

Abstract
Objectives:
Arterial stiffness is recognized as an important predictor of cardiovascular disease (CVD) morbidity and mortality, independent of traditional CVD risk factors. Given that arterial tissue is not easily accessible, most gene expression studies on arterial stiffness have been conducted on animals or on patients who have undergone by-pass surgeries. In order to obtain a deeper understanding of early changes of arterial stiffness, this study compared transcriptome profiles between healthy adults with high and low arterial stiffness.
Method:
The sample included twenty healthy female adults without CVD. Arterial stiffness was measured by carotid-femoral pulse wave velocity, the ‘gold-standard’ measure of central arterial stiffness. Peripheral blood samples collected to PAXgene™ RNA tubes were used for RNA sequencing (RNA-seq). The potential confounding effects of age, body mass index, and mean arterial pressure were controlled for in RNA-seq analysis. To validate RNA-seq results, quantitative real-time PCR (qRT-PCR) was performed for 6 selected genes.
Results:
The findings demonstrated that genes including CAPN9, IL32, ERAP2, RAB6B, MYBPH, and miRNA626 were down-regulated, and that MOCS1 gene was up-regulated among the people with higher arterial stiffness. qRT-PCR showed that the changes of CAPN9, IL32, ERAP2, and RAB6B were in concordance with RNA-Seq data, and confirmed the validity of the gene expression profiles obtained by RNA-seq analysis.
Conclusion:
Previous studies have suggested the potential roles of CAPN9, IL32, and ERAP2 in structural changes of the arterial wall through up-regulation of metalloproteinases. However, the current study showed that CAPN9, IL32, and ERAP2 were down-regulated in the individuals with higher arterial stiffness, compared with those with lower arterial stiffness. The unexpected directions of expression of these genes may indicate an effort to maintain vascular homeostasis during increased arterial stiffness among healthy individuals. Further studies are guaranteed to investigate the roles of CAPN9, IL32, and ERAP2 in regulating arterial stiffness in people with and without CVD.
Keywords: RNA-seq, transcriptome, arterial stiffness, pulse wave velocity
Introduction
Arterial stiffness is recognized as a hallmark of arterial aging, and strongly predicts cardiovascular disease (CVD) morbidity and mortality, independent of traditional CVD risk factors (1). Animal and human studies suggest that progressive degradation of the medial wall, characterized by elastin fatigue and increased fragmentation of collagen fibers, might be an important determinant of arterial stiffness (2). These structural changes, in addition to functional changes such as inflammation and endothelial dysfunction as well as impairment of nitric oxide synthase, appear to contribute to arterial stiffness (3). A loss of elastic properties of the arterial wall results in early reflection of the blood pressure wave, which is central to the pathogenesis of isolated systolic hypertension and left ventricular hypertrophy (4).
Propagation of the pressure wave along the arterial tree is faster in stiff arteries; therefore, determining pulse wave velocity (PWV) is accepted as a noninvasive and reproducible method to measure arterial stiffness (5). PWV can be determined by the distance the pulse travels between two arterial sites (D) divided by pulse transit time (t) or as PWV (m/s) = D (meters) / t (seconds). Carotid-femoral PWV(cfPWV) is considered the ‘gold-standard’ measure of central arterial stiffness because it measures along the aorto-iliac pathway, and the thoracic and abdominal aortas make the largest contribution to the arterial buffering function (6). Many studies suggest that cfPWV has a significant prognostic value for hypertension (7), left ventricular hypertrophy (8), coronary artery disease, and stroke (9). This noninvasive measure of arterial stiffness made the assessment of CVD risk feasible not only in patient groups but also in the general population.
While existing genome-wide studies report candidate gene polymorphisms that have a moderate to substantial heritable component (10), gene expression analyses are particularly useful for obtaining a deeper understanding of the changes at transcriptome level. Given that arterial tissue is not easily accessible, most gene expression studies on arterial stiffness have been conducted on animals or on patients who have undergone by-pass surgeries. Again, studies have rarely looked at gene expression profiles with regard to arterial stiffness in healthy individuals. Recently, there is increasing evidence that the expression pattern of peripheral blood cells reflects gene expression changes in the vascular wall, suggesting that gene expression profiling of blood could be a useful tool to explore the molecular mechanisms of arterial disease (11–14). Studies have shown that the genes implicated in the underlying mechanisms of arterial stiffness are expressed in blood cells. Only two studies have examined the relationship between arterial stiffness and gene expression in peripheral blood. These used real-time reverse transcription polymerase chain reaction (qRT-PCR) for only a few selected genes, including angiopoietin-1 (Ang-1) and Ang-2 (15), and thioredoxin (TXN) (16), suggesting that those genes may play significant roles in increased arterial stiffness. It has been reported that RNA-stabilized whole blood samples are more robust and reliable for gene expression profiling than RNA isolation from peripheral blood mononuclear cells (17). The advantages of RNA-sequencing (RNA-seq) over microarrays have been well documented and include higher specificity and sensitivity for enhanced detection of differential expression (18).
Although use of cfPWV has been recommended for early detection of vascular damage and CVD risk, most studies using cfPWV have focused on identifying pathological mechanisms of arterial stiffness in CVD patient groups. Examining arterial stiffness in individuals without CVD facilitates exclusion of potential confounding effects of existing conditions. In order to better understand (patho)physiological mechanisms of arterial stiffness, we compared differently regulated genes between two groups of healthy female adults – those with high and those with low arterial stiffness measured by cfPWV. We performed RNA-seq on peripheral whole blood samples. We subsequently validated the RNA-seq results using quantitative real time PCR (qRT-PCR).
