

Abstract
Fluorescent antibodies are a workhorse of biomedical science, but fluorescence multiplexing has been notoriously difficult due to spectral overlap between fluorophores. We recently established proof-of-principal for fluorescence Multiplexing using Spectral Imaging and Combinatorics (MuSIC), which uses combinations of existing fluorophores to create unique spectral signatures for increased multiplexing. However, a method for labeling antibodies with MuSIC probes has not yet been developed. Here, we present a method for labeling antibodies with MuSIC probes. We conjugate a DBCO-Peg5-NHS ester linker to antibodies, a single stranded DNA “docking strand” to the linker, and finally, hybridize two MuSIC-compatible, fluorescently-labeled oligos to the docking strand. We validate the labeling protocol with spin-column purification and absorbance measurements. We demonstrate the approach using (i) Cy3, (ii) Tex615, and (iii) a Cy3-Tex615 combination as three different MuSIC probes attached to three separate batches of antibodies. We created single, double, and triple positive beads that are analogous to single cells by incubating MuSIC probe-labeled antibodies with protein A beads. Spectral flow cytometry experiments demonstrate that each MuSIC probe can be uniquely distinguished, and the fraction of beads in a mixture with different staining patterns are accurately inferred. The approach is general and might be more broadly applied to cell type profiling or tissue heterogeneity studies in clinical, biomedical, and drug discovery research.
Introduction
Ultraviolet-to-infrared fluorescence is a bedrock of experimental science, particularly the biomedical sciences. However, multiplexing—the simultaneous analysis of multiple fluorophores in a single sample, is severely limited by spectral overlap1–4, where excitation and/or emission spectra of fluorescent probes share broad wavelength domains. Spectral overlap limits most standard fluorescence assays to 2–4 readouts at a time. Yet, many applications would benefit from increased fluorescence multiplexing capabilities; one example is cancer. Tumor heterogeneity is multi-dimensional, including spatial variation in cell type, driver mutation profiles, protein expression, and oxygen/metabolic gradients5–10. As a result, there are hundreds of markers that have an impact on a tumor’s evolution, fitness, and drug sensitivity5,11,12.
Current sequencing methods can reach high levels of multiplexing and have been used in cancer diagnosis and prognosis13–15. Yet, the now somewhat standard biopsy- or homogenized tissue-based deep DNA or mRNA sequencing, and now increasingly single-cell sequencing16–18, largely do not allow for spatial resolution. However, some recent sequencing-based methods can provide spatial in situ data19–22. Sequential fluorescence in situ hybridization (seqFISH+) is capable of transcriptome-wide imaging in single cells but has challenges in scaling to large numbers of cells or large areas of tissue sections. Slide-seq, alternatively, made mRNA sequencing compatible with tissue section imaging over large spatial scales with ~10 um resolution23. Although powerful advances, such sequencing methods cannot yet fully capture the heterogeneity of tissue samples, which includes single and subcellular resolution and molecules other than mRNA (i.e., DNA, proteins, post-translational modifications, etc.). On the other hand, antibody-based imaging can access multiple molecule types at single and subcellular resolution while also spanning physiologically relevant length scales. Therefore, increased antibody multiplexing capabilities remain highly complementary to these sequencing-based methods.
There have been many recent advances for increased antibody-based multiplexing with single cell and subcellular spatial resolution, most of which use standard “filter-based” instrumentation that robustly allow imaging 2–4 fluorescence colors simultaneously. A widely adopted strategy is repeated rounds of staining, imaging, and bleaching of fluorophores24–27. By performing multiple cycles of 2–4 color imaging, these methods drastically increase fluorescent multiplexing capabilities (up to 60 analytes). Multiplexed fluorescence microscopy (MxIF) was the first but requires proprietary and expensive equipment / reagents24. Cyclic Immunofluorescence (CyCIF) is similar in principle but uses inexpensive reagents and standard equipment26,28. Similar to MxIF and CyCIF, Iterative indirect immunofluorescence imaging (4i) uses cycles of imaging but leverages fluorophore-conjugated secondary antibodies rather than fluorophore-conjugated primary antibodies as in the above techniques, allowing the use of “off-the-shelf” primary antibodies25. Another method that uses staining and bleaching cycles is co-detection by indexing (CODEX)27, but it differs from the above methods as it uses DNA-conjugated antibodies and sequencing-like methods to multiplex. While these cyclic methods have significantly expanded multiplexing capability, a primary limitation is the number of rounds of imaging that are possible before sample degradation begins to occur. Additionally, the length of time each round takes to complete, multiplied by the number of rounds, can make these methods excessively time-consuming.
Another way to achieve higher degrees of antibody multiplexing is by labeling antibodies with isotopically pure rare earth metals, such as in imaging mass cytometry (IMC)29 and multiplexed ion beam imaging (MIBI)30. IMC and MIBI can respectively image 32 and 40 analytes simultaneously from a tissue sample. The use of mass spectrometry for quantification makes these techniques easier to multiplex compared to ones that use fluorescence, as they are not limited by spectral overlap. However, these methods use a laser or ion beam to ablate the sample, destroying the sample and preventing further analysis or use, including cyclic methods as above. Additionally, the specialized equipment and reagents required for these techniques can be more expensive than standard fluorescence microscopes and antibodies, making them not as widely available.
The fluorescence-based techniques that were previously described use “filter-based” imaging that lumps emission wavelengths together and thus restricts multiplexing to 2–4 channels, but some have instead used spectral imaging that measures emission intensity with much finer wavelength resolution. Fluorescence emission follows the principle of linear superposition, meaning that the emission spectra of a mixture of fluorophores can be cast as a sum of contributions from individual probes using a matrix equation. Solving this matrix equation for the levels of individual probes, given the spectra of the mixture and each isolated probe, is called unmixing. These “hyperspectral” techniques have been used to image up to seven analytes simultaneously in tissue sections3,31–34. CLASI-FISH (combinatorial labeling and spectral imaging - fluorescence in situ hybridization), which builds upon traditional spectral imaging, classifies up to 15 microbe types using probe combinations35. One constraint of CLASI-FISH is that probes must be spatially segregated for demultiplexing. Spectrally resolved fluorescence lifetime imaging microscopy (sFLIM)36 combines spectral imaging with fluorescence lifetime information and can multiplex nine antibodies simultaneously.
We recently developed an approach called Multiplexing using Spectral Imaging and Combinatorics (MuSIC), which leverages currently available fluorophores along with the power of combinatorics to increase the number of available probes for simultaneous staining37. MuSIC probes are created using Förster resonance energy transfer (FRET)-producing fluorophore combinations, which results in a unique probe emission spectrum that is linearly independent from that of the individual fluorophores that make up the combination, enabling unmixing. Our previous work, based on simulation, suggested that MuSIC may increase simultaneous fluorescence multiplexing capabilities ~4–5 fold37. Proof-of-principal experimental studies that focused on a small range of excitation wavelength space have shown that nine MuSIC probes can be accurately unmixed, which should increase when the full range is used. Moreover, MuSIC is compatible with cyclic imaging methods, which would allow more analytes to be measured per cycle, increasing multiplexing capabilities even further. MuSIC differs from CLASI-FISH in that it is not limited by spatial segregation.
Methods to conjugate MuSIC probes to antibodies have not yet been developed. Our previous work showed that standard primary-amine-based conjugation of two fluorophores to the same antibody does not produce a high enough FRET efficiency to create robust MuSIC probes38. Here, we report a fluorescent oligo-based labeling approach to conjugate MuSIC probes to antibodies. A DBCO-Peg5-NHS ester molecule (the linker) is used to attach an azide modified oligo (the docking strand) to the antibody. Fluorescent oligos hybridized to the docking strand bring the fluorophores into FRET-compatible distances. Mixtures of antibody-conjugated MuSIC probes using (i) Cy3, (ii) Tex615, and (iii) a Cy3-Tex615 combination were analyzed and accurately unmixed using spectral flow cytometry as a proof-of-principle. These oligo-based MuSIC probes are compatible with the wide range of clinical, biomedical, and drug discovery applications that currently use fluorescent antibodies and spectral imaging.
Results
Probe design and labeling process.
A fundamental component of the Multiplexing using Spectral Imaging and Combinatorics (MuSIC) approach is that combinations of fluorophores exhibiting FRET create a unique emission spectrum that is linearly independent from the individual fluorophores in the combination. Thus, creating MuSIC probes on antibodies requires combinations of fluorophores to be stably associated with antibodies with spatial proximity sufficient for FRET. To achieve this, we started from a prior description of antibody-oligo labeling39 (Figure 1a). First, a DBCO (dibenzocyclooctyne)-PEG5-NHS ester molecule (referred to as the linker) is attached to the antibody. The NHS ester group at the end of the linker reacts with available NH2 groups on the surface of the antibody. From here, a 55 bp DNA oligo with a 5’ azide modification (referred to as the docking strand) is added to the complex. The azide reacts with the DBCO group of the linker via copper-free click chemistry, creating an antibody-linker-docking strand conjugate. The PEG5 group is included in the linker to increase the water solubility of the DBCO group and provide space between the antibody and the docking strand39. Finally, 20 bp oligos with 5’ or 3’ fluorophore modifications (referred to as the donor and acceptor strands, respectively) are added to the antibody-linker-docking strand conjugate solution. When the donor and acceptor strands hybridize to the docking strand, the two fluorophores are in close physical proximity to enable FRET. The final product of these reactions should be an antibody labeled with a MuSIC probe. An in-depth view of the linker and oligo complex is shown in Figure 1b.
