

Abstract
The autophagic ubiquitin-like protein LC3 functions through interactions with LC3-interaction regions (LIRs) of other autophagy proteins including autophagy receptors, which stands out as a promising protein-protein interaction (PPI) target for the intervention of autophagy. Post-translational modifications like acetylation of Lys49 on the LIR-interacting surface could disrupt the interaction, offering an opportunity to design covalent small molecules interfering the interface. Through screening covalent compounds, we discover a small molecule modulator of LC3A/B that covalently modifies LC3A/B protein at Lys49. Activity-based protein profiling (ABPP) based evaluations reveal that a derivative molecule DC-LC3in-D5 exhibits a potent covalent reactivity and selectivity to LC3A/B in HeLa cells. DC-LC3in-D5 compromises LC3B lipidation in vitro and in HeLa cells, leading to deficiency in the formation of autophagic structures and autophagic substrate degradation. DC-LC3in-D5 could serve as a powerful tool for autophagy research as well as for therapeutic interventions.
Keywords: LC3, covalent modification, post-translational modification, autophagy
Graphical Abstract
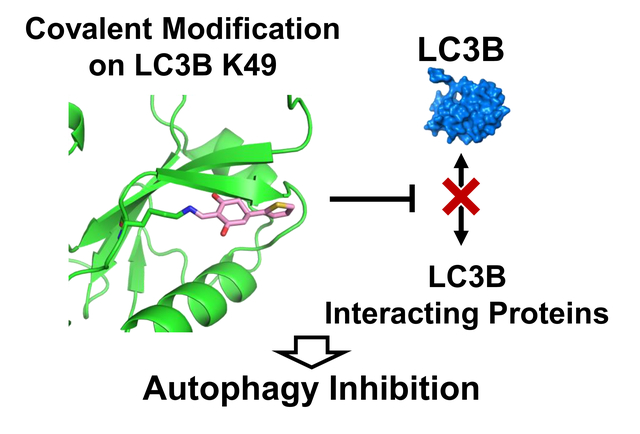
Through screening on covalent probe library, we discovered small molecule DC-LC3in covalently targeting LC3A/B on the Lys49 which could be modificated by acetylation. After crystal sturcture evaluation and further optimization, a more potent probe DC-LC3in-D5 was discovered and showed high selectivity to LC3A/B among proteome. DC-LC3in-D5 could inhibit autophagy by attenuating LC3B lipidation, which subsequently reduces the extent of autophagic structure formation and later substrate degradation.
Introduction
Autophagy is a dynamic process wherein eukaryotic cells remove dispensable or toxic cytoplasmic materials through lysosomal degradation. Central to autophagy is the formation of double-membraned vesicles known as autophagosomes. Cellular components are sequestered into autophagosomes, which then fuse with lysosomes to generate autolysosomes for degradation [1]. Human diseases, including but not limited to cancers, metabolic diseases, immune disorders, and neurodegenerative diseases, are related to disorder of autophagy[2].
The autophagy machinery is composed of several proteins including the ubiquitin-like modifier ATG8 in yeast [3]. To date, at least 8 orthologues of ATG8 have been identified in mammals. In humans, there are three LC3s (LC3A, LC3B, and LC3C), two GABARAPs (GABARAP and GABARAPL1) and one GATE-16 (also called GABARAPL2) protein [4]. All these homologies of Atg8 are thought to function in autophagic process, and they are often referred to generally as ATG8. ATG8 proteins are biosynthesized as precursors. Upon processing by ATG4, ATG8 loses C-terminal residues, making a glycine residue the C-terminus. The processed ATG8 is referred to as ATG8-I (similar LC3-I and GABARAP-I). ATG8-I undergoes a ubiquitination-like enzymatic reaction cascade to be covalently attached to the lipid molecule phosphatidylethanolamine (PE) of the autophagosomal membranes. ATG8-I is first activated by the E1 enzyme ATG7 to form the ATG7~ATG8 thioester intermediate and then transferred to the E2 enzyme ATG3. ATG3 recognizes PE and transfer the thioester bonded ATG8 to PE. ATG8-PE is referred to as ATG8-II (LC3-II, GABARAP-II). The final step of the PE-conjugation is greatly enhanced by the E3 factor ATG12~ATG5 conjugate[5].
PE-conjugated ATG8 proteins serve as binding platforms for autophagy proteins and autophagy receptors through protein-peptide interactions[6]. These interactors contain ATG8-interacting motif (AIM) or LC3-interacting region (LIR) [7]–a linear motif described as W/F-X-X-L/I/V (X is any types of amino acids). Typically, LIRs adopt an extended conformation and each of the hydrophobic residues at their first and forth positions dock into a dedicated pocket of LC3[8]. Functional LIR motifs are present in autophagosome-biogenesis and maturation-related components, such as ULK1[9] or PLEKHM1[10], as well as in cargo receptors, such as p62 and NBR1[11], that participate in selective autophagy [7, 12]. The LIR-LC3 interaction is required for binding of a substantial fraction of ATG8-interacting proteins to ATG8 to regulate autophagy [13]. Several peptides generated and modified from LIR sequences bind ATG8 with high affinities (), blocking autophagy when expressed in cells[14], strengthening that the ATG8-LIR interaction is a potential target to modulate ATG8 function as well as autophagy. Recently, it was reported that LC3A and LC3B LIR-interaction interface are druggable by a small molecule inhibitor novobiocin though with weak potency[15]. Autophagy-tethering compounds (ATTECs) like HTT-LC3 or lipid droplets-LC3 linker compounds which could bind with LC3 were also reported but they showed no inhibition on autophagy[16]. We were thus prompted to conduct a screen exploiting the ATG8-LIR interaction in an effort to identify much more potent ATG8 inhibitors to modulate autophagy.
It is well acknowledged that flat interacting surfaces between proteins are difficult to target and modifications of covalent probes may help overcome the disadvantages of designing protein-protein interaction inhibitors[17]. Interestingly, the Lys49 and Lys51 residues of LC3B located around interacting interface was reported to undergo endogenous acetylation and deacetylation modifications in cells[18]: acetylation of Lys49 and Lys51 in LC3B prevents interactions with proteins such as p62 and ATG7 and also leads to the accumulation of LC3B-I and to inhibition of autophagy[19]. Lys49 and Lys51 are located close to the LIR docking site. Modification on these amino acid side chains may cause steric hindrance that prevents LC3 from binding to its LIR-containing partners. These data provide a rationale that the covalent modification of the LIR-LC3 interaction interface might interfere with autophagy.
Thus, we set out to find covalent probes targeting LC3 PPI surface through screening on a compound library constituting diverse probes with lysine-targeting covalent warheads. We established a fluorescence polarization (FP) based high-throughput screening assay and identified a potent hit. It covalently reacted with the ε-amino side chain of Lys49 of LC3B. We determined the crystal structure of LC3B modified by one of its analogues and confirmed the covalent interaction. Further chemical modification of the compound led to DC-LC3in-D5 with improved potency and DC-LC3in-D5 showed selectivity to LC3A/B among proteome. Modification of cellular LC3B by our DC-LC3in-D5 blocked the lipidation of LC3B by blocking interaction between ATG7 and LC3B, inhibited autophagosome formation and autophagic substrate degradation, and resulted in cellular autophagy inhibition. The molecules modulating autophagy by covalent modification of LC3 on theLIR-LC3 interface may be useful for LC3 function and autophagy research.
Results and Discussion
Identification of LC3 modulators using high-throughput screen based on covalent probes library.
As mentioned above, the acetylation of Lys49 and Lys51 on LC3B may cause steric hindrance that prevents LC3B from binding its LIR-containing partners. Pursuing this, we aligned the apo LC3B crystal structure with other LC3B-LIR peptide complex structures, and their alignment indicated that Lys49 and Lys51 are located near the LIR interaction surface (Figure 1A). Thus, chemical modifications on these amino acid side chains may block the LIR-LC3 interactions. To examine whether covalent probes can react with these lysine residues, the pKa of all lysine residues in LC3B as an indicator for their reactivities, since lysine residues that are ready to act as nucleophiles should have a low pKa value[20] to support a nucleophilic reaction with covalent probes. Among the 9 lysine residues in LC3B, Lys49 has the lowest pKa value, which suggests that this is the most reactive residue. (Figure 1B). Therefore, it is reasonable to target LIR-interaction surface by screening covalent probes modifying lysine residues especially Lys49 on LC3B.
