

Abstract
TLR4 signaling via endotoxemia in macrophages promotes macrophage transition to the inflammatory phenotype through NLRP3 inflammasome activation. This transition event has the potential to trigger acute lung injury (ALI). However, relatively little is known about the regulation of NLRP3 and its role in the pathogenesis of ALI. Here we interrogated the signaling pathway activated by CD38, an ectoenzyme expressed in macrophages, in preventing ALI through suppressing NLRP3 activation. Wild-type and Cd38-knockout (Cd38−/−) mice were used to assess inflammatory lung injury, and isolated macrophages were used to delineate underlying TLR4 signaling pathway. We showed that CD38 suppressed TLR4 signaling in macrophages by inhibiting Bruton’s tyrosine kinase (Btk) through the recruitment of Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2) and resulting in the dephosphorylation of activated Btk. Cd38−/− mice show enhanced lung polymorphonuclear leukocyte extravasation and severe lung injury. LPS- or polymicrobial sepsis-induced mortality in Cd38−/− mice were markedly augmented compared with wild types. CD38 in macrophages functioned by inhibiting Btk activation through activation of SHP2 and resulting dephosphorylation of Btk, and thereby preventing activation of downstream targets NF-κB and NLRP3. Cd38−/− macrophages displayed markedly increased activation of Btk, NF-κB, and NLRP3, whereas in vivo administration of the Btk inhibitor ibrutinib (a Food and Drug Administration-approved drug) prevented augmented TLR4-induced inflammatory lung injury seen in Cd38−/− mice. Our findings together show upregulation of CD38 activity and inhibition of Btk activation downstream of TLR4 activation as potential strategies to prevent endotoxemic ALI.
Keywords: NLRP3 inflammasome, protein tyrosine phosphatase SHP2, acute lung injury
CD38 is a type II transmembrane glycoprotein expressed in macrophages (1). CD38 consists of a long carboxy-terminal catalytic extracellular domain and a short amino-terminal cytoplasmic domain (1). CD38 can function both as a receptor and an ectoenzyme (2, 3) with its catalytic activity and signaling properties independent of each other (2, 3). CD38 converts NAD+ to cyclic-ADP-ribose (cADPR) and hydrolyzes cADPR to produce ADP-ribose (ADPR) (4). CD38 regulates Ca2 + influx in T and B cells through cADPR production (5, 6). CD38-mediated cADPR production also regulates intracellular Ca2 + release in neutrophils via the ryanodine receptor (7). In T-lymphocytes, CD38 signaling is mediated through association of its short cytosolic domain and Src homology-2 domain containing signaling proteins such as Lyn, a member of the Src family tyrosine kinases (3, 8). CD38 activation though cross linking in B-lymphocytes with agonistic antibodies induced Bruton’s tyrosine kinase (Btk)-dependent B cell proliferation and maturation through activation of NF-κB signaling (3).
TLR4 signaling induced by LPS in lung macrophages is causally linked to the pathogenesis of ALI (9). However, little is known about the role of CD38 activation of TLR4 signaling of NF-κB in macrophages as a determinant of ALI. Since lung macrophages upon shifting their phenotype transition to inflammatory or antiinflammatory cells that can either induce or suppress inflammatory lung injury (10), we addressed the question of whether CD38 regulates TLR4 signaling and contributes to the pathogenesis of ALI. We demonstrate that CD38 negatively regulated TLR4 signaling in macrophages and thus modulated lung inflammation and ALI and reduced mortality in mice. The mechanism of CD38-mediated protection involved preventing the activation of Btk, a nonreceptor protein tyrosine kinase containing an N-terminal Pleckstrin homology domain, Tec homology domain, Src homology (SH), SH3 and SH2 domains. Upon TLR4 activation of CD38-knockout (Cd38−/−) macrophages, we observed intensified activation of Btk, NF-κB, and NLRP3 inflammasome. In vivo administration of the Btk selective inhibitor ibrutinib prevented TLR4-induced lung injury similar to that seen in Cd38−/− mice. Expression of Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2), which negatively regulates TLR4 signaling and NLRP3 inflammasome activation, functioned by dephosphorylation and inactivation of Btk. Thus, CD38 expressed in macrophages downregulates TLR4 signaling and prevents ALI through blockade of Btk activation, suggesting Btk as a potential anti-ALI target.
Methods
Antibodies and Other Reagents
The antibodies and other reagents used for experiments were listed in Table 1.
Table 1.
Antibodies and Other Reagents
| Antibodies | Vendors | Catalog Numbers |
|---|---|---|
| CD38 | Protein Tech | 60006-1-Ig |
| GFP | Protein Tech | 50430-2AP |
| HA | Protein Tech | 51064-2-AP |
| Flag | Protein Tech | 66008-2-Ig |
| Phospho-IKKβ | Cell Signaling | 2078S |
| IKKβ | Cell Signaling | 2684S |
| Phospho-IkBα | Cell Signaling | 2859T |
| IkBα | Cell Signaling | 9242S |
| phospho-Btk | Cell Signaling | D9T6H |
| Btk | Santa Cruz | 7F12H4 sc-81159 |
| iNOS | Santa Cruz | sc-8310 |
| NLRP3 | Adipogen | AG-20B-0014-C100 |
| mIL-1β | RD Systems | AF-401-NA |
| SHP2 | Cell Signaling | CS3752s |
| pan-anti-ubiquitin | Cell Signaling | 3933S |
| Other Reagents | ||
| LPS | Sigma | L3012 |
| Src inhibitor PP1 | Calbiochem | CAS172889-26-8 |
| Ibrutinib | Cell Signaling | 16483 |
| C-terminal GFP-tagged Btk plasmid | Addgene | 51463 |
| HA-tagged CD38 plasmid | Addgene | 542152 |
| CalPhos transfection kit | ClonTech | 631312 |
| ADPR ELISA Kit | Trevigen, Inc | 4520-096-K |
| PCR Primers | Integrated DNA Tech | Custom |
Definition of abbreviation: SHP2 = Src homology-2 domain containing protein tyrosine phosphatase-2.
Mice
All mice were housed at the University of Illinois Animal Care Facility in accordance with institutional guidelines and guidelines of the National Institutes of Health. Veterinary care of the mice and the related animal experiments was approved by the University of Illinois Animal Resources Center. Cd38−/− mice (11) originally generated on a B6;139p2 background and have been backcrossed to C57BL/6J for 12 generations, and Cd38+/+ (wild-type [WT]) mice were obtained from Jackson Laboratory.
Lung Injury Assessment in Mice
Age-matched WT and Cd38−/− mice of either sex received indicated concentrations of a single dose of LPS (E. coli 011:B4) intraperitoneally (12). For histology, 5-μm paraffin-embedded sections prepared from the lungs were stained with hematoxylin and eosin (13). For myeloperoxidase (MPO) assays, lungs were perfused with PBS to remove all blood and then used to measure MPO activity (13). In vivo mouse lung vascular leak (permeability) was assessed by measuring the uptake of Evans blue dye conjugated with albumin (EBA) as described (14). Polymicrobial sepsis was induced by cecal ligation and puncture (CLP) with an 18-gauge needle as described (15). For survival studies, mice were monitored four times daily.
BAL Fluid
BAL fluid from anesthetized mice was collected (10) and used for measurement of cytokines through ELISA kit (ebiosciences) per manufacturer’s instructions.
Macrophages
Alveolar macrophages (AMs) and bone marrow-derived macrophages (BMDMs) were prepared as we described (10).
Bone Marrow Transplantation
Lethal irradiation was performed as described previously (16). At 8 hours after irradiation, bone marrow cells from Cd38−/− female mice (1 × 107 bone marrow cells/mouse) or Cd38+/+ (WT) female mice injected retroorbitally into the recipient male Cd38+/+ (WT) mice. After 3 weeks, the bone marrow transplantation efficiency was assessed using qualitative PCR for the male-specific SRY gene in recipient mice blood cells. Six weeks after reconstitution, mice were used for the experiments.