Figure 1:
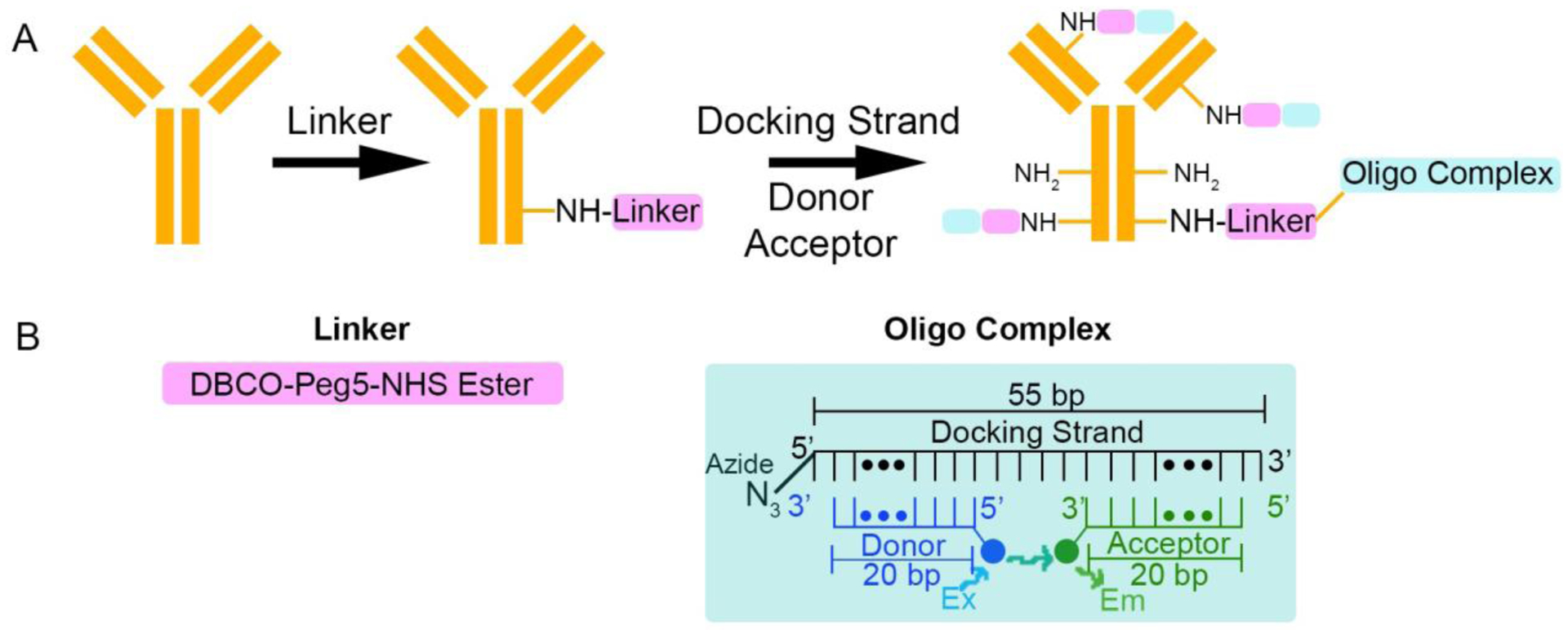
Labeling antibodies with oligo-based MuSIC probes. (A) Labeling schematic for MuSIC probes. First, the linker is added to the antibody by reacting the NHS ester on the linker with the NH2 group on the antibody. Then the docking strand is added and reacts with the linker via copper-free click chemistry. Lastly, the donor and acceptor strands are annealed to the docking strand to form the oligo complex. The linker can attach to the antibody at multiple NH2 sites, allowing an increased degree of labeling. (B) Detailed versions of the linker and the oligo complex
Attaching the linker to the antibody.
We developed the protocol around labeling 50 μg of IgG, although it is scalable in either direction. The linker is added to the antibody in 60 molar excess, as the linker will react with multiple free amine sites on the surface of the antibody, and the extent of reaction is not certain, but it is desired to maximize the degree of labeling. After incubation, unattached linker needs to be separated from the antibody-linker conjugate. To do this, we used Amicon Ultra 100 kDa molecular weight cut-off (MWCO) filters (Figure 2a). The antibody has a molecular weight of ~150 kDa and the linker has a molecular weight of 0.7 kDa, so once the solution is spun and washed, any linker that does not attach to the antibody will freely flow through the column (Figure 2b). In order to verify that all unattached linker was removed, retentate absorbances were measured at 309 nm, where the linker strongly absorbs39 (Fig. S1), for samples containing the antibody alone, the linker alone, and then antibody and linker together. Results show that the linker is predominantly in the retentate only when the antibody is present (Figure 2c). The degree of labeling was estimated to be ~9+/− 0.57 molecules of linker/antibody based on absorbance measurements (see Methods and Figure S1). These results demonstrate that the antibody and linker can stably associate and that unattached linker can be effectively removed from solution.
Figure 2:
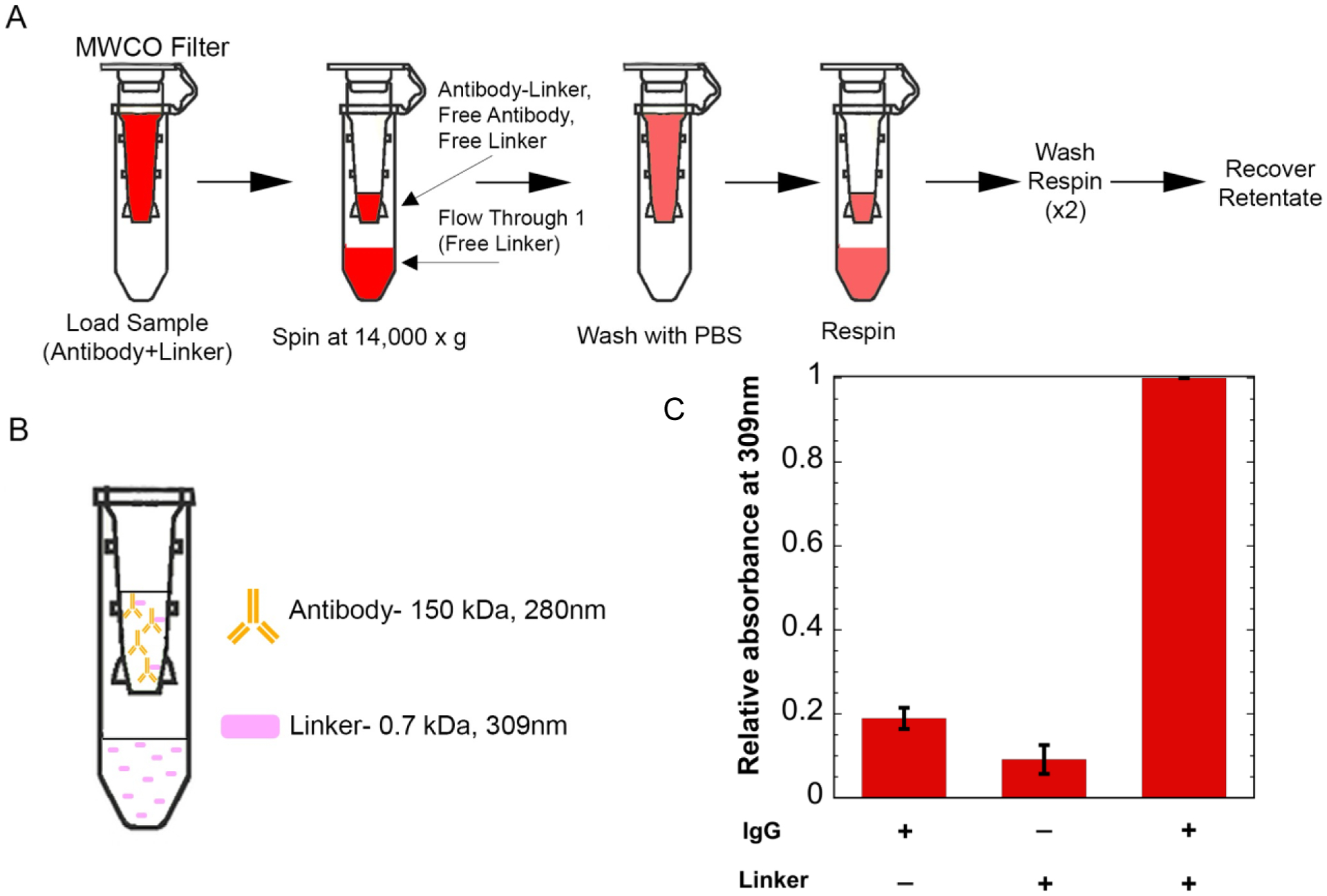
Adding linker to antibody. (A) Separating free linker using 100 kDa molecular weight cut-off (MWCO) filters. The antibody and linker are incubated, then the sample is added to the molecular weight cut-off filters. The filters are spun to separate unattached linker and then go through a series of washes. Finally, the retentate is recovered. (B) Expected separation of components after spin and wash steps. (C) Retentate absorbances at 309nm. Results show an increased signal at 309nm when the linker is in the presence of the antibody.
