

ABSTRACT
Cefepime is the second most common cephalosporin used in U.S. hospitals. We aim to develop and validate a cefepime population pharmacokinetic (PK) model and integrate it into a precision dosing tool for implementation. Two data sets (680 patients) were used to build the cefepime PK model in Pmetrics, and three data sets (34 patients) were used for the validation. A separate application data set (115 patients) was used for the implementation and validation of a precision dosing tool. The model support points and covariates were used to generate the optimal initial dose (OID). Cefepime PK was described by a two-compartment model including weight and creatinine clearance (CrCl) as covariates. The median rate of elimination was 0.30 h−1 (adults) and 0.96 h−1 (children), the central volume of distribution was 13.85 L, and the rate of transfer from the central to the peripheral compartments was 1.22 h−1 and from the peripheral to the central compartments was 1.38 h−1. After integration in BestDose, the observed versus predicted cefepime concentration fit using the application data set was excellent (R2 > 0.98), and the median difference between what was observed and what BestDose predicted on a second occasion was 4%. For the OID, cefepime at a 0.5- to 1-g 4-h infusion every 8 to 24 h (q8 to 24 h) with a CrCl of <70 mL/min was needed to achieve a target range of free trough:MIC 1 to 4 at a MIC of 8 mg/L, while continuous infusion was needed for higher CrCl and weight values. In conclusion, we developed and validated a cefepime model for clinical application. The model was integrated in a precision dosing tool for implementation, and the median concentration prediction bias was 4%. The OID algorithm was provided.
KEYWORDS: cefepime, pharmacokinetics, precision dosing
INTRODUCTION
A 2015 survey of antimicrobial use in U.S. hospitals identified third- and fourth-generation cephalosporins as the most common antibiotics administered to patients. Of these, cefepime was the second most common cephalosporin, with a rate of 3.5% of all antimicrobials administered (1). Cefepime is widely used in intensive care unit (ICU) patients due to its wide spectrum of activity encompassing Gram-negative and Gram-positive organisms. Appropriate dosing of beta-lactams, such as cefepime, is essential for therapeutic effect, especially in ICU patients with septic shock, where reduced time to effective antimicrobial therapy is associated with a mortality benefit (2). Beta-lactam antibacterial efficacy is best predicted by the percentage of time free drug concentrations remain above a pathogen’s MIC (fT>MIC). There are several factors that may alter patients’ beta-lactam pharmacokinetic (PK) profiles and subsequent pharmacodynamic (PD) effects, including sickness severity, organ dysfunction, and bacterial MIC (3).
Integration of therapeutic drug monitoring (TDM) into clinical practice has the potential to improve patient outcomes through optimization of dosing based on factors relevant to the respective individual patient and pathogen. Proposed beta-lactam PK/PD targets include 60% fT>MIC and 100% fT>1–5×MIC in critically ill patients (4, 5). Beta-lactam dosing regimens are typically selected based on recommendations provided in the package insert, and these doses may fail to achieve PK/PD targets, especially in ICU patients (6, 7). As a result, there is a need for an individualized approach to optimize beta-lactam dosing, especially in ICU patients. The purpose of this study was to develop a cefepime population PK model using multiple patient data sets, validate the model in a precision dosing tool, and generate optimal initial doses (OID) based on the model and patients’ characteristics.
RESULTS
Population characteristics.
A total of 680 patients and 1,633 cefepime plasma samples were included in the original data set to develop the population PK model. Table 1 summarizes the demographics of these patients; 56% were males and 5% were children.
TABLE 1.
Demographics of patients included in the PK model development and validationa
| Characteristic | Model development from: |
Model validation from: |
|||
|---|---|---|---|---|---|
| Reed (8) | UF Health | Roberts (7) | Sime (9) | Whited (10) | |
| No. of patients/samples | 36/511 | 644/1,122b | 13/26 | 12/48 | 9/93 |
| Population | Neonatal/pediatric ICU | Adult ICU | Adult ICU | Cancer with febrile neutropenia | Cancer with febrile neutropenia |
| Age (yrs) | 3.9 (4.7) | 55 (19) | 56 (41–64)c | 57 (52–67)c | 54 (10) |
| No. male | 21 (58) | 362 (56) | 9 (69) | 9 (75) | 5 (56) |
| Wt (kg) | 16 (16) | 85 (33) | 76 (14) | 89 (32) | 83 (8) |
| Wt median (range) | 11 (4–75) | 79 (30–247) | 76 (55–105) | 82 (64–194) | 85 (69–94) |
| CrCl (mL/min) | 68 (39) | 111 (73) | 105 (77) | 76 (14) | 149 (36) |
| CrCl median (range) | 56 (21–188) | 97 (7–609) | 89 (10–289) | 72 (61–106) | 127 (109–220) |
| Cefepime regimen | 50 mg/kg q8h over 30 min | 0.5–6 g/dayd | 6 g/day (5–6)c | 2 g q8h over 30 min | 2 g q8h over 30 min |
Data presented as mean (SD) or n (%) unless indicated. CrCl, creatinine clearance; ICU, intensive care unit; UF, University of Florida; q8h, every 8 h.
