

Abstract
Haploinsufficiency of the progranulin (PGRN)‐encoding gene (GRN) causes frontotemporal lobar degeneration (GRN‐FTLD) and results in microglial hyperactivation, TREM2 activation, lysosomal dysfunction, and TDP‐43 deposition. To understand the contribution of microglial hyperactivation to pathology, we used genetic and pharmacological approaches to suppress TREM2‐dependent transition of microglia from a homeostatic to a disease‐associated state. Trem2 deficiency in Grn KO mice reduced microglia hyperactivation. To explore antibody‐mediated pharmacological modulation of TREM2‐dependent microglial states, we identified antagonistic TREM2 antibodies. Treatment of macrophages from GRN‐FTLD patients with these antibodies led to reduced TREM2 signaling due to its enhanced shedding. Furthermore, TREM2 antibody‐treated PGRN‐deficient microglia derived from human‐induced pluripotent stem cells showed reduced microglial hyperactivation, TREM2 signaling, and phagocytic activity, but lysosomal dysfunction was not rescued. Similarly, lysosomal dysfunction, lipid dysregulation, and glucose hypometabolism of Grn KO mice were not rescued by TREM2 ablation. Synaptic loss and neurofilament light‐chain (NfL) levels, a biomarker for neurodegeneration, were further elevated in the Grn/Trem2 KO cerebrospinal fluid (CSF). These findings suggest that TREM2‐dependent microglia hyperactivation in models of GRN deficiency does not promote neurotoxicity, but rather neuroprotection.
Keywords: frontotemporal lobar degeneration, lysosomes, microglia, neurodegeneration, progranulin
Subject Categories: Immunology, Neuroscience
TREM2‐dependent hyperactivated microglia retain neuroprotective functions in the neurodegenerative disorder GRN‐FTLD.

Introduction
Neurodegenerative diseases are currently incurable and novel therapeutic strategies are desperately required. Besides disease‐defining protein deposits (Aguzzi & Haass, 2003), microgliosis is observed in almost all neurodegenerative diseases (Ransohoff, 2016). Microgliosis can be detrimental (Heneka et al, 2013; Hong et al, 2016a). However, recent findings strongly suggested that certain microglial responses to brain pathology may also be neuroprotective (Deczkowska et al, 2020; Lewcock et al, 2020). This is based on the identification of variants in genes predominantly or exclusively expressed in microglia within the brain that increase the risk for late‐onset Alzheimer’s disease (LOAD) and other neurodegenerative disorders (Efthymiou & Goate, 2017). Protective microglial functions became particularly evident upon functional investigations of coding variants found within the triggering receptor expressed on myeloid cells 2 (TREM2) gene, which can increase the risk for LOAD and other neurogenerative disorders including frontotemporal dementia‐like syndromes (Guerreiro et al, 2013; Jonsson et al, 2013). These TREM2 variants reduce lipid ligand binding, lipid and energy metabolism, chemotaxis, survival/proliferation, phagocytosis of cellular debris, and potentially other essential microglial functions (Deczkowska et al, 2020; Lewcock et al, 2020). Moreover, a loss of TREM2 function locks microglia in a dysfunctional homeostatic state (Keren‐Shaul et al, 2017; Krasemann et al, 2017; Mazaheri et al, 2017; Nugent et al, 2020), in which they are unable to respond to pathological challenges by inducing a disease‐associated mRNA signature.
Disease‐associated microglia (DAM) respond to amyloid pathology by clustering around amyloid plaques where they exhibit a protective function by encapsulating the protein deposits via a barrier function (Yuan et al, 2016) that promotes amyloid plaque compaction (Ulrich et al, 2014; Wang et al, 2016; Meilandt et al, 2020) and reduces de novo seeding of amyloid plaques (Parhizkar et al, 2019). TREM2 is therefore believed to be a central target for therapeutic modulation of microglial functions (Deczkowska et al, 2020; Lewcock et al, 2020). A number of agonistic anti‐TREM2 antibodies were recently developed (Cheng et al, 2018; Cignarella et al, 2020; Price et al, 2020; Schlepckow et al, 2020; Wang et al, 2020; Ellwanger et al, 2021; Fassler et al, 2021), which either enhance cell surface levels of signaling‐competent TREM2 by blocking TREM2 shedding and/or crosslinking TREM2 receptors to stimulate downstream signaling via Syk phosphorylation. In preclinical studies, these antibodies boost protective functions of microglia as shown by enhanced amyloid β‐peptide and myelin clearance, reduced amyloid plaque load, improved memory in models of amyloidosis, and supported axon regeneration and remyelination in models of demyelinating disorders such as multiple sclerosis (Cheng et al, 2018; Cignarella et al, 2020; Lewcock et al, 2020; Price et al, 2020; Schlepckow et al, 2020; Wang et al, 2020; Bosch‐Queralt et al, 2021; Ellwanger et al, 2021; Fassler et al, 2021).
Although increased TREM2 may be protective in AD patients (Ewers et al, 2019), in other neurodegenerative diseases, microglia may be overactivated and become dysfunctional (Heneka et al, 2013; Hong et al, 2016a; Ransohoff, 2016). Therefore, in these contexts, antagonistic TREM2 antibodies may display therapeutic benefit through dampening microglial hyperactivation. A well‐described example of a neurodegenerative disorder where microglia are hyperactivated is GRN‐associated frontotemporal lobar degeneration (GRN‐FTLD) with TDP‐43 (transactive response DNA‐binding protein 43 kDa) deposition caused by progranulin (PGRN) deficiency (Baker et al, 2006; Cruts et al, 2006; Gotzl et al, 2019). In models of GRN‐FTLD‐associated haploinsufficiency, hyperactivation of microglia is evident, as demonstrated by an increased disease‐associated mRNA signature as well as strongly increased 18‐kDa translocator protein positron emission‐tomography ((TSPO)‐PET) signals in mouse models (Martens et al, 2012; Lui et al, 2016; Krabbe et al, 2017; Gotzl et al, 2019; Huang et al, 2020; Marschallinger et al, 2020; Zhang et al, 2020). This is the opposite phenotype of Trem2 knockout (KO) microglia, which are locked in a homeostatic state (Keren‐Shaul et al, 2017; Kleinberger et al, 2017; Krasemann et al, 2017; Mazaheri et al, 2017; Gotzl et al, 2019; Nugent et al, 2020). Hyperactivation of microglia is also observed in the brain of GRN‐FTLD patients (Lui et al, 2016; Woollacott et al, 2018; Gotzl et al, 2019). PGRN is a secreted protein, which is also transported to lysosomes (Hu et al, 2010; Zhou et al, 2015), where it appears to control activity of hydrolases, such as cathepsins and glucocerebrosidase (GCase) (Gotzl et al, 2016, 2018; Beel et al, 2017; Ward et al, 2017; Paushter et al, 2018; Arrant et al, 2019; Butler et al, 2019b; Logan et al, 2021). Total loss of PGRN results in a lysosomal storage disorder (Smith et al, 2012; Almeida et al, 2016). A potential synergistic contribution of lysosomal dysfunction and hyperactivated microglia to the disease pathology and specifically to the deposition of TDP‐43 in neurons is likely but currently not understood (Kao et al, 2017; Ward et al, 2017; Huang et al, 2020; Logan et al, 2021).
To determine whether hyperactivation of microglia and its pathological consequences in Grn KO mice are dependent on aberrant TREM2 signaling, we sought to reduce the microglial activation status by crossing them to Trem2 KO mice. This reduced the expression of DAM genes, suggesting that negative modulation of TREM2 signaling may be exploited to lower microglial activation in neuroinflammatory disorders. In analogy to the agonistic 4D9 TREM2 antibody developed earlier in our laboratory (Schlepckow et al, 2020), we therefore generated monoclonal antibodies with opposite, namely antagonistic, properties. Such antibodies blocked lipid ligand‐induced TREM2 signaling, reduced signaling‐competent cell surface TREM2 in GRN‐FTLD patient‐derived macrophages, and concomitantly increased shedding of TREM2, which resulted in enhanced release of soluble TREM2 (sTREM2). Therefore, TREM2 antagonist antibodies inhibit receptor function via multiple mechanisms of action. In genetically engineered human‐induced pluripotent stem cell‐derived (iPSC) microglia‐lacking PGRN, the antagonistic antibodies reduced expression of the majority of candidate genes of the DAM signature, however, they failed to restore lysosomal function. Similarly, in Grn/Trem2 double‐knockout (Double KO) mice, lysosomal dysfunction was not rescued. Moreover, pathological features such as reduced 2‐deoxy‐2‐[18F]fluoro‐d‐glucose (FDG) uptake, disturbed lipid metabolism, and abnormal microglial morphology were not ameliorated. Strikingly, synapse loss was exacerbated, and neurofilament light chain (NfL), a sensitive fluid biomarker of neurodegeneration (Meeter et al, 2016; Rohrer et al, 2016; Preische et al, 2019), was also not reduced but unexpectedly increased in the cerebrospinal fluid (CSF). These findings therefore suggest that against common expectations, hyperactivated microglia may retain at least some TREM2‐dependent neuroprotective activities.
Results
Trem2 KO dampens hyperactivation of microglia in PGRN‐deficient mice
PGRN and TREM2 deficiency results in opposite microglial activation states (Gotzl et al, 2019). To determine if reduction in TREM2 signaling can ameliorate hyperactivation of PGRN‐deficient microglia, we crossed Grn KO mice (Kayasuga et al, 2007) to Trem2 KO mice (Turnbull et al, 2006) and performed TSPO‐PET imaging using established protocols (Liu et al, 2015; Kleinberger et al, 2017). TSPO‐PET imaging in mice is rather specific for microglial activation, as plexin treatment and ablation of TREM2, both of which reduce microglia numbers, strongly reduce the TSPO signal (Xiang et al, 2021). In line with our earlier findings (Gotzl et al, 2019), we confirmed a strong increase in the TSPO‐PET signal in the brains of Grn KO mice when compared to WT (P < 0.01) (Figs 1A and B, and EV5A). We also confirmed reduced TSPO expression in the brain of Trem2 KO mice (P < 0.03) (Figs 1A and B, and EV5A), consistent with our initial findings in TREM2 loss‐of‐function models (Kleinberger et al, 2017; Gotzl et al, 2019). Consistent with the above‐described goal to dampen hyperactivation of microglia, investigation of Double KO mice (Figs 1A and B, and EV5A) indicated a balanced TSPO expression without a significant difference when compared to WT (P = 0.945) and a reduction in TSPO expression relative to Grn KO mice (P < 0.05) (Figs 1A and B, and EV5A).
Figure 1. TSPO‐PET imaging indicates rescue of microglial hyperactivation in Double −/− mice.
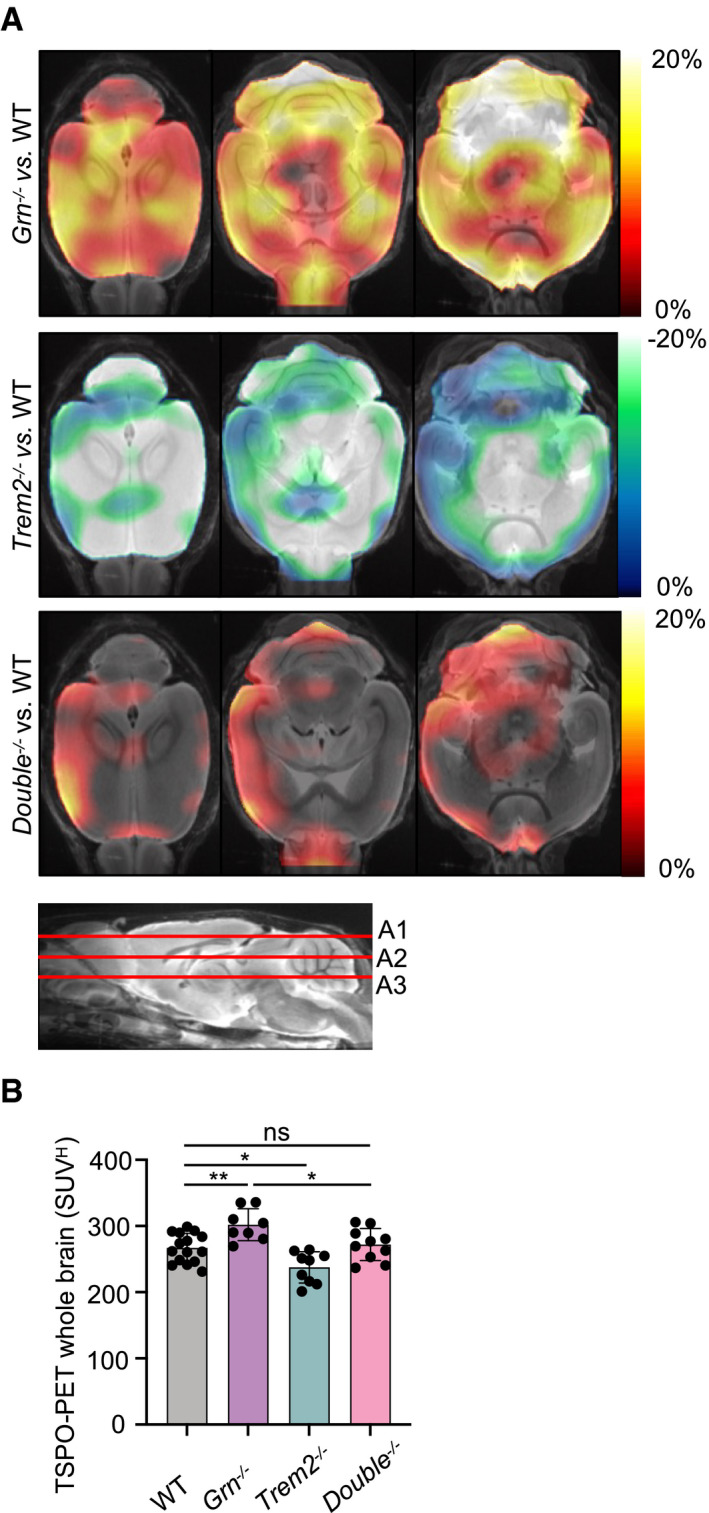
- Axial slices as indicated below (A1, A2, and A3) show %‐TSPO‐PET differences between Grn −/−, Trem2 −/−, or Double −/− mice and WT at the group level. Images adjusted to an MRI template indicate increased microglial activity in the brain of Grn −/− mice (hot color scale), compensated microglial activity in the brain of Double −/− mice, and decreased microglial activity in the brain of Trem2 −/− mice (cold color scale), each in contrast against age‐matched WT mice.
- Scatter plot illustrates individual mouse TSPO‐PET values derived from a whole‐brain volume of interest. A total of 8–15 female mice per group at an average age of 11.1 ± 1.6 months (Grn −/− (n = 8), Trem2 −/− (n = 9), Double −/− (n = 10), and WT (n = 15)). Data represent mean ± SD. For statistical analysis, one‐way ANOVA with Tukey post hoc test was used. Statistical significance was set at *P < 0.05; **P < 0.01; ns, not significant.
Source data are available online for this figure.
Figure EV5. Group‐level PET images without MRI template overlay.
- TSPO‐PET images depict increased microglial activity in Grn −/− mice, reduced microglial activity in Trem2 −/− mice, and similar levels of microglial activity in Double −/− and WT mice.
- FDG‐PET images indicate similar reduction in glucose uptake in Grn −/−, Trem2 −/−, and Double −/− in comparison to WT mice.