Attaching the docking strand to the antibody.
We added the docking strand to the antibody-linker retentate from the previous step in 6 molar excess to the antibody to account for multiple labeling sites. After incubation, unattached docking strand needs to be separated from the antibody-linker-docking strand conjugate. Similar to above, we use Amicon Ultra 100 kDa MWCO filters (Figure 3a). Since the docking strand is only 17 kDa, it should freely flow through the columns if it is not attached to the antibody-linker conjugate (Figure 3b). In order to evaluate whether unattached docking strand is removed, retentate absorbances were measured at 260 nm, as this is where the docking strand strongly absorbs. Results show that the docking strand can be seen in the retentate when in the presence of the antibody and the linker, as expected. However, a strong retentate signal was also seen for the docking strand when in the presence of only the antibody, without the linker (Figure 3c). The cause for the strong docking strand signal in the retentate without the linker present is unknown, but before proceeding, we wanted to understand whether the docking strand was stably bound to the antibody without the linker present or whether it could be removed with further washing via an orthogonal separation mechanism.
Figure 3:
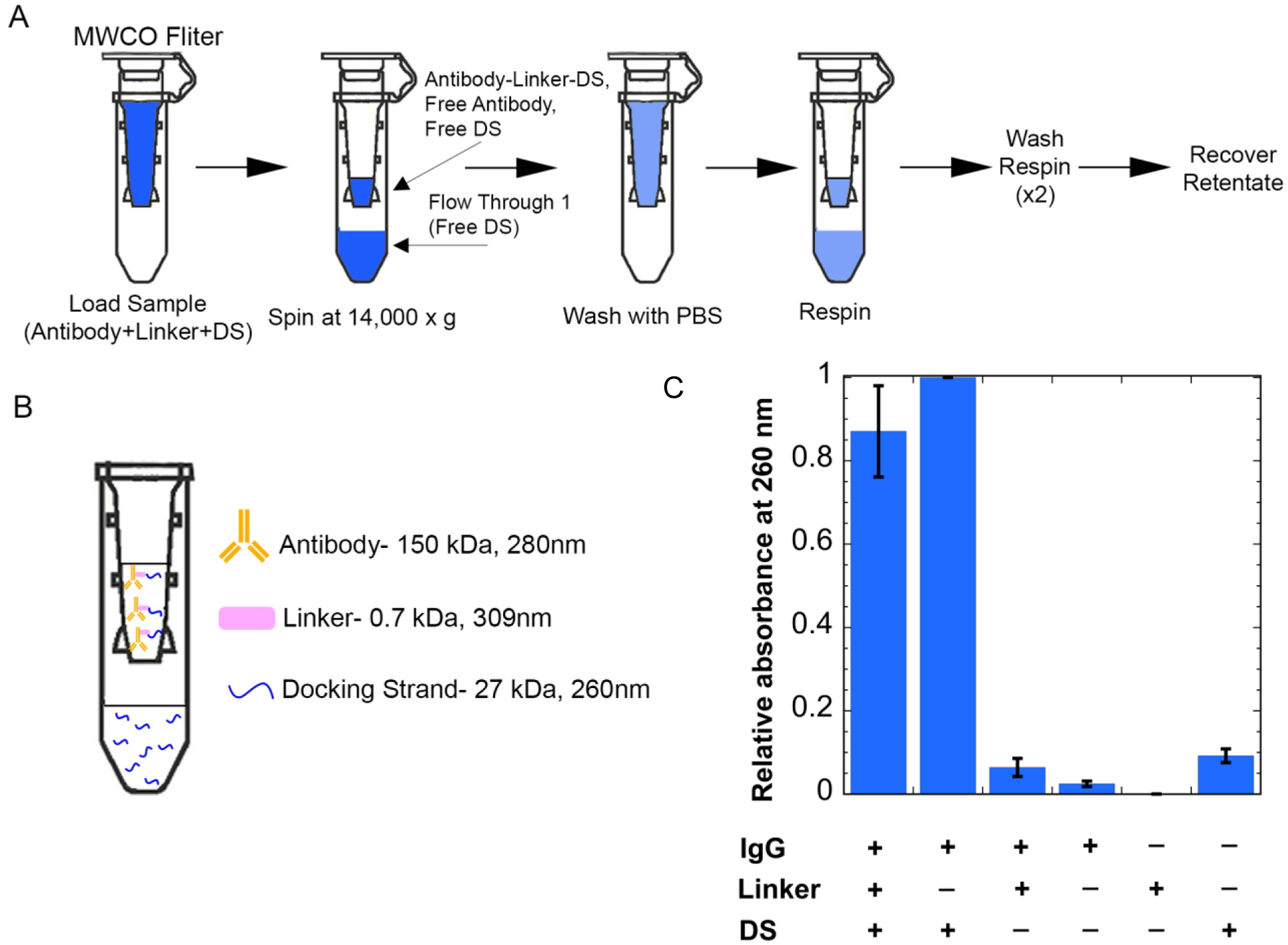
Adding docking strand (DS) to antibody-linker. (A) Separating free DS using 100 kDa MWCO centrifugal filters. The DS and antibody-linker conjugate are incubated, then the sample is added to the molecular weight cut-off filters. (B) Expected separation of components after spin and wash steps. (C) Retentate absorbances at 260nm. Results show an increased signal at 260nm when the DS is in the presence of the antibody-linker conjugate. An increased signal can also be seen for the case of just the DS and antibody, which is accounted for in later steps.
The docking strand requires the linker to be stably associated with the antibody.
To determine whether the docking strand could stably bind to the antibody without the linker, we used protein A dynabeads. The beads should strongly and selectively bind to the antibody, and anything attached to the antibody will also be bound to the beads. We generated samples with and without linker containing antibody, docking strand, and a donor strand with the fluorophore Atto 488 (for measurement). The supernatant containing any non-stably attached reagents can be removed by washing when the solution is placed on a magnet (Figure 4a). Atto 488 fluorescence was measured to evaluate whether the docking strand could stably associate with the antibody without the linker. The bead-based nature of the experiment precluded reliable absorbance assays as used previously; consequently, we are not able to estimate the degree of labeling for the docking strand on the antibody. The fluorescence signal for samples without the linker present was comparable to the signals of the controls where no fluorophore was present, while when the linker was present, a significant fluorescence signal was observed (Figure 4b). We conclude that the linker is needed for the antibody to be stably associated with the docking strand, and subsequently, fluorophore-labeled donor or acceptor strands.
