, the main source of cellular reducing power. Because reducing conditions help to avoid mutations during DNA synthesis (Chen et al, 2007), the additional NADPH might maintain genome stability and aid replication restart after recovery from stress. Second, the reducing cellular environment can relieve the damage caused by hydroperoxyl radicals produced by HU (Huang et al, 2016; Singh & Xu, 2016), which quench a tyrosyl radical in the catalytic core of RNR (Eklund et al, 2001). This mechanism also explains the underlying mechanism of SNF1 in antioxidative stress reported previously (Rabinovitch et al, 2017; Ren & Shen, 2019). Third, SNF1 promotes the rapid formation of RPA‐coated ssDNA, and thus prompt activation of Mec1. We show here that SNF1 is activated within seconds after exposure to HU and a previous study showed activation within 5 min after glucose deprivation (Bendrioua et al, 2014). Such rapid kinetics makes it suitable for the immediate response to HU upstream of Mec1, which is currently regarded as the first kinase in the replication checkpoint signaling pathway. Unlike mec1 and rad53 alleles, snf1∆ cells are not so sensitive to low levels of HU, implicating that SNF1‐mediated rapid activation of the replication checkpoint may become more important when cells suffer severe replication stress.
HU inhibits RNR, and thus the production of dNTPs from R5P. Activation of the PPP by SNF1 generates not only NADPH, which favors binding of RPA to ssDNA and activation of the replication checkpoint, but also R5P, the precursor of dNTPs, which are required to relieve replication stress. Our evidence indicates that SNF1 functions by an NADPH‐dependent and R5P‐independent mechanism to induce the replication checkpoint. Neither supplementation with R5P nor RNR overexpression rescues the checkpoint defects due to the absence of SNF1. Only some special reductants can substitute for the role of SNF1 in RPA recruitment and Mec1–Rad53 checkpoint activation. Reducing power defines a rate‐limiting determinant in a fast‐mode activation of the DNA replication checkpoint to maintain genome stability, and thus provides a direct molecular link between metabolism and genome integrity in addition to nucleotide biogenesis.
In addition to inducing ZWF1 expression, SNF1 may rewire carbon metabolism and cellular redox by other mechanisms to fasten the DNA replication checkpoint activation in response to HU. In animals, for example, the ortholog of SNF1, AMPK, and its activator, the kinase LKB1, inhibit glycolysis (Faubert et al, 2014), which may also help to divert glucose to other pathways including the PPP. Another mechanism whereby SNF1 might produce NADPH to promote the DNA replication checkpoint is through fatty acid metabolism. In solid tumor cells of animals, AMPK maintains NADPH levels by reducing NADPH consumption due to fatty acid synthesis and increasing NADPH production by fatty acid oxidation (Jeon et al, 2012). Consistent with the existence of alternative mechanisms for generating NADPH in yeast, we note that cells, in which the ZWF1 gene is deleted, are hardly sensitive to HU.
Besides its canonical role as a master energy controller regulating catabolism and anabolism in response to nutrients, we show here that SNF1 is also a critical player in maintaining cellular redox homeostasis and genome integrity. SNF1 is activated by binding to AMP, which accumulates in response to energy stress. The molecular details of how SNF1 is activated upon replication stress remain to be determined. Our finding that SNF1 phosphorylates the C‐terminal domain of Mig1 (Ssn6 interaction domain, on T489 and S495) in response to replication stress and regulatory domain of Mig1 (on S222, S278, S311, and S381) in response to carbohydrate stress may provide a clue. This differential phosphorylation of Mig1 also induces the expression of distinct target genes involved in the PPP and carbon utilization. These findings reveal that, on the one hand, SNF1 integrates different signals into one signaling pathway and, on the other hand, divides the work to deal with different types of environmental challenges. SNF1 directly links cell metabolism and redox state to the DNA replication checkpoint, and thus to genome maintenance, providing new insights into the integration of these fundamental cellular processes.