Method
Study subjects
The sample was selected from an original study (under review) conducted by the authors. In that study, a convenience sample of 85 female adults was recruited from Charlottesville, VA. Written informed consent was obtained from all patients for being included in the study. All the measurements and blood samples used in this report were obtained during the original study. The purpose of the original study was to evaluate changes of arterial stiffness after stress induction. Gender differences in stress levels have been reported. To minimize heterogeneity of the sample, only females were included. Arterial stiffness increases with age. To exclude cardiovascular conditions that may be aggravated by age, female adults aged between 18 to 55 years were included. Exclusion criteria included known history of CVD (that may affect arterial stiffness), hypercholesterolemia as screened by fasting lipid profile, taking medications known to affect hemodynamics (e.g., antihypertensives, thyroid hormones, or steroids), irregular heart rhythm that prohibits measuring cfPWV, and being unable to fast for ~6 hours. Among the original participants, forty subjects who demonstrated hemodynamic changes after stress induction were selected. From this pool, 20 subjects were selected from the pre-test (pre-stress induction) data for RNA-seq data analysis, where 10 were from the upper quartile and another 10 were from the lower quartile of cfPWV.
Standardization of subject conditions
Study procedures complied with the European Society of Cardiology’s recommendations to standardize subject conditions when calculating cardiovascular measures (19,20). All subjects were asked to abstain from rigorous physical activity for one day prior to their appointment, to fast overnight beginning the midnight before the data collection, and to consume the identical breakfast of cereal (35g), milk (8oz), and orange juice (8oz) followed by fasting until data collection was completed in the afternoon.
Measures of physiological factors
Body mass index (BMI).
After changing into a hospital gown, undressing except for undergarments, participants’ height and weight were measured using an Accu-Hite wall-mounted stadiometer (WA, US) and a Withings electronic scale (PA, US). These measures were used to calculate BMI (weight (kg) / [height (m)]2).
Blood pressure (BP).
Subjects rested for 10 minutes lying on a bed. Then we measured BP using Vital Signs Monitor 300 Series (WelchAllyn, US). To measure BP, the midsection circumference of the dominant upper-arm was measured with a tape measure (21), and the proper sized cuff was selected accordingly. Using Vital Signs Monitor 300 Series (WelchAllyn, US), systolic and diastolic BP were measured on the dominant arm. Mean arterial pressure (MAP) was calculated by using the formula, MAP = (systolic BP + 2 [diastolic BP])/3.
Arterial stiffness.
Arterial stiffness was assessed by measuring carotid-femoral pulse wave velocity (PWV) using the SphygmoCor system (AtCor Medical, AU). While an electrocardiogram was being recorded, each carotid and femoral waveform was acquired by applying a pressure sensitive transducer (tonometer) on carotid and femoral sites. The transit time of the pulse from the left ventricle to the carotid artery (t1) and the transit time of the pulse from the left ventricle to the femoral artery (t2) were calculated by identifying the location of the foot of the pulse waves. The distance from the suprasternal notch to the carotid artery site (d_carotid) and the distance from the suprasternal notch to the femoral artery site (d_femoral) were measured on the body using a tape measure. PWV (m/s) reflects the difference in distances from the suprasternal notch to two arterial sites (d_femoral - d_carotid) divided by the mean time difference (t2 - t1) (19).
Biological sample collection
Pain and anxiety from phlebotomy may cause hemodynamic changes; thus, we drew blood as the last step of data collection. Peripheral blood (2.5 ml) was collected to a PAXgene™ RNA tube containing red blood cell lysis buffer and an RNA stabilizing solution (QIAGEN, Frederick, MD). The collected blood was stored at −80°C until ready for RNA extraction.
RNA extraction, library preparation and sequencing
We extracted RNA from whole blood using the PAXgene™ Blood RNA system (QIAGEN). The quality of the RNA samples was confirmed using an Agilent 4200 Tapestation (Agilent Technologies, US) while the quantity of the RNA was measured using a Qubit (Life Technologies, US). All of the samples used for this study had an excellent purity (A260/A280≥1.9; A260/A230≥2) and showed no visible signs of degradation (RIN ≥ 9). We used a TruSeq Stranded mRNA library prep kit (Illumina, San Diego, US) to generate mRNA-seq libraries. These kits generated high quality cDNA libraries for sequencing by fragmentizing the RNA species, ligating adapters, and performing reverse transcription. We indexed (or barcoded) the samples allowing individual libraries to be pooled in equi-molar amounts across all lanes, minimizing potential technical bias of run variation.
Bioinformatic Analysis of RNA-seq Data
For bioinformatics analysis, we performed quality check for the fastq reads using FastQC, version 0.11.7. Poor quality reads and adapter sequences were filtered out by running CutAdapt(V2.5). We used RNA-SeQC for quality control specific to RNA-seq. We assessed total number of reads, depth of reads, average read length, average coverage across the gene, number of genes identified, PCR duplication rate, ribosomal content, and exon/intron representation to confirm the quality of the library and sequencing. We aligned the raw reads to GRCh38 reference genome using STAR, version 2.6.1a. We counted number of reads mapped to genes using htseq, version 0.11.0. We performed differential gene expression analysis using DESeq2 (V1.22.2) in R (V3.5.2 Bioconductor (V3.8). The gene count table was imported to DESeq2. The distribution of the reads was modeled as a negative binomial distribution with the mean and the variance estimated from the data. Age, BMI, and MAP were controlled as potential covariates in designing the differential expression testing. P-values were calculated by using Wald test. Multiple testing correction is performed with Benjamini-Hochberg’s False Discovery Rate (FDR) adjusted by Independent Hypothesis Weighting method accounting for the age, BMI, and MAP as its covariate variables. Cutoff value of 0.05 was used on the final adjusted p-value.