Figure 1. Identification of LC3 modulators using high-throughput screen based on covalent probes library.
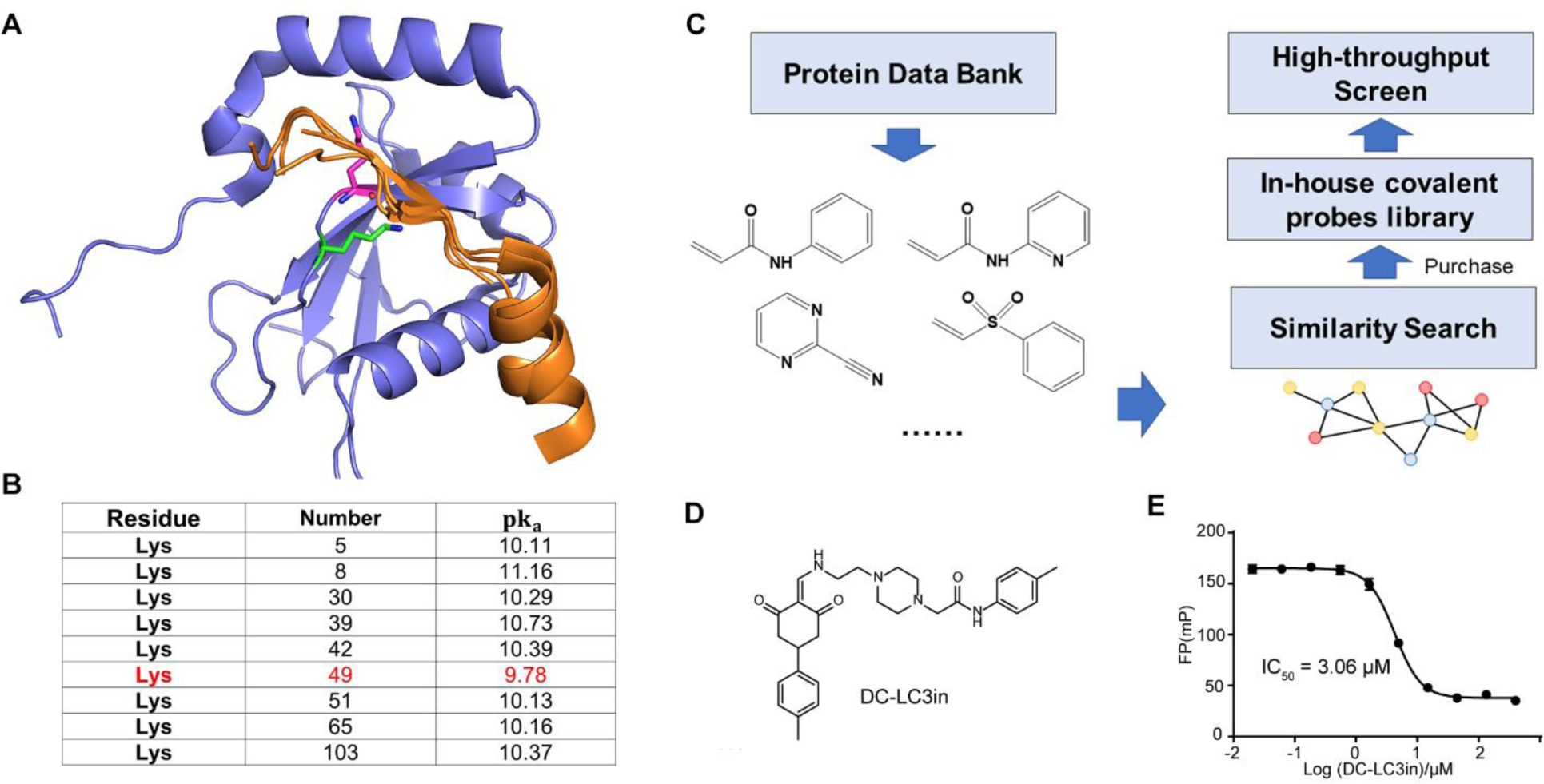
(A) Post-translational sites of Lys49 and Lys51on LC3B (PDB ID: 3VTU) are located near LIR peptides. Lys49 is represented in green and Lys51 is represented in magenta. FYCO1 (PDB ID: 5WRD), FUNDC1 (PDB ID: 5GMV), and p62 (PDB ID: 2ZJD) LIR peptides are aligned and are shown in orange. (B) pKa of lysine residues in LC3B predicted by PROPKA. Entry, PDB ID 3VTU; forcefield, CHARMM. (C) Construction of in-house covalent probes library. Covalent warheads that may react with lysine residues were collected from Protein Data Bank (PDB) and similarity search was performed to find probes containing these covalent warheads. (D-E) Chemical structure of the most potent inhibitor (“DC-LC3in”), and determination of the half maximal inhibitory concentration of this compound for the protein-peptide complex by FP assay.
To identify covalent probes that may target lysine residues on the LIR-LC3 interface, we constructed a covalent probe library for high-throughput screen (Figure 1C and Table S1). Covalent warhead motifs were collected from the PDB (Protein Data Bank) database and commercial compound libraries. Then, we performed a similarity search to explore diverse covalent probes. About 2000 probes with potential covalent warheads were obtained to construct our in-house covalent probe library.
We established a fluorescence polarization (FP)-based high-throughput screening assay to support screening for covalent probes that target theLIR-LC3 interface and disrupt the LIR-LC3 interaction, using LC3B protein, which is the best studied LC3, and a LIR-containing peptide (LBP2) derived from the LC3-interacting protein p62. Finally, the screen yielded seven hits having IC50 values less than 50μM (Figure 1D and Table S2). The most potent one, DC-LC3in, disrupted the LC3B-LBP2 interaction with an IC50 of 3.06μM (Figure 1E).
The small molecule DC-LC3in covalently binds to LC3B.
To determine whether the newly identified hit DC-LC3in directly interacts with LC3, we first evaluated the thermal stability change of the LC3B protein in the presence of these molecules using a modified differential scanning fluorimetry (DSF) method [21]. DC-LC3in significantly stabilized LC3B (i.e., led to a large increase in the melting temperature, Tm, Figure 2A), doing so in a concentration-dependent manner. These data are indicative of direct binding for DC-LC3in; in contrast, only very weak or no impact was observed for the other hits from the screen (Table S2). Later, nine DC-LC3in analogues were tested and eight of them showed both direct LC3 binding and disruption of the LC3-LBP2 interaction (Table S3).
Figure 2. DC-LC3in binds to LC3B and covalently modifies Lysine 49.