Materials and Methods
Antibodies and reagents
Antibodies and reagents used in the study are listed as following: Mouse anti‐FLAG (Sigma‐Aldrich, F1804), Mouse anti‐Rad53 (Abcam, Ab16859), Rabbit anti‐γ‐H2A (Abcam, Ab15083), Rabbit anti‐Rfa1 (Agrisera, as07214), Mouse anti‐Orc2 (Abcam, ab31930), Rat anti‐Tubulin (Abcam, ab6160), Mouse anti‐His (ORIGENE, TA15088),
Mouse anti‐GST (huaxingbio, HX1807), Rabbit anti‐MYC was a gift from Dr. Qun He, Rabbit anti‐Phospho‐AMPKα Thr172 (Cell Signaling Technology, 2535S), Rabbit anti‐Phospho‐(Ser/Thr) AMPK Substrate (Cell Signaling Technology, 5759S) was a gift from Dr. Cong Yi, Rabbit anti‐GFP (Proteintech, 50430‐2‐AP), HRP‐conjugated anti‐Mouse (Sigma‐Aldrich, A4416), HRP‐conjugated anti‐Rabbit (Sigma‐Aldrich, A6154), HRP‐conjugated anti‐Rat (Sigma‐Aldrich, A9037). 4‐Hydroxy‐TEMPO, 4,5‐Dihydroxy‐1,3‐benzenedisulfonic acid disodium salt monohydrate, (±)‐6‐Hydroxy‐2,5,7,8‐tetramethylchromane‐2‐carboxylic acid, Formaldehyde, Glucose‐6‐phosphate Dehydrogenase from baker's yeast, Hydroxyurea, NADP (disodium salt), Phenazine ethosulfate, Ribose‐5‐phosphate, RNaseA, Sorbitol, ssDNA, Sucrose and Thiazolyl Blue Tetrazolium Bromide were purchased from Sigma‐Aldrich. Albumin, Bovine (BSA), Alpha‐ketoglutaric acid free acid, and Glycine were purchased from AMRESCO. Poly FLAG Peptide Lyophilized Powder and Alpha‐factor Peptide were purchased from Scilight Peptide. Enhanced BCA Protein Assay Kit and NADPH were purchased from Beyotime. Anti‐FLAG (DYKDDDK) Affinity Gel was purchased from Bimake. D‐Glucose 6‐phosphate disodium salt hydrate was purchased from Solarbio. FastStart Universal SYBR Green Master was purchased from Roche. GoScript® Reverse Transcriptase and Recombinant RNasin Ribonuclease Inhibitor were purchased from Promega. Protein G Sepharose Beads were purchased from GE healthcare. Proteinase K was purchased from MP Biomedicals.
Yeast strains, plasmids, and manipulations
The Saccharomyces cerevisiae strains and plasmids used in this study are listed in Appendix Tables S1 and S2, respectively. Strains without plasmids were usually grown in rich medium (YPD) containing 1% yeast extract, 2% peptone, and 2% glucose (or 5% where specifically indicated), and strains with plasmids were cultured in synthetic complete dropout medium (SC medium). According to the requirement, cells were synchronized in G1 phase by adding α‐factor mating pheromone, whereas the S‐phase cells were obtained by releasing into fresh medium. Nocodazole was added to the culture to arrest cells in G2/M. All cell cycle experiments were validated by flow cytometry.
Yeast metabolomic measurements
Metabolic samples were prepared as previously described with slight modifications (Campbell et al, 2020). Briefly, strains cultured overnight were transferred into the fresh medium at an initial concentration of 0.1 measured at 600 nm (OD600), until an optical density reached 0.6. Hydroxyurea was added to a final concentration of 200 mM and incubated for 60 min (200 rpm, 30°C). Cells were harvested using Beckman Coulter Avanti JXN‐26 (4,000 rpm, 4°C). Using 200‐ml precooled PBS, cells were washed twice to remove the residual media. Cell pellets were frozen to quench cellular enzyme activities and then maintained at −80°C until processed. To remove proteins and obtain metabolic extracts, we added 500‐μl precooled (−20°C) 75% methanol containing Terbutaline and Brobutero and 250‐μl precooled glass beads (−20°C) into frozen pellets and broken three times through mini‐bead beater for 30 s and 3‐min interval precooling followed by centrifugation at 4°C. A quantity of 500 μl of 75% methanol was added again to wash glass beads. Therefore, totally 1‐ml metabolic extracts were generated. The extracts were concentrated using Eppendorf Concentrator plus. Concentrated extracts were re‐dissolved by 50% methanol. The resulting extract was analyzed by two separate reverse phases (RP)/LC‐MS methods (ACQUITY UPLCI‐Class High‐performance liquid chromatography) with positive or negative ion mode. Additionally, quality control (QC) samples were generated by mixing a small volume of each experimental sample to serve as a technical replicate through the data set and monitor instrument performance as well as aiding toward chromatographic alignment.
The raw data were processed using Xcalibur software (Thermo Scientific, USA). Mass spectrometric features were extracted using Progenesis QI (Waters, UK) and processed according to the retention time, mass‐to‐charge ratio (m/z), and peak area. Multivariate statistics analyses, including principal components analysis (PCA) and partial least squares‐discriminant analysis (PLS‐DA), were performed following the instruction of SIMCA‐P 13.0 software (Umetrics). A simplified two‐dimensional projection procedure facilitated the simultaneous comparison of a large number of mass spectrometric data.
