

PURPOSE
Interpretation of genomic variants in tumor samples still presents a challenge in research and the clinical setting. A major issue is that information for variant interpretation is fragmented across disparate databases, and aggregation of information from these requires building extensive infrastructure. To this end, we have developed Genome Nexus, a one-stop shop for variant annotation with a user-friendly interface for cancer researchers and clinicians.
METHODS
Genome Nexus (1) aggregates variant information from sources that are relevant to cancer research and clinical applications, (2) allows high-performance programmatic access to the aggregated data via a unified application programming interface, (3) provides a reference page for individual cancer variants, (4) provides user-friendly tools for annotating variants in patients, and (5) is freely available under an open source license and can be installed in a private cloud or local environment and integrated with local institutional resources.
RESULTS
Genome Nexus is available at https://www.genomenexus.org. It displays annotations from more than a dozen resources including those that provide variant effect information (variant effect predictor), protein sequence annotation (Uniprot, Pfam, and dbPTM), functional consequence prediction (Polyphen-2, Mutation Assessor, and SIFT), population prevalences (gnomAD, dbSNP, and ExAC), cancer population prevalences (Cancer hotspots and SignalDB), and clinical actionability (OncoKB, CIViC, and ClinVar). We describe several use cases that demonstrate the utility of Genome Nexus to clinicians, researchers, and bioinformaticians. We cover single-variant annotation, cohort analysis, and programmatic use of the application programming interface. Genome Nexus is unique in providing a user-friendly interface specific to cancer that allows high-performance annotation of any variant including unknown ones.
CONCLUSION
Interpretation of cancer genomic variants is improved tremendously by having an integrated resource for annotations. Genome Nexus is freely available under an open source license.
INTRODUCTION
The past decade of molecular profiling of tumor samples, including national and international projects such as The Cancer Genome Atlas (TCGA),1 institutional clinical genomics initiatives such as MSK-IMPACT,2 and cross-institutional consortia such as AACR Project GENIE,3 has revealed a complex landscape of genetic variants across cancer types. There is a tremendous range in the number of mutations identified in individual cancer samples, from less than a handful of mutations per sample in some tumor samples to several thousand mutations in protein-coding genes per sample in others. Not all of these mutations are biologically functional or clinically relevant. Identifying the few functionally relevant mutations that contribute to tumor initiation and progression (drivers) among the many nononcogenic mutations (passengers) thus presents a major challenge in cancer research and in the clinical care of patients with cancer.
CONTEXT
Key Objective
To improve annotation and interpretation of genomic variants in cancer.
Knowledge Generated
Genome Nexus is a comprehensive resource integrating annotations from more than a dozen sources relevant to cancer. The annotations can be accessed through a performant application programming interface and an intuitive user interface, supporting various use cases of variant interpretation. It is freely available under an open source license and can be installed in a private cloud or local environment and integrated with local institutional resources.
Relevance
Genome Nexus is the main annotation service for the popular cancer genomics tool cBioPortal, which serves thousands of users daily. Now, it is offered as a standalone tool for annotation, allowing researchers, clinicians, and genomic infrastructure developers to leverage it directly in their own workflows.
One of the main challenges that a cancer researcher or physician faces when interpreting variants is that variant information is fragmented across disparate databases. Many individual resources exist that aid in the interpretation of cancer variants, from variant prevalence,4-6 functional impact prediction,7-9 to manually curated biologic and clinical implications.10-12 After collecting information from these various resources, combining and sorting them poses another challenge as the presentation of variant information is often inconsistent among different resources. These challenges are amplified in large cancer genomics research projects when thousands to millions of cancer variants need to be programmatically annotated in a high-throughput manner.
To address these challenges, there is a need to build a comprehensive resource that aggregates cancer-relevant information from individual resources, standardizes and organizes them consistently, and exposes them through unified interfaces to support annotation and interpretation of a single variant, prioritization of variants in a given patient, and analysis of variants across a cohort. It is essential that such a resource has a high-performance application programming interface (API) that supports automated real-time annotation of cancer variants at scale. It is also desirable to have an intuitive user interface (UI) for querying, visualizing, and prioritizing sources of information for variant interpretation on the basis of their functional and clinical relevance to cancer. Although various efforts exist in this space,13-19 they each have limitations: some lack API support or do not have a user-friendly web interface and others lack cancer-specific information or do not support novel variants not previously annotated, ie, variants that are unique to a sample (see the comparison of current tools in Table A1).
Here, we present Genome Nexus, a comprehensive one-stop resource for annotating and interpreting variants in human cancer (Fig 1). Genome Nexus aggregates variant information from a large number of sources that are relevant to cancer research and clinical care and integrates them into a scalable MongoDB NoSQL database. The unified information is served to researchers and clinicians through a user-friendly website20 for interpreting variants and a performant REST API for programmatic annotation of cancer variants. API clients and command-line tools, such as a MAF file annotator, are also provided for high-throughput variant annotation in a cohort. The source code of Genome Nexus and associated tools is available at GitHub21 under the open source MIT License. The tool can be installed in a public or private cloud or local environment at any institution. Genome Nexus is currently used by the cBioPortal for Cancer Genomics22-24 for annotating all mutation files (MAFs), and it serves thousands of daily users for real-time variant interpretation.
FIG 1.
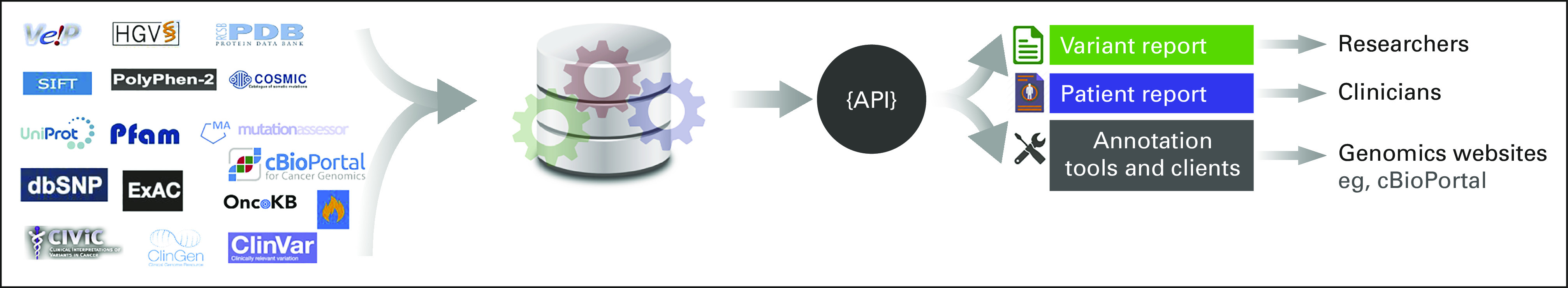
Genome Nexus overview. Genome Nexus is a comprehensive one-stop resource for annotating and interpreting variants in human cancer. The API allows for easy integration into variant and patient reports as well as existing genomics websites such as cBioPortal. API, application programming interface.