ADPR and Ca2+ Influx
Macrophages in culture with or without H2O2 stimulation were lysed, and the lysates ADRP concentrations at picogram levels were measured using HT PARP in vivo pharmacodynamic ELISA kit II from Trevigen, Inc. Ca2 + influx in macrophages was determined as we described previously (12).
Transfection
HEK293 cells expressing TLR4 were transfected with plasmid DNA (2 μg/ml) using CalPhos mammalian transfection kit from ClonTech laboratories. At 72 hours after transfection, cells were used for experiments.
Quantitative Real-Time PCR
Total RNA was isolated from lung tissue and reverse-transcribed with oligo(dT) primers and SuperScript reverse transcriptase (Invitrogen). The cDNA obtained was mixed with SYBR Green PCR mix (AB Applied Biosystems). An ABI prism 7000 was used for quantitative PCR. GAPDH expression served as an internal control. The following mouse primers were used: SHP2 forward, 5′-GGCAAAGTTTCTCACCAGCA-3′, and reverse, 3′-TCCAGGATCTCAAGCTGCAA-5′; GAPDH forward, 5′-ACCCAGAAGACTGTGGATGG-3′, and reverse, 5′-CACATTGGGGGTAGGAACAC-3′.
IB and IP
IB and IP experiments were performed as described (17).
Statistics
Results were analyzed by an unpaired two-tailed Student’s t-test and log-rank test. Differences in mean values were considered significant at a P value of less than 0.05.
Results
CD38 Deletion Enhances Mortality, Inflammatory Lung Injury, Lung Polymorphonuclear Leukocyte Accumulation and Generation of Proinflammatory Cytokines in Response to Endotoxin
Western blot analysis showed that CD38 protein expression was absent in Cd38−/− mouse lungs (Figure 1A). To determine whether CD38 plays a role in sepsis, we injected a lethal dose of LPS (20 mg/kg intraperitoneally) to WT and Cd38−/− mice and found that Cd38−/− mice exhibited significantly greater mortality after LPS challenge than WT mice (Figure 1B). We also used another model of sepsis involving CLP, in which Cd38−/− mice exhibited greater mortality to sepsis (Figure 1C) as well as increased bacteremia compared with WT mice (Figure 1D), indicating defective bacterial killing. Histopathological examination of lungs from WT and Cd38−/− mice challenged with LPS at indicated time-points up to 48 hours (Figure 1E) showed that Cd38−/− mice exhibited greater increase in MPO (reflecting augmented lung polymorphonuclear leukocyte [PMN] infiltration) (Figure 1F), lung transvascular albumin permeability (EBA) (Figure 1G), generation of inflammatory cytokines, IFN-γ, TNF-α, and IL-6 (Figures 1H–1J), and decreased generation of antiinflammatory cytokines, IL-4 and IL-10 (Figures 1K and 1L) at 6 and 48 hours after LPS.
Figure 1.

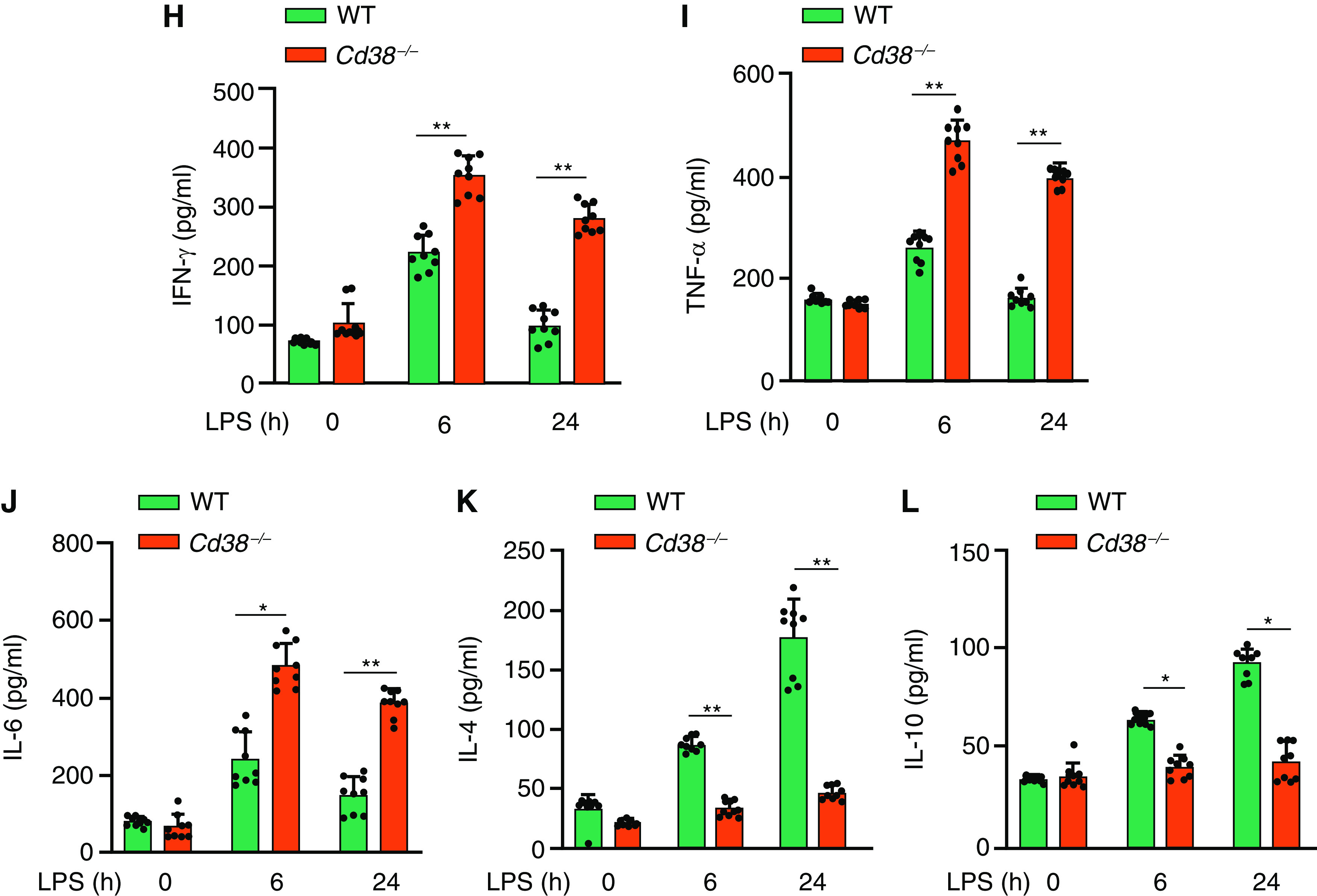
CD38 deletion in mice enhances mortality, inflammatory lung injury, lung polymorphonuclear leukocyte (PMN) accumulation, and generation of proinflammatory cytokines in response to endotoxin. (A) IB showing expression of CD38 in lung tissue of wild-type (WT) and Cd38−/− mice. (B) Survival of age- and weight-matched Cd38+/+ (WT) and Cd38−/− mice challenged with a lethal dose of LPS (intraperitoneally, 20 mg/kg). (C) Survival of age- and weight-matched Cd38+/+ and Cd38−/− mice challenged with cecal litigation and puncture (CLP). (D) Blood collected 24 hours after CLP from WT and Cd38−/− mice was cultured on blood agar, and number of colonies were counted to assess bacteremia. Bar graph represents the number of colonies forming unit per ml of blood in WT and Cd38−/− mice. n = 6 per genotype. **P < 0.001 WT versus Cd38−/− mice. (E) Hematoxylin and eosin staining of lung sections from WT and Cd38−/− mice challenged with LPS (5 mg/kg body weight, intraperitoneally) for 0, 6, and 48 hours. Scale bars, 100 μm. (F and G) WT and Cd38−/− mice challenged with LPS (10 mg/kg, intraperitoneally); lungs harvested at indicated time points were used for myeloperoxidase (MPO) activity to assess PMN influx (F) or lung transvascular albumin permeability (Evans blue dye conjugated with albumin [EBA] uptake) (G). n = 6 mice per treatment per group. (H–L) Concentrations of proinflammatory cytokines (TNF-α, IL-6, and IFN-γ) and antiinflammatory cytokines (IL-10 and IL-4) measured in BAL fluid of mice challenged with LPS (10 mg/kg, intraperitoneally). n = 8 mice per treatment per group. *P < 0.05 and **P < 0.01 versus WT mice. Br = bronchus; CFU = colony forming unit; v = vessel.
