

Abstract
Background
D‐penicillamine is used for patients with primary biliary cirrhosis due to its hepatic copper decreasing and immunomodulatory potentials. The results from randomised clinical trials have been inconsistent.
Objectives
To systematically review the beneficial and harmful effects of D‐penicillamine for patients with primary biliary cirrhosis.
Search methods
We identified trials through electronic searches of The Cochrane Hepato‐Biliary Group Controlled Trials Register (September 2003), The Cochrane Central Register of Controlled Trials on The Cochrane Library (Issue 3, 2003), MEDLINE (January 1966 to September 2003), EMBASE (January 1980 to September 2003), The Chinese Biomedical CD Database (January 1979 to August 2003), and LILACS (1982 to 2003); through manual searches of bibliographies; and by contacting authors of the trials and pharmaceutical companies.
Selection criteria
We included randomised clinical trials comparing D‐penicillamine with placebo/no intervention or other control intervention irrespective of language, year of publication, and publication status.
Data collection and analysis
Two reviewers independently assessed the methodological quality of the trials and extracted data, validated by a third reviewer. The primary outcomes were 1) mortality and 2) a combination of those who died or underwent liver transplantation. We analysed dichotomous outcomes as relative risk (RR) with 95% confidence interval (CI) by a fixed effect model and a random effects model. We investigated sources of heterogeneity by subgroup analyses and tested the robustness of our findings by sensitivity analyses.
Main results
We included seven trials randomising 706 patients with primary biliary cirrhosis. D‐penicillamine compared with placebo/no intervention tended to increase mortality (RR 1.34, 95% CI 1.09 to 1.64, fixed; RR 1.46, 95% CI 0.85 to 2.50, random). However, there was substantial heterogeneity. No significant differences were detected regarding the risks of mortality or liver transplantation, pruritus, liver complications, progression of liver histological stage, or the levels of liver biochemical variables (except alanine aminotransferase). D‐penicillamine versus placebo/no intervention significantly increased the risk of adverse events (RR 3.11, 95% CI 2.33 to 4.16, fixed; RR 4.18, 95% CI 1.38 to 12.69, random).
Authors' conclusions
D‐penicillamine did not appear to reduce the risk of mortality, but significantly increased the occurrences of adverse events in patients with primary biliary cirrhosis. We do not support the use of D‐penicillamine for patients with primary biliary cirrhosis.
Plain language summary
D‐penicillamine did not reduce the risk of mortality of patients with primary biliary cirrhosis but increased the occurrences of adverse events
Primary biliary cirrhosis is an uncommon, chronic liver disease of unknown etiology. D‐penicillamine, a cupruretic drug, has been tested in randomised clinical trials and is used to treat patients with primary biliary cirrhosis. After combining results from seven trials, D‐penicillamine did not appear to improve survival of patients. D‐penicillamine was associated with a four‐time increase of adverse events. There were no significant differences between D‐penicillamine and placebo/no intervention with respect to clinical changes, liver histology, and liver biochemistry.
Background
Primary biliary cirrhosis is an uncommon chronic progressive liver disease of unknown etiology. Ninety per cent of patients with primary biliary cirrhosis are females, and the majority are diagnosed after the age of 40 years (James 1981). Primary biliary cirrhosis is classically defined on the basis of the triad: antimitochondrial antibodies, found in over 95 per cent patients with primary biliary cirrhosis (Fregeau 1989; Lacerda 1995; Invernizzi 1997; Turchany 1997; Mattalia 1998); abnormal liver function tests that are typically cholestatic (raised activity of alkaline phosphatases are the most frequently seen abnormality); and characteristic liver histological changes (Scheuer 1967) without extrahepatic biliary obstruction (Kaplan 1996). Patients may either be diagnosed during a symptomatic phase (with common symptoms as pruritus, fatigue, jaundice, liver enlargement, signs of portal hypertension, sicca complex, and scleroderma‐like lesions) when survival is decreased or during an asymptomatic phase when the prognosis is relatively favourable (Beswick 1985; Balasubramaniam 1990). However, 40 to 100 per cent of the asymptomatic patients will subsequently develop symptoms of primary biliary cirrhosis (Nyberg 1989; Metcalf 1996; Prince 2000).
Although the etiology remains unknown, primary biliary cirrhosis is in many respects analogous to the graft‐versus‐host syndrome in which the immune system is sensitised to foreign proteins. Most primary biliary cirrhosis patients have increased expression of class II human leukocyte antigen (HLA) histocompatibility on bile duct cells (Ballardini 1984; Van den Oord 1986). The bile duct epithelium in these patients is infiltrated with cytotoxic T‐cells (Yamada 1986). Lacrimal and pancreatic glands, for example, with a high concentration of HLA class II antigens on their epithelium, may be involved in the disease process (Epstein 1982).
Patients with primary biliary cirrhosis are administered many drugs. Ursodeoxycholic acid, a bile acid, is the most extensively used drug (Verma 1999). However, a meta‐analysis and a systematic Cochrane review were unable to demonstrate any significant effect of ursodeoxycholic acid on mortality or liver transplantation (Goulis 1999; Gluud 2002). Over the years, a number of other drugs have been evaluated for primary biliary cirrhosis. Attempts to treat primary biliary cirrhosis using immune‐modulating and other agents such as azathioprine (Heathcote 1976; Christensen 1985), prednisolone (Mitchison 1992), chlorambucil (Hoofnagle 1986), cyclosporine (Wiesner 1990), colchicine (Warnes 1987; Vuoristo 1995; Poupon 1996), or methotrexate (Kaplan 1991; Lindor 1995) have resulted in clinical effects that have not led to widespread acceptance of these drugs in patients with primary biliary cirrhosis (Kaplan 1994).
D‐penicillamine is a cupruretic drug known for its efficacy in treating Wilson's disease (Sternlieb 1964; Deiss 1971). Primary biliary cirrhosis is also associated with increased hepatic levels of copper. Therefore, the major rationale for evaluating D‐penicillamine in primary biliary cirrhosis was its ability to induce cupruresis. In addition, D‐penicillamine has other pharmacologic actions of potential benefit, including antifibrogenic effect, ability to decrease circulating immune complexes, and inhibitory effect on lymphocyte function (Nimni 1972; Epstein 1979; Lipsky 1980). There are about 2,500,000 patients with primary biliary cirrhosis in the world (Kim 2000). At least 2.8 per cent of these patients are probably being treated with D‐penicillamine according to UK experience (Verma 1999). This means that about 70,000 primary biliary cirrhosis patients around the world may receive D‐penicillamine as treatment. This figure may even be larger as we think that physicians in UK are conservative ‐ at least when compared to other European physicians regarding interventions for alcoholic liver disease (Gluud 1993).
Conflicting reports concerning the effects of D‐penicillamine in the treatment of primary biliary cirrhosis have been published. Earlier reports showed that D‐penicillamine was a promising drug, improving survival in patients with primary biliary cirrhosis and having relatively few side‐effects (Triger 1980; Epstein 1981; Taal 1983). Several later studies showed that D‐penicillamine did decrease hepatic levels of copper, but it did not have a beneficial effect on symptoms related to primary biliary cirrhosis, hepatic biochemistries, histologic progression, or survival. In addition, D‐penicillamine was associated with up to a 46 per cent incidence of major toxicity, most commonly proteinuria, allergic drug reaction, and rarely bone marrow depression (Matloff 1982; Neuberger 1985; Dickson 1985; Bodenheimer 1985). We have been unable to identify meta‐analyses or systematic reviews on the beneficial and harmful effects of D‐penicillamine in patients with primary biliary cirrhosis.
Objectives
To systematically assess the beneficial and harmful effects of D‐penicillamine in patients with primary biliary cirrhosis.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised clinical trials irrespective of language, year of publication, and publication status. We excluded studies using quasi‐randomisation (eg, allocation by date of birth).
Types of participants
Patients with primary biliary cirrhosis, ie, patients having at least two of the following: elevated serum activity of alkaline phosphatases (or other markers of intrahepatic cholestasis), and/or a positive result for serum mitochondrial antibody, and/or liver biopsy findings diagnostic for or compatible with primary biliary cirrhosis.
Types of interventions
D‐penicillamine at any dose compared with placebo, no intervention, another active drug, or other dose of D‐penicillamine. Co‐interventions were allowed as long as both intervention arms of the randomised clinical trial received similar co‐interventions.
Types of outcome measures
The primary outcome measures were:
Mortality.
A combination of mortality or liver transplantation.
The secondary outcome measures were:
Liver transplantation.
Pruritus: number of patients without improvement of pruritus and/or pruritus score.
Fatigue: number of patients without improvement of fatigue and/or fatigue score.
Liver complications: number of patients developing variceal bleeding, ascites, hepatic encephalopathy, jaundice, hepato‐renal syndrome, or sicca complex.
Liver biopsy findings: deterioration of liver histological stage or score.
Liver biochemistry: serum (s)‐bilirubin; s‐alkaline phosphatases; s‐gamma‐glutamyltransferase; s‐aspartate aminotransferase; s‐alanine aminotransferase; s‐albumin; s‐cholesterol (total); plasma immunoglobulin M, etc.
Adverse events. The adverse event is defined as any untoward medical occurrence in a patient in either of the two arms of the included randomised clinical trials, which did not necessarily have a causal relationship with the treatment, but did, however, result in a dose reduction, discontinuation of treatment, or registration of the advent as an adverse event/side effect. The adverse events are subdivided into non‐serious adverse events as well as serious adverse events according to the ICH‐GCP guidelines (ICH‐GCP 1997). A serious adverse event is any event that leads to death, is life‐threatening, requires inpatient hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or congenital anomaly/birth defect, or any important medical event which may jeopardize the patient or requires intervention to prevent it.
Quality of life: a broad concept that includes physical functioning (ability to carry out activities of daily living such as self‐care and walking around), psychological functioning (emotional and mental well‐being), social functioning (social relationships and participation in social activities), and perception of health, pain, and overall satisfaction with life.
Cost‐effectiveness: the estimated costs connected with the interventions were to be weighed against any possible health gains.
Search methods for identification of studies
Relevant randomised clinical trials were identified by searching The Cochrane Hepato‐Biliary Group Controlled Trials Register (September 2003), The Cochrane Central Register of Controlled Trials on The Cochrane Library (Issue 3, 2003), MEDLINE (January 1966 to September 2003), and EMBASE (January 1980 to September 2003), The Chinese Biomedical CD Database (January 1979 to August 2003), and LILACS (1982 to 2003). See 'Appendix 1' for the search strategies applied to the individual electronic databases.
Further trials were identified by reading the reference lists of the identified studies. We wrote to the principal authors of the identified randomised clinical trials and to researchers active in the field to inquire about additional randomised clinical trials they might know of. We also wrote to the pharmaceutical companies that sponsored D‐penicillamine in the identified trials in order to obtain any unidentified or unpublished randomised clinical trial.