A total of 198 patients had peak and trough samples drawn in the same dosing interval.
Median (interquartile range).
Range.
PK model.
Cefepime was well described by a two-compartment model with the normalized weight allometrically scaled as a covariate on the central volume of distribution (Vd) and the total elimination rate constant (ke), and the normalized creatinine clearance (CrCl) and age group as covariates on ke:
where Peds stands for pediatric group, with a value of 1 if the patient is pediatric or 0 if adult. Table 2 summarizes the PK parameter estimates. Figure 1 shows the observed versus population and individual predicted concentrations, and Fig. S2 in the supplemental material shows the visual predictive checks and weighted residual error plots. The model generated 81 support points (Fig. S3). Figure S4 shows the observed versus population and individual predicted cefepime concentration for the validation data set.
TABLE 2.
Summary of the cefepime population pharmacokinetic parameter estimatesa
| Parameter | Median | 95% credibility interval | Mean | SD | Shrinkage (%) |
|---|---|---|---|---|---|
| ke, h−1 | |||||
| Adults | 0.30 | 0.24–0.37 | 0.37 | 0.29 | 57.94 |
| Pediatrics | 0.96 | 0.88–1.30 | 1.22 | 0.73 | 79.72 |
| Vd (L) | 13.85 | 12.73–17.88 | 18.33 | 10.37 | 64.69 |
| kcp (h−1) | 1.22 | 0.78–1.66 | 1.93 | 2.67 | 64.76 |
| kpc (h−1) | 1.38 | 1.07–1.76 | 2.10 | 2.79 | 78.83 |
kcp, rate of transfer from the central to the peripheral compartment; ke, elimination rate constant; kpc, rate of transfer from the peripheral to the central compartment; Vd, central volume of distribution.
FIG 1.
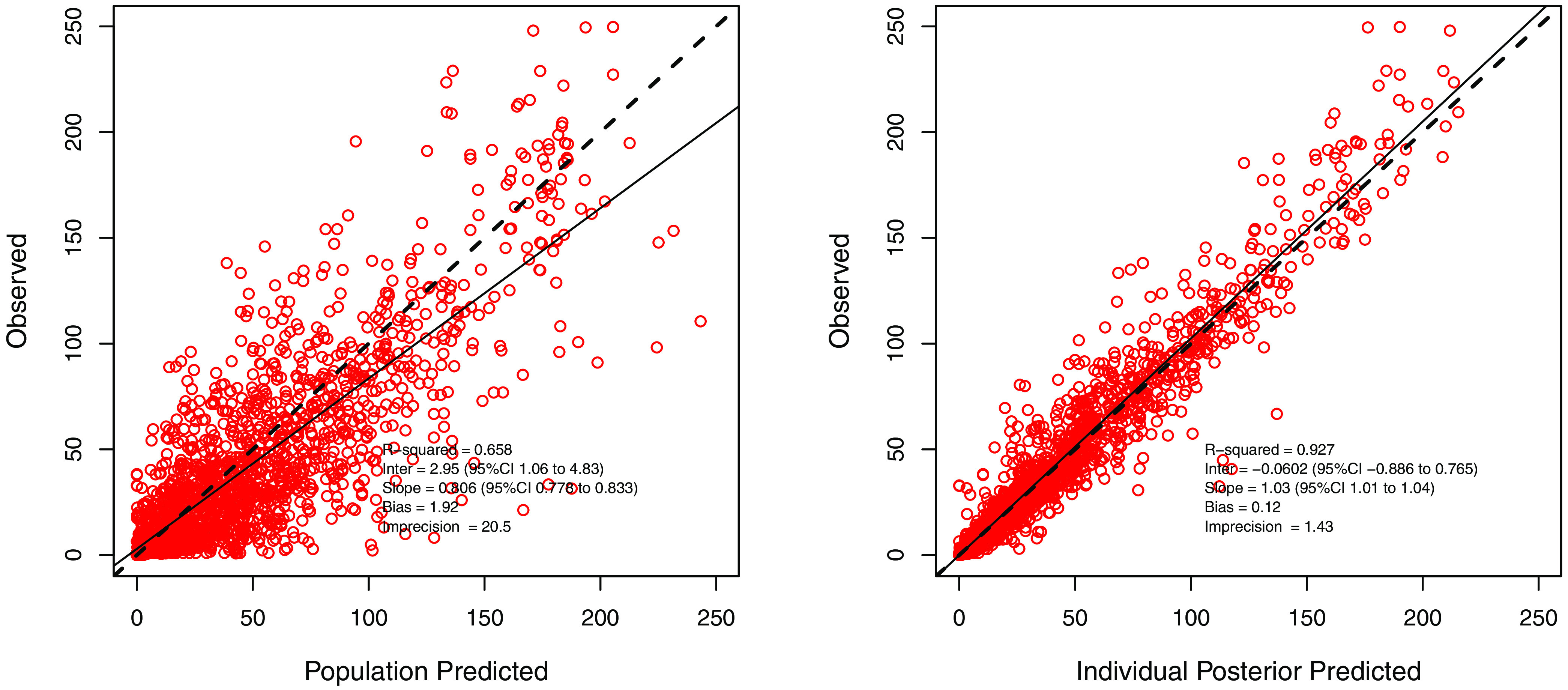
Observed versus population and individual predicted concentrations for the cefepime pharmacokinetic model.
