

PURPOSE
High tumor mutation burden (TMB) in many cancer types is associated with the production of tumor-specific neoantigens, a favorable outcome and response to immune checkpoint blockade (ICB) therapy. Besides mutation-derived neoantigens, aberrant intron retention also produces tumor neopeptides that could trigger an immune response. The relationship between intron-retention–derived tumor neoantigens (IR-neoAg) and clinical outcomes in pancreatic cancer remains uncertain. Here, we quantify IR-neoAg in pancreatic cancer and evaluate whether IR-neoAg load might serve as a biomarker for selecting patients who may benefit from ICB therapy.
METHODS
We developed a computational approach to estimate patient-specific IR-neoAg load from transcriptome data available in The Cancer Genome Atlas pancreatic cancer cohort. Associations between IR-neoAg load and patient overall survival were evaluated using Kaplan-Meier estimates and Cox regression. Differential expression of immune checkpoint and HLA-I genes was evaluated in tumors with high IR-neoAg load.
RESULTS
High IR-neoAg load predicted better overall survival in pancreatic cancer, although no association was found for TMB. IR-neoAg load remained a significant prognostic factor after adjusting for patient age, sex, tumor stage and grade, and TMB. Moreover, pancreatic tumors with both high IR-neoAg load and high HLA-I gene expression had similar gene expression profiles as other tumor types that showed response to anti–programmed cell death protein 1 therapy.
CONCLUSION
IR-neoAg load is associated with favorable survival in pancreatic cancer. These findings provide strong evidence for considering IR-neoAgs when selecting patients who might benefit from ICB therapy.
INTRODUCTION
Advances in immune checkpoint blockade (ICB) therapy using antibodies that block programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) have resulted in remarkable clinical responses in a wide variety of patients with cancer.1-3 Response to ICB therapy correlates with high tumor mutation burden (TMB) and the cell surface display of tumor neoantigens by major histocompatibility complex (MHC) molecules, which is critical for T-cell recognition and immune-mediated killing of tumor cells.4-7
CONTEXT
Key Objective
Our aim was to use a bioinformatics approach to predict tumor-specific neoantigens that are derived from retained intron sequences in publicly available transcriptome data. We asked whether the level of intron-retention neoantigens (IR-neoAg) could predict a favorable outcome in patients with pancreatic cancer.
Knowledge Generated
We found that the IR-neoAg load is an independent predictor of overall survival in patients with pancreatic cancer. In addition, a subset of tumors with high IR-neoAg load and high HLA class-I gene expression showed an immune response consisting of infiltrating cytotoxic T cells, and also had transcriptome profiles similar to immune checkpoint blockade therapy–responsive tumors.
Relevance
RNA sequencing data from patient tumor samples can be used to evaluate intron retention and HLA-I gene expression. These two tumor markers could be potentially important for selecting patients with pancreatic cancer who might benefit from immune checkpoint blockade therapy.
Pancreatic cancer consists of two categories, namely cancers developing from the exocrine cells that make up the exocrine glands and ducts of the pancreas and cancers that develop from cells in the endocrine glands. More than 95% of pancreatic cancers arise from exocrine cells, with pancreatic ductal adenocarcinoma (PDAC) being the most common histologic type, as well as one of the deadliest with a 5-year survival rate < 8%.8 PDAC is considered an immune-privileged tumor and to date, ICB therapy has not shown efficacy in patients with PDAC, nor affected overall survival (OS).9,10 One explanation for why PDAC responds poorly to ICB therapy is a low TMB.11 However, recent studies have shown that some PDAC tumors do express neoantigens and exhibit T-cell infiltration.12,13 These findings imply that some patients are able to generate an antitumor immune response and, therefore, may benefit from ICB therapy.
In addition to somatic DNA mutations, aberrant RNA transcripts can be a major source of neopeptides when retained introns are translated and degraded through the nonsense-mediated decay mechanism.14 Intron retention (IR) commonly occurs in a wide variety of cancer transcriptomes, including PDAC, compared with normal tissues.15,16 Additionally, high IR levels in PDAC appears to be an independent predictor of tumor progression.17 Therefore, we hypothesized that in the presence of low TMB, aberrant IR-induced neoantigens (IR-neoAg) contribute to immune-mediated clearance of pancreatic cancer cells.
To begin to test this hypothesis, we herein used in silico prediction on RNA-sequencing data from two large independent cohorts of patients with pancreatic cancer from The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) pancreatic cancer cohorts to determine whether IR-neoAg load was associated with longer OS for these patients. We also estimated tumor-infiltrating immune cell proportions and determined the association of IR-neoAgs with various tumor lymphocyte populations. In addition, we investigated the association of IR-neoAg load with expression of immune checkpoint genes and HLA genes encoding the MHC class-I molecules in pancreatic cancer. Finally, we compared gene expression profiles in pancreatic cancer with those of melanoma tumors that responded to anti–PD-1 checkpoint therapy. The results from this study could be useful in selecting patients with pancreatic cancer who might benefit from ICB therapy.
METHODS
Pancreatic Cancer and Normal Pancreas Data Sets
RNA-seq data from TCGA-PAAD (N = 178) with clinical and pathologic characteristics were downloaded from the Genomic Data Commons.18 RNA-seq data from the ICGC Pancreatic Cancer cohorts (ICGC-PDAC-AU, N = 81) were downloaded from the ICGC data portal.19 One sample each from TCGA-PAAD and ICGC-PDAC-AU cohorts was missing survival event time and was excluded from survival analyses. RNA-seq data from normal pancreas samples (n = 68) were retrieved from genotype-tissue expression (GTEx) projects.20 Four PDAC microarray data sets, GSE15471, GSE16515, GSE28735, and GSE62452,21-24 were downloaded from the GEO database, and the Bioconductor affy package was used for raw data processing and normalization.