Serial‐dilution analyses (Spot assay)
Equal amounts of starting overnight cells (0.2 OD600) were serially diluted fivefold and then spotted onto appropriate medium. Plates were cultured at 30°C for 48 h, or otherwise indicated, and then photographed.
Cell lysates and Western blots
For western blotting analysis, strains were grown and 5 OD600 cells were collected. Protein extracts were prepared using the trichloroacetic acid (TCA) precipitation and probed for specific antibodies. The Rad53, γ‐H2A, FLAG, and Tubulin protein levels were detected with mouse anti‐Rad53 (Abcam, ab16859, 1:1,000), rabbit anti‐γ‐H2A (Abcam, ab15083, 1:1,000), mouse anti‐FLAG (Sigma‐Aldrich, F1804, 1:2,000), and rat anti‐Tubulin (Abcam, ab6160, 1:10,000), respectively. HRP‐conjugated anti‐Mouse (Sigma‐Aldrich, A4416‐1 ml, 1:10,000), anti‐Rabbit (Sigma‐Aldrich, A6154‐1 ml, 1:10,000), or anti‐Rat IgG (Sigma‐Aldrich, A9037‐1 ml, 1:10,000) serve as the secondary antibodies, respectively.
Immunoblots for SNF1 activity
To prevent activation of Snf1 during processing (Wilson et al, 1996), we killed cells before centrifugation by boiling for 3 min. Protein extracts were prepared as previously described (Orlova et al, 2008). The Snf1‐13MYC, Snf1 T210 protein levels were detected with rabbit anti‐MYC (1:10,000) and rabbit anti‐Phospho‐AMPKα Thr172 (1:1,000), respectively.
Recombinant protein purification
DNA encoding Mig1 88‐C (amino acids 88 to 504) was amplified by PCR and cloned into pGEX‐6p‐1 vector. pGEX‐6p‐1‐mig1 88‐C‐2A was constructed by quick point‐mutant PCR. Expression plasmids were transformed into E. coli expression strains (ED3 pLysS). A single transformation colony was inoculated into a 50‐ml flask containing 25 ml of Luria–Bertani broth (LB) supplemented with ampicillin (50 mg/ml) and chloramphenicol (34 μg/ml). The inoculated culture was grown overnight at 37°C. The following day, 10 ml of the overnight culture was used to inoculate a 2‐l flask containing 1 l of LB, supplemented with antibiotics as described above. Cultures were grown at 37°C until the optical density (OD600) reached 0.6–0.8. Then, 0.1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) was added and recombinant proteins were induced at 30°C for 2 h. Cells were harvested by centrifugation and the pellets were stored at −80°C. The cell pellet arising from 1 l of culture was resuspended in 30 ml of Buffer A: 50 mM Tris‐HCl pH 7.4, 500 mM NaCl, 0.1% Tween‐20, supplemented with proteinase inhibitor PMSF and lysed by sonication (1 s on 3 s off, on ice), then cell debris and precipitated material were removed by high‐speed centrifugation at 20,000 rpm for 1 h at 4°C. The supernatants were incubated with 100 μl GST beads for 2 h at 4°C. Finally, 20 mM GSH was used to elute Mig1 C or 2A proteins.
Chromatin fractionation
To examine chromatin‐bound and non‐chromatin‐bound protein levels, we fractionated cells as previously described with minor modifications (Quan et al, 2015). Briefly, 20 OD600 cells were collected and washed once with precooled 1 × PBS. Cells were suspended with 1 ml PSB buffer and strewed for 10 min at room temperature. After spheroplasting using lyticase in SB buffer, cells were washed once with SWB buffer. Cells were lysed for whole‐cell extracts (WCE) in EB buffer containing 0.25% Triton X‐100. WCE were gently spread on the 30% sucrose and the chromatin‐bound fraction (Chromatin) was isolated after centrifugation for 15 min at 14,000 rpm, and the supernatant was carefully removed. EBX buffer suspended the precipitation as the chromatin non‐chromatin‐bound fraction (non‐Chromatin). Proteins were detected using rabbit anti‐Rfa1 (Agrisera, as07214, 1:10,000), rat anti‐tubulin (Abcam, ab6160, 1:10,000), and mouse anti‐Orc2 (Abcam, ab31930, 1:1,000).
Buffers for chromatin fractionation:
PSB buffer: 100 mM PIPES/KOH pH 9.4, 10 mM DTT, 0.1% NaN3;
SB buffer: 50 mM KHPO4 pH 7.4, 600 mM Sorbitol, 10 mM DTT;
SWB buffer: 50 mM HEPES/KOH pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 400 mM Sorbitol;
EB buffer: 50 mM HEPES/KOH pH 7.5, 150 mM NaCl, 2.5 mM MgoAc, 0.1 mM ZnoAc, 2 mM NaF, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 1 μg/ml pepstatin A, 10 mM benzamidine.