Precision dosing application.
Table 3 summarizes the patient demographics included in the precision dosing application. Figure 2 shows the observed versus predicted cefepime concentration based on multiple model (MM) and interacting multiple model (IMM) fit. Out of 115 patients, 25 were resampled after a subsequent dose, producing an additional 38 cefepime samples. These additional samples were used for evaluating the model prediction. The median (interquartile range [IQR]) percentage of difference between measured cefepime concentration and what BestDose forecasted in the second TDM occasion was +4% (–25% to +26%) (coefficient of determination [R2] = 0.33; root mean square error [RMSE] = 20.93). One outlier sample had a percentage difference of +627% (Fig. 3).
TABLE 3.
Patient demographics in the precision dosing application data set from UF Healtha
| Characteristic | Mean (SD) or n (%) |
|---|---|
| No. of patients/samplesb | 115/181 |
| Population | ICU |
| Age (yrs) | 54 (21) |
| Pediatrics | 6 (5) |
| Wt (kg) | 82 (28) |
| Wt median (range) | 80 (4.5–172) |
| CrCl (mL/min) | 118 (78) |
| CrCl (mL/min) median (range) | 103 (14–431) |
| Cefepime doses | 0.22–3 g per dosec |
Data presented as mean (SD) or n (%) unless indicated. CrCl, creatinine clearance; ICU, intensive care unit; UF, University of Florida.
A total of 25 patients had peak and trough samples drawn in the same dosing interval.
Range.
FIG 2.
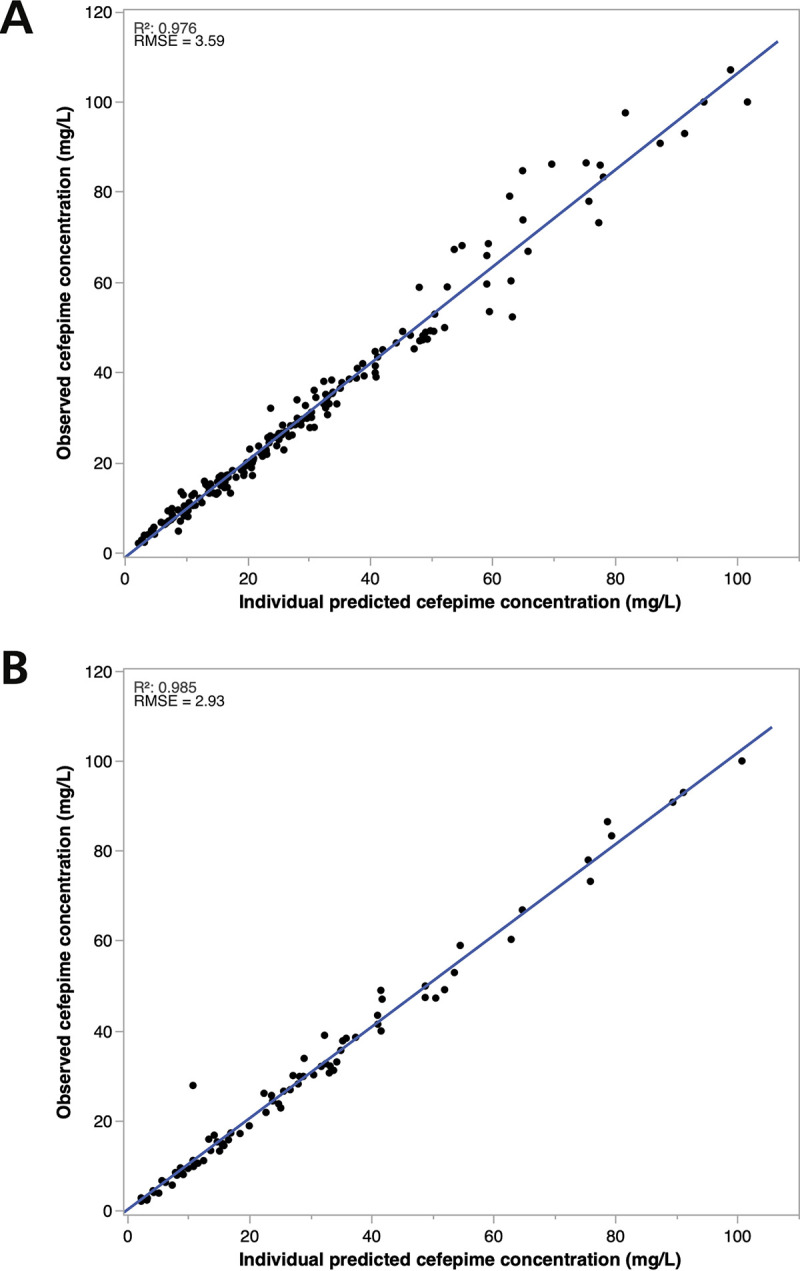
(A and B) Cefepime observed versus BestDose predicted concentration in the precision dosing external data set using (A) MM and (B) IMM Bayesian approach. R2, coefficient of determination; RMSE, root mean square error.