Identification of IR Events
Raw fastq files were aligned to GRCh38 reference genome using STAR (v.2.7.2).25 The exon sets for each protein-coding gene were reannotated using GTF files (Gencode.v32). The union of each exon set was used to define introns as the interval between exon sets (Data Supplement). Exons and introns were quantified using uniquely mapped reads on the basis of the reannotated GTF file using HTseq.26 IR events were further filtered using the following criteria: (1) both the intron region and its flanking exon regions had read counts > 10 and (2) the transcripts per million ratios of the intron to flanking exons was > 0.05 and < 0.5. These filters allowed identification of IR events that had expression levels that were comparable with the flanking exons in the messenger RNA transcripts that were composed of a mixture of normal and aberrant splicing products. IR events that were also observed in at least 25% of normal pancreas RNA-seq data sets from GTEx20 were filtered to obtain the final set of pancreatic tumor-specific IR events.
IR Neoantigen Prediction
To obtain the set of neopeptides derived from retained introns, the open reading frames of the upstream exons were extended into introns until the first stop codon. The translated peptides were segmented into fragments of 8-11 amino acids that contained at least one intron-encoded amino acid. We estimated the binding affinity of each IR-derived neopeptide with specific MHC class-I molecules for each sample using NetMHCpan (v.4.1).27 Each patient's HLA genotype was deduced from RNA-seq data using arcasHLA (v1.1).28 NetMHCpan compares raw prediction scores to a set of random natural peptides to calculate the % rank, which provides robust binding metrics.27 Neopeptides with % rank < 0.5 were defined as strong binders, as recommended by NetMHCpan, and were considered as IR-neoAgs in this study.
Tumor Immune Cell Proportions and Prediction of Immunotherapy Response
The relative proportion of 22 types of tumor-infiltrating immune cells were inferred from bulk RNA-seq expression data using the online CIBERSORT application,29 with parameters set as LM22 signatures and 100 permutations. We used SubMap30 to evaluate the similarity between global gene expression patterns of TCGA-PAAD tumors with either high or low IR-neoAgs and samples from immunotherapy-treated melanoma patients.31 The mapping information generated by SubMap identifies subclasses common to two independent data sets and calculates the probability that they share similar biologic properties. Default parameters were used.32
Differential Expression and Pathway Enrichment Analysis
Salmon (v1.2.1) was used to quantify the gene expression levels from RNA-seq data,33 using the reference annotation GRCh38 (gencode.v32). Differentially expressed genes among patient groups were identified with the limma package in R.34 The clusterProfiler package was used to test the pathway enrichment significance.35 P values were adjusted using the Benjamini-Hochberg method.36
Statistical Analysis
All analyses and visualization were performed in R (v.4.0.2). Kaplan-Meier survival estimates and Cox proportional hazard analysis were performed with log-rank test and hazard ratio (HR) to compare patient groups, using the Survival package.37 Mann-Whitney-Wilcoxon test was used to compare differences in the value distribution between groups.38
RESULTS
IR Is a Potential Source of Neoantigens in PDAC
We first surveyed PDAC primary tumors in the TCGA SpliceSeq database to examine the frequency of IR compared with other alternative splicing events. We found that IR accounted for 16.5% of all alternative splicing events, which was second behind skipped exon (56%) and more common than alternative 5′- and 3′-splice site and mutually exclusive exons (13.7%, 12.1%, and 0.7%, respectively). Using Kyoto Encyclopedia of Genes and Genomes gene set enrichment analysis, we also found that the spliceosome pathway was significantly upregulated in PDAC compared with adjacent normal tissue (Data Supplement). These findings are consistent with those of Wang et al,16 who first reported the frequency of different alternative splicing events and dysregulation of the spliceosome machinery using Affymetrix exon array data from PDAC tissues.
Next, we investigated the immunogenic potential of IR-derived neopeptides using RNA-seq data from 178 pancreatic cancer patient samples in the TCGA-PAAD cohort. An overview of the data processing steps in our computational pipeline to identify IR events and IR-neoAgs is diagrammed in Figure 1A. IR events were first identified in 68 normal pancreas GTEx RNA-seq data sets. We found a total of 5,927 IR events that occurred in at least 25% of the GTEx samples (Data Supplement). After filtering these normal IR events, there were an average of 600 tumor-specific IR events per sample. Each patient's HLA genotype was further deduced from RNA-seq data (Data Supplement) and used to predict the number of IR-neoAgs in their tumor sample. A total of 171,526 IR-neoAgs were predicted with an average IR-neoAg load of 963 (range 43-4,284) per tumor sample (Data Supplement). The number of IR-neoAgs per tumor was strongly correlated with the number of IR events (Spearman correlation rho = 0.93, P < .001, Fig 1B). Interestingly, the predicted number of IR-neoAgs presented by each HLA type was not related to HLA allele frequency (Fig 1C). For example, HLA-A02:01 showed the highest allele frequency (24.2%), but this allele was only predicted to present 11,014 (6.4%) of the IR-neoAg peptides. By contrast, HLA-B07:02 had a lower allele frequency (8.7%) but was predicted to present 23,481 (13.7%) IR-neoAgs. These findings suggest that a patient's HLA genotype is an important factor in predicting immunogenicity of potential IR-neoAgs.
FIG 1.