These findings suggest that DAM gene expression patterns as observed in Grn KO mice may be partially rescued in Double KO mice. To test this, we isolated microglia from adult mouse brains. Microglial mRNA of all three mouse lines was analyzed using a customized nCounter panel (NanoString Technologies), which includes 65 genes that previously showed opposite expression levels in Grn and Trem2 KO mice (Mazaheri et al, 2017; Gotzl et al, 2019). Gene expression levels were normalized against the geometric mean of four housekeeping genes, including Asb10, Cltc, Hprt1, and Tubb5. In accordance with our previous findings (Gotzl et al, 2019), candidate genes of the DAM signature such as ApoE, Cd22, Ly9, Clec7a, Spp1, and Olfr110 were massively upregulated in Grn KO microglia while these genes were suppressed in the Trem2 KO microglia (Fig 2A–E). In the Double KO microglia, expression of the DAM signature genes Olfr110, Spp1, and Clec7a is fully rescued to WT expression levels and others, such as Ly9, Cd22, and ApoE, are at least partially reduced compared to the Grn KO (Fig 2D and E). These data thus provide direct evidence that in Double KO mice, the molecular signature of microglia is shifted away from a DAM state toward a homeostatic state.
Figure 2. Loss of TREM2 reduces the DAM signature of Grn −/− mice.
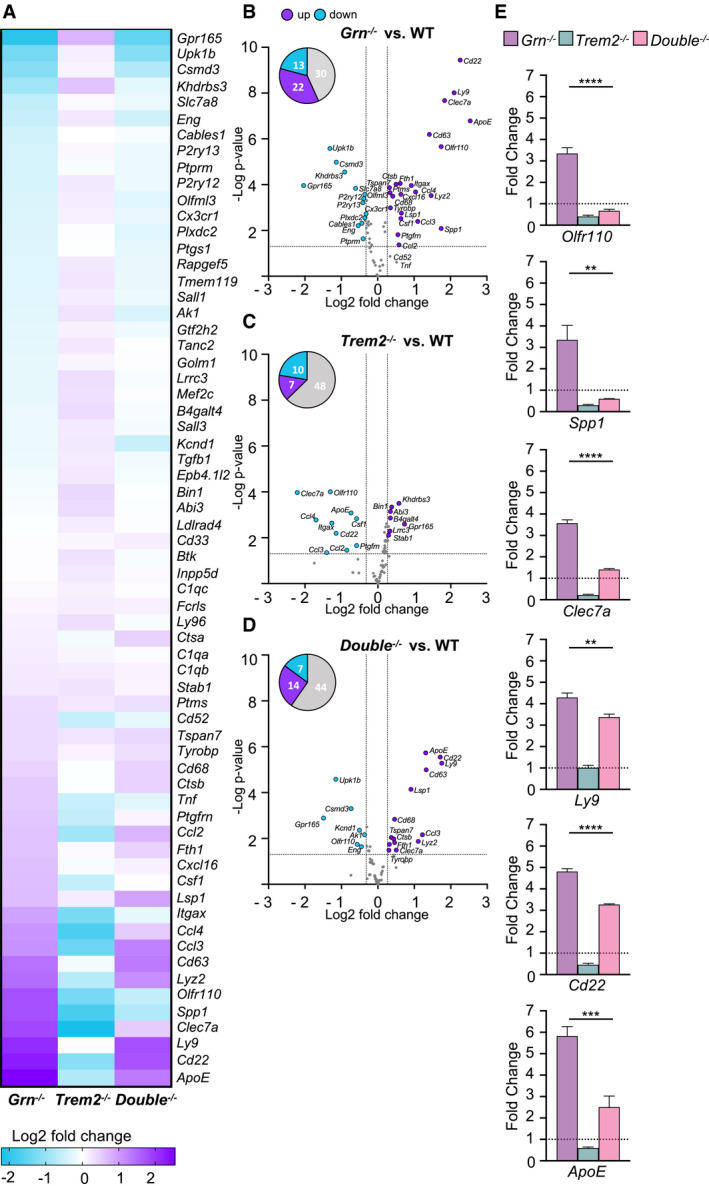
- Heatmap of 65 DAM‐associated gene transcripts analyzed by NanoString in FCRLS‐ and CD11b‐positive Grn −/− (n = 6), Trem2 −/− (n = 5), and Double −/− (n = 6) microglia in comparison to WT (n = 7) microglia isolated from 6‐month‐old male mice. The expression‐corrected and housekeeping gene normalized RNA counts for each gene and sample were normalized to the mean value of WT followed by a log2 transformation.
- Volcano plot presentation of the differently expressed transcripts in FCRLS‐ and CD11b‐positive Grn −/− (n = 6) in comparison to WT (n = 6) microglia isolated from 6‐month‐old male mice. A total of 35 of 65 analyzed genes are significantly changed more than 20%, with 22 genes upregulated (purple) and 13 genes downregulated (blue).
- Volcano plot presentation of the differently expressed transcripts in FCRLS‐ and CD11b‐positive Trem2 −/− (n = 5) in comparison to WT (n = 6) microglia isolated from 6‐month‐old male mice. A total of 17 of 65 analyzed genes are significantly changed more than 20%, with 7 genes upregulated (purple) and 10 genes downregulated (blue).
- Volcano plot presentation of the differently expressed transcripts in FCRLS‐ and CD11b‐positive Double −/− (n = 6) in comparison to WT (n = 6) microglia isolated from 6‐month‐old male mice. A total of 21 of 65 analyzed genes are significantly changed more than 20%, with 14 genes upregulated (purple) and 7 genes downregulated (blue).
- Expression profiles of selected DAM genes, whose mRNA levels are rescued in Double −/− (n = 6) versus Grn −/− (n = 6) microglia. mRNA expression normalized to the mean of the WT cohort. Data represent mean ± SEM.
Data information: For statistical analysis in B–D, the unpaired, two‐tailed student’s t‐test was performed, and in E, one‐way ANOVA with Dunnett’s post hoc test was used to compare Grn −/−, Trem2 −/−, and Double −/− microglia. Statistical significance was set at **P < 0.01; ***P < 0.001; ****P < 0.0001.
Source data are available online for this figure.
Antagonistic TREM2 antibodies decrease cell surface TREM2 and reduce ligand‐induced Syk signaling in monocyte‐derived patient macrophages
Antagonist TREM2 antibodies were generated by immunizing rodents with human TREM2 extracellular domain (ECD)‐Fc fusion protein and performing single B‐cell sequencing on peripheral lymphoid tissues. Antibodies that bound specifically to human TREM2 were evaluated for functional impact to TREM2 signaling. Antagonistic antibodies were identified by their ability to block TREM2‐dependent lipid ligand‐induced activation of p‐Syk on HEK293 cells overexpressing TREM2/DAP12 (Fig EV1). Cells were dosed with three different concentrations of liposomes, and antagonistic antibody 1 (Ab1) and antagonistic antibody 2 (Ab2), which were found to block phosphatidylserine (PS)‐induced p‐Syk activity (Fig EV1A). Both antibodies bind to an epitope in the IgV domain between amino acids 30 and 63 of human TREM2 (Figs 3A and EV1B). These selected antibodies were reformatted onto an effectorless human hIgG1‐LALAPG backbone, and demonstrated high affinity for cell surface TREM2 (0.38 nM EC50 Ab1 and 0.18 nM EC50 Ab2 in cell binding) and high affinity to human TREM2 ECD protein binding via surface plasmon resonance (0.21 nM Ab1 and 4.5 nM Ab2) (Fig EV1C–G). Ligand blocking activity was further validated in human monocyte‐derived macrophages, which were treated in a dose–response format with antibodies in the presence of PS‐containing liposomes to determine the potency of Ab1 and Ab2 to block liposome‐induced TREM2‐mediated p‐Syk signaling (Figs 3B and EV1C).
Figure EV1. Generation and characterization of TREM2 antagonistic antibodies.

-
AAlphaLISA quantification of p‐Syk levels in HEK293 cells overexpressing TREM2‐DAP12 upon treatment with Ab1 or Ab2 with three doses of liposomes (EC10: 0.0189527 mg/ml, EC25: 0.0633607 mg/ml, or EC50: 0.211731 mg/ml). PBS was used as a negative control. Individual data points are shown (n = 2 independent experiments).
-
BBiochemical binding data of TREM2 IgV peptides to antagonistic antibodies Ab1 and Ab2, as well as isotype control. Positive data represent binding level above a threshold of 2*LLOD (lower limit of detection), from an average of three independent experiments.
-
CTable of Ab1 and Ab2 with Biacore binding affinities, cell binding affinities, and liposome signaling (p‐Syk) inhibition (EC50 and maximal inhibition) in human macrophage (n = 3 independent experiments).
-
D, ECell‐binding dose–response curves generated by FACS in HEK293 cells overexpressing TREM2/DAP12 versus parental lines shown as mean fluorescence intensity (MFI).
-
F, GBiacore binding measurements of immobilized antagonist antibody to three concentrations of recombinant hTREM2‐ECD.
Figure 3. Human antagonistic TREM2 antibodies rescue elevated membrane‐bound TREM2 levels and reduce p‐Syk in primary human macrophages isolated from PGRN mutation carriers.

- Schematic presentation of human TREM2 with the identified binding site of antagonistic antibodies Ab1 and Ab2 (purple) in the Ig‐like V‐type domain. Light purple indicates the overlapping amino acid sequence of the two peptides, which are bound by Ab1 and Ab2 (see also EV1B). The disease‐associated Y38C and R47H mutations are indicated in yellow. Created with BioRender.com.
- AlphaLISA‐mediated quantification of p‐Syk in human macrophages with a dose titration treatment of Ab1 and Ab2 with or without liposomes for 5 min. IC50 and maximal inhibition (max) are indicated by a dotted line. Data represent the mean ± SEM (n = 3 independent experiments).
- ELISA‐mediated quantification confirms reduced PGRN serum levels in GRN mutation carriers versus healthy controls. PGRN was measured by ELISA in technical triplicates and normalized to serum levels of healthy controls. Data points indicate individual patients (GRN‐FTLD) and healthy controls (HC).
- Western blot of TREM2 in lysates and conditioned media of cultured human macrophages isolated from GRN‐FTLD patients and HC. Mature (mTREM2), immature (imTREM2), and soluble TREM2 (sTREM2) are indicated. Calnexin was used as loading control.
- Quantification of mTREM2 expression levels in lysates of cultured human macrophages isolated from GRN‐FTLD patients (data shown in D). mTREM2 levels were normalized to HC (n = 4). Data points indicate individual patients.
- ELISA‐mediated quantification of sTREM2 in conditioned media of human macrophages isolated from GRN‐FTLD patients and HC (n = 3). sTREM2 could not be measured in conditioned media of human macrophages isolated from patient #3 due to low overall cell yield. Data points indicate individual patients and HC.
- Western blot of TREM2 in lysates and media of cultured human macrophages isolated from GRN‐FTLD #1 and HC #1 upon treatment with Ab1 and Ab2. An isotype antibody was used as a negative control. ADAM protease inhibition (GM) does not further increase mTREM2 levels in GRN‐FTLD patients. Equal amounts of protein were loaded. GAPDH was used as loading control.
- Quantification of mTREM2 expression normalized to HC (n = 4) (data shown in G). Data points indicate individual patients.
- ELISA‐mediated quantification of sTREM2 in conditioned media of human macrophages isolated from GRN‐FTLD patients (n = 3). sTREM2 could not be measured in conditioned media of human macrophages isolated from patient #3 due to low overall cell yield. Data points indicate individual patients.
- AlphaLISA‐mediated quantification of p‐Syk levels in human macrophages upon treatment with Ab1 and Ab2 with liposomes for 60 min (n = 3). An isotype antibody was used as a negative control. Data points indicate individual patients. Isolated material from patient #3 did not yield enough cells to perform this experiment.
Data information: Data represent mean ± SEM. For statistical analysis of patient samples in comparison to HC, the unpaired, two‐tailed student’s t‐test was performed. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant
Source data are available online for this figure.
Next, we tested if antagonistic TREM2 antibodies are capable of reducing TREM2 signaling in GRN‐FTLD patient‐derived macrophages. To do so, we identified four patients with low PGRN plasma levels (Fig 3C) and confirmed heterozygous GRN loss‐of‐function mutation (Fig EV2A and B). We then generated monocyte‐derived macrophages from peripheral blood samples of these patients and healthy volunteers. Western blot analysis revealed that macrophages of GRN mutation carriers show significantly enhanced levels of mature TREM2 as compared to healthy controls (Fig 3D and E). Although GRN mutation carriers express more mature TREM2 than healthy controls, sTREM2 in the conditioned media was not significantly altered (Fig 3D and F). Since evidence exists that shedding of TREM2 terminates cell autonomous signaling in myeloid cells (Kleinberger et al, 2014; Schlepckow et al, 2017, 2020; Thornton et al, 2017), these findings suggest that macrophages from GRN‐FTLD patients exhibit increased TREM2 signaling, which occurs in conjunction with the microglial hyperactivation phenotype observed in vitro and in vivo (Gotzl et al, 2019).
Figure EV2. GRN‐FTLD patients.
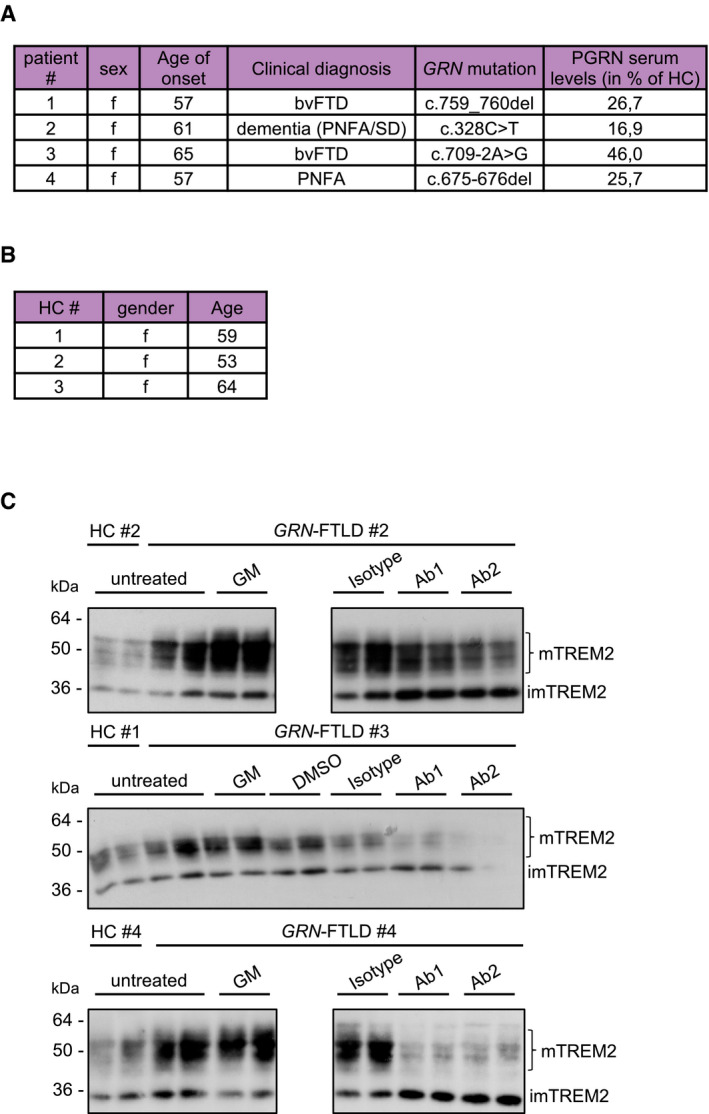
- Clinical data and mutation status of the four identified GRN mutation carriers and healthy controls. Mutation status of patients #1–3 was already confirmed before. The mutation status of patient #4 was detected by sequencing of exons including flanking sequences.
- Clinical data of healthy controls (HC).