Data collection and analysis
The meta‐analyses were performed following the published protocol and the recommendations given by the Cochrane Reviewers' Handbook (Alderson 2003).
Trials selection Identified trials were listed and two contributors (YG and SLF) independently evaluated whether the trials fulfilled the inclusion criteria. Excluded trials were listed in 'Characteristics of excluded studies' with the reasons for exclusion. Disagreements were resolved by discussion.
Data extraction YG and SLF independently extracted data onto a standard paper form, and CG validated the data extraction. We wrote to the authors of the included trials and asked them to specify the data of interest if those data were not reported clearly in their reports.
Assessment of methodological quality of included trials The methodological quality of the randomised clinical trials was assessed using four components (Schulz 1995; Moher 1998; Kjaergard 2001):
Generation of the allocation sequence
Adequate, if the allocation sequence was generated by a computer or random number table. Drawing of lots, tossing of a coin, shuffling of cards, or throwing dice will be considered as adequate if a person who was not otherwise involved in the recruitment of participants performed the procedure;
Unclear, if the trial was described as randomised, but the method used for the allocation sequence generation was not described;
Inadequate, if a system involving dates, names, or admittance numbers were used for the allocation of patients. These studies are known as quasi‐randomised and were excluded from the present review.
Allocation concealment
Adequate, if the allocation of patients involved a central independent unit, on‐site locked computer, identically appearing numbered drug bottles or containers prepared by an independent pharmacist or investigator, or sealed envelopes;
Unclear, if the trial was described as randomised, but the method used to conceal the allocation was not described;
Inadequate, if the allocation sequence was known to the investigators who assigned participants or if the study was quasi‐randomised. Such studies were excluded from the present review.
Blinding (or masking)
Adequate, if the trial was described as double blind and the method of blinding involved identical placebo or active drug;
Unclear, if the trial was described as double blind, but the method of blinding was not described;
Not performed, if the trial was not double blind.
Follow‐up
Adequate, if the numbers and reasons for dropouts and withdrawals in all intervention groups were described or if it was specified that there were no dropouts or withdrawals;
Unclear, if the report gave the impression that there had been no dropouts or withdrawals, but this was not specifically stated;
Inadequate, if the number or reasons for dropouts and withdrawals were not described.
Characteristics of patients Number of patients randomised; patient inclusion and exclusion criteria; mean (or median) age; sex ratio; number of patients lost to follow‐up; drop‐outs; withdrawals.
Characteristics of interventions Type, dose, and form of D‐penicillamine intervention; type of intervention in the control group and collateral interventions; trial duration.
Characteristics of outcomes All outcomes were extracted from each included trial.
We analysed mortality and/or liver transplantation at maximum follow‐up. We analysed other outcomes, which were repeatedly observed on patients (like liver biochemistry, clinical symptoms, etc.) at maximum follow‐up.
Statistical methods We intended to include parallel group and cross‐over trials. For cross‐over trials, we only intended to include data from the first period. We used the statistical package (RevMan Analyses 1.0.2) provided by The Cochrane Collaboration. We presented dichotomous data as relative risk (RR) with 95% confidence interval (CI) and continuous outcome measures by weighted mean differences (WMD) with 95% CI. All analyses for primary outcomes were performed according to the intention‐to‐treat principle, which means that participants in the trials should have been analysed in the groups to which they were randomised, regardless of whether they received or adhered to the allocated intervention.
We examined intervention effects by using both a random effects model (DerSimonian 1986) and a fixed effect model (DeMets 1987) with the significant level set at P‐value ≤ 0.05. If the results of the two analyses led to the same conclusion, we presented only the results of the fixed effect analysis. In case of discrepancies of the two models, we reported the results of both models. We explored the presence of statistical heterogeneity by chi‐squared test with significance set at P‐value ≤ 0.10 and measured the quantities of heterogeneity by I2. However, due to possible few anticipated trials and the relative large number of outcomes going to be assessed, we interpreted significant results with caution.
Subgroup analysis and sensitivity analysis We performed subgroup analyses, in which trials were grouped according to the methodological quality of the included trials, dosage of D‐penicillamine, and duration of treatment and follow‐up. The high methodological quality was confined to adequate generation of the allocation sequence, allocation concealment, blinding, and follow‐up. The difference between the estimates of two subgroups was estimated according to Altman 2003.
Regarding the primary outcome measure, ie, mortality, we included patients with incomplete or missing data in the sensitivity analyses by imputing them (Hollis 1999):
Available case analysis: data on only those whose results are known, using as denominator the total number of patients who completed the trial;
Assuming poor outcome: dropouts from both the D‐penicillamine and control groups had the primary outcomes;
Assuming good outcome: none of the dropouts from the D‐penicillamine and control groups had the primary outcomes;
Extreme case favouring D‐penicillamine: none of the dropouts from the D‐penicillamine‐group but the dropouts from the control group had the primary outcomes;
Extreme case favouring control: all dropouts from the D‐penicillamine‐group but none from the control group had the primary outcomes.
For secondary outcomes, we adopted 'available case analysis'. Therefore, in the review, the number of patients in the denominator changed according to the secondary outcomes investigated.
Bias exploration Funnel plot was used to provide a visual assessment of whether treatment estimates are associated with study size. The performance of the available methods of detecting publication bias and other biases (Begg 1994; Egger 1997; Macaskill 2001) vary with the magnitude of the treatment effect, the distribution of study size, and whether a one‐ or two‐tailed test is used (Macaskill 2001). Therefore, we used the most appropriate method, which has a good trade‐off in the sensitivity and specificity, based on characteristics of the trials included in this review.
Results
Description of studies
We identified a total of 178 references through electronic searches of The Cochrane Hepato‐Biliary Group Controlled Trials Register (n = 26), The Cochrane Central Register of Controlled Trials on The Cochrane Library (n = 28), MEDLINE (n = 29), EMBASE (n = 51), The Chinese Biomedical CD Database (n = 43), and LILACS (n = 1). We excluded 143 duplicates and clearly irrelevant references by reading abstracts. Accordingly, 35 references were retrieved for further assessment. Of these, we excluded three because they were non‐randomised clinical studies or observational studies. The remaining 32 references referred to seven randomised clinical trials involving 706 patients with primary biliary cirrhosis, which fulfilled our inclusion criteria ('Characteristics of included studies' table). The year of publication of these trials ranged from year 1981 to 1985. The Bassendine 1982 trial was published as an abstract only, while the other six trials were published as full papers.
The mean age of the patients was about 51 years. The majority of the patients were females (female/male: 495/53) in the trials reporting gender distribution. The Bassendine 1982 trial and the Taal 1983 trial did not report the baseline histological status of primary biliary cirrhosis. Data from the other five trials showed that more patients had stage III or IV than stage I or II (stage III or IV /stage I or II: 443/168).
Of the seven trials, six trials compared D‐penicillamine with placebo/no intervention. One trial compared 750 mg/day D‐penicillamine with 250 mg/day D‐penicillamine (Bodenheimer 1985). The Bassendine 1982 trial had three groups of comparisons: D‐penicillamine 1g/day, 250 mg/day, and no intervention group. We extracted data from the group of 1g/day versus no intervention, which was the most commonly used dosage. All of the remaining five trials employed placebo as control intervention. The D‐penicillamine dosage of 1g/day was applied in four trials (Bassendine 1982; Matloff 1982; Taal 1983; Dickson 1985), 1.2 g/day in the Neuberger 1985 trial, and 0.6 g/day in the Epstein 1981 trial. The duration of treatment and follow‐up varied from 1.5 to 9 years.
Risk of bias in included studies
Generation of the allocation sequence was adequate in one trial (Dickson 1985) and unclear in the other six. Allocation concealment was adequate in two trials (Dickson 1985; Matloff 1982) and unclear in the other five. Blinding was adequate in five trials, was considered inadequate in the Neuberger 1985 trial, and was not performed in the Bassendine 1982 trial. It should be noted that the description of the control in the trials reporting double blinding was not sufficient since all the trials claiming to be double blind only stated the use of identical in appearance placebo tablets, but did not address smell and taste. Follow‐up was adequate in five trials, but considered inadequate in two trials (Bodenheimer 1985; Epstein 1981). In total, 90 patients (17%) were lost to follow‐up: 79 patients in D‐penicillamine and 11 in control group. In the Neuberger 1985 trial, 35 (36%) patients in the D‐penicillamine group and 7 (8%) in the control group were lost to follow‐up. None of the trials reported a sample size estimate. No trials reported that they used intention‐to‐treat analyses. Overall, only the Dickson 1985 trial was viewed as a high methodological quality trial, ie, having adequate generation of allocation, allocation concealment, blinding, and follow‐up.
Effects of interventions
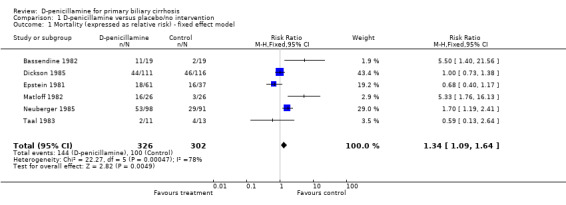
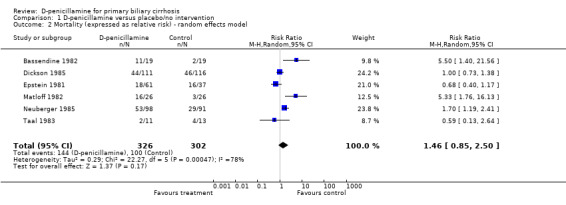
D‐penicillamine versus placebo/no intervention Mortality Six trials (628 patients) provided data to estimate the risk of mortality of D‐penicillamine versus placebo/no intervention (Comparison 01‐01; Comparison 01‐02). The mortality risk was 1.46 (95% CI 0.85 to 2.50) by the random effects model and 1.34 (95% 1.09 to 1.64) by the fixed effect model. The trials had significant heterogeneity (I2 = 77.5%).
We performed sensitivity analyses to assess the impact of missing data. The 'assuming poor outcome' showed a significant harmful effect of D‐penicillamine on mortality. However, the 'assuming good outcome' analyses did not detect a significant difference of mortality between D‐penicillamine and placebo/no intervention (RR 0.95, 95% CI 0.71 to 1.26). The 'extreme case favouring control' showed a significant harmful effect of D‐penicillamine on mortality (RR 1.92, 95% CI 1.51 to 2.43, fixed, RR 2.13, 95%CI 1.16 to 3.90, random). The 'extreme case favouring D‐penicillamine' showed a significant beneficial effect of D‐penicillamine (RR 0.66, 95% CI 0.51 to 0.86). We also performed 'available case analysis', in which we did not find a significant difference between D‐penicillamine and placebo (RR 1.08, 95% 0.82 to 1.43).