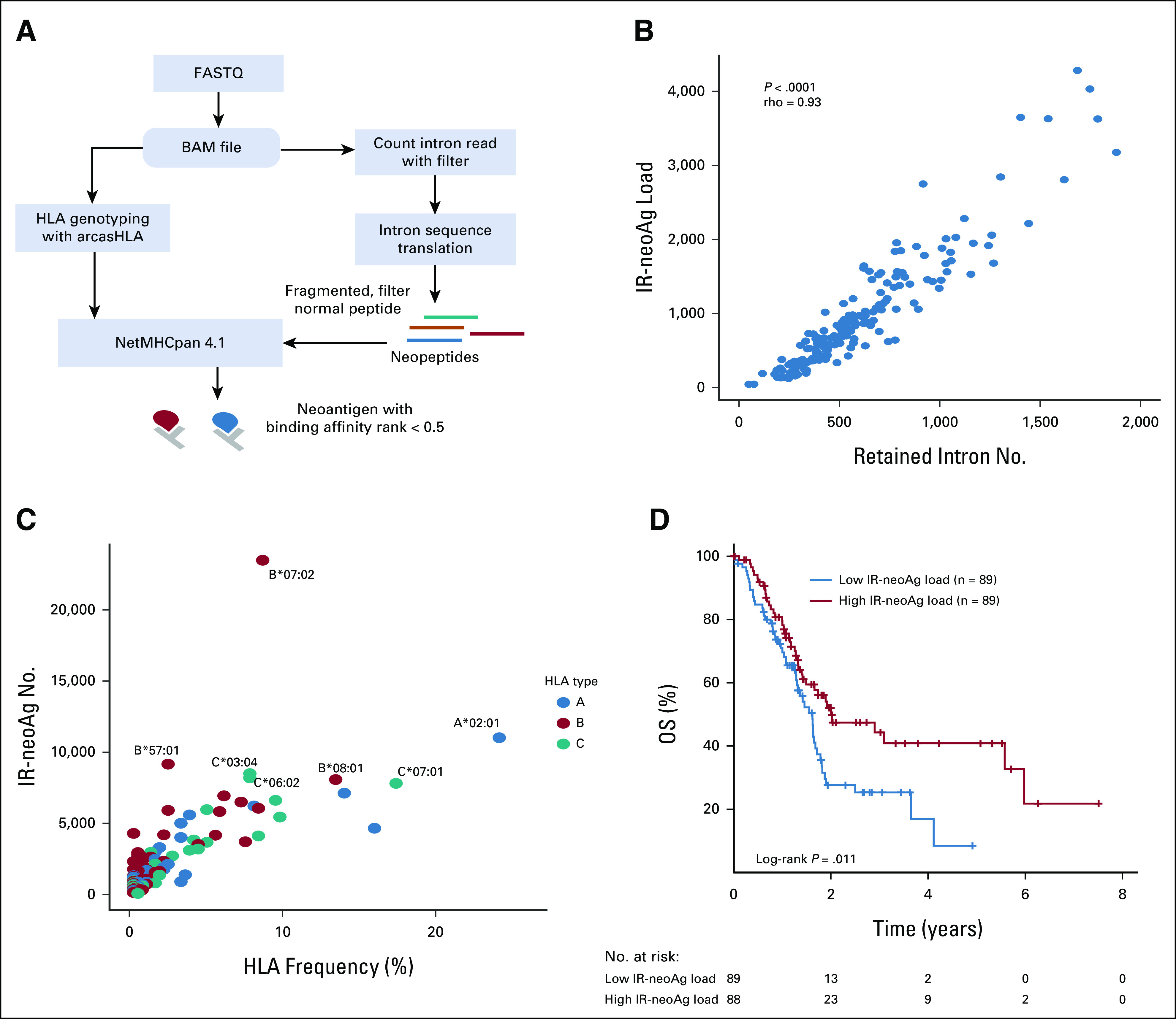
Intron-retention neoantigen predicts favorable survival. (A) Workflow for identifying IR-neoAgs from RNA-seq data. (B) Scatter plot showing the correlation between the number of IR events and the IR-neoAg load in the TCGA-PAAD RNA-seq data. Each dot represents an individual patient (N = 178). (C) Scatter plot showing HLA allele frequency in the TCGA-PAAD data set versus the number of IR-neoAgs each HLA allele can potentially present. Each dot represents an individual allele type. (D) Kaplan-Meier curves for OS of groups with high and low tumor IR-neoAg load in the TCGA-PAAD cohort. A risk table is displayed below the plot. IR-neoAg, intron-retention neoantigens; OS, overall survival.
IR-neoAg Load Is an Independent Prognostic Factor for Pancreatic Cancer
TCGA-PAAD tumors (N = 178) were divided into high and low IR-neoAg groups on the basis of the median IR-neoAg number. The high and low IR-neoAg groups were not significantly different with respect to age, sex, tobacco smoking, or for clinical features such as tumor stage, grade, or microsatellite instability (Table 1). However, the median OS time for patients with high IR-neoAg load was 24 months compared with 19 months for those with low IR-neoAg load (Kaplan-Meier log-rank test, P = .011; Fig 1D). The association between high IR-neoAg load and survival was independent of clinicopathologic factors (P = .008, multivariate Cox regression analysis; Table 2). The HR was 0.55, indicating that high IR-neoAg load reduced the risk of poor outcome by 45%. High IR-neoAg load was also associated with longer survival in the ICGC PDAC cohort (N = 81), although these findings did not reach statistical significance likely because of the smaller sample size (Data Supplement).
TABLE 1.