- Western blot analysis of TREM2 using lysates of cultured human macrophages isolated from GRN mutation carriers (patients #2 to #4) (A) and healthy control upon treatment with Ab1 and Ab2. An isotype antibody was used as a negative control. ADAM protease inhibition (GM) does not further increase mTREM2 levels in PGRN mutation carriers. Equal amounts of protein were loaded.
Macrophages from GRN‐FTLD patients and healthy controls were then treated with Ab1 and Ab2 for 24 h. TREM2 levels in cell lysates revealed that both antibodies reduced mature TREM2, whereas an isotype control antibody had no effect (Figs 3G and H, and EV2C). The reduction in mature membrane‐bound TREM2 was accompanied by an increase in sTREM2 in conditioned media (Fig 3G and I). Thus, in line with the data shown in Figs 3A and B, and EV1A–G, both antibodies reduce signaling‐competent mature TREM2 and increase TREM2 shedding. To further demonstrate that TREM2 signaling can be modulated by TREM2 antagonistic antibodies in patient‐derived macrophages, we quantified Syk signaling. This demonstrated that both antagonistic antibodies reduce p‐Syk in liposome‐stimulated macrophages, suggesting that antagonistic TREM2 antibodies may be capable of modulating TREM2 hypersignaling in microglia in a beneficial manner (Fig 3J).
Antagonistic TREM2 antibodies reduce hyperactivation of PGRN‐deficient human microglia
To corroborate and extend our findings in human myeloid cells, we aimed to test modulation of TREM2 via the antagonistic antibodies in human‐induced pluripotent stem cell (iPSC)‐derived microglia (hiMGL). For this purpose, we generated GRN KO iPSC by targeting exon 2 using our established CRISPR genome‐editing pipeline (Weisheit et al, 2020; see methods for details). We deeply phenotyped GRN KO iPSC to confirm loss of PGRN protein expression, maintenance of pluripotency, clonality, as well as absence of unintended on‐ and off‐target effects and chromosomal abnormalities (Weisheit et al, 2021) (Figs 4A and EV3A–G; Appendix Fig S1). As expected, GRN KO hiMGL increased expression of TREM2 (Fig 4B and C) and showed consequently elevated levels of sTREM2 (Fig 4D). PGRN‐deficient hiMGL were treated with the antagonistic TREM2 antibodies as described above. Consistent with the antibody mechanism of action, antagonistic antibodies increased secretion of sTREM2 (Fig 4E). In line with this finding, both antagonistic antibodies reduced p‐Syk signaling (Fig 4F). Moreover, both antibodies not only reduced the phagocytic activity of WT hiMGL (Fig 4G), but also ameliorated the pathologically increased phagocytic activity of PGRN‐deficient hiMGL (Fig 4H), indicating that they dampen the activation state of PGRN‐deficient hiMGL. To further extend these findings, we asked if the antagonistic antibodies could also correct the transcriptional signature of hyperactivated hiMGL. Therefore, we used a customized nCounter panel (NanoString Technologies) analyzing gene expression of 82 microglia‐related genes and 8 housekeeping genes of WT and PGRN‐deficient hiMGL treated with the two antagonistic antibodies or isotype control (Fig 5A). Gene expression levels in each sample were normalized against the geometric mean of five housekeeping genes including CLTC, HPRT1, RPL13A, TBP and PPIA. DAM genes, such as APOE, SPP1, GPNMB, CSF1, LGALS3, CCL3, LPL, TREM2, ITGAX and CD68, were all significantly upregulated in PGRN‐deficient hiMGL compared to WT hiMGL (Fig 5A and B). In addition, expression of genes associated with lysosomal dysfunction was also significantly upregulated (CTSD, NPC2, and CD68). Both antagonistic TREM2 antibodies significantly modulated the mRNA signature of PGRN‐deficient hiMGL toward a more homeostatic state (Fig 5A–D). Upregulation of TREM2 in PGRN‐deficient hiMGL was completely corrected by treating the cells with either antagonist antibody (Fig 5E). Upregulation of DAM genes was completely (GPNMB, LGALS3, SPP1, CSF1, CCL3, LPL, and ITGAX) or at least partially (APOE and CCL2) rescued, while downregulation of the homeostatic marker P2RY12 was reversed by antibody treatment (Fig 5E). Thus, TREM2 modulation with antagonistic antibodies ameliorates hyperactivation of microglia.
Figure 4. Antagonistic TREM2 antibodies enhance TREM2 shedding and reduce TREM2‐dependent signaling and phagocytosis in PGRN‐deficient iPSC‐derived human microglia (hiMGL).

- Western blot of PGRN in whole‐cell lysates of WT and GRN −/− hiMGL. Actin was used as loading control.
- Western blot of TREM2 in whole‐cell lysates of WT and GRN −/− hiMGL. Mature (mTREM) and immature TREM2 (imTREM2) are indicated. Actin was used as loading control.
- Quantification of mTREM2 expression in whole‐cell lysates of WT and GRN −/− hiMGL (data shown in B). mTREM2 levels were normalized to WT (n = 3, biological replicates).
- ELISA‐mediated quantification of sTREM2 in conditioned media of WT and GRN −/− hiMGL (n = 3, biological replicates).
- ELISA‐mediated quantification of sTREM2 in conditioned media of GRN −/− hiMGL upon treatment with Ab1 and Ab2 (20 μg/ml, 40 μg/ml) (n = 3, biological replicates). An isotype antibody (10 μg/ml) was used as a negative control.
- AlphaLISA‐mediated quantification of p‐Syk levels in GRN −/− hiMGL upon treatment with Ab1 and Ab2 (5 μg/ml, 10 μg/ml) with liposomes (1 mg/ml) for 5 min. (n = 8, biological replicates).
- Uptake assay for fluorescent myelin in WT hiMGL. Phagocytosis of myelin significantly decreased upon treatment with TREM2 antagonistic antibodies Ab1 and Ab2 (n = 4, biological replicates).
- Uptake assay for fluorescent myelin. GRN −/− hiMGL phagocytose significantly more myelin as compare to WT hiMGL. This is reversed upon treatment with TREM2 antagonistic antibodies Ab1 and Ab2 (n = 4, biological replicates).
Data information: Data represent mean ± SEM. For statistical analysis in C and D, the unpaired, two‐tailed student’s t‐test was performed, in E and F, one‐way ANOVA with Dunnett’s post hoc test, and in G and H, one‐way ANOVA with Tukey’s post hoc was used to compare untreated, Ab1, and Ab2 (20 µg/ml and 40µg/ml) conditions to the isotype‐treated condition. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; and ns, not significant.
Source data are available online for this figure.
Figure EV3. Generation and characterization of GRN −/− iPSC line.
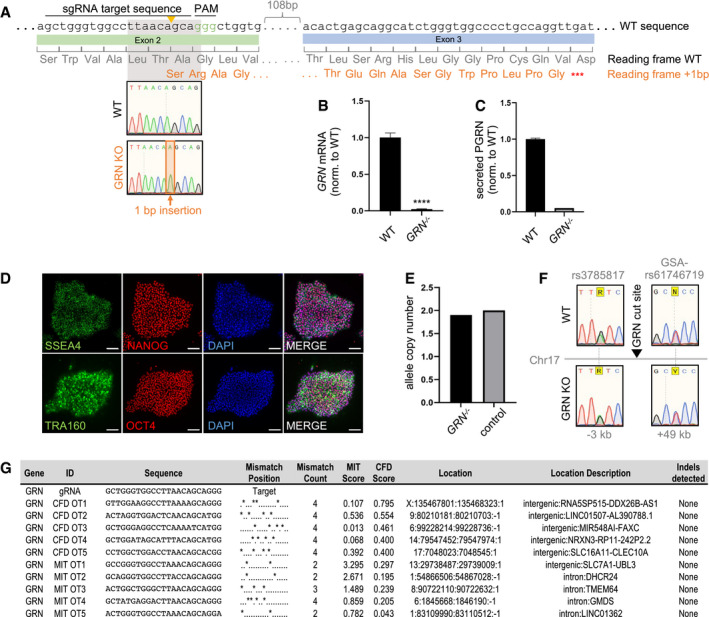
-
AGRN knockout generation strategy: GRN was targeted in exon 2 by a sgRNA (target and PAM sequence indicated), leading to a one base pair insertion in the GRN −/− line. The resulting frameshift exposes a nearby stop codon.
-
BGRN mRNA transcript levels in WT and GRN −/− hiMGL normalized to WT, as measured by qPCR (n = 3, biological replicates).
-
CELISA‐mediated quantification of secreted PGRN in conditioned media of WT and GRN −/− hiMGL (n = 3, biological replicates).
-
DImmunofluorescence analysis of pluripotency markers SSEA4, NANOG, TRA160, and OCT 4 with DAPI in GRN −/− iPSCs. Scalebars = 100 μm.
-
E, FAnalysis of CRISPR‐mediated on‐target effects by qgPCR quantitation of allele copy number (E) and Sanger sequencing of SNPs near the edited locus in WT and GRN −/− iPSC lines (F) shows maintenance of both alleles after editing.
-
GList of top five most similar off‐target sites ranked by the CFD and MIT prediction scores, respectively. No off‐target editing was detected by Sanger sequencing.
Data information: Data represent the mean ± SD. For statistical analysis, the unpaired, two‐tailed student’s t‐test was performed. Statistical significance was set at ****P < 0.0001, and ns, not significant.
Figure 5. Antagonistic TREM2 antibodies reduce hyperactivation of PGRN‐deficient hiMGL.

- Expression of all analyzed gene transcripts in GRN −/− hiMGL treated with control isotype antibody, Ab1, or Ab2 in comparison to WT hiMGL. Data show the mean of four individual treatments and NanoString measurements. The mRNA counts for each gene were normalized to the mean value of all WT samples followed by a log2 transformation.
- Volcano plot presentation of the differently expressed transcripts in GRN −/− hiMGL treated with isotype compared to WT hiMGL. Genes with more than 20% significantly changed expression are marked in purple (upregulated) or blue (downregulated).
- Volcano plot presentation of the differently expressed transcripts in GRN −/− hiMGL treated with Ab1 comparison to WT hiMGL. Genes with more than 20% significantly changed expression are marked in purple (upregulated) or blue (downregulated).
- Volcano plot presentation of the differently expressed transcripts in GRN −/− hiMGL treated with Ab2 comparison to WT hiMGL. Genes with more than 20% significantly changed expression are marked in purple (upregulated) or blue (downregulated).
- Transcript levels of DAM gene transcripts significantly altered in GRN −/− hiMGL, treated with Ab1 or Ab2 in comparison to isotype treatment from the data set in A, and normalized to the mean of the WT hiMGL samples (n = 4, biological replicates).
- Transcript levels of CTSD, NPC2, and CD68 of WT and GRN −/− hiMGL untreated, treated with isotype control, and Ab1 or Ab2 in comparison to WT hiMGL from the data set in A normalized to the mean of the WT hiMGL samples (n = 4, biological replicates).
- Catalytic activity of cathepsin D (CatD) in untreated WT and GRN −/− hiMGL or GRN −/− hiMGL treated with isotype control, Ab1 or Ab2 (20 μg/ml, 40 μg/ml), as measured by a CatD in vitro activity assay (n = 3, biological replicates).
Data information: Data represent mean ± SEM. For statistical analysis in B–D, the unpaired, two‐tailed student’s t‐test was performed, in E, one‐way ANOVA with Dunnett’s post hoc test was used to compare Ab1 and Ab2 (20 µg/ml and 40 µg/ml) conditions to the isotype‐treated condition, and in F and G, one‐way ANOVA with Dunnett`s post hoc test was used to compare Ab1‐, Ab2‐ (20 µg/ml and 40 µg/ml), and isotype‐treated condition to WT cells. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, *****P < 0.00001, and ns, not significant.
Source data are available online for this figure.
Reduced TREM2 signaling does not rescue lysosomal dysfunction
Next, we searched for a rescue of lysosomal phenotypes in PGRN‐deficient hiMGL. In contrast to the profound rescue of the homeostatic and disease‐associated mRNA signatures upon treatment with the two antagonistic antibodies (Fig 5A–E), we did not observe a significant rescue of increased gene expression patterns associated with lysosomal dysfunction upon PGRN deficiency, like CTSD, NPC2, and CD68 mRNA expression (Fig 5A–D and F). Antagonistic antibodies also failed to rescue elevated cathepsin D (CatD) activity in PGRN‐deficient hiMGL (Fig 5G).
In total brain lysates of 14‐month‐old Grn KO and Double KO mice, CatD single chain (sc) and heavy chain (hc) were both increased without a reduction in Double KO mice (Fig 6A–C). Furthermore, the catalytic activity of CatD, which was increased in Grn KO mice in an age‐dependent manner (Fig 6D and E), was also not rescued by the additional loss of TREM2 (Fig 6E), suggesting that lysosomal dysfunction of Grn KO mice cannot be rescued by TREM2 modulation. To further support this, we investigated Double KO brains for the accumulation of lipofuscin, an autofluorescent lipopigment, found in Grn KO and several lysosomal storage disorders (Gotzl et al, 2014). In line with the failure of the Double KO to rescue lysosomal hyperactivity, lipofuscin accumulation was not reduced upon loss of TREM2 in Grn KO mice, although remarkably almost no lipofuscin was observed in single Trem2 KO mice via mechanisms that have yet to be investigated (Fig 6F and G).
Figure 6. Abolishing TREM2 signaling does not rescue lysosomal dysfunction in Grn −/− mice.

-
AWestern blot of CatD in total brain lysates from 14‐month‐old female WT, Grn −/−, Trem2 −/−, and Double −/− mice. CatD maturation variants are indicated (sc: single chain; hc: heavy chain; n = 3).
-
B, CQuantification of CatD variants in A normalized to WT (n = 3 per genotype).
-
D, ECatalytic activity of CatD in brain lysates from female 6‐month‐old (n = 4 per genotype) (D) or 14‐month‐old (n = 3 per genotype) (E) Grn −/−, Trem2 −/−, and Double −/− mice normalized to WT.
-
FImmunohistochemical analysis of lipofuscin (green) in coronal brain sections. Representative images of thalamus are shown. Scalebars = 50 μm.
-
GQuantification of lipofuscin autofluorescence. Five images per mouse were taken, and means were normalized to WT samples (n = 3 per genotype, female).
Data information: Data represent mean ± SEM. For statistical analysis, one‐way ANOVA with Tukey’s post hoc test of Grn −/−, Trem2 −/−, and Double −/− was used. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, and ns, not significant.
Source data are available online for this figure.
Loss of TREM2 does not rescue lysosomal lipid dyshomeostasis in Grn KO mice
Previous studies have examined the impact of either Trem2 or Grn deletion on the lipidome of mouse brain. In the case of Trem2, no significant lipid changes were observed in Trem2 KO mouse brain at baseline, although upon cuprizone challenge, a striking accumulation of cholesterol esters and various sphingolipids was revealed (Nugent et al, 2020). In Grn KO mice, lipid metabolism is altered (Evers et al, 2017; Marschallinger et al, 2020) and a recent study described an age‐independent deficit in levels of the lysosomal lipid bis(monoacylglycerol)phosphate (BMP) that was accompanied by an age‐dependent accumulation of the GCase substrate glucosylsphingosine (GlcSph) (Logan et al, 2021). To determine whether deletion of Trem2 on the Grn KO background has any effect on the composition of the brain lipidome, we performed targeted lipidomic analysis using LCMS on 6‐month‐old WT, Grn KO, Trem2 KO, and Double KO mouse brain homogenates (Fig 7). As previously described (Nugent et al, 2020), the Trem2 KO showed no significant differences in brain lipid content relative to WT mice (Fig 7B), while the Grn KO as well as the Double KO showed a significant decrease in several BMP species as well as an increase in GlcSph (Fig 7A, C and E–G), which is consistent with previous data (Logan et al, 2021). Consistent with previous findings (Jian et al, 2016; Arrant et al, 2019; Zhou et al, 2019; Logan et al, 2021) and the increased accumulation of the GCase substrate GlcSph, we found a significant decrease in the GCase activity in Grn KO mice and Double KO (Fig 7H). Importantly, genetic interaction analysis demonstrated no statistically significant difference in the levels of any analyte in the Double KO brain compared to the Grn KO alone (Fig 7D). Thus, ablation of TREM2 fails to correct abnormal lysosomal function and lipid metabolism in PGRN‐deficient mice.