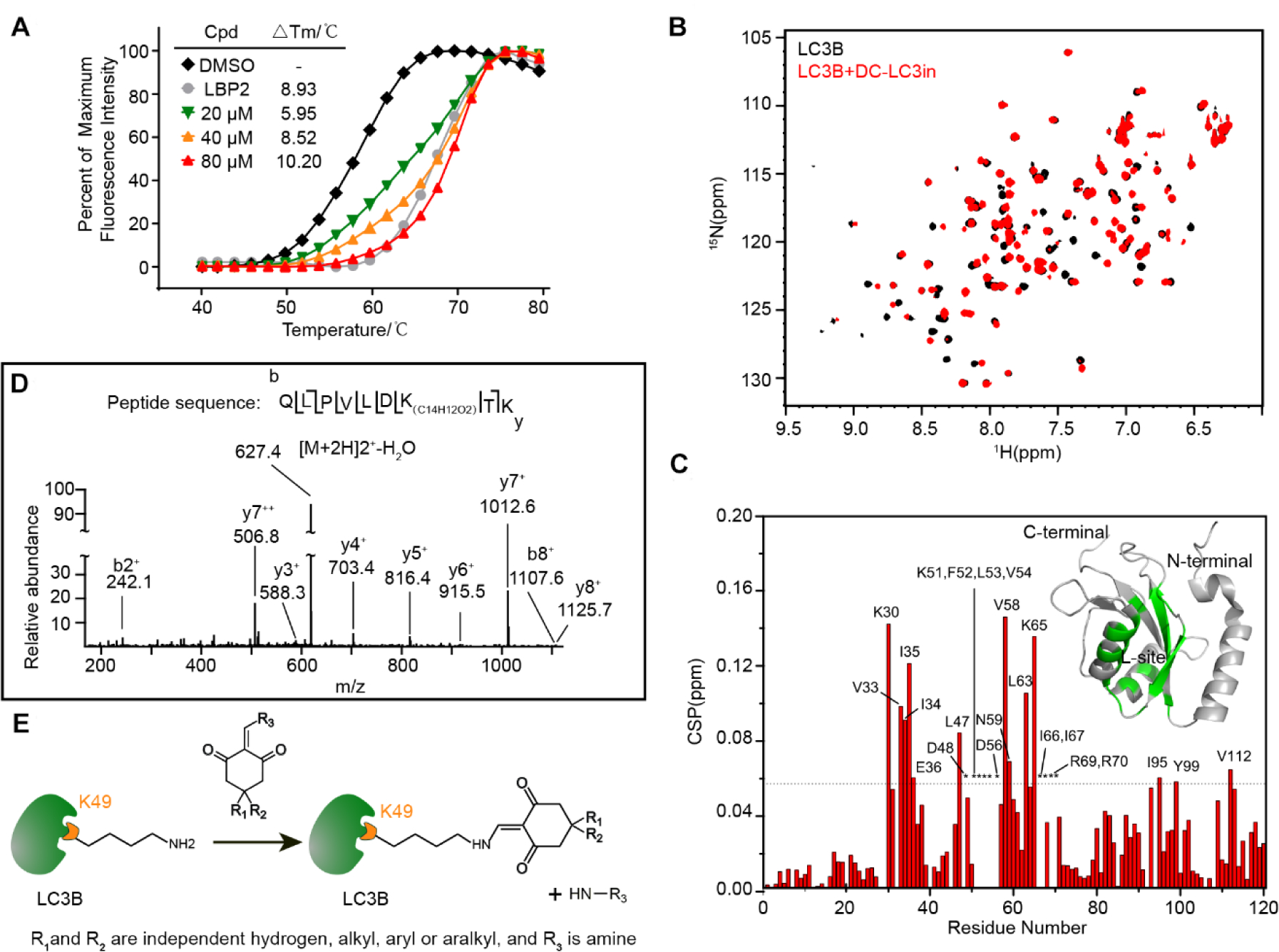
(A) Change in thermodynamic stability of LC3B upon binding with DC-LC3in and LBP2. The △Tm was calculated as the difference vs. the DMSO control sample. (B) Overlapped 1H-15N HSQC spectra for LC3B in the presence (red) or absence (black) of DC-LC3in at a 1:1 probe:protein ratio. (C) Chemical shift perturbation (CSP) analysis for LC3B DC-LC3in binding, and structural mapping of LC3B based on the CSP data. Residues with CSP values over the mean ± SD (dashed line) or those with attenuated resonance signals (indicated by asterisk) are labeled. Residues perturbed significantly or attenuated upon the binding of DC-LC3in are colored (green) on a 3D cartoon structure of LC3B (PDB ID 1V49). (D) HPLC-nESI MS/MS scan of the fragmented precursor ion of an LC3B peptide (m/z = 627.4), demonstrating that LC3B was covalently modified by DC-LC3in at Lysine 49. The difference in the detected peptide fragment mass vs. the theoretically predicted m/z for modified peptide corresponds to the monoisotopic mass for a C14H12O2 isomer. (E) Proposed model for the reaction between DC-LC3in analogs and LC3B’s Lys49 residue.
The interaction between DC-LC3in and LC3B was also validated by a 2D nuclear resonance experiment using an 1H-15N labeled LC3B protein. As expected, a number of HN cross-peaks in the HSQC spectrum of LC3B showed obvious chemical shift changes or resonance signal attenuations upon addition of the ligand (Figure 2B), results clearly demonstrating the binding of DC-LC3in with LC3B. Subsequent chemical shift perturbation (CSP) analysis indicated that the affected residues, including Phe52, Leu53, Val 54, Val58, Leu63, Ile66, and Ile67 (Figure 2C), were mainly hydrophobic and were located around the L-site of LIR-LC3 interface, which indicates that DC-LC3in may block the LIR-LC3interaction.
To evaluate whether DC-LC3in could covalently modifies LC3B, we used mass spectrometry to assess the covalent bonding of DC-LC3in and LC3B. MS/MS scan of the fragmented precursor ions revealed a covalent modification at residue Lys49 rather than Lys51 of LC3B when incubating DC-LC3in with LC3B (Figure 2D). Furthermore, DC-LC3in was able to covalently modify the Lys49 of EGFP-LC3B overexpressed in HeLa cells (Figure S1). Two analogues of DC-LC3in (a1 and a2) also covalently bound to LC3B at Lys49 (Figure S2). Modification of covalent probes on Lys49 is close to the LIR-LC3 interface of LC3B, consisting with the perturbed region detected in 2D NMR experiments.
Together, these results revealed that DC-LC3in could modify Lys49 covalently and bind around the LIR-LC3 interface. The reaction between Lys49 of LC3B and the compound DC-LC3in or its analogues is likely to be a nucleophilic substitution reaction, involving an addition with the ε-amino group of Lys49 followed by elimination of the amine group of the compounds, reforming the enamine with Lys49 covalently bound (Figure 2E).
Crystal structure of LC3B in complex with a DC-LC3in analogue.
To verity detailed LC3B-inhibitor interactions and help optimize more potent probes, we resolved the structure of LC3B (2–119) in complex with a4 (Table S3), an analogue of DC-LC3in at 1.6 Å resolution (PDB ID 7ELG, for diffraction and refinement statistics see Table S4). The electron density map of the bound a4 showed continuous density between the ε-amino group of Lys49 and the vinyl group of a4, indicating the formation of a covalent bond between them (Figure 3A), which strongly supports the proposed reaction (Figure 2E). Compound a4 attached to LC3B showed extraordinary electrostatic complementarity with the LIR-LC3 interacting surface of LC3B (Figure 3B): the complex was stabilized by cation-π interaction between the attached thiophene ring and the ionized Lys30 (calculated using H++17). Additionally, three hydrogen bonds formed between a4 and surrounding residues Leu53, Lys51, and Arg70, which also contributed to the stabilization of the complex (Figure 3C).
Figure 3. Crystallographic structure studies of LC3B and biochemical assays with mutant variants reveal the specific covalent reaction.