Quantitative RT‐qPCR
Cells were collected in demand and total RNA extracts were prepared using a commercial TRIZOL reagent (CoWin Biosciences) following the manufacturer’s instructions. For reverse transcription‐PCR (RT‐PCR) analysis, reverse transcription with 1 μl oligo (dT) primer was performed with 2 μg of total RNA, 1 mM dNTPs, 0.5 μl RT, and 1 μl RNasin for 45 min at 42°C, which was followed by a 10‐min heat inactivation at 72°C. Real‐time quantitative PCR amplification was performed using SYBR‐Green Mix (Roche) on QuantStudio 6 Flex system (Life).
Primers for RT‐qPCR
ACTIN forward: 5’‐ CCCAGGTATTGCCGAAAGAATGC‐3’
ACTIN reverse: 5’‐TTTGTTGGAAGGTAGTCAAAGAAGCC‐3’
ZWF1 forward: 5’‐CGACACAGATGAAGGCTTCG‐3’
ZWF1 reverse: 5’‐TCTCTACGATTACACGGGTG‐3’
Chromatin immunoprecipitation for next‐generation sequencing (ChIP‐seq)
Cells were lysed in 800 μl of lysis buffer (50 mM HEPES KOH pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.1% Na‐deoxycholate, 0.1% SDS) with zirconia beads using a bead beater. Chromatin was sheared by sonication to fragments of 200–500 bp. Cell lysates and glass beads were separated by centrifugation (1,000 g 4°C). One percent of the extract was taken as input and the remainder was used for immunoprecipitation with anti‐MYC antibody for overnight and followed by 30 min incubation with protein G sepharose beads. Beads were subsequently washed in 1× lysis buffer, 1× lysis buffer with 500 mM NaCl, 1× wash buffer, and 1× TE pH 8.0. Elution of immunoprecipitated complexes was achieved with 1% SDS + 0.1 mol/l NaHCO3, proteins were degraded with a final concentration of 1 μg/μl Proteinase K (4 h, 45°C) and crosslinks reversed (8 h, 65°C). DNA was purified using phenol–chloroform extraction. DNA concentration was determined using Qubit 3.0 fluorometer (Invitrogen) and Qubit dsDNA HS assay kit (Invitrogen, Cat# Q32854).
Next‐generation sequencing (NGS)
Immunoprecipitated DNA and input DNA were sequenced by Illumina GAIIx (one sample per lane). 75‐bp paired‐end runs were performed using a TruSeq SBS Kit v5 (Illumina). For the sequencing, DNA library was prepared using VAHTS™ Universal DNA Library Prep Kit for Illumina® V3 (Vazyme Biotech Co., Ltd) reagent set for Illumina and the multiplex oligos.
ChIP‐seq data analysis
After trimming adapter sequences and low‐quality reads by trimmomatic (v0.39) (Bolger et al, 2014) with option “LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 HEADCROP:2 AVGQUAL:30 MINLEN:36”, FastQC (v0.11.8) was performed on trimmed reads to confirm high‐quality sequences (Brown et al, 2017). Clean reads were then aligned to the yeast Saccharomyces cerevisiae genome (R64‐1‐1) using bowtie2 (v2.3.5.1) (Langmead & Salzberg, 2012) with default parameters. Reads with low mapping quality (MAPQ score < 10) were removed using samtools (v1.3.1) (Li et al, 2009). PCR duplicate reads were also removed using the MarkDuplicates utility within Picard 2.18.29 (http://broadinstitute.github.io/picard/). For normalization and visualization, the sorted, filtered.bam files from each sample were converted to bigwig format using bamCoverage program with parameters “‐bs 50,‐‐normalizeUsing RPKM” in deepTools v3.3.0 (Ramirez et al, 2016). The normalized ChIP‐seq signals for a scaled region representing each of the ARS consensus sequence (ACS) plus/minus 5 kb were compiled using the computeMatrix program from deepTools package. The visualization was conducted using the software R (v 3.5.1; www.r‐project.org). Peak calling on ChIP‐seq data was performed using MACS2 (v2.1.2) (Zhang et al, 2008) callpeak with P‐value lower than 0.01. The MAnorm program (Shao et al, 2012) was applied to identify differential peaks between wild type and snf4Δ, using a fold change threshold of 2 and P‐value threshold of 0.0001.