Clinical and Pathologic Characteristics of TCGA-PAAD Data Set
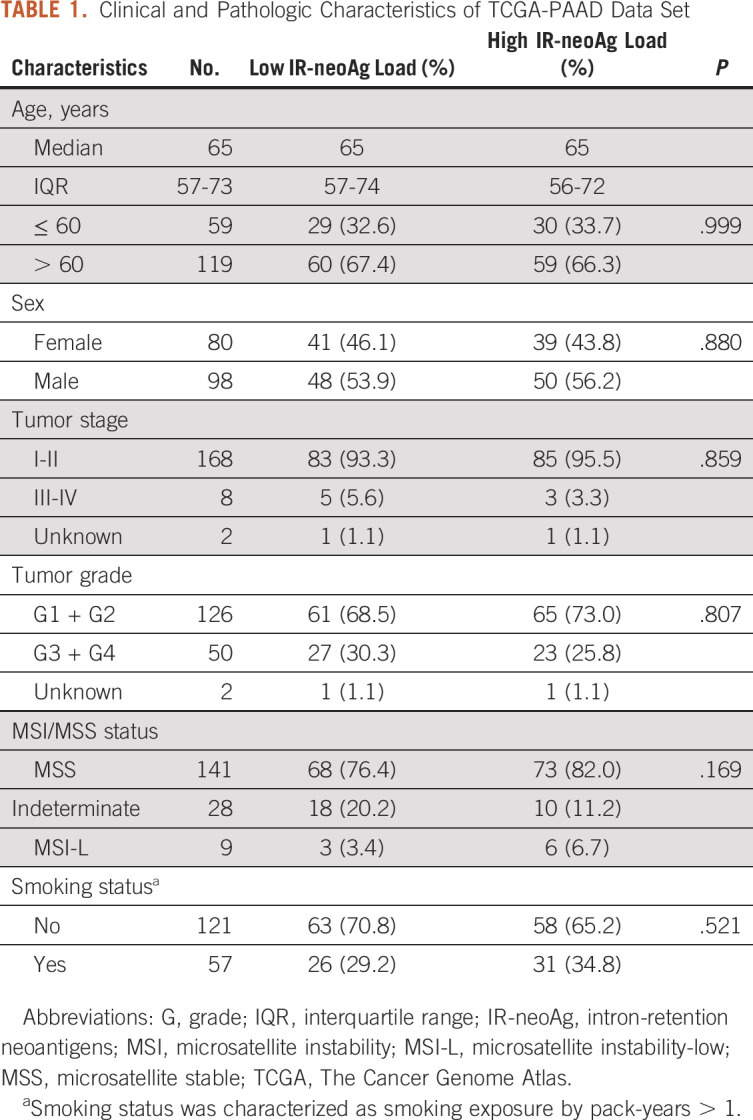
TABLE 2.
Univariate and Multivariate Cox Regression Analyses of Overall Survival in TCGA-PAAD Patients

Our findings agree with those of Tan et al,17 who showed that IR was associated with PDAC patient outcome in a curated subset of the TCGA-PAAD cohort (n = 150), which excluded non-PDAC tumors such as acinar cell carcinoma, pancreatic neuroendocrine tumors (PanNET), benign neoplasms, and tumors with < 1% neoplastic cellularity.39 Since our initial survival analysis was performed using all 178 TCGA-PAAD samples with RNA-seq data, we repeated the multivariate Cox regression analysis on the curated set of 150 PDAC samples and considered tumor purity as a covariate. Our results showed that high IR-neoAg load remained significantly associated with longer OS (HR, 0.605; 95% CI, 0.377 to 0.971; P = .037; Data Supplement). These results, taken together, support IR and IR-neoAg load as independent predictors of pancreatic cancer progression and patient survival.
IR-neoAg Load Is Associated With Features of Tumor Immune Response
To test whether high IR-neoAg load was accompanied by immune cell infiltration in the TCGA cohort, we used a deconvolution algorithm to estimate the relative percentages and activation states of 22 types of immune cells from bulk RNA-seq data.29 Seven cell types showed statistically significant differences between high and low IR-neoAg load tumors (Fig 2A). There were more memory B cells (P = .014), follicular helper T (Tfh) cells (P = .011), and naive CD4+ T cells (P = .044) in tumors with high IR-neoAg load. By contrast, these tumors had fewer resting mast cells (P = .002), resting dendritic cells (P = .011), M1-macrophages (P = .013), and neutrophils (P = .003). However, no significant correlation was observed between IR-neoAg load and the relative percentage of CD8+ T cells or other cytotoxic immune cell types (Data Supplement). Although the overall percentage of infiltrating immune cells was low, higher levels of memory B and Tfh cells are generally associated with better prognosis and cancer immunotherapy response.40-42 These findings imply that IR-neoAg load may affect the immune cell composition of the tumor microenvironment (TME).
FIG 2.
