

To the Editor — The COVID-19 pandemic is the first health crisis characterized by large amounts of genomic data1. Computational infrastructure can be a bottleneck for data analysis, amplifying global inequalities in ability to track SARS-CoV-2 evolution. This is an issue even in developed countries, as computational infrastructure requires expertise in resource procurement, configuration and maintenance. Commercial computational clouds do not fully address the problem because these resources must still be configured and funded. Furthermore, commercial clouds are predominantly US-based and many countries have policies making payments to foreign providers impractical. In developing countries, research computing infrastructure is rare and researchers often cannot afford commercial cloud-based computation. Here, we present the COVID-19 effort by the Galaxy Project, which pools free worldwide public computational infrastructure, making the analysis of deep sequencing data accessible to anyone while also providing an analytical framework for global pathogen genomic surveillance based on raw sequencing-read data.
Despite the existence of well designed and validated SARS-CoV-2 data analysis approaches2,3, the ad hoc4 nature of their application often complicates the integration and comparison of analysis results. Public computational infrastructure (XSEDE, ELIXIR and Nectar Cloud in the United States, European Union and Australia, respectively) coupled with existing open-source software offers a solution to SARS-CoV-2 analytics challenges. However, glue is required to bind these resources into a unified platform for managing users, allocating storage and pairing analysis tools with appropriate computational resources. Such a platform is best not developed by a single principal investigator, group or institution, but rather supported by an international community of users, developers and educators.
We have developed a two-stage platform (Fig. 1) housed on three public Galaxy instances5 in the United States (http://usegalaxy.org), the European Union (http://usegalaxy.eu) and Australia (http://usegalaxy.org.au) and capable of supporting hundreds of thousands of complex analyses per month. Anyone can run effectively unlimited computation with 250 Gb (expandable) of disk space. The COVID-19 Galaxy Project comprises two stages (Fig. 1): the software components of stage 1—mature utilities for quality control, mapping, assembly and allelic variant (AV) calling—run entirely in Galaxy and are distributed via the BioConda project6; the software components of stage 2 are snippets of code for data transformation, exploration and visualization running within standard web-browser-based notebook environments. Stage 1 produces variant lists whereas stage 2 uses notebooks to perform descriptive analyses of datasets. In addition, an interactive dashboard is available that tracks temporal AV dynamics. (See https://covid19.galaxyproject.org for data, workflows, notebooks, dashboard and our ongoing automated tracking of large-scale genomic surveillance projects.)
Fig. 1 |. Analysis flow for calling SARS-CoV-2 variants using Galaxy.
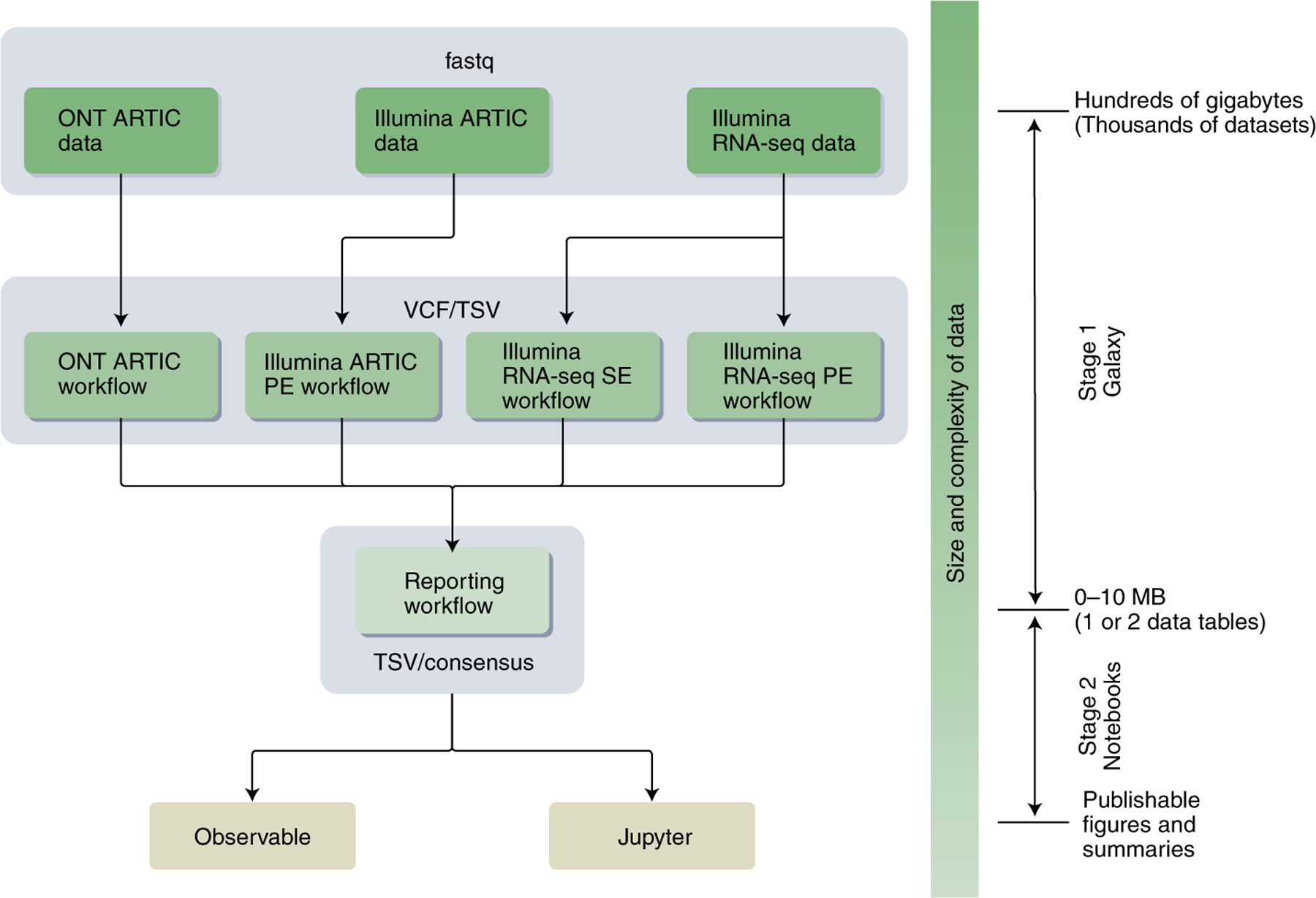
ONT, Oxford Nanopore Technologies; VCF, variant call format; TSV, tab-separated values; PE, paired end; SE, single end. For more information, see https://covid19.galaxyproject.org.
Four primary analysis workflows (Supplementary Table 1) support the identification of SARS-CoV-2 AVs from deep-sequencing reads via the production of annotated AVs through a series of steps including quality control, trimming, mapping, deduplication, AV calling and filtering. Their output is processed by the Reporting and Consensus workflows (Supplementary Table 1) to generate standardized data tables describing AVs along with consensus genome sequences. These are further processed to summarize and visualize the data using interactive notebooks.