Myeloid Cell-expressed CD38 Negatively Regulates TLR4-mediated Lung Inflammation
To study whether CD38 deficiency in myeloid cells was responsible for the intensified lung inflammatory response described above, we lethally irradiated WT and Cd38−/− mice for subsequent myeloid cell transplantation. Cd38−/− cells were transplanted into irradiated WT mice. PCR results showed effective bone marrow reconstitution of Cd38−/− myeloid cells (Figure 2A). WT mice reconstituted with Cd38−/− myeloid cells exhibited mortality rates similar to non-Cd38−/− reconstituted control mice (Figure 2B vs. Figure 1B). Lung neutrophil sequestration in myeloid Cd38−/− reconstituted mice was greater than in control mice but similar to nonreconstituted control Cd38−/− mice (Figures 2C and 2D). Thus, CD38 signaling in myeloid cells functions to suppress TLR4-induced mortality and inflammatory lung injury.
Figure 2.
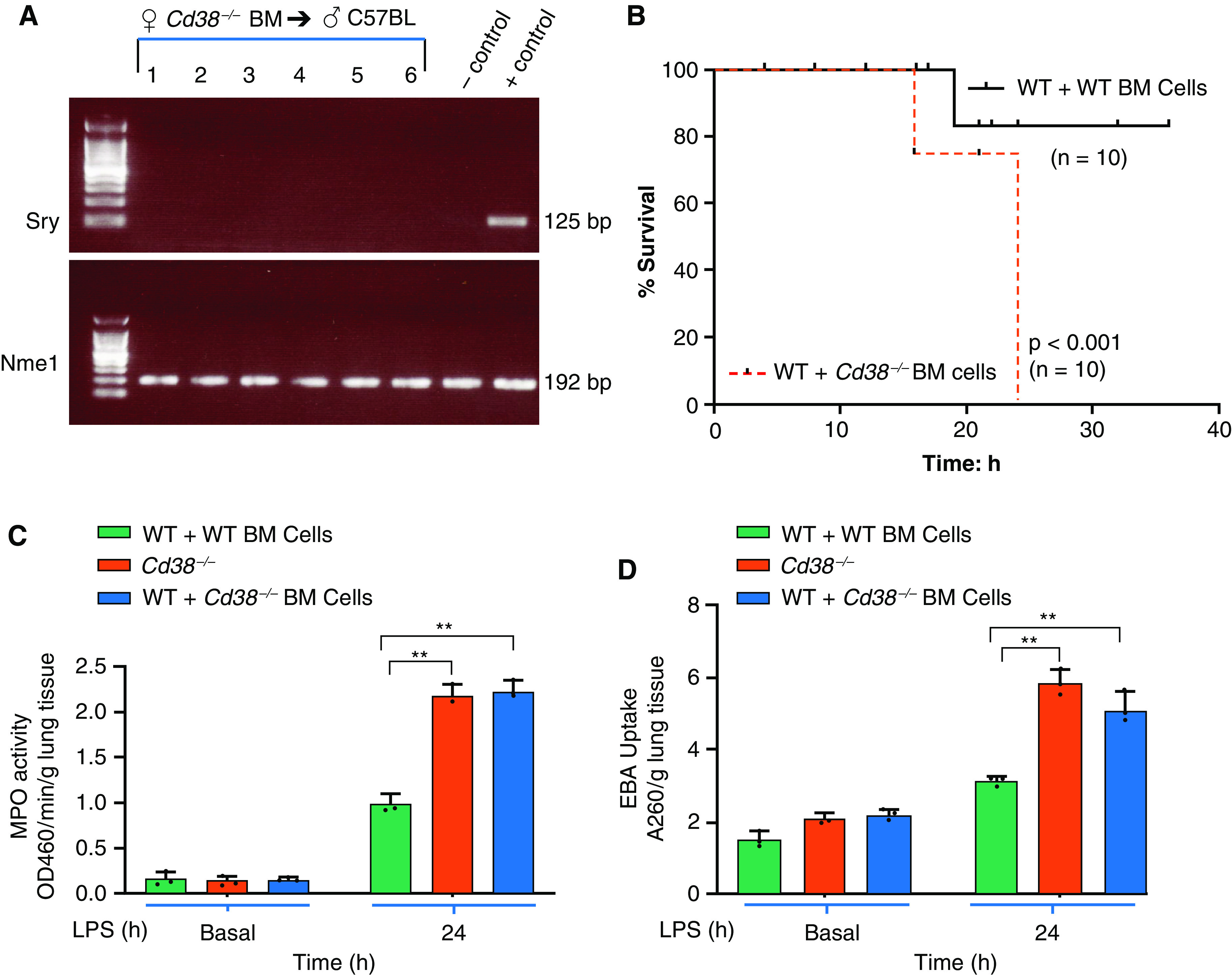
Myeloid cell-expressed CD38 negatively regulates TLR4-mediated lung inflammation. (A) At 3 weeks following bone marrow (BM) cells transplantation (female Cd38−/− BM → male WT mice or female Cd38+/+ BM → male WT mice). Recipient mouse blood collected by tail nick was used for DNA isolation and PCR. Sry gene (male specific) and autosomal gene Nme1 were determined to assess the reconstitution of BM cells. Left lane, DNA ladder markers; lanes 1–6, individual recipient samples used for PCR analysis. (B) Chimeric mice (6 weeks after transplantation) were challenged with a lethal dose of LPS (20 mg/kg, intraperitoneally), and mortality was monitored. (n = 10 mice per group). (C and D) Cd38−/− and chimeric (Cd38+/+ BM →WT and Cd38−/− BM →WT) mice were challenged with LPS (10 mg/kg, intraperitoneally), and the lungs harvested were used to measure MPO activity and EBA uptake (n = 6 mice per group). **P < 0.001 versus WT mice.
CD38 Deficiency Has No Effect on Macrophage ADPR Production and in Thereby Amplifying Inflammatory Lung Injury
CD38-mediated cADPR production gates the redox-sensitive nonselective cation channel, TRPM2, in macrophages (18, 19). TRPM2 activation in turn mediated Ca2 + influx in macrophages and protected lungs from endotoxin-induced injury in mice (19). Thus, we determined whether differences in CD38-catalyzed ADPR generation in macrophages (BMDMs) from control and Cd38−/− mice might explain the augmented inflammatory lung injury response seen in Cd38−/− mice. Since H2O2 generation downstream of TLR4 induced ADPR production in macrophages (19), we first measured basal as well as H2O2-induced ADPR release in BMDMs obtained from WT and Cd38−/− mice. We observed no significant difference in basal and H2O2-induced ADPR release in WT and Cd38−/− macrophages (Figure 3A). Furthermore, we showed no differences in ADPR release in BMDMs from WT and Trpm2−/− mice (Figure 3B). In addition, TRPM2-mediated Ca2 + influx was essentially similar in BMDMs of WT and Cd38−/− mice (Figure 3C). Thus, any difference in CD38-catalyzed products in macrophages are not likely responsible for the augmented TLR4-induced lung inflammatory injury in Cd38−/− mice.