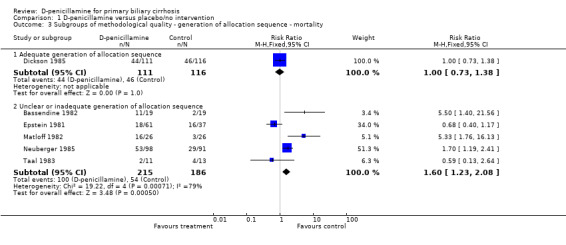
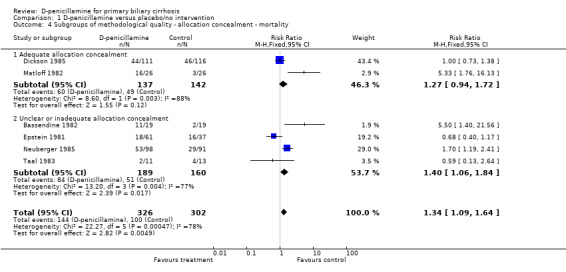
We performed subgroup analyses according to different methodological quality, dosages of D‐penicillamine, duration of treatment and follow‐up, and histological stages (Comparison 01‐05 to 11). The estimate of intervention effect were significantly different in the subgroup analyses of generation of allocation sequence (P = 0.03), allocation concealment (P = 0.04), blinding (P = 0.007), and follow‐up (P = 0.008). The subgroup analyses stratifying the trials into three dosages of D‐penicillamine (1.2 g/day, 1 g/day, or 0.6 g/day) did not show a clear increasing trend towards harmful effects of D‐penicillamine along with increased dosage (Comparison 01‐09), although the lowest dose had the lowest harm profile. The trial using dosage of 0.6 g/day showed a significant difference from the trials with 1 g/day (P = 0.04) and with 1.2 g/day (P = 0.005), while the comparison between 1 g/day and 1.2 g/day did not achieve significance. The risks of mortality in the trials with short‐term treatment and follow‐up (shorter than three years) had a significant difference with the trials with long‐term treatment and follow‐up (longer than three years) (P = 0.003).
Mortality or liver transplantation Only one trial (Neuberger 1985) reported the number of patients who underwent liver transplantation (RR 0.93, 95% CI 0.06 to 14.63). Accordingly, the relative risk of mortality or liver transplantation was 1.33 (95% CI 1.09 to 1.63) in the fixed effect model and 1.45 (95% CI 0.85 to 2.48) in the random effects model (Comparison 01‐13,01‐14).
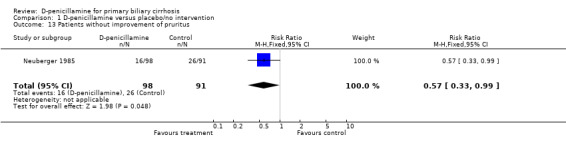
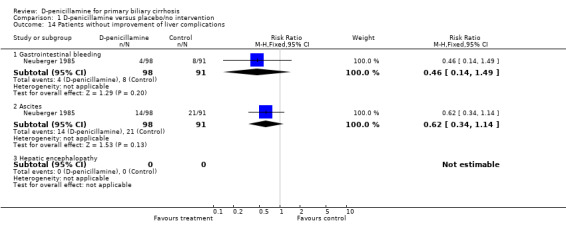
Pruritus, fatigue, and liver complications Neuberger 1985 observed a marginal beneficial effect of D‐penicillamine on pruritus (RR 0.57, 95% CI 0.33 to 0.99) (Comparison 01‐15). Evidence about fatigue was not located. For liver complication, no significant differences were found with respect to gastrointestinal bleeding and ascites (Comparison 01‐16)
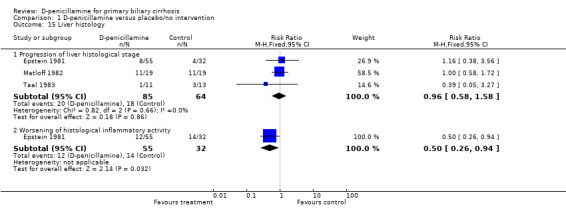
Liver histological and biochemical outcomes Data from three trials with 149 patients estimated the effects of D‐penicillamine on liver histology (Epstein 1981; Matloff 1982; Taal 1983). D‐penicillamine did not retard the progression of liver histological stage (RR 0.96, 95% CI 0.58 to 1.58) but D‐penicillamine had a significant beneficial effect on inflammatory activity in the Epstein 1981 trial (RR 0.50, 95% CI 0.26 to 0.94, one trial).
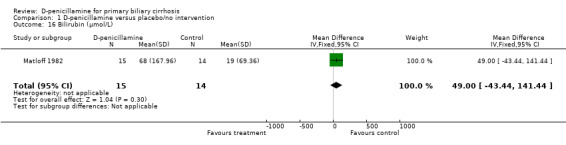
Matloff 1982 provided data on liver biochemical outcomes presented as mean changes from values for each patient before randomisation and showed no significant differences between D‐penicillamine and placebo except for alanine aminotransferase.
Adverse events All the seven trials reported adverse events in both groups. In the D‐penicillamine group, 139 (43%) patients had adverse events (types of adverse events in Table 1) versus 44 (15%) patients treated with placebo/no intervention (RR 4.18, 95% CI 1.38 to 12.69, random; RR 3.11, 95% CI 2.33 to 4.16, fixed, I2 = 93.2%) (Comparison 01‐23, 24). In the sensitivity analysis after excluding the Taal 1983 trial, which had the smallest sample size (24 patients)and the highest placebo response rate (85 per cent), the RR changed to 3.69 (95% CI 2.62 to 5.19) and I2 went down to 49.7%. We were unable to distinguish between serious and non‐serious adverse events due to insufficient reporting.
1. Adverse events in the included trials.
| Trials | D‐penicillamine | Control |
| Bassendine 1982 | Proteinuria, rash, 'lupus' syndrome, myasthenia, thrombocytopenia. | None. |
| Dickson 1985 | Hypersensitivity, cytopenia, arthralgias, linchen planus, loss of taste, proteinuria. | Cytopenia, arthralgias, linchen planus, dysgeusia, proteinuria. |
| Epstein 1981 | Rashes, proteinuria, neutropenia. | None. |
| Matloff 1982 | Goodpasture‐like syndrome, myasthenia, proteinuria, linchen planus, arthralgias, splenomegaly, rash, loss of taste, stomatitis. | Proteinuria. |
| Neuberger 1985 | Rash, proteinuria, thrombocytopenia, arthralgia, gastrointestinal upset, leucopenia, asthma, pemphigoid, loss of taste, psychosis, palpitations, non‐compliance. | Proteinuria, gastrointestinal upset, headaches, non‐compliance, neurological complications. |
| Taal 1983 | Exanthema, gastrointestinal upset, loss of taste. | Exanthema, gastrointestinal upset. |
| Bodenheimer 1985 | In 750 mg/day (high‐dose) group: Fever, rash, arthralgia, loss of taste, mouth ulcers, nausea, haemolysis, thrombocytopenia, neutropenia, pulmonary fibrosis, albuminuria, neuropathy. In 250 mg/day (low‐dose) group: Fever, rash, arthralgia, loss of taste, mouth ulcers, thrombocytopenia, neutropenia, pulmonary fibrosis, albuminuria, neuropathy. |
Quality of life and cost‐effectiveness None of the trials examined specific quality‐of‐life scales or outcomes regarding cost‐effectiveness. High‐dose D‐penicillamine versus low‐dose D‐penicillamine In the Bassendine 1982 trial, the risk of mortality tended to be lower with a high‐dose than with a low‐dose D‐penicillamine, although this difference is not significant (RR 0.26, 95% CI 0.06 to 1.05). More patients in the high‐dose group than in the low‐dose group tended to develop adverse events (RR 1.99, 95% CI 0.81 to 4.89). The Bodenheimer 1985 trial only reported the total number of deaths in the two groups, and more patients in the high‐dose group had improvement of histological progression than in the low‐dose group.
Bias exploration We did not perform funnel plot analysis and did not apply the three statistical methods to detect publication bias and other biases because the power of those would have been low and inconsistent because of the small number of included trials.
Discussion
We found that D‐penicillamine tended to have a detrimental effect on mortality of patients with primary biliary cirrhosis. The meta‐analysis also showed that the use of D‐penicillamine significantly increased the occurrences of adverse events.
Our systematic review on D‐penicillamine versus placebo/no intervention analysed only six trials involving 628 patients. This is a low number of patients (Ioannidis 2001). None of the trials reported a sample size estimate. The loss during follow‐up was relatively high in the D‐penicillamine group. The methodological trial quality was generally low, which makes it hard to interpret this sample of trials. Generally, low methodological quality trials overestimate significantly intervention effects (Schulz 1995; Moher 1998; Kjaergard 2001). If the same overestimation is valid for the present sample of trials, the prospects for D‐penicillamine for primary biliary cirrhosis look even worse, ie, the harmful effects could be even larger. On the other hand, we cannot preclude that such low‐doses D‐penicillamine may have beneficial effects because only a few trials have been performed with low‐doses. In addition, most of the trials have shorter follow‐up than the estimated median survival of 10 to 15 years (Prince 2002). Therefore, it is difficult to detect a significant difference on mortality.
Heterogeneity is an important aspect of a meta‐analysis. Heterogeneity can occur because of an artefact of the summary measures used and of trial design features such as duration of follow‐up, reliability of outcome measures, or methodological quality of the trial. It may also be due to real variations in the treatment effect, such as the underlying risk of the patients in the different trials, intervention timing or intensity, co‐intervention, or the outcome measurement and timing. Although the ideal way to study causes of true variations is within trials rather than between, in most situations we had to do with a trial level investigation in the present meta‐analyses (Glasziou 2002).
Regarding mortality, we found 'severe' heterogeneity (Higgins 2002) across the trials and also discrepancy between the fixed effect analysis and the random effects analysis. In the fixed effect analysis, we detected a significant harmful effect of D‐penicillamine, while in the random effects analysis, no significant difference was found. Due to the low number of trials, which did not allow us to perform a meaningful meta‐regression, we performed subgroup analyses according to the methodological quality, dosage of D‐penicillamine, and duration of treatment and follow‐up. Bearing in mind the observational nature of the subgroup analyses, we found that only the unclear/inadequate follow‐up tended to underestimate the beneficial effects of D‐penicillamine. This finding is in contrast to previous studies (Schulz 1995; Moher 1998; Kjaergard 2001), probably because too low number of trials were included to perform any meaningful subgroup analyses.
We found that D‐penicillamine had no significant effect on reducing the risk of mortality compared to placebo/no intervention. The pooled estimate from high quality trials also support this finding. The analyses of the four scenarios, which took the impact of missing data into consideration, showed that patients taking D‐penicillamine were more likely to have higher risk of mortality compared to patients taking placebo or getting no intervention. The 'assuming poor outcome' showed the significantly harmful effect of D‐penicillamine, while the 'assuming good outcome' did not catch any significant difference between D‐penicillamine and placebo/no intervention.