ChIP‐qPCR analysis
ChIP was performed according to procedures previously described with slight modifications (Li et al, 2019a). Briefly, cells were synchronized at G1 phase by α‐factor and then released into a fresh medium containing 200 mM HU for the indicated time to collect more HU‐stalled replication forks. Cells were crosslinked with 1% formaldehyde (Sigma‐Aldrich, F1635‐500 ml) at room temperature for 17 min and quenched with 125 mM glycine (Ameresco, 0617‐1 kg) for 5 min. Cell extracts were prepared and DNA was sheared to an average size of 200–500 bp using a Bioruptor. Endogenous Rfa1 proteins carrying a 13MYC tag were precipitated by an anti‐MYC antibody overnight at 4°C. The immune complexes were harvested by the addition of 50 μl of protein G sepharose beads. Formaldehyde crosslinks were reversed by incubation at 65°C for 4 h, followed by protease K treatment at 42°C for 1 h. Then, co‐precipitated genomic DNA was purified using phenol–chloroform extraction and subjected to quantitative real‐time PCR SYBR‐Green Mix on QuantStudio 6 Flex system (Life).
Primers for ChIP‐qPCR
ARS607 forward: 5’‐TGCCGCACGCCAAACATTGC‐3’
ARS607 reverse: 5’‐CGGCTCGTGCATTAAGCTTG‐3’
ARS607 + 14kb forward: 5’‐CTCTTCATCACTGGAGTCCT‐3’
ARS607 + 14kb reverse: 5’‐CGGCTGTCATGCCAAGATGC‐3’
ARS305 forward: 5’‐AGCAAGACCGGCCAGTTTGA‐3’
ARS305 reverse: 5’‐GCACTTTGATGAGGCTCTAGCAA‐3’
ARS305 ‐ 12kb forward: 5’‐GCGGAAGTCTTTGCAACTGATATG‐3’
ARS305 ‐ 12kb reverse: 5’‐TGCTTGATTTCTTCGCAGTATTGG‐3’
In vitro SNF1 kinase assay
The SNF1 kinase complex containing SNF1‐5FLAG or snf1‐KD‐5FLAG (K84RT210A) was purified from yeast cells treated by 200 mM HU for 20 min. In vitro kinase assays were performed as previously described (Yi et al, 2017). A quantity of 60‐µl reaction mixture contains about 20 ng Snf1‐5FLAG or snf1‐KD‐5FLAG as a kinase, 2 mg GST‐Mig1 88‐C WT or 2A as a substrate. 1 × kinase buffer was diluted from 10 × store buffer (250 mM HEPES pH 7.5, 100 mM MgCl2, 500 mM NaCl, 20 mM DTT, 1 mM EDTA, 1 mM EGTA, and 10 mM ATP). Reaction mixtures were incubated at 30°C for 30 min, and the reaction was stopped by adding 15 µl 5 × SDS loading buffer and boiling for 5 min. Samples were resolved by 8% SDS‐PAGE and analyzed by immunoblotting using anti‐Phospho‐(Ser/Thr) AMPK Substrate antibody and anti‐Phospho‐AMPKα Thr172.
Quantification of cellular NADPH: NADP+ ratio
Cellular NADPH/NADP+ measurement was performed as previously described with minor modifications (Wang et al, 2016). Briefly, cells were cultured in 40 ml YPD from an initial OD600 of 0.2 to a final OD600 of 1.5. Then, a concentration of 200 mM HU was added for the indicated time. Cells were harvested (4,000 rpm, 3 min) and washed twice using 200‐ml precooled PBS. Cell pellets were frozen in liquid nitrogen for 1 min to quench the enzyme activities. Then, cells were resuspended in 300 μl of 200‐mM NaOH for NADPH extraction, or 300 μl of 200 mM HCl for NADP+ extraction to be multi‐gelated by liquid nitrogen. The extracts were neutralized by adding 300 μl of 100 mM HCl (for NADPH extraction) or 300 μl of 100 mM NaOH (for NADP+ extraction). The cellular debris was removed by centrifugation at 14,000 rpm for 15 min. The concentration of NADPH or NADP+ was quantified using a sensitive enzymatic cycling assay in a reaction buffer containing 100 μl of 1.0 M bicine buffer (pH 8.0), 100 μl of 100 mM glucose‐6‐phosphate, 100 μl of 40 mM EDTA (pH 8.0), 100 μl of 4.2 mM MTT (3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2H‐tetrazolium bromide), 200 μl of 16.6 mM PES (phenazine ethosulfate), and 300 μL of ddH2O, and incubated at 30°C for 10 min. Then, 50‐μl neutralized extract and 50 μl of yeast glucose‐6‐phosphate dehydrogenase (50 U/ml, Sigma‐Aldrich, G7877‐250U) were added to start reaction. The absorb strength at 570 nm was recorded for 12 min at 45‐s intervals at 30°C. The standard curves of NADPH and NADP+ were performed as the manner using a gradient concentration of NADPH or NADP+ instead of the neutralized extract. Additionally, to rule out the difference of extract strength, protein concentrations were determined using Enhanced BCA Protein Assay Kit (Beyotime, P0010S).