Figure 4:
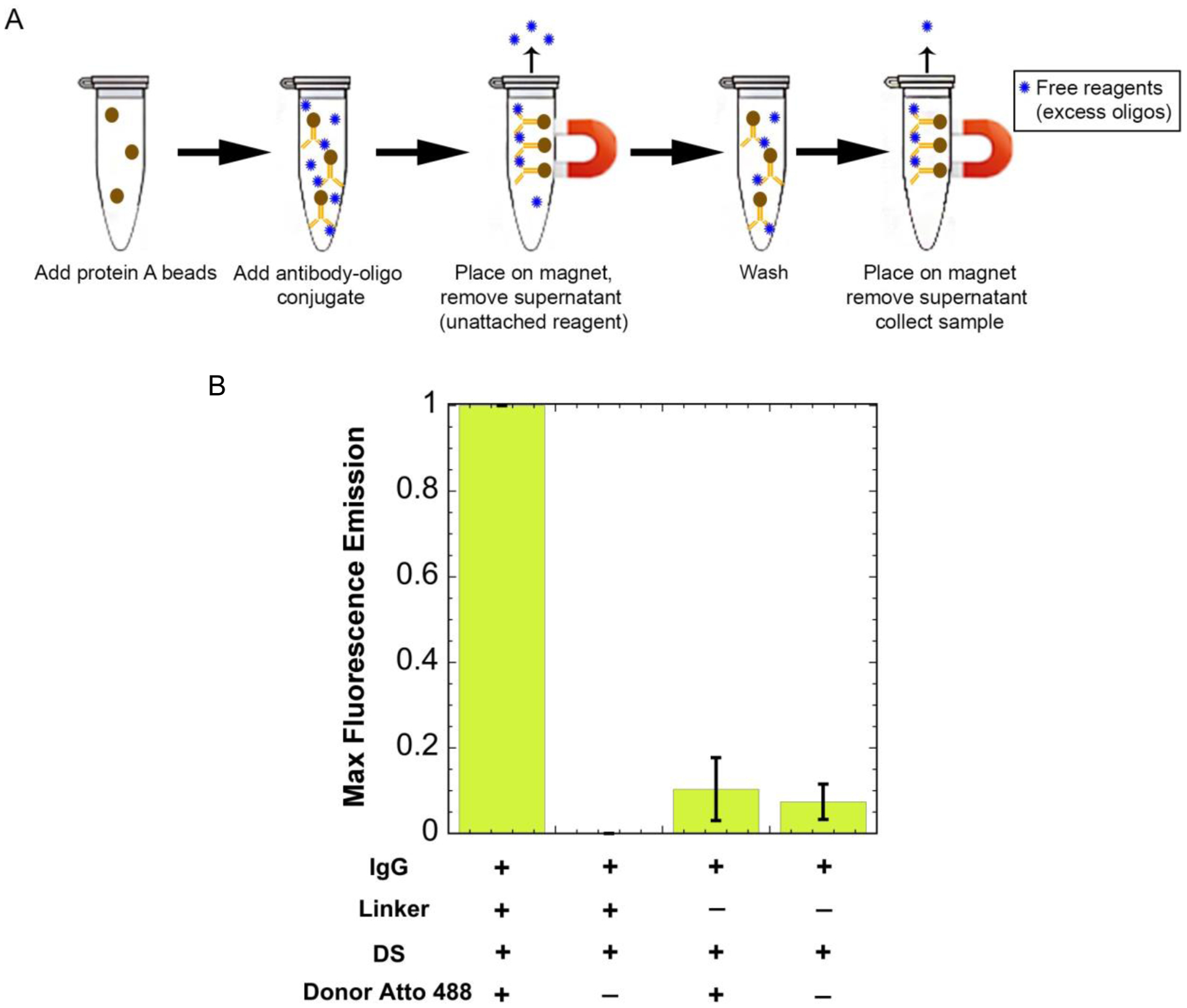
Separating free reagents using protein A beads. (A) The antibody-oligo conjugate (red Y with a blue circle attached) is added to the protein A beads (brown circle) and is incubated with rotation for 10 minutes. It is then placed on a magnet, pulling the beads out of solution, and the supernatant containing free reagents (unattached blue circles) is removed. The final product is collected containing the antibody-oligo conjugate. (B) Maximum fluorescence intensity values when excited at 450nm. Results show an increased fluorescence signal for the donor when the linker is added. Without the linker, the fluorescence signal for the donor is the same intensity as the background fluorescence.
Obtaining a donor and acceptor pair that produces FRET when co-hybridized to the docking strand.
As mentioned above, MuSIC probes must have donor and acceptor pairs that exhibit FRET, such that the combination probe has a unique spectral signature. To test if a donor and acceptor pair exhibits FRET, the emission spectra of solutions containing (i) just the donor, (ii) just the acceptor, (iii) the donor and the acceptor, and (iv) the donor and acceptor co-hybridized to the docking strand were analyzed using a plate reader (Figure 5a). We used 488 nm excitation, a common laser line in multiple assay types. The pair of Cy3 (donor) and Tex615 (acceptor) showed a much larger, red-shifted emission peak when excited at 488 nm and co-hybridized to the docking strand, as compared to the case without docking strand, indicating strong FRET (Figure 5a). These results show that this donor and acceptor pair would be a suitable MuSIC probe candidate, i.e., a donor and acceptor strand hybridized to the antibody-linker-docking strand conjugate.
Figure 5:
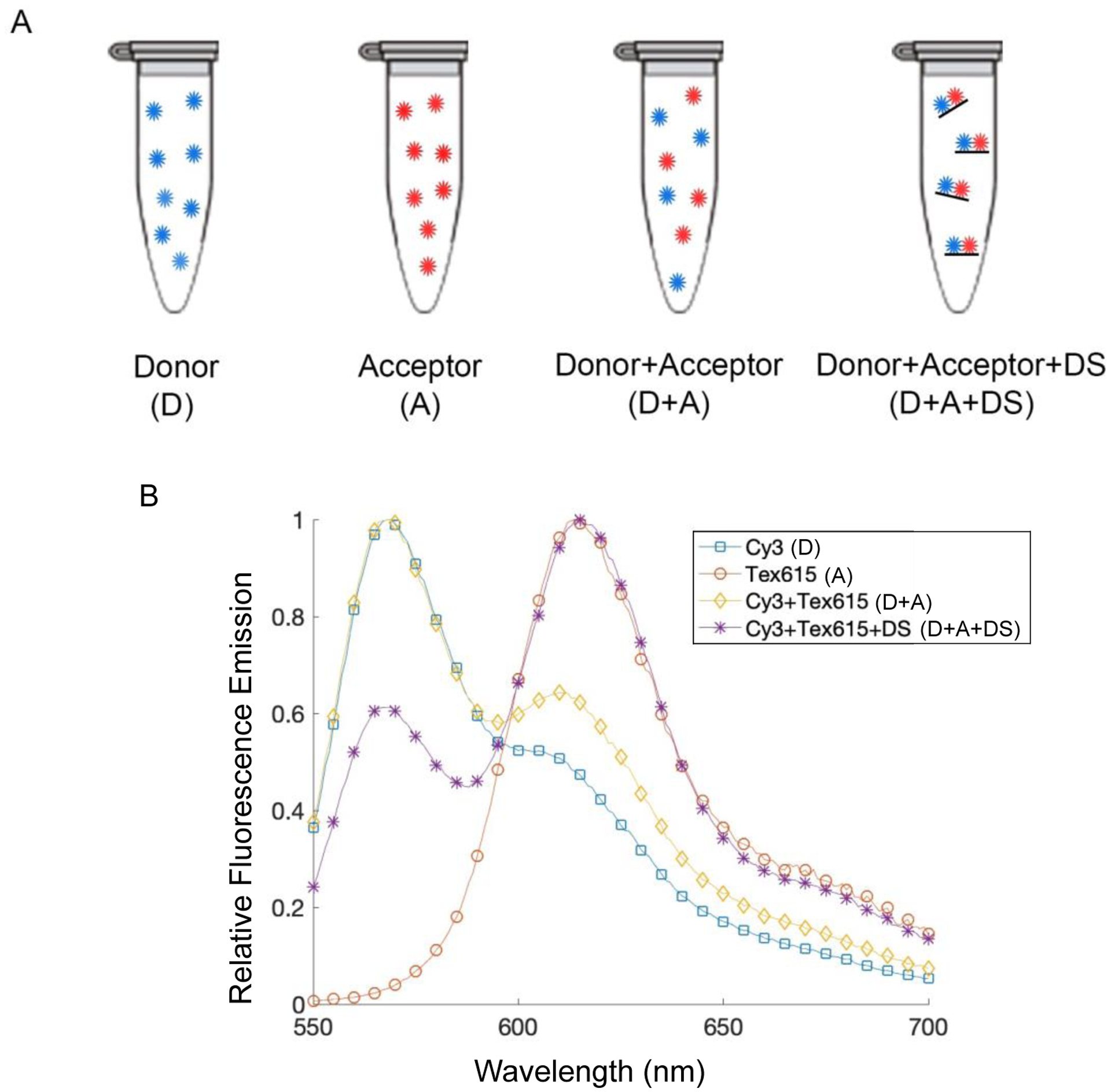
Donor and acceptor fluorophore pair, Cy3 and Tex615. (A) Experimental setup for testing fluorophore combination. The tubes contain the donor alone (blue circles), the acceptor alone (red circles), the donor and acceptor free in solution together, and then the donor and acceptor bound to the docking strand (black line). We expect only the sample with the DS shows significant FRET. DS: docking strand. (B) Fluorescence emission spectra when excited at 488 nm. An increased acceptor emission peak is seen when the donor and acceptor are annealed to the docking strand, indicating increased FRET.
Application to flow cytometry for event classification.
While there are many potential applications of MuSIC probe-labeled antibodies, we set out to obtain proof-of-principle data using spectral flow cytometry. Namely, we wanted to understand whether we could (i) robustly classify events as containing a particular combination of MuSIC probes and (ii) estimate the proportion of events having a particular probe staining pattern (Fig. 6a). This is analogous to cell type classification assays such as peripheral blood mononuclear cell (PBMC) analysis40,41. Three antibody batches with different probes were created: probe 1-donor Cy3 and acceptor Cy3; probe 2-donor Tex615 and acceptor 615; and probe 3-donor Cy3 and acceptor Tex615. Because Cy3 and Tex615 produce FRET when co-hybridized to the docking strand, probes with this combination of fluorophores can be thought of as a different “color” from the probes with the individual fluorophores of the combination. Once the antibodies with either MuSIC probe 1, 2, or 3 are created, they are incubated with protein A dynabeads to be analyzed using the flow cytometer. Each bead is similar to a single “cell.” One or more antibody type (i.e., with probes 1, 2, or 3) can be conjugated to the same set of beads. For example, incubating beads with two antibody types creates “double positive beads (cells).” In the following set of experiments, we made single positive beads (one antibody type conjugated to one bead set), double positive beads (two antibody types conjugated to one bead set), and triple positive beads (three antibody types conjugated to one bead set) (Figure 6a). This is related to (i) above. We also make mixtures of these different bead sets. This is related to (ii) above. For analysis, we use simple quadrant gates on bivariate plots to classify beads as negative, single positive, or double positive, and additionally, estimate the proportion of beads that fall into each category (Figure 6b). Populations of single positive beads are observed in R1 and R4, populations of double positive beads are observed in R2, and the negative population of beads is observed in R3. Triple positive bead classification is done by further gating on double positive populations.