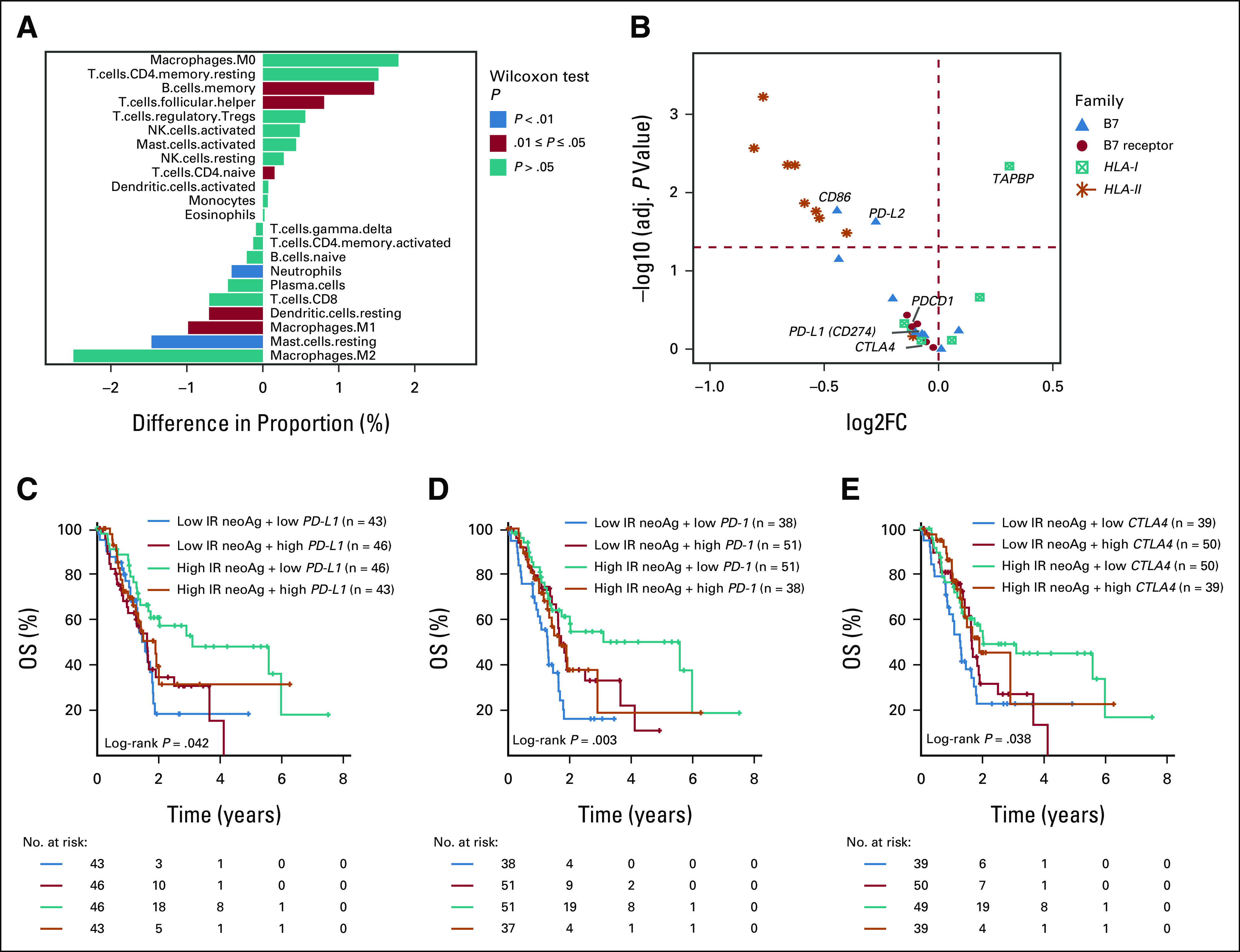
IR-neoAg load is associated with immune features in the TCGA-PAAD cohort. (A) Comparison of the percentage of tumor-infiltrated immune cells between tumors with high and low IR-neoAg load. A positive value along the x-axis represents a higher and a negative value represents a lower proportion of cells in the high IR-neoAg samples. P values were calculated using the Mann-Whitney-Wilcoxon test. (B) Volcano plot showing the expression differences of immune-related genes between the high and low IR-neoAg load tumors. Symbols to the right of the vertical dashed line represent genes with higher expression and symbols on the left represent genes with lower expression in high IR-neoAg tumors. Symbols above the red dashed line represent genes with adjusted P-values < .05. Kaplan-Meier overall survival curves for four patient groups stratified by IR-neoAg load and the gene expression levels of (C) PD-L1, (D) PD-1, and (E) CTLA-4. A risk table is displayed below the plot. CTLA-4, cytotoxic T-lymphocyte antigen 4; IR-neoAg, intron-retention neoantigens; OS, overall survival; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1.
To better understand the molecular differences that might contribute to OS between pancreatic cancers with high and low IR-neoAg load, we compared the expression levels of a collection of genes related to immune cell response, consisting of nine B7 ligand genes, five B7 receptor genes, six MHC-I genes, and nine MHC-II genes. We found that 12 of 14 B7 ligand and receptor genes had lower expression in samples with high IR neoAg load, with CD86 and PD-L2 reaching statistical significance (Fig 2B). In addition, all nine MHC-II genes were expressed at lower levels in the high IR-neoAg group, with eight of nine reaching statistical significance. Only one MHC-I pathway gene, TAPBP, showed significant differential expression between tumors with high and low IR-neoAg load. Thus, longer OS in patients with high IR-neoAg load tumors could be partially explained by low expression levels of immune coinhibitory genes that dampen effector T-cell responses.
IR-neoAg Load Together With Immune Checkpoint Gene Expression Levels Are Associated With OS
The expression level of immune checkpoint genes, such as PD-L1, is associated with ICB response and survival outcomes in multiple cancers.43-45 Therefore, we asked whether the correlation of IR-neoAg load and patient survival was associated with the expression levels of immune checkpoint genes. To address this question, we stratified TGCA-PAAD samples into four groups by IR-neoAg load and immune checkpoint gene expression levels (median as the cutoffs, Data Supplement). Kaplan-Meier survival analysis revealed that patients with high IR-neoAg load tumors and low PD-L1 gene expression (Fig 2C) had the longest survival time compared with the other groups. Similar findings were observed for the combinations of high IR-neoAg load and low PD-1 (Fig 2D) or low CTLA-4 (Fig 2E) gene expression. Additional Kaplan-Meier survival curves comparing these four patient groups stratified by IR-neoAg load and expression of other inhibitory checkpoint genes are shown in the Data Supplement. Taken together, our results suggest that although most pancreatic cancers have an immune-privileged phenotype, a subset of patients with high tumor IR-neoAg and low expression of coinhibitory genes may be able to generate a spontaneous antitumor immune response that could potentially restrain tumor progression and increase patient survival. Further analysis showed that all eight PanNET patients in the TCGA-PAAD cohort fell into this category and were among the patients with the longest OS.