FIG 3.

Boxplot for the percentage of difference between measured and BestDose future-predicted cefepime concentration. One outlier sample (not shown) had a percentage difference of +627%.
OID generation and validation.
Tables S1 and S2 summarize the OID targeting free trough (fCmin):MIC 1 to 4 for adults and children, respectively. As one could expect, the optimal dosage increased with both body weight and renal function. In adults with weight ranging from 60 to 100 kg, extended infusion was optimal only in patients with a CrCl between 50 and 70 mL/min. With further increases in weight and/or CrCl, continuous infusion (CI) most consistently achieved target exposure. For a given dosage regimen, a priori probability to achieve the target interval varied with weight and CrCl (57% to 90%) in subjects with a CrCl of ≥20 mL/min.
TABLE S3 shows the target attainment results obtained from the simulation of the OID and the approved dose in the validation data set. The OID approach was successful in maximizing attainment of the target interval and reducing both under- and overexposure, as well as overall variability. Using the package insert dose, 62% were within, 24% below, and 15% above the target, while using the OID, 76% were within, 18% below, and 6% above the target.
DISCUSSION
We presented a cefepime population PK model developed from the largest data set thus far reported. We combined a previously published pediatric data set (8) with a University of Florida (UF) clinical data set to build the model, validated the model with three published data sets (7, 9, 10), integrated it into a precision dosing software, and favorably evaluated it in yet another independent data set. Also, we provided a comprehensive, initial dosing algorithm to optimize drug exposure in each patient. Although our model was not as rich as the models generated by planned PK studies, having a large number of patients enriched the model with support points. Both the MM and IMM Bayesian methods fit the data very well in BestDose, and IMM was slightly superior. This is consistent with the previous work on amikacin and vancomycin using the same software (11). Given the continuous changes in ICU patients’ PK parameters, the MM predictions for the subsequent TDM occasions were good (i.e., the median percentage difference between actual and predicted MM was +4%). The predictions may improve in future studies if the changes in PK parameters can be correlated with other variables in ICU patients (e.g., fluid input/output).
In the nonparametric modeling, the probability distribution of the PK parameter values is discrete. When estimating individual PK parameters, the entire discrete joint distribution of parameters is used as a prior, rather than using the mean and standard deviation (12). Then, the probabilities of the support points, each containing a set of PK parameters, will be updated such that the ones that best fit the data become more probable, while those poorly fitting the data become less probable or zero (MM approach) (Fig. S1). Such tools may increase the accuracy of estimated PK profiles in the clinical setting as shown previously with different antibiotics (13–16).
Two previous PK models used part of the data sets in our model. The first included pediatrics and UF data sets (patients admitted between 2016 and 2018) to a build cefepime two-compartment model and simulate different regimens to assess 100% fT>MIC and fT>4×MIC attainment. The rate constants were similar to those in our model, but the Vd was lower with a median population parameter estimate of 6.71 L (17). The increase in the Vd in the new model might be due to the addition of more adult patients who might have larger Vd than the pediatric patients. The second model was built using the pediatrics and validation data sets in our model. The data from Whited et al. (10) and part of that from Reed et al. (8) were used for model building, while the remaining of Reed’s data along with that of Roberts et al. and Sime et al. (7, 9) were used for validation. This was a two-compartment model with ke estimated as both renal and nonrenal elimination (18). The population parameter estimates generated by this model were similar to those of our model. In this paper, we have expanded the UF data set and added more than 400 patients to enrich the model in order to be able to apply it to a larger number of patients using precision dosing software.
Poor target attainment early in therapy was reported with the current regimens of beta-lactams. In a prospective ICU study, 100 patients with CrCl of >60 mL/min had their beta-lactam concentration measured within the first 3 to 5 days of therapy; 80% of those patients had subtherapeutic beta-lactam trough (Cmin), including cefepime (19). In another ICU study, 80 patients received one dose of beta-lactam and had their exposure evaluated. A total of 19 patients received cefepime at 2 g, and only 3 achieved 70% fT>4×MIC (20). This highlights the need for better initial, individualized regimens based on patients’ characteristics. Also, the current recommended dose adjustments of cefepime are overly simplistic, as they only consider impaired renal function. In addition, dosage reductions are applied at arbitrary cutoff values of renal function, irrespective of the real covariate-parameter relationships. Considering that the relationship between ke and CrCl in adults is linear, it is suboptimal to select a single CrCl threshold, such as 30 or 60 mL/min, for population-based dose changes. Using OID algorithm, we provided a more continuous dosing approach including both CrCl and weight, which also provides useful information with the probability of achieving the goal (i.e., Cmin of 10 to 40). Clinicians should be aware that these are still empirical doses based on population estimates and are not guaranteed to achieve the optimal individual exposure. This is shown in the validation fit (Fig. S4), where the population predictions were not as good as the individual predictions. Consequently, TDM and model-informed precision dosing should follow to individualize therapy. As the cefepime toxicity target is not well defined yet, clinicians should exercise caution in giving large doses to their patients without the availability of the TDM at their institutions. This might be the case in many health institutions globally which do not have access to an in-house cefepime assay, and the turnaround time for sending the samples to another lab may be inconvenient. As a result, the availability of the cefepime assay with results available in a reasonable time is important to promptly adjust therapy.