Figure 7. Reduced TREM2 signaling does not rescue dysregulated lipids in Grn −/− mice.
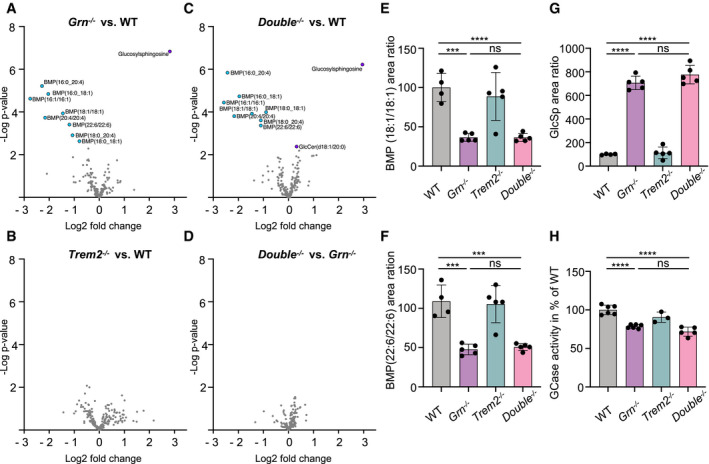
-
A–DVolcano plot presentation of lipids and metabolites upregulated (purple) or downregulated (blue) in total brain homogenates from 6‐month‐old male Grn −/− (A, n = 5), Trem2 −/− (B, n = 5), and Double −/− (C, n = 5) mice in comparison to WT (n = 4), and Double −/− in comparison to Grn −/− mice (D). Counts for each sample were normalized to the mean value of WT followed by a log2 transformation (n = 4–5 per genotype). Analyte values were adjusted with an FDR < 10% to exclude type I errors in null hypothesis testing.
-
E–GAbundance of BMP species and glucosylsphingosine (GlcSph) in total brain of 6‐month‐old Grn −/−, Trem2 −/−, Double −/−, and WT mice (n = 4–5 per genotype).
-
HGlucocerebrosidase (GCase) activity in whole‐brain lysates from 6‐month‐old male Grn −/−, Trem2 −/−, Double −/−, and WT mice. The linear increase in fluorescence signal was measured and then normalized to WT mice (n = 3–6 per genotype).
Data information: Data represent mean ± SEM. For statistical analysis, one‐way ANOVA with Tukey’s post hoc test of Grn −/−, Trem2 −/−, and Double −/− was used. Statistical significance was set at ***P < 0.001; ****P < 0.0001, and ns, not significant.
Source data are available online for this figure.
Enhanced brain pathology in Double KO mice suggests a neuroprotective function of hyperactivated microglia
Aged Grn KO mice show enhanced synaptic pruning (Lui et al, 2016; Zhang et al, 2020). To investigate if abolishing TREM2 signaling mitigates a neurodegenerative phenotype in 14‐month‐old Grn KO mice, we analyzed the thalamic synaptic density using synaptophysin (SPH) and the vesicular GABA transporter (VGAT) as a marker for presynaptic density (Fig 8A–C). Interestingly, immunohistochemical staining of the thalamic region revealed a reduction in SPH in Trem2 KO, which was even more robust in Double KO mice (Fig 8A and B). Western blot analysis confirmed reduced total SPH levels in Trem2 KO (Jadhav et al, 2020) and Double KO mice (Fig 8D and E). Similarly, immunostainings against VGAT revealed a further reduction in Double KO mice compared to WT, Grn KO, and Trem2 KO (Fig 8A and C).
Figure 8. Loss of TREM2 does not prevent synapse loss and activated microglia morphology.

- Immunohistochemical analysis of synaptophysin (SPH, pink) and VGAT (yellow) in coronal brain sections. Representative images of thalamus are shown. Scalebars = 50 μm.
- Quantification of SPH‐positive area. Three images per mouse were taken, and means were normalized to WT samples (n = 3 per genotype, female).
- Quantification of VGAT‐positive area. Three images per mouse were taken, and means were normalized to WT samples (n = 3 per genotype, female).
- Western blot of SPH in RIPA lysates from 14‐month‐old female WT, Grn −/−, Trem2 −/−, and Double −/− mice. Actin was used as loading control.
- Quantification of SPH protein levels in D normalized to WT (n = 3).
- Morphological analysis of cortical microglia. Representative maximum‐intensity projections of confocal z‐stack images showing IBA1+ microglial cells of female WT, Grn −/−, Trem2 −/−, and Double −/− mice (scalebar = 50 µm). Arrows point to individual microglia, which are shown as three‐dimensional reconstruction, scalebar = 10 µm.
- Morphological differences in cortical microglia from WT, Grn −/−, Trem2 −/−, and Double −/− mice shown by branch volume, sphericity score, branch length, and the number of branch nodes. Statistical analysis of group difference for the morphological scores “Branch volume” (auc = 0.72), “Sphericity score” (auc = 0.82), “Branch length” (auc = 0.69), and “Number of branch nodes” (auc = 0.80) was performed using the Wilcoxon rank‐sum test with continuity correction and Bonferroni post hoc correction for multiple testing in R (version 4.0.3). Two images per mouse (n = 3 per genotype, female) were analyzed, each data point represents one microglia cell. Median and interquartile range are displayed.
Data information: Data represent mean ± SEM. For statistical analysis in B‐C and E, one‐way ANOVA with Tukey’s post hoc test of Grn −/−, Trem2 −/−, and Double −/− was used. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, and ns, not significant.
Source data are available online for this figure.
To obtain additional information on the activation status of Double KO microglia, we determined microglial morphology. We extracted morphological features in WT, Grn KO, Trem2 KO, and Double KO animals after 3D reconstruction of IBA1+ microglia from confocal z‐stack images (Fig 8F and G). Microglial cells from Grn KO and Double KO animals showed a significantly decreased score for “branch volume,” “number of branch nodes,” and less pronounced for “branch length,” as well as a significantly increased score for “sphericity,” which is associated with an increased activation state of microglia (Heindl et al, 2018). In contrast, the morphological scores for Trem2 KO animals were comparable to WT. Thus, although the transcriptional signature of hyperactivated PGRN‐deficient microglia is partially rescued by the loss of TREM2, the morphological and immunohistochemical analysis indicates that Double KO microglia neither rescue Grn KO microglial morphology nor synapse loss.
To further test if reduction in microglial hyperactivation fails to ameliorate secondary neurodegeneration, we analyzed the concentrations of NfL, a fluid biomarker for neuronal damage (Meeter et al, 2016; Rohrer et al, 2016; Preische et al, 2019), in the CSF of 6‐month‐old (Fig 9A) and 14‐month‐old mice (Fig 9B). In line with previous findings (Zhang et al, 2020), NfL was increased in PGRN‐deficient mice, whereas no change was observed in Trem2 KO animals as compared to WT mice (Fig 9A and B). Surprisingly, we found a striking increase in NfL in the 14‐month‐old Double KO mice (Fig 9B), suggesting a protective role of TREM2‐dependent microglial hyperactivation in PGRN‐deficient mice. To further elucidate which genes and pathological pathways may be affected by eliminating TREM2 in Grn KO mice, we isolated mRNA from total brain of all three mouse models after 6 and 14 months of age and searched for changes in mRNA expression using the nCounter Neuropathology panel (NanoString Technologies) (Werner et al, 2020). The Neuropathology panel with 770 genes included was specifically designed to analyze neurodegenerative phenotypes in mouse models and allows investigating six fundamental themes of neurodegeneration, namely neurotransmission, neuron–glia interaction, neuroplasticity, cell structure integrity, neuroinflammation, and metabolism. Analysis of total brain mRNA confirmed rescue of the age‐dependent Grn KO‐associated DAM signature in the Double KO mice (Fig EV4A–G, Appendix Fig S2A and B) and revealed no significant upregulation of genes associated with neuroinflammation like Gfap, Tnf, or Tnfrsf11b or genes associated with synaptic pruning, such as the complement factors (C1qc, C1qa, and C1qb) compared to WT mice (Fig EV4G). Although differences in the transcriptomic signature of male and female microglia were overserved, as described earlier (Villa et al, 2018), rescue of gene expression associated with neuroinflammation and synaptic pruning was evident in both sexes (Fig EV4G, Appendix Fig S3). Pathway analysis in Grn KO mice revealed the highest increases in “autophagy,” “activated microglia,” “angiogenesis,” and “disease association” associated clusters with further increase during aging (Fig 9C). These four pathways score very low in Trem2 KO mice, again confirming opposite effects of the two single‐gene deletions. In the 6‐month‐old and in the 14‐month‐old cohort, all four pathways score is lower in the Double KO than in the single Grn KO. Three of these pathways, namely “activated microglia,” “angiogenesis,” and “disease association,” are even downregulated in the 6‐month‐old Double KO cohort compared to WT. However, other pathways like “neuronal cytoskeleton,” “tissue integrity,” and “transmitter synthesis and storage” and “transmitter response and uptake,” are most heavily affected in the 6‐month‐old Double KO, which is consistent with enhanced neuropathological phenotypes. When looking into individual genes, we found that strikingly, compared to single Grn KO and WT mice, only two genes were significantly altered and in fact downregulated more than 20% in the 6‐month‐old Double KO. These include the transcription factor Npas4 (Neuronal PAS domain protein 4), which regulates activation of genes involved in the excitatory–inhibitory balance and is known to exert neuroprotective activities (Spiegel et al, 2014; Fu et al, 2020) (Figs 9D and EV4C), and Grin3b (Perez‐Otano et al, 2016), a glutamate receptor subunit (Figs 9D and EV4C). In the 14‐month‐old cohort Npas4 expression is not significantly altered in Double KO mice compared to WT or Grn KO mice, although still reduced in single Grn KO mice (Fig 9D). In comparison to single Grn KO and WT mice, only four genes were significantly downregulated more than 20% in the Double KO mice. Interestingly, three of these, namely Ninj2, Ugt8a, and Plnxb3, are exclusively expressed in oligodendrocytes, suggesting major deficits in myelination in Double KO mice (Fig 9E) as recently reported for Trem2 KO (Wu et al, 2021).
Figure 9. Hyperactivation of microglia in Grn −/− mice is not deleterious.

- Immunoassay‐based quantification of neurofilament light‐chain (NfL) protein levels in CSF of 6‐month‐old Grn −/− (n = 4), Trem2 −/− (n = 4), Double −/− (n = 4), and WT (n = 5) male mice.
- Immunoassay‐based quantification of NfL levels in CSF of 14‐month‐old Grn −/−, Trem2 −/−, Double −/−, and WT female mice (n = 3 per genotype).
- Neuropathology NanoString panel analysis of total brain mRNA expression of 6‐month‐old and 14‐month‐old Grn −/−, Trem2 −/−, Double −/−, and WT male mice based on NanoString advanced analysis R‐script included in the panel (6‐month‐old mice: n = 4, 14‐month‐old mice: n = 3).
- Transcript levels of all significantly changed genes in Double −/− versus Grn −/− brain mRNA of 6‐month‐old and 14‐month‐old mice analyzed in C. Grin3b were under detection limit in the 14‐month‐old cohort. Transcript expression is normalized to the mean of the WT cohort.
- The same cohort of mice scanned for TSPO‐PET was additionally scanned for FDG‐PET. Axial slices as indicated show %‐FDG‐PET differences among Grn −/−, Trem2 −/−, and Double −/− (all cold color scales) when compared to WT at the group level. Images were adjusted to an MRI template.
- Bar graph illustrates individual FDG‐PET values derived from a whole‐brain volume of interest. Data represent mean ± SD. A total of 8–15 female mice per group at an average age of 10.7 ± 1.5 months (Grn −/− n = 8, Trem2 −/− n = 10, Double −/− n = 10, WT n = 15) were used.
Data information: Data in A, B, and D represent mean ± SEM. For statistical analysis in A–B, two‐way ANOVA with Dunnett’s post hoc test was used, in D, F, the unpaired, two‐tailed student's t‐test was performed. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, and ns, not significant.
Source data are available online for this figure.
Figure EV4. Differential gene expression in Grn −/− and Trem2 −/− is partially rescued in Double KO mice.

- Volcano plot presentation of the differently expressed transcripts in brains of 6‐month‐old male Grn −/− mice compared to WT brain mRNA (n = 4 per genotype). A total of 17 of 752 analyzed genes are significantly changed more than 20%, with 11 genes upregulated (purple) and 6 genes downregulated (blue).
- Volcano plot presentation of the differently expressed genes in brain mRNA of 6‐month‐old male Trem2 −/− mice in comparison to WT brain mRNA (n = 4 per genotype). A total of 16 of 752 analyzed genes are significantly changed by more than 20%, with 4 genes upregulated (purple) and 12 genes downregulated (blue).
- Volcano plot presentation of the differently expressed genes in brain mRNA of 6‐month‐old male Double −/− mice in comparison to WT brain mRNA (n = 4 per genotype). A total of 25 of 752 analyzed genes are significantly changed by more than 20%, with 8 genes upregulated (purple) and 17 genes downregulated (blue).
- Volcano plot presentation of the differently expressed transcripts in brains of 14‐month‐old male Grn −/− mice compared to WT brain mRNA (n = 3 per genotype). A total of 49 of 752 analyzed genes are significantly changed more than 20%, with 44 genes upregulated (purple) and 5 genes downregulated (blue).
- Volcano plot presentation of the differently expressed genes in brain mRNA of 14‐month‐old male Trem2 −/− mice in comparison to WT brain mRNA (n = 3 per genotype). A total of 7 of 752 analyzed genes are significantly changed by more than 20%, with 7 downregulated (blue).
- Volcano plot presentation of the differently expressed genes in brain mRNA of 14‐month‐old male Double −/− mice in comparison to WT brain mRNA (n = 3 per genotype). A total of 21 of 752 analyzed genes are significantly changed by more than 20%, with 8 genes upregulated (purple) and 13 genes downregulated (blue).
- Transcript levels of selected significantly rescued genes in Double −/− versus Grn −/− brain mRNA of 6‐month‐old (n = 4 per genotype) and 14‐month‐old mice (n = 3 per genotype). Transcript expression is normalized to the mean of the WT cohort.
Data information: Data represent mean ± SEM. For statistical analysis in G, the unpaired, two‐tailed student’s t‐test was performed to compare Grn −/−, Trem2 −/−, and Double −/− whole‐brain mRNA expression. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, and ns, not significant.
Although gene expression analysis of the Double KO suggests dampening of the pathological DAM signature, enhanced synaptic loss and increased NfL in CSF of Double KO indicate that neurodegeneration of PGRN‐deficient mice may not be improved by the additional knockout of TREM2 but instead increased even further. We therefore investigated if the additional loss of TREM2 in PGRN‐deficient mice affects deficits in glucose uptake in vivo. To determine brain cerebral uptake rates of glucose in Double KO in vivo, we performed 2‐[18F]fluoro‐d‐glucose PET (FDG‐PET). We confirmed a reduced cerebral glucose uptake in Grn KO (P < 0.05) and Trem2 KO (P < 0.0001) mice compared to WT (Figs 9E and F, and EV5B), as described in previous studies (Kleinberger et al, 2017; Gotzl et al, 2019). However, we still observed similar decreased glucose uptake in Double KO mice when compared to WT (P < 0.0001) (Figs 9E and F, and EV5B), revealing a consistently decreased glucose uptake between Grn KO, Trem2 KO, or Double KO mice and WT mice. Together, all our findings indicate that reducing hyperactivation of microglia does not ameliorate lysosomal dysfunction of PGRN‐deficient mice but may even promote neurodegeneration.