METHODS
Categorization and Integration of Existing Annotation Resources
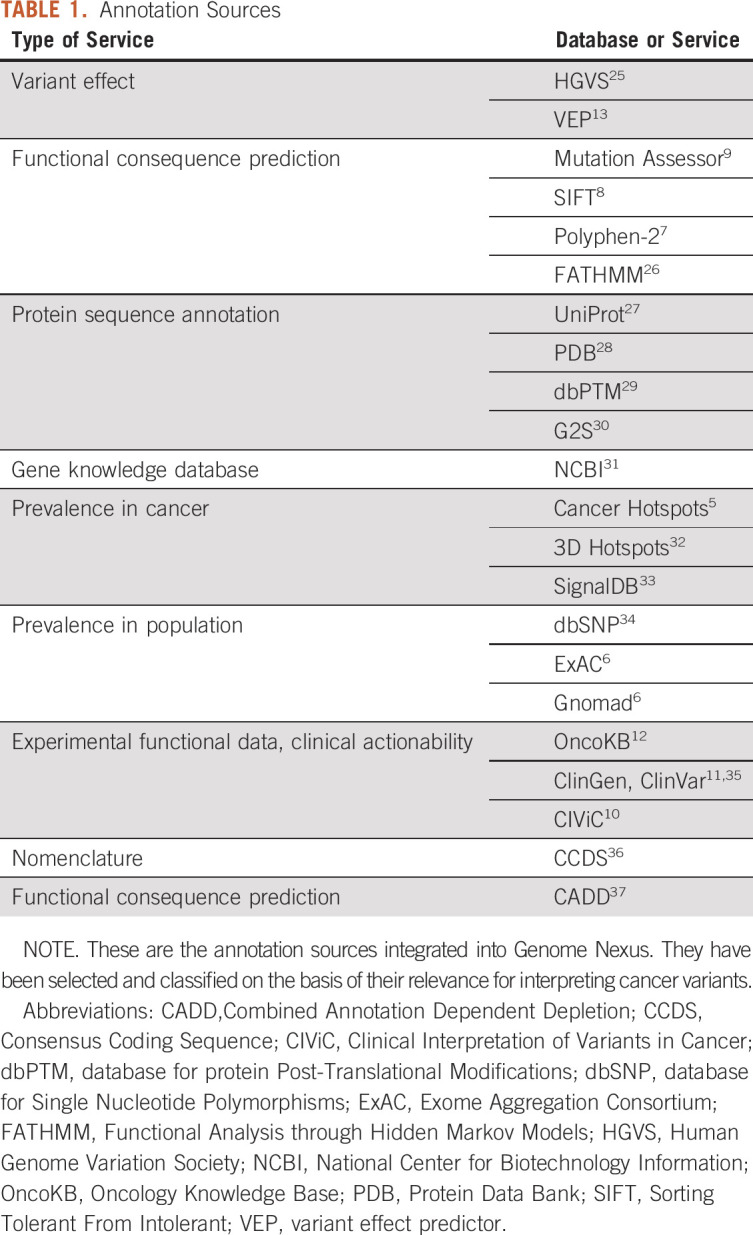
Genome Nexus retrieves variant information from a variety of resources that are used for variant annotation and interpretation. These resources have been selected and classified on the basis of their relevance for interpreting cancer variants. For a complete overview, see Table 1. Annotations retrieved from these sources are heterogeneous. They span a great range of different types of annotation, from biologic annotation and statistical data to clinical guidelines, and they are provided in different data formats. Insufficient standardization and nomenclature in reporting affect gene identifiers and isoforms (some resources use gene names, and others protein names, with a variety of different isoforms and identifiers) and variant notations. To address these challenges, we unify gene, transcript, and protein identifiers to Ensembl IDs and follow HGVS nomenclature25 for variant annotation.
TABLE 1.
Annotation Sources
A High-Performance Web API for Variant Annotation
Genome Nexus consists of three main components: a database, a REST API, and a website (Fig 2). The MongoDB database can be accessed through a REST API, which is written in Java. The website's front end is a single-page app built using React that connects to the REST API. The database stores two types of variant annotation sources: (1) static and (2) dynamic. The static ones are simple lookups where no computation needs to be performed, for example, gnomAD and dbSNP. These can be stored in their entirety in the database. The annotations are versioned, and scripts have been developed to update them as new versions become available. Dynamic sources can compute annotations for new variants on the fly, examples of which are variant effect predictor (VEP) and Mutation Assessor. These cannot be stored in their entirety in the database or precomputed because the number of possible unique variants is infinite. Therefore, these annotations are obtained from the respective annotation sources on the fly and cached in the database. The cache is cleared whenever new versions of the annotation source become available.
FIG 2.

Technical architecture overview. The left side of the figure shows the full stack from the UIs at the top to the database at the bottom. The right side describes the stack in more detail. The top shows several ways that end users can access the annotations including the Genome Nexus Website, cBioPortal, potential other genomics websites, and the command-line interface. The middle shows what programmatic clients the end UIs leverage to request annotations from the REST API, and the bottom details the annotation retrieval from the database and external annotation sources. API, application programming interface; MAF, Mutation Annotation Format; REST, Representational State Transfer; UI, user interface; VCF, Variant Call Format.
Both annotation sources can be accessed in the same manner through the REST API. VEP's output is used as the core variant JSON format. Annotations from other sources are integrated by extending this core JSON data (Fig 3C). The data can be queried by common variant nomenclatures and identifiers, including HGVS notation (genomic, cDNA, or protein change), dbSNP identifiers, and COSMIC identifiers. Different versions of genome assemblies (GRCh37 and GRCh38) are supported. The API specification of the REST API follows the OpenAPI format, which allows one to auto-generate clients in a variety of programming languages including JavaScript, R, Python, and Java (Fig 2). Both the front end for Genome Nexus and cBioPortal use these clients. Documentation is available that illustrates how to use the Python and R clients in the form of Jupyter Notebooks. In addition, there are command-line interfaces available to annotate MAF and VCF files using Genome Nexus.