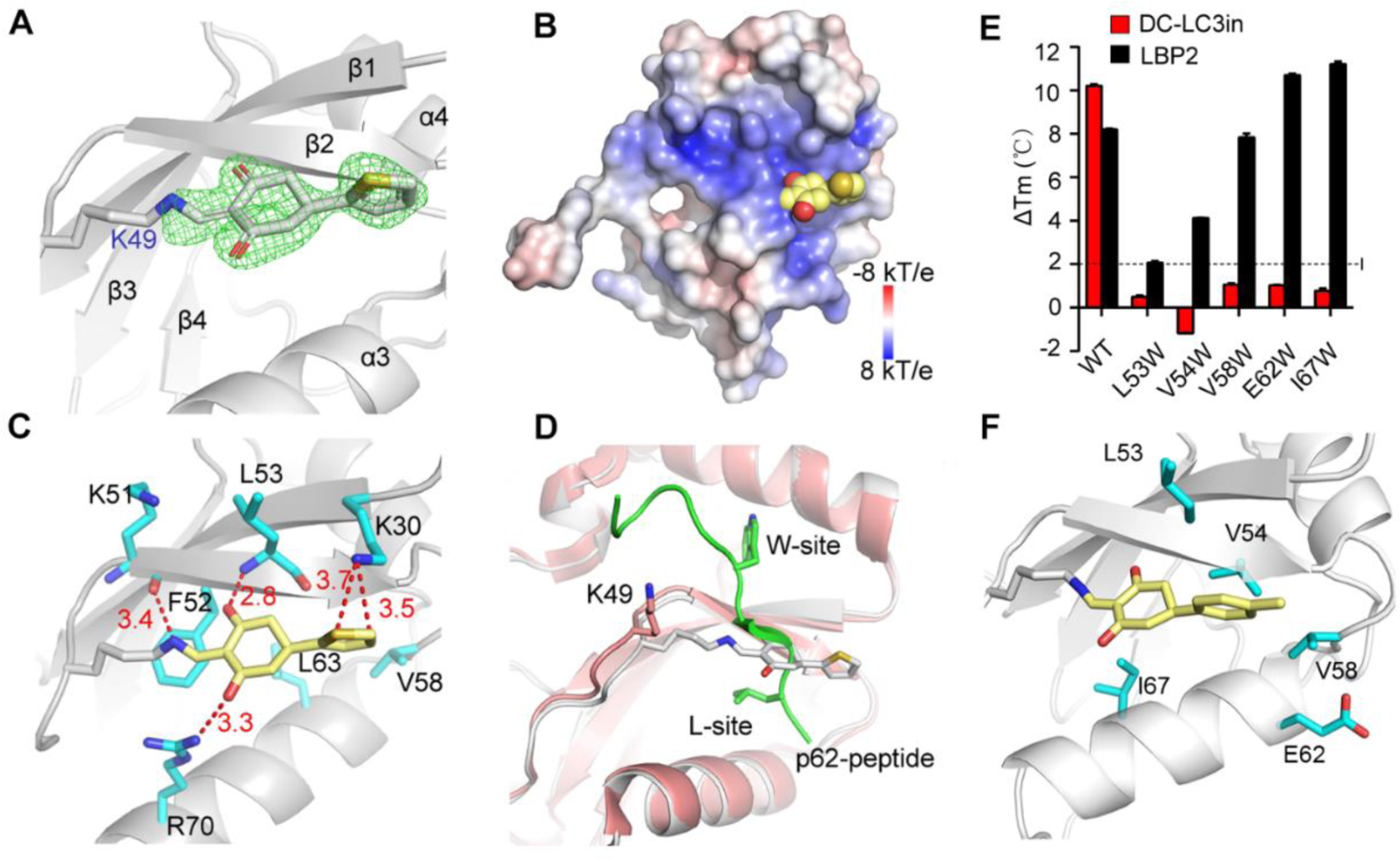
(A) X-ray crystallographic structure of LC3B covalently modified by a4 at residue Lys49, with Fo-Fc omit map (green) contoured at 3.0 σ within 1.6 Å of ligand (PDB ID 7ELG). (B) Electrostatic surface of LC3B covalently modified by a4. The ligand is shown as a CPK model (Carbon, yellow; Oxygen, red; Sulfur, orange). (C) Interactions of the modified residue K49 with surrounding residues (red dash lines; distances in Å). (D) Superimposition of LC3B-p62 complex (salmon and green, PDB ID 2ZJD) with a4 modified LC3B (gray) indicating that a4-modified K49 can block binding of the p62 peptide with LC3B. (E) Differential scanning fluorimetry based Tm Comparison of wild type LC3B and five mutant variants wherein single residue have been mutated to tryptophan. (F) Representation of the five residues around Lys49 in the modeled DC-LC3in-modified LC3B structure.
To better understand how the modification of Lys49 by this compound blocks the binding of LIR motifs to LC3B, we aligned our newly determined complex structure with a reported LC3B-p62 peptide complex [22]. As shown in Figure 3D, the superposed structure revealed a clear steric clash between the peptide and the bound a4, which suggested that LC3B modified with a4 cannot undergo its normal binding interaction with LIR peptides like LBP2.
Next, to test whether DC-LC3in also binds with LC3B as a4, several other residues around Lys49 in the pocket were mutated to tryptophan individually to introduce steric hindrance, and their impacts on DC-LC3in binding were explored. Compared to LC3BWT, the L53W, V54W, V58W, E62W, and I67W variants displayed no obvious increase in protein Tm upon incubation with DC-LC3in (Figure 3E and Table S5), suggesting that the change of amino acids surrounding Lys49 indeed interfered with the DC-LC3in and LC3B interaction. To better illustrate these results, we generated model structures of a DC-LC3in-modified LC3 complex using the Maestro module in Schrodinger18 based on our a4 modified structure (Figure 3F). According to the model, the L53W, V54W, V58W, E62W, and I67W mutations each induce clear steric hindrance for DC-LC3in occupation, preventing DC-LC3in from reacting with Lys49. Overall, these data demonstrated that DC-LC3in could form covalent bond with Lys49 in LIR-LC3 interacting surface on LC3B, which may prevent binding of LIR peptides.
Potent molecule DC-LC3in-D5 binds with LC3B in a two-step manner.
Further chemical optimization of DC-LC3in led to a more potent molecule DC-LC3in-D5 (also abbreviated as D5 in figures), which achieved a 13-fold higher activity (IC50 value of 200 nM, Figure 4A) and did not bind with LC3B K49R (Figure S3)—with a Kinact/Ki value of 0.36 μM−1min−1 (Figure 4B). DC-LC3in-D9 (also abbreviated as D9 in figures), which showed no inhibitory activity against the LC3B-LBP2 interaction and had no LC3 binding ability (Figure 4A), was synthesized and used as a negative control. Then we incubated DC-LC3in-D5 with purified LC3B protein (Figure 4C) or a peptide truncated from LC3B with Lys49 and Lys51 (marked as red) included (Figure 4D and Figure S3). DC-LC3in-D5 only covalently modified Lys49 in the folded LC3B protein like DC-LC3in rather than unfolded peptide, demonstrating that DC-LC3in-D5 has a low possibility to react with lysine residues randomly. Because DC-LC3in-D5 displayed reversible affinity to LC3B before the formation of covalent bond as illustrated by Kinact/Ki (Figure 4B), combined with the selective covalent reaction with folded LC3B protein, we considered that DC-LC3in-D5 binds with LC3B in a two-step manner.
Figure 4. The potent inhibitor DC-LC3in-D5 selectively reacts with LC3A/B both in vitro and in cells.
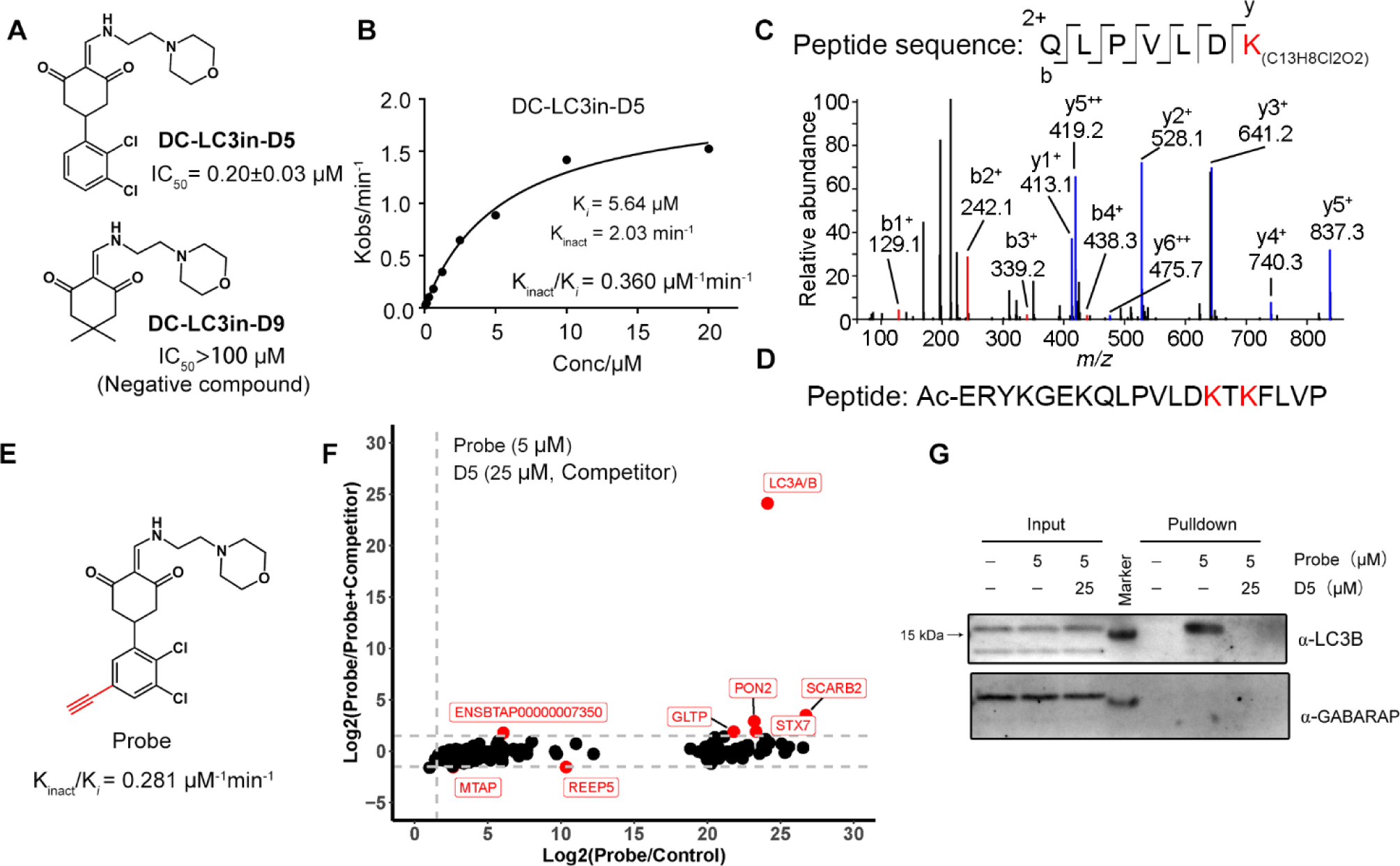
(A) Chemical structures and inhibitory activities (assessed via FP) for the improved compound DC-LC3in-D5 and the negative control DC-LC3in-D9. (B) Determination of Ki and Kinact values of DC-LC3in-D5 by FP assays. (C) HPLC-nESI MS/MS results of DC-LC3in-D5 incubated with LC3B protein. (D) Sequence of the LC3B peptide with Lys49 and several lysine residues included. (E) Chemical structure of the probe used in the ABPP experiment; the probe is a synthesized alkynylated variant of DC-LC3in-D5. (F)Probe capturing targets were determined by MS. The relative intensity ratio of probe/control and probe/probe+competitor (DC-LC3in-D5) were calculated (The 0 intensity is set as 100). The 327 significant proteins in ANOVA (see Method) were plotted (p < 0.01). The threshold of Log2(Intensity ratio) is 1.5 (grey dashed lines). Data downstream analysis details are in Figure S4. Raw data are available via ProteomeXchange with identifier PXD026874. (G) Western blot analysis of the samples from (F).