Discussion
PGRN and the proteolytically derived granulin peptides may have important lysosomal functions, as exemplified by the identification of homozygous loss‐of‐function GRN mutations, which are causative for NCL (Smith et al, 2012). Accumulating evidence suggests that PGRN /granulins directly or indirectly regulate the activity of lysosomal enzymes such as CatD (Beel et al, 2017; Valdez et al, 2017; Zhou et al, 2017; Butler et al, 2019a, 2019b; Huang et al, 2020), GCase (Jian et al, 2016; Arrant et al, 2019; Zhou et al, 2019), and HexA (Chen et al, 2018). The last two enzymes are involved in sphingolipid degradation, a process regulated by the lysosomal phospholipid BMP, which is stabilized by PGRN (Logan et al, 2021). PGRN may affect lysosome acidification and thereby lysosomal enzyme activity (Tanaka et al, 2017; Logan et al, 2021). We and others have shown that PGRN deficiency results in upregulation of several lysosomal enzymes (Gotzl et al, 2014, 2018; Klein et al, 2017; Huang et al, 2020; Root et al, 2021). However, it remained unclear if microglial hyperactivation observed in PGRN‐deficient microglia contributes to or is a consequence of lysosomal dysfunction. Activated microglia are found in late stages of many neurodegenerative diseases including AD and FTLD, and are believed to be deleterious by promoting synaptic pruning and neuronal cell death (Heneka et al, 2013; Hong et al, 2016a, 2016b). Specifically, FTLD patients suffering from GRN haploinsufficiency show pathological hyperactivation of microglia as measured by TSPO‐PET (Martens et al, 2012; Gotzl et al, 2019; Marschallinger et al, 2020; Zhang et al, 2020). Similarly, mice lacking PGRN exhibit hyperactivation of microglia as indicated by an enhanced DAM signature including TREM2 and galectin 3, an increased TSPO signal, and increased phagocytic and synaptic pruning activity (Lui et al, 2016; Gotzl et al, 2019; Zhang et al, 2020). We therefore asked if the pathological outcome of PGRN deficiency may be promoted by TREM2‐dependent microglial overaction. To address this question, we sought to reduce TREM2‐dependent signaling by two independent strategies: genetic loss‐of‐function and pharmacological inhibition with antagonist antibodies. To achieve the former, we crossed Trem2 KO mice to the Grn KO to generate a Double KO model. For the latter approach, we identified TREM2 antagonistic antibodies, which negatively regulate TREM2 by increasing surface receptor shedding and preventing lipid ligand‐induced signaling of the co‐receptor DAP12. Both approaches successfully dampened several aspects of TREM2‐dependent microglial activation. However, although reduction in TREM2 signaling by two independent approaches rescued microglial hyperactivation to some extent, this was not sufficient to ameliorate lysosomal deficits, dysregulation of lysosomal lipids, synapse loss/neurodegeneration, and reduced glucose uptake. These findings demonstrate in this model that microglial hyperactivation is secondary to the primary loss of lysosomal function caused by PGRN deficiency. Surprisingly, inhibition of TREM2 function results in elevated markers for neurodegeneration and synapse loss in Double KO animals. Our extensive gene expression analyses do not suggest that the total loss of TREM2 function in Grn KO mice causes additional neurotoxicity, for example, by supporting pro‐inflammatory microglial responses. Instead, the fact that the additional loss of TREM2 leads to increased brain pathology indicates that TREM2‐regulated microglial activation states may not necessarily be deleterious but protective. We suggest that hyperactivated microglia, for example, in Grn KO mice, resemble the previously described DAM2 microglia or may develop into them by even further increasing their DAM signature (Keren‐Shaul et al, 2017). Consistently, fully activated DAM2 microglia were recently described to be particularly protective in a mouse model for amyloidosis and tau pathology (Lee et al, 2021). This is very surprising since chronically activated microglia, as observed in PGRN loss‐of‐function models and mouse models for amyloid and tau pathology, would have been expected to exert significant damage within the brain, for example, by induction of the inflammasome (Heneka et al, 2018). However, our findings together with those by Lee et al (2021) rather suggest that TREM2‐dependent chronic activation is protective, which may have implications for therapeutic attempts employing modulation of TREM2 activity by agonistic antibodies (Deczkowska et al, 2020; Lewcock et al, 2020). In that regard, the nomenclature used for describing diverse microglial states, namely homeostatic, disease associated, and hyperactivated, may need to be reconsidered. We find these terms misleading, as they indicate that homeostatic microglia are beneficial, whereas disease‐associated or ‐hyperactivated microglia are deleterious. Importantly, the brain environment and pathological context is important for ascribing microglial state and associated functions and requires a deeper understanding beyond transcriptional characterization to elucidate the overall impact to brain function and disease. However, one may describe these fundamentally different states of microglia as “surveilling” versus “responding” microglia to capture activities that occur during normal versus pathological contexts. The term “responding” would implicate that these microglia exert protective effects.
Protective microglial functions are promoted by enhancing TREM2 signaling with agonist TREM2 antibodies (Lewcock et al, 2020). All currently described agonistic TREM2 antibodies act via similar mechanisms by inhibiting shedding and directly activating TREM2, and therefore increasing functional receptor on the cell surface (Lewcock et al, 2020). Notably, all known agonistic TREM2 antibodies bind to the stalk region close to the cleavage site by ADAM10/17 (Schlepckow et al, 2017; Lewcock et al, 2020). In contrast, the two antagonistic antibodies described here bind in the IgV‐fold between amino acids 30 and 63 (Fig EV1A). Interestingly, this domain of TREM2 harbors a number of AD‐ and FTLD‐associated sequence variants (Colonna & Wang, 2016). The R47H variant increases AD risk and affects TREM2‐dependent microglial proliferation, lipid metabolism, and microgliosis. Similarly, the FTLD‐associated Y38C variant causes a loss of function by misfolding and retention of TREM2 within the endoplasmic reticulum (Kleinberger et al, 2014). Antagonistic antibodies therefore appear to bind at a functionally critical region and may displace natural ligands (e.g., lipids), thus preventing induction of TREM2 signaling, in addition to promoting TREM2 shedding.
Finally, our findings also contribute to addressing the long‐standing question of whether microglia can dynamically reverse their activation state in both directions, from homeostatic to DAM and back again. Our findings strongly indicate that even hyperactivated microglia can switch back to homeostatic microglia. The fact that one can influence microglial activation in both directions with TREM2‐modulating antibodies is another example for the tremendous dynamics of microglia, and offers the opportunity for therapeutic fine tuning of microglial activity.
Taken together, eliminating TREM2 function and thus reducing hyperactivation by two independent approaches do not rescue lysosomal dysfunction caused by GRN deficiency, but rather exacerbates pathological endpoints characteristic for neurodegeneration, including elevation of CSF NfL and reduced transcription of the neuroprotective transcription factor Npas. Thus, despite common assumptions, these results suggest that hyperactivated microglia can retain TREM2‐dependent protective functions.
Materials and Methods
Animal experiments and mouse brain tissue
All animal experiments were performed in accordance with German animal welfare law and approved by the government of upper Bavaria. Mice were kept under standard housing conditions including standard pellet food and water provided ad libitum. Mice were sacrificed by CO2 inhalation or deep/lethal anesthesia followed by PBS perfusion. Brain tissue was obtained from male and female of the following mouse strains: C57BL/6J Grn (Kayasuga et al, 2007) and Trem2 knockout line (Turnbull et al, 2006). To minimize mouse numbers and to reduce variability due to sex differences, both male and female cohorts were used but separately analyzed. PET scans and CSF withdrawal were performed under the animal license: ROB 55.2‐2532. Vet_02‐18‐32.
Isolation, differentiation, and culture of human primary monocytes
Human primary monocytes were isolated from whole blood using Sepmate tubes (StemCell Technologies, #85450) in combination with RosetteSep Human Monocyte Enrichment Cocktail (StemCell Technologies, #15068) according to the manufacturer’s protocol. Briefly, fresh blood was collected into EDTA‐coated collection tubes and stored at room temperature (RT) until further processing for maximal 6 h. EDTA was added to a final concentration of 1 mM and tubes were mixed by inversion. Fifty µl/ml blood of RosetteSep cocktail was added and samples for incubated for 20 min at RT. Cells were separated on a density gradient using Ficoll‐Paque PLUS (ThermoFisher, #11768538). After centrifugation isolated cells were washed with PBS supplemented with 2% FCS. Leftover red blood cells were lysed using ACK lysing buffer (ThermoFisher, #11509876) for 2 min at RT. Subsequently, cells were washed two times with PBS supplemented with 2% FCS. Cells were counted using Trypan blue as a viability dye and 1 × 106 cells were plated in 10 cm dishes in 10‐ml RPMI medium supplemented with 10% FCS, 10% NEAA, 10% l‐glutamine, 10% sodium pyruvate, and M‐CSF with a final concentration of 50 ng/ml. Forty‐eight hours after isolation, 1‐ml medium with 500 ng/ml M‐CSF was added to the cells. Five days after isolation, cells were washed with PBS and scraped. Cells were counted as described above and plated in either 96‐well plates at a density of 5 × 104 cells/well in 100 μl or in 12‐well plates at a density of 3 × 105 cells/well in 600 μl RPMI medium supplemented with 10% FCS, 10% NEAA, 10% l‐glutamine, 10% sodium pyruvate, and M‐CSF with a final concentration of 50 ng/ml.
Generation and maintenance of GRN KO iPSC lines
iPSC experiments were performed in accordance with all relevant guidelines and regulations. Female iPSC line A18944 was purchased from ThermoFisher (#A18945). iPSCs were grown in Essential 8 Flex Medium (ThermoFisher, #A2858501) on VTN‐coated (ThermoFisher, #A14700) cell culture plates at 37°C with 5% CO2 and split twice a week as small clumps after a 5 min incubation in PBS/EDTA. Prior to electroporation, iPSCs were split to single cells after a 10 min incubation in PBS/EDTA to Geltrex‐coated (ThermoFisher, #A1413302) plates and cultured in StemFlex Medium (ThermoFisher, #A3349401) containing 10 mM ROCK inhibitor (Selleck‐chem S1049) for 2 days. iPSCs were transfected by electroporation as described earlier (Kwart et al, 2017) with modifications. Briefly, 2 million cells were harvested with Accutase (ThermoFisher, #A1110501), resuspended in 100 ml cold BTXpress electroporation solution (VWR International GmbH, #732‐1285) with 20 mg Cas9 (pSpCas9(BB)‐2A‐Puro (PX459) V2.0 (gift from Feng Zhang; Addgene plasmid #62988; http://n2t.net/addgene:62988; RRID: Addgene_62988 (Ran et al, 2013)) and 5 mg sgRNA cloned into the BsmBI restriction site of the MLM3636 plasmid (gift from Keith Joung, Addgene plasmid #43860; http://n2t.net/addgene:48360; RRID: Addgene_43860). Cells were electroporated with two pulses at 65 mV for 20 ms in a 1 mm cuvette (ThermoFisher, #15437270). After electroporation, cells were transferred to Geltrex‐coated 10 cm plates and grown in StemFlex Medium containing 10 mM ROCK inhibitor until visible colonies appeared. Cells expressing Cas9 were selected by 350 ng/ml Puromycin dihydrochloride (VWR International GmbH, #J593) for 3 consecutive days starting 1 day after electroporation (Steyer et al, 2018). Single‐cell clone colonies were then picked and analyzed by RFLP assay, using NEB enzyme MwoI for the GRN KO, and Sanger sequencing as previously described (Kwart et al, 2017).
CRISPR/Cas9 genome editing
Design and preparation of editing reagents and quality control of edited iPSCs was performed as described previously (Weisheit et al, 2020, 2021). Briefly, we used CRISPOR (http://crispor.tefor.net (Concordet & Haeussler, 2018)) to select guide RNAs and determine putative off‐target loci. We chose gRNAs targeting exon 2 of GRN, as it is present in most splice isoforms and a frameshift would affect large parts of the coding region. We also ensured presence of nearby stop codons in alternate reading frames in the sequence after the cut site. Successful knockout was confirmed on mRNA level by qPCR, and on protein level by Western blot using RIPA lysate and ELISA using conditioned media, respectively. For quality control of edited iPSC clones, we checked absence of off‐target effects by PCR amplifying and Sanger sequencing the top five hits based on MIT and CFD scores on CRISPOR. We also confirmed absence of on‐target effects such as large deletions and loss of heterozygosity using qgPCR and nearby SNP sequencing (Weisheit et al, 2021). Finally, we also ensured pluripotency by immunofluorescence staining for typical markers OCT4, NANOG, SSEA4, and TRA160, and chromosomal integrity by molecular karyotyping (LIFE & BRAIN GmbH). One clonal cell line passed the described quality controls and was further differentiated into hiMGL.
Differentiation of human iPSC‐derived Microglia (hiMGL)
We differentiated hiMGL from iPSCs as described (Abud et al, 2017) with modifications to improve efficiency and yield: When iPSCs were 70–90% confluent, they were split 1:100–200 onto GelTrex‐coated six‐well plates for the HPC differentiation using EDTA to get around ~20 small colonies per well. Cells were fed with 2 ml of HemA medium (HPC differentiation kit, StemCell Technologies) on day 0 and half‐fed with 1 ml on day 2. Media were switched to 2 ml of HemB on day 3 with half‐feeds on days 5 and 7 and 1 ml added on top on day 10. On day 12, HPCs were collected as non‐adherent cells to either freeze or continue with the microglia differentiation. HPCs were frozen at 1 million cells per ml in BamBanker (FUJIFILM Wako Chemicals). They were then thawed directly onto GelTrex‐coated six‐well plates with 1 million cells evenly distributed among six wells in 2‐ml iMGL media with 25 ng/ml M‐CSF, 100 ng/ml IL‐34, and 50 ng/ml TGF‐β added fresh. One milliliter of media was added every other day. During the microglia differentiation, the cells were split 1:2 every 6–8 days depending on confluency. We did not use CD200 and CX3CL1 for final differentiation, as this did not alter hiMGL gene expression in NanoString analysis. hiMGL were used for experiments on day 16 of the differentiation. A very similar differentiation protocol was published recently (McQuade et al, 2018).
Antagonist antibody generation and verification
Antibody generation was carried out by performing single B‐cell sequencing on lymphoid tissues from rodents immunized full‐length human TREM2 ectodomaine (ECD)‐Fc protein (AbCellera Inc.). Antibodies were screened based on binding to human TREM2, and clones of interest were reformatted onto human effectorless human IgG1‐LALAPG backbones for material generation and further evaluation of cell‐binding potency and functional impact to TREM2 signaling. Antagonists were identified by their ability to block lipid ligand‐induced activation of p‐Syk on HEK293 cells overexpressing TREM2‐DAP12.
Affinity determination and binding kinetics
Human TREM2‐binding affinities of anti‐TREM2 antibodies were determined by surface plasmon resonance using a Biacore 8K instrument. Biacore Series S CM5 sensor chip was immobilized with a mixture of two monoclonal mouse anti‐Fab antibodies (Human Fab capture kit from GE Healthcare) to capture antibodies for binding measurements. In order to measure human TREM2‐binding affinities of anti‐TREM2 antibodies, serial threefold dilutions of recombinant human TREM2‐ECD protein were injected at a flow rate of 30 μl/min for 300 s followed by 600 s dissociation in HBS‐EP+ running buffer (GE, #BR100669). A 1:1 Languir model of simultaneous fitting of kon and koff was used for antigen‐binding kinetics analysis.