Figure 6:
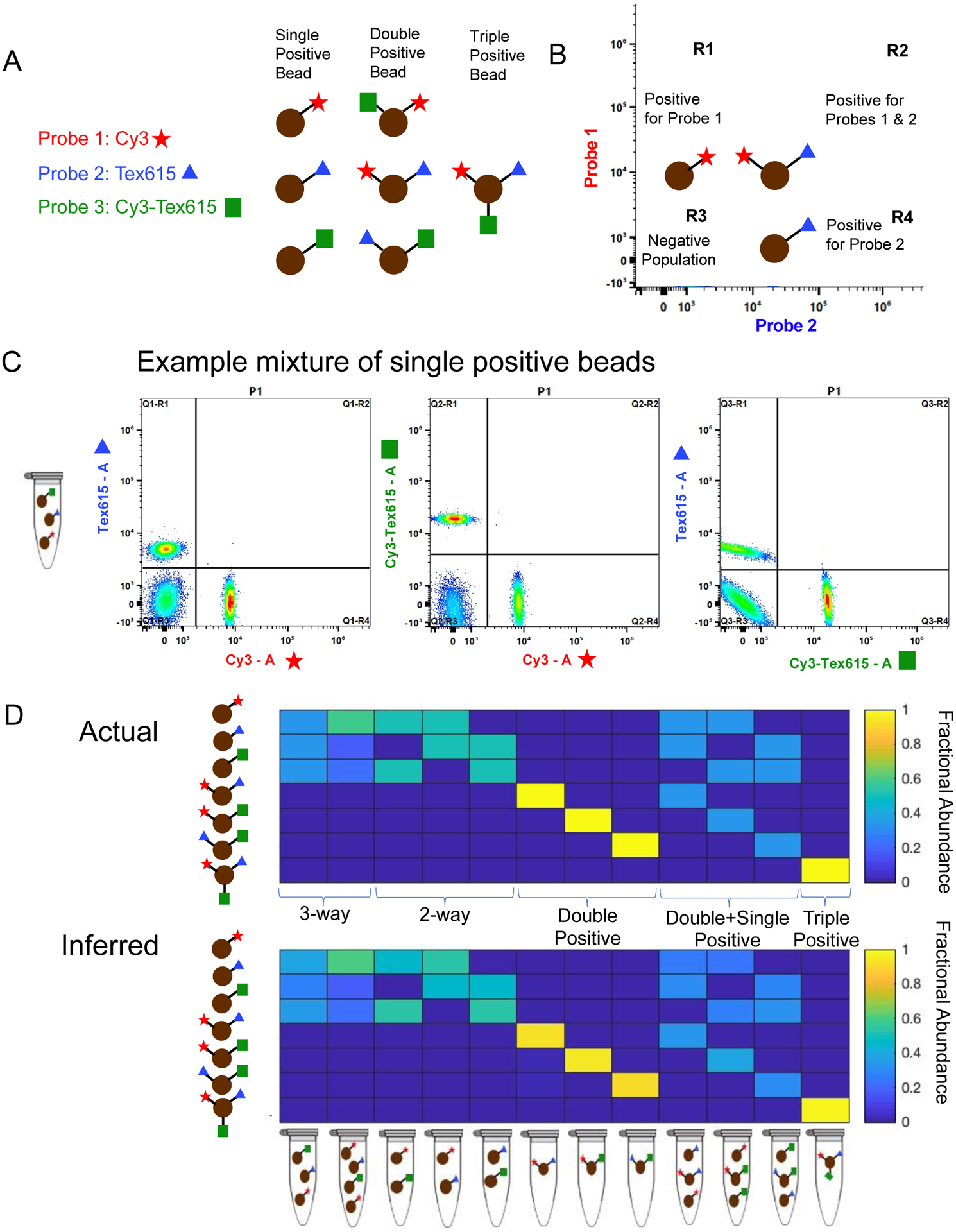
Spectral flow cytometry setup and results. (A) Experimental setup for the three-probe mixture ((1) Cy3 (red star), (2) Tex615 (blue triangle), and (3) Cy3-Tex615 (green square)) using single positive, double positive, and triple positive beads (brown circles). (B) Gating strategy for the populations of beads in the three-probe mixture. (C) Unmixing populations of single-labeled beads in a three-way equimolar mixture of probes Cy3, Tex615, and Cy3-Tex615 using spectral flow cytometry. The plots show unmixing results of Tex615 compared to Cy3 (left), Cy3-Tex615 compared to Cy3 (middle), and Tex615 compared to Cy3-Tex615 (right). (D) Comparing actual amounts of each probe in the mixture (top panel) to the inferred or calculated amounts of each probe in the mixture (bottom panel). The composition of each mixture is shown below the bottom panel.
First, we made an equal 3-way mixture from single positive bead sets and analyzed it by spectral flow cytometry. Unmixing results showed relatively equal amounts of each bead type in the mixture, demonstrating that single positives could be robustly classified (Figure 6c, 6d first column). We also tested a single positive mixture containing more probe 1 beads than probe 2 or 3 beads, and unmixing results showed relatively similar compositions compared to the known compositions (Figure 6d second column). We then investigated if various mixtures of single, double, and triple positive beads could be accurately unmixed (Figure 6d). Overall, results demonstrate robust classification of bead type, as well as accurate estimation of the relative abundance of each bead type. (Figure 6d-compare actual to inferred heatmaps). We conclude that MuSIC probe-labeled antibodies as generated here can be used in spectral flow cytometry applications for cell type classification and proportion estimation.
Discussion
Here we established a method to conjugate two fluorophores to an antibody in a way that enables FRET between them (if they are compatible). The use of combinations of fluorophores that exhibit FRET creates unique emission spectral signatures that can be used for multiplexing via the MuSIC approach. Antibodies are labeled with combinations of fluorophores by combining a “linker” and DNA oligos. The linker is used to covalently attach a “docking strand” oligo to the antibody. Separate “donor” and “acceptor” strands then hybridize to the docking strand. The donor and acceptor strand oligos place the fluorophores at a specified distance from one another on the antibody. Absorbance data suggested a degree of labeling of ~9+/− 0.57 linker/antibody molecules. We validated the approach using three different MuSIC probes (Cy3, Tex615, and a Cy3-Tex615 combination) attached to three separate mixtures of antibodies. MuSIC probe-labeled antibodies attached to protein A beads served as surrogate single, double, and triple positive cells for testing via spectral flow cytometry. Spectral flow cytometry experiments demonstrated that each MuSIC probe can be uniquely differentiated by accurately determining compositions of bead mixtures.
While the focus here was using MuSIC probe labeled antibodies with spectral flow cytometry, they are also compatible in principle with spectral imaging. Several methods that increase image multiplexing capabilities use a stain/strip technique, which involves cycles of staining, imaging, and bleaching24,26,28,42. These methods have improved multiplexing abilities by ~10 fold over standard single-round 4-color imaging. The use of MuSIC probes is in principle compatible with the cyclic methods, which would expand the number of probes that can be used per round of imaging using spectral scanning microscopes. Current cyclic methods on average use 10 rounds of four-color imaging and our previous simulation studies suggested that ~25 MuSIC probes might be accurately unmixed37. Therefore, the use of MuSIC probes may allow 10 rounds of 25 color imaging, thus increasing multiplexing capabilities by roughly another six-fold. However, spectral emission scanning microscopes are certainly not as pervasive and filter-based microscopes currently. Angle-tuned emission filters for wavelength scanning may help to make such technology more accessible43. Such microscopes also commonly have white light lasers for tunable excitation wavelengths and a potentially large number of channels, which would further empower multiplexing capabilities via MuSIC approaches.