Author contributions
Lili Li: Conceptualization; Data curation; Software; Investigation; Visualization; Writing – original draft; Writing – review and editing. Jie Wang: Investigation. Zijia Yang: Investigation. Yiling Zhao: Investigation. Hui Jiang: Investigation; Writing – review and editing. Luguang Jiang: Methodology; Writing – review and editing. Wenya Hou: Resources; Writing – review and editing. Risheng Ye: Writing – review and editing. Qun He: Resources; Writing – review and editing. Martin Kupiec: Supervision; Funding acquisition; Writing – review and editing. Brian Luke: Supervision; Writing – review and editing. Qinhong Cao: Resources; Funding acquisition. Zhi Qi: Supervision; Funding acquisition; Methodology; Writing – original draft. Zhen Li: Supervision; Funding acquisition; Methodology. Huiqiang Lou: Conceptualization; Supervision; Funding acquisition; Visualization; Writing – original draft; Writing – review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization, LL and HL; Methodology, ZQ, LJ, and ZL; Investigation, LL, YZ, JW, ZY, and HJ; Resources, WH, QH, and QC; Writing‐Original Draft, HL, LL, and ZQ; Writing‐Review & Editing, HL, LL, WH, QH, RY, BL, and MK; Visualization, LL, HL, LJ, and ZQ; Supervision, HL, ZQ, ZL, BL, and MK; Funding Acquisition, HL, ZQ, ZL, QC, and MK.
Disclosure statement and competing interests
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We thank Drs. Marc Wold from the University of Iowa for the plasmid encoding RPA (p11td‐scRPA), Qing Li from Peking University for help with RPA purification, Cong Yi from Zhejiang University for an AMPK substrate antibody, Yu Shen from Shandong University and Xiaoming Bao from Qilu University of Technology for help with NADPH/NADP+ measurement and Acc1 plasmids, Yan He from China Agricultural University for NGS data analysis, Carol Featherstone of Plume Scientific Communication Services (France) for professional editing services, and Drs. Marco Foiani, Philippe Pasero, and members of the Lou lab for helpful discussions and comments on the manuscript. This work was supported by the National Natural Science Foundation of China Grants 32161133015, 31630005, 31770084, and 31771382, 32101039, and 31670762; the National Key R&D Program of China 2019YFA0903900, Beijing Municipal Natural Science Foundation 5204036, the Israel Science Foundation, the Israel Cancer Research Fund and the Minerva Center for In‐lab Evolution, Open Research Fund of the National Center for Protein Sciences at Peking University in Beijing, and Project Program of the State Key Laboratory of Agrobiotechnology 2019SKLAB1‐7.
The EMBO Journal (2022) 41: e108290.
Data availability
The metabolomics dataset generated during this study is available through a MetaboLights database entry (https://www.ebi.ac.uk/metabolights/) with the study identifier MTBLS1402. The ChIP‐seq raw data in this study have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number CRA005117 that are publicly accessible at http://ngdc.cncb.ac.cn/gsa. Other data that support the findings of this study are available from the corresponding authors upon request.
References
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 3: 958–965 [DOI] [PubMed] [Google Scholar]
- Awasthi P, Foiani M, Kumar A (2015) ATM and ATR signaling at a glance. J Cell Sci 128: 4255–4262 [DOI] [PubMed] [Google Scholar]
- Bendrioua L, Smedh M, Almquist J, Cvijovic M, Jirstrand M, Goksor M, Adiels CB, Hohmann S (2014) Yeast AMP‐activated protein kinase monitors glucose concentration changes and absolute glucose levels. J Biol Chem 289: 12863–12875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Cortez D, Lopes M (2020) The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 21: 633–651 [DOI] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell 66: 801–817 [DOI] [PubMed] [Google Scholar]
- Bochkareva E, Korolev S, Bochkarev A (2000) The role for zinc in replication protein A. J Biol Chem 275: 27332–27338 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Pirrung M, McCue LA (2017) FQC Dashboard: integrates FastQC results into a web‐based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33: 3137–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnelli S, Tripodi F, Nicastro R, Cirulli C, Tedeschi G, Pagliarin R, Alberghina L, Coccetti P (2013) Snf1/AMPK promotes SBF and MBF‐dependent transcription in budding yeast. Biochim Biophys Acta 1833: 3254–3264 [DOI] [PubMed] [Google Scholar]
- Campbell K, Westholm J, Kasvandik S, Di Bartolomeo F, Mormino M, Nielsen J (2020) Building blocks are synthesized on demand during the yeast cell cycle. Proc Natl Acad Sci USA 117: 7575–7583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yang X, Shen Y, Hou J, Bao X (2018) Screening phosphorylation site mutations in yeast acetyl‐CoA carboxylase using malonyl‐CoA sensor to improve malonyl‐CoA‐derived product. Front Microbiol 9: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Odstrcil EA, Tu BP, McKnight SL (2007) Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science 316: 1916–1919 [DOI] [PubMed] [Google Scholar]