The subgroup analysis showed that the risk of mortality seemed to increase by dosage. This observation, however, was not supported by the Bassendine 1982 trial, where the patients taking high dose of D‐penicillamine had lower risk of mortality than patients on low dose. Since the ideal way to study causes of true variation is within trials rather than between, and the purpose and nature of this meta‐analysis was not to study the dose‐response, the relationship between the effect of D‐penicillamine and dosage is not clear.
It is presumed that high‐risk groups will have more to gain from an intervention and may therefore experience sufficient benefit to outweigh the harms. Whether the severity of primary biliary cirrhosis was related to the treatment effect of D‐penicillamine is not confirmed in this review. There was lack of trials to be included and also the possible relationship was not indicated in many of the trials.
Only Neuberger et al reported the number of patients having clinical changes (Neuberger 1985), which revealed that there were no significant differences on the state of pruritus, gastrointestinal bleeding, or ascites between D‐penicillamine and placebo. Although the remaining trials did not report the exact data, they all claimed that no consistent clinical improvement in either the D‐penicillamine or placebo group had been found (Dickson 1985; Matloff 1982; Taal 1983).
Data from three trials enabled us to meta‐analyse the effects of D‐penicillamine on liver histology and we found that the rate of liver histological progression neither favoured D‐penicillamine nor favoured placebo/no intervention (Epstein 1981; Matloff 1982; Taal 1983). There is a significant beneficial effect of D‐penicillamine regarding histological inflammatory activity (Epstein 1981). However, the effect is only marginally significant and based on only one trial with a small sample size of patients.
The report by Matloff et al allowed us to extract data on liver biochemical variables, which resulted in no significant differences except for alanine aminotransferase (Matloff 1982). This finding was replicated in the Neuberger 1985 trial in which alanine aminotransferase was the only significant difference among the various liver biochemical variables. Epstein 1981and Bassendine 1982 found a beneficial effect of D‐penicillamine in reducing the levels of aspartate aminotransferase and immunoglobulin. Dickson 1985 did not detect any significant effect, and Taal 1983 found that D‐penicillamine significantly decreased immunoglobulin M and G levels. Thus, the inconsistent findings across the trials weakened the conclusion of beneficial effect of D‐penicillamine on liver biochemical variables at large.
Six out of seven trials reported on adverse events and showed that the risk of adverse events in the D‐penicillamine group was, on average, four times higher than the placebo/no intervention group both in random effects and fixed effect models. Most of the adverse events were proteinuria, gastrointestinal upset, rash, cytopenia, etc.
In the meta‐analyses of adverse events, we also found severe heterogeneity across the trials. Although the results from the fixed effect and random effects models indicated that the use of D‐penicillamine highly increased the occurrences of adverse events, investigation for sources of heterogeneity was necessary. We found that I2 decreased to zero (no statistical heterogeneity) when changing the RR to the odds ratio (OR). However, the selection of a summary measure on the basis of minimising heterogeneity is a somewhat data derived approach since it generates spurious, over‐optimistic findings. It is theoretically possible that important sources of heterogeneity could be missed if the strategy of using the summary with the smallest heterogeneity statistic is universally applied (Deeks 2001a; Deeks 2002). Considering that the selection of a summary measure being argued on the grounds of consistency of effect, ease of interpretation, and mathematical properties, we left RR as the summary measure in the analysis of adverse events.
For meta‐analyses of RR, the proportional weights, given to trials estimating the same effect with the same sample size, increase with increasing event rates. The relationship becomes particularly strong when the event rates are above 50 per cent (Deeks 2001b). In this respect, we scrutinized the event rates in the included trials and we noticed that the Taal 1983 trial had the smallest sample size (24 patients), but surprisingly the highest placebo response rate, 85 per cent. It was offered the second most weight, 23 per cent in the analysis of RR, whereas the weight of 2 per cent was used in the analysis of OR. Hence, we performed a sensitivity analysis excluding the Taal 1983 trial, and it resulted in the RR of 3.69 (95% CI 2.62 to 5.19) with the acceptable moderate heterogeneity (I2 = 49.7%). Therefore, our conclusion, that the use of D‐penicillamine was associated with the increase of adverse events, was consolidated.
Authors' conclusions
Implications for practice.
D‐penicillamine did not significantly reduce the risk of mortality of patients with primary biliary cirrhosis. Furthermore, we found a significant increase of adverse events when comparing patients taking D‐penicillamine with those on placebo/no intervention. Hence, we are against using D‐penicillamine for patients with primary biliary cirrhosis.
Implications for research.
We do not recommend further randomised clinical trials aiming at establishing the value of D‐penicillamine in the treatment of primary biliary cirrhosis, at least not with the dosages employed in previous trials. The possibility that low doses may offer beneficial effects cannot be excluded. Investigators ought to report their trials according to the CONSORT Statement (www.consort‐statement.org).
What's new
| Date | Event | Description |
|---|---|---|
| 17 October 2008 | Amended | Converted to new review format. |
Acknowledgements
We primarily extend our acknowledgements to the patients who took part in and the investigators who designed and conducted the reviewed trials. We also thank Bodil Als‐Nielsen, Ronald L. Koretz, Luigi Pagliaro and Rosa Simonetti as well as the peer reviewers for valuable comments to an earlier draft. Furthermore, Dimitrinka Nikolova, Nader Salasshahri, and Styrbjørn Birch, all from The Cochrane Hepato‐Biliary Group, are thanked for expert assistance during the preparation of this review.
Appendices
Appendix 1. Search Strategies
| Database | Period | Search strategy |
| The Cochrane Hepato‐Biliary Group Controlled Trials Register | September 2003 | #1= 'PRIMARY BILIARY CIRRHOSIS' and 'D‐PENICILLAMINE' |
| The Cochrane Central Register of Controlled Trials on The Cochrane Library | Issue 3, 2003 | #1 = LIVER CIRRHOSIS BILIARY: MESH #2 = primary and biliary and cirrhosis #3 = primary biliary cirrhosis #4 = pbc #5 = #1 or #2 or #3 or #4 #6 = PENICILLAMINE: MESH #7 = CHELATING AGENTS: MESH #8 = penicillamine #9 = chelating next agent* #10 = #6 or #7 or #8 or #9 #11 = #5 and #10 |
| MEDLINE | January 1966 to September 2003 | #1 = Liver‐Cirrhosis‐Biliary: MESH #2 = primary and biliary and cirrhosis #3 = primary biliary cirrhosis #4 = PBC #5 = #1 or #2 or #3 or #4 #6 = Penicillamine: MESH #7 = Chelating‐Agents: MESH #8 = penicillamine #9 = chelating agent* #10 = #6 or #7 or #8 or #9 #11 = #5 and #10 #12 = random* or placebo* or blind* or meta‐analysis #13 = #11 and #12 |
| EMBASE | January 1980 to September 2003. | #1 = primary‐biliary‐cirrhosis: MESH #2 = biliary‐cirrhosis: MESH #3 = primary and biliary and cirrhosis #4 = primary biliary cirrhosis #5 = PBC #6 = #1 or #2 or #3 or #4 or #5 #7 = penicillamine: MESH #8 = chelating‐agent: MESH #9 = penicillamine #10 = chelating agent* #11 = #7 or #8 or #9 or #10 #12 = #6 and #11 #13 = random* or placebo* or blind* or meta‐analysis #14 = #12 and #13 |
| Chinese Biochemical CD Database | January 1979 to August 2003 | #1 = Liver‐Cirrhosis‐Biliary: MESH #2 = primary and biliary and cirrhosis #3 = primary biliary cirrhosis #4 = PBC #5 = #1 or #2 or #3 or #4 #6 = Penicillamine: MESH #7 = Chelating‐Agents: MESH #8 = penicillamine #9 = chelating agent* #10 = #6 or #7 or #8 or #9 #11 = #5 and #10 #12 = random* or placebo* or blind* or meta‐analysis #13 = #11 and #12 |
| LILACS | 1982 to 2003 | #1 = (primary and biliary and cirrhosis) or (primary biliary cirrhosis) #2 = primary biliary cirrhosis #3 = penicillamine |
Data and analyses
Comparison 1. D‐penicillamine versus placebo/no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality (expressed as relative risk) ‐ fixed effect model | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.09, 1.64] |
| 2 Mortality (expressed as relative risk) ‐ random effects model | 6 | 628 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.85, 2.50] |
| 3 Subgroups of methodological quality ‐ generation of allocation sequence ‐ mortality | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Adequate generation of allocation sequence | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.73, 1.38] |
| 3.2 Unclear or inadequate generation of allocation sequence | 5 | 401 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.60 [1.23, 2.08] |
| 4 Subgroups of methodological quality ‐ allocation concealment ‐ mortality | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.09, 1.64] |
| 4.1 Adequate allocation concealment | 2 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.94, 1.72] |
| 4.2 Unclear or inadequate allocation concealment | 4 | 349 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [1.06, 1.84] |
| 5 Subgroups of methodological quality ‐ blinding ‐ mortality | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.09, 1.64] |
| 5.1 Adequate blinding | 4 | 401 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.83, 1.39] |
| 5.2 Unclear or blinding not performed | 2 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.38, 2.72] |
| 6 Subgroups of methodological quality ‐ follow‐up ‐ mortality | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.09, 1.64] |
| 6.1 Adequate follow‐up | 5 | 530 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [1.20, 1.87] |
| 6.2 Unclear or inadequate follow‐up | 1 | 98 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.40, 1.17] |
| 7 Subgroups of dosage ‐ mortality | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 D‐penicillamine 1.2 g/day | 1 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.70 [1.19, 2.41] |
| 7.2 D‐penicillamine 1 g/day | 4 | 341 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.04, 1.84] |
| 7.3 D‐penicillamine 0.6 g/day | 1 | 98 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.40, 1.17] |
| 8 Subgroups of treatment and follow‐up duration ‐ mortality | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.09, 1.64] |
| 8.1 Long‐term treatment and long‐term follow‐up | 3 | 514 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.93, 1.43] |
| 8.2 Short‐term treatment and short‐term follow‐up | 3 | 114 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.37 [1.70, 6.66] |
| 9 Subgroups of PBC histological stage ‐ mortality | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 PBC stage III or IV | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.63, 1.15] |
| 9.2 PBC stage I or II | 1 | 27 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10 Sensitivity analyses ‐ mortality | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Available patient course analysis | 6 | 525 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.82, 1.43] |
| 10.2 Assuming poor outcome | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.09, 1.64] |
| 10.3 Assuming good outcome | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.71, 1.26] |
| 10.4 Extreme case favouring D‐penicillamine | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.51, 0.86] |
| 10.5 Extreme case favouring control | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.92 [1.51, 2.43] |
| 11 Mortality or liver transplantation ‐ fixed effect model | 6 | 628 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [1.09, 1.63] |
| 12 Mortality or liver transplantation ‐ random effects model | 6 | 628 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [0.85, 2.48] |
| 13 Patients without improvement of pruritus | 1 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.33, 0.99] |
| 14 Patients without improvement of liver complications | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 Gastrointestinal bleeding | 1 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.14, 1.49] |
| 14.2 Ascites | 1 | 189 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.34, 1.14] |
| 14.3 Hepatic encephalopathy | 0 | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Liver histology | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 Progression of liver histological stage | 3 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.58, 1.58] |
| 15.2 Worsening of histological inflammatory activity | 1 | 87 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.26, 0.94] |
| 16 Bilirubin (µmol/L) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | 49.0 [‐43.44, 141.44] |
| 17 Alkaline phosphatases (IU/L) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐62.50 [‐294.67, 169.67] |
| 18 Aspartate aminotransferase (IU/L) | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐38.0 [‐79.82, 3.82] |
| 19 Alanine aminotransferase (IU/L) | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐45.0 [‐75.11, ‐14.89] |
| 20 Albumin (g/dL) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐0.5 [‐1.04, 0.04] |
| 21 Adverse event ‐ fixed effect model | 6 | 617 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.08 [2.31, 4.11] |
| 22 Adverse event ‐ random effects model | 6 | 617 | Risk Ratio (M‐H, Random, 95% CI) | 3.98 [1.43, 11.04] |
| 23 Adverse event ‐ excluding Taal 1983 trial | 5 | 593 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.69 [2.62, 5.19] |
1.1. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 1 Mortality (expressed as relative risk) ‐ fixed effect model.