Figure 3.

(A and B) CD38 deficiency has no effect on ADP-ribose production in macrophages and in amplifying inflammatory lung injury. BM-derived macrophages (BMDMs) from WT and Cd38−/− mice (A) or BMDMs from WT and Trpm2−/− mice (B) were used for H2O2 (300 μM)-induced ADP-ribose generation. (C) BMDMs from WT and Cd38−/− mice were used to measure H2O2 (300 μM)-induced Ca2 + entry. Results shown are representative of three separate experiments. ADPR = ADP ribose; FLIPR = fluorescent image plate reader; ns = not significant.
CD38 Deficiency in Macrophages Augments TLR4-induced Btk Activation to Amplify Inflammatory Lung Injury
Btk-deficient macrophages previously showed impaired polarization into the inflammatory phenotype while exhibiting enhanced immunosuppression in response to LPS (20). We thus determined whether CD38 mitigates TLR4 activation through a Btk-dependent mechanism in macrophages. We observed that LPS challenge caused time-dependent phosphorylation of Btk in BMDMs from WT within 5 minutes of LPS exposure whereas Btk phosphorylation was increased further in Cd38−/− BMDMs (Figure 4A). Btk differs from Src-related tyrosine kinases in its absence of a negative regulatory phosphorylation site (21). Src-mediated phosphorylation of Btk at Y551 enhanced the catalytic activity of Btk through autophosphorylation of Y223 (22). Thus, we determined the effects of Src inhibition on LPS-induced phosphorylation of Btk and observed that it prevented Btk phosphorylation in response to LPS (Figure 4B), indicating that Src lies upstream of Btk in the TLR4 signaling pathway in macrophages.
Figure 4.
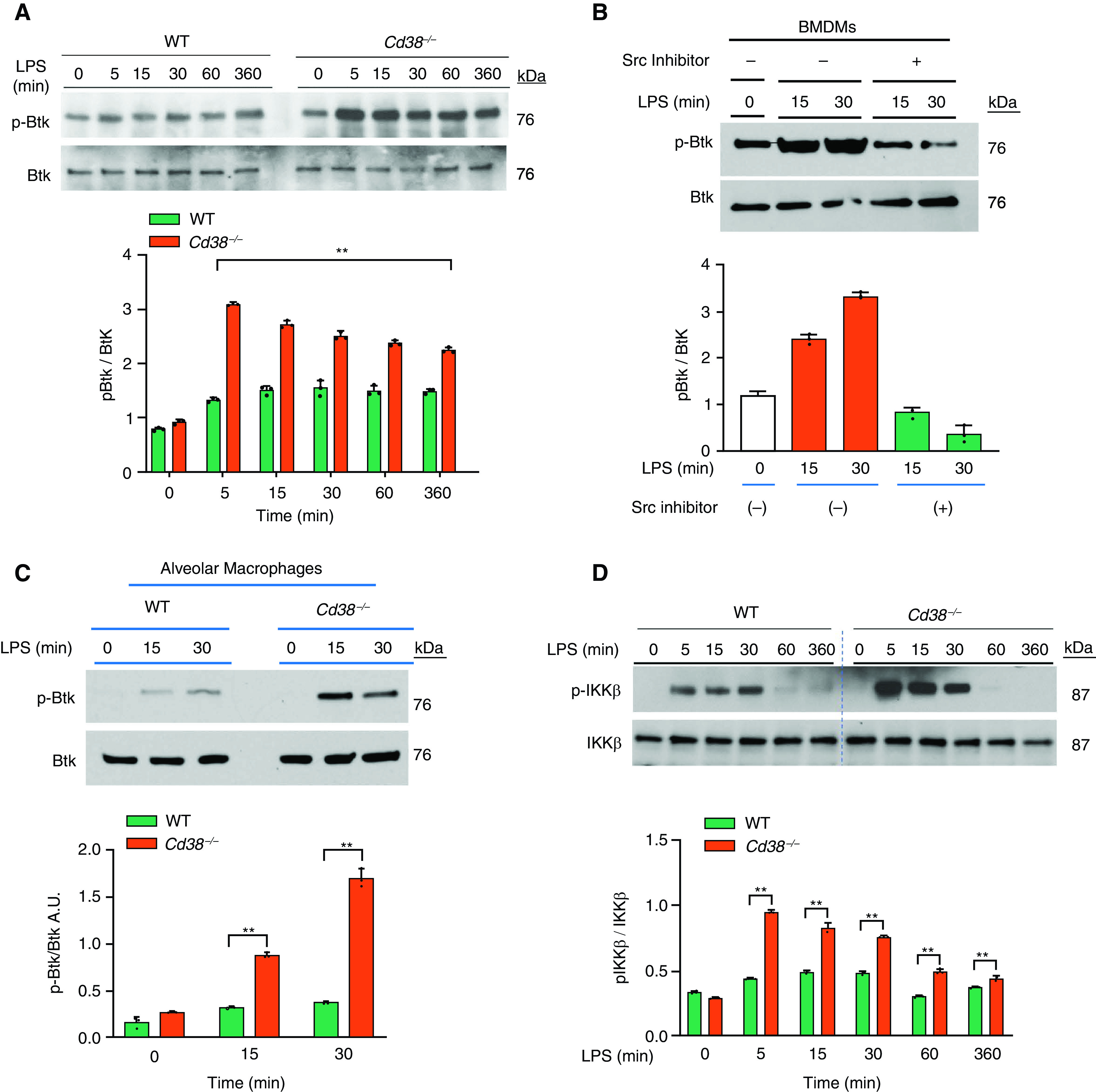
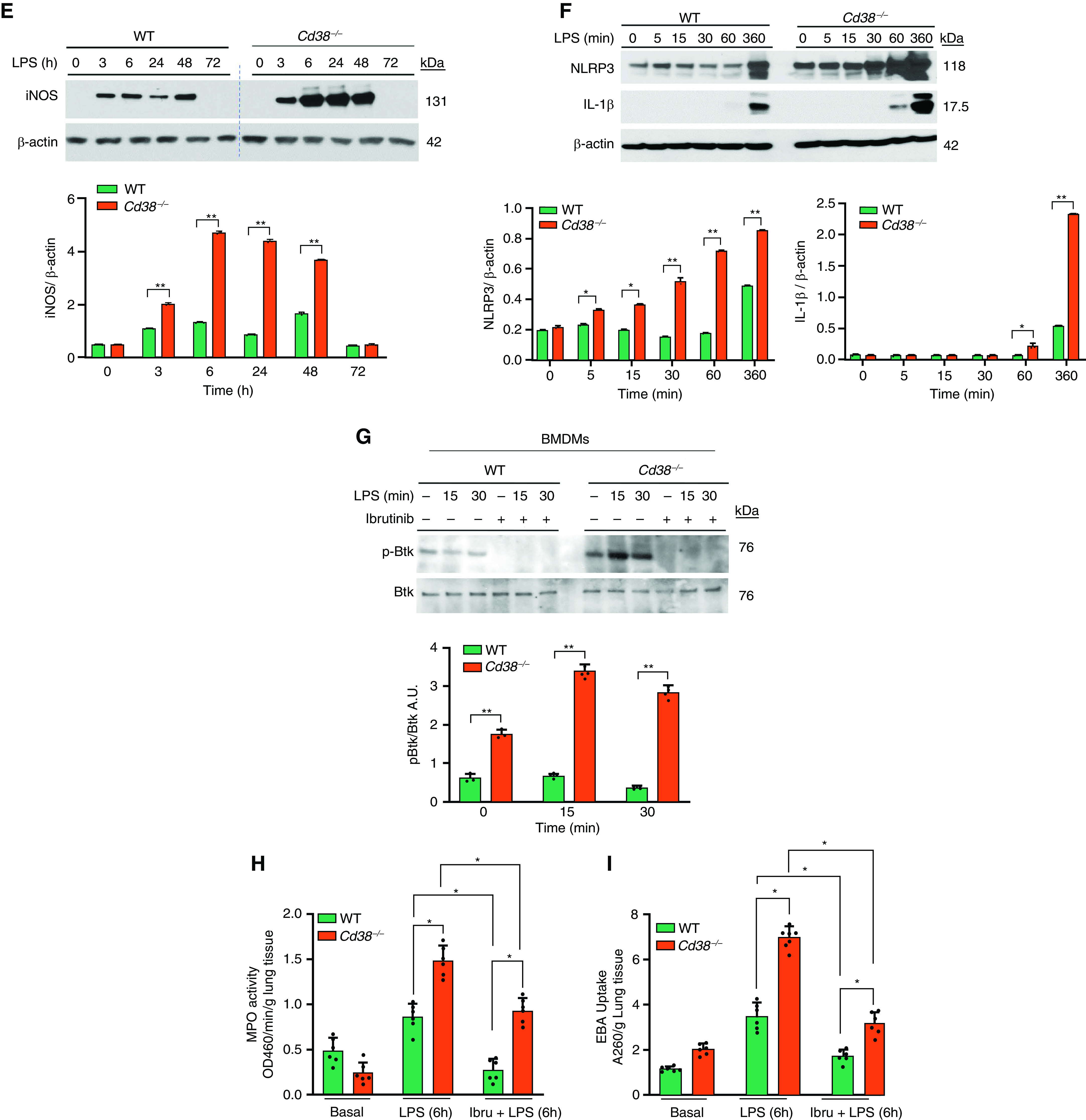
CD38 deficiency augments TLR4-induced Bruton’s tyrosine kinase (Btk) activation in macrophages as well as inflammatory lung injury. (A) IB analysis of LPS (1 μg/ml)-induced tyrosine phosphorylation of Btk (p-Btk) in BMDMs from WT and Cd38−/− mice. Below, quantification of results above, presented as the ratio of phosphorylated Btk to total Btk. (B) BMDMs from Cd38−/− mice were pretreated with Src inhibitor PP1 (10 μM) for 30 minutes and then LPS-induced tyrosine p-Btk as above. (C) IB analysis showing LPS-induced phosphorylation of Btk in alveolar macrophages from WT and Cd38−/− mice. (D) IB showing LPS-induced NF-κB activation measured by IκB kinase β (IKKβ) phosphorylation in BMDMs from WT and Cd38−/− mice. Below, quantification of results above, presented as the ratio of phosphorylated IKKβ to total IKKβ. (E) IB analysis of NF-κB target gene iNOS expression in BMDMs from WT and Cd38−/− mice in response to LPS. Below, quantification of results, presented as the ratio of iNOS to β-actin. (F) IB showing LPS-induced expression of NLRP3 and mature IL-1β in BMDMs of WT and Cd38−/− mice. Below, quantification of results, presented as the ratio of NLRP3 to β-actin or IL-1β to β-actin. (A–F) Data are means ± SEM of three independent experiments. (G) IB showing Btk inhibitor (ibrutinib; 1 μM, 30 min before treatment) prevents LPS-induced Btk phosphorylation in BMDMs. (H and I) Ibrutinib (6 mg/kg intraperitoneally) or dimethylsulfoxide (vehicle) was given daily for 7 days. Mice were challenged with LPS (10 mg/kg intraperitoneally) on the 8th day, and lungs harvested were used for MPO activity and EBA uptake. n = 6 mice per treatment per group. *P < 0.01 versus WT mice and **P < 0.001 versus Cd38−/− mice at LPS 6 hours A.U. = arbitrary unit; Ibru = ibrutinib; iNOS = inducible nitric oxide synthase.