IR-neoAg Load and HLA-I Expression Identify a Subgroup of Tumors That Have Similar Gene Expression Patterns as Tumors That Respond to ICB Therapy
Tumor neoantigens are critical mediators of host immune response and immunotherapy treatment efficacy. In addition, MHC class-I expression is essential for neoantigen presentation. Because ICB therapy has not been used routinely for patients with PDAC, we used SubMap, a subclass mapping method, to determine whether TCGA-PAAD tumors with high IR-neoAg load and high HLA-I gene expression levels shared similar transcriptomic profiles with ICB therapy–responsive melanoma tumors.31 We calculated an HLA-I score for each TCGA-PAAD tumor sample by averaging the expression value of the three major HLA genes, HLA-A, -B, and -C (Data Supplement). We then divided the samples into two groups on the basis of the number of IR-neoAgs and the HLA-I score. One group contained samples with higher than the median IR-neoAg and HLA-I score, denoted as IR-neoAghi-HLA-Ihi (n = 43) and the other group consisted of all other tumors (n = 135). Notably, SubMap analysis indicated that the gene expression profiles of the IR-neoAghi-HLA-Ihi group showed significant similarity with the subset of melanoma tumors that were responsive to anti–PD-1 immunotherapy (Fig 3A, Bonferroni-corrected P = .028). However, no significant similarities were observed between the TCGA-PAAD samples with IR-neoAghi alone or combinations of IR-neoAghi with PD-1, PD-L1, or CTLA-4 expression and the anti–PD-1 responsive melanoma samples (data not shown). Notably, 93% of the IR-neoAghi-HLA-Ihi tumors (40 of 43) were classified as PDAC; three tumors were excluded from the curated 150 TCGA-PAAD set, which included two samples with low neoplastic cellularity and one tumor that did not arise from the pancreas. We also found that CD8+ T cells, as well as the total number of tumor-infiltrating immune cells, were both significantly higher in IR-neoAghi-HLA-Ihi samples compared with the other tumors (Fig 3B, Wilcoxon test, P < .001). Furthermore, this set of IR-neoAghi-HLA-Ihi tumors had significantly higher expression of PD-1 and PD-L1 genes (Fig 3C), which likely reflects the higher proportion of CD8+ T cells and other cytotoxic immune cells. Together, these results indicate that patients with pancreatic cancer with IR-neoAghi-HLA-Ihi tumors may represent a group that is more likely to respond to anti–PD-1 treatment.
FIG 3.
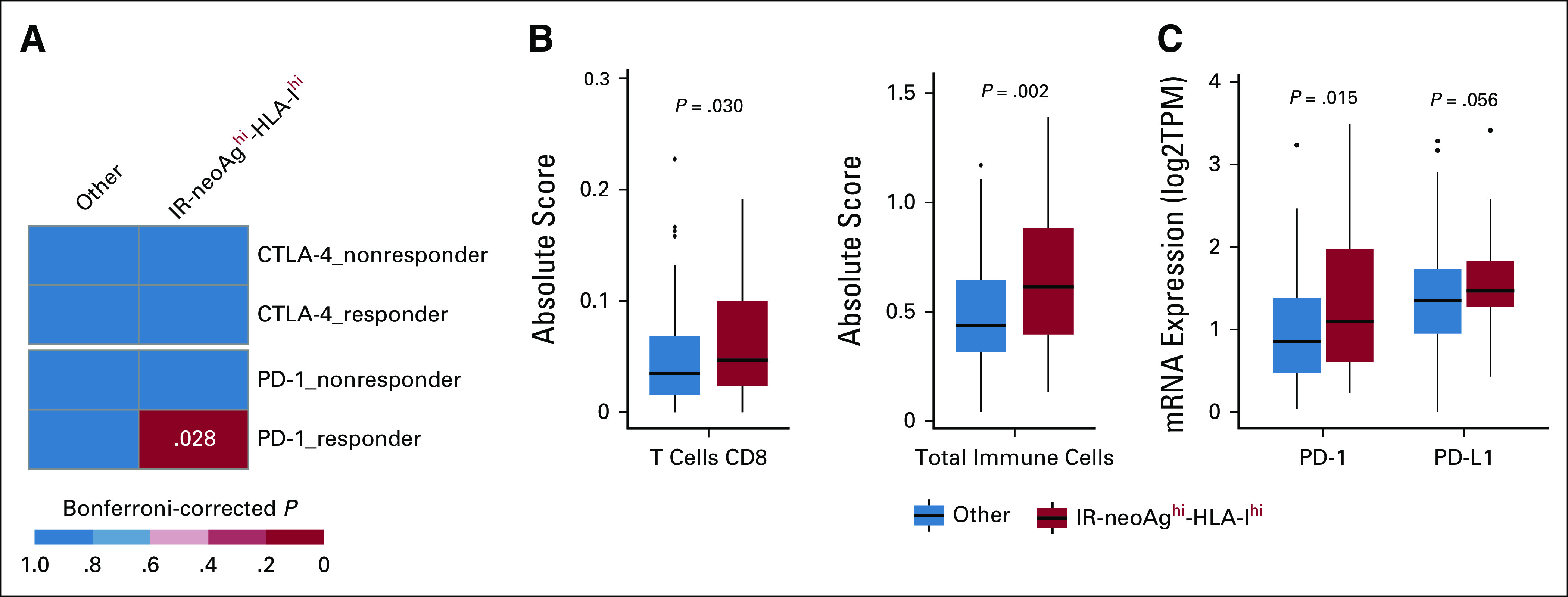
High IR-neoAg and high HLA class-I expression identify pancreatic cancers with similarities to tumors responsive to immune checkpoint blockade therapy. (A) Submap analysis comparing two groups from the TCGA-PAAD cohort (IR-neoAghi-HLA-Ihi [n = 43], and all Others [n = 135]) and four groups from an ICB-treated melanoma patient data set (anti–PD-1 responders [n = 12], anti–PD-1 nonresponders [n = 14], anti–CTLA-4 responders [n = 8], and anti–CTLA-4 nonresponders [n = 28]). Bonferroni-corrected P values were calculated in the SubMap program. (B) Box plot representation of the proportion of CD8+ T cells and total tumor-infiltrated immune cells between tumors with IR-neoAghi-HLA-Ihi and all other tumors in the TCGA-PAAD data set. Absolute score is estimated as the median expression level of all genes in the signature matrix divided by the median expression level of all genes in the mixture and is used to scale the relative cell