Construction of pathway-gene-process network.
To identify dysregulated biological pathways and networks related to arterial stiffness, we performed pathway analysis using Ingenuity Pathway Analysis (Ingenuity® Systems, Redwood City, CA). Genes after filtering by FDR<=0.05 were used as input for pathway testing. Significance value of the pathways fitting was computed by right-tailed Fisher’s exact test.
RNA-seq validation with real time-PCR
To validate RNA-seq results, a two-step qRT-PCR was performed to quantify the RNA transcript level for 6 selected genes among the significant genes found in the RNA-seq, which included CAPN9, ERAP2, IL32, MOCS1, MYBPH, and RAB6B. Two reference genes, B2M and Tbp, were used as housekeeping genes for the purposes of normalization. Taqman real-time PCR assays were purchased from Fisher Scientific/Invitrogen and validated using human reference RNA. First, total RNA prepared in the same way as for RNA-seq was subjected to reverse transcription to make cDNA. Quantitative RT-PCR was performed in a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA) following the manufacturer’s recommended protocol for Taqman qPCR assays. The data collected as the cycle threshold numbers were normalized by the geometric means of the housekeeping genes, and then used to derive the fold changes and p-values using the delta-delta Ct comparative expression method (22).
Results
Mean age of the study participants was 33.45 (SD 13.71) years. Mean BMI was 22.48 (SD 3.67) kg/m2 and mean arterial pressure (MAP) was 76.35 (SD 8.32) mmHg. Groups with high arterial stiffness had significantly higher age (p=0.003) and MAP (p=0.002) compared with those with low arterial stiffness (Table 1).
Table 1.
Characteristics of study subjects
| All | High AS group n=10 |
Low AS group n=10 |
p-value | |
|---|---|---|---|---|
| Age (year) | 33.45±13.71 | 42.60±14.41 | 24.30±1.83 | 0.003 |
| BMI (kg/m2) | 22.48±3.67 | 23.19±2.75 | 21.77±4.44 | 0.403 |
| MAP (mmHg) | 76.35±8.32 | 81.60±8.45 | 71.10±3.69 | 0.002 |
| cfPWV (m/s) | 6.31±1.61 | 7.67±1.13 | 4.95±0.28 | 0.000 |
BMI = body mass index; MAP = mean arterial pressure; cfPWV = carotid-femoral pulse wave velocity; AS = arterial stiffness.
We conducted a one-way ANCOVA to compare the difference of arterial stiffness between two groups with high or low arterial stiffness after controlling for age, BMI, and MAP. There remained a significant difference in mean cfPWV between the two groups; F (1, 15) = 15.54, p=0.001 (Table 2).
Table 2.
Group difference in cfPWV after controlling for age, BMI, and MAP
| Source | Sum of Squares | df | Mean Square | F | Sig. |
|---|---|---|---|---|---|
| Age (year) | 2.486 | 1 | 2.486 | 7.359 | .016 |
| BMI (kg/m2) | .039 | 1 | .039 | .116 | .738 |
| MAP (mmHg) | .929 | 1 | .929 | 2.750 | .118 |
| GROUP | 5.249 | 1 | 5.249 | 15.537 | .001 |
| Error | 5.068 | 15 | .338 |
BMI = body mass index; MAP = mean arterial pressure; cfPWV = carotid-femoral pulse wave velocity; GROUP = higher vs. lower cfPWV; R2 = .897 (Adjusted R2 = .869)
The Illumina’s NextSeq 500 sequencer generated around 30 million paired end reads with read length of 75 bp (2 × 75 bp) per sample/library by multiplexing all samples across all sequencing runs. The significantly up- and down-regulated genes associated with high arterial stiffness are delineated in Tables 3 and 4.
Table 3.
Assessment of up-regulated genes
| Gene title | Coding/non-coding | Log2(Fold-change) | Adjusted p-value (FDR) | |
|---|---|---|---|---|
| MOCS1 | Molybdenum Cofactor Synthesis 1 | Protein coding gene | 1.82 | 5.8 × 10−3 |
| LOC400655 | Uncharacterized LOC400655 | non-coding RNA | 25.67 | 7.80E-06 |
Table 4.
Assessment of down-regulated genes
| Gene title | Coding/non-coding | Log2(Fold-change) | Adjusted p-value (FDR) | |
|---|---|---|---|---|
| CAPN9 | Calpain 9 | Protein coding gene | −29.87 | 2.11E-33 |
| ERAP2 | Endoplasmic Reticulum Aminopeptidase 2 | Protein coding gene | −1.69 | 8.00E-05 |
| IL32 | Interleukin 32 | Protein coding gene | −0.78 | 0.0314 |
| MYBPH | Myosin Binding Protein H | Protein Coding gene | −1.74 | 7.1 × 10−3 |
| RAB6B | A member of the small Rab (G-protein) family | Protein Coding gene | −1.15 | 0.0260 |
| GLRA4 | Glycine Receptor Alpha 4 | Protein coding gene | −29.92 | 1.21E-35 |
| SEC14L6 | SEC14 Like Lipid Binding 6 | Protein Coding gene | −15.89 | 2.26E-11 |
| LINC00337 | Long Intergenic Non-Protein Coding RNA 337 | non-coding RNA | −14.68 | 4.83E-07 |
| LOC101927666 | Uncharacterized LOC101927666 | non-coding RNA | −24.18 | 0.8 × 10−3 |
| MIR626 | MicroRNA 626 | non-coding RNA | −29.99 | 9.26E-07 |
Six genes including CAPN9, IL32, ERAP2, RAB6B, MYBPH, and MOCS1 were selected for verification of gene expression by real time-PCR (Table 5). The direction of expression of these genes as quantified by real time-PCR were consistent with the results obtained by RNA-seq analysis. Given high CT levels and variability in qPCR measurements, CAPN9 showed a clear tendency to significance. The changes of other genes including IL32, ERAP2, and RAB6B were also found to be in concordance with RNA-Seq data, and thereby confirmed the validity of the gene expression profiles obtained by RNA-seq analysis.