In isolated alveolar macrophages from WT and Cd38−/− mice, we also observed that LPS-induced phosphorylation of Btk was augmented in macrophages from Cd38−/− mice as compared with WT mice (Figure 4C). We next determined LPS-induced NF-κB activation by measuring IκB kinase-β (IKKβ) phosphorylation and observed that NF-κB activation was augmented in BMDMs from Cd38−/− mice as compared with WT BMDMs (Figure 4D). Additionally, we observed time-dependent increased expression of the NF-κB target genes, iNOS and NLRP3, in BMDMs from Cd38−/− mice as compared with WT BMDMs (Figure 4E). Next, we showed that CD38 deficiency in macrophages interfered with NLRP3 inflammasome-mediated release of mature IL-1β. Furthermore, NLRP3-mediated IL-1β release in LPS-challenged macrophages was markedly increased in Cd38−/− BMDMs as compared with controls (Figure 4F). These findings together support the key role of CD38 in augmenting Btk activation downstream of TLR4 in macrophages.
To address whether the augmented Btk signaling was responsible for the augmented TLR4-induced ALI seen in Cd38−/− mice, we determined the effects of Btk inhibition on LPS-induced lung inflammatory response in WT and Cd38−/− mice. We observed that the Food and Drug Administration-approved Btk inhibitor, ibrutinib, blocked LPS-induced Btk activation in BMDMs from WT and Cd38−/− mice (Figure 4G).
Next, to address whether uncontrolled Btk activation in vivo contributed to the intensified lung inflammation in Cd38−/− mice, we treated both WT and Cd38−/− mice with the Btk inhibitor, ibrutinib (6 mg/kg intraperitoneally), daily for 7 days (23). Drug pretreatment prevented LPS-induced inflammatory lung injury in WT mice assessed by measuring PMN influx into lungs and pulmonary transvascular albumin permeability (EBA uptake) (Figures 4H and 4I). Interestingly, in Cd38−/− mice, LPS-induced PMN influx and permeability were substantially increased as compared with untreated controls (Figures 4H and 4I). These results together show the requirement of CD38 in preventing TLR4-induced inflammatory lung injury through suppression of Btk activation in macrophages.
CD38/SHP2 Complex Formation Inhibits TLR4-induced Btk Activation and Inflammatory Lung Injury
SHP2, encoded by PTPN11 gene (24), negatively regulates inflammatory signaling originating from TLR4 as well as NLRP3 inflammasome (25, 26). Since the cytosolic region of CD38 binds to the SH2 domain containing signaling molecules, which regulate cellular signaling in myeloid cells (8), we surmised that CD38 binding to the SH2 domain of SHP2 negatively regulates inflammatory responses in macrophages. Here we expressed Btk in HEK293 cells stably expressing TLR4 and measured LPS-induced phosphorylation of Btk and IκB kinase activation by measuring the phosphorylation of IκBα. We observed equal SHP2 expression in HEK293 cells expressing TLR4 in all groups (Figure 5A). However, LPS stimulation resulted in phosphorylation of Btk (Figure 5A) and IκBα (Figure 5B), whereas coexpression of CD38 with Btk abolished LPS-induced phosphorylation of Btk (Figure 5A) and IκBα (Figure 5B).
Figure 5.