FIG 3.
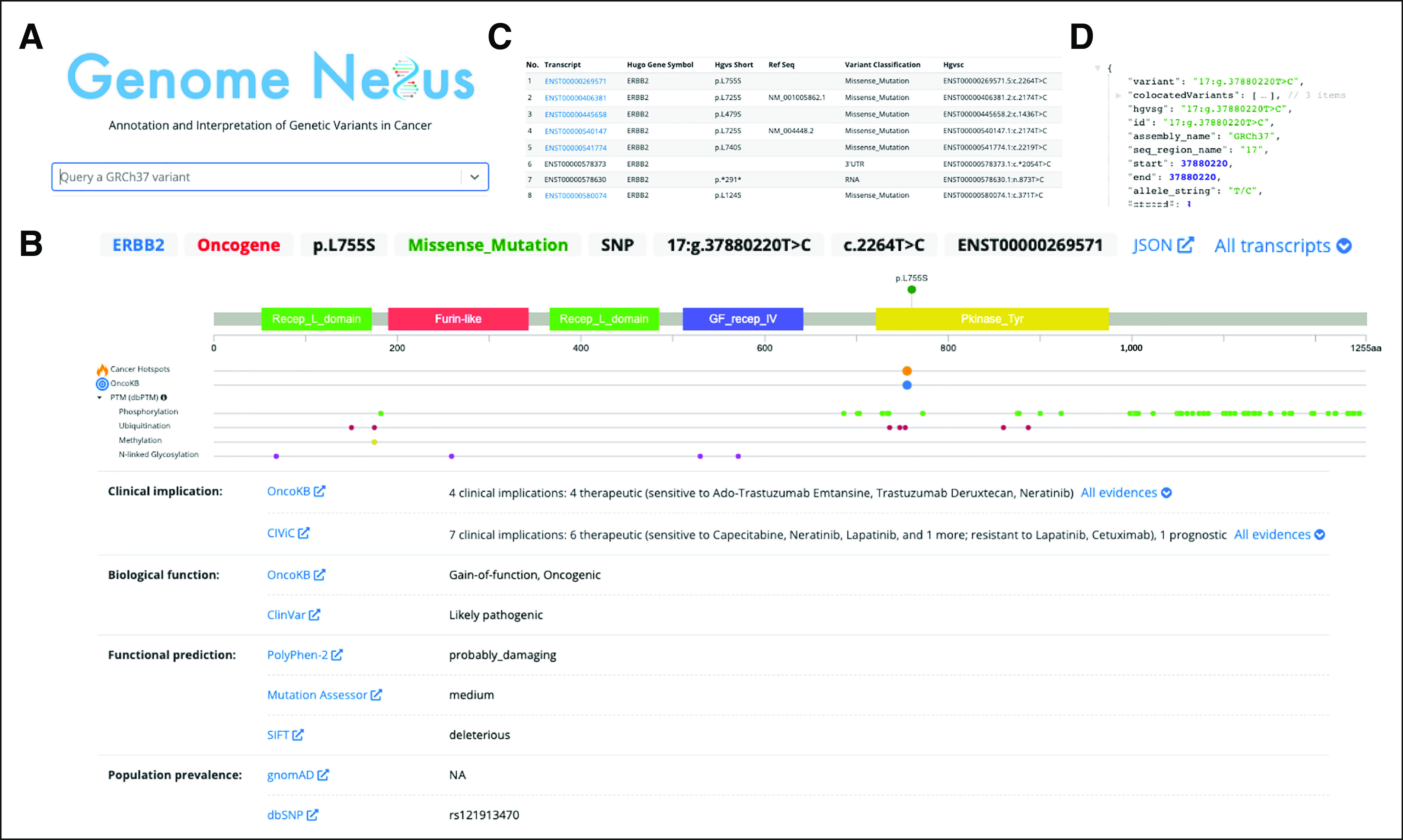
Interpretation of a single cancer variant using Genome Nexus. The ERBB2 L755S variant is used as an example. The homepage allows one to search for (A) a variant, which then leads the user to (B) the Variant Page. This page includes a lollipop diagram, showing the domains (PFAM) and annotations from several other resources (Cancer Hotspots, OncoKB, dbPTM, Uniprot, and variant effect predictor). Each of these annotation sources is hyperlinked when hovering and can be investigated in more detail. Below the lollipop plot, there is a summary of information for each annotation source. They are grouped by higher-level categories, such as clinical implication, biologic function, functional prediction, and population prevalence. The displayed transcript is ENST00000269571. One can investigate any transcript for ERBB2 by clicking on (C) the All transcripts dropdown. The raw data from the application programming interface can be seen in (D). CIViC, Clinical Interpretation of Variants in Cancer; dbPTM, database for protein Post-Translational Modifications; dbSNP, database for Single Nucleotide Polymorphisms; NA, not applicable; OncoKB, Oncology Knowledge Base; PTM, Post-Translational Modification; SIFT, Sorting Tolerant From Intolerant.
A Scalable Cloud Deployment Architecture
Genome Nexus is fully containerized to facilitate easy deployment to any cloud provider. Both the Genome Nexus Java Spring Boot app and the Mongo database with preloaded data can be found on Docker Hub.38 It is possible to deploy VEP containers as well, to have a setup that does not rely on the public VEP service. Configuration files are provided for both Docker Compose and Kubernetes. For a single node setup with relatively little traffic, the Docker Compose configuration is a good option. For a high-traffic, high-reliability setup with multiple nodes, one can use the Kubernetes configuration files, which are used in production for the public instance of Genome Nexus.
RESULTS
To demonstrate the utility of Genome Nexus, we describe several use cases: (1) annotating a single variant, (2) cohort analysis in cBioPortal, (3) annotating a list of variants in a mutations file, and (4) using the API programmatically.