Table 5.
Real time-PCR results of selected genes
| Genes | Fold change | p-value | Avg CT |
|---|---|---|---|
| MOCS1 | 1.33 | 0.575 | 32.06 |
| MYBPH | −1.36 | 0.341 | 32.27 |
| IL32 | −1.61 | 0.009 | 23.96 |
| ERAP2 | −1.69 | 0.018 | 26.51 |
| RAB6B | −2.18 | 0.005 | 30.78 |
| CAPN9 | −7.94 | 0.052 | 38.15 |
Ingenuity pathway analysis showed that down-regulated genes including CAPN9, IL32, ERAP2, RAB6B, and MYBPH are included in the pathway of post-translational modification, protein degradation, and protein synthesis (Figure 1).
Figure 1. Pathway-gene-process network.
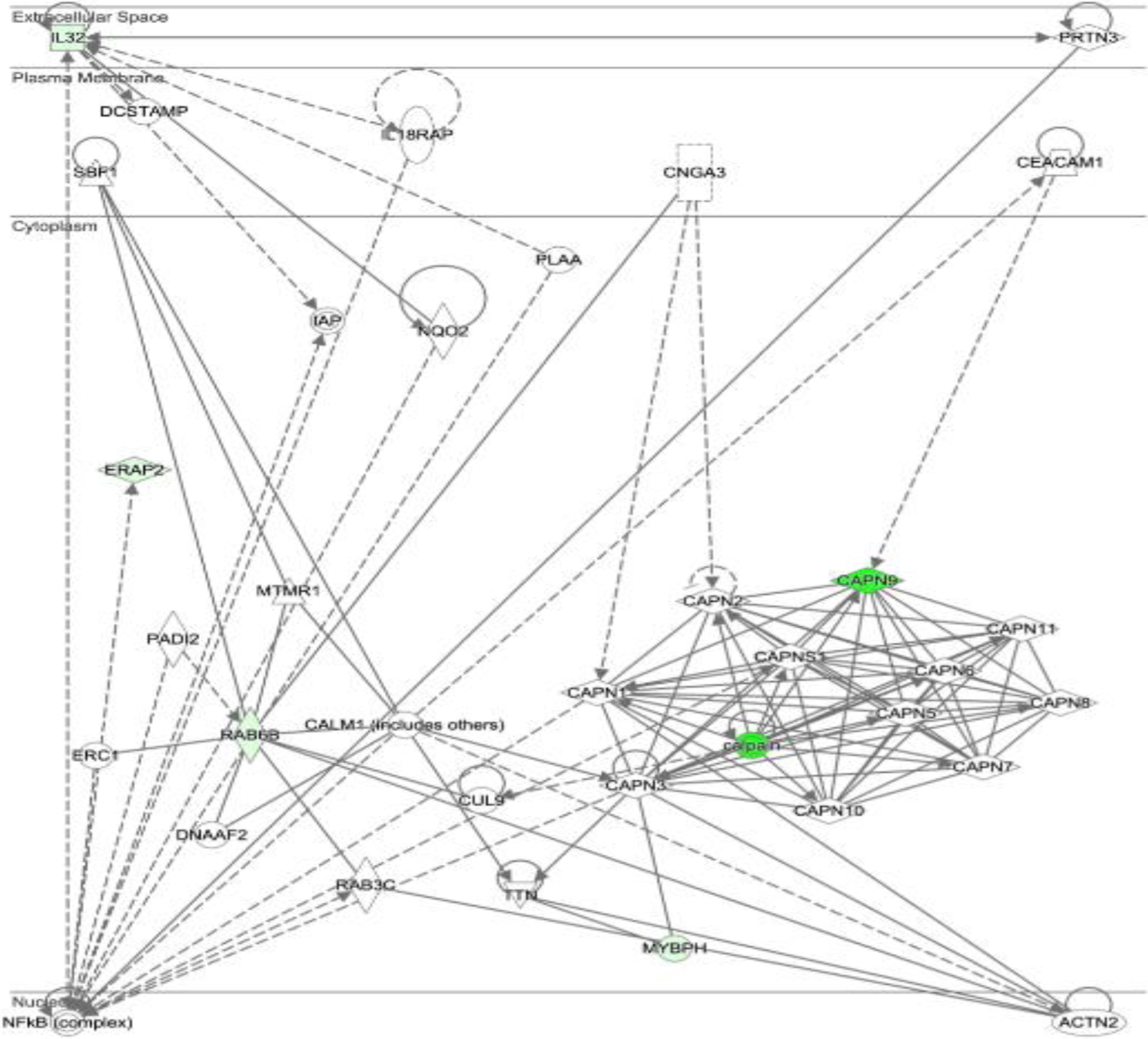
Downregulated genes (CAPN9, IL32, ERAP2, RAB6B, and MYBPH) are shown in green (the darker, the more down-regulated expression).