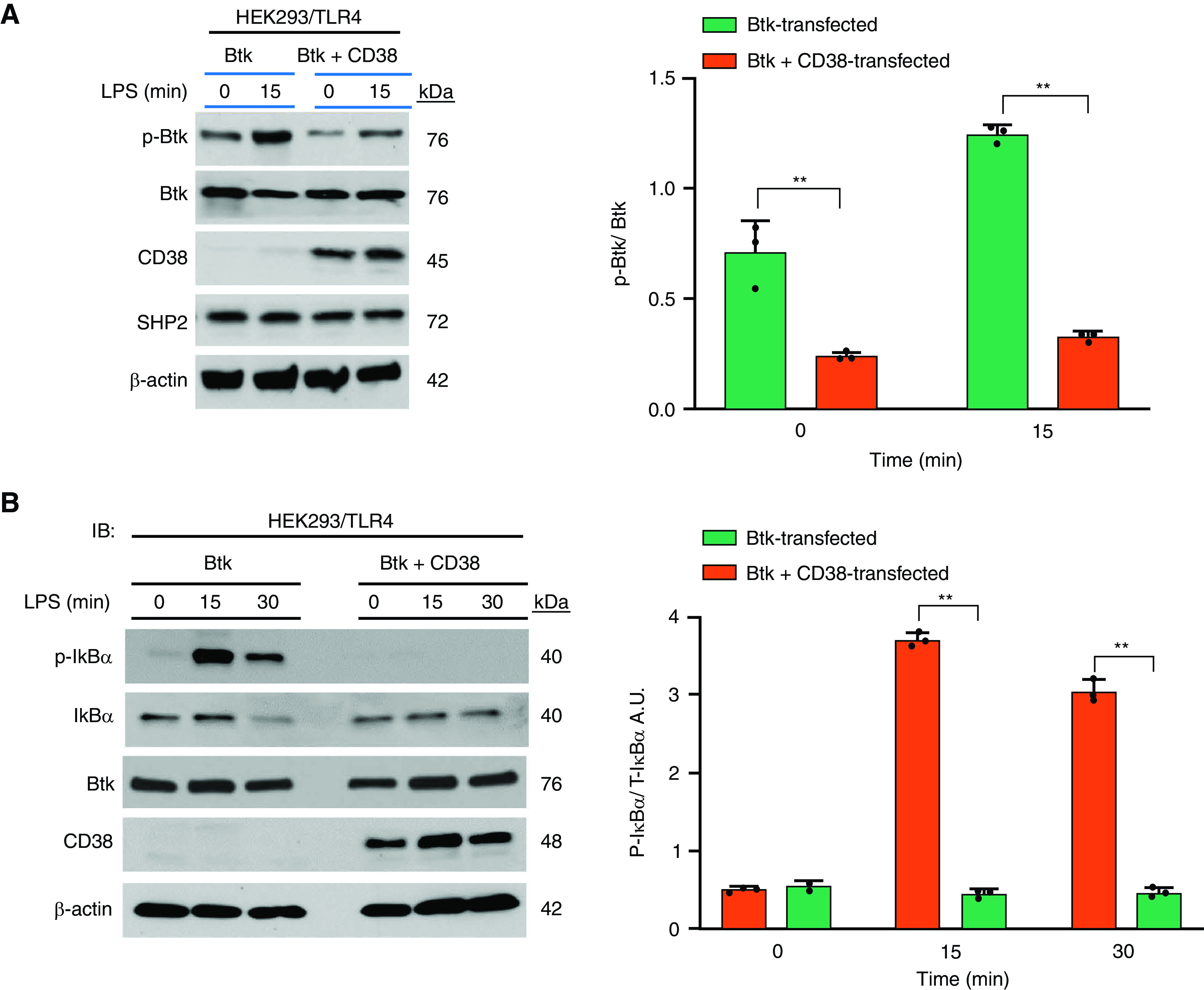

Formation of CD38 SHP2 signaling complex inhibits TLR4-induced Btk activation and inflammatory lung injury. (A and B) HEK293/TLR4 cells were transfected with Btk alone or Btk plus CD38. After 72 hours, cells were challenged with LPS for indicated time periods and used for IB analysis to determine phosphorylation of Btk (A) and IκBα (B). β-actin was used for loading control. Bar graph represents the quantified results. Data are means ± SEM of three independent experiments. P < 0.001, Btk versus Btk plus CD38 transfected. (C) HEK293/TLR4 cells were transfected with Btk alone or cotransfected with Btk plus CD38. After 72 hours of transfection, cells were lysed and IPed with GFP antibody (Btk C-terminal GFP-tagged). IPed samples were IBed for the expression of SHP2 and Btk (upper panel 2). The lower panels show CD38 and β-actin in total cell lysate. Below, quantified results, means ± SEM from three independent experiments showing SHP2 binding to Btk in the presence of CD38. **P < 0.001. (D) BMDMs and lung tissue (LT) from WT or Cd38−/− mice were used for IB analysis to determine SHP2 expression. LT, n = 4 mice per genotype; n = 4 mice per genotype used for the isolation of bone marrow cells. Data are means ± SEM. P < 0.001, WT versus Cd38−/− mice. (E) SHP2 mRNA expression in lungs of WT and Cd38−/− mice under basal condition. n = 8 mice per genotype. (F) BMDMs pretreated with or without MG-132 (10 μM) for 2 hours, and lysates were IPed using SHP2 antibody. The IPed samples were IBed with pan-antiubiquitin antibody. Data are representative of three independent experiments. (G) Role of macrophage-expressed CD38 in regulating TLR4-induced acute lung injury. In myeloid cells, LPS ligation of TLR4 activates Btk (27) through Src-mediated phosphorylation of Btk on Y551, which triggers IKK activation to promote expression of NF-κB target genes including inflammatory cytokines and NLRP3 protein (left). Furthermore, activated Btk physically associates with NLRP3 and phosphorylates NLRP3 and ASC (not shown) to promote caspase-1-mediated release of mature IL-1β (right). Production of proinflammatory cytokines and release of mature IL-1β contribute to the pathogenesis of acute lung injury. Under basal conditions, SHP2 interacts with plasma membrane localized CD38 in macrophages. However, TLR4 signaling induces translocation of SHP2/CD38 complex to Btk signalosome, where SHP2 dephosphorylates Btk to inactivate Btk and prevent uncontrolled release of inflammatory cytokines, NLRP3 activation, and inflammatory lung injury. ASC = apoptosis-associated speck-like protein containing a CARD; IKK = IκB kinase; NLRP3 = nod-like receptor family, pyrin domain containing 3; SHP2 = Src homology-2 domain containing protein tyrosine phosphatase 2; Ub = ubiquitin.
Next, we challenged with LPS control HEK293/TLR4 cells, HEK293/TLR4 cells expressing Btk, or HEK293/TLR4 cells expressing Btk plus CD38, and cell lysates were immunoprecipitated with Btk and blotted with SHP2 antibody to assess endogenous SHP2 binding to the Btk signaling complex. We observed LPS-induced minimal binding of SHP2 to Btk complex in HEK293/TLR4 expressing Btk (Figure 5C), whereas in HEK293/TLR4 expressing Btk plus CD38, the LPS-induced binding of SHP2 with Btk signaling complex was substantially increased (Figure 5C), indicating that CD38 promoted SHP2 binding to the Btk signaling complex and thus was crucial for SHP2-mediated dephosphorylation that inactivated Btk.
To address the functional significance of CD38/SHP2 signaling complex, we measured SHP2 expression in BMDMs and lung tissue from WT and Cd38−/− mice. We observed SHP2 protein expression was markedly reduced in Cd38−/− mice (Figure 5D), whereas SHP2 mRNA expression was similar in both genotypes (Figure 5E). To gain insights into the mechanism of CD38 stabilization of SHP2, we determined whether CD38 binding to SHP2 prevented SHP2 degradation via the proteasomal pathway. We thus measured ubiquitylation of SHP2 using BMDMs from WT and Cd38−/− mice and observed augmented ubiquitylation and degradation of SHP2 in BMDMs from Cd38−/− mice (Figure 5F). Thus, CD38 interacted with SHP2 to prevent ubiquitylation-mediated degradation of SHP2. Based on these findings, we propose a model (Figure 5G) showing a pivotal role of macrophage-expressed CD38 signaling in modulating inflammatory lung injury.