To further increase fluorescent multiplexing capabilities using the MuSIC approach, additional combinations of fluorophores are needed. The FRET efficiency of a fluorophore combination is dependent on the physical distance between the two fluorophores based on the Förster radius, which is dependent on the spectral properties of the pair. Some fluorophore pairs may require different distances between the two fluorophores in order to optimize FRET efficiency. This distance between the fluorophores can be varied by using different length docking strands which have varying numbers of spacer base pairs—the nucleotides in the middle between the donor and acceptor strand binding sites. Thus, we expect future solutions will use different length docking strands for different fluorophore combinations in the march towards a larger palate of antibody-compatible MuSIC probes. Additionally, in this paper, we demonstrated unmixing of MuSIC probes using a two-laser spectral flow cytometer (488nm and 638 nm). The number of useful MuSIC probe combinations can be further increased by using a spectral flow cytometer with five excitation lasers (355 nm, 405 nm, 488nm, 561 nm, and 638nm—Cytek Aurora). We are currently screening large sets of fluorescent oligos for MuSIC-probe suitability for a 3-laser and 5-laser setup.
While MuSIC probes may be useful for multiple flow cytometry applications, one of which in particular is immune profiling44–46. Flow cytometry-based immune profiling has limited multiplexing to roughly a dozen analytes (depending on the capabilities of the instrument) as a result of spectral overlap47,48. Mass cytometry has been transformative for immune profiling49–51, but is slower than flow cytometry and is destructive, so it prevents further use of the cells after analysis47. The use of MuSIC probes for immune profiling via flow cytometry may allow for increased multiplexing for deep immune profiling on par with mass cytometry while also being fast (more than 10,000 cells/second rather than about 1,000 as with mass cytometry52) and non-destructive. This could open up avenues of increased throughput for monitoring immune responses across large patient cohorts, as well as the isolation of rare cell types alone or in specified combinations that would otherwise not be possible.
We conclude that oligo-based approaches are a robust and modular way to create MuSIC probe-labeled antibodies. Future work needs to expand the MuSIC probe palette, as well as expand to larger antibody panels for flow cytometry or other spectral fluorescence applications. This would enable broader applications for advancing our understanding of microbial communities53 such as gut and skin microbiomes54,55, cancer research and clinical diagnostics, host-pathogen interactions, developmental biology, and many other areas of life science research where more highly multiplexed single and sub-cellular resolution of antibody-target readouts is informative.
Methods
Adding the linker to the antibody
This and the below procedures were developed around labeling 50 μg of IgG but are compatible with scaling up or down. In our case, normal Rabbit IgG (ThermoFisher Cat: 31235) is combined with DBCO-Peg5-NHS Ester (linker; 10 mM in DMSO; Click Chemistry Tools Cat: 1378531-80-6) in 20 molar excess (50 μg of Rabbit IgG and 4.6 μg of linker). This is brought to a volume of 100μl with PBS and allowed to incubate for 30 minutes at 25°C. After incubation, the solution is added to an Amicon Ultra 0.5ml 100 kDa centrifugal filter (Fisher Scientific Cat: UFC5100BK) and spun for 5 minutes at 14,000 × g. The filter is then placed into a new tube, and PBS is added to the top of the filter in order to bring the total volume back to 100 μl and is spun again for 5 minutes at 14,000 × g. This wash step is repeated twice more (three total). Finally, the filter is flipped upside down and placed in a clean tube and spun for 1 minute at 1000 × g to collect the retentate. The retentate absorbance is measured at 309nm, where the linker strongly absorbs, and 280nm, where the antibody strongly absorbs, using a NanoDrop spectrophotometer (Thermo Scientific).
Adding the docking strand to the antibody-linker conjugate
The docking strand (Integrated DNA technologies-Table 1) is added to the antibody-linker retentate from the previous step in 6 molar excess to the original amount of antibody (2 nmoles of docking strand). The volume is brought up to 100 μl with PBS and incubated at 4°C overnight. The sample is then placed in an Amicon Ultra 0.5ml 100 kDa centrifugal filter and spun for 5 minutes at 14,000 × g. Once this spin is completed, the filter is placed into a new tube, and PBS is added to the top of the filter in order to bring the total volume back to 100 μl and is spun again for 5 minutes at 14,000 × g. This wash step is repeated twice. Finally, the filter is flipped upside down and placed in a clean tube and spun for 1 minute at 1000 × g to collect the retentate. The retentate absorbance is measured at 309nm and 280nm as above, and also at 260nm, where the docking strand strongly absorbs light, using a NanoDrop spectrophotometer (Thermo Scientific).
Table 1:
Oligo Sequences
| Component | Sequence |
|---|---|
| Docking Strand | 5’ – Azide - GTG TAG TTC AGG TCA AGA CAT CGT GCG ACC AGT CAG CAT GAG ACT CAT TGG TGC G -3’ |
| Donor Strand | 3’- C AAG TCC AGT TCT GTA GCA C - Fluorophore- 5’ |
| Acceptor Strand | 3’ - Fluorophore - CA GTC GTA CTC TGA GTA AC – 5’ |
Degree of Labeling
To generate calibration curves for concentrations of the antibody, linker, and docking strand, absorbance measurements were taken using a NanoDrop spectrophotometer (Thermo Scientific) for known concentrations of the antibody, linker, and docking strand at 309, 280, and 260 nm. Five-point, 2-fold serial dilutions were used to generate samples for the calibration curve. A least-squares line of best fit (MATLAB) is generated to estimate absorbance extinction coefficients based on Beer’s law (1) for each component at 309, 280, and 260nm.
| (1) |
Here, A is the absorbance of the solution, ε is the extinction coefficient, L is the length of the path traveled by light (1 mm), and c is the concentration of the solution. From here, a system of three simultaneous equations are solved in Matlab using the function vpasolve to estimate molar concentrations of the antibody (a), linker (l), and docking strand (DS) given absorbance measurements at 260, 280, and 309 nm from a mixture (M).
| (2) |
| (3) |
| (4) |
The degree of labeling for the linker to antibody could be calculated from the above-estimated concentrations. However, due to the nature of the spin column-based separation, some unreacted linker will remain. This amount of residual linker can be calculated based on mole balance, and we used this calculation to correct the degree of labeling as follows, where n is the number of washes, cl0 is the initial concentration of the linker before washing, Vr is the volume of the retentate (μl) after washes, Vw is the wash volume (μl), and Vrf is the volume of the final retentate (μl).
The concentration of the residual linker is subtracted from the calculated linker concentration to determine the concentration of the linker that is attached to the antibody in the retentate. The degree of labeling is then calculated as the ratio of this adjusted linker concentration to that of the antibody concentration.
Adding the donor and acceptor strands to the antibody-linker-docking strand conjugate
A 20 bp oligo with a 5’ fluorophore modification (donor strand) and a 20 bp oligo with a 3’ fluorophore modification (acceptor strand) (each 100 μM in water, Integrated DNA technologies) are added in equimolar amounts (2 nmoles each) to the antibody-linker-docking strand retentate and brought up to 100 μl with PBS. Sequences are shown in Table 1. This solution is allowed to incubate for 15 minutes at 25°C in the dark. When testing the necessity of the linker, the donor strand with an Atto 488 modification was added to the antibody-linker-docking strand retentate. To make the different probes, Probe 1 consists of equimolar amounts of the donor strand with a Cy3 modification and the acceptor strand with a Cy3 modification (each 2 nmoles), Probe 2 consists of equimolar amounts of the donor strand with a Tex615 modification and the acceptor strand with a Tex615 modification (each 2 nmoles), and Probe 3 consists of equimolar amounts of the donor strand with a Cy3 modification and the acceptor strand with a Tex615 modification (each 2 nmoles).
Choosing donor and acceptor pairs
To test the donor and acceptor fluorophore pair of Cy3 and Tex615, four samples are created: (1) donor strand with a 5’ Cy3 modification and acceptor strand with a 3’ Tex615 modification (each 100 μM in water) are added in equimolar amounts (0.2 nmoles), (2) The donor, acceptor, and docking strands are added in equimolar amounts (0.2 nmoles), (3) 0.2 nmoles of the donor strand, (4) 0.2 nmoles of the acceptor strand. All samples are brought to 50 μl with PBS. The samples (oligos in solution) are loaded into a black 96 well plate (Fisher Scientific Cat: 655900), and fluorescence emission spectra are assayed with a Synergy MX microplate reader (Biotek). Parameters are set to a slit width of 9nm, a 10-second shake prior to reading, taking readings from the top, and an excitation wavelength of 488 nm. The emission start ranges are 50nm greater than the excitation wavelength.
Incubating labeled antibodies with protein A dynabeads
The MuSIC-probe labeled antibodies from above were suspended in 200 μl of 0.02% (2 μl/10ml) Tween 20 (Fisher Scientific Cat: 9005-64-5) in PBS and added to 50 μl of protein A dynabeads (Fisher Scientific Cat: 10 001 D—33 μg of initially added IgG; 100 μg batch makes three incubations). For making double positive beads, both probes are simultaneously added to 50 μl of protein A dynabeads. This solution is allowed to incubate for 10 minutes with rotation in the dark. After incubation, the solution is placed on a magnet, the supernatant is removed, and the bead-antibody complex is resuspended in 200 μl PBS with 0.02% Tween-20 (Fisher Scientific Cat: BP337–100). The solution is then placed back on the magnet, and the supernatant is again removed and is resuspended in PBS.