Discussion
Although arterial stiffness is recognized as one of the most important contributors to CVD, molecular mechanisms involved in arterial stiffness remain unclear. We believe that this is the first study reporting genes that are differentially expressed in the peripheral blood of healthy female adults. This study demonstrates that CAPN9, IL32, ERAP2, and RAB6B were differentially expressed between two groups with high and low levels of arterial stiffness, after controlling for influence of age, BMI, and MAP.
We found Calpain9 (CAPN9) to be one of the most significantly down-regulated genes. Calpains are the family of Ca2+- activated cysteine proteases. These neutral proteases (Calpains) have received considerable attention because intracellular protein degradation is an important mechanisms for growth and atrophy of tissues (23–25). Calpain1 and Calpain2 are the commonly evaluated members of this family, and it is strongly suggested that they may be involved in the degradation of the endogenous inhibitor of nuclear factor (NF)-kappa (NF-kB), which in turn increases the activation of NF-κB (26). NF-kB plays a critical role in the expression of proinflammatory genes including cytokines and chemokines (27). Evidence from previous studies proposes that the activities of Calpains are involved in the inflammation process and may serve as novel molecular targets for inflammation-associated diseases such as atherosclerosis (25,28). Furthermore, a previous study reported the significant role of Calpain inhibitors in inhibiting matrix metalloproteinase 2 (MMP2) activity (29). MMPs are a family of proteinases that degrade extracellular matrix proteins such as elastin and collagen (30). In particular, MMP2 has been associated with arterial stiffness in healthy subjects (31) and those with chronic kidney disease (32). It was reported that calpain inhibitors (calpain inhibitor III, ALLN, and PD-150606) have significant pharmacological activity as inhibitors of MMP2 enzymatic activity (29). Although Calpain1 & Calpain2 are often reported, Calpain9 is rarely reported in previous studies of arterial stiffness, and available studies suggest that Calpain9 is associated with gastric cancer. Although the function of Calpain9 is not known yet, it is recognized that the proteins within a family may have similar functions within the cell (33). If that is the case here, then it is contradictory to the expectation that in our study, the expression of Calpain9 is 29-fold down-regulated in persons with high arterial stiffness compared with low arterial stiffness. Although the study participants had significantly higher arterial stiffness after controlling for age, BMI, and MAP, they were healthy with no known CVD. Several mechanisms appear to jointly contribute to increases in arterial stiffening (34). It can be speculated that this result indicates defensive mechanisms to counteract increased arterial stiffness in this group of people who do not possess other impaired mechanisms such as increased activity of the renin-angiotensin-aldosterone system.
The IPA pathway analysis suggests that cellular mechanics could be dysregulated by Calpain protease. This pathway includes four other genes (IL32, ERAP2, RAB6B, and MYBPH) that were significantly dysregulated along with the CAPN9 gene in this study. Interleukin32 (IL32), a pro-inflammatory cytokine, is known to induce other pro-inflammatory cytokines and also to increase the expression of chemokine and soluble vascular cell adhesion molecules (sVCAM-1) (35). Another in-vitro study also demonstrated that IL32 facilitated cell migration and invasion, and this process was mediated through activation of the NF-κB signaling pathway (36). The same study further demonstrated that up-regulation of IL32 could induce up-regulation of MMP2 and MMP9 (36), that have been associated with arterial stiffness in healthy subjects (31) and in patients with essential hypertension (37) and chronic kidney disease (38). By promoting the synthesis of MMPs, IL32 may also contribute to plaque instability and rupture during the later stages of atherosclerosis (39). Previous evidence suggest that IL32 is involved in chronic low grade but persistent inflammation (35). Recent reports further suggest a crucial role of IL32 in chronic inflammatory diseases including rheumatoid arthritis, chronic obstructive pulmonary disease, inflammatory bowel disease, as well as cardiovascular disease (35). No published study has thus far examined the levels of IL32 in regard to arterial stiffness; our study shows unexpected association between the down-regulation of IL32 gene in people with higher arterial stiffness. Further studies are needed to examine the regulation of IL32 gene in individuals with no CVD, as these may increase our understanding in the roles of IL32 in the development of CVD.
The RNA-seq analysis in this study identified ERAP2 and RAB6B that are dysregulated in persons with high arterial stiffness but are rarely reported in the literature. The endoplasmic reticulum aminopeptidase 2 gene (ERAP2) encodes a zinc metalloaminopeptidase of M1 protease family. ERAP2 is also known as leukocyte-derived arginine aminopeptidase. Recently, a study reported the structure and mechanism of action of homologous zinc metallopeptidases, ERAP1, but the function and mechanisms of ERAP 2 are not well studied (40). Given that arterial stiffness is characterized by protein rearrangement, potential role of this metalloaminopeptidase enzyme in arterial stiffness may contribute to our understanding the mechanism of arterial stiffness. RAB6B is a member of the small Rab (G-protein) family which plays roles as regulators of vesicle traffic (41). While our study showed significant down-regulation of RAB6B in the group with high arterial stiffness, no previous evidence has been published that recognizes a potential role of RAB6B in vascular biology.
Taken together, the present study, for the first time, provides an analysis comparing the whole transcriptomic profiles between healthy female adults with high and low arterial stiffness. Many previous studies have demonstrated that arterial stiffness is increased by elastase activity of matric metalloproteinases (MMPs) which are the family of calcium-dependent zin-containing endopeptidases (37,38,42). Our study suggests that Calpain9 (Ca2+- activated neutral proteases), IL32, and ERAP2 (zinc metalloaminopeptidase) may also be involved in structural changes of arterial wall. Downregulation of these genes in the healthy individuals with higher levels of arterial stiffness guarantees further studies to investigate potential roles of these genes in vascular homeostasis and disease progress.
A limitation of this study is the relatively small sample size. Along with potential statistic errors and residual effects, a small sample size may limit the generalizability of the study findings. Since the study included only females, further exploration is needed in men. Another limitation of the study was an inability to compare mRNA expression among groups with equal basic characteristics. In an effort to reduce the confounding effect of inter-individual variation in mRNA expression, we purposely controlled the common risk factors of arterial stiffness; however, other uncontrolled risk factors may still exist that affect arterial stiffness and gene expression.
Conclusion
Arterial stiffness is an important determinant of CVD, the leading cause of morbidity and mortality worldwide (43). Early intervention is of great importance in reducing disease burden. Identifying useful biomarkers of arterial stiffness is considered critical in order to prevent and reduce CVD burden. We examined gene expression in a community-based sample of healthy female adults using RNA-seq technology, and explored actively dysregulated genes in women with different levels of arterial stiffness. The findings illustrate that CAPN9, IL32, ERAP2, and RAB6B are significantly down-regulated in individuals with higher levels of arterial stiffness. Given previous evidence on the potential roles of CAPN9, IL32, and ERAP2 in altering vascular structure and function, the unexpected directions of the expression of these genes may indicate an internal effort to maintain vascular homeostasis during increased arterial stiffness in healthy individuals. The current study may provide a mechanistic insight into the regulation of arterial stiffness in healthy individuals. The important next step is to further verify the study findings with a greater number of samples and examine the translation stage of gene transcripts in protein levels. Such studies will support the endeavor to further understand the mechanisms of arterial stiffness and identify therapeutic targets, and will advance the knowledge necessary to decrease societal CVD burden.
Sources of Funding
This study was supported by the K23NR016215 grant from the National Institutes of Health/National Institute of Nursing Research.
Footnotes
Disclosures
The authors do not have any credit or conflicts of interest to disclose.
Human subjects/informed consent statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from all patients for being included in the study.
Contributor Information
Jeongok Logan, University of Virginia, School of Nursing, 202 Jeanette Lancaster Way, Charlottesville, VA 22903, United States.
Sijung Yun, National Institute of Health, Co-founder and Chief Technology Officer of ZtoMed, Inc, Yotta Biomed, LLC. 8908 Ewing Dr., Bethesda, MD 20817.
Yongde Bao, University of Virginia, Microbiology, Immunology, and Cancer Biology, PO Box 800734, Jordan Hall, 1070, Charlottesville, VA 22908.
Emily Farber, Center for Public Health Genomics, School of Medicine, University of Virginia, Charlottesville, VA, 22908.
Charles R. Farber, Departments of Public Health Sciences and Biochemistry and Molecular Genetics, Center for Public Health Genomics, University of Virginia, Public Health Sciences, OMS 3817B, PO Box 800717, Charlottesville, VA 22908, United States.
References
- 1.Sethi S, Rivera O, Oliveros R, Chilton R. Aortic stiffness: pathophysiology, clinical implications, and approach to treatment. Integr Blood Press Control [Internet]. 2014. [cited 2019 Aug 27];7:29–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24910511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinn U, Tomlinson LA, Cockcroft JR. Arterial stiffness. JRSM Cardiovasc Dis [Internet]. 2012. Sep 30 [cited 2019 Aug 27];1(6). Available from: http://www.ncbi.nlm.nih.gov/pubmed/24175072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Bussel BC, Schouten F, Henry RM, Schalkwijk CG, de Boer MR, Ferreira I, et al. Endothelial dysfunction and low-grade inflammation are associated with greater arterial stiffness over a 6-year period. Hypertens (Dallas, Tex 1979) [Internet]. 2011. Oct [cited 2019 Aug 27];58(4):588–95. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21859964 [DOI] [PubMed] [Google Scholar]
- 4.Sethi S, Rivera O, Oliveros R, Chilton R. Aortic stiffness: pathophysiology, clinical implications, and approach to treatment. Integr Blood Press Control. 2014;7:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackenzie IS, Wilkinson IB, Cockcroft JR. Assessment of arterial stiffness in clinical practice. QJM [Internet]. 2002. Feb 1 [cited 2019 Aug 27];95(2):67–74. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11861952 [DOI] [PubMed] [Google Scholar]
- 6.Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J [Internet]. 2006. Sep 25 [cited 2019 Jun 18];27(21):2588–605. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17000623 [DOI] [PubMed] [Google Scholar]
- 7.Oh YS. Arterial stiffness and hypertension. Clin Hypertens [Internet]. 2018. Dec 1 [cited 2019 Aug 27];24(1):17. Available from: https://clinicalhypertension.biomedcentral.com/articles/10.1186/s40885-018-0102-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Chowienczyk PJ, Spector TD, Jiang B. Relation of arterial stiffness to left ventricular structure and function in healthy women. Cardiovasc Ultrasound [Internet]. 2018. Dec 25 [cited 2019 Jun 28];16(1):21. Available from: https://cardiovascularultrasound.biomedcentral.com/articles/10.1186/s12947-018-0139-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mattace-Raso FUS, van der Cammen TJM, Hofman A, van Popele NM, Bos ML, Schalekamp MADH, et al. Arterial Stiffness and Risk of Coronary Heart Disease and Stroke. Circulation [Internet]. 2006. Feb 7 [cited 2019 Aug 27];113(5):657–63. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.105.555235 [DOI] [PubMed] [Google Scholar]
- 10.Lacolley P, Challande P, Osborne-Pellegrin M, Regnault V. Genetics and pathophysiology of arterial stiffness. Cardiovasc Res [Internet]. 2008. Dec 20 [cited 2019 Aug 27];81(4):637–48. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19098299 [DOI] [PubMed] [Google Scholar]
- 11.Aziz H, Zaas A, Ginsburg GS. Peripheral blood gene expression profiling for cardiovascular disease assessment. Genomic Med [Internet]. 2007. Sep 27 [cited 2019 Aug 27];1(3–4):105–12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18923935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laguna JC, Alegret M. Regulation of gene expression in atherosclerosis: insights from microarray studies in monocytes/macrophages. Pharmacogenomics [Internet]. 2012. Mar [cited 2019 Aug 27];13(4):477–95. Available from: https://www.futuremedicine.com/doi/10.2217/pgs.12.9 [DOI] [PubMed] [Google Scholar]
- 13.Masud R, Shameer K, Dhar A, Ding K, Kullo IJ. Gene expression profiling of peripheral blood mononuclear cells in the setting of peripheral arterial disease. J Clin Bioinforma [Internet]. 2012. Mar 12 [cited 2019 Aug 27];2(1):6. Available from: http://jclinbioinformatics.biomedcentral.com/articles/10.1186/2043-9113-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinnaeve PR, Donahue MP, Grass P, Seo D, Vonderscher J, Chibout S-D, et al. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. Reitsma PH, editor. PLoS One [Internet]. 2009. Sep 14 [cited 2019 Aug 27];4(9):e7037. Available from: https://dx.plos.org/10.1371/journal.pone.0007037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marketou ME, Kontaraki JE, Tsakountakis NA, Zacharis EA, Kochiadakis GE, Arfanakis DA, et al. Arterial stiffness in hypertensives in relation to expression of angiopoietin-1 and 2 genes in peripheral monocytes. J Hum Hypertens [Internet]. 2010. May 14 [cited 2019 Aug 27];24(5):306–11. Available from: http://www.nature.com/articles/jhh200995 [DOI] [PubMed] [Google Scholar]
- 16.Detchaporn P, Kukongviriyapan U, Prawan A, Jetsrisuparb A, Greenwald SE, Kukongviriyapan V. Altered vascular function, arterial stiffness, and antioxidant gene responses in pediatric thalassemia patients. Pediatr Cardiol [Internet]. 2012. Oct 14 [cited 2019 Aug 27];33(7):1054–60. Available from: http://link.springer.com/10.1007/s00246-012-0225-8 [DOI] [PubMed] [Google Scholar]
- 17.Debey-Pascher S, Hofmann A, Kreusch F, Schuler G, Schuler-Thurner B, Schultze JL, et al. RNA-Stabilized Whole Blood Samples but Not Peripheral Blood Mononuclear Cells Can Be Stored for Prolonged Time Periods Prior to Transcriptome Analysis. J Mol Diagnostics [Internet]. 2011. Jul [cited 2019 Aug 27];13(4):452–60. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21704280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao S, Fung-Leung W-P, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. Zhang S-D, editor. PLoS One [Internet]. 2014. Jan 16 [cited 2019 Aug 27];9(1):e78644. Available from: https://dx.plos.org/10.1371/journal.pone.0078644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J [Internet]. 2006. Nov 25 [cited 2019 Jul 9];27(21):2588–605. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehl254 [DOI] [PubMed] [Google Scholar]
- 20.De Backer G, Ambrosioni E, Borch-Johnsen K, Brotons C, Cifkova R, Dallongeville J, et al. European guidelines on cardiovascular disease prevention in clinical practice. Third Joint Task Force Of European and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of eight societies and by invited experts). Arch Mal Coeur Vaiss [Internet]. 2004. Oct [cited 2019 Sep 11];97(10):1019–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16008181 [PubMed] [Google Scholar]
- 21.Maxwell M, Schroth P, Waks A, Karam M, Dornfeld L. ERROR IN BLOOD-PRESSURE MEASUREMENT DUE TO INCORRECT CUFF SIZE IN OBESE PATIENTS. Lancet [Internet]. 1982. Jul [cited 2019 Jun 22];320(8288):33–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673682911631 [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods [Internet]. 2001. Dec [cited 2019 Sep 13];25(4):402–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1046202301912629 [DOI] [PubMed] [Google Scholar]
- 23.Croall DE, DeMartino GN. Calcium-activated neutral protease (calpain) system: structure, function, and regulation. Physiol Rev [Internet]. 1991. Jul [cited 2019 Sep 12];71(3):813–47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2057527 [DOI] [PubMed] [Google Scholar]
- 24.Vanderklish PW, Bahr BA. The pathogenic activation of calpain: a marker and mediator of cellular toxicity and disease states. Int J Exp Pathol [Internet]. 2000. Oct 10 [cited 2019 Sep 12];81(5):323–39. Available from: http://doi.wiley.com/10.1111/j.1365-2613.2000.00169.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji J, Su L, Liu Z. Critical role of calpain in inflammation. Vol. 5, Biomedical Reports. Spandidos Publications; 2016. p. 647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Z, Andersson R. NF-kappaB activation and inhibition: a review. Shock [Internet]. 2002. Aug [cited 2019 Sep 12];18(2):99–106. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12166787 [DOI] [PubMed] [Google Scholar]