DC-LC3in-D5 exhibits a high cellular selectivity for LC3A/B.
To globally assess potential DC-LC3in-D5 reactivity at a proteome scale, we conducted activity-based protein profiling (ABPP) in HeLa cells using a synthesized probe with an alkyne group added to DC-LC3in-D5 (Figure 4E). In another treatment, DC-LC3in-D5 was used to compete with that synthesized ABPP probe. LC3A/B stood out as the most prominent target among the probe-labeled proteins together because LC3A/B share the same peptide sequence detected by mass spectrum. (Figure 4F, analysis details in Figure S5). Other LC3 homologues were not found in the labeled and competed-off protein list. As a negative probe showing no activity in vitro, DC-LC3in-D9 did not bind with target in competing assay (Figure S6). Immunoblotting for the same samples of MS also showed that DC-LC3in-D5 did not bind with GABARAP (Figure 4G), which was further proved by CETSA (Cellular Thermal Shift Assay) (Figure S7). This is also consistent with result of in vitro GABARAP-LBP2 fluorescence polarization assay (Figure S8). We further confirmed that DC-LC3in-D5 did not bind with PON2 or SCARB2 which were also enriched in MS results (Figure S7B–C). Together, these observations strongly suggest that DC-LC3in-D5 is highly selective for LC3 (LC3A/B) in cells, making it a suitable tool to explore the biology of LC3 as well as to modulate autophagy.
Modification of LC3B with DC-LC3in-D5 disrupts the ATG7-LC3B interaction and blocks LC3B lipidation.
It was previously reported that acetylation of Lys49 and Lys51 on LC3B blocks the ATG7-LC3B interaction[19a]. Additionally, Saccharomyces cerevisiae Atg7 recognizes Atg8 through its C-terminal tail’s binding with Atg8-AIM (Atg8 family-interacting motif)[23], which implies that human ATG7 may also interact with LC3B through an LIR-LC3 interaction. We therefore assumed that DC-LC3in-D5 covalent binding with Lys49 on the LC3B LIR-LC3 interface may affect the ATG7-LC3B interaction. Pull-down assays wherein EGFP-LC3B and N-terminal FLAG-tagged ATG7 were overexpressed in HEK293T cells showed that DC-LC3in-D5 could indeed block the LC3B-ATG7 interaction (Figure 5A). The negative control probe D9 did not block the interaction.
Figure 5. Modification with DC-LC3in-D5 disrupts ATG7-LC3B interaction in HeLa cells and slows down LC3B lipidation in vitro.
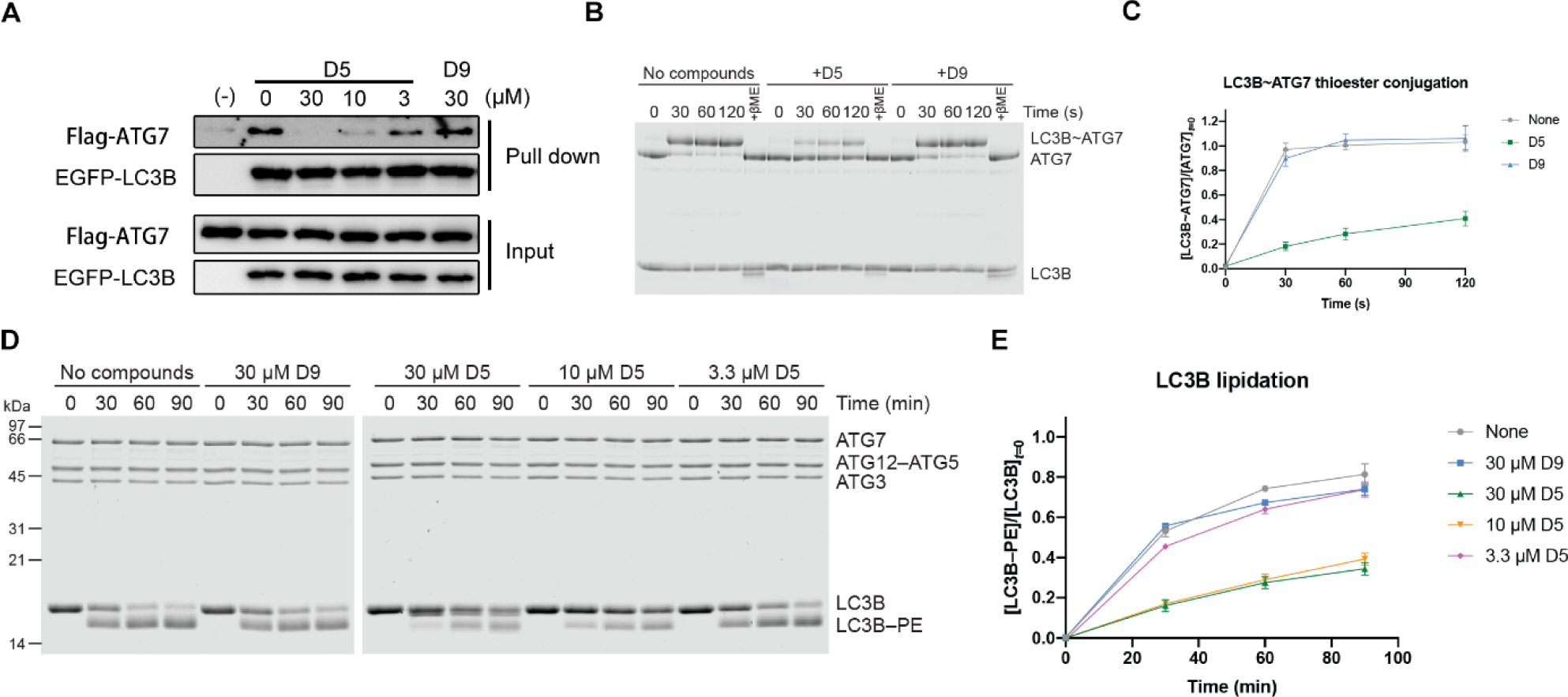
(A) DC-LC3in-D5 blocks ATG7-LC3B interaction in EGFP pull-down assay. (B) In vitro ATG7-LC3B conjugation assay performed with purified proteins and compounds as indicated. The compound:LC3B ratios were 3:1. (C) The quantifications of (B). The ATG7-conjugation ratios of LC3B were calculated in independent experiments (right, ±s.d.; n=3). (D) In vitro LC3B lipidation assay. The concentration of LC3B was 10μM. (E) The quantification of (D). The PE-modification ratio of LC3B was measured in independent lipidation experiments (right, ±s.d.; n=6).