Analyzing probe mixtures using Cytek Aurora flow cytometer
Mixtures of bead-conjugated probes are analyzed using a Cytek Aurora spectral flow cytometer with 488nm and 638nm lasers. First, beads with single probes are assayed as reference controls. The events to record is set to 5,000, the stopping time is set to 10,000 sec, and the stopping volume is set to 3,000 μl. For samples containing mixtures of bead types or double-positive beads, the events to record are set to 15,000, the stopping time is set to 10,000 sec, and the stopping volume is set to 3,000 μl. Once mixtures have been analyzed, the SpectroFlo software (Cytek) is used to first gate single beads with forward and side scatter, and then to unmix and report (i) the amount of each probe on every bead that was analyzed and (ii) the fraction of each bead type in each mixture of bead types.
Supplementary Material
Acknowledgments:
We thank Allon Klein at HMS Systems Biology for helpful discussion related to oligo-based labeling and Chloe Worthy for help with illustrations.
Funding Information:
MRB received funding from Clemson University and the NIH/NCI Grant R21CA196418. MEM received funding from the Department of Education Grant P200A180076.
References
- (1).Zimmermann T Spectral Imaging and Linear Unmixing in Light Microscopy. In Microscopy Techniques: −/−; Rietdorf J, Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2005; pp 245–265. 10.1007/b102216. [DOI] [PubMed] [Google Scholar]
- (2).Provenzano Paolo P.; Rueden Curtis T.; Trier Steven M.; Yan Long; Ponik Suzanne M.; Inman David R.; Keely Patricia J.; Eliceiri Kevin W.. Nonlinear Optical Imaging and Spectral-Lifetime Computational Analysis of Endogenous and Exogenous Fluorophores in Breast Cancer. J. Biomed. Opt 2008, 13 (3), 1–11. 10.1117/1.2940365. [DOI] [PubMed] [Google Scholar]
- (3).Tsurui H; Nishimura H; Hattori S; Hirose S; Okumura K; Shirai T Seven-Color Fluorescence Imaging of Tissue Samples Based on Fourier Spectroscopy and Singular Value Decomposition. J. Histochem. Cytochem 2000, 48 (5), 653–662. 10.1177/002215540004800509. [DOI] [PubMed] [Google Scholar]
- (4).Yang Chenying; Hou Vivian W.; Nelson Leonard Y.; Seibel Eric J.. Mitigating Fluorescence Spectral Overlap in Wide-Field Endoscopic Imaging. J. Biomed. Opt 2013, 18 (8), 1–14. 10.1117/1.JBO.18.8.086012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Graf JF; Zavodszky MI Characterizing the Heterogeneity of Tumor Tissues from Spatially Resolved Molecular Measures. PLoS ONE 2017, 12 (11), 1–20. 10.1371/journal.pone.0188878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Alizadeh AA; Aranda V; Bardelli A; Blanpain C; Bock C; Borowski C; Caldas C; Califano A; Doherty M; Elsner M; et al. Toward Understanding and Exploiting Tumor Heterogeneity. Nat. Med 2015, 21, 846–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).O’Connor JPB; Rose CJ; Waterton JC; Carano RAD; Parker GJM; Jackson A Imaging Intratumor Heterogeneity: Role in Therapy Response, Resistance, and Clinical Outcome. Clin. Cancer Res 2015, 21 (2), 249–257. 10.1158/1078-0432.CCR-14-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sottoriva A; Spiteri I; Piccirillo SGM; Touloumis A; Collins VP; Marioni JC; Curtis C; Watts C; Tavaré S Intratumor Heterogeneity in Human Glioblastoma Reflects Cancer Evolutionary Dynamics. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (10), 4009–4014. 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gerlinger M; Rowan AJ; Horswell S; Math M; Larkin J; Endesfelder D; Gronroos E; Martinez P; Matthews N; Stewart A; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med 2012, 366 (10), 883–892. 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Landau DA; Carter SL; Stojanov P; McKenna A; Stevenson K; Lawrence MS; Sougnez C; Stewart C; Sivachenko A; Wang L; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152 (4), 714–726. 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Turashvili G; Brogi E Tumor Heterogeneity in Breast Cancer. Front. Med 2017, 4, 227. 10.3389/fmed.2017.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Esposito A; Criscitiello C; Locatelli M; Milano M; Curigliano G Liquid Biopsies for Solid Tumors: Understanding Tumor Heterogeneity and Real Time Monitoring of Early Resistance to Targeted Therapies. Pharmacol. Ther 2016, 157, 120–124. 10.1016/j.pharmthera.2015.11.007. [DOI] [PubMed] [Google Scholar]
- (13).Meldrum C; Doyle MA; Tothill RW Next-Generation Sequencing for Cancer Diagnostics: A Practical Perspective. Clin. Biochem. Rev 2011, 32 (4), 177–195. [PMC free article] [PubMed] [Google Scholar]
- (14).Bagger FO; Probst V Single Cell Sequencing in Cancer Diagnostics. In Single-cell Sequencing and Methylation: Methods and Clinical Applications; Yu B, Zhang J, Zeng Y, Li L, Wang X, Eds.; Springer Singapore: Singapore, 2020; pp 175–193. 10.1007/978-981-15-4494-1_15. [DOI] [PubMed] [Google Scholar]
- (15).Müllauer L Next Generation Sequencing: Clinical Applications in Solid Tumours. Memo - Mag. Eur. Med. Oncol 2017, 10 (4), 244–247. 10.1007/s12254-017-0361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Alemany A; Florescu M; Baron CS; Peterson-Maduro J; van Oudenaarden A Whole-Organism Clone Tracing Using Single-Cell Sequencing. Nature 2018, 556 (7699), 108–112. 10.1038/nature25969. [DOI] [PubMed] [Google Scholar]
- (17).Wen W; Su W; Tang H; Le W; Zhang X; Zheng Y; Liu X; Xie L; Li J; Ye J; et al. Immune Cell Profiling of COVID-19 Patients in the Recovery Stageby Single-Cell Sequencing. Cell Discov. 2020, 6 (1), 31. 10.1038/s41421-020-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gomes T; Teichmann SA; Talavera-López C Immunology Driven by Large-Scale Single-Cell Sequencing. Trends Immunol. 2019, 40 (11), 1011–1021. 10.1016/j.it.2019.09.004. [DOI] [PubMed] [Google Scholar]
- (19).Mader S; Pantel K Liquid Biopsy: Current Status and Future Perspectives. Oncol. Res. Treat 2017, 40 (7–8), 404–408. 10.1159/000478018. [DOI] [PubMed] [Google Scholar]
- (20).Chen M; Zhao H Next-Generation Sequencing in Liquid Biopsy: Cancer Screening and Early Detection. Hum. Genomics 2019, 13 (1), 34. 10.1186/s40246-019-0220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Iwahashi N; Sakai K; Noguchi T; Yahata T; Matsukawa H; Toujima S; Nishio K; Ino K Liquid Biopsy-Based Comprehensive Gene Mutation Profiling for Gynecological Cancer Using CAncer Personalized Profiling by Deep Sequencing. Sci. Rep 2019, 9 (1), 10426. 10.1038/s41598-019-47030-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Eng C-HL; Lawson M; Zhu Q; Dries R; Koulena N; Takei Y; Yun J; Cronin C; Karp C; Yuan G-C; Cai L Transcriptome-Scale Super-Resolved Imaging in Tissues by RNA SeqFISH+. Nature 2019, 568 (7751), 235–239. 10.1038/s41586-019-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rodriques SG; Stickels RR; Goeva A; Martin CA; Murray E; Vanderburg CR; Welch J; Chen LM; Chen F; Macosko EZ Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363 (6434), 1463–1467. 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gerdes MJ; Sevinsky CJ; Sood A; Adak S; Bello MO; Bordwell A; Can A; Corwin A; Dinn S; Filkins RJ; et al. Highly Multiplexed Single-Cell Analysis of Formalin-Fixed, Paraffin-Embedded Cancer Tissue. Proc. Natl. Acad. Sci 2013, 110 (29), 11982–11987. 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gut G; Herrmann MD; Pelkmans L Multiplexed Protein Maps Link Subcellular Organization to Cellular States. Science 2018, 361 (6401), eaar7042–eaar7042. 10.1126/science.aar7042. [DOI] [PubMed] [Google Scholar]