1.2. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 2 Mortality (expressed as relative risk) ‐ random effects model.
1.3. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 3 Subgroups of methodological quality ‐ generation of allocation sequence ‐ mortality.
1.4. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 4 Subgroups of methodological quality ‐ allocation concealment ‐ mortality.
1.5. Analysis.
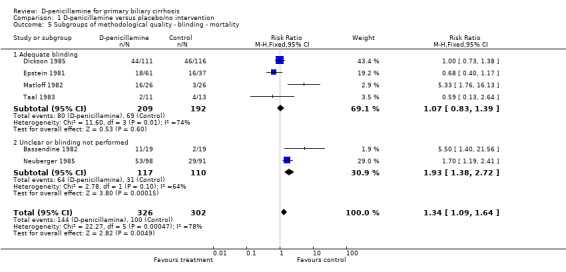
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 5 Subgroups of methodological quality ‐ blinding ‐ mortality.
1.6. Analysis.
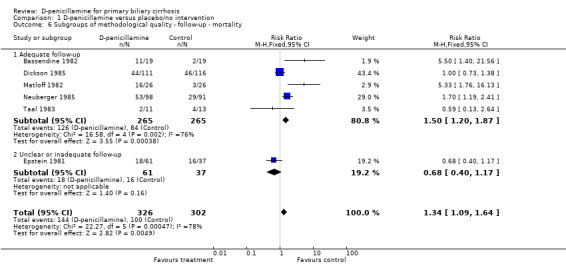
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 6 Subgroups of methodological quality ‐ follow‐up ‐ mortality.
1.7. Analysis.
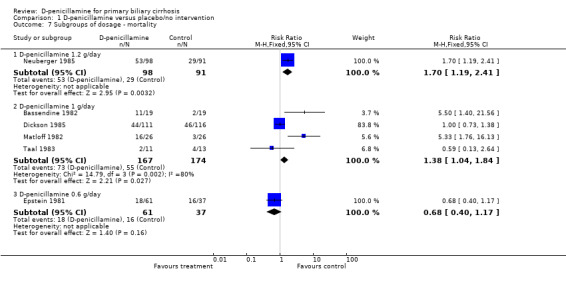
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 7 Subgroups of dosage ‐ mortality.
1.8. Analysis.
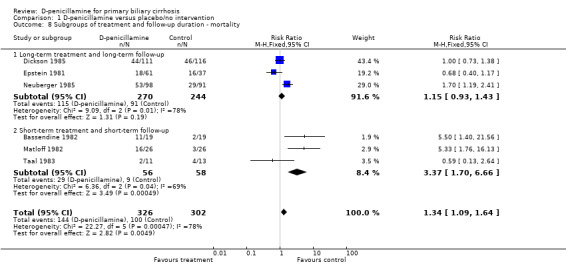
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 8 Subgroups of treatment and follow‐up duration ‐ mortality.
1.9. Analysis.
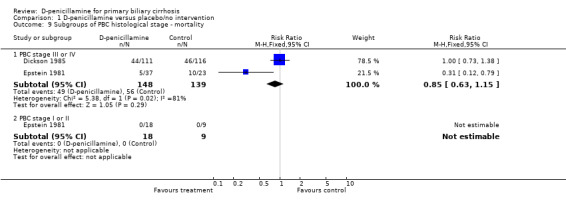
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 9 Subgroups of PBC histological stage ‐ mortality.
1.10. Analysis.
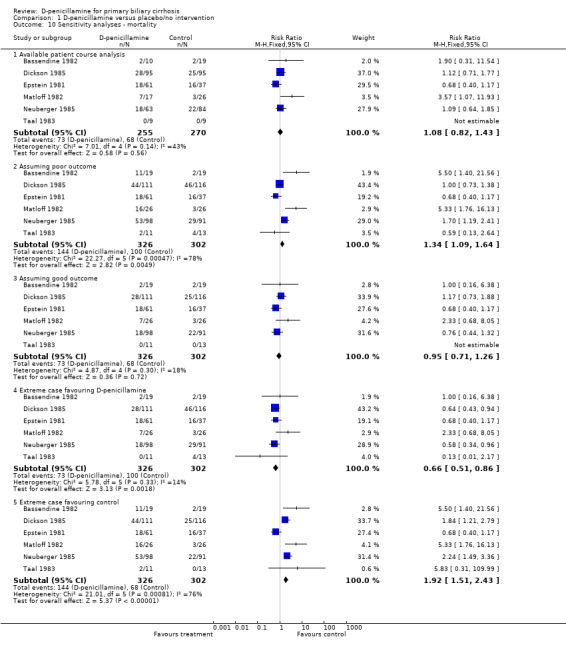
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 10 Sensitivity analyses ‐ mortality.
1.11. Analysis.
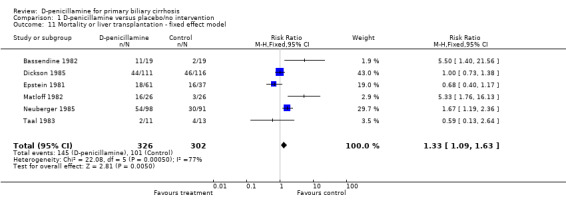
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 11 Mortality or liver transplantation ‐ fixed effect model.
1.12. Analysis.
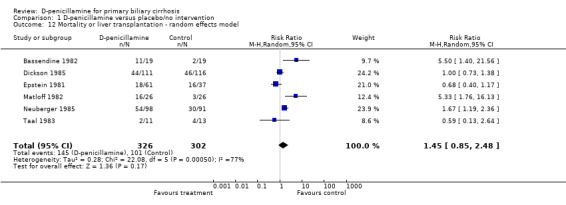
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 12 Mortality or liver transplantation ‐ random effects model.
1.13. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 13 Patients without improvement of pruritus.
1.14. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 14 Patients without improvement of liver complications.
1.15. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 15 Liver histology.
1.16. Analysis.
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 16 Bilirubin (µmol/L).
1.17. Analysis.
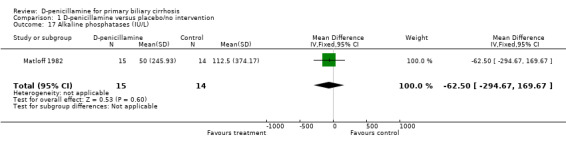
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 17 Alkaline phosphatases (IU/L).
1.18. Analysis.
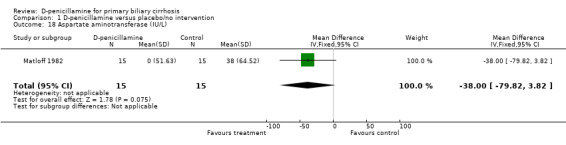
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 18 Aspartate aminotransferase (IU/L).
1.19. Analysis.
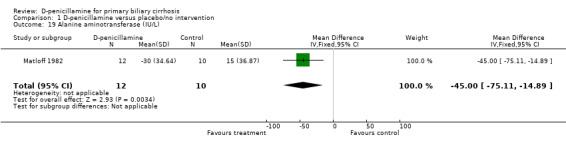
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 19 Alanine aminotransferase (IU/L).
1.20. Analysis.
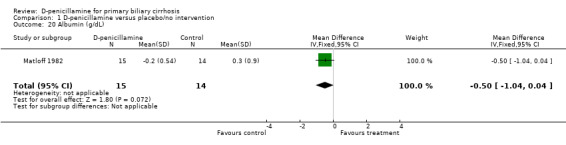
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 20 Albumin (g/dL).
1.21. Analysis.
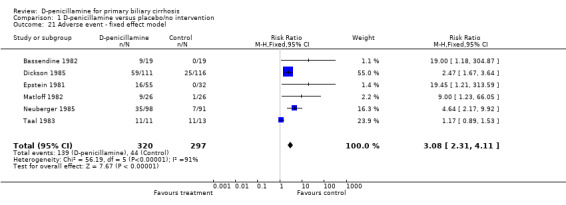
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 21 Adverse event ‐ fixed effect model.
1.22. Analysis.
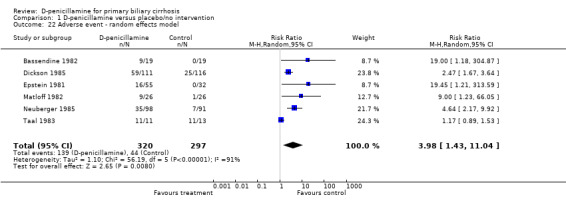
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 22 Adverse event ‐ random effects model.
1.23. Analysis.
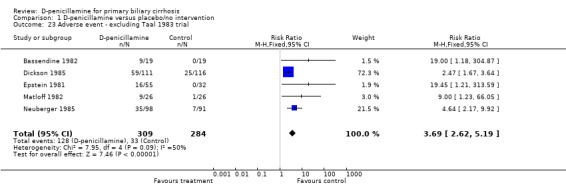
Comparison 1 D‐penicillamine versus placebo/no intervention, Outcome 23 Adverse event ‐ excluding Taal 1983 trial.
Comparison 2. High‐dose D‐penicillamine versus low‐dose D‐penicillamine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.06, 1.05] |
| 2 Patients without improvement of liver histological progression | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.31, 1.29] |
| 3 Adverse event | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.99 [0.81, 4.89] |
2.1. Analysis.
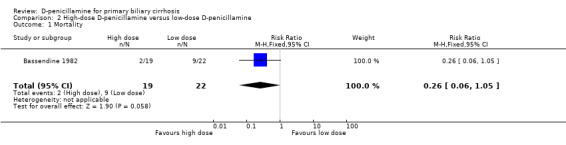
Comparison 2 High‐dose D‐penicillamine versus low‐dose D‐penicillamine, Outcome 1 Mortality.
2.2. Analysis.
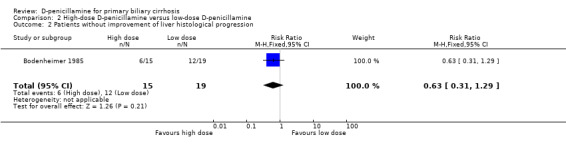
Comparison 2 High‐dose D‐penicillamine versus low‐dose D‐penicillamine, Outcome 2 Patients without improvement of liver histological progression.
2.3. Analysis.
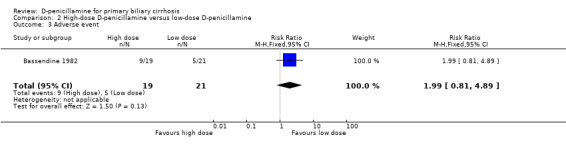
Comparison 2 High‐dose D‐penicillamine versus low‐dose D‐penicillamine, Outcome 3 Adverse event.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bassendine 1982.
| Methods | Generation of allocation sequence: unclear. Allocation concealment: unclear. Blinding: not performed. Follow‐up: adequate, four patients in the high‐dose D‐penicillamine, five patients in the low‐dose D‐penicillamine, and none in control were lost to follow‐up. | |
| Participants | Country: UK. Mean age: not reported. Female/Male: not reported. PBC stage status: not reported. | |
| Interventions | D‐penicillamine 1g/day (n = 19) D‐penicillamine 250 mg/day (n = 22) No intervention (n = 19) Mean period of follow‐up: 37 months. Analysed duration of trial: 3 years. | |
| Outcomes | 1. Mortality. 2. Liver biochemical variables. 3. Adverse events. | |
| Notes | 1. Side effects required withdrawal of D‐penicillamine in nine patients. 2. It was only published as an abstract. 3. Correspondence sent to the author on 2 December 2003. No reply was received by 20 June 2004. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Bodenheimer 1985.
| Methods | Generation of allocation sequence: unclear. Allocation concealment: unclear. Blinding: adequate, identical placebo. Follow‐up: inadequate, 26 patients in both groups were lost to follow‐up. | |
| Participants | Place: USA. Mean age: 52 in high‐dose group and the same in low‐dose group. Female/Male:51/5. PBC stage status: 10 with stage I or II in high‐dose group; 8 with stage I or II in low‐dose group. 20 with stage III or IV in high dose group; 18 with stage III or IV in low dose group. Inclusion criteria: 1. A history of chronic cholestatic liver disease. 2. Liver biopsies were compatible with PBC. | |
| Interventions | High‐dose group (n = 30): 250 mg/day increased gradually until 750 mg/day was achieved. Low‐dose group (n = 26): 250 mg/day. Mean period of treatment and follow‐up: three years. | |
| Outcomes | 1. Mortality data (only total number for two groups). 2. Liver test results. 3. Liver biopsy findings. 4. Adverse effects. | |
| Notes | 1. Liver test results were analysed as logarithms due to log‐normal distribution of data and reported as per cent change. Therefore, it is not possible for us to extract the data. 2. Correspondence sent to the author on 2 December 2003. Reply was received on 13 February 2004. No additional information were added. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | D ‐ Not used |
Dickson 1985.
| Methods | Generation of allocation sequence: adequate, a table of random numbers. Allocation concealment: adequate, a central pharmacist. Blinding: adequate, identical placebo. Follow‐up: adequate, 24 patients in the D‐penicillamine and no patient in the placebo group withdrew from this trial. | |
| Participants | Country: USA. Mean age: not reported, but 43% patients in D‐penicillamine and 54% in placebo not older than 50 years. Female/Male: 200/27. PBC stage status: 3 and 4. Inclusion criteria: 1. Established liver disease of more than six months' duration. 2. Raised alkaline phosphatases more than 2.5 times the normal level. 3. AMA titer greater than 1:10. 4. Liver biopsy diagnostic of, or consistent with PBC. | |
| Interventions | D‐penicillamine 250 mg/day for 2 weeks increased by 250 mg/day every 2 weeks until 1 g/day (n = 111) Placebo (the administration same as D‐penicillamine) (n = 116) Median period of follow‐up: 5 years. Analysed duration of trial: 10 years. | |
| Outcomes | 1. Survival analysis. 2. Clinical and biochemical changes. 3. Histologic results. 4. Toxicity. | |
| Notes | 1. Survival data at 5 years were available to be extracted only. 2. Correspondence sent to the author on 2 December 2003. No reply was received by 20 June 2004. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Epstein 1981.
| Methods | Generation of allocation sequence: unclear. Allocation concealment: unclear. Blinding: adequate, identical placebo. Follow‐up: inadequate. | |
| Participants | Country: UK. Mean age: not reported, instead, median age: 52 years in D‐penicillamine, 54 years in placebo. Female/Male: not reported. PBC stage status: 18 with stage 1 or 2 and 37 with stage 3 or 4 in D‐penicillamine; 9 with stage 1 or 2 and 23 with stage 3 or 4 in placebo. Inclusion criteria: 1. Liver test pointed to cholestasis. 2. AMA test positive. 3. Normal extrahepatic bile ducts by cholangiography. 4. Liver histology either diagnostic of or highly suggestive of PBC. | |
| Interventions | D‐penicillamine: over 8 to 10 weeks from 150 mg/day to 600 mg/day (n = 61). Placebo (n = 37). Median period follow‐up: 33 months. Analysed duration of trial: 6 years. | |
| Outcomes | 1. Survival data. 2. Liver biochemical variables. 3. Liver histology. | |
| Notes | 1. The trial has recruited 98 patients, but data on adverse events were only reported for 87 patients (55 in D‐penicillamine and 32 in placebo group). 2. Because of expected withdrawals due to D‐penicillamine drug reactions, the randomisation was weighted to allow a 3:2 ratio of D‐penicillamine to placebo treated patients. 2. Correspondence sent to the author on 2 December 2003. No reply was received by 20 June 2004. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Matloff 1982.
| Methods | Generation of allocation sequence: unclear. Allocation concealment: adequate, a study monitor. Blinding: adequate, identical placebo. Follow‐up: adequate, nine patients in the D‐penicillamine group and no patients in the placebo group were lost to follow‐up. | |
| Participants | Country: USA. Mean age: 51.5 years in D‐penicillamine, 51.5 years in placebo. Female/Male: 48 /4. PBC stage status: 14 patients with advanced disease in D‐penicillamine, 13 in placebo. Inclusion criteria: 1. A history of chronic cholestatic liver disease. 2. Raised alkaline phosphatases. 3. patent extrahepatic bile ducts. 4. Liver specimen diagnostic of or consistent with primary biliary cirrhosis. 5. AMA test positive. | |
| Interventions | D‐penicillamine 1g/day (n = 26). Placebo: identical placebo (n = 26). Total treatment duration: 28 months. | |
| Outcomes | 1. Survival data. 2. Liver histology. 3. Liver biochemical variables. 4. Adverse events. | |
| Notes | 1. Because a high incidence of side effects was noted in the first 39 patients, the last 13 patients were begun on a dose of 250 mg per day, which was gradually increased to 1g per day over a six‐week period. 2. Correspondence sent to the author on 2 December 2003. Reply was received on 19 December 2003. No additional information was added. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Low risk | A ‐ Adequate |
Neuberger 1985.
| Methods | Generation of allocation sequence: unclear. Allocation concealment: unclear. Blinding: inadequate. Follow‐up: adequate, 35 patients in the D‐penicillamine and seven patients in the placebo group were lost to follow‐up. | |
| Participants | Country: UK, Spain, Denmark. Mean age: not reported. Female/Male: 173/16. PBC stage status: 12% patients in D‐penicillamine and 15% in placebo with stage 1; 40% in D‐penicillamine and 37% in placebo with stage 2; 24% in D‐penicillamine and 21% in placebo with stage 3; 24% in D‐penicillamine and 27% in placebo with stage 4. Inclusion criteria: 1. A clinical and histological picture compatible with that of primary biliary cirrhosis. 2. Raised alkaline phosphatase in the absence of evidence of extrahepatic biliary obstruction. | |
| Interventions | D‐penicillamine 1.2 g/day, increased from 300 mg by 300 mg each fortnight until 1.2 g (n = 98) Placebo, taken in the same way (n = 91). Analysed duration of trial: 4 years. | |
| Outcomes | 1. Clinical features. 2. Liver biochemical variables. 3, Liver histology. 4. Survival data. 5. Adverse events. | |
| Notes | 1. It was an international multicentre (3 centres) trial. 2. Correspondence with the author 2 December 2003. Reply was received on 3 December 2003. No additional information was added. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
Taal 1983.
| Methods | Generation of allocation sequence: unclear. Allocation concealment: unclear. Blinding: adequate, identical placebo. Follow‐up: adequate, 2 in the D‐penicillamine and 4 in the placebo group were lost to follow‐up. | |
| Participants | Country: Netherlands. Median age: 51 years in D‐penicillamine, 48 years in placebo. Female/Male: 23/1. PBC stage status: not reported. Inclusion criteria: 1. Raised serum alkaline phosphatases. 2. AMA test positive. 3. A liver biopsy showing lymphoplasmacellular infiltrates with destruction of interlobular bile ducts or a paucity of bile ducts, and no demonstrable abnormalities of the extrahepatic bile ducts on the cholangiogram. 4. Only symptomatic patients (fatigue, pruritus, and/or jaundice). | |
| Interventions | D‐penicillamine 1 g/day (increased from 250 mg every month until 1 g for the first 6 months. After that, decreased to 500 mg/day for the remaining 6 months (n = 11). Placebo taken in the same way (n = 13). Duration of treatment: 1 year Duration of post‐treatment follow‐up: 0.5 year. Analysed duration of trial: 1.5 years. | |
| Outcomes | 1. Survival data. 2. PBC‐related symptoms. 3. Liver biochemical variables. 4. Liver histological variables. 5. Adverse events. | |
| Notes | 1. It involved two centres. 2. Correspondence sent to the author on 2 December 2003. Reply was received on 3 December 2003. No additional information was added. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment? | Unclear risk | B ‐ Unclear |
PBC: primary biliary cirrhosis AMA: antimitochondrial antibody
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Gupta 1982 | An observational study, examing for three years the serum levels of immune complexes from 88 patient with primary biliary cirrhosis, treated with D‐penicillamine. |
| Savolainen 1983 | Non‐randomised clinical study. |
| Triger 1980 | Non‐randomised clinical study. |
Contributions of authors
YG performed the searches, selected trials for inclusion, wrote to authors and pharmaceutical companies, performed data extraction and data analyses, and drafted the protocol and the systematic review. SLF modified the search strategy, extracted data, and revised the protocol and the systematic review. CG formulated the idea of this review and revised the protocol, solved discrepancy of data extraction, validated data analyses, and revised the review.