Discussion
Here we determined the role of the ectozyme, CD38, expressed in the macrophage plasmalemma in regulating the inflammatory function of macrophages and in thereby influencing the pathogenesis of inflammatory lung injury induced by endotoxemia. The results based on using both Cd38−/− macrophages obtained from Cd38−/− mice and mouse model of LPS-induced inflammatory lung injury demonstrated strikingly that CD38 functioned as a negative regulator of TLR4 signaling.
In these studies, TLR4 was activated by its natural ligand, LPS, known to induce inflammatory lung injury (9, 13). We used the knockout macrophages and mice to identify the novel role of CD38 in macrophages in regulating the inflammatory lung injury response to LPS. We observed surprisingly markedly augmented LPS-induced inflammatory lung injury in Cd38−/− mice as defined by multiple indices: increased neutrophil sequestration in lungs, increased leakiness of lung microvessels (indicating vascular endothelial injury), release of macrophage-generated proinflammatory cytokines (while simultaneously reduced generation of antiinflammatory cytokines), and markedly increased LPS-induced mortality in CD38-deleted mice.
We demonstrated that the significantly augmented proinflammatory response in CD38-deleted macrophages was the result of increased TLR4-induced Btk phosphorylation in macrophages. Phosphorylation of Btk in WT macrophages was evident within 5 minutes of LPS exposure and persisted over hours, whereas Btk phosphorylation was increased to an even greater degree in Cd38−/− macrophages. The magnitude of Btk activation appeared to be a crucial determinant of augmented inflammatory lung injury following deletion of CD38. Inhibition of Btk with ibrutinib (23) prevented the amplification of the inflammatory response in both CD38-deleted macrophages and mice, indicating the involvement of Btk in modulating or negatively regulating the response. These results are consistent with findings that Btk-deficient macrophages show impaired polarization into the inflammatory phenotype while exhibiting enhanced immunosuppression in response to LPS (20). Thus, our results showed that CD38 mitigated TLR4 activation through Btk phosphorylation in macrophages.
A key question is what the mechanism of Btk phosphorylation is. Our results shed light on the important relationship between Src kinase and Btk (22). Src-mediated phosphorylation of Btk at Y551 enhanced the catalytic activity of Btk through autophosphorylation of Y223 (22). On determining the effects of Src inhibition on LPS-induced phosphorylation of Btk, we observed inhibition of Btk phosphorylation in response to LPS, suggesting that Src activation occurring downstream of TLR4 signaling regulated Btk phosphorylation and thereby the activation state of macrophages. Furthermore, we observed that deletion of CD38 in macrophages had the opposite effect; that is, Btk phosphorylation was enhanced as compared with WT macrophages, indicating that CD38 regulation of Btk activation was responsible for the altered macrophage polarity and augmented inflammatory lung injury in mice.
It is known that TLR4/LPS-mediated inflammatory lung injury is dependent on NF-κB activation (28). Thus, we also addressed whether phospho-Btk modulated NF-κB activation in macrophages. Consistent with our hypothesized model, we observed that CD38 activation negatively regulated NF-κB activation. This was evident from the augmented activation of NF-κB in macrophages from Cd38−/− mice as compared with WT as well as time-dependent augmented increases in the expression of NF-κB target genes, iNOS and of NLRP3, in Cd38−/− macrophages as compared with WT. Importantly, CD38 deficiency in macrophages also enhanced the NLRP3-generated release of mature IL-1β, which is essential in the pathogenesis of inflammatory lung injury as recently shown by us (13). As lung inflammatory and macrophage responses were augmented with deletion of CD38, these findings support the key role of CD38 in suppressing Btk and NF-κB activation downstream of TLR4 signaling in macrophages. They also support the usefulness of blocking activated Btk in patients with acute respiratory distress syndrome as a potentially fructuous therapeutic approach.
CD38 was originally identified as an ectoenzyme that synthesized cADPR from NAD+ and hydrolyzed cADPR into ADPR (4). Both cADPR and ADPR are known to mobilize intracellular Ca2 + (5, 18, 19). However, the augmentation effect of CD38 deletion shown in the present studies could not be ascribed to this totemic catalytic function of CD38 in generating ADPR and cADPR (4) and the activation of the cation channel TRPM2 in macrophages (18, 19). We previously showed that TRPM2 activation by this mechanism mediated Ca2 + influx in macrophages and protected lungs from LPS-induced injury in mice (19). In the present study, we demonstrated that TRPM2-mediated Ca2 + influx was essentially the same in macrophages from WT and Cd38−/− mice. Thus, any differences in generation of the CD38-catalyzed products in macrophage cADPR and ADPR and enhanced Ca2 + signaling via the TRPM2 channel cannot explain the severe augmentation of TLR4-induced inflammatory lung injury seen in Cd38−/− mice. Since we observed that ADPR production was not different between these two genotypes, it is possible that in macrophages activation of poly-ADPR polymerases consumes NAD+ to produce poly-ADPR and thus ADPR is released from poly-ADPR hydrolysis by the poly-ADPR glycohydrolase (29).
Btk physically interacts with the NLRP3 inflammasome activation complex and is thereby believed to mediate caspase 1-dependent release of mature IL-β and cause inflammation (30, 31). Importantly, suppression of Btk expression by siRNA or inhibition of Btk rescued rodents from ALI induced by polymicrobial sepsis (32), hemorrhagic shock (33), and lethal influenza virus infection (34). Furthermore, it has been shown that treatment with Btk inhibitors attenuated inflammatory responses and improved lung function in hospitalized SARS-CoV2-infected patients (35). Therefore, to ascertain whether unchecked Btk activation in vivo was responsible for the intensified inflammatory lung injury seen in Cd38−/− mice, we treatedCd38−/− mice with the Btk inhibitor ibrutinib (6 mg/kg intraperitoneally) daily for 7 days (23). This regimen prevented LPS-induced inflammatory lung injury in WT mice assessed by measuring PMN influx and pulmonary transvascular albumin permeability (EBA uptake). Interestingly, LPS-induced PMN influx and permeability were substantially reduced in Cd38−/− mice treated with ibrutinib compared with untreated control (only LPS-challenged) Cd38−/− mice. These results further support our contention of the crucial requirement of CD38 in preventing TLR4-induced inflammatory lung injury through suppression of Btk activation in macrophages.
An important question arises regarding how CD38 negatively regulates Btk to suppress the activation of macrophages and inflammatory lung injury. Since CD38 may regulate cellular signaling through its cytosolic domain binding to SH2 domain-containing proteins (8), we investigated the possibility that CD38 in association with SH2 domain-containing signaling proteins functioned to dampen Btk activation in macrophages. The SH2 domain containing cytosolic protein tyrosine phosphatase SHP2 was shown to negatively regulate TLR signaling pathways (25, 26). Studies using macrophage-specific conditional SHP2 knockout mouse also showed that SHP2 was required to negatively regulate NLRP3-mediated release of mature IL-1β in macrophages (26). Thus, we investigated the possibility that CD38 negatively regulated Btk activation through binding to SHP2. We showed that CD38 coexpression with Btk prevented LPS-induced activation of Btk and NF-κB in cells stably expressing TLR4. We also showed that SHP2 binding to the Btk signaling complex upon ligation of TLR4 with LPS. When CD38 was coexpressed with Btk, SHP2 binding to Btk signaling complex was markedly increased upon TLR4 activation with LPS, indicating that CD38 is required for optimal functioning of SHP2. These findings are consistent with our observation of intensified LPS-induced Btk phosphorylation (activation) in macrophages from Cd38−/− mice.