fractions to absolute abundances. (C) Box plot representation of the tumor expression levels of immune checkpoint genes (PD-1 and PD-L1) in TCGA-PAAD tumors with IR-neoAghi-HLA-Ihi and all others. Box plots represent median and IQR. Upper whisker extends to the third quartile plus 1.5× IQR. Lower whisker extends to the first quartile minus 1.5× IQR. P values were determined using Wilcoxon test. CTLA-4, cytotoxic T-lymphocyte antigen 4; IQR, interquartile range; IR-neoAg, intron-retention neoantigens; mRNA, messenger RNA; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; TPM, transcripts per million.
DISCUSSION
We have shown that tumor-specific IR-neoAg load is an independent predictor of OS in patients with pancreatic cancer. In addition, we found that the subset of tumors with the combination of high IR-neoAg load and high HLA-I gene expression had transcriptome profiles with significant similarities to melanoma tumors that were responsive to anti–PD-1 therapy. This subset of IR-neoAghi-HLAhi tumors showed higher numbers of tumor-infiltrating immune cells, including CD8+ T cells. Collectively, our findings suggest that IR-neoAg load identifies patients with better prognosis, and together with HLA-I expression levels, could be a useful biomarker for selecting patients who may benefit from ICB therapy.
TMB has emerged as a biomarker of response to ICB therapy because it is a source of tumor-specific neoantigens that are targets of activated immune cells. TMB and neoantigen load have been shown to correlate with patient response to ICB therapy in several cancer types.1,46-48 However, pancreatic cancer is characterized by a low TMB that frequently does not meet the threshold defined by clinical trials for ICB benefit.39,49-52 Like TMB, intron retention is also a source of tumor neoantigens that can be presented by MHC-I.14,53 Thus, in cancers with low TMB, IR-neoAgs could be a potentially important biomarker for selecting patients who might benefit from ICB therapy.
Importantly, additional factors other than neoantigens influence the ability of T cells to recognize and kill tumor cells. For example, the TME is an important factor in the poor responsiveness of PDAC to ICB therapy. PDAC TME is highly desmoplastic because of the presence of cancer-associated fibroblasts and a dense extracellular matrix,54 which impedes drug delivery. Because most patients with PDAC present with advanced disease, combination therapies will undoubtedly be necessary for overcoming resistance and improving immunotherapy strategies. We provide evidence that IR-neoAgs may aid in advancing these efforts by providing a new tool for selecting patients for participation in future clinical trials.
ACKNOWLEDGMENT
This study used The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) pancreatic cancer data set. The authors acknowledge the efforts of the TCGA and ICGC research consortiums to provide the fundamental resource for our study.
Kelvin P. Lee
Research Funding: Bristol Myers Squibb
Xiongbin Lu
Consulting or Advisory Role: Pharscin Pharma
Research Funding: Vera Bradley Foundation for Breast Cancer Research
Patents, Royalties, Other Intellectual Property: I am the coinventor on one awarded patent and four pending international patent applications (Inst)
No other potential conflicts of interest were reported.
SUPPORT
Supported by Indiana University Grand Challenge Precision Health Initiative.