- 27.Lawrence T The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol [Internet]. 2009. Dec [cited 2019 Sep 12];1(6):a001651. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20457564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyazaki T, Koya T, Kigawa Y, Oguchi T, Lei XF, Kim-Kaneyama J ri, et al. Calpain and atherosclerosis. Vol. 20, Journal of Atherosclerosis and Thrombosis. 2013. p. 228–37. [DOI] [PubMed] [Google Scholar]
- 29.Ali MAM, Stepanko A, Fan X, Holt A, Schulz R. Calpain inhibitors exhibit matrix metalloproteinase-2 inhibitory activity. Biochem Biophys Res Commun [Internet]. 2012. Jun 22 [cited 2019 Sep 12];423(1):1–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22575511 [DOI] [PubMed] [Google Scholar]
- 30.Androulakis E, Tousoulis D, Papageorgiou N, Latsios G, Siasos G, Stefanadis C. The Role of Matrix Metalloproteinases in Essential Hypertension. Curr Top Med Chem [Internet]. 2012. Apr 1 [cited 2019 Sep 12];12(10):1149–58. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22519445 [DOI] [PubMed] [Google Scholar]
- 31.Yasmin, McEniery CM, Wallace S, Dakham Z, Pulsalkar P, Pusalkar P, et al. Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arterioscler Thromb Vasc Biol [Internet]. 2005. Feb [cited 2019 Sep 12];25(2):372. Available from: https://www.ahajournals.org/doi/10.1161/01.ATV.0000151373.33830.41 [DOI] [PubMed] [Google Scholar]
- 32.Chung AWY, Yang HHC, Kim JM, Sigrist MK, Chum E, Gourlay WA, et al. Upregulation of Matrix Metalloproteinase-2 in the Arterial Vasculature Contributes to Stiffening and Vasomotor Dysfunction in Patients With Chronic Kidney Disease. Circulation [Internet]. 2009. Sep 1 [cited 2019 Sep 12];120(9):792–801. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19687355 [DOI] [PubMed] [Google Scholar]
- 33.Alberts B, Bray D, Hopkin K, Johnson A, Lewis J, Raff MC, et al. Essential cell biology. Fourth edi. 2014. Unit 2: How Do Cells Decode Genetic Information in. [Google Scholar]
- 34.Lyle AN, Raaz U. Killing Me Unsoftly: Causes and Mechanisms of Arterial Stiffness. Arterioscler Thromb Vasc Biol [Internet]. 2017. Feb [cited 2019 Sep 13];37(2):e1–11. Available from: https://www.ahajournals.org/doi/10.1161/ATVBAHA.116.308563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Damen MSMA, Popa CD, Netea MG, Dinarello CA, Joosten LAB. Interleukin-32 in chronic inflammatory conditions is associated with a higher risk of cardiovascular diseases. Atherosclerosis [Internet]. 2017. Sep [cited 2019 Sep 13];264:83–91. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021915017311802 [DOI] [PubMed] [Google Scholar]
- 36.Zeng Q, Li S, Zhou Y, Ou W, Cai X, Zhang L, et al. Interleukin-32 contributes to invasion and metastasis of primary lung adenocarcinoma via NF-kappaB induced matrix metalloproteinases 2 and 9 expression. Cytokine [Internet]. 2014. Jan [cited 2019 Sep 13];65(1):24–32. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1043466613007084 [DOI] [PubMed] [Google Scholar]
- 37.Tan J, Hua Q, Xing X, Wen J, Liu R, Yang Z. Impact of the metalloproteinase-9/tissue inhibitor of metalloproteinase-1 system on large arterial stiffness in patients with essential hypertension. Hypertens Res [Internet]. 2007. Oct [cited 2019 Sep 12];30(10):959–63. Available from: http://www.nature.com/doifinder/10.1291/hypres.30.959 [DOI] [PubMed] [Google Scholar]
- 38.Chung AWY, Yang HHC, Kim JM, Sigrist MK, Chum E, Gourlay WA, et al. Upregulation of matrix metalloproteinase-2 in the arterial vasculature contributes to stiffening and vasomotor dysfunction in patients with chronic kidney disease. Circulation [Internet]. 2009. Sep 1 [cited 2019 Sep 16];120(9):792–801. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.109.862565 [DOI] [PubMed] [Google Scholar]
- 39.Heinhuis B, Popa CD, van Tits BLJH, Kim S-H, Zeeuwen PL, van den Berg WB, et al. Towards a role of interleukin-32 in atherosclerosis. Cytokine [Internet]. 2013. Oct [cited 2019 Sep 13];64(1):433–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23727326 [DOI] [PubMed] [Google Scholar]
- 40.Papakyriakou A, Stratikos E. The Role of Conformational Dynamics in Antigen Trimming by Intracellular Aminopeptidases. Front Immunol [Internet]. 2017. Aug 7 [cited 2019 Sep 16];8:946. Available from: http://journal.frontiersin.org/article/10.3389/fimmu.2017.00946/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Saez I, Tcherniuk S, Kozielski F. The structure of human neuronal Rab6B in the active and inactive form. Acta Crystallogr D Biol Crystallogr [Internet]. 2006. Jul 1 [cited 2019 Sep 13];62(Pt 7):725–33. Available from: http://scripts.iucr.org/cgi-bin/paper?S0907444906015319 [DOI] [PubMed] [Google Scholar]
- 42.Yasmin, McEniery CM, Wallace S, Dakham Z, Pulsalkar P, Pusalkar P, et al. Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arterioscler Thromb Vasc Biol [Internet]. 2005. Feb [cited 2019 Sep 16];25(2):372. Available from: https://www.ahajournals.org/doi/10.1161/01.ATV.0000151373.33830.41 [DOI] [PubMed] [Google Scholar]
- 43.Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of Cardiovascular Events and All-Cause Mortality With Arterial Stiffness. J Am Coll Cardiol [Internet]. 2010. Mar 30 [cited 2019 Jun 28];55(13):1318–27. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20338492 [DOI] [PubMed] [Google Scholar]