ATG7-LC3 thioester bond formation is a key step in the ATG7-conjugation process. We next evaluated whether disruption of the ATG7-LC3B interaction leads to disability of the thioester bond formation. In vitro ATG7-LC3B conjugation assays showed that the modification with DC-LC3in-D5 impairs ATG7-LC3B thioester bond formation (Figure 5B–C).
Considering that ATG7-conjugation, the first step of LC3 lipidation process, is inhibited by modification with DC-LC3in-D5, we then wanted to test if DC-LC3in-D5 could block LC3B lipidation. Results of in vitro lipidation assays showed that DC-LC3in-D5 slows down LC3B lipidation (Figure 5D–E), suggesting that disrupting the protein-protein interaction between LC3B and ATG7 by modification of DC-LC3in-D5 Lys49 inhibits LC3B lipidation.
DC-LC3in-D5 as autophagy inhibitor.
We showed that DC-LC3in-D5 can block LC3B lipidation. To assess if DC-LC3in-D5 has any effects on autophagy, HeLa cells were starved to induce autophagy after a pre-treatment incubation with DC-LC3in-D5 or DC-LC3in-D9 (CM, complete medium; EBSS, Earle’s Balanced Salt Solution). Immunoblotting showed that cells pre-treated with DC-LC3in-D5 accumulated significant more p62 than DMSO-treated control samples, indicating that the substrate degradation was inhibited by DC-LC3in-D5 (Figure 6A). Additionally, DC-LC3in-D5 attenuated LC3-I/II lipidation in cells exposed to autophagy inducing conditions (Figure 6A) No impact on p62 levels or LC3B lipidation was observed in cells treated with the inactive negative control analog DC-LC3in-D9 (Figure 6A).
Figure 6. DC-LC3in-D5 as autophagy inhibitor.
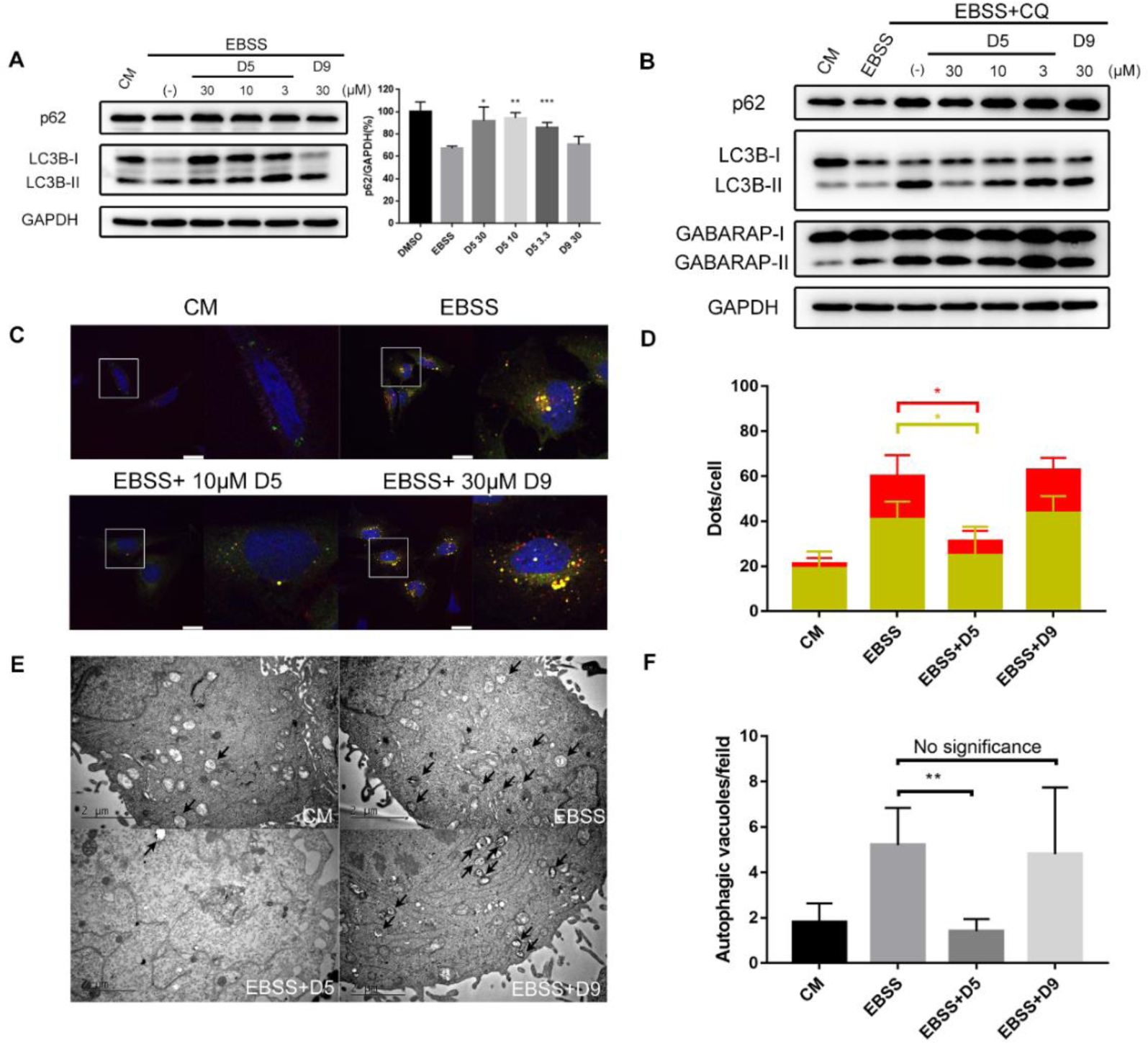
(A) Western blot analysis of autophagy markers in HeLa cells cultured in starvation conditions. To induce autophagy, EBSS was used for another 4 hrs after 16 hrs treatment with the indicated concentrations of inhibitors. Quantification of p62/GAPDH intensity (right, ±s.d.; n=3). *p < 0.05, **p < 0.01, ***p<0.001. (B) Western blot analysis of LC3B and GABARAP lipidation in HeLa cells. To induce autophagy and block LC3-II degradation, EBSS or EBSS combined with CQ was used for another 4 hrs after the 16 hrs treatment with the indicated concentrations of inhibitors. (C) Tandem reporter of autophagic flux. Cells were pre-treated with DC-LC3in-D5 or DC-LC3in-D9 (10 μM) for 16 hrs, followed by co-treatment with EBSS for 4 hrs. Yellow dots indicate autophagosomes. Red dots indicated autolysosomes. Scale bars, 25μm. (D) Quantification of autophagosomes and autolysosomes was performed in different fields randomly (right, ±s.d.; n≥4). *p < 0.05. (E) TEM of HeLa cells treated in the indicated conditions. Cells were pre-treated with DC-LC3in-D5 or DC-LC3in-D9 (10 μM) for 16 hrs, followed by co-treatment with EBSS for 4 hrs. Scale bars, 2 μm. (F) Quantification of autophagic vacuoles in (E) (±s.d.; n=5) **p < 0.01.
It has been reported that lipidation of LC3 is associated with the formation of autophagic structures such as autophagosomes and autolysosomes[24]. Therefore, we investigated the effects of DC-LC3in-D5 on autophagic flux and autophagosome formation using tandem TurboRFP-EGFP-LC3B. However, as DC-LC3in-D5 affects LC3B lipidation, we expected that LC3B would not be appropriate as an autophagosome marker. GABARAP, whose lipidation is unaffected by DC-LC3in-D5 in vitro (Figure S9), may represent a substitute alternative to label autophagosomes and autophagolysosomes. To further validate that DC-LC3in-D5 did not inhibit GABARAP lipidation in cells, we used CQ (chloroquine) to prevent LC3-II degradation. The result showed that DC-LC3in-D5 can disrupt LC3B lipidation. In contrast, there was little change in the lipidation status of GABARAP (Figure 6B).
We then constructed HeLa cell lines in which tandem TurboRFP-EGFP-GABARAP is overexpressed. Using confocal microscopy, we counted the numbers of yellow and red GABARAP puncta representing autophagosomes and autophagolysosomes, respectively. Both numbers decreased upon incubations with DC-LC3in-D5, suggesting that modification of LC3A/B by DC-LC3in-D5 partially impair autophagosome formation (Figure 6C–D). We next used transmission electron microscopy (TEM) to observe cellular substructures including autophagosomes and autolysosomes. DC-LC3in-D5 significantly decreased the number of autophagic vesicles induced by starvation (Figure 6E–F), underscoring the autophagy inhibition effect of DC-LC3in-D5. Notably, DC-LC3in-D5 showed little cellular toxicity, with GI50 values over 100 μM in several human cell lines (Table S6). We conclude that modification of DC-LC3in-D5 on LC3A/B blocks autophagy by attenuating LC3B lipidation, which subsequently reduces the extent of autophagic structure formation and later substrate degradation.