Interpreting a Single Variant—ERBB2 L755S
A common use case for clinicians and researchers is to gather information about a single variant and interpret it in the disease context. Genome Nexus allows users to search a variant and access, visualize, and analyze the integrated information described above through an intuitive web interface that is specifically designed for interpreting variants in cancer. Figure 3 shows an example of the interpretation of ERBB2 L755S using Genome Nexus. First, the user searches for a variant (Fig 3A), which takes them to the Variant Page (Fig 3B) where all annotations about ERBB2 L755S from various sources are presented and organized on the basis of their relevance to cancer. From the lollipop diagram of ERBB2, one can see that the variant occurs in the kinase domain, that it is a mutational hotspot in cancer, and that it is oncogenic and a biomarker of drug sensitivity on the basis of both OncoKB and CIViC. There are also ubiquitination and phosphorylation sites adjacent to the variant. One can see that several sources are in agreement, that is, both the functional prediction sources, PolyPhen-2 and SIFT, predict this event to have the highest impact on their respective scales. OncoKB also annotates this variant as oncogenic and as gain-of-function. At first sight, there seems to be discordance between the population databases dbSNP and gnomAD. There is an ID for dbSNP, but not for gnomAD. Using the link to directly go to dbSNP helps clarify this discrepancy: there are no frequency data in dbSNP, indicating that the variant is not a common germline variant. For each gene, the canonical transcript is annotated by default, in this case ENST00000269571. One can, however, investigate any transcript for ERBB2 in addition to the default one by clicking on the All transcripts dropdown (Fig 3D). A common issue is that people refer to variants only by their amino acid change, which can differ across transcripts. For example, in two RefSeq IDs (NM_001005862.1 and NM_004448.2), the amino acid change is L725S instead of L755S. To further investigate a different transcript, one can click on the ID and see the lollipop representation and other associated annotations. Finally, to get the data for each annotation source in a format easily used in any programming language, one can click on the JSON link to retrieve the raw representation from the API in JSON.
Interpreting Variants in a Cohort in cBioPortal
A common use case in cancer genomics is to analyze all mutations in a gene that occurred in a patient cohort (Fig 4), for example, to study the landscape of somatic driver mutations to the gene in the cohort. Although one can identify potential driver mutations on the basis of their frequencies (ie, hotspot mutations), less frequent mutations are more difficult to interpret and therefore would require additional annotations for their interpretation. Figure 4 shows an example of the analysis of 799 EGFR mutations in a cohort of 10,945 patients sequenced using the MSK-IMPACT2 assay and visualized in the cBioPortal. All mutations are annotated by the Genome Nexus API in real time when the mutations are visualized, providing relevant information from various sources to help users interpret the mutations (Figs 4A-4E). In this example, 548 mutations occur in identified cancer mutational hotspots in EGFR.5 The most frequent EGFR mutations in this cohort (eg, L858R, T790M, and E746_A750del) occur in the kinase domain. Five hundred eighty-six mutations were annotated as oncogenic or likely oncogenic (driver) by OncoKB, including the majority of hotspot mutations (540) and 46 less frequent mutations. The remaining mutations (213) were annotated as likely neutral or of unknown significance. Figure A1 shows another example of analysis of 74 CCND1 (cyclin D1) mutations in TCGA samples. Missense mutations in 19 samples were identified as hotspot mutations on residues T286 and P287. As shown in Figure A1, T286 is a ubiquitination site and phosphorylation site. It is known that T286 ubiquitination is dependent on its phosphorylation, and mutations of T286 reduce ubiquitination and subsequent degradation of the oncoprotein.39 Since the kinase GSK-3β, which phosphorylates T286, is proline-directed kinase,40 mutations of P287 will inhibit the phosphorylation of T286 by disrupting the interaction between GSK-3β and cyclin D1. In addition to the two hotspots, OncoKB annotated five nonsense mutations and two in-frame deletions as drivers, all of which are near the C-terminal end adjacent to T286. This implies, consistent with the literature, that disrupting the ubiquitination of CCND1 is the predominant mechanism in tumors to stabilize this oncoprotein.
FIG 4.
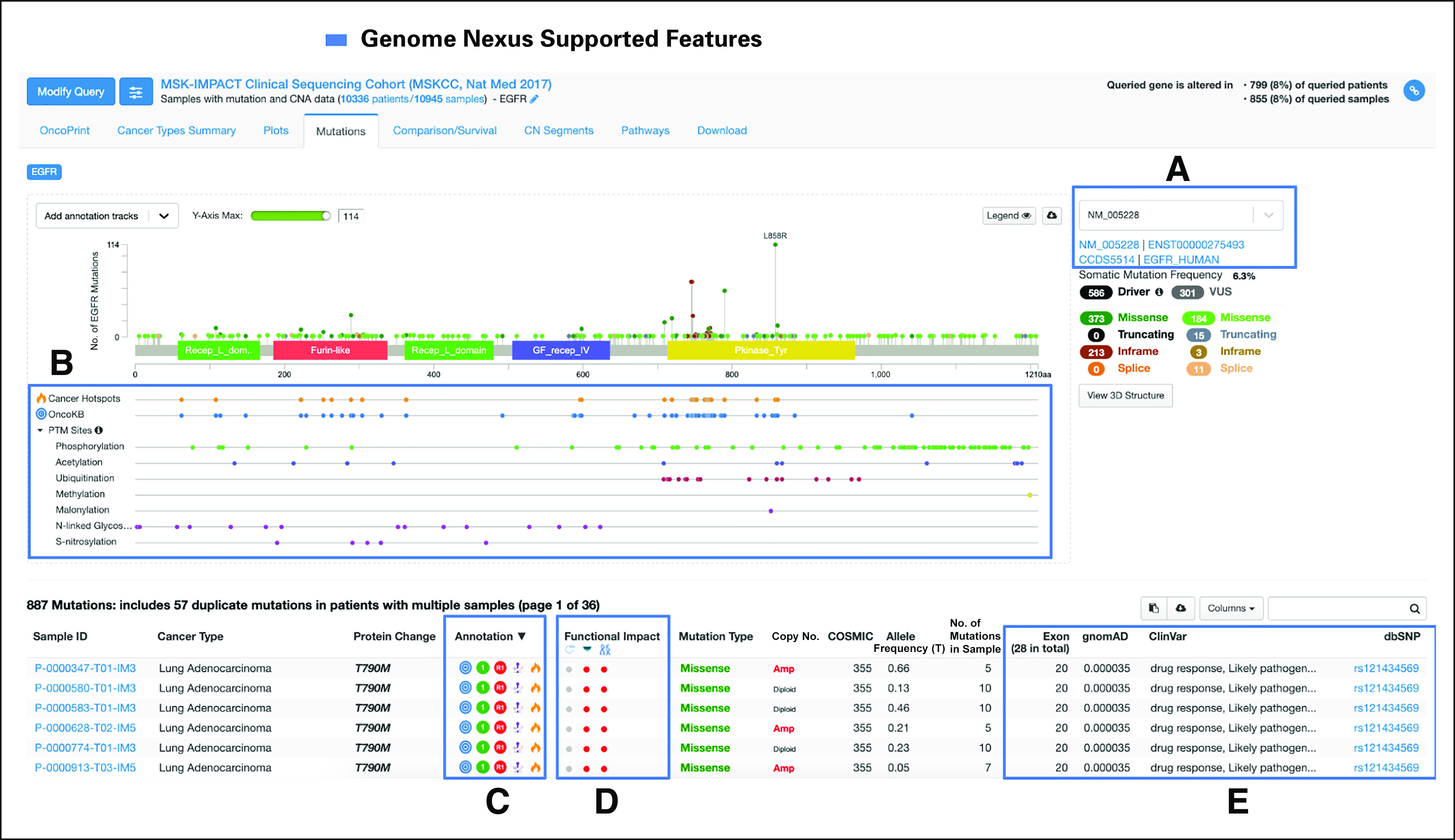
Interpreting variants in a cohort in cBioPortal (EGFR). (A) Users can switch between different transcripts and see how the mutations affect their transcript of choice. (B) Protein-level annotations, such as PTM sites, can be seen along with the protein representation of the transcript. In addition, one can see (C-E) many of the variant-specific annotations provided by Genome Nexus as columns in the mutation table, including Hotspots, Functional Impact, gnomAD, and ClinVar. dbSNP, database for Single Nucleotide Polymorphisms; OncoKB, Oncology Knowledge Base; PTM, Post-Translational Modification; VUS, Variant of Unknown Significance.
Annotating Mutations in a User-Supplied Cohort
Genome Nexus allows annotating a user-supplied cohort via a command-line interface. This is useful when trying to annotate larger data sets or when looking to annotate cohorts programmatically. Common mutation formats in both MAF and VCF are supported. The output is a MAF file with all desired annotations from Genome Nexus included in separate columns. Optional parameters can be used to choose which annotations to include or exclude.
In addition to the command-line tool, there is a web interface to annotate mutations. This offers the same functionality as described in the section of cohort analysis in cBioPortal, but on a user-supplied cohort instead. In Figure 5, mutations in a cohort of the lung, breast, and ovarian cancer samples from the TCGA are supplied. Genome Nexus annotates the mutations and then displays lollipop diagrams for each gene. Navigating through each of them shows that there are 122 mutations in EGFR, 85 in BRCA1, 113 in BRCA2, and 136 in PTEN. Using the search field to filter the mutations in the lollipop diagram helps to compare mutational patterns in the lollipop across the protein. For instance, the ratio of truncating mutations in BRCA1 is much higher in serous ovarian cancer than in invasive breast carcinoma and lung adenocarcinoma.
FIG 5.
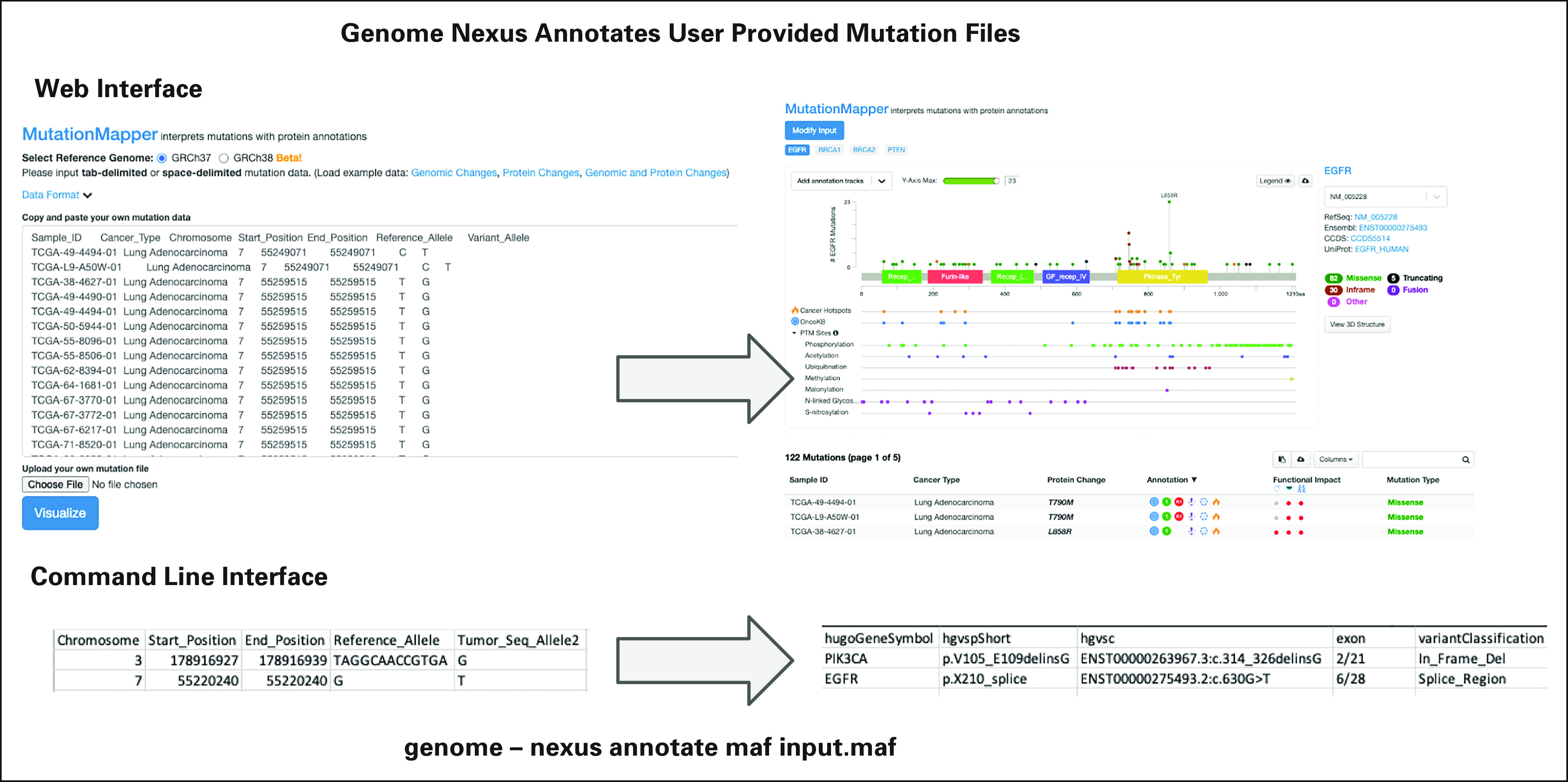
Annotating mutations in a user-supplied cohort. At the top, mutations in a cohort are supplied through the web interface. Genome Nexus annotates the mutations, and subsequently, lollipop diagrams for each gene are displayed. At the bottom, a mutations file is supplied to the command-line client, which outputs a mutations file with several new columns, including protein change, exon, and variant classification. OncoKB, Oncology Knowledge Base; PTM, Post-Translational Modification.