Epitope mapping of antagonist TREM2 antibodies
Biotinylated polypeptides for human Trem2 IgV domain (Sequences in Fig EV1B) were purchased from Elim Biopharmaceuticals, Inc. N‐terminal cysteine was added to peptides to enable maleimide–thiol conjugation of biotin. The lyophilized biotinylated peptides were reconstituted in 20 mM Tris buffer at pH 8.0. Antibody binding to TREM2 IgV domain peptides was detected using a sandwich ELISA. Briefly, a 96‐well half‐area ELISA plate was coated with streptavidin overnight at 4°C. The following day, biotinylated TREM2 IgV peptides diluted to 1 µM in 1% BSA/PBS were added to the plate and incubated for 1 h. Antibodies diluted to 120 nM in 1% BSA/PBS were then added and incubated for 1 h. Antibodies bound to peptide were detected with anti‐Human IgG‐HRP secondary antibody diluted in 1% BSA/PBS. Plates were developed with the addition of TMB substrate and stopped by the addition of 2N sulfuric acid. Absorbance at 450 nm was measured on the Synergy Neo2 plate reader (Biotek). A positive signal was identified as an absorbance value above twofold of lower limit of detection (defined as average of blank + threefold SD of blank).
Detection of anti‐TREM2 antibody cell binding by flow cytometry
HEK293‐overexpressing human TREM2 (HEK293‐H6) and HEK293‐overexpressing GFP were harvested using 0.05% trypsin and incubated at 37°C for 2 h. All cells were centrifuged and washed in FACS buffer (PBS + 0.5% BSA) twice. Mixed cells were resuspended in FACS buffer at a density of 106 cells/ml per cell line. The mixed cell lines were seeded at 100,000 cells per well in a 96‐well V‐bottom plate and incubated for 20 min at RT. After incubation, the cells were centrifuged and incubated with anti‐TREM2 antibodies in a dose titration from 0 to 300 nM for 45 min on ice. After incubation, cells were centrifuged and washed with FACS buffer three times. The cells were then incubated with secondary antibody (Alexa Fluor 647 AffiniPure F(ab’)2 fragment goat anti‐human IgG (H + L), Jackson ImmunoResearch Laboratories, #109‐606‐088, 1:800 dilution), for 30 min on ice without exposure to light. After incubation, the cells were washed with FACS buffer three times, resuspended in 100 μl of FACS buffer, and analyzed by flow cytometry (BD FACSCanto II, San Jose, CA), for which 50,000 events were obtained for each sample. Mean fluorescence intensity (MFI) per cell was calculated by FLowJO software and used for generation of dose–response binding curve.
Antibody treatment
Eight hours after seeding the cells, they were treated with anti‐human TREM2 antibodies (Fig EV1C). Antibodies were diluted in RPMI medium and added to the cells with a final concentration of 20 µg/ml. As control for TREM2 shedding, cells were treated with GM6001 (25 µM, Enzo Life Sciences), or DMSO as a vehicle control. hiMGL were seeded in six‐well plates with 400,000 cells/well. Eight hours after seeding, antibodies were diluted in iMGL media and added at a concentration of either 20 or 40 µg/ml. Isotype or TREM2 antibodies were added at a concentration of 40 µg/ml and 24 h after antibody treatment, medium and cells were harvested as previously described.
Small animal PET/MRI
All rodent PET procedures followed an established standardized protocol for radiochemistry, acquisition times, and post‐processing (Brendel et al, 2016; Overhoff et al, 2016), which was transferred to a novel PET/MRI system.
All mice were scanned with a 3T Mediso nanoScan PET/MR scanner (Mediso Ltd) with a single‐mouse imaging chamber. A 15‐min anatomical T1 MR scan was performed at 15 min after [18F]‐FDG injection or at 45 min after [18F]‐GE180 injection (head receive coil, matrix size 96 × 96 × 22, voxel size 0.24 × 0.24 × 0.80 mm³, repetition time 677 ms, echo time 28.56 ms, and flip angle 90°). PET emission was recorded at 30–60 min p.i. ([18F]‐FDG) or at 60–90 min p.i. ([18F]‐GE‐180). PET list‐mode data within 400–600 keV energy window were reconstructed using a 3D iterative algorithm (Tera‐Tomo 3D, Mediso Ltd) with the following parameters: matrix size 55 × 62 × 187 mm³, voxel size 0.3 × 0.3 × 0.3 mm³, eight iterations, six subsets. Decay, random, and attenuation correction were applied. The T1 image was used to create a body–air material map for the attenuation correction. We studied PET images of Grn KO mice (n = 8), Trem2 KO mice (n = 10 or n = 9), Double KO mice (n = 10), and WT mice (n = 15), all female at an average age of 10.9 ± 1.6 months or 11.1 ± 1.6 months, as indicated in the figure legends. Normalization of injected activity was performed by the previously validated myocardium correction method (Deussing et al, 2018) for [18F]‐GE‐180 TSPO‐PET and by standardized uptake value (SUV) normalization for [18F]‐FDG‐PET. Groups of Grn KO, Trem2 KO, and Double KO mice were compared against WT mice by calculation of %‐differences in each cerebral voxel. Finally, [18F]‐TSPO‐PET and [18F]‐FDG‐PET values deriving from a whole‐brain VOI (Kleinberger et al, 2017) were extracted and compared between groups of different genotypes by a one‐way ANOVA including Tukey post hoc correction.
CSF collection
Mice were fully anesthetized via an intraperitoneal injection of medetomidine (0.5 mg/kg) + midazolam (5 mg/kg) + fentanyl (0.05 mg/kg). CSF was collected as described previously (Lim et al, 2018). Briefly, subcutaneous tissue and musculature were removed to expose the meninges overlying the cisterna magna. A glass capillary with a trimmed tip (inner diameter is approximately 0.75 mm) was used to puncture the membrane, and CSF was allowed to flow into the capillary for approximately 10 min. After collection, CSF was centrifuged at 1,000 g for 10 min, assessed macroscopically for blood contamination, aliquoted (5 μl) in propylene tubes, snap‐frozen in liquid nitrogen, and stored at −80°C until use.
CSF neurofilament light‐chain analysis
NfL levels were quantitatively determined in CSF samples using the Simoa NF‐light Advantage kit (Quanterix, #103186) following the manufacturer’s instructions. CSF samples were diluted 1:10 in sample dilution buffer and mixed with Simoa detector reagent and bead reagent, following an incubation at 30°C for 30 min, shaking at 800 rpm. Plates were washed with Simoa washing buffer A and SBG reagent from the kit was added. Following a 10 min incubation at 30°C and shaking at 800 rpm, plates were washed twice and sample beads were resuspended in Simoa wash buffer B. NfL concentrations were measured after a 10 min drying at RT using the Simoa DH‐1 analyzer (Quanterix).
Gene expression profiling of total brain
Adult mice were perfused transcardially with PBS and dissected brains were snap frozen in liquid nitrogen. Snap frozen brains were mechanically powdered in liquid nitrogen. Total RNA was isolated using the RNeasy Mini kit (Qiagen, #74104) and 60 ng of total RNA per sample was subjected to gene expression profiling using the nCounter® Neuropathology panel from NanoString (NanoString Technologies). Gene expression levels in each sample were normalized against the geometric mean of four housekeeping genes including Asb10, Cltc, Hprt1, and Tubb5 using the nSolver Analysis Software, version 4.0. Gusb was excluded because of significant changes in Grn KO and Double KO mice.
Gene expression profiling of primary microglia
CD11b+ and FCRL+ primary microglia were isolated from adult mouse brain. Mice were perfused transcardially and brains were collected into ice‐cold HBSS (ThermoFisher, #14175095). Brain tissue was mechanically dissociated into single‐cell suspension using Potter‐Elvehjem homogenizers with PTFE Pestle. Microglia cell pellets were resuspended in 70% Percoll and overlayed with equal volumes of 40% Percoll. Microglia were enriched at the interface of 70% (v/v) to 40% (v/v) Percoll after centrifugation (at 18°C, 300g for 30 min; slow acceleration and deceleration: 3) (Mazaheri et al, 2017). Microglia were collected, filtered through 100 μm cell strainers, and washed with blocking buffer (0.2% BSA in HBSS). Cells were then consecutively stained with FCRLs monoclonal rat antibody (Butovsky et al, 2014) (30 min), goat anti‐rat APC antibody (Biogegend, # 405407) (20 min), and Cd11b PeCy7 antibody (BD, #553142) (20 min) on ice. Cells were then washed and resuspended in 0.5 ml blocking buffer and subjected to cell sorting. Sorted CD11b+ and FCRL+ cells were pelleted by centrifugation and snap frozen in liquid nitrogen, stored at −80°C until further use. Following total cell lysis in 1:3 diluted RLT buffer (Quiagen, RNeasy Mini Kit), 10,000 cells in 4 µl volume were subjected to gene expression profiling with the nCounter® customized panel from NanoString (NanoString Technologies). We generated an nCounter panel for analyzing gene expression of 65 microglial activation‐related genes including five (Asb10, Cltc, Hprt1 Tubb5, and Gusb) housekeeping genes. Gene expression levels in each sample were normalized against the geometric mean of four housekeeping genes using the nSolver Analysis Software, version 4.0. Gusb was excluded because of significant changes in Grn KO and Double KO mice.
Gene expression profiling of human hiMGL
hiMGL were seeded into 12‐well plates and incubated for 3 h at 37°C (5% CO2). Thereafter, microglia were treated with TREM2 antagonistic antibodies and isotype control for 24 h. After treatment, cells were collected and RNA was isolated using the E.Z.N.A HP Total RNA kit (Omega Bio‐Tek) according to the manufacturer’s instructions. Following isolation, RNA quality was determined using a 4200 TapeStation (Agilent) and gene expression profiling with the nCounter® customized panel from NanoString (NanoString Technologies) was performed. We generated an nCounter panel for analyzing gene expression of 82 microglial‐related genes and 8 housekeeping genes. Gene expression levels in each sample were normalized against the geometric mean of five housekeeping genes including CLTC, HPRT1, RPL13A, TBP, and PPIA using the nSolver Analysis Software, version 4.0. CALR, TUBB5, and YWAHZ were excluded because of significant changes in Grn KO and WT microglia.
Lipid analysis by liquid chromatography‐mass spectrometry (LCMS)
Sample preparation for LCMS
For LCMS sample preparation, 10 mg of brain powder prepared from whole‐brain–powered homogenates was mixed with 400 ml of methanol spiked with internal standards and homogenized with a 3 mm tungsten carbide bead (shaken at 25 Hz for 30 s). The methanol fraction was then isolated via centrifugation (20 min at 4°C, 14,000 g, followed by transfer of supernatant to a 96‐well plate, and 1 h incubation at −20°C followed by an additional 20 min centrifugation (4,000 g at 4°C)) and transferred to glass vials for LCMS analysis. For analysis of a GlcCer/GalCer panel, an aliquot of the methanol fraction was dried under N2 gas and then resuspended in 100 ml of 92.5/5/2.5 CAN/IPA/H2 (MS grade) with 5 mM ammonium formate (MS grade) and 0.5% formic acid (MS grade).
Unless otherwise noted, relative quantification of lipids and metabolites was performed using the Shimadzu Nexera X2 LC system (Shimadzu Scientific Instrument) coupled to Sciex QTRAP 6500+ mass spectrometer (Sciex).
Lipidomic analysis
For each analysis, 5 µl of sample was injected on a BEH C18 1.7 µm, 2.1 × 100 mm column (Waters Corporation), using a flow rate of 0.25 ml/min at 55°C. Mobile phase A consisted of 60:40 acetonitrile/water (v/v); and mobile phase B consisted of 90:10 isopropyl alcohol/acetonitrile (v/v). These buffers were fortified with 10 mM ammonium formate with 0.1% formic acid (positive ionization) or with 10 mM ammonium acetate (negative ionization). The gradient was programmed as follows: 0.0–8.0 min from 45% B to 99% B, 8.0–9.0 min at 99% B, 9.0–9.1 min to 45% B, and 9.1–10.0 min at 45% B. Source settings were as follows: curtain gas at 30 psi; collision gas was set at medium; ion spray voltage at 5,500 V (positive mode) or 4,500 V (negative mode); temperature at 250°C (positive mode) or 600°C (negative mode); ion source gas 1 at 55 psi; and ion source gas 2 at 60 psi. Data acquisition was performed using Analyst 1.6.3 (Sciex) in multiple reaction monitoring mode (MRM). Area ratios of endogenous metabolites and surrogate internal standards were quantified using MultiQuant 3.02 (Sciex).
Protein analysis and Western blotting
Cell pellets obtained from human primary monocytes, cultured hiMGL, or aliquots of powdered frozen brains were lysed in Triton lysis buffer (150 mM NaCl, 50 mM Tris‐HCL, pH 7.6, 2 mM EDTA, 1% Triton X‐100) supplemented with protease inhibitor (Sigma‐Aldrich). Lysates were incubated on ice for 30 min and then centrifuged at 17,000 g for 15 min at 4°C. For sequential biochemical protein extraction of soluble, less soluble, and insoluble proteins, brain powder was lysed in high salt (HS) buffer (0.5 M NaCl, 10 mM Tris‐HCL pH 7.5, 5 mM EDTA, 1 mM DTT, 10% sucrose), then RIPA buffer (150 mM NaCl, 20 mM Tris‐HCL pH7.4, 1% NP‐40, 0.05% Triton X‐100, 0.5% sodium‐desoxycholate, 2.5 mM EDTA), followed by urea buffer (30 mM Tris‐HCL pH 8.5, 7 M urea, 2 M thiourea) as described previously (Gotzl et al, 2014). For membrane preparation of hiMG, pellets were resuspended with hypotonic buffer (10 mM Tris, pH 7.4, 1 mM EDTA, pH 8.0, 1 mM EGTA, pH 8.0) and incubated on ice for 30 min, vortexed every 10 min, followed by a freeze–thaw cycle, and centrifuged at 17,000 g for 45 min at 4°C. The supernatants were collected (cytosolic fraction) and the pellet (membrane fraction) resuspended in STEN lysis buffer and incubated on ice for 20 min. Insoluble proteins were pelleted at 17,000 g for 20 min at 4°C and the supernatant (membrane fraction) was collected and used for further analysis. Protein concentrations were determined using the BCA protein assay (Pierce, ThermoFisher). Equal amounts of protein adjusted to lysis buffer were mixed with Laemmli sample buffer supplemented with β‐mercaptoethanol. Proteins were separated by SDS‐PAGE and transferred onto polyvinylidene difluoride membranes (Immobilon‐P, Merck Millipore). Proteins of interest were detected using the following primary antibodies: goat anti‐TREM2 (R&D Systems, Inc., #AF1828), rabbit anti‐PGRN (ThermoFisher, #40‐3400), goat anti‐CatD (R&D, #AF1029), mouse anti‐SPH (abcam, # ab8049), mouse anti‐βActin (Sigma, #A5316), mouse anti‐GAPDH (Invitrogen, #AM4300), and rabbit anti‐Calnexin (Stressgene, #SPA‐860) followed by incubation with horseradish peroxidase‐conjugated secondary antibodies and ECL Plus substrate (ThermoFisher, Pierce ECL Plus Western Blotting Substrates). For quantification, images were taken with a Luminescent Image Analyzer LAS‐4000 (Fujifilm Life Science, Tokyo, Japan) and evaluated with the Multi GaugeV3.0 software (Fujifilm Life Science, Tokyo, Japan).