Next, to address the functional relevance of CD38/SHP2 signaling complex in macrophages, we measured SHP2 protein expression and observed that SHP2 protein was nearly absent in Cd38−/− mice. The basis for this may be augmented ubiquitylation and degradation of SHP2 in macrophages from Cd38−/− mice as compared WT mice. These results together support the model that CD38 interacts with SHP2, which thereby prevents ubiquitylation-mediated degradation of SHP2 in macrophages.
Footnotes
Supported by National Institutes of Health grants, T32-HL007829, P01-HL077806, R01-HL122157, and R01-HL156965.
Author Contributions: J.F., C.T., and A.B.M. designed the studies. J.F., Y.T., A.M., M.M., and S.N. performed the experiments. J.F. and C.T. performed data analysis. J.F. prepared figures for the manuscript. C.T. and A.B.M. wrote the manuscript.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0272OC on October 27, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Lund FE, Cockayne DA, Randall TD, Solvason N, Schuber F, Howard MC. CD38: a new paradigm in lymphocyte activation and signal transduction. Immunol Rev . 1998;161:79–93. doi: 10.1111/j.1600-065x.1998.tb01573.x. [DOI] [PubMed] [Google Scholar]
- 2. Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, et al. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom . 2013;84:207–217. doi: 10.1002/cyto.b.21092. [DOI] [PubMed] [Google Scholar]
- 3. Vences-Catalán F, Santos-Argumedo L. CD38 through the life of a murine B lymphocyte. IUBMB Life . 2011;63:840–846. doi: 10.1002/iub.549. [DOI] [PubMed] [Google Scholar]
- 4. Hogan KA, Chini CCS, Chini EN. The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging, and metabolic diseases. Front Immunol . 2019;10:1187. doi: 10.3389/fimmu.2019.01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guse AH, da Silva CP, Berg I, Skapenko AL, Weber K, Heyer P, et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature . 1999;398:70–73. doi: 10.1038/18024. [DOI] [PubMed] [Google Scholar]
- 6. Berg I, Potter BV, Mayr GW, Guse AH. Nicotinic acid adenine dinucleotide phosphate (NAADP(+)) is an essential regulator of T-lymphocyte Ca(2+)-signaling. J Cell Biol . 2000;150:581–588. doi: 10.1083/jcb.150.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med . 2001;7:1209–1216. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 8. Cho YS, Han MK, Choi YB, Yun Y, Shin J, Kim UH. Direct interaction of the CD38 cytoplasmic tail and the Lck SH2 domain. Cd38 transduces T cell activation signals through associated Lck. J Biol Chem . 2000;275:1685–1690. doi: 10.1074/jbc.275.3.1685. [DOI] [PubMed] [Google Scholar]
- 9. Aggarwal NR, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol . 2014;306:L709–L725. doi: 10.1152/ajplung.00341.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nepal S, Tiruppathi C, Tsukasaki Y, Farahany J, Mittal M, Rehman J, et al. STAT6 induces expression of Gas6 in macrophages to clear apoptotic neutrophils and resolve inflammation. Proc Natl Acad Sci USA . 2019;116:16513–16518. doi: 10.1073/pnas.1821601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato I, Yamamoto Y, Fujimura M, Noguchi N, Takasawa S, Okamoto H. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J Biol Chem . 1999;274:1869–1872. doi: 10.1074/jbc.274.4.1869. [DOI] [PubMed] [Google Scholar]
- 12. Mittal M, Nepal S, Tsukasaki Y, Hecquet CM, Soni D, Rehman J, et al. Neutrophil activation of endothelial cell-expressed TRPM2 mediates transendothelial neutrophil migration and vascular injury. Circ Res . 2017;121:1081–1091. doi: 10.1161/CIRCRESAHA.117.311747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity . 2018;49:56–65.e4. doi: 10.1016/j.immuni.2018.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DebRoy A, Vogel SM, Soni D, Sundivakkam PC, Malik AB, Tiruppathi C. Cooperative signaling via transcription factors NF-κB and AP1/c-Fos mediates endothelial cell STIM1 expression and hyperpermeability in response to endotoxin. J Biol Chem . 2014;289:24188–24201. doi: 10.1074/jbc.M114.570051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc . 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao YY, Gao XP, Zhao YD, Mirza MK, Frey RS, Kalinichenko VV, et al. Endothelial cell-restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J Clin Invest . 2006;116:2333–2343. doi: 10.1172/JCI27154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soni D, Regmi SC, Wang DM, DebRoy A, Zhao YY, Vogel SM, et al. Pyk2 phosphorylation of VE-PTP downstream of STIM1-induced Ca2+ entry regulates disassembly of adherens junctions. Am J Physiol Lung Cell Mol Physiol . 2017;312:L1003–L1017. doi: 10.1152/ajplung.00008.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol . 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, et al. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol . 2011;13:29–34. doi: 10.1038/ni.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ní Gabhann J, Hams E, Smith S, Wynne C, Byrne JC, Brennan K, et al. Btk regulates macrophage polarization in response to lipopolysaccharide. PLoS One . 2014;9:e85834. doi: 10.1371/journal.pone.0085834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Desiderio S. Role of Btk in B cell development and signaling. Curr Opin Immunol . 1997;9:534–540. doi: 10.1016/s0952-7915(97)80107-0. [DOI] [PubMed] [Google Scholar]
- 22. Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science . 1996;271:822–825. doi: 10.1126/science.271.5250.822. [DOI] [PubMed] [Google Scholar]
- 23. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun . 2015;6:7360. doi: 10.1038/ncomms8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev . 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 25. An H, Zhao W, Hou J, Zhang Y, Xie Y, Zheng Y, et al. SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity . 2006;25:919–928. doi: 10.1016/j.immuni.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 26. Guo W, Liu W, Chen Z, Gu Y, Peng S, Shen L, et al. Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat Commun . 2017;8:2168. doi: 10.1038/s41467-017-02351-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem . 2003;278:26258–26264. doi: 10.1074/jbc.M301484200. [DOI] [PubMed] [Google Scholar]
- 28. Fan J, Ye RD, Malik AB. Transcriptional mechanisms of acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2001;281:L1037–L1050. doi: 10.1152/ajplung.2001.281.5.L1037. [DOI] [PubMed] [Google Scholar]
- 29. Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol . 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 30. Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front Immunol . 2017;8:1454. doi: 10.3389/fimmu.2017.01454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weber ANR, Bittner ZA, Shankar S, Liu X, Chang TH, Jin T, et al. Recent insights into the regulatory networks of NLRP3 inflammasome activation. J Cell Sci . 2020;133:jcs248344. doi: 10.1242/jcs.248344. [DOI] [PubMed] [Google Scholar]
- 32. Zhou P, Ma B, Xu S, Zhang S, Tang H, Zhu S, et al. Knockdown of Burton’s tyrosine kinase confers potent protection against sepsis-induced acute lung injury. Cell Biochem Biophys . 2014;70:1265–1275. doi: 10.1007/s12013-014-0050-1. [DOI] [PubMed] [Google Scholar]
- 33. Liu X, Zhang J, Han W, Wang Y, Liu Y, Zhang Y, et al. Inhibition of BTK protects lungs from trauma-hemorrhagic shock-induced injury in rats. Mol Med Rep . 2017;16:192–200. doi: 10.3892/mmr.2017.6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Florence JM, Krupa A, Booshehri LM, Davis SA, Matthay MA, Kurdowska AK. Inhibiting Bruton’s tyrosine kinase rescues mice from lethal influenza-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2018;315:L52–L58. doi: 10.1152/ajplung.00047.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roschewski M, Lionakis MS, Sharman JP, Roswarski J, Goy A, Monticelli MA, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol . 2020;5:eabd0110. doi: 10.1126/sciimmunol.abd0110. [DOI] [PMC free article] [PubMed] [Google Scholar]