AUTHOR CONTRIBUTIONS
Conception and design: Chuanpeng Dong, Yunlong Liu
Financial support: Yunlong Liu
Collection and assembly of data: Chuanpeng Dong, Edward Dong
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/cci/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Kelvin P. Lee
Research Funding: Bristol Myers Squibb
Xiongbin Lu
Consulting or Advisory Role: Pharscin Pharma
Research Funding: Vera Bradley Foundation for Breast Cancer Research
Patents, Royalties, Other Intellectual Property: I am the coinventor on one awarded patent and four pending international patent applications (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Rizvi NA, Hellmann MD, Snyder A, et al. : Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124-128, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahmer JR, Tykodi SS, Chow LQ, et al. : Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455-2465, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji RR, Chasalow SD, Wang L, et al. : An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 61:1019-1031, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodman AM, Castro A, Pyke RM, et al. : MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med 12:45, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valero C, Lee M, Hoen D, et al. : The association between tumor mutational burden and prognosis is dependent on treatment context. Nat Genet 53:11-15, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samstein RM, Lee CH, Shoushtari AN, et al. : Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51:202-206, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hendriks LE, Rouleau E, Besse B: Clinical utility of tumor mutational burden in patients with non-small cell lung cancer treated with immunotherapy. Transl Lung Cancer Res 7:647-660, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orth M, Metzger P, Gerum S, et al. : Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat Oncol 14:141, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amin S, Baine MJ, Meza JL, et al. : The association of the sequence of immunotherapy with the survival of unresectable pancreatic adenocarcinoma patients: A retrospective analysis of the National Cancer Database. Front Oncol 10:1518, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Principe DR, Korc M, Kamath SD, et al. : Trials and tribulations of pancreatic cancer immunotherapy. Cancer Lett 504:1-14, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maleki Vareki S: High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 6:157, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balachandran VP, Luksza M, Zhao JN, et al. : Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551:512-516, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hegde S, Krisnawan VE, Herzog BH, et al. : Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell 37:289-307.e9, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smart AC, Margolis CA, Pimentel H, et al. : Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 36:1056-1058, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dvinge H, Bradley RK: Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7:45, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Dumartin L, Mafficini A, et al. : Splice variants as novel targets in pancreatic ductal adenocarcinoma. Scientific Rep 7:2980, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan DJ, Mitra M, Chiu AM, et al. : Intron retention is a robust marker of intertumoral heterogeneity in pancreatic ductal adenocarcinoma. NPJ Genom Med 5:55, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutter C, Zenklusen JC: The Cancer Genome Atlas: Creating lasting value beyond its data. Cell 173:283-285, 2018 [DOI] [PubMed] [Google Scholar]
- 19.Waddell N, Pajic M, Patch AM, et al. : Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518:495-501, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.GTEx Consortium : The Genotype-Tissue Expression (GTEx) project. Nat Genet 45:580-585, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Badea L, Herlea V, Dima SO, et al. : Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 55:2016-2027, 2008 [PubMed] [Google Scholar]
- 22.Pei H, Li L, Fridley BL, et al. : FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16:259-266, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang G, Schetter A, He P, et al. : DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS One 7:e31507, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang S, He P, Wang J, et al. : A novel MIF signaling pathway drives the malignant character of pancreatic cancer by targeting NR3C2. Cancer Res 76:3838-3850, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobin A, Davis CA, Schlesinger F, et al. : STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29:15-21, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anders S, Pyl PT, Huber W: HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31:166-169, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynisson B, Alvarez B, Paul S, et al. : NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 48:W449-W454, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orenbuch R, Filip I, Comito D, et al. : arcasHLA: High-resolution HLA typing from RNAseq. Bioinformatics 36:33-40, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman AM, Liu CL, Green MR, et al. : Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12:453-457, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoshida Y, Brunet JP, Tamayo P, et al. : Subclass mapping: Identifying common subtypes in independent disease data sets. PLoS One 2:e1195, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roh W, Chen PL, Reuben A, et al. : Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med 9:eaah3560, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen R, Li P, Li B, et al. : Identification of distinct immune subtypes in colorectal cancer based on the stromal compartment. Front Oncol 9:1497, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patro R, Duggal G, Love MI, et al. : Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417-419, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ritchie ME, Phipson B, Wu D, et al. : limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu G, Wang LG, Han Y, et al. : clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284-287, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y: Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. J R Stat Soc B 57:289-300, 1995 [Google Scholar]
- 37.Goel MK, Khanna P, Kishore J: Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res 1:274-278, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dexter F: Wilcoxon-Mann-Whitney test used for data that are not normally distributed. Anesth Analg 117:537-538, 2013 [DOI] [PubMed] [Google Scholar]
- 39.Raphael BJ, Hruban RH, Aguirre AJ, et al. : Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32:185-203.e13, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin X, Ye L, Wang X, et al. : Follicular helper T cells remodel the immune microenvironment of pancreatic cancer via secreting CXCL13 and IL-21. Cancers (Basel) 13:3678, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruno TC: New predictors for immunotherapy responses sharpen our view of the tumour microenvironment. Nature 577:474-476, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sautès-Fridman C, Petitprez F, Calderaro J, et al. : Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 19:307-325, 2019 [DOI] [PubMed] [Google Scholar]
- 43.Ahn S, Lee JC, Shin DW, et al. : High PD-L1 expression is associated with therapeutic response to pembrolizumab in patients with advanced biliary tract cancer. Sci Rep 10:12348, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel SP, Kurzrock R: PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 14:847-856, 2015 [DOI] [PubMed] [Google Scholar]
- 45.Maleki Vareki S, Garrigos C, Duran I: Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev Oncol Hematol 116:116-124, 2017 [DOI] [PubMed] [Google Scholar]
- 46.Snyder A, Makarov V, Merghoub T, et al. : Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371:2189-2199, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Rooij N, van Buuren MM, Philips D, et al. : Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 31:e439-e442, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yarchoan M, Hopkins A, Jaffee EM: Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 377:2500-2501, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415-421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chalmers ZR, Connelly CF, Fabrizio D, et al. : Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9:34, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan TA, Yarchoan M, Jaffee E, et al. : Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann Oncol 30:44-56, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stenzinger A, Allen JD, Maas J, et al. : Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer 58:578-588, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Apcher S, Daskalogianni C, Lejeune F, et al. : Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proc Natl Acad Sci USA 108:11572, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kleeff J, Korc M, Apte M, et al. : Pancreatic cancer. Nat Rev Dis Primers 2:16022, 2016 [DOI] [PubMed] [Google Scholar]