Programmatic Access to the API
The use cases described above are examples that leverage the Genome Nexus API programmatically. Tool builders can do the same thing by using one of the OpenAPI clients generated in their preferred programming language. We provide documentation on using the OpenAPI-generated clients41 and notebook examples describing how to perform an analysis that leverages the API. The high-performance API allows any bioinformatics application to connect and perform variant annotation.
Using container technologies such as Docker and Kubernetes, the Genome Nexus API can be easily installed on local servers or in a cloud environment. This will ensure that sensitive patient variant data can stay within institutional firewalls when required.
DISCUSSION
The development of Genome Nexus originated from the increasing need to annotate and interpret cancer variants in cBioPortal with information from various sources. In 2015, we carefully reviewed existing tools available in the space and concluded that none would satisfy the requirements of cBioPortal, specifically (1) comprehensive annotations relevant and specific to cancer, (2) a performant API to support real-time annotation for thousands of users, and (3) an API architecture that can be easily extended to incorporate new sources. The development of Genome Nexus started shortly after that and picked up speed in 2017. Since 2019, the Genome Nexus API has been supporting all annotation needs of the data import pipeline (batch annotation) and the UI (real-time annotation) of cBioPortal. Although the development of Genome Nexus originally stemmed from the needs of cBioPortal, it was designed from the beginning as a standalone tool and resource. With its performant API, flexible architecture, and cloud-friendly deployment, Genome Nexus is ready to support variant annotation needs for bioinformatics pipelines and tools beyond cBioPortal.
Moreover, geared with a user-friendly interface to query and interpret variants, the public instance of Genome Nexus20 is positioned to become a reference resource for variant annotation and interpretation. We would like to emphasize that Genome Nexus was not developed to replace interpretation by experts (cancer researchers and clinicians) but rather to support them. It was designed to organize and visualize variant information in a form that can be quickly understood by users and therefore help improve and accelerate their interpretation of variants. For example, when there is conflicting information from different sources, it is especially important for users to carefully interpret the provided information in the context of their use cases with their domain knowledge and expertise.
Genome Nexus is under active development, and there are still many challenges and limitations that need to be addressed. First of all, Genome Nexus relies on variant information from external resources (Table 1), whose quality and stability can vary. The quality of information was ensured by 1) careful review of the resource's annotations for common variants in cancer or 2) the resource being in use by the cancer genomics community for many years. As the stability of these resources varies, it is inevitable that some of them go offline from time to time. The caching implementation alleviates this issue (the annotation will be retrieved from the cache first even if a resource is offline), but does not fully address it for new variants. For some web services, we, therefore, either import a local copy of the data (PFAM) or run a Docker image of the service (VEP). When possible, we include multiple resources for the same type of annotation, for example, OncoKB and CIViC for clinical interpretation and Uniprot and dbPTM for PTM sites, so that when one service is down, others can be used. For future work, we will improve detection and error handling of unstable services.
A second related challenge around aggregating many resources is versioning. For clinical decision support systems, it is essential to have controlled data updates. Resources that are imported or run locally are already versioned. In an ideal scenario, one would be able to run a local instance for all annotation sources, but this is unfortunately not always possible. The difficulty with external resources that are pulled on the fly and cached is that the version might change over time. We, therefore, plan to add timestamps, such that users will be able to determine when the annotation was generated.
Third, there is a lack of standardization at the API level across annotation sources, which requires developers to write a lot of custom code to handle each individual source. We plan to alleviate this burden by evolving the software architecture to an extensible plug-in framework such that anyone can write API plug-ins to integrate other resources of interest for the open source community and proprietary annotation sources for their local installations.
Besides the plans to address the previous challenges mentioned, we also aim to extend Genome Nexus beyond its current scope. Genome Nexus currently only supports single-nucleotide variants and insertions and deletions. We plan to expand it to support other alteration types such as structural variants and copy number alterations. In addition, we would like to support model organisms in the future.
ACKNOWLEDGMENT
We are grateful to have received valuable feedback on Genome Nexus' user experience from Ritika Kundra, Yichao Sun, Ramya Madupuri, Linda Bosch, and Kim Monkhorst.
APPENDIX
FIG A1.