Neurotoxicity might be a concern with increased cefepime exposure. Currently published data in this area are mainly retrospective with trough-only sampling. Different cefepime trough thresholds were suggested in these studies to be associated with neurotoxicity, including 22, 38, and 49 mg/L (21–23). More investigations are needed in this area, including well-defined diagnosis criteria and a better plasma and/or cerebrospinal fluid sampling scheme to assess the best PK/PD predictor of this event (24, 25).
We included a large number of patients and samples in the PK model, which was validated using multiple data sets and applied to an additional data set using TDM software. However, there are a number of limitations. First, the majority of the data contributed to the model were sparse TDM data, which resulted in elevated shrinkage of the PK parameters. However, that may have affected the individual predictions which were still reasonable in the validation (Fig. S4) given that most of the patients in the model-building data set had at least Cmin measured, which was the concentration we targeted in our individual predictions. Second, only the total concentration was measured, and the unbound fraction was assumed. Third, the number of pediatric patients in the model was low. Finally, while intrapatient variability may have been fitted in the past, future changes in the PK parameters were not possible to predict (26). Future efforts should address these limitations to improve the predictions and utility of precision dosing tools.
Conclusions.
We described cefepime population PK using a large data set mainly from the ICU. The model was integrated into precision dosing software to fit a new data set and showed good prediction ability. Optimal initial dosing recommendations in this work are based on modeling and simulation to maximize the probability of achieving prespecified target concentrations. Such dosing recommendations have not been otherwise validated in retrospective or prospective studies. Hence, if dosing outside the approved regimens in the package insert or institutional precedent, the appropriate cefepime plasma concentration should be verified by TDM and further dose individualization if needed.
MATERIALS AND METHODS
Study population.
This was a PK study combining six cefepime data sets.
PK model development data sets. The first data set presented pediatric patients between 2 months and 18 years old who received cefepime at 50 mg/kg intravenously (i.v.) over 30 min every 8 h. Samples were drawn at times 0, 0.5, 0.75, 1, 2, 4, 6, and 8 h after starting cefepime infusion (8).
The second data set was an update of a previously published one which was based on a retrospective chart review of adult ICU patients admitted to UF Health between 2016 and 2018 (17). The data set was updated to include adult ICU patients admitted to UF Health between 2016 and 2019. Patients who received cefepime and had a cefepime concentration reported were included. Cefepime was administered as 2 g i.v. every 8 h over 30 min, 3 to 4 h, or 6-g CI. Cefepime dose was adjusted based on renal function and the measured concentrations. The recommended blood sampling times were 1 h after the end of infusion and before the next dose within the same dosing interval.
Model validation data sets. The third data set was from a prospective international study which included adult ICU patients who received beta-lactams, including cefepime. The median (IQR) cefepime daily dose was 6 g (5, 6). Plasma samples were collected midinterval and before the next dose (7).
The fourth data set included adult patients with hematological malignancy who developed febrile neutropenia and were initiated on cefepime. Cefepime was administered as 2 g i.v. every 8 h over 30 min. Five plasma samples were drawn, two after the third dose at 60% of the dosing interval and a trough; the next two were drawn similarly after the sixth dose, and one sample was drawn before the tenth dose (9).
The fifth data set had adult patients with hematological malignancy and hematopoietic cell transplant who received cefepime 2 g i.v. every 8 h over 30 min for the treatment of febrile neutropenia. Blood samples were collected after ≥2 days into therapy at times 0, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, and 8 h after the start of cefepime infusion (10).
Precision dosing application data set. Similar to the second data set, the sixth data set included ICU patients admitted to UF Health between 2016 and 2019 who received cefepime therapy and had available cefepime concentrations. This data set was independent from others and was used with precision dosing software to evaluate the predictive accuracy and precision of the PK model we developed using the previously mentioned data sets. Also, this data set was used to validate the OID generated using the PK model and assess the target attainment.
For all the data sets, patients on renal replacement therapy were excluded. Data collected included age, sex, weight, serum creatinine, cefepime regimens, and serum concentrations. CrCl was calculated using the Cockcroft-Gault equation (27), using total body weight and 0.85 correction factor in females, to find an acceptable renally based descriptor of cefepime elimination as reported previously in similar models (17, 18). Institutional review boards at all the involved sites reviewed and approved the corresponding studies, and informed consent was obtained from participants or legal guardians (prospective studies) or waived (retrospective studies).
Population pharmacokinetics.
The actual dosing and sampling times were used in the data sets to calculate the elapsed time, and no assumption was made about reaching steady state. The nonparametric adaptive grid (NPAG) algorithm in the Pmetrics v1.9.7 R package was used to build the cefepime PK model. NPAG builds nonparametric population PK models by creating support points, with each point comprising a set of estimates for all PK parameters in the model and the associated probability (weight) of that set of estimates, with no underlying assumption of PK parameter value probability distributions. This property is suitable for characterizing populations expected to have outliers with extreme parameter values, such as ICU patients (28). One- and two-compartment models were evaluated, and the best-fit model was chosen. We tested for the weight, CrCl, and age group covariates by adding them to PK parameters in a forward stepwise fashion. If the CrCl at the time of the first dose was not available, the next closest CrCl value was used. On certain occasions when the CrCl is not available with subsequent doses because serum creatinine is not measured, Pmetrics will linearly extrapolate between available values or, if there are no further values, carry the last value forward. The final models were compared based on the Akaike information criterion, R2 of observed versus population and individual predicted concentration plots, bias, and imprecision. The assay error (standard deviation [SD]) was accounted for using error polynomial as a function of observed concentration (SD = C0 + [C1 × observed concentration]) using C0 (intercept) and C1 (slope) values of 1 and 0.1, respectively. A gamma multiplicative error model value was set to 2 to estimate the magnitude of additional noise or model misspecification (error = SD × gamma) (29). The final model gamma was estimated to be 1.78, with a value of 1 indicating no additional source of error. The model was validated by using it as a prior with zero cycles and the three external data sets (see “Model Validation Data Sets”). The observed versus predicted plots were evaluated for the acceptability of the model predictions.
Precision dosing application.
We used BestDose (www.lapk.org), software for individualized drug dosing, to test the predictive ability of our model against the external data set from UF Health. BestDose combined the cefepime PK model with the data from each patient in the external data set to update the model to an individualized, Bayesian posterior version, which comprises the most likely probability distribution of support points for each patient. The predicted cefepime concentration-time profile is the weighted average across all support points (i.e., MM design) in the posterior distribution (Fig. S1) (30, 31). In addition, BestDose includes the IMM Bayesian approach, which is specifically useful in ICU patients with changes in their PK parameters during therapy. This approach allows the posterior distribution to change from the previous distribution each time a new drug concentration is obtained (31). BestDose was used to fit the cefepime model to the data using the MM approach for samples in each separate dosing occasion and using the IMM approach for all samples in different dosing occasions. For patients who had cefepime concentrations from more than one dosing occasion, the prediction of cefepime concentration was evaluated using the MM fit from the previous occasion.
Optimal initial dosing simulations.
The final model was used to calculate the initial dose that maximizes the a priori probability of achieving a concentration target of efficacy, as described elsewhere (32). Using the Pseudomonas aeruginosa susceptibility breakpoint of 8 mg/L, the efficacy target was Cmin between 10 and 40 mg/L (corresponding to fCmin of 8 of 32 mg/L and fCmin:MIC 1 to 4). Optimal doses were derived for a large range of body weight and creatinine clearance values. The method is described in detail in the supplemental material.
ACKNOWLEDGMENTS
We thank all the clinical pharmacists, nurses, and physicians who assisted in obtaining and assessing cefepime concentrations as part of clinical care. We acknowledge the University of Florida Integrated Data Repository (IDR) and the UF Health Office of the Chief Data Officer for providing the analytic data set for this project.
Additionally, the research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under University of Florida Clinical and Translational Science Awards UL1 TR000064 and UL1TR001427. J.A. Roberts acknowledges funding from the Australian National Health and Medical Research Council for a Centre of Research Excellence (APP1099452) and a Practitioner Fellowship (APP1117065), as well as an Advancing Queensland Clinical Fellowship.
Disclaimer for J. Liu: This article reflects the views of the author and should not be construed to represent the FDA’s views or policies.
M.H. Scheetz had a contract with Allecra. M.N. Neely developed the BestDose software, and his lab has received a small beta use fee from some users. Other authors have no conflict of interest to disclose.
No external funding was received to conduct this study.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Magill SS, O’Leary E, Ray SM, Kainer MA, Evans C, Bamberg WM, Johnston H, Janelle SJ, Oyewumi T, Lynfield R, Rainbow J, Warnke L, Nadle J, Thompson DL, Sharmin S, Pierce R, Zhang AY, Ocampo V, Maloney M, Greissman S, Wilson LE, Dumyati G, Edwards JR, Frank L, Godine D, Martin B, Parker E, Pasutti L, Friedman S, Jones A, Kosmicki T, Fisher J, Maslar A, Meek J, Melchreit R, Badrun F, Fiore A, Fridkin SK, Morabit SL, Perry LA, Perlmutter R, Vaeth E, Gross A, Harper J, Pattee B, Rahmathullah N, Baumbach J, Sievers M, Concannon C, Felsen C, Emerging Infections Program Hospital Prevalence Survey Team, et al. 2021. Antimicrobial use in US Hospitals: comparison of results from emerging infections program prevalence surveys, 2015 and 2011. Clinical Infectious Diseases 72:1784–1792., 10.1093/cid/ciaa373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D. 2006. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 34:1589–1596. 10.1097/01.CCM.0000217961.75225.E9. [DOI] [PubMed] [Google Scholar]
- 3.Roberts JA, Abdul-Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, Hope WW, Farkas A, Neely MN, Schentag JJ, Drusano G, Frey OR, Theuretzbacher U, Kuti JL, International Society of Anti-Infective Pharmacology and the Pharmacokinetics and Pharmacodynamics Study Group of the European Society of Clinical Microbiology and Infectious Diseases. 2014. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis 14:498–509. 10.1016/S1473-3099(14)70036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10. 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- 5.Abdul-Aziz MH, Alffenaar J-WC, Bassetti M, Bracht H, Dimopoulos G, Marriott D, Neely MN, Paiva J-A, Pea F, Sjovall F, Timsit JF, Udy AA, Wicha SG, Zeitlinger M, De Waele JJ, Roberts JA, Infections in the ICU and Sepsis Working Group of International Society of Antimicrobial Chemotherapy (ISAC). 2020. Antimicrobial therapeutic drug monitoring in critically ill adult patients: a position paper. Intensive Care Med 46:1127–1153. 10.1007/s00134-020-06050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdulla A, Dijkstra A, Hunfeld NG, Endeman H, Bahmany S, Ewoldt TM, Muller AE, van Gelder T, Gommers D, Koch BC. 2020. Failure of target attainment of beta-lactam antibiotics in critically ill patients and associated risk factors: a two-center prospective study (EXPAT). Crit Care 24:1–12. 10.1186/s13054-020-03272-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts JA, Paul SK, Akova M, Bassetti M, De Waele JJ, Dimopoulos G, Kaukonen K-M, Koulenti D, Martin C, Montravers P, Rello J, Rhodes A, Starr T, Wallis SC, Lipman J, Roberts JA, Lipman J, Starr T, Wallis SC, Paul SK, Margarit Ribas A, De Waele JJ, De Crop L, Spapen H, Wauters J, Dugernier T, Jorens P, Dapper I, De Backer D, Taccone FS, Rello J, Ruano L, Afonso E, Alvarez-Lerma F, Gracia-Arnillas MP, Fernandez F, Feijoo N, Bardolet N, Rovira A, Garro P, Colon D, Castillo C, Fernado J, Lopez MJ, Fernandez JL, Arribas AM, Teja JL, Ots E, Carlos Montejo J, Catalan M, DALI Study, et al. 2014. DALI: defining antibiotic levels in intensive care unit patients: are current β-lactam antibiotic doses sufficient for critically ill patients? Clinical Infect Dis 58:1072–1083. 10.1093/cid/ciu027. [DOI] [PubMed] [Google Scholar]
- 8.Reed MD, Yamashita TS, Knupp CK, Veazey JM Jr, Blumer JL. 1997. Pharmacokinetics of intravenously and intramuscularly administered cefepime in infants and children. Antimicrob Agents Chemother 41:1783–1787. 10.1128/AAC.41.8.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sime FB, Roberts MS, Tiong IS, Gardner JH, Lehman S, Peake SL, Hahn U, Warner MS, Roberts JA. 2015. Adequacy of high-dose cefepime regimen in febrile neutropenic patients with hematological malignancies. Antimicrob Agents Chemother 59:5463–5469. 10.1128/AAC.00389-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whited L, Grove M, Rose D, Rhodes NJ, Scheetz MH, O’Donnell JN, Neeb J, Thoele K, Jones DR, Lowe C, Moore D, Kiel PJ. 2016. Pharmacokinetics of cefepime in patients with cancer and febrile neutropenia in the setting of hematologic malignancies or hematopoeitic cell transplantation. Pharmacotherapy 36:1003–1010. 10.1002/phar.1807. [DOI] [PubMed] [Google Scholar]
- 11.Goutelle S, Alloux C, Bourguignon L, Van Guilder M, Neely M, Maire P. 2021. To estimate or to forecast? Lessons from a comparative analysis of four Bayesian fitting methods based on nonparametric models. Ther Drug Monit 43:461–471. 10.1097/FTD.0000000000000879. [DOI] [PubMed] [Google Scholar]
- 12.Bustad A, Terziivanov D, Leary R, Port R, Schumitzky A, Jelliffe R. 2006. Parametric and nonparametric population methods: their comparative performance in analysing a clinical dataset and two Monte Carlo simulation studies. Clin Pharmacokinet 45:365–383. 10.2165/00003088-200645040-00003. [DOI] [PubMed] [Google Scholar]