Sources of support
Internal sources
Copenhagen Trial Unit, Centre for Clinical Intervention Research, Denmark.
External sources
No sources of support supplied
Declarations of interest
None known. We have no affiliations or financial contracts with companies producing the drugs examined in this review.
Edited (no change to conclusions)
References
References to studies included in this review
Bassendine 1982 {published data only}
- Bassendine MF, Macklon AF, Mulcahy R, James OFW. Controlled trial of high and low dose D‐penicillamine (DP) in primary biliary cirrhosis (PBC): results at three years (abstract). Gut 1982;23:A909. [Google Scholar]
- Macklon AF, Bassendine MF, James OFW. Controlled trial of D‐penicillamine in primary biliary cirrhosis: incidence of side effects and relation to dose (IASL abstract). Hepatology 1982;2:166. [Google Scholar]
Bodenheimer 1985 {published data only}
- Bodenheimer HC, Charland C, Thayer WR, Schaffner F, Staples PJ. Immunologic effects of penicillamine in patients with primary biliary cirrhosis. Hepatology 1983;3:845. [Google Scholar]
- Bodenheimer HC, Colette CJr, Thayer WR, Schaffner FJr, Staples PJ. Effects of penicillamine on serum immunoglobulins and immune complex‐reactive material in primary biliary cirrhosis. Gastroenterology 1985;88:412‐7. [DOI] [PubMed] [Google Scholar]
- Bodenheimer HC, Schaffner F, Sternlieb I, Klion FM, Vernace S, Pezzullo J. A prospective clinical trial of D‐penicillamine in the treatment of primary biliary cirrhosis. Hepatology 1985;5(6):1139‐42. [DOI] [PubMed] [Google Scholar]
- Schaffner F, Sternlieb I, Sachs H. A two dose level randomized double blind controlled trial of penicillamine in primary biliary cirrhosis. Hepatology 1982;2(5):168. [Google Scholar]
Dickson 1985 {published data only}
- Deering TB, Dickson ER, Fleming CR, Geall MG, McCall JT, Baggenstoss AH. Effect of D‐penicillamine on copper retention in patients with primary biliary cirrhosis. Gastroenterology 1977;72:1208‐12. [PubMed] [Google Scholar]
- Dickson ER. The syndrome of primary biliary cirrhosis. Journal of Rheumatology 1981;8(Suppl 7):121‐3. [PubMed] [Google Scholar]
- Dickson ER, Fleming TR, Wiesner RH, Baldus WP, Fleming CR, Ludwig J, et al. Trial of penicillamine in advanced primary biliary cirrhosis. New England Journal of Medicine 1985;312(16):1011‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Dickson ER, Wiesner RH, Baldus WP, Fleming CR, Ludwig JL. D‐penicillamine improves survival and retards histologic progression in primary biliary cirrhosis (abstract). Gastroenterology 1982;82:1225. [Google Scholar]
- Locke GR, Therneau TM, Lugwig J, Dickson ER, Lindor KD. Time course of histological progression in primary biliary cirrhosis. Hepatology 1996;23:52‐6. [DOI] [PubMed] [Google Scholar]
- Powell FC, Dickson ER. Primary biliary cirrhosis and lichen planus. Journal of the American Academy of Dermatology 1983;9(4):540‐5. [DOI] [PubMed] [Google Scholar]
- Reed M. Penicillaine therapy 'encouraging' in primary biliary cirrhosis study. JAMA 1982;248:11‐2. [PubMed] [Google Scholar]
Epstein 1981 {published data only}
- Epstein O, Cook DG, Jain S, McIntyre N, Sherlock S. D‐penicillamine and clinical trials in PBC (AASLD abstract). Hepatology 1984;4:1032. [Google Scholar]
- Epstein O, Cook DG, Jain S, Sherlick S. D‐penicillamine in primary biliary cirrhosis (PBC) ‐ an untested (and untestable?) treatment (abstract). Gut 1984;25:A1134. [Google Scholar]
- Epstein O, Jain S, Lee RG, Cook DG, Boss AM, Scheuer PJ, Scherlock S. D‐penicillamine treatment improves survival in primary biliary cirrhosis (abstract). Gut 1981;22:A433. [DOI] [PubMed] [Google Scholar]
- Epstein O, Jain S, Lee RG, Cook DG, Boss AM, Scheuer PJ, et al. D‐penicillamine treatment improves survival in primary biliary cirrhosis. Lancet 1981;1:1275‐7. [DOI] [PubMed] [Google Scholar]
- Epstein O, Villiers DD, Jain S, Potter B, Thomas H, Sherlock S. Reduction of immune complexes and immunoglobulins induced by D‐penicillamine in primary biliary cirrhosis. The New England Journal of Medicine 1979;300:274‐8. [DOI] [PubMed] [Google Scholar]
- Epstein O, Villiers DD, Jain S, Potter BJ, Thomas HC, Sherlock S. Effect of penicillamine on immune complexes and immunoglobulins in primary biliary cirrhosis (PBC) (abstract). Gut 1978;19:A994. [DOI] [PubMed] [Google Scholar]
- Jain S, McGee JO'D, Scheuer PJ, Samourian S, Sherlock S. A controlled trial of D‐penicillamine therapy in primary biliary cirrhosis and chronic active hepatitis (abstract). Digestion 1976;14:523. [Google Scholar]
- Jain S, Scheuer PJ, Samourian S, McGee J, Sherlock S. A controlled trial of D‐penicillamine therapy in primary biliary cirrhosis. Lancet 1977;16:831‐4. [DOI] [PubMed] [Google Scholar]
- Jain S, Scheur PJ, Samourian S, McGee JO'D, Sherlock S. A controlled trial of D‐penicillamine therapy in primary biliary cirrhosis (abstract). Gut 1976;17:822. [PMC free article] [PubMed] [Google Scholar]
Matloff 1982 {published data only}
- Matloff DS, Alpert E, Resnick RH, Kaplan MM. A prospective trial of D‐penicillamine in primary biliary cirrhosis. New England Journal of Medicine 1982;306(6):319‐26. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Matloff DS, Resnick RH, Alpert E, Kaplan M. D‐penicillamine does not alter the course of primary biliary cirrhosis (abstract). Clinical Research 1979;27:579A. [Google Scholar]
Neuberger 1985 {published data only}
- Neuberger J, Christensen E, Popper H, Portmann B, Caballeri J, Rodes J, et al. D‐penicillamine in primary biliary cirrhosis: preliminary results of an international trial (EASL abstract). Liver 1984;4:G31. [Google Scholar]
- Neuberger J, Christensen E, Popper H, Portmann B, Caballeri J, Rodes J, et al. D‐penicillamine in primary biliary cirrhosis: preliminary results of an international trial (abstract). Gut 1983;24:A968. [Google Scholar]
- Neuberger J, Christensen E, Portmann B, Caballeria J, Rodes J, Ranek L, et al. Double blind controlled trial of D‐penicillamine in patients with primary biliary cirrhosis. Gut 1985;26(2):114‐9. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Taal 1983 {published data only}
- Taal BG, Schalm SW. Cryoglobulins in primary biliary cirrhosis: prevalence and modulation by immunosuppressive therapy. Zeitschrift für Gastroenterologie 1985;23:228‐34. [PubMed] [Google Scholar]
- Taal BG, Schalm SW. Prednisone plus D‐penicillamine, D‐penicillamine and placebo compared in primary biliary cirrhosis syndrome (abstract). Gastroenterology 1981;80:1351. [Google Scholar]
- Taal BG, Schalm SW, Kate FWJ, Henegouwen GPB, Brandt KH. Low therapeutic value of D‐penicillamine in a short‐term prospective trial in primary biliary cirrhosis. Liver 1983;3:345‐52. [DOI] [PubMed] [Google Scholar]
- Taal BG, Schalm SW, Kate FWJ, Berge Henegouwen GP, Brandt KH. A double‐blind controlled trial of D‐penicillamine for PBC: the dose dependent effect (abstract) [Een dubbelblind onderzoek met een controlegroep met D‐penicillamine bij primaire biliaire cirrose: van de dosis afhankelijke effecten]. Nederlands Tijdschrift voor Geneeskunde 1982;126:547. [Google Scholar]
References to studies excluded from this review
Gupta 1982 {published data only}
- Gupta RC, Dickson ER, McDuffie FC, Baggenstoss AH. Immune complexes in primary biliary cirrhosis: high prevalence of circulating immune complexes in patients with associated autoimmune features. American Journal of Medicine 1982;73(2):192‐8. [DOI] [PubMed] [Google Scholar]
Savolainen 1983 {published data only}
- Savolainen ER, Miettinen TA, Pikkarainen P, Salaspuro MP, Kivirikko KI. Enzymes of collagen synthesis and type III procollagen aminopropeptide in the evaluation of D‐penicillamine and medroxyprogesterone treatments of primary biliary cirrhosis. Gut 1983;24:136‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Triger 1980 {published data only}
- Triger DR, Manifold IH, Cloke P, Underwood JCE. D‐penicillamine in primary biliary cirrhosis: two year results of a single centre, double‐blind controlled trial. Gut 1980;21(9):A919‐20. [Google Scholar]
Additional references
Alderson 2003
- Alderson P, Green S, Higgins JPT, editors. Cochrane Reviewers' Handbook 4.2.1[updated December 2003]. The Cochrane Library 2004, Issue 1. [Google Scholar]
Altman 2003
- Altman DG, Bland JM. Interaction revisited: the difference between two estimates. BMJ 2003;326:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Balasubramaniam 1990
- Balasubramaniam K, Grambsch PM, Wiesner RH, Lindor KD, Dickson ER. Diminished survival in asymptomatic primary biliary cirrhosis: a prospective study. Gastroenterology 1990;98:1567‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ballardini 1984
- Ballardini G, Mirakian R, Bianchi FB, Pisi E, Doniach D, Bottazzo GF. Aberrant expression of HLA‐DR antigens on bile duct epithelium in primary biliary cirrhosis: relevance to pathogenesis. Lancet 1984;ii:1009. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Begg 1994
- Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics 1994;50(4):1088‐1101. [PubMed] [Google Scholar]
Beswick 1985
- Beswick DR, Klatskin G, Boyer JL. Asymptomatic primary biliary cirrhosis: a progress report on long‐term follow‐up and natural history. Gastroenterology 1985;89:267‐71. [MEDLINE: ] [PubMed] [Google Scholar]
Christensen 1985
- Christensen E, Neuberger J, Crowe J, Altman DG, Popper H, Portmann B, et al. Beneficial effect of azathioprine and prediction of prognosis in primary biliary cirrhosis. Final results of an international trial. Gastroenterology 1985;89:1084‐91. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Deeks 2001a
- Deeks JJ, Altman DG. Effect measures for meta‐analysis of trials with binary outcomes. In: Egger M, Davey Smith G, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐Analysis in Context. Second Edition. London: BMJ Books, 2001. [Google Scholar]
Deeks 2001b
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examing heterogeneity and combing results from several studies in meta‐analysis. In: Egger M, Davey Smith G, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐analysis in Context. Second Edition. London: BMJ Books, 2001. [Google Scholar]
Deeks 2002
- Deeks JJ. Issues in the selection of a summary statistic for meta‐analysis of clinical trials with binary outcomes. Statistics in Medicine 2002;21:1575‐1600. [DOI] [PubMed] [Google Scholar]
Deiss 1971
- Deiss A, Lynch RE, Lee GR, Cartwright GE. Long term therapy of Wilson's disease. Annals of Internal Medicine 1971;75:57‐65. [DOI] [PubMed] [Google Scholar]
DeMets 1987
- DeMets DL. Methods of combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Smith GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ 1997;315(7109):629‐34. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Epstein 1979
- Epstein O, Villiers D, Jain S, Potter BJ, Thomas HC, Sherlock S. Reduction of immune complexes and immunoglobulins induced by D‐penicillamine in primary biliary cirrhosis. New England Journal of Medicine 1979;300:274‐8. [DOI] [PubMed] [Google Scholar]
Epstein 1982
- Epstein O, Chapman RWG, Lake‐Bakaar G, Foo AY, Rosalki SB, Sherlock S, et al. The pancreas in primary biliary cirrhosis and primary sclerosing cholangitis. Gastroenterology 1982;83(6):1177‐82. [PubMed] [Google Scholar]
Fregeau 1989
- Fregeau D, Water J, Danner D, Ansart T, Coppel R, Gershwin M. Antimitochondrial antibodies of primary biliary cirrhosis recongnize dihydrolipoamide acyltransferase and inhibit enzyme function of the branched chain alpha ketoacid dehydrogenase complex. Journal of Immunology 1989;142(11):3815‐20. [BC86215 Hominidae] [PubMed] [Google Scholar]
Glasziou 2002
- Glasziou PP, Sanders SL. Investigating causes of heterogeneity in systematic reviews. Statistics in Medicine 2002;21:1503‐11. [DOI] [PubMed] [Google Scholar]
Gluud 1993
- Gluud C, Afroudakis AP, Caballeria J, Laskus T, Morgan M, Rueff B, et al. Diagnosis and treatment of alcoholic liver disease in Europe ‐ First Report by the Gastroenterology Across Frontiers Panel. Gastroenterology International 1993;6:221‐30. [Google Scholar]
Gluud 2002
- Gluud C, Christensen E. Ursodeoxycholic acid for primary biliary cirrhosis. The Cochrane Library 2002, Issue 2. [DOI] [PubMed] [Google Scholar]
Goulis 1999
- Goulis J, Leandro G, Burroughs AK. Randomised controlled trials of ursodeoxycholic‐acid therapy for primary biliary cirrhosis: a meta‐analysis. Lancet 1999;354:1053‐60. [DOI] [PubMed] [Google Scholar]
Heathcote 1976
- Heathcote J, Ross A, Sherlock S. A prospective controlled trial of azathioprine in primary biliary cirrhosis. Gastroenterology 1976;70(5 Pt. 1):656‐60. [MEDLINE: ] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21:1539‐58. [DOI] [PubMed] [Google Scholar]
Hollis 1999
- Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. British Medical Journal 1999;319:670‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hoofnagle 1986
- Hoofnagle JH, Davis GL, Schafer DF, Peters M, Avigan MI, Pappas SC, et al. Randomized trial of chlorambucil for primary biliary cirrhosis. Gastroenterology 1986;91(6):1327‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Code of Federal Regulation & ICH Guidelines. Media: PAREXEL BARNETT, 1997. [Google Scholar]
Invernizzi 1997
- Invernizzi P, Crosignani A, Battezzati PM, Covini G, De‐Valle G, Larghi A, et al. Comparison of the clinical features and clinical course of antimitochondrial antibody‐positive and negative primary biliary cirrhosis. Hepatology 1997;25(5):1090‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ioannidis 2001
- Ioannidis JPA, Lau J. Evolution of treatment effects over time: empirical insight from recursive cumulative metaanalyses. Proceedings of the National Academy of Sciences of the United States of America 2001;98(3):831‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
James 1981
- James O, Macklon AF, Waston AJ. Primary biliary cirrhosis ‐ a revised clinical spectrum. Lancet 1981;1(8233):1278‐81. [DOI] [PubMed] [Google Scholar]
Kaplan 1991
- Kaplan MM, Knox TA. Treatment of primary biliary cirrhosis with low‐dose weekly methotrexate. Gastroenterology 1991;101(5):1332‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kaplan 1994
- Kaplan MM. Primary biliary cirrhosis ‐ a first step in prolonging survival. New England Journal of Medicine 1994;330(19):1386‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kaplan 1996
- Kaplan MM. Primary biliary cirrhosis. New England Journal of Medicine 1996;335(21):1570‐80. [DOI] [PubMed] [Google Scholar]
Kim 2000
- Kim WR, Lindor KD, Locke GR 3rd, Therneau TM, Homburger HA, Batts KP, et al. Epidemiology and natural history of primary biliary cirrhosis in a U.S. community. Gatroenterology 2000;119:1631‐6. [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodological quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI] [PubMed] [Google Scholar]
Lacerda 1995
- Lacerda MA, Ludwig J, Dickson ER, Jorgensen RA, Lindor KD. Antimitochondrial antibody‐negative primary biliary cirrhosis. American Journal of Gastroenterology 1995;90(2):247‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Lindor 1995
- Lindor KD, Dickson ER, Jorgensen RA, Anderson ML, Wiesner RH, Gores GJ, et al. The combination of ursodeoxycholic acid and methotrexate for patients with primary biliary cirrhosis: the results of a pilot study. Hepatology 1995;22(4 Pt 1):1158‐62. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Lipsky 1980
- Lipsky PE, Ziff M. Inhibition of human helper T cell function in vitro by D‐penicillamine and copper sulfate. Journal of Clinical Investigation 1980;65:1069‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Macaskill 2001
- Macaskill P, Walter SD, Irwig L. A comparison of methods to detect publication bias in meta‐analysis. Statistics in Medicine 2001;20:641‐54. [DOI] [PubMed] [Google Scholar]
Mattalia 1998
- Mattalia A, Quaranta S, Leung PS, Bauducci M, Van‐de‐Water J, Calvo PL. Characterization of antimitochondrial antibodies in health adults. Hepatology 1998;27(3):656‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Metcalf 1996
- Metcalf J, Mitchison H, Palmer J, Jones D, Bassendine M, James O. Natural history of early primary biliary cirrhosis. Lancet 1996;348(9039):1399‐402. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mitchison 1992
- Mitchison HC, Palmer JM, Bassendine MF, Watson AJ, Record CO, James OF. A controlled trial of prednisolone treatment in primary biliary cirrhosis. Three‐year results. Journal of Hepatology 1992;15(3):336‐44. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Nimni 1972
- Nimni ME, Deshmukh K, Gerth N. Collagen defect induced by D‐penicillamine. Nature 1972;240:220‐1. [DOI] [PubMed] [Google Scholar]
Nyberg 1989
- Nyberg A, Loof L. Primary biliary cirrhosis: clinical features and outcome, with special reference to asymptomatic disease. Scandinavian Journal of Gastroenterology 1989;24(1):57‐64. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Poupon 1996
- Poupon RE, Huet PM, Poupon R, Bonnand AM, Nhieu JT, Zafrani ES, et al. A randomized trial comparing colchicine and ursodeoxycholic acid combination to ursodeoxycholic acid in primary biliary cirrhosis. Hepatology 1996;24(5):1098‐103. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Prince 2000
- Prince M, Jones D, Metcalf J, Craig W, James O. Symptom development and prognosis of initially asymptomatic PBC. Hepatology 2000;32(4 Pt2):171A. [Google Scholar]
Prince 2002
- Prince M, Chetwynd A, Newman W, Metcalf JV, James OFW. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow‐up for up to 28 years. Gatroenterology 2002;123:1044‐51. [DOI] [PubMed] [Google Scholar]
Scheuer 1967
- Scheuer P. Primary biliary cirrhosis. Proceedings of the Royal Society of Medicine 1967;60(12):1257‐60. [MEDLINE: ] [PMC free article] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sternlieb 1964
- Sternlieb I, Scheinberg IH. Penicillamine therapy for hepatolenticular degeneration. JAMA 1964;189:748‐54. [DOI] [PubMed] [Google Scholar]
Turchany 1997
- Turchany JM, Uibo R, Kivik T, Van‐de‐Water J, Prindiville T, Coppel RL, et al. A study of antimitochondrial antibodies in a random population in Estonia. American Journal of Gastroenterology 1997;92(1):124‐6. [MEDLINE: ] [PubMed] [Google Scholar]
Van den Oord 1986
- Oord JJ, Sciot R, Desmet VJ. Expression of MHC products by normal and abnormal bile duct epithelium. Journal of Hepatology 1986;3(3):310‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Verma 1999
- Verma A, Jazrawi RP, Ahmed HA, Northfield TC. Prescribing habits in primary biliary cirrhosis: a national survey. European Journal of Gastroenterology and Hepatology 1999;11(8):817‐20. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Vuoristo 1995
- Vuoristo M, Farkkila M, Karvonen AL, Leino R, Lehtola J, Makinen J, et al. A placebo‐controlled trial of primary biliary cirrhosis treatment with colchicine and ursodeoxycholic acid [see comments]. Gastroenterology 1995;108(5):1470‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Warnes 1987
- Warnes TW, Smith A, Lee FI, Haboubi NY, Johnson PJ, Hunt L. A controlled trial of colchicine in primary biliary cirrhosis: trial design and preliminary report. Journal of Hepatology 1987;5(1):1‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wiesner 1990
- Wiesner RH, Ludwig J, Lindor KD, Jorgensen RA, Baldus WP, Homburger HA, et al. A controlled trial of cyclosporine in the treatment of primary biliary cirrhosis. New England Journal of Medicine 1990;322(20):1419‐24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Yamada 1986
- Yamada G, Hyodo I, Tobe K, Mizuno M, Nishihara T, Kobayashi T, et al. Ultrastructural immunocytochemical analysis of lymphocytes infiltrating bile duct epithelia in primary biliary cirrhosis. Hepatology 1986;6(3):385‐91. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Gong 2007
- Gong Y, Klingenberg L, Gluud C. D‐penicillamine vs placebo/no intervention in patients with primary biliary cirrhosis. Evidence‐Based Gastroenterology 2007;8(2):41‐2. [Google Scholar]