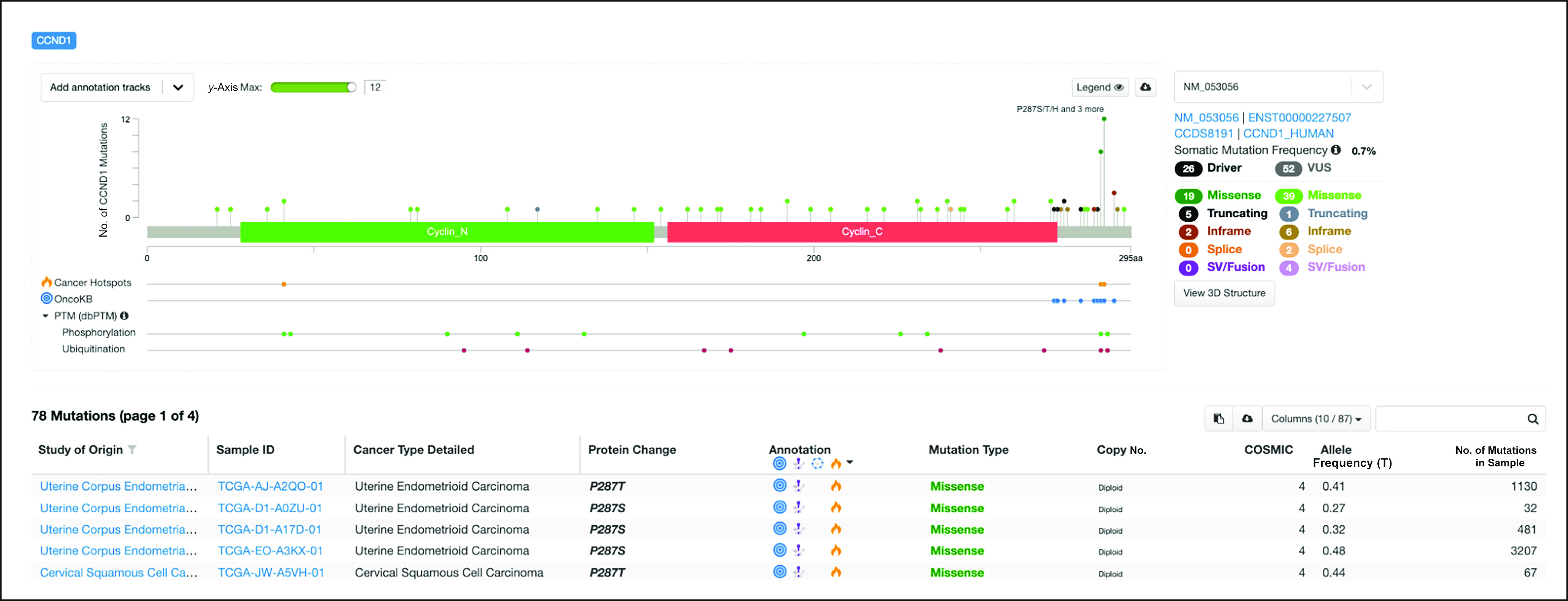
Interpreting CCND1 mutations in the TCGA cohort. The example shows 74 CCND1 (cyclin D1) mutations in the TCGA PanCan Atlas cohort. Missense mutations in 19 samples were identified as hotspot mutations on residues T286 and P287. The former is a ubiquitination site and phosphorylation site. In addition to the two hotspots, OncoKB annotated five nonsense mutations and two in-frame deletions as drivers, all of which are near the C-terminal end adjacent to T286. OncoKB, Oncology Knowledge Base; PTM, Post-Translational Modification; SV, Structural Variant; TCGA, The Cancer Genome Atlas; VUS, Variant of Unknown Significance.
TABLE A1.
Feature Comparison of Variant Annotation Services

Robert Sheridan
Stock and Other Ownership Interests: Agios, Alnylam (I), Alphabet (I), Amazon Inc, Amgen (I), Biogen (I), Bluebird Bio, Bristol Myers Squibb (I), Clementia Pharmaceuticals (I), General Electric Company, Johnson & Johnson (I), Merck (I), Northwest Biotherapeutics (I), Pfizer (I), Portola Pharmaceuticals, Viatris, Organon & Co, ClearPoint Neuro Inc, 2Seventy Bio Inc
Angelica Ochoa
Stock and Other Ownership Interests: Infinity Pharmaceuticals, AstraZeneca/MedImmune
Avery Wang
Stock and Other Ownership Interests: Gilead Sciences, CVS, Brickell Biotech, Zomedica, Bristol Myers Squibb
Adam Abeshouse
Stock and Other Ownership Interests: Ligand Pharmaceuticals
Cyriac Kandoth
Employment: Thermo Fisher Scientific (I)
Stock and Other Ownership Interests: Massive Bio
Sander Rodenburg
Consulting or Advisory Role: The Hyve
Sander Tan
Employment: myTomorrows
Sjoerd van Hagen
Stock and Other Ownership Interests: Galapagos NV
Remond J. A. Fijneman
Research Funding: Merck BV (Inst), Personal Genome (Inst), Delfi Diagnostics (Inst), Cergentis (Inst)
Patents, Royalties, Other Intellectual Property: several patents pending (Inst)
Gerrit A. Meijer
Stock and Other Ownership Interests: crcBioscreen BV
Research Funding: Exact Sciences (Inst), Sysmex (Inst), Sentinel Diagnostics (Inst), Personal Genome Diagnostics (Inst), Hartwig Medical Foundation (Inst)
Patents, Royalties, Other Intellectual Property: Several patents pending (Inst)
No other potential conflicts of interest were reported.
SUPPORT
Supported by Marie-Josée and Henry R. Kravis Center for Molecular Oncology, The Fund for Innovation in Cancer Informatics (to J.G. and N.S.), and National Cancer Institute Cancer Center Core Grant No. P30-CA008748.