- Choi J‐H, Lindsey‐Boltz LA, Kemp M, Mason AC, Wold MS, Sancar A (2010) Reconstitution of RPA‐covered single‐stranded DNA‐activated ATR‐Chk1 signaling. Proc Natl Acad Sci USA 107: 13660–13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BW, Kohanski MA, Simmons LA, Winkler JA, Collins JJ, Walker GC (2009) Hydroxyurea induces hydroxyl radical‐mediated cell death in Escherichia coli . Mol Cell 36: 845–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desany BA, Alcasabas AA, Bachant JB, Elledge SJ (1998) Recovery from DNA replicational stress is the essential function of the S‐phase checkpoint pathway. Genes Dev 12: 2956–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande I, Seeber A, Shimada K, Keusch JJ, Gut H, Gasser SM (2017) Structural basis of Mec1‐Ddc2‐RPA assembly and activation on single‐stranded DNA at sites of damage. Mol Cell 68: 431–445 [DOI] [PubMed] [Google Scholar]
- Dubacq C, Chevalier A, Mann C (2004) The protein kinase Snf1 is required for tolerance to the ribonucleotide reductase inhibitor hydroxyurea. Mol Cell Biol 24: 2560–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund H, Uhlin U, Färnegårdh M, Logan DT, Nordlund P (2001) Structure and function of the radical enzyme ribonucleotide reductase. Prog Biophys Mol Biol 77: 177–268 [DOI] [PubMed] [Google Scholar]
- Estruch F, Treitel MA, Yang X, Carlson M (1992) N‐terminal mutations modulate yeast SNF1 protein kinase function. Genetics 132: 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ et al (2014) Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF‐1α. Proc Natl Acad Sci USA 111: 2554–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Branzei D (2017) S‐phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell Mol Life Sci 74: 2361–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT (2010) ATM activation by oxidative stress. Science 330: 517–521 [DOI] [PubMed] [Google Scholar]
- Hardie DG (2014) AMP‐activated protein kinase: maintaining energy homeostasis at the cellular and whole‐body levels. Annu Rev Nutr 34: 31–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedbacker K, Carlson M (2008) SNF1/AMPK pathways in yeast. Front Biosci 13: 2408–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Zhou Z, Elledge SJ (1998) The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94: 595–605 [DOI] [PubMed] [Google Scholar]
- Huang ME, Facca C, Fatmi Z, Baille D, Benakli S, Vernis L (2016) DNA replication inhibitor hydroxyurea alters Fe‐S centers by producing reactive oxygen species in vivo . Sci Rep 6: 29361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, Gasser SM, Shimada K (2013) Replication checkpoint: tuning and coordination of replication forks in s phase. Genes 4: 388–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S‐M, Chandel NS, Hay N (2012) AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485: 661–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Dibitetto D, Smolka MB (2019) DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J 38: e101801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Xu Z, Xu J, Zuo L, Yu C, Zheng PU, Gan H, Wang X, Li L, Sharma S et al (2018) Rtt105 functions as a chaperone for replication protein A to preserve genome stability. EMBO J 37: e99154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Jin X, Sharma S, Liu X, Zhang J, Niu Y, Li J, Li Z, Zhang J, Cao Q et al (2019a) Mck1 defines a key S‐phase checkpoint effector in response to various degrees of replication threats. PLoS Genet 15: e1008136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yao C‐F, Xu F‐J, Qu Y‐Y, Li J‐T, Lin Y, Cao Z‐L, Lin P‐C, Xu W, Zhao S‐M et al (2019b) APC/C(CDH1) synchronizes ribose‐5‐phosphate levels and DNA synthesis to cell cycle progression. Nat Commun 10: 2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Xu Z, Leng H, Zheng P, Yang J, Chen K, Feng J, Li Q (2017) RPA binds histone H3–H4 and functions in DNA replication‐coupled nucleosome assembly. Science 355: 415–420 [DOI] [PubMed] [Google Scholar]
- Maréchal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5: a012716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi‐Patil VM, Burgers PM (2009) A tale of two tails: activation of DNA damage checkpoint kinase Mec1/ATR by the 9‐1‐1 clamp and by Dpb11/TopBP1. DNA Repair 8: 996–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova M, Barrett L, Kuchin S (2008) Detection of endogenous Snf1 and its activation state: application to Saccharomyces and Candida species. Yeast 25: 745–754 [DOI] [PubMed] [Google Scholar]