- 13.Felton TW, Roberts JA, Lodise TP, Van Guilder M, Boselli E, Neely MN, Hope WW. 2014. Individualization of piperacillin dosing for critically ill patients: dosing software to optimize antimicrobial therapy. Antimicrob Agents Chemother 58:4094–4102. 10.1128/AAC.02664-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Praet A, Bourguignon L, Vetele F, Breant V, Genestet C, Dumitrescu O, Doleans-Jordheim A, Reix P, Goutelle S. 2021. Population pharmacokinetic modeling and dosing simulations of tobramycin in pediatric patients with cystic fibrosis. Antimicrob Agents Chemother 65:e00737-21. 10.1128/AAC.00737-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramos-Martín V, Neely MN, Padmore K, Peak M, Beresford MW, Turner MA, Paulus S, López-Herce J, Hope WW. 2017. Tools for the individualized therapy of teicoplanin for neonates and children. Antimicrob Agents Chemother 61:e00707-17. 10.1128/AAC.00707-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neely MN, Kato L, Youn G, Kraler L, Bayard D, van Guilder M, Schumitzky A, Yamada W, Jones B, Minejima E. 2018. Prospective trial on the use of trough concentration versus area under the curve to determine therapeutic vancomycin dosing. Antimicrob Agents Chemother 62:e02042-17. 10.1128/AAC.02042-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Shaer MH, Neely MN, Liu J, Cherabuddi K, Venugopalan V, Rhodes NJ, Klinker K, Scheetz MH, Peloquin CA. 2020. Population pharmacokinetics and target attainment of cefepime in critically ill patients and guidance for initial dosing. Antimicrob Agents Chemother 64:e00745-20. 10.1128/AAC.00745-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J, Neely M, Lipman J, Sime F, Roberts JA, Kiel PJ, Avedissian SN, Rhodes NJ, Scheetz MH. 2020. Development of population and Bayesian models for applied use in patients receiving cefepime. Clin Pharmacokinet 59:1027–1036. 10.1007/s40262-020-00873-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huttner A, Von Dach E, Renzoni A, Huttner BD, Affaticati M, Pagani L, Daali Y, Pugin J, Karmime A, Fathi M, Lew D, Harbarth S. 2015. Augmented renal clearance, low β-lactam concentrations and clinical outcomes in the critically ill: an observational prospective cohort study. Int J Antimicrob Agents 45:385–392. 10.1016/j.ijantimicag.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 20.Taccone FS, Laterre P-F, Dugernier T, Spapen H, Delattre I, Witebolle X, De Backer D, Layeux B, Wallemacq P, Vincent J-L, Jacobs F. 2010. Insufficient β-lactam concentrations in the early phase of severe sepsis and septic shock. Crit Care 14:R126–R129. 10.1186/cc9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boschung-Pasquier L, Atkinson A, Kastner LK, Banholzer S, Haschke M, Buetti N, Furrer DI, Hauser C, Jent P, Que YA, Furrer H, Babouee Flury B. 2020. Cefepime neurotoxicity: thresholds and risk factors. A retrospective cohort study. Clin Microbiol Infect 26:333–339. 10.1016/j.cmi.2019.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Lau C, Marriott D, Schultz HB, Gould M, Andresen D, Wicha SG, Alffenaar JW, Penm J, Reuter SE. 2021. Assessment of cefepime toxicodynamics: comprehensive examination of pharmacokinetic/pharmacodynamic targets for cefepime-induced neurotoxicity and evaluation of current dosing guidelines. Int J Antimicrobial Agents 58:106443. 10.1016/j.ijantimicag.2021.106443. [DOI] [PubMed] [Google Scholar]
- 23.Lamoth F, Buclin T, Pascual A, Vora S, Bolay S, Decosterd LA, Calandra T, Marchetti O. 2010. High cefepime plasma concentrations and neurological toxicity in febrile neutropenic patients with mild impairment of renal function. Antimicrob Agents Chemother 54:4360–4367. 10.1128/AAC.01595-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhodes NJ, Kuti JL, Nicolau DP, Neely MN, Nicasio AM, Scheetz MH. 2016. An exploratory analysis of the ability of a cefepime trough concentration greater than 22 mg/L to predict neurotoxicity. J Infect Chemother 22:78–83. 10.1016/j.jiac.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Al-Shaer MH, Peloquin CA. 2021. Using precision dosing to minimize cefepime-induced neurotoxicity: the challenge of targets. J Infect Chemother 27:929–930. 10.1016/j.jiac.2021.02.020. [DOI] [PubMed] [Google Scholar]
- 26.Abrantes JA, Jönsson S, Karlsson MO, Nielsen EI. 2019. Handling interoccasion variability in model‐based dose individualization using therapeutic drug monitoring data. Br J Clin Pharmacol 85:1326–1336. 10.1111/bcp.13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cockcroft DW, Gault H. 1976. Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41. 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 28.Neely M, van Guilder M, Yamada W, Schumitzky A, Jelliffe R. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jelliffe RW. 2012. Some comments and suggestions concerning population pharmacokinetic modeling, especially of digoxin, and its relation to clinical therapy. Ther Drug Monit 34:368–377. 10.1097/FTD.0b013e31825c88bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jelliffe R, Bayard D, Milman M, Van Guilder M, Schumitzky A. 2000. Achieving target goals most precisely using nonparametric compartmental models and “multiple model” design of dosage regimens. Ther Drug Monit 22:346–353. 10.1097/00007691-200006000-00018. [DOI] [PubMed] [Google Scholar]
- 31.Bayard DS, Jelliffe RW. 2004. A Bayesian approach to tracking patients having changing pharmacokinetic parameters. J Pharmacokinet Pharmacodyn 31:75–107. 10.1023/B:JOPA.0000029490.76908.0c. [DOI] [PubMed] [Google Scholar]
- 32.Boidin C, Bourguignon L, Cohen S, Roger C, Lefrant JY, Roberts JA, Allaouchiche B, Lepape A, Friggeri A, Goutelle S. 2019. Amikacin initial dose in critically ill patients: a nonparametric approach to optimize a priori pharmacokinetic/pharmacodynamic target attainments in individual patients. Antimicrob Agents Chemother 63:e00993-19. 10.1128/AAC.00993-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aac.02046-21-s0001.pdf, PDF file, 0.7 MB (703.6KB, pdf)