- (26).Lin JR; Fallahi-Sichani M; Chen JY; Sorger PK Cyclic Immunofluorescence (CycIF), A Highly Multiplexed Method for Single-Cell Imaging. Curr. Protoc. Chem. Biol 2016, 8 (4), 251–264. 10.1002/cpch.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Goltsev Y; Samusik N; Kennedy-Darling J; Bhate S; Hale M; Vazquez G; Black S; Nolan GP Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174 (4), 968–981.e15. 10.1016/j.cell.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lin JR; Izar B; Wang S; Yapp C; Mei S; Shah PM; Santagata S; Sorger PK Highly Multiplexed Immunofluorescence Imaging of Human Tissues and Tumors Using T-CyCIF and Conventional Optical Microscopes. eLife 2018, 7, 1–46. 10.7554/eLife.31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Giesen C; Wang HAO; Schapiro D; Zivanovic N; Jacobs A; Hattendorf B; Schüffler PJ; Grolimund D; Buhmann JM; Brandt S; et al. Highly Multiplexed Imaging of Tumor Tissues with Subcellular Resolution by Mass Cytometry. Nat. Methods 2014, 11 (4), 417–422. 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- (30).Angelo M; Bendall SC; Finck R; Hale MB; Hitzman C; Borowsky AD; Levenson RM; Lowe JB; Liu SD; Natkunam Y; et al. Multiplexed Ion Beam Imaing (MIBI) of Human Breast Tumors. Nat. Med 2014, 20 (4), 436–442. 10.1038/nm.3488.Multiplexed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Haraguchi T; Shimi T; Koujin T; Hashiguchi N; Hiraoka Y Spectral Imaging Fluorescence Microscopy. Genes Cells 2002, 7 (9), 881–887. 10.1046/j.1365-2443.2002.00575.x. [DOI] [PubMed] [Google Scholar]
- (32).Lu G; Fei B Medical Hyperspectral Imaging: A Review. J. Biomed. Opt 2014, 19 (1), 010901–010901. 10.1117/1.jbo.19.1.010901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Martin ME; Wabuyele MB; Chen K; Kasili P; Panjehpour M; Phan M; Overholt B; Cunningham G; Wilson D; DeNovo RC; Vo-Dinh T Development of an Advanced Hyperspectral Imaging (HSI) System with Applications for Cancer Detection. Ann. Biomed. Eng 2006, 34 (6), 1061–1068. 10.1007/s10439-006-9121-9. [DOI] [PubMed] [Google Scholar]
- (34).Akbari H; Halig L; Schuster DM; Fei B; Osunkoya A; Master V; Nieh P; Chen G Hyperspectral Imaging and Quantitative Analysis for Prostate Cancer Detection. J. Biomed. Opt 2012, 17 (7), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Valm AM; Mark Welch JL; Borisy GG CLASI-FISH: Principles of Combinatorial Labeling and Spectral Imaging. Spec. Issue Fluoresc. Situ Hybrid. FISH 2012, 35 (8), 496–502. 10.1016/j.syapm.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Niehörster T; Löschberger A; Gregor I; Krämer B; Rahn H-J; Patting M; Koberling F; Enderlein J; Sauer M Multi-Target Spectrally Resolved Fluorescence Lifetime Imaging Microscopy. Nat. Methods 2016, 13, 257–257. [DOI] [PubMed] [Google Scholar]
- (37).Holzapfel HY; Stern AD; Bouhaddou M; Anglin CM; Putur D; Comer S; Birtwistle MR Fluorescence Multiplexing with Spectral Imaging and Combinatorics. ACS Comb. Sci 2018, 20 (11), 653–659. 10.1021/acscombsci.8b00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Holzapfel HY; Birtwistle MR Creating Complex Fluorophore Spectra on Antibodies Through Combinatorial Labeling. Transl. Sci 2016, 2 (3), e03. [PMC free article] [PubMed] [Google Scholar]
- (39).Gong H; Holcomb I; Ooi A; Wang X; Majonis D; Unger MA; Ramakrishnan R Simple Method To Prepare Oligonucleotide-Conjugated Antibodies and Its Application in Multiplex Protein Detection in Single Cells. Bioconjug. Chem 2016, 27 (1), 217–225. 10.1021/acs.bioconjchem.5b00613. [DOI] [PubMed] [Google Scholar]
- (40).Varn FS; Tafe LJ; Amos CI; Cheng C Computational Immune Profiling in Lung Adenocarcinoma Reveals Reproducible Prognostic Associations with Implications for Immunotherapy. OncoImmunology 2018, 7 (6), e1431084. 10.1080/2162402X.2018.1431084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Goswami S; Walle T; Cornish AE; Basu S; Anandhan S; Fernandez I; Vence L; Blando J; Zhao H; Yadav SS; et al. Immune Profiling of Human Tumors Identifies CD73 as a Combinatorial Target in Glioblastoma. Nat. Med 2020, 26 (1), 39–46. 10.1038/s41591-019-0694-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Trindade CJ; McDonough E; Hanson J; Walter Rodriguez B; Roper N; Gasmi B; Roque C; Gebregziabher M; Ylaya K; Fetsch P; et al. Utilization of Novel Highly Multiplexed Immunofluorescence Microscopy Technology to Understand Immunological Tumor Microenvironments in Small Cell Lung Carcinoma Patients Receiving Combination PD-L1 and PARP Inhibition Therapy. J. Clin. Oncol 2019, 37 (15_suppl), e14289–e14289. 10.1200/JCO.2019.37.15_suppl.e14289. [DOI] [Google Scholar]
- (43).Yu K; Liu Y; Yin J; Bao J A Novel Angle-Tuned Thin Film Filter with Low Angle Sensitivity. Opt. Laser Technol 2015, 68, 141–145. 10.1016/j.optlastec.2014.11.022. [DOI] [Google Scholar]
- (44).Daud AI; Loo K; Pauli ML; Sanchez-Rodriguez R; Sandoval PM; Taravati K; Tsai K; Nosrati A; Nardo L; Alvarado MD; et al. Tumor Immune Profiling Predicts Response to Anti–PD-1 Therapy in Human Melanoma. J. Clin. Invest 2016, 126 (9), 3447–3452. 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Koutsakos M; Sekiya T; Chua BY; Nguyen THO; Wheatley AK; Juno JA; Ohno M; Nomura N; Ohara Y; Nishimura T; et al. Immune Profiling of Influenza-Specific B- and T-Cell Responses in Macaques Using Flow Cytometry-Based Assays. Immunol. Cell Biol 2020, n/a (n/a). 10.1111/imcb.12383. [DOI] [PubMed] [Google Scholar]
- (46).Vacchi E; Burrello J; Di Silvestre D; Burrello A; Bolis S; Mauri P; Vassalli G; Cereda CW; Farina C; Barile L; et al. Immune Profiling of Plasma-Derived Extracellular Vesicles Identifies Parkinson Disease. Neurol. - Neuroimmunol. Neuroinflammation 2020, 7 (6), e866. 10.1212/NXI.0000000000000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Landhuis E S Ingle-Cell Approaches to Immune Profiling. Nature 2018, 557 (7706), 595–597. 10.1038/d41586-018-05214-w. [DOI] [PubMed] [Google Scholar]
- (48).Spitzer MH; Nolan GP Mass Cytometry: Single Cells, Many Features. Cell 2016, 165 (4), 780–791. 10.1016/j.cell.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bengsch B; Ohtani T; Herati RS; Bovenschen N; Chang K-M; Wherry EJ Deep Immune Profiling by Mass Cytometry Links Human T and NK Cell Differentiation and Cytotoxic Molecule Expression Patterns. Mass Cytom. Methods Appl 2018, 453, 3–10. 10.1016/j.jim.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Böttcher C; Fernández-Zapata C; Schlickeiser S; Kunkel D; Schulz AR; Mei HE; Weidinger C; Gieß RM; Asseyer S; Siegmund B; et al. Multi-Parameter Immune Profiling of Peripheral Blood Mononuclear Cells by Multiplexed Single-Cell Mass Cytometry in Patients with Early Multiple Sclerosis. Sci. Rep 2019, 9 (1), 19471. 10.1038/s41598-019-55852-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wang W; Su B; Pang L; Qiao L; Feng Y; Ouyang Y; Guo X; Shi H; Wei F; Su X; Yin J; Jin R; Chen D High-Dimensional Immune Profiling by Mass Cytometry Revealed Immunosuppression and Dysfunction of Immunity in COVID-19 Patients. Cell. Mol. Immunol 2020, 17 (6), 650–652. 10.1038/s41423-020-0447-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Li L; Yan S; Lin B; Shi Q; Lu Y Chapter Eight - Single-Cell Proteomics for Cancer Immunotherapy. In Advances in Cancer Research; Broome A-M, Ed.; Academic Press, 2018; Vol. 139, pp 185–207. 10.1016/bs.acr.2018.04.006. [DOI] [PubMed] [Google Scholar]
- (53).Amann R; Fuchs BM Single-Cell Identification in Microbial Communities by Improved Fluorescence in Situ Hybridization Techniques. Nat. Rev. Microbiol 2008, 6 (5), 339–348. 10.1038/nrmicro1888. [DOI] [PubMed] [Google Scholar]
- (54).Tropini C; Earle KA; Huang KC; Sonnenburg JL The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe 2017, 21 (4), 433–442. 10.1016/j.chom.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Brandwein M; Steinberg D; Meshner S Microbial Biofilms and the Human Skin Microbiome. Npj Biofilms Microbiomes 2016, 2 (1), 3. 10.1038/s41522-016-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.