Conclusion
LC3s are core functional proteins during autophagy, yet no potent LC3-inhibiting small molecules have been reported to date. Starting with the concept for chemical modification by covalent probe to regulate target functions, a high-throughput screen followed by biochemical studies were performed. We identified a hit compound DC-LC3in and its analogues as covalent modulators of LC3B. Crystallographic and MS results revealed the formation of a covalent bond between the compound and lysine 49 of LC3B, explaining its mechanism of action. Further chemical optimization led to the DC-LC3in-D5 with improved potency. Treatment of cells with DC-LC3in-D5 resulted in disruption of LC3B lipidation, inhibition of autophagic vesicle formation, and accumulation of p62. Reactivity studies proved that DC-LC3in-D5 binds with LC3B in a two-step manner by calculating kinetic parameters. ABPP experiment confirmed that DC-LC3in-D5 is highly selective for LC3A/B. Further, DC-LC3in-D5 only reacted with Lys49 of the folded and biologically active LC3B protein but not the Lys49 from the LC3B peptide, raising the necessity of specific pocket environment around Lys49 for covalent reaction. Though Lys49 is conserved among human ATG8 family, other ATG8 proteins like GABARAP may have different pocket environment, which could explain the selectivity of DC-LC3in-D5. All of these demonstrated that DC-LC3in-D5 is a selective and potent LC3A/B covalent inhibitor which could interfere autophagy.
Post-translational modifications (PTMs) on proteins regulate various biological processes like autophagy[25], immunity[26] and tumorgenesis[27]. Modifications on specific amino acids implies that chemical reactions around these sites may be favorable. Combining huge PTM information databases and growing covalent compound libraries, it is possible to help discovering more potent molecules targeting specific sites or overcoming challenges of designing PPI disrupting molecules. Covalent modifications of molecules may regulate targets’ functions like natural PTMs to induce inhibition, degradation, extending the methods to interfere targets artificially[28].
The research of covalent probes, such as inhibitors targeting KRAS G12C[29], RhoA[30], BTK[31], or EGFR[32], have been advancing during recent years. Among numerous covalent probes, chemical probes that selectively modify lysine residues are under prosperous research[33]. The lysine reactivity and ligandability in the human proteome has been profiled to identify much more lysine probes[34]. What’s more, it is reported that disrupting Protein-protein Interaction (PPI) by lysine-directed probes is much favorable[35]. Considering that DC-LC3in-D5 which could disrupt LC3 protein interactions reacts with one lysine ε-amino group of LC3 in a selective manner, its covalent warhead may be helpful for the design and optimization of lysine-targeted probes especially blocking Protein-protein interactions. The reaction between DC-LC3in-D5 and Lys49 in a specific microenvironment also deserves detailed research to help computational prediction and mechanism study of lysine targeting covalent reactions.
Given the great therapeutic potential of modulating autophagy, much effort has been made in the identification and development of compounds that can induce or suppress autophagy[36]. Autophagic inhibition has conferred benefits such as overcoming treatment resistance [37] and enhancing anti-HCV therapy [38]. Two studies also showed the efficiency of autophagy inhibitor in the combination treatment of RAS-driven cancers[39]. However, the development of potent and selective autophagy inhibitors has remained largely elusive: serious side effects have been observed due to the off-target effects of existing drugs (such as chloroquine and hydroxychloroquine). DC-LC3in-D5 may contribute to anti-HCV or combination treatments in cancer through inhibiting autophagy.
LC3 also plays roles in non-autophagic pathways, such as LC3-Associated Phagocytosis (LAP). In tumor model the vesicles surrounded by LC3 help engulf apoptosis tumor cells in macrophages[40]. LAP could be disrupted if LC3 lipidation is inhibited, which made macrophages in the tumor microenvironment more lethal for killing tumor cells[41]. In addition, it has been reported that STING, an innate immune signaling protein, binds to LC3[42] and its activation leads to autophagy induction[43], which results in STING degradation and terminates innate immunity signal[44]. We envision that the LC3 inhibitors we have identified may assist dissecting these non-autophagic roles of LC3.
Supplementary Material
Acknowledgements
We thank Dr. Jun O. Liu and Prof Wei. Liu for the helpful editing and critical comments. We are extremely grateful to National Centre for Protein Science Shanghai (Shanghai Science Research Center, Protein Expression and Purification system) for their instrument support and technical assistance. We thank the staffs from BL17U1 and BL19U1 beamlines at Shanghai Synchrotron Radiation Facility (SSRF), for assistance during data collection.
We also gratefully acknowledge the financial supports from National Natural Science Foundation of China (21820102008 to H.J; 81625022, 91853205, 81821005 to C.L.; 31270830, 21572038 to Y.D.), Chinese Academy of Science grant (CASIMM0120184015 to C.L.), Science and Technology Commission of Shanghai Municipality (18431907100 to H.J.; 19XD1404700 to C.L.) and National Institutes of Health grant (GM092740 to T.O.).
References
- [1].Galluzzi L, Pietrocola F, Levine B, Kroemer G, Cell 2014, 159, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Choi AM, Ryter SW, Levine B, N Engl J Med 2013, 368, 651–662; [DOI] [PubMed] [Google Scholar]; b) Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G, Nat Rev Drug Discov 2017, 16, 487–511; [DOI] [PubMed] [Google Scholar]; c) Levine B, Mizushima N, Virgin HW, Nature 2011, 469, 323–335; [DOI] [PubMed] [Google Scholar]; d) Menzies FM, Fleming A, Rubinsztein DC, Nat Rev Neurosci 2015, 16, 345–357. [DOI] [PubMed] [Google Scholar]
- [3].a) Ohsumi Y, Cell research 2014, 24, 9–23; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Levine B, Klionsky DJ, Developmental Cell 2004, 6, 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Shpilka T, Weidberg H, Pietrokovski S, Elazar Z, Genome Biol 2011, 12, 226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y, Pei Y, Wan B, Guo J, Yu L, Biochemical and Biophysical Research Communications 2006, 339, 437–442; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) He H, Dang Y, Dai F, Guo Z, Wu J, She X, Pei Y, Chen Y, Ling W, Wu C, Zhao S, Liu JO, Yu L, J Biol Chem 2003, 278, 29278–29287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y, Journal of Biological Chemistry 2007, 282, 37298–37302; [DOI] [PubMed] [Google Scholar]; b) Otomo C, Metlagel Z, Takaesu G, Otomo T, Nature Structural & Molecular Biology 2013, 20, 59–66. [DOI] [PubMed] [Google Scholar]
- [6].a) Wild P, McEwan DG, Dikic I, Journal of Cell Science 2014, 127, 3–9; [DOI] [PubMed] [Google Scholar]; b) Behrends C, Sowa ME, Gygi SP, Harper JW, Nature 2010, 466, 68–76. [DOI] [PubMed] [Google Scholar]
- [7].Wild P, McEwan DG, Dikic I, J Cell Sci 2014, 127, 3–9. [DOI] [PubMed] [Google Scholar]
- [8].a) Noda NN, Ohsumi Y, Inagaki F, FEBS Letters 2010, 584, 1379–1385; [DOI] [PubMed] [Google Scholar]; b) Nakatogawa H, Ichimura Y, Ohsumi Y, Cell 2007, 130, 165–178. [DOI] [PubMed] [Google Scholar]