To illustrate the platform’s utility and scalability, we refer the reader to two large SARS-CoV-2 Illumina datasets (PRJNA622837, 619 samples from early SARS-CoV-2 transmission in the Boston area7; and PRJEB37886, ~100,000 samples analyzed as of the time of writing from the COVID-19 Genomics UK (COG-UK) genomic surveillance effort8) detailed in Supplementary Tables 1–3 and Supplementary Figs. 1–3. Analysis on COVID-19 Galaxy Project resources provides insights into co-occurrence patterns, presence of mutations defining variants of concern (https://cov-lineages.github.io/lineages-website/global_report.html), and intersection with sites under selection, including non-random associations among common low-frequency AVs that may reflect shared intra-host dynamics (Supplementary Fig. 1 and Supplementary Table 2). It can also highlight the emergence of mutations interfering with binding of polyclonal antibodies9 (for example, COG-UK data in Supplementary Fig. 2), suggesting possible intra-host dynamics. These and other interactive notebooks and dashboards on the platform could identify AVs that warrant closer monitoring as the pandemic continues.
Our system is designed to encourage scalable collaborative worldwide genomic surveillance to identify and respond to emerging variants. By relying on raw read data rather than assembled genomes and allowing every result to be traced back to its raw data, it goes a step beyond current surveillance efforts. Specifically, it enables surveillance of intra-patient minor AV frequencies—a distinction that could yield early warnings of epidemiological conditions conducive to the emergence of variants with altered pathogenicity, vaccine sensitivity or drug resistance.
Supplementary Material
Acknowledgements
The authors are grateful to the broader Galaxy community for their support and software development efforts. This work is funded by NIH grants U41 HG006620 and NSF ABI grant 1661497. Usegalaxy.eu is supported by the German Federal Ministry of Education and Research grants 031L0101C and de.NBI-epi to B.G. Galaxy and HyPhy integration is supported by NIH grant R01 AI134384 to A.N. Usegalaxy.org.au is supported by Bioplatforms Australia and the Australian Research Data Commons through funding from the Australian Government National Collaborative Research Infrastructure Strategy. The hyphy.org development team is supported by NIH grant R01GM093939. Usegalaxy.be is supported by the Research Foundation-Flanders (FWO) grant I002919N and the Flemish Supercomputer Center (VSC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-021-01069-1.
References
- 1.Hodcroft EB et al. Nature 591, 30–33 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Quick J et al. Nat. Protoc 12, 1261–1276 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grubaugh ND et al. Genome Biol. 20, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker D et al. PLoS Pathog. 16, e1008643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jalili V et al. Nucleic Acids Res. 48 W1, W395–W402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grüning B et al. Nat. Methods 15, 475–476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemieux J et al. Science 10.1126/science.abe3261 (2021). [DOI] [Google Scholar]
- 8.du Plessis L et al. Science 371, 708–712 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greaney AJ et al. Cell Host Microbe 29, 463–476.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.