AUTHOR CONTRIBUTIONS
Conception and design: Ino de Bruijn, Xiang Li, Benjamin Gross, Robert Sheridan, Avery Wang, Hongxin Zhang, Ananthan Sadagopan, Cyriac Kandoth, Sander Rodenburg, Pieter Lukasse, Sjoerd van Hagen, Remond J. A. Fijneman, Gerrit A. Meijer, Nikolaus Schultz, Jianjiong Gao
Collection and assembly of data: Ino de Bruijn, Xiang Li, Selcuk Onur Sumer, Angelica Ochoa, Manda Wilson, Emily Zhang, Alice Thum, Ananthan Sadagopan, Zachary Heins, Sander Rodenburg, Nikolaus Schultz, Jianjiong Gao
Data analysis and interpretation: Ino de Bruijn, Xiang Li, Selcuk Onur Sumer, Hongxin Zhang, Aaron Lisman, Adam Abeshouse, Zachary Heins, Sander Tan, Nikolaus Schultz, Jianjiong Gao
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/cci/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Robert Sheridan
Stock and Other Ownership Interests: Agios, Alnylam (I), Alphabet (I), Amazon Inc, Amgen (I), Biogen (I), Bluebird Bio, Bristol Myers Squibb (I), Clementia Pharmaceuticals (I), General Electric Company, Johnson & Johnson (I), Merck (I), Northwest Biotherapeutics (I), Pfizer (I), Portola Pharmaceuticals, Viatris, Organon & Co, ClearPoint Neuro Inc, 2Seventy Bio Inc
Angelica Ochoa
Stock and Other Ownership Interests: Infinity Pharmaceuticals, AstraZeneca/MedImmune
Avery Wang
Stock and Other Ownership Interests: Gilead Sciences, CVS, Brickell Biotech, Zomedica, Bristol Myers Squibb
Adam Abeshouse
Stock and Other Ownership Interests: Ligand Pharmaceuticals
Cyriac Kandoth
Employment: Thermo Fisher Scientific (I)
Stock and Other Ownership Interests: Massive Bio
Sander Rodenburg
Consulting or Advisory Role: The Hyve
Sander Tan
Employment: myTomorrows
Sjoerd van Hagen
Stock and Other Ownership Interests: Galapagos NV
Remond J. A. Fijneman
Research Funding: Merck BV (Inst), Personal Genome (Inst), Delfi Diagnostics (Inst), Cergentis (Inst)
Patents, Royalties, Other Intellectual Property: several patents pending (Inst)
Gerrit A. Meijer
Stock and Other Ownership Interests: crcBioscreen BV
Research Funding: Exact Sciences (Inst), Sysmex (Inst), Sentinel Diagnostics (Inst), Personal Genome Diagnostics (Inst), Hartwig Medical Foundation (Inst)
Patents, Royalties, Other Intellectual Property: Several patents pending (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Hoadley KA, Yau C, Hinoue T, et al. : Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173:291-304.e6, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zehir A, Benayed R, Shah RH, et al. : Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23:703-713, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.AACR Project GENIE Consortium : AACR project GENIE: Powering precision medicine through an International Consortium. Cancer Discov 7:818-831, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forbes SA, Beare D, Gunasekaran P, et al. : COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43:D805-D811, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang MT, Bhattarai TS, Schram AM, et al. : Accelerating discovery of functional mutant alleles in cancer. Cancer Discov 8:174-183, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lek M, Karczewski KJ, Minikel EV, et al. : Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285-291, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adzhubei I, Jordan DM, Sunyaev SR: Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet, 2013. Chapter 7:Unit7.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar P, Henikoff S, Ng PC: Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073-1081, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Reva B, Antipin Y, Sander C: Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res 39:e118, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffith M, Spies NC, Krysiak K, et al. : CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet 49:170-174, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landrum MJ, Lee JM, Benson M, et al. : ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44:D862-D868, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakravarty D, Gao J, Phillips SM, et al. : OncoKB: A precision oncology knowledge base. JCO Precis Oncol 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLaren W, Gil L, Hunt SE, et al. : The ensembl variant effect predictor. Genome Biol 17:122, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xin J, Mark A, Afrasiabi C, et al. : High-performance web services for querying gene and variant annotation. Genome Biol 17:91, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funcotator—GATK. https://gatk.broadinstitute.org/hc/en-us/articles/360037224432-Funcotator
- 16.Wang K, Li M, Hakonarson H: ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cingolani P, Platts A, Wang LL, et al. : A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80-92, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamborero D, Rubio-Perez C, Deu-Pons J, et al. : Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med 10:25, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douville C, Carter H, Kim R, et al. : CRAVAT: Cancer-related analysis of variants toolkit. Bioinformatics 29:647-648, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genome Nexus. https://genomenexus.org
- 21.Genome Nexus Source Code. https://github.com/genome-nexus
- 22.Gao J, Aksoy BA, Dogrusoz U, et al. : Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerami E, Gao J, Dogrusoz U, et al. : The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401-404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The cBioPortal for Cancer Genomics. https://cbioportal.org
- 25.den Dunnen JT, Dalgleish R, Maglott DR, et al. : HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37:564-569, 2016 [DOI] [PubMed] [Google Scholar]
- 26.Shihab HA, Gough J, Cooper DN, et al. : Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34:57-65, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The UniProt Consortium : UniProt: The universal protein knowledgebase. Nucleic Acids Res 45:D158-D169, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman HM, Westbrook J, Feng Z, et al. : The protein data bank. Nucleic Acids Res 28:235-242, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee T-Y, Huang H-D, Hung J-H, et al. : dbPTM: an information repository of protein post-translational modification. Nucleic Acids Res 34:D622-D627, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Sheridan R, Sumer SO, et al. : G2S: A web-service for annotating genomic variants on 3D protein structures. Bioinformatics 34:1949-1950, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown GR, Hem V, Katz KS, et al. : Gene: A gene-centered information resource at NCBI. Nucleic Acids Res 43:D36-D42, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J, Chang MT, Johnsen HC, et al. : 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med 9:4, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srinivasan P, Bandlamudi C, Jonsson P et al. : The context-specific role of germline pathogenicity in tumorigenesis. Nat Genet 53:1577-1585, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherry ST, Ward MH, Kholodov M, et al. : dbSNP: The NCBI database of genetic variation. Nucleic Acids Res 29:308-311, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gelb BD, Cavé H, Dillon MW, et al. : ClinGen's RASopathy Expert Panel consensus methods for variant interpretation. Genet Med 20:1334-1345, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pruitt KD, Harrow J, Harte RA, et al. : The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 19:1316-1323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rentzsch P, Witten D, Cooper GM, et al. : CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47:D886-D894, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Genome Nexus Docker Containers. https://hub.docker.com/r/genomenexus/
- 39.Santra MK, Wajapeyee N, Green MR: F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 459:722-725, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diehl JA, Cheng M, Roussel MF, et al. : Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12:3499-3511, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Genome Nexus API Documentation. https://docs.genomenexus.org/api
- 42.myvariant.info. http://myvariant.info/
- 43.VEP. https://useast.ensembl.org/info/docs/tools/vep/index.html
- 44.Ensembl VEP REST. https://rest.ensembl.org/#VEP
- 45.Open CRAVAT. https://opencravat.org/
- 46.Funcotator. https://gatk.broadinstitute.org/hc/en-us/articles/360035889931-Funcotator-Information-and-Tutorial
- 47.ANNOVAR. https://annovar.openbioinformatics.org/en/latest/
- 48.SnpEff. https://pcingola.github.io/SnpEff/se_introduction/
- 49.Cancer genome interpreter. https://www.cancergenomeinterpreter.org/home
- 50.Allele registry. http://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/landing