ELISA‐based quantification of sTREM2 and PGRN
sTREM2 in conditioned media was quantitated using the Meso Scale Discovery Platform as described previously (Schlepckow et al, 2020). Briefly, streptavidin‐coated small‐spot 96‐well plates were blocked overnight at 4°C, incubated with 0.125 μg/ml biotinylated polyclonal goat anti‐human TREM2 capture antibody (R&D, #BAF1828). After washing, plates were incubated with samples and standards for 2 h at RT. If cells were antibody treated, samples and standards were previously mixed 9:1 with denaturing buffer (200 mM Tris‐HCL pH 6.8, 4% (w/v) SDS, 40% (v/v) glycerol, 2% (v/v) β‐mercaptoethanol, 50 mM EDTA), and boiled at 95°C for 5 min to dissociate and denature TREM2 antibodies‐bound antibodies. Plates were washed before incubation for 1 h at RT with 1 μg/ml mouse anti‐human TREM2 antibody (SantaCruz Biotechnology, B‐3 SCBT‐373828). After washing, plates were incubated with a SULFO‐TAG‐labeled anti‐mouse secondary antibody (MesoScaleDiscovery, R32AC‐5) for 1 h at RT. After additional washing steps, 1× Meso Scale Discovery Read buffer was added and the light emission at 620 nm after electrochemical stimulation as measured with a Meso Scale Discovery Sector Imager 2400 reader.
PGRN levels were determined using a previously described protocol (Gotzl et al, 2019) using the following antibodies: a biotinylated polyclonal goat anti‐human PGRN antibody (R&D, #BAF2420) at 0.2 μg/ml as capture antibody, a mouse anti‐human PGRN antibody (R&D, #MAB2420) as detection antibody, and a SULFO‐TAG‐labeled anti‐mouse (MesoScaleDiscovery, #R32AC‐5) as secondary antibody.
GRN gene sequencing
All 12 coding exons including intron boundaries of the GRN gene were amplified by PCR. For PCR reaction, the Q5 polymerase was used according to the manufacturer’s protocol. PCR products were then subjected to Sanger sequencing and sequences were compared to the healthy control sequence. A two base pair deletion in exon 6 was detected using forward primer GGGCCTCATTGACTCCAAG and reverse primer GTGGTGTAAGCGGTACCCTC.
p‐Syk AlphaLISA
Phosphorylated SYK (p‐Syk) was measured using the AlphaLISA SureFire Ultra p‐Syk Assay kit (PerkinElmer, #ALSU‐PSYK‐A‐HV) following the manufacturer’s instructions. Briefly, differentiated human macrophages were plated in 100 µl media at a density of 50,000 cells/well. hiMGL were plated in iMGL media at a density of 30,000 cells/well, in 96‐well plates, and incubated overnight at 37°C in a cell culture incubator. Plates were then washed three times with HBSS and 50 µl of liposome (1 mg/ml)/antibody (20 µg/ml) mix was added to the cells. Following an incubation at 37°C for 1 h for macrophages, or 5 min for hiMGL, treatment solutions were removed and cells were lysed with 40 µl lysis buffer supplemented with protease inhibitor mix (Sigma) and phosphatase inhibitor (PhosSTOP, Roche) for 30 min at 4°C. Equal volumes of lysate were then subjected to analysis using an EnSpire Multimode Plate Reader (PerkinElmer).
Liposome preparation
POPC/POPS (7:3) liposomes at 10 mg/ml were prepared as follows: 7 mg POPC and 3 mg POPS were dissolved in chloroform followed by thorough evaporation of solvent. The lipid mixture was then resuspended in 1 ml HBSS, and suspensions were extruded using 100 nm polycarbonate membranes (Whatman, #800309) and a LiposoFast extruder device (Sigma‐Aldrich) to generate large unilamellar vesicles.
Cathepsin activity assay
hiMGL cell pellets or powdered mouse brain tissue was used for cathepsin D fluorescence‐based activity assays (Abnova) as described previously (Gotzl et al, 2018). Mouse brain tissue was homogenized using precellys lysing kit (Bertin Instruments, #P000933‐LYSK0‐A).
GCase activity assay
Brain powder was lysed in GCase lysis buffer (150 mM NaCl, 20 mM Tris‐HCl (pH 7.5), 1% Triton X‐100, 1 mM EDTA, and 1 mM EGTA) and protein concentrations were determined using the BCA assay. Lysates were adjusted to 2 mg/ml. Lysates were diluted 12.5‐fold in GCase activity buffer (100 mM Phosphate Citrate buffer pH 5.2, 0.5% Sodium Taurocholate, 0.25% Triton‐X 100) and 4‐Methylumbelliferyl β‐D‐glucopyranoside stock solution (30 mM; Sigma‐Aldrich, M3633‐1G, stock solution in DMF) was diluted threefold in the GCase activity buffer. Ninety microliter of the diluted lysates and 10 µl of the diluted 4‐Methylumbelliferyl β‐D‐glucopyranoside were added to a 96‐well plate. Plates were incubated for 15 min at 37°C. Signal intensities were measured continuously for 1 h (Ex 365 nm/Em 455 nm).
Phagocytosis assays
Microglial phagocytosis was determined using the IncuCyte S3 Live‐Cell Analysis System (Sartorius). hiMGL cells were plated in 96‐well plates at 30% confluency. Cells were incubated at 37°C and the confluency (pre‐treatment) of each well was determined with the IncuCyte S3 3 h after seeding. Thereafter, TREM2 antagonistic antibodies and isotype control were added at 20 and 40 µg/ml. Eighteen hours after antibody treatment, pHrodo‐labeled myelin (5 µg/ml) was added to the hiMGL cells and images of fluorescence and phase were captured at 4× in the IncuCyte S3 live cell imager every 15 min. Using IncuCyte 2020B software (Satorius), image masks for phase and fluorescent signal (phagocytosis of pHrodo‐labelled myelin) were acquired, and the fluorescent signal was normalized to cell confluency (cell body area), which was measured before the antibody treatment.
pHrodo labeling of myelin
Myelin was labeled with amine reactive pHrodoTM Red succinimidyl ester (ThermoFisher) for 45 min at RT (protected from light). Labeled myelin was washed with PBS and either directly used or stored in aliquots at −80°C.
Immunohistochemistry and image acquisition
Mice were transcardially perfused with PBS and brains were dissected into two hemispheres. One hemisphere was snap frozen and stored at −80°C until further use. The other hemisphere was immersion fixed for 24 h in 4% paraformaldehyde, washed with PBS, and incubated 30% sucrose for 48 h for cryoprotection. After freezing, brains were cut into 50 μm or 100 μm coronal sections using a vibratome (Leica Biosystems), collected in PBS, and stored at 4°C until further use. For visualizing lipofuscin, SPH or VGAT, 50 μm free‐floating sections were stained with primary antibodies (mouse anti‐VGAT (Synaptic Systems, #131011C3, or rabbit anti‐SPH (Abcam, #ab32594)) in 5% donkey serum in PBS overnight at 4°C with slow agitation. After washing, tissue sections were incubated with corresponding secondary antibodies for 3 h at RT. After washing, tissue sections were stained with DAPI for 10 min at RT and mounted onto slides using ProlongTM Gold Antifade reagent (ThermoFisher, #P36961). For morphological analysis of microglia, 100 μm sections were blocked with goat serum blocking buffer (2% goat serum, 0.05% Tween 20 in 0.01 M PBS, pH 7.2–7.4) and stained with anti‐IBA1 in primary antibody buffer (1% bovine serum albumin, 0.1% gelatin from cold water fish skin, 0.5% Triton X‐100 in 0.01 M PBS, pH 7.2–7.4) and anti‐rabbit IgG‐coupled Alexa Flour 555 in secondary antibody buffer (0.05% Tween 20 in 0.01 M PBS, pH 7.2–7.4). After washing, tissue sections were stained with DAPI for 10 min at RT and mounted onto slides using ProlongTM Gold Antifade reagent. Images were acquired using a LSM800 Zeiss confocal microscope and the ZEN 2011 software package (blue edition). For lipofuscin analysis, five images were taken per slide using a 20× objective at 2,048 × 2,048 pixel resolution. For SPH and VGAT analysis, three images were taken per slide using a 20× objective at 2,048 × 2,048 pixel resolution. Total fluorescence above background was quantified using the Fiji software (ImageJ, Version 1.0).
Automated analysis of microglia morphology
For morphological analysis of microglia, two z‐stack images per animal (n = 3) were recorded with a 40× objective in a resolution of 1,024 × 1,024 pixels (x–y‐pixel size = 0.15598 µm) and a slice distance (z) of 0.4 µm. The raw confocal z‐stacks were then analyzed using the Microglia Morphology Quantification Tool (MMQT) for automated analysis of microglial morphology as previously described (Heindl et al, 2018). The algorithm was run in MATLAB (Version R2016b). To identify the most discriminating features, a receiver operating characteristic (ROC) analysis was performed in R (version 4.0.3) for calculating the area under the curve (auc) between the groups “WT” and “Double KO.” Statistical analysis of group difference for the morphological scores, “Branch volume” (auc = 0.72), “Sphericity score” (auc = 0.82), “Branch length” (auc = 0.69), and “Number of branch nodes” (auc = 0.80) was performed using the Wilcoxon rank‐sum test with continuity correction and Bonferroni post hoc correction for multiple testing in R (version 4.0.3).
Statistical analysis
Data were analyzed using GraphPad Prism 9. If no other test of significance is indicated, for statistical analysis of two groups of samples, the unpaired, two‐tailed student’s t‐test was performed. For comparison of more than two groups, one‐way ANOVA and Dunnett’s or Tukey’s post hoc test were used. Statistical significance was set at *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
Author contributions
CH, AC, and AR conceived the study and analyzed the results. CH wrote the manuscript with further input from AR, AC, GDP, KMM, JWL, SR, and DP. AR performed and analyzed Western Blots, ELISAs, enzyme activity assays, mRNA isolation, NanoString experiments, and immunofluorescence on all mouse samples. AR isolated and performed all experiments of human‐derived macrophages and analyzed hiMGL NanoString data. With supervision of DP, SR generated and validated GRN KO hiPSC, differentiated into hiMGL, and performed and analyzed Western Blots, ELISAs, and enzyme activity assays. JK and GK helped to establish hiMGL cell differentiation. KMM and BVL discovered, generated, and validated antagonistic TREM2 antibodies. TL and JS performed lipidomic analysis. MAV and SHa performed NanoString and phagocytosis assays on hiMGL cells. JGn, KW, AZ, and MB conducted, performed, and analyzed PET imaging. SHe performed automated analysis on microglia morphology, with supervision of AL. IP performed CSF isolation. JKG helped to establish mouse lines. JL, KB, JD‐S, EW, and LR identified FTLD patients and performed sequencing analysis. BN performed NfL measurements. The synopsis image was created with BioRender.com.
Disclosure statement and competing interests
CH collaborates with Denali Therapeutics, participated on one advisory board meeting of Biogen, and received a speaker honorarium from Novartis and Roche. CH is chief advisor of ISAR Bioscience. KMM, BVL, TL, JS, JWL, and GDP are employees and shareholders of Denali Therapeutics. DP is a scientific advisor of ISAR Bioscience. MB received speaker honoraria from GE healthcare, Roche, and LMI and is an advisor of LMI.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198) (to CH and DP), a Koselleck Project HA1737/16‐1 (to CH), BR4580/1‐1 (to MB), the Helmholtz‐Gemeinschaft Zukunftsthema “Immunology and Inflammation” (ZT‐0027) (to CH), Alzheimer’s Association ADSF‐21‐831213‐C (to CH and DP), Vascular Dementia Research Foundation (to DP), and the donors of the ADR AD2019604S, a program of the BrightFocus Foundation (to DP). AR is supported by a Ph.D. stipend from the Hans and Ilse Breuer Foundation. MB was supported by the Alzheimer Forschung Initiative e.V (grant number 19063p). The authors like to thank Michael Heide and Oliver Weigert (Core Facility “Digital Single Molecule/NanoString Technologies”; Deutsches Konsortium für Translationale Krebsforschung, Partner Site München, Labor für Experimentelle Leukämie‐ und Lymphom‐Forschung (ELLF)) for supporting NanoString measurements, Ludovico Cantuti‐Castelvetri and Mikael Simons for preparing labeled myelin, and Jane Hettinger and Alba Simats for performing CSF withdrawal. LC/MS‐based lipidomics analyses were supported by Sonnet Davis. Antibody discovery, material generation, and characterization of TREM2 antagonist antibodies were supported by Josh Park, Do Jin Kim, Yaneth Robles, Rachel Prorok, Steve Lianoglou, Cathal Mahon, and Tina Giese from Denali Therapeutics. The authors like to thank Dieter Edbauer for providing GRN‐sequencing primers. Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2022) 41: e109108.
Contributor Information
Kathryn M Monroe, Email: monroe@dnli.com.
Dominik Paquet, Email: dominik.paquet@med.uni-muenchen.de.
Anja Capell, Email: anja.capell@med.uni-muenchen.de.
Christian Haass, Email: christian.haass@dzne.de.
Data availability
The accession number for the Neuropathology NanoString data reported in this paper is Gene Expression Omnibus GSE181135 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181135 (6‐month‐old cohort) and GSE185510 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE185510 (14‐month‐old cohort).
References
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C et al (2017) iPSC‐derived human microglia‐like cells to study neurological diseases. Neuron 94: 278–293.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, Haass C (2003) Games played by rogue proteins in prion disorders and Alzheimer's disease. Science 302: 814–818 [DOI] [PubMed] [Google Scholar]
- Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I (2016) Portuguese family with the co‐occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging 41: 200 e201–200 e205 [DOI] [PubMed] [Google Scholar]
- Arrant AE, Roth JR, Boyle NR, Kashyap SN, Hoffmann MQ, Murchison CF, Ramos EM, Nana AL, Spina S, Grinberg LT et al (2019) Impaired beta‐glucocerebrosidase activity and processing in frontotemporal dementia due to progranulin mutations. Acta Neuropathol Commun 7: 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering‐Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S et al (2006) Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442: 916–919 [DOI] [PubMed] [Google Scholar]
- Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, Saftig P, Van Damme P (2017) Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum Mol Genet 26: 2850–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch‐Queralt M, Cantuti‐Castelvetri L, Damkou A, Schifferer M, Schlepckow K, Alexopoulos I, Lutjohann D, Klose C, Vaculciakova L, Masuda T et al (2021) Diet‐dependent regulation of TGFbeta impairs reparative innate immune responses after demyelination. Nat Metab 3: 211–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel M, Probst F, Jaworska A, Overhoff F, Korzhova V, Albert NL, Beck R, Lindner S, Gildehaus F‐J, Baumann K et al (2016) Glial activation and glucose metabolism in a transgenic amyloid mouse model: a triple‐tracer PET study. J Nucl Med 57: 954–960 [DOI] [PubMed] [Google Scholar]
- Butler VJ, Cortopassi WA, Argouarch AR, Ivry SL, Craik CS, Jacobson MP, Kao AW (2019a) Progranulin stimulates the in vitro maturation of pro‐cathepsin D at acidic pH. J Mol Biol 431: 1038–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler VJ, Cortopassi WA, Gururaj S, Wang AL, Pierce OM, Jacobson MP, Kao AW (2019b) Multi‐granulin domain peptides bind to pro‐cathepsin D and stimulate its enzymatic activity more effectively than progranulin in vitro. Biochemistry 58: 2670–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE et al (2014) Identification of a unique TGF‐beta‐dependent molecular and functional signature in microglia. Nat Neurosci 17: 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Jian J, Hettinghouse A, Zhao X, Setchell KDR, Sun Y, Liu CJ (2018) Progranulin associates with hexosaminidase A and ameliorates GM2 ganglioside accumulation and lysosomal storage in Tay‐Sachs disease. J Mol Med 96: 1359–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Danao J, Talreja S, Wen P, Yin J, Sun N, Li C‐M, Chui D, Tran D, Koirala S et al (2018) TREM2‐activating antibodies abrogate the negative pleiotropic effects of the Alzheimer's disease variant Trem 2(R47H) on murine myeloid cell function. J Biol Chem 293: 12620–12633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cignarella F, Filipello F, Bollman B, Cantoni C, Locca A, Mikesell R, Manis M, Ibrahim A, Deng LI, Benitez BA et al (2020) TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol 140: 513–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Wang Y (2016) TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 17: 201–207 [DOI] [PubMed] [Google Scholar]
- Concordet JP, Haeussler M (2018) CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46: W242–W245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin J‐J et al (2006) Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 442: 920–924 [DOI] [PubMed] [Google Scholar]
- Deczkowska A, Weiner A, Amit I (2020) The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell 181: 1207–1217 [DOI] [PubMed] [Google Scholar]
- Deussing M, Blume T, Vomacka L, Mahler C, Focke C, Todica A, Unterrainer M, Albert NL, Lindner S, von Ungern‐Sternberg B et al (2018) Coupling between physiological TSPO expression in brain and myocardium allows stabilization of late‐phase cerebral [(18)F]GE180 PET quantification. NeuroImage 165: 83–91 [DOI] [PubMed] [Google Scholar]
- Efthymiou AG, Goate AM (2017) Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwanger DC, Wang S, Brioschi S, Shao Z, Green L, Case R, Yoo D, Weishuhn D, Rathanaswami P, Bradley J et al (2021) Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 118: e2017742118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers BM, Rodriguez‐Navas C, Tesla RJ, Prange‐Kiel J, Wasser CR, Yoo KS, McDonald J, Cenik B, Ravenscroft TA, Plattner F et al (2017) Lipidomic and transcriptomic basis of lysosomal dysfunction in progranulin deficiency. Cell Rep 20: 2565–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M, Franzmeier N, Suárez‐Calvet M, Morenas‐Rodriguez E, Caballero MAA, Kleinberger G, Piccio L, Cruchaga C, Deming Y, Dichgans M et al (2019) Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer's disease. Sci Transl Med 10.1126/scitranslmed.aav6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler M, Rappaport MS, Cuno CB, George J (2021) Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer's disease models. J Neuroinflammation 18: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Guo O, Zhen Z, Zhen J (2020) Essential functions of the transcription factor Npas4 in neural circuit development, plasticity, and diseases. Front Neurosci 14: 603373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzl JK, Brendel M, Werner G, Parhizkar S, Sebastian Monasor L, Kleinberger G, Colombo AV, Deussing M, Wagner M, Winkelmann J et al (2019) Opposite microglial activation stages upon loss of PGRN or TREM2 result in reduced cerebral glucose metabolism. EMBO Mol Med 11: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzl JK, Colombo AV, Fellerer K, Reifschneider A, Werner G, Tahirovic S, Haass C, Capell A (2018) Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice. Mol Neurodegener 13: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzl JK, Lang CM, Haass C, Capell A (2016) Impaired protein degradation in FTLD and related disorders. Ageing Res Rev 32: 122–139 [DOI] [PubMed] [Google Scholar]
- Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E et al (2014) Common pathobiochemical hallmarks of progranulin‐associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol 127: 845–860 [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindl S, Gesierich B, Benakis C, Llovera G, Duering M, Liesz A (2018) Automated morphological analysis of microglia after stroke. Front Cell Neurosci 12: 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira‐Saecker A, Griep A, Axt D, Remus A, Tzeng T‐C et al (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493: 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, McManus RM, Latz E (2018) Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19: 610–621 [DOI] [PubMed] [Google Scholar]
- Hong S, Beja‐Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA et al (2016a) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dissing‐Olesen L, Stevens B (2016b) New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36: 128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM (2010) Sortilin‐mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68: 654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Modeste E, Dammer E, Merino P, Taylor G, Duong DM, Deng Q, Holler CJ, Gearing M, Dickson D et al (2020) Network analysis of the progranulin‐deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol Commun 8: 163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav VS, Lin PBC, Pennington T, Di Prisco GV, Jannu AJ, Xu G, Moutinho M, Zhang J, Atwood BK, Puntambekar SS et al (2020) Trem2 Y38C mutation and loss of Trem2 impairs neuronal synapses in adult mice. Mol Neurodegener 15: 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian J, Zhao S, Tian Q‐Y, Liu H, Zhao Y, Chen W‐C, Grunig G, Torres PA, Wang BC, Zeng B et al (2016) Association between progranulin and gaucher disease. EBioMedicine 11: 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao AW, McKay A, Singh PP, Brunet A, Huang EJ (2017) Progranulin, lysosomal regulation and neurodegenerative disease. Nat Rev Neurosci 18: 325–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayasuga Y, Chiba S, Suzuki M, Kikusui T, Matsuwaki T, Yamanouchi K, Kotaki H, Horai R, Iwakura Y, Nishihara M (2007) Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav Brain Res 185: 110–118 [DOI] [PubMed] [Google Scholar]
- Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, David E, Baruch K, Lara‐Astaiso D, Toth B et al (2017) A Unique microglia type associated with restricting development of Alzheimer's disease. Cell 169: 1276–1290.e17 [DOI] [PubMed] [Google Scholar]
- Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, Strittmatter SM (2017) Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia‐related phenotypes in progranulin‐deficient mice. Neuron 95: 281–296.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Brendel M, Mracsko E, Wefers B, Groeneweg L, Xiang X, Focke C, Deussing M, Suarez‐Calvet M, Mazaheri F et al (2017) The FTD‐like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J 36: 1837–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez‐Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger‐Weinzierl A, Mazaheri F et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6: 243ra286 [DOI] [PubMed] [Google Scholar]
- Krabbe G, Minami SS, Etchegaray JI, Taneja P, Djukic B, Davalos D, Le D, Lo I, Zhan L, Reichert MC et al (2017) Microglial NFkappaB‐TNFalpha hyperactivation induces obsessive‐compulsive behavior in mouse models of progranulin‐deficient frontotemporal dementia. Proc Natl Acad Sci USA 114: 5029–5034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z et al (2017) The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47: 566–581.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwart D, Paquet D, Teo S, Tessier‐Lavigne M (2017) Precise and efficient scarless genome editing in stem cells using CORRECT. Nat Protoc 12: 329–354 [DOI] [PubMed] [Google Scholar]
- Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, Rezzonico MG, Imperio J, Lalehzadeh G, Huntley MA et al (2021) Trem2 restrains the enhancement of tau accumulation and neurodegeneration by beta‐amyloid pathology. Neuron 109: 1283–1301 e1286 [DOI] [PubMed] [Google Scholar]
- Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, Haass C (2020) Emerging microglia biology defines novel therapeutic approaches for Alzheimer's disease. Neuron 108: 801–821 [DOI] [PubMed] [Google Scholar]
- Lim NK, Moestrup V, Zhang X, Wang WA, Moller A, Huang FD (2018) An improved method for collection of cerebrospinal fluid from anesthetized mice. J Vis Exp 10.3791/56774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Le KX, Park MA, Wang S, Belanger AP, Dubey S, Frost JL, Holton P, Reiser V, Jones PA et al (2015) In vivo detection of age‐ and disease‐related increases in neuroinflammation by 18F‐GE180 TSPO MicroPET imaging in wild‐type and Alzheimer's transgenic mice. J Neurosci 35: 15716–15730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan T, Simon JS, Rana A, Cherf GM, Srivastava A, Davis SS, Yoon Low RL, Chiu C, Fang M, Huang F et al (2021) Rescue of a lysosomal storage disorder caused by BMP deficiency in Grn loss‐of‐function with a brain penetrant progranulin biologic. Cell 184: 4651–4668.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui H, Zhang J, Makinson S, Cahill M, Kelley K, Huang H‐Y, Shang Y, Oldham M, Martens L, Gao F et al (2016) Progranulin Deficiency Promotes Circuit‐Specific Synaptic Pruning By Microglia Via Complement Activation. Cell 165: 921–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, Pluvinage JV, Mathur V, Hahn O, Morgens DW et al (2020) Lipid‐droplet‐accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 23: 194–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min S‐W, Gan LI, Finkbeiner S, Huang EJ et al (2012) Progranulin deficiency promotes neuroinflammation and neuron loss following toxin‐induced injury. J Clin Invest 122: 3955–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trumbach D, Wurst W et al (2017) TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep 18: 1186–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, Blurton‐Jones M (2018) Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol Neurodegener 13: 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeter LH, Dopper EG, Jiskoot LC, Sanchez‐Valle R, Graff C, Benussi L, Ghidoni R, Pijnenburg YA, Borroni B, Galimberti D et al (2016) Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 3: 623–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilandt WJ, Ngu H, Gogineni A, Lalehzadeh G, Lee SH, Srinivasan K, Imperio J, Wu T, Weber M, Kruse AJ et al (2020) Trem2 deletion reduces late‐stage amyloid plaque accumulation, elevates the Abeta42:Abeta40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer's mouse model. J Neurosci 40: 1956–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D et al (2020) TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105: 837–854.e9 [DOI] [PubMed] [Google Scholar]
- Overhoff F, Brendel M, Jaworska A, Korzhova V, Delker A, Probst F, Focke C, Gildehaus F‐J, Carlsen J, Baumann K et al (2016) Automated spatial brain normalization and hindbrain white matter reference tissue give improved [(18)F]‐Florbetaben PET quantitation in Alzheimer's model mice. Front Neurosci 10: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, Ghasemigharagoz A, Katzmarski N et al (2019) Loss of TREM2 function increases amyloid seeding but reduces plaque‐associated ApoE. Nat Neurosci 22: 191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushter DH, Du H, Feng T, Hu F (2018) The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol 136: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Otano I, Larsen RS, Wesseling JF (2016) Emerging roles of GluN3‐containing NMDA receptors in the CNS. Nat Rev Neurosci 17: 623–635 [DOI] [PubMed] [Google Scholar]
- Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, Gräber S, Kuder‐Buletta E, LaFougere C, Laske C et al (2019) Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med 25: 277–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BR, Sudduth TL, Weekman EM, Johnson S, Hawthorne D, Woolums A, Wilcock DM (2020) Therapeutic Trem2 activation ameliorates amyloid‐beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J Neuroinflammation 17: 238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2016) How neuroinflammation contributes to neurodegeneration. Science 353: 777–783 [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Woollacott IOC, Dick KM, Brotherhood E, Gordon E, Fellows A, Toombs J, Druyeh R, Cardoso MJ, Ourselin S et al (2016) Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology 87: 1329–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root J, Merino P, Nuckols A, Johnson M, Kukar T (2021) Lysosome dysfunction as a cause of neurodegenerative diseases: lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis 154: 105360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H, Haass C (2017) An Alzheimer‐associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med 9: 1356–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlepckow K, Monroe KM, Kleinberger G, Cantuti‐Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B et al (2020) Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med 12: e11227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole S, Staropoli J et al (2012) Strikingly different clinicopathological phenotypes determined by progranulin‐mutation dosage. Am J Hum Genet 90: 1102–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, Greenberg ME (2014) Npas4 regulates excitatory‐inhibitory balance within neural circuits through cell‐type‐specific gene programs. Cell 157: 1216–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer B, Bu Q, Cory E, Jiang K, Duong S, Sinha D, Steltzer S, Gamm D, Chang Q, Saha K (2018) Scarless genome editing of human pluripotent stem cells via transient puromycin selection. Stem Cell Rep 10: 642–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Suzuki G, Matsuwaki T, Hosokawa M, Serrano G, Beach TG, Yamanouchi K, Hasegawa M, Nishihara M (2017) Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum Mol Genet 26: 969–988 [DOI] [PubMed] [Google Scholar]
- Thornton P, Sevalle J, Deery MJ, Fraser G, Zhou Y, Stahl S, Franssen EH, Dodd RB, Qamar S, Gomez Perez‐Nievas B et al (2017) TREM2 shedding by cleavage at the H157–S158 bond is accelerated for the Alzheimer's disease‐associated H157Y variant. EMBO Mol Med 9: 1366–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM‐2 attenuates macrophage activation. J Immunol 177: 3520–3524 [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM (2014) Altered microglial response to Abeta plaques in APPPS1‐21 mice heterozygous for TREM2. Mol Neurodegener 9: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D (2017) Progranulin‐mediated deficiency of cathepsin D results in FTD and NCL‐like phenotypes in neurons derived from FTD patients. Hum Mol Genet 26: 4861–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, Lolli F, Marcello E, Sironi L, Vegeto E et al (2018) Sex‐specific features of microglia from adult mice. Cell Rep 23: 3501–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, Ward M, Siddiqui O, Paul R, Gilfillan S et al (2020) Anti‐human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med 10.1084/jem.20200785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S et al (2016) TREM2‐mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME, Chen R, Huang H‐Y, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P et al (2017) Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med 10.1126/scitranslmed.aah5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisheit I, Kroeger JA, Malik R, Klimmt J, Crusius D, Dannert A, Dichgans M, Paquet D (2020) Detection of deleterious on‐target effects after HDR‐mediated CRISPR editing. Cell Rep 31: 107689 [DOI] [PubMed] [Google Scholar]
- Weisheit I, Kroeger JA, Malik R, Wefers B, Lichtner P, Wurst W, Dichgans M, Paquet D (2021) Simple and reliable detection of CRISPR‐induced on‐target effects by qgPCR and SNP genotyping. Nat Protoc 16: 1714–1739 [DOI] [PubMed] [Google Scholar]
- Werner G, Damme M, Schludi M, Gnorich J, Wind K, Fellerer K, Wefers B, Wurst W, Edbauer D, Brendel M et al (2020) Loss of TMEM106B potentiates lysosomal and FTLD‐like pathology in progranulin‐deficient mice. EMBO Rep 21: e50241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woollacott IOC, Bocchetta M, Sudre CH, Ridha BH, Strand C, Courtney R, Ourselin S, Cardoso MJ, Warren JD, Rossor MN et al (2018) Pathological correlates of white matter hyperintensities in a case of progranulin mutation associated frontotemporal dementia. Neurocase 24: 166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Shao W, Todd TW, Tong J, Yue M, Koga S, Castanedes‐Casey M, Librero AL, Lee CW, Mackenzie IR et al (2021) Microglial lysosome dysfunction contributes to white matter pathology and TDP‐43 proteinopathy in GRN‐associated FTD. Cell Rep 36: 109581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Wind K, Wiedemann T, Blume T, Shi Y, Briel N, Beyer L, Biechele G, Eckenweber F, Zatcepin A et al (2021) Microglial activation states drive glucose uptake and FDG‐PET alterations in neurodegenerative diseases. Sci Transl Med 13: eabe5640 [DOI] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 92: 252–264 [DOI] [PubMed] [Google Scholar]
- Zhang J, Velmeshev D, Hashimoto K, Huang Y‐H, Hofmann JW, Shi X, Chen J, Leidal AM, Dishart JG, Cahill MK et al (2020) Neurotoxic microglia promote TDP‐43 proteinopathy in progranulin deficiency. Nature 588: 459–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Feng T, Pardon CM, Mendoza CS, Hu F (2017) Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol 134: 151–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Pagan MD, Kim D, Nunez Santos M, Lieberman RL, Overkleeft HS, Sun Y, Smolka MB, Hu F (2019) Progranulin deficiency leads to reduced glucocerebrosidase activity. PLoS One 14: e0212382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Bastos de Oliveira F, Qi X, Brown WJ, Smolka MB, Sun Y, Hu F (2015) Prosaposin facilitates sortilin‐independent lysosomal trafficking of progranulin. J Cell Biol 210: 991–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]