- [9].Grunwald DS, Otto NM, Park JM, Song D, Kim DH, Autophagy 2020, 16, 600–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, de Stegmann DM, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I, Molecular Cell 2015, 57, 39–54. [DOI] [PubMed] [Google Scholar]
- [11].Kirkin V, Lamark T, Sou Y-S, Bjørkøy G, Nunn JL, Bruun J-A, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat J-P, Øvervatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T, Molecular Cell 2009, 33, 505–516. [DOI] [PubMed] [Google Scholar]
- [12].Birgisdottir AB, Lamark T, Johansen T, J Cell Sci 2013, 126, 3237–3247. [DOI] [PubMed] [Google Scholar]
- [13].a) Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I, EMBO Rep 2010, 11, 45–51; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T, Mol Cell 2009, 33, 505–516; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ho K-H, Chang H-E, Huang W-P, Autophagy 2014, 5, 461–471; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nakatogawa H, Ohbayashi S, Sakoh-Nakatogawa M, Kakuta S, Suzuki SW, Kirisako H, Kondo-Kakuta C, Noda NN, Yamamoto H, Ohsumi Y, J Biol Chem 2012, 287, 28503–28507; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nobuo HK, Noda1 N, Nakatogawa Hitoshi, Satoo Kenji,, Wakana Adachi JI, Fujioka Yuko, Ohsumi Yoshinori, and Inagaki Fuyuhiko, Genes to Cells 2008, 13, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li JC, Zhu RC, Chen KY, Zheng H, Zhao HY, Yuan CZ, Zhang H, Wang C, Zhang MJ, Nature Chemical Biology 2018, 14, 778–787. [DOI] [PubMed] [Google Scholar]
- [15].Hartmann M, Huber J, Kramer JS, Heering J, Pietsch L, Stark H, Odadzic D, Bischoff I, Fürst R, Schröder M, Akutsu M, Chaikuad A, Dötsch V, Knapp S, Biondi RM, Rogov VV, Proschak E, Journal of Medicinal Chemistry 2021, 64, 3720–3746. [DOI] [PubMed] [Google Scholar]
- [16].a) Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T, Ma L, Gao C, Yang Y, Sun Y, Wang J, Sun X, Lu C, Difiglia M, Mei Y, Ding C, Luo S, Dang Y, Ding Y, Fei Y, Lu B, Nature 2019, 575, 203–209; [DOI] [PubMed] [Google Scholar]; b) Fu Y, Chen N, Wang Z, Luo S, Ding Y, Lu B, Cell Research 2021, 10.1038/s41422-021-00532-7. [DOI] [PubMed] [Google Scholar]
- [17].Fu C, Zhu X, Xu P, Li Y, Onco Targets Ther 2019, 12, 609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Popelka H, Klionsky DJ, FEBS J 2015, 282, 3474–3488; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McEwan DG, Dikic I, Trends Cell Biol 2011, 21, 195–201; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hamaï A, Codogno P, Science Signaling 2012, 5, pe29–pe29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, Liu B, Chang C, Zhou T, Lippincott-Schwartz J, Liu W, Mol Cell 2015, 57, 456–466; [DOI] [PubMed] [Google Scholar]; b) Song T, Su H, Yin W, Wang L, Huang R, FEBS Lett 2019, 593, 414–422. [DOI] [PubMed] [Google Scholar]
- [20].Pettinger J, Jones K, Cheeseman MD, Angew Chem Int Ed Engl 2017, 56, 15200–15209. [DOI] [PubMed] [Google Scholar]
- [21].Simeonov A, Expert Opin Drug Discov 2013, 8, 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M, J Biol Chem 2008, 283, 22847–22857. [DOI] [PubMed] [Google Scholar]
- [23].Noda Nobuo N., Satoo K, Fujioka Y, Kumeta H, Ogura K, Nakatogawa H, Ohsumi Y, Inagaki F, Molecular Cell 2011, 44, 462–475. [DOI] [PubMed] [Google Scholar]
- [24].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T, The EMBO Journal 2000, 19, 5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wani WY, Boyer-Guittaut M, Dodson M, Chatham J, Darley-Usmar V, Zhang J, Lab Invest 2015, 95, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu J, Qian C, Cao X, Immunity 2016, 45, 15–30. [DOI] [PubMed] [Google Scholar]
- [27].a) Bode AM, Dong Z, Nat Rev Cancer 2004, 4, 793–805; [DOI] [PubMed] [Google Scholar]; b) Ko P-J, Dixon SJ, EMBO reports 2018, 19, e46666. [DOI] [PubMed] [Google Scholar]
- [28].Meng F, Liang Z, Zhao K, Luo C, Med Res Rev 2021, 41, 1701–1750. [DOI] [PubMed] [Google Scholar]
- [29].Janes MR, Zhang J, Li L-S, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, Feng J, Chen JH, Li S, Li S, Long YO, Thach C, Liu Y, Zarieh A, Ely T, Kucharski JM, Kessler LV, Wu T, Yu K, Wang Y, Yao Y, Deng X, Zarrinkar PP, Brehmer D, Dhanak D, Lorenzi MV, Hu-Lowe D, Patricelli MP, Ren P, Liu Y, Cell 2018, 172, 578–589.e517. [DOI] [PubMed] [Google Scholar]
- [30].Sun Z, Zhang H, Zhang Y, Liao L, Zhou W, Zhang F, Lian F, Huang J, Xu P, Zhang R, Lu W, Zhu M, Tao H, Yang F, Ding H, Chen S, Yue L, Zhou B, Zhang N, Tan M, Jiang H, Chen K, Liu B, Liu C, Dang Y, Luo C, Adv Sci (Weinh) 2020, 7, 2000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liang C, Tian D, Ren X, Ding S, Jia M, Xin M, Thareja S, European Journal of Medicinal Chemistry 2018, 151, 315–326. [DOI] [PubMed] [Google Scholar]
- [32].Jia Y, Yun C-H, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys P-Y, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong K-K, Jänne PA, Eck MJ, Nature 2016, 534, 129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Cuesta A, Taunton J, Annu Rev Biochem 2019, 88, 365–381; [DOI] [PubMed] [Google Scholar]; b) Pettinger J, Jones K, Cheeseman MD, Angewandte Chemie International Edition 2017, 56, 15200–15209. [DOI] [PubMed] [Google Scholar]
- [34].Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, Cravatt BF, Nature Chemistry 2017, 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].a) Akçay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, Su Q, Nature Chemical Biology 2016, 12, 931–936; [DOI] [PubMed] [Google Scholar]; b) Ueda T, Tamura T, Kawano M, Shiono K, Hobor F, Wilson AJ, Hamachi I, Journal of the American Chemical Society 2021, 143, 4766–4774. [DOI] [PubMed] [Google Scholar]
- [36].a) Ha J, Kim J, Expert Opin Ther Pat 2016, 26, 1273–1289; [DOI] [PubMed] [Google Scholar]; b) Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G, Nature Reviews Drug Discovery 2017, 16, 487–511. [DOI] [PubMed] [Google Scholar]
- [37].Guo JY, Xia B, White E, Cell 2013, 155, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fang S, Su J, Liang B, Li X, Li Y, Jiang J, Huang J, Zhou B, Ning C, Li J, Ho W, Li Y, Chen H, Liang H, Ye L, Sci Rep 2017, 7, 44039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].a) Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, Burrell LD, Lum DH, Whisenant JR, Gilcrease GW 3rd, Cavalieri CC, Rehbein KM, Cutler SL, Affolter KE, Welm AL, Welm BE, Scaife CL, Snyder EL, McMahon M, Nat Med 2019, 25, 620–627; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, Tomar G, Papke B, Hobbs GA, Yan L, Hayes TK, Diehl JN, Goode GD, Chaika NV, Wang Y, Zhang GF, Witkiewicz AK, Knudsen ES, Petricoin EF 3rd, Singh PK, Macdonald JM, Tran NL, Lyssiotis CA, Ying H, Kimmelman AC, Cox AD, Der CJ, Nat Med 2019, 25, 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR, Journal of Molecular Biology 2017, 429, 3561–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada-Romero E, Kalkavan H, Johnson MDL, Natarajan S, Turnis ME, Finkelstein D, Opferman JT, Gawad C, Green DR, Cell 2018, 175, 429–441.e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu D, Wu H, Wang CG, Li YJ, Tian HB, Siraj S, Sehgal SA, Wang XH, Wang J, Shang YL, Jiang ZF, Liu L, Chen Q, Cell Death and Differentiation 2019, 26, 1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, Chen ZJ, Nature 2019, 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Prabakaran T, Bodda C, Krapp C, Zhang B.-c., Christensen MH, Sun C, Reinert L, Cai Y, Jensen SB, Skouboe MK, Nyengaard JR, Thompson CB, Lebbink RJ, Sen GC, van Loo G, Nielsen R, Komatsu M, Nejsum LN, Jakobsen MR, Gyrd-Hansen M, Paludan SR, The EMBO Journal 2018, 37, e97858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.