- Pardo B, Crabbé L, Pasero P (2016) Signaling pathways of replication stress in yeast. FEMS Yeast Res 17: 1–11 [DOI] [PubMed] [Google Scholar]
- Park JS, Wang M, Park SJ, Lee SH (1999) Zinc finger of replication protein A, a non‐DNA binding element, regulates its DNA binding activity through redox. J Biol Chem 274: 29075–29080 [DOI] [PubMed] [Google Scholar]
- Pearl LH, Schierz AC, Ward SE, Al‐Lazikani B, Pearl FMG (2015) Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 15: 166–180 [DOI] [PubMed] [Google Scholar]
- Pessina S, Tsiarentsyeva V, Busnelli S, Vanoni M, Alberghina L, Coccetti P (2010) Snf1/AMPK promotes S‐phase entrance by controlling CLB5 transcription in budding yeast. Cell Cycle 9: 2189–2200 [DOI] [PubMed] [Google Scholar]
- Quan Y, Xia Y, Liu L, Cui J, Li Z, Cao Q, Chen XS, Campbell JL, Lou H (2015) Cell‐cycle‐regulated interaction between Mcm10 and double hexameric Mcm2‐7 is required for helicase splitting and activation during S phase. Cell Rep 13: 2576–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St‐Pierre J, Jones RG (2017) AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep 21: 1–9 [DOI] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T (2016) deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Shen H‐M (2019) Critical role of AMPK in redox regulation under glucose starvation. Redox Biol 25: 101154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA (2017) The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18: 622–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A, Hegnauer AM, Hustedt N, Deshpande I, Poli J, Eglinger J, Pasero P, Gut H, Shinohara M, Hopfner K‐P et al (2016) RPA mediates recruitment of MRX to forks and double‐strand breaks to hold sister chromatids together. Mol Cell 64: 951–966 [DOI] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan GC, Orkin SH, Waxman DJ (2012) MAnorm: a robust model for quantitative comparison of ChIP‐Seq data sets. Genome Biol 13: R16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Xu Y‐J (2016) The cell killing mechanisms of hydroxyurea. Genes 7: 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FC, Davies SP, Wilson WA, Carling D, Hardie DG (1999) The SNF1 kinase complex from Saccharomyces cerevisiae phosphorylates the transcriptional repressor protein Mig1p in vitro at four sites within or near regulatory domain 1. FEBS Lett 453: 219–223 [DOI] [PubMed] [Google Scholar]
- Tittel‐Elmer M, Alabert C, Pasero P, Cobb JA (2009) The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J 28: 1142–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treitel MA, Carlson M (1995) Repression by SSN6‐TUP1 is directed by MIG1, a repressor/activator protein. Proc Natl Acad Sci USA 92: 3132–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treitel MA, Kuchin S, Carlson M (1998) Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae . Mol Cell Biol 18: 6273–6280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, You JS, Lee SH (2001) Role of zinc‐finger motif in redox regulation of human replication protein A. Antioxid Redox Signal 3: 657–669 [DOI] [PubMed] [Google Scholar]
- Wang X, Liang Z, Hou J, Bao X, Shen Y (2016) Identification and functional evaluation of the reductases and dehydrogenases from Saccharomyces cerevisiae involved in vanillin resistance. BMC Biotechnol 16: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WA, Hawley SA, Hardie DG (1996) Glucose repression/derepression in budding yeast: SNF1 protein kinase is activated by phosphorylation under derepressing conditions, and this correlates with a high AMP:ATP ratio. Curr Biol 6: 1426–1434 [DOI] [PubMed] [Google Scholar]
- Wold MS (1997) Replication protein A: a heterotrimeric, single‐stranded DNA‐binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem 66: 61–92 [DOI] [PubMed] [Google Scholar]
- Yang X, Peng X, Huang J (2018) Inhibiting 6‐phosphogluconate dehydrogenase selectively targets breast cancer through AMPK activation. Clin Transl Oncol 20: 1145–1152 [DOI] [PubMed] [Google Scholar]
- Yazinski SA, Zou L (2016) Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet 50: 155–173 [DOI] [PubMed] [Google Scholar]
- Yi C, Tong J, Lu P, Wang Y, Zhang J, Sun C, Yuan K, Xue R, Zou B, Li N et al (2017) Formation of a Snf1‐Mec1‐Atg1 module on mitochondria governs energy deprivation‐induced autophagy by regulating mitochondrial respiration. Dev Cell 41: 59–71 [DOI] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF (2010) Checkpoint‐dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340 [DOI] [PubMed] [Google Scholar]
- Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R (2001) The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J 20: 3544–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Thompson CB (2019) Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 20: 436–450 [DOI] [PMC free article] [PubMed] [Google Scholar]