

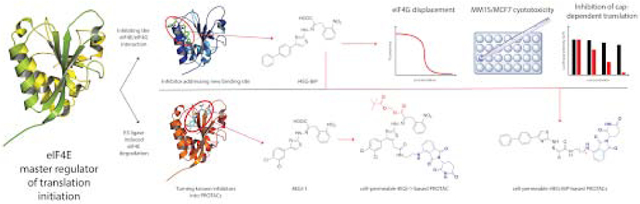
Abstract
The eukaryotic translation initiation factor 4E (eIF4E) is the master regulator of cap-dependent protein synthesis. Overexpression of eIF4E is implicated in diseases such as cancer, where dysregulation of oncogenic protein translation is frequently observed. eIF4E has been an attractive target for cancer treatment. Here we report a high-resolution X-ray crystal structure of eIF4E in complex with a novel inhibitor (i4EG-BiP) that targets an internal binding site, in contrast to the previously described inhibitor, 4EGI-1, which binds to the surface. We demonstrate that i4EG-BiP is able to displace the scaffold protein eIF4G and inhibit the proliferation of cancer cells. We provide insights into how i4EG-BiP is able to inhibit cap-dependent translation by increasing the eIF4E-4E-BP1 interaction while diminishing the interaction of eIF4E with eIF4G. Leveraging structural details we designed proteolysis targeted chimeras (PROTACs) derived from 4EGI-1 and i4EG-BiP and characterized these on biochemical and cellular levels. We were able to design PROTACs capable of binding eIF4E and successfully engaging Cereblon, which targets proteins for proteolysis. However, these initial PROTACs did not successfully stimulate degradation of eIF4E, possibly due to competitive effects from 4E-BP1 binding. Our results highlight challenges of targeted proteasomal degradation of eIF4E that must be addressed by future efforts.
Graphical Abstarct
INTRODUCTION
Cap-dependent translation in eukaryotes is initiated when eIF4E binds to the m7GTP cap of mRNAs, a rate-limiting step that results in the formation of the eIF4F complex, which is comprised of eIF4E, the DEAD-box RNA helicase eIF4A, and the scaffold protein eIF4G (1-7). eIF4E’s interaction with eIF4G facilitates loading of the 40S small ribosomal unit onto the mRNA, triggering scanning for the start codon. eIF4E binding proteins (4E-BPs) compete with eIF4G for binding to eIF4E, and successful binding of 4E-BPs abolishes cap-dependent translation, making 4E-BPs important regulators of this process. This competition is regulated by the 4E-BP phosphorylation state. Upon phosphorylation by mTORC1, 4E-BP isoforms disassociate from eIF4E, freeing it to then engage with eIF4G and form the eIF4F complex (8) (Figure 1A). The critical role of eIF4E in cancer was first identified when overexpression was observed to cause tumorigenic transformation of fibroblasts (9). eIF4E has since been found to be overexpressed in a number of cancer types, including breast (10), non-Hodgkin lymphoma (11) and head and neck (12).
Figure 1.

A. Assembly of the translation initiation complex eIF4F, which consists of eIF4E, eIF4G and eIF4A. eIF4E is negatively regulated by hypo-phosphorylated 4E-BP, which in turn is regulated by mTORC1. B. Structures of the small molecule inhibitors 4EGI-1 and i4EG-BiP. C. Fluorescence polarization assay for displacement of eIF4G peptide from eIF4E. Both 4EGI-1 and i4EG-BiP are capable of displacing eIF4G with similar affinities.
The structure of eIF4E resembles a hand with a palm consisting of β-strands and dorsally positioned α-helices (13, 14). The m7GTP cap binds tightly to the palm region and is stabilized by interactions with four tryptophan sidechains. Both eIF4G and 4E-BPs engage eIF4E, in part, through conserved motifs with the consensus sequence YX4LΦ, where Φ is a hydrophobic amino acid. Structural studies have revealed that the conserved 4G/4E-BP binding motif binds to the dorsal surface, opposite of the cap-binding surface (15, 16). Studies have shown crosstalk between the cap-binding and 4G/4E-BP binding events with binding at one site affecting the affinity of the other (17). All the published structures of eIF4E in complex with minimal binding epitopes from eIF4G or 4E-BPs are nearly identical and only provided information about the eIF4G and 4E-BP peptides engaging eIF4E. However, structures with larger fragments of eIF4G or 4E-BP in complex with eIF4E have been recently determined (15, 16). These structures show that eIF4G and 4E-BPs use interfaces beyond their minimal consensus sequence to engage eIF4E.
Despite this structural data, the atomic detail of how 4E-BPs outcompete eIF4G is still not fully known. Although the canonical binding helix is conserved between eIF4G and 4E-BPs, the residues beyond the consensus sequence are not, and in the case of 4E-BPs, this non-consensus sequence harbors several phosphorylation sites (15, 16, 18-20). It has been reported that phosphorylation of T37 and T46 of 4E-BP2 induces folding of the protein into a four β-strand folded domain that sequesters the eIF4E binding motif (21). More recently, it was found that hyper-phosphorylation of the C-terminal intrinsically disordered phosphor-sites of 4E-BP2 stabilizes the β-stranded folded domain, further reducing 4E-BP2 binding to eIF4E (22). Dislodging eIF4G by binding of either 4E-BPs or a small molecule inhibitor would stop cap-dependent translation and this latter approach could be used for the therapeutic treatment of eIF4E-mediated pathogenic dysregulation of translation.
4EGI-1 was the first identified small molecule inhibitor of the eIF4E-eIF4G interaction, binding to eIF4E with low micromolar affinity (IC50 = 57 ± 1 μM (23, 24)). 4EGI-1 has been shown to be effective in arresting proliferation in a number of cancer cell lines (25-28). The crystal structure of 4EGI-1 in complex with eIF4E reported in 2014 (PDB: 4TPW) (24) showed that 4EGI-1 bound to eIF4E at a site that is distinctly different from the primary binding site of the eIF4G/4E-BP consensus sequence or the m7GTP cap binding site. In the crystal structure, the binding of 4EGI-1 induces a conformational change in helix α1 of eIF4E, thus leading to the hypothesis that 4EGI-1 is an allosteric inhibitor. Another small molecule inhibitor that displaces eIF4G from eIF4E is 4E1RCat was discovered with a high-throughput screen using a time resolved (TR)-FRET based assay (29). There is no high-resolution structures of 4E1RCat bound to eIF4E. Analogues of 4EGI-1 co-crystallized by our group reveal binding at the same site as 4EGI-1 (24). This common binding site is proximal to a cavity on the surface of eIF4E.
In an effort to increase the efficacy of 4EGI-1, we previously synthesized and screened multiple analogues of this compound. Systematic variation of the di-chlorophenyl "head", the nitrophenyl "tail" or the thiazole core did not produce analogs with significantly increased binding affinity to eIF4E. Removal of the hydrazone linker consistently reduced binding affinity. In contrast, certain significant changes in the aromatic "head" could be accommodated without significant loss in binding affinity. For example, an analog (4EGI-1A, Supplementary Figure-2) containing an additional cyclohexyl ring had similar to 4EGI-1 activity in an FP assay measuring displacement of an eIF4G peptide from eIF4E and was a part of the original publication(23). We then synthesized a head-head dimer of 4EGI-1 which was active, and 4EGI-dimer displayed a slightly better Kd than 4EGI-1 in the FP assay. We then synthesized i4EG-BiP, containing a biphenyl instead of cyclohexyl-phenyl "head"(here BiP denotes the biphenyl moiety in the scaffold), as a control for a series of "head-to-head" dimer series such as 4EGI-dimer (Supplementary Figure-2). We serendipitously discovered that i4EG-BiP binds to a new internal cavity on eIF4E, near the 4EGI-1 binding site.
We were also interested in leveraging information from high-resolution structures of 4EGI-1 and i4EG-BiP bound to eIF4E to engineer PROteolysis-TArgeting Chimeras (PROTACs) in an effort to trigger specific degradation of eIF4E as an alternative to traditional small molecule inhibitors that target protein-protein interfaces (30-35). PROTACs are heterobifunctional molecules in which a small molecule ligand is conjugated to an E3 ligase ligand with a linker, frequently PEG- or carbon-based. PROTACs take advantage of the host E3 ubiquitin ligase machinery to induce targeted proteasomal degradation of the protein of interest. Studies with PROTACs based on promiscuous kinase degraders have shown that ligands with weak binding (KD > 10 μM) can still be turned into potent degraders (30, 36). Kaur et al. previously attempted to employ PROTAC strategies against eIF4E using derivatized cap analogues coupled to lenalidomide or von Hippel-Lindau (VHL) ligands (37). Bn7GDP linked to lenalidomide was shown to be an effective eIF4E binder in vitro with an affinity of 50 μM, but all tested compounds failed to degrade eIF4E in cellular assays. This was discovered to be due to insufficient cell permeability, a known problem for highly negatively charged cap analogues. Our secondary goal in this study was therefore to generate cell-permeable degraders of eIF4E based on our inhibitors 4EGI-1 and i4EG-BiP. Here, we present the structural and biochemical characterization of i4EG-BiP and our PROTAC derivatives of 4EGI-1 and i4EG-BiP. Since there are quite a few acronyms and abbreviations we have included a table (Supplementary Table-2) in the supplementary section to ease readership.
RESULTS
Discovery of i4EG-BiP
In an effort to increase the efficacy of 4EGI-1, we synthesized and screened multiple analogues of 4EGI-1. The analogues synthesized included variations of the di-chlorophenyl moiety, the nitrophenyl moiety or the thiazole core. However, none of these compound variations improved the binding affinity. Amongst the synthesized compounds were also dimeric versions of 4EGI-1, mirrored along the dichlorophenyl moiety. One of the intermediates from this synthesis was the biphenyl compound i4EG-BiP (Figure 1B). This compound caught our attention due to its similar eIF4G displacement properties when compared to 4EGI-1 (Figure 1C); i4EG-BiP displaces eIF4G peptide from eIF4E with an IC50 value of 68 ± 2 μM, which is similar to that of 4EGI-1. Since 4EGI-1 binds on the surface of eIF4E, we were intrigued as to how the extra aromatic ring would be stabilized, so to answer this question, we solved the structure of i4EG-BiP in complex with eIF4E by X-ray crystallography.
The structure of eIF4E bound to i4EG-BiP
The eIF4E structure in complex with i4EG-BiP was resolved by molecular replacement to a resolution of 1.9 Å (Figure 2A). The refined model allowed us to unambiguously locate the position of the ligand in the electron density map (Figure 2B). i4EG-BiP binds to a cavity on the surface of eIF4E that is near the 4EGI-1 binding site, opening up possibilities for the development of a new class of eIF4E inhibitors. The structure of the protein itself has a striking similarity to that of eIF4E bound to 4EGI-1 (PDB ID 4TPW). The unit cell is comprised of a dimer with two copies of palm-like eIF4E molecules arranged at 180° orientations relative to each other around a pseudo-symmetry axis positioned between the two dorsal surfaces. m7GTP, which constitutes the cap structure of mRNA, is bound to the palm of eIF4E. However, only one copy of the protein (chain A) is occupied by the i4EG-BiP ligand, while the binding site on the other copy (chain B) is empty. This is similar to the case of 4EGI-1, where we found the ligand bound to only one of the two proteins in the unit cell. Furthermore, as observed in the case of 4EGI-1, the 3-10 helix between residues S82 and L85 has melted into a loop while the α1 helix (residues H78-L85) is extended by one turn upon engagement of i4EG-BiP. This conformational change induced by i4EG-BiP is seen in chain A, but is obviously not present in chain B, which lacks the small molecule.
Figure 2.

A. Structure of eIF4E bound to i4EG-BiP. The asymmetric unit consists of two cap-bound eIF4E structures, of which one shows density for the small molecule. B. i4EG-BiP density is unambiguously located between helix α1 and α2. C. Surface representation of the co-crystal structure shows the cavity into which i4EG-BiP binds.
i4EG-BiP fits well within its binding cavity on the surface of eIF4E, burying 334 Å2 of the eIF4E surface area, yet leaving room to expand the small molecule near the thiazole moiety, which is proximal to a deep cavity as shown in Figure 2C. There is an estimated 200 Å2 of additional surface area available that could be leveraged to increase the binding affinity of i4EG-BiP. The i4EG-BiP binding mode to eIF4E is distinct from any previously described eIF4E binder. The compound engages the pocket between helix α1 and helix α2 (Figure 3A). The biphenyl moiety is buried deeply while the carboxylic-acid and the nitrophenyl functionalities are solvent exposed. The compound engages eIF4E using hydrophobic interactions with residues L45, L75, I79, L93 and L134. Edge-to-face π-π interactions are formed between the biphenyl moiety and Y76, Y91 and W130. Hydrogen bonds are formed between the nitro group of i4EG-BiP and the amide side chain of Q80 as well as between the hydrazone NH of i4EG-BiP and the backbone carbonyl oxygen of Q80. The carboxylic acid moiety of i4EG-BiP forms a hydrogen bond with the NH backbone of L85 (Figure 3C), instead of the salt-bridge with K49, which represents the strongest interaction found for 4EGI-1 (Figure 3D). Upon binding of i4EG-BiP, the α1 helix is extended by the amino acids Q80, L81, S82 and S83. These four residues constitute a loop that faces the α2 helix in other eIF4E structures without a small molecule inhibitor. This observed helix extension is needed to create room for the thiazole, hydrazone and nitrophenyl moieties of i4EG-BiP and the formation of hydrogen bonds between Q80 and the ligand. It is unclear why the same helix extension is observed with the binding of 4EGI-1, since in this case the structural rearrangement neither enables new protein interactions nor does it serve to avoid steric clashes. This helix extension does, however, appear to be a crucial factor in eIF4G displacement which is discussed in greater detail later.
Figure 3.

A. Interaction profile of i4EG-BP. eIF4E is depicted in light blue, i4EG-BP is depicted in salmon and the side chains of eIF4E involved in the interaction are depicted in blue. B. Interaction profile of 4EGI-1. eIF4E is depicted in light blue, 4EGI-1 is depicted in salmon and the side chains of eIF4E involved in the interaction are depicted in blue. C. i4EG-BP makes hydrophobic interactions with most residues of helix α1 and α2. Hydrogen bonds are observed between the small molecule and the side chains of Gln80 and Leu85. D. 4EGI-1 makes primarily hydrophobic interactions with different residues than those interacting with i4EG-BP. A salt-bridge and hydrogen bond between the small molecule and Lys49 is also formed. E. Depiction of eIF4E residues within 4 Å of i4EG-BiP.
i4EG-BiP is able to displace eIF4G peptide from eIF4E
To assess the ability of inhibitors binding to eIF4E to disrupt the eIF4E-eIF4G interaction, we used a previously reported fluorescence polarization (FP) assay (23). This assay leverages the fact that a free fluorescently labelled eIF4G peptide has a molecular correlation time that is vastly different from when this peptide is bound to eIF4E, thereby allowing displaced (free) peptide to be distinguished from bound peptide. In this displacement assay, increasing concentrations of compounds were titrated against eIF4E bound to a fluorescently labelled eIF4G peptide. Compounds that can displace the eIF4G peptide led to a reduction in FP signals. Our results show that i4EG-BiP was able to displace the eIF4G peptide from eIF4E with an IC50 of 68 ± 2 μM, which is similar to that observed for 4EGI-1 (Figure 1C).
i4EG-BiP impairs viability in MCF7 and MM1S cells
To test the activity of i4EG-BiP in cells, we performed time-dependent CS-BLI cell viability assays (38) in two different cell lines. MCF7 and MM1S cells were chosen as representative cell lines due to reported sensitivity to eIF4E ablation in the DepMap database (39). Cells were treated once with serial dilutions of either 4EGI-1 or i4EG-BiP and assayed at 24, 48 or 72 h (Figure 4A). 24 h of treating MCF7 cells with 4EGI-1 resulted in an IC50 of 71 μM, which decreased to 43 μM at 48 h and 35 μM at 72 h, thus resulting in a 2-fold decrease in viability as compared to 24 h. i4EG-BiP treatment inhibited cell viability of MCF7 cells over a treatment period of 24 h with an IC50 value of 59 μM, which decreased to 27 μM at 48 h and 18 μM when treatment is prolonged for 72 h. This corresponds to a decrease in viability of more than 3-fold from 72 h treatment as compared to 24 h. In MM1S cells, we did not observe a significant time-dependent effect with either compound. The IC50 values for 4EGI-1 were 23 μM for a treatment period of 24 h, 19 μM for 48 h and 16 μM for 72 h. i4EG-BiP was less active in MM1S cells, with IC50 values of 67 μM after treatment for 24 h, 62 μM for 48 h and 56 μM for 72 h. This data indicates that i4EG-BiP affects the viability of MCF7 to a similar extent to 4EGI-1. In MM1S cells i4EG-BiP is less active compared to 4EGI-1, while both compounds do not exert significant time dependency in their cytotoxic effects.
Figure 4.

A. Cell viability assay of 4EGI-1 and i4EG-BiP in two different cell lines (MCF7 and MM1S) as a function of concentration over 24 h, 48 h and 72 h. Error bars represent standard deviation and in all cases n=3. B. A dual luciferase assay for 4EGI-1 and i4EG-BiP shows a greater decrease in cap-dependent translation with increasing compound concentration for i4EG-BiP. C. Western blot of an eIF4E pull down with m7-GDP functionalized agarose beads in the absence and presence of increasing concentrations of i4EG-BiP. With increasing concentrations of i4EG-BiP, the eIF4E/eIF4G ratio increases, while the eIF4E/4E-BP ratio decreases.
i4EG-BiP inhibits cap-dependent translation better than 4EGI-1 in cellular assays
To test the inhibition of cap-dependent protein translation by i4EG-BiP in cells, we transfected into HEK293 cells a plasmid that produces a bicistronic transcript containing a 5’-Firefly luciferase followed by an IRES and a 3’-Renilla luciferase. The Firefly luciferase therefore reports on cap-dependent translation while the Renilla luciferase reports on cap-independent translation. The day after transfection, cells were treated with increasing concentrations of compounds and following a 3-h incubation, Firefly and Renilla luciferase activity were measured by chemiluminescence. The interaction between eIF4E and eIF4G is essential for cap-dependent translation, but not IRES-dependent translation, and therefore only the Firely luciferase activity should be affected by compound treatment. Interestingly, we found a 65% reduction in the Firefly to Renilla (L/R) luciferase activity ratio with i4EG-BiP treatment while treatment with 4EGI-1 resulted in a considerably lower 15% L/R ratio reduction (Figure 4B). This data indicates that i4EG-BiP inhibits cap-dependent translation to a greater extent than 4EGI-1.
i4EG-BiP disrupts the eIF4E-eIF4G and strengthens the eIF4E- 4E-BP1 interactions
We assessed the interaction between eIF4E and its binding partners eIF4G and 4E-BP1 in pull-down experiments using a simplified cap-analogue (m7GDP-agarose) resin (40). HeLa cell lysates were incubated with either DMSO as a negative control or increasing concentrations of i4EG-BiP. After incubation of the mixture with m7GDP agarose resin, beads were washed, followed by elution of bound proteins and analysis by Western Blot. As can be seen in Figure 4C, the amount of eIF4G relative to eIF4E decreases with increasing amounts of i4EG-BiP, while the amount of 4E-BP1 increases. These results suggest that the extension of the α1-helix in eIF4E, caused by binding of i4EG-BiP, is detrimental for eIF4G binding to eIF4E, but does not adversely impact 4E-BP1 binding.
Design and synthesis of the 4EGI-1-based PROTAC d4E-1
Next, we decided to leverage the structural information we had to design eIF4E-targeting degrader molecules (PROTACs). The idea was to deal a double-blow to inhibit translation, one by blocking the eIF4E-eIF4G interaction, and the other by degrading eIF4E. We first assessed the structure of 4EGI-1 for functional groups to which could be coupled a flexible linker for the attachment of lenalidomide. The carboxylic acid moiety appeared to be ideal for linkage; however, it is also involved in a crucial interaction with eIF4E (Figure 3D). We therefore decided to attach the linker to the free carbon of the thiazole moiety, which is surface exposed in the co-crystal structure (PDB ID: 4TPW). Based on the simulated modelling of eIF4E bound to 4EGI-1 in complex with Cereblon (CRBN) bound to lenalidomide (PDB ID: 4CI2), we decided to synthesize the first test compound with a 4-carbon linker (Figure 5A).
Figure 5.

A. Docking of Lenalidomide bound to Cereblon (PDB ID: 4CI2) and eIF4E bound to 4EGI-1 (PDB ID: 4TPW). Surface exposed parts of the small molecule can be arranged in proximity without steric clashes. The target degrader molecule derived from this docking pose (d4E-1) is depicted on the right. B. Synthesis of the double protected 4EGI-1 building block 7.
The synthetic challenge was to introduce the acetic acid functionality to the thiazole ring and choose a selective protection strategy for the resulting two carboxylic acids. Therefore, succinic anhydride was reacted with 1,2-dichloro benzene in a Friedel Crafts acylation reaction to produce the phenyl-4-oxobutanoic acid 1 with 41 % yield (Figure 5B). The carboxylic acid was protected using iodomethane as a methylation agent to quantitively yield the methyl ester 2. Bromination of 2 in the alpha-keto position produced the phenyl-3-bromo-4-oxobutanoate 3 with 96 % yield. The hydrazine functionalized thiazole 4 was obtained by reacting this product with thiosemicarbazide. Due to the high reactivity of 4 it was used without further purification. To finish the synthesis is the 4EGI-1 analogue, the nitrophenyl-2-oxopropanoic acid 5 was created by hydrolysis of an oxazol-5-one derivative, which was obtained from reacting 2-Nitrobenzaldehyde with acetyl glycine. The overall yield of this 2-step reaction was 70 %. Then, the acid was protected using tert-butyl acetate to form the tert-butyl ester 6 with 79 % yield. The crude hydrazineylthiazol 4 was then coupled to the tert-butyl ester 6 to form an E/Z-isomeric mixture of the protected 4EGI-1 building block 7 with 63 % yield.
As a starting point, we chose to synthesize the first PROTAC as a 4EGI-1 analogue linked to a 2-phenoxyacetamide derivative of thalidomide, as described for dBET1, a BET bromodomain degrader developed by Winter et al.(33). To achieve this, 4-Hydroxythalidomide 8 was synthesized by reacting 3-Hydroxypththalic anhydride and 3-Aminopiperidine-2,6-dione in glacial acetic acid with 96 % yield (Figure 6A). The linker was prepared using a benzyloxycarbonyl (CBz) protected diamine, which is more economical than the previously used Boc protected variant(33). Benzyl (4-aminobutyl)carbamate was reacted with 2-Chloroacetyl chloride to produce CBz-protected 2-Chloroamide 9 in 91 % yield, which was subsequently transformed into the more reactive 2-lodoamide 10 via Finkelstein reaction in a quantitative manner. 10 was then coupled to 4-Hydroxythalidomde 8 to yield the CBz-protected Thalidomide linker, which was deprotected using hydrogen gas and a Palladium black catalyst to give the free Thalidomide linker 11 in 41 % yield.
Figure 6.
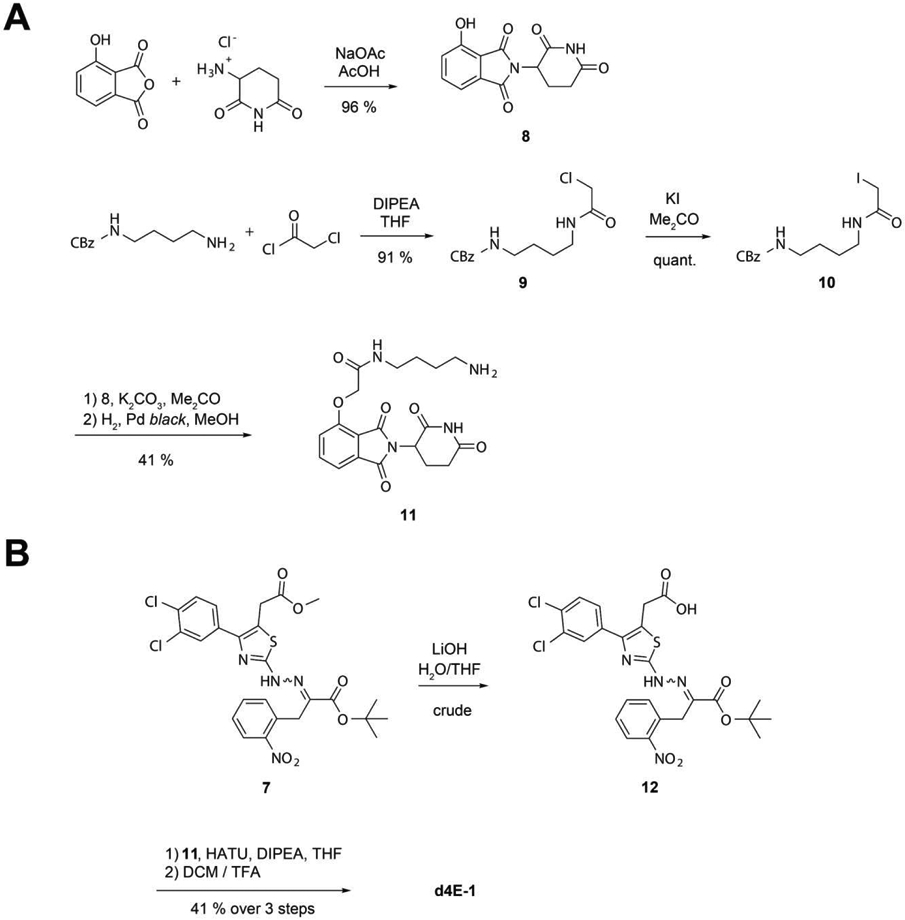
A. Synthesis of the 2-phenoxyacetamide thalidomide linker 11. B. Endgame of the synthesis of target structure d4E-1.
To selectively deprotect the acid moiety on the thiazole ring and generate the final PROTAC d4E-1, methyl ester hydrolysis of the tert-butyl protected 4EGI-1 building block 7 was performed using lithium hydroxide at 4 °C to create the ready-to-couple 4EGI-1 derivative 14 with 84 % yield (Figure 6B). The final product d4Ei-1 was then generated with 49 % yield over two steps using HATU-mediated coupling of the 4EGI-1 derivative 14 with the Thalidomide linker 5 and subsequent tert-butyl deprotection using 12 % trifluoroacetic acid in dichloromethane.
d4E-1 interacts with eIF4E, but does not engage Cereblon in cells
After successful synthesis of the 4EGI-1 based PROTAC d4E-1, we next wanted to analyse binding of the bifunctional molecule to its targets: eIF4E and Cereblon.
Binding to eIF4E and displacing the eIF4G:
To test whether d4E-1 can displace eIF4G in a similar manner compared to 4EGI-1, the compound was tested for its ability to displace a fluorescently tagged eIF4G peptide in an FP assay (Figure 8A). At the three measured concentrations (33, 100 and 300 μM), both d4E-1 and 4EGI-1 displaced the 4G peptide in a similar manner. We therefore concluded that the binding affinity of our PROTAC compound is comparable to that of the parent compound, 4EGI-1.
Figure 8.

A. A fluorescence polarization assay shows that d4E-1 displaces the eIF4G peptide in a manner comparable to 4EGI-1. B. Cellular Cereblon engagement assay used to elucidate intracellular activities of d4E-1 and d4E-6.
Cellular assay to evaluate the ability of 4EGI-1 degraders to engage Cereblon:
Next, we wanted to assess the cellular CRBN engagement of d4E-1. We used a previously described assay (41, 42) in which compounds are tested for their ability to rescue dBET6-induced (a CRBN-dependent BET bromodomain degrader) degradation of BRD4BD2 by competing for binding to CRBN. Flp293T cells stably expressing a BRD4BD2-GFP fusion protein and an mCherry reporter were co-treated with dBET6 at 100 nM and compounds in dose response, with lenalidomide used as a positive control. The GFP/RFP signal ratio was quantified using an Acumen laser scanning cytometer (TTP Labtech). Active compounds were identified by an increased GFP/mCherry ratio resulting from inhibition of BRD4BD2-GFP degradation by dBET6. To our surprise, d4E-1 did not prevent degradation of BRD4BD2 in a dose dependent manner (results shown later). We hypothesized that this could be due to the net negative charge of d4E-1, which could hinder cellular uptake in a similar manner to the cap-based degraders previously reported (37). Therefore, our next goal was to synthesize an optimized prodrug version of d4E-1.
Design, synthesis and CRBN engagement of a 4EGI-1-based prodrug PROTAC
To negate the hypothesized negative effects from the negative charge on the carboxylic acid, a commonly applied prodrug approach was employed. In this approach, the carboxylate is modified with a protective group that can be cleaved off in cells after uptake. We chose to use a pivaloyloxymethyl ester as a prodrug unit, a moiety that is also used in prodrug versions of ampicillin (pivampicillin) (43) or butyric acid (AN-9) (44), for example. In addition to the prodrug strategy, arylamine linkage to thalidomide was used as these CRBN ligands reduce synthetic efforts significantly. To further improve cellular uptake, the 2-nitrophenyl moiety was removed from the 4EGI-1 scaffold, as this functionality reduces cellular uptake (results from i4EG-BiP PROTAC analogues shown later).
The arylamine linked thalidomide derivative 15 was synthesized as previously described by others (45).To make the free acid of the modified 4EGI-1 PROTAC, the hydrazine derivative 4 was reacted with pyruvic acid and subsequently protected using tert-butylacetate (Figure 7). HATU-mediated amide coupling with 15 and acidic deprotection with 12 % trifluoroacetic acid in dichloromethane yielded the free acid of the 4EGI-1-prodrug PROTAC d4E-6. Despite repeated efforts, the purity of the d4E-6 free acid could not be improved to satisfying levels, hence the crude product was reacted with pivaloyloxymethyl iodide to generate pure d4E-6 with an overall yield of 31 % over 5 steps.
Figure 7.
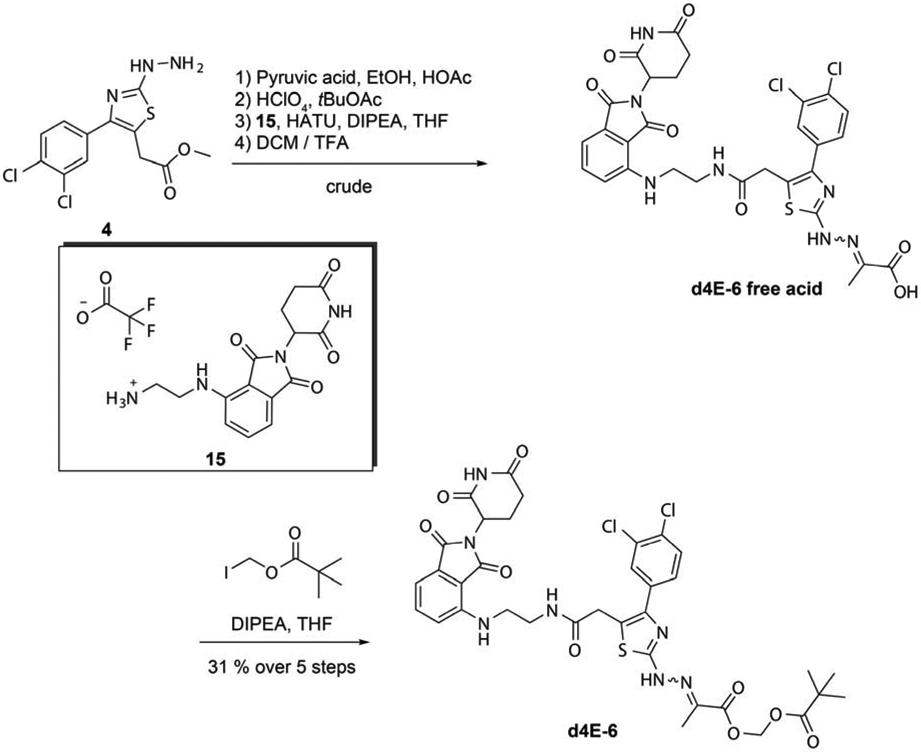
Synthesis of the 4EGI-1 prodrug PROTAC d4E-6 by POMylation of the free acid 4EGI-1 analog with iodomethyl pivalate.
Using the same assay for cellular CRBN engagement, we evaluated d4E-6 and its parent compound. While the free acid was not capable of rescuing dBET6-induced degradation of BRD4 (data not shown), the prodrug did with an IC50 value of ~ 20 μM (Figure 8B). These encouraging results supported our previous hypothesis that the net negative charge in our 4EGI-1-based PROTAC d4E-1 was root cause for its lack of activity.
Design and synthesis of the i4EG-BiP-based PROTACs (d4E-2 to d4E-5)
To optimize and expand the spectrum of possible eIF4E degraders, we switched to using i4EG-BiP as a scaffold for new PROTACs. In this case, the carboxylic acid moiety is not involved in the same crucial interaction found with 4EGI-1 (Figure 3C and D). Therefore, we hypothesised that direct linking of the acid to a thalidomide linker should not result in a significant loss of affinity, while beneficially eliminating the net negative charge of the product. Furthermore, the structure suggests that the 2-nitrophenyl moiety plays a minor role for interaction. On the basis of this observation, we decided to include structures that lack this functional group. We tried to further improve CRBN binding by switching from phthalimide ether to arylamine phthalimide. This driven by the fact the field developed improvements in linker design which encouraged us to opt for arylamine phthalimides instead of the phthalimide ether used for d4E-1 (46). Regarding chirality of the thalidomide analogs it is well established that the stereocenter interconverts in vivo (47).
Starting from 1-([1,1'-biphenyl]-4-yl)-2-bromoethan-1-one, the crude reactive hydrazine derivative 13 was synthesized using thiosemicarbazide (Figure 9A). i4EG-BiP and the i4EG-BiP derivative lacking the 2-nitrophenyl moiety 14 were generated using 2-nitrophenyl pyruvic acid and pyruvic acid with 63 % and 57 % yield, respectively. The arylamine linked thalidomide derivatives 15, 16 and 17 were synthesized as previously described by others (45) (Figure 9B). As before, HATU-mediated amide coupling was used to generate the PROTACs d4E-2, d4E-3, d4E-4 and d4E-5 with 43 %, 39 %, 40 % and 43 % yields.
Figure 9.

A. Structure of analyzed i4EG-BP-based PROTACs. B. Cellular Cereblon engagement assay shows intracellular activity for d4E-2 and d4E-3. C. Cell viability data for PROTACs in HeLa cells (triplicates over 24 h). D. Cellular Cereblon engagement assay used to elucidate intracellular activities of d4E-2 and d4E-3. E. Representative western blot of eIF4E degradation by d4E-4 and d4E-6 at 10 μM compound concentration.
i4EG-BiP-based degraders engage Cereblon in cells
To test whether the new PROTACs were able to engage CRBN in cells, we tested two model compounds to evaluate the effects of the 2-nitrophenyl moiety on cellular uptake: d4E-2 (4-carbon linker, with no nitrophenyl), and d4E-3 (4-carbon linker, nitrophenyl included). The best molecule tested was d4E-2 with an IC50 value of 1.6 μM. In contrast, d4E-3 was approximately 8-fold less active (with an IC50 value of 13 μM), suggesting the nitrophenyl moiety had a negative effect on activity (Figure 9D). Based on these findings, we tested three nitrophenyl-free compounds with varying carbon linker lengths (d4E-2, d4E-4 and d4E-5) for cytotoxicity in HeLa cells (Figure 9C). The efficacy of the tested compounds decreased with increasing linker length, but more strikingly all PROTACs displayed strong cytotoxic effects, even at rather low concentrations, with d4E-4 and d4E-5 killing over 50 percent of the cells at the lowest concentration measured (10 μM).
After these promising initial biochemical and cellular results, we proceeded to test our two best compounds (d4E-4 and d4E-6) for their potential to induce eIF4E degradation in HEK293T cells. We performed two experiments: 1) treatment of HEK293T cells with PROTAC concentrations varying between 0.1-10 μM for a period of 24 h, and 2) treatment of the same number of HEK293T cells with 1 μM PROTAC for varying lengths of time, ranging from 0.5 h to 24 h. Unfortunately, neither experiment showed significant degradation of eIF4E, as assessed by immunoblotting (representative Western Blot shown in Figure 9E). However, we did observe that eIF4E levels varied at different time points for both compound as well as DMSO treated cells. This suggests that a cellular response may be counteracting eIF4E degradation. To test whether eIF4E was being ubiquitinated in the first place, HEK293T cells were incubated with 1 μM PROTACs in the presence of 100 nM Bortezomib, a known inhibitor of the 26S proteasome (48). We compared eIF4E ubiquitination in these cells to those from cells treated with Bortezomib and a DMSO control; however, we were not able to detect any significant differences (data not shown), suggesting that our PROTACs did not induce CRBN-mediated ubiquitination of eIF4E.
We performed quantitative proteomics experiments to measure the global change in protein expression resulting from treatment with d4E-2 relative to DMSO control. Proteomics experiments have confirmed that d4E-2 is engaging CRBN in cells as we see downregulation of C2H2 zinc finger targets, SALL4, ZNF692 and ZNF827, which are commonly degraded as a result of the IMiD CRBN-binding handle of the degrader (58)(Supplementary Figure-3). We also observed that d4E-2 did not downregulate eIF4E.
Solution NMR studies confirm binding modes of the i4EG-BiP-based degraders to eIF4E are similar to that of the parental compound
Due to these unexpected results, we wanted to confirm that the derivatization of i4EG-BiP with thalidomide did not affect the binding of the degraders to eIF4E. To do this, we used solution-state nuclear magnetic resonance (NMR) spectroscopy as an additional method to identify the binding site of i4EG-BiP on eIF4E and compare it with the i4EG-BiP-based PROTAC d4E-4 (Figure 10A and C). Chemical shift perturbations (CSPs) were observed when 15N-labelled GB1-eIF4E was titrated with i4EG-BiP or d4E-4. Significant CSPs were observed in both cases at the binding interface identified from the co-crystal structure (Figure 10B and D). A molecular docking simulation of d4E-4 was carried out with QuickVina 2 (49), which is based on AutoDock Vina (50). The obtained docking score was −8.2 kcal/mol. CSPs plotted onto the structure of eIF4E bound to i4EG-BiP as well as the docked structure of d4E-4 reveal significant CSPs located in similar regions for both small molecules (Figure 10E and F). Although the overall CSP patterns look similar for i4EG-BiP and d4E-4, indicating similar binding modes, some additional CSPs were observed towards the N- and C-termini of the protein when d4E-4 was added. This could originate from non-specific binding events due to the 3-fold excess of inhibitor that was used in these experiments, in order to obtain significant CSPs. Alternatively, these additional CSPs could be the result of conformational changes induced by ligand binding. In particular, the C-terminal residues constitute the cap-binding region and there have been previous reports of crosstalk between eIF4G binding to the dorsal surface and cap binding to the palm of eIF4E. This may be the result of dynamic loops in the cap-binding region, which have been observed in multiple conformations in the various published crystal structures.
Figure 10.

A. 15N-1H-HSQC overlay of GB1-eIF4E in the absence (blue) and presence (red) of i4EG-BP. B. Chemical shift perturbation (CSP) plot of changes induced by i4EG-BP as a function of residue number. The solid line represents the mean value of all CSPs, the dashed line represents the mean value plus one standard deviation of all CSPs. C. 15N-1H-HSQC overlay of GB1-eIF4E in the absence (blue) and presence (orange) of d4E-4. D. CSP plot of changes induced by d4E-4 as a function of residue number. The solid line represents the mean value of all CSPs, the dashed line represents the mean value plus one standard deviation of all CSPs.E. CSPs plotted onto the eIF4E-i4EG-BP structure: Highlighted in orange are residues with CSPs > mean + one standard deviation, highlighted in red are residues with CSPs > mean + two standard deviations. F. CSPs plotted onto the docked eIF4E-d4E-4 structure. Highlighted in orange are residues with CSPs > mean + one standard deviation, highlighted in red are residues with CSPs > mean + two standard deviations.
DISCUSSION
The cap-dependent translation machinery represents a relatively underexplored area of vulnerability for targeted therapeutics in cancer, with many genes (eIF4A, eIF4E, eIF4G) listed as sensitive in DepMap. Inhibiting the protein-protein interaction of eIF4E with eIF4G and concomitantly disrupting the translation initiation complex has been shown to be an attractive therapeutic option. Inhibitors of the eIF4E-eIF4G interaction have been shown to inhibit translation, and exhibit antiproliferative and putative antitumor activities (51, 52).
The previously described 4EGI-1 and i4EG-BiP, introduced here, bind to different allosteric sites on eIF4E, but both result in a similar conformational change (extension of the helix α1 by one turn) which in turn displaces eIF4G. i4EG-BiP binds to an internal cavity of eIF4E, rather than on the surface as done by 4EGI-1. There is distinct possibility that an endogenous metabolite could also engage this i4EG-BiP cavity, thus modulating translation, but such a molecule has yet to be identified. We found that 4EGI-1 and i4EG-BiP could displace eIF4G but not 4E-BP1 in cells, although both eIF4G and 4E-BP1 share the same consensus binding motif. We posit that extension of the helix resulting from engagement of the small molecules hinder binding of eIF4G due to a steric clash. Specifically, the side chain of Ser-82 in eIF4E will clash with a conserved Leu-641 in eIF4G that is a part of the extended interface. In the case of 4E-BP1, this steric clash would not occur. Instead, we predict the complex with eIF4E would be stabilized by a putative hydrogen bond between the sidechain of Ser-82 in eIF4E and the backbone of Ser-83 from 4E-BP1 (Supplementary movie M1). This model in which the small molecule would inhibit the translation activator eIF4G and stabilize the inhibitor 4E-BP1 presents a two-fold attack on translation initiation. Since the inhibitory effect stems from a similar allosteric change, both 4EGI-1 and i4EG-BiP, although binding to different sites, have similar ability to displace the eIF4G/4E-BP consensus peptide and exhibit similar cellular activity. Exactly how the extension of the helix-α1 displaces the eIF4G/4E-BP consensus peptide, which lacks the extended binding region of the full-length proteins, still remains an open question.
To further improve the potency of the inhibitors 4EGI-1 and i4EG-BiP, we designed PROTAC versions of the two molecules. Here, we employed 4EGI-1 and the novel inhibitor i4EG-BiP as scaffolds to create cell-permeable thalidomide conjugates that bind to eIF4E and engage Cereblon. Engagement of Cereblon, as well as VHL, with eIF4E ligands has been previously attempted by Kaur, et al. using cap analogues. Even though they observed binding to eIF4E by their PROTACs, target degradation was not achieved. The authors attributed this failure to the net negative charge of the m7GDP-based compounds (37). Here we show that the design of a neutral PROTAC based on 4EGI-1 using a prodrug approach and neutral PROTACs based on i4EG-BiP using the carboxylic acid moiety as a linking point seems to alleviate this hurdle on efficacy. Using cellular Cereblon engagement assays, we were able to show a concentration-dependent cell-based effect of our compounds.
However, we were not able to detect any targeted cellular degradation of eIF4E. This could, in part, be due to the relatively low binding affinity of the parent compounds, 4EGI-1 and i4EG-BiP. Using medicinal chemistry approaches to improve the binding affinity of i4EG-BiP might help to improve the PROTAC strategy for our i4EG-BiP-based molecules. This is particularly possible as there is unoccupied space on eIF4E in the vicinity of the i4EG-BiP binding site which can be leveraged to improve molecule affinity. The failed degradation of eIF4E could also be explained by increased binding of 4E-BP1 to eIF4E as a result of eIF4E binding 4EGI-1 or i4EG-BiP. Increased binding of 4E-BP to eIF4E could result from conformational changes in eIF4E upon small molecule binding, which could increase the affinity for 4E-BP, and/or the fact that the competing binding protein, eIF4G, is no longer able to engage eIF4E. It has been previously shown that ubiquitination of eIF4E at K159 is orchestrated by the E3 ubiquitin ligase CHIP, which leads to subsequent proteasomal degradation, and furthermore that this ubiquitination can be blocked by overexpression of 4E-BP1 (53). Therefore, increased 4E-BP1 binding as a result of 4EGI-1 or i4EG-BiP binding could protect eIF4E from ubiquitination and stabilize the protein. It should however be noted that in our current study the ubiquitination is performed in a Cereblon-dependent manner, which might not have the same inhibition response to 4E-BP as CHIP. Future studies will be needed to address this possible obstructive role of 4E-BP on targeted degradation of eIF4E. If indeed the association of 4E-BP prevents the ubiquitination of eIF4E, an interesting approach would be to use 4E1RCat as a scaffold to create eIF4E targeting PROTACs, since this inhibitor has been shown to displace both eIF4G and 4E-BP1 binding to eIF4E (29). Utilizing the synthetical method we provide in this manuscript to link the carboxylic acid moiety of 4E1RCat to lenalidomide could circumvent both cell-permeability hurdles as well as evasion of eIF4E ubiquitination by enhanced 4E-BP1 binding.
Blocking cap-dependent translation provides an attractive opportunity for future efforts to target cancer cells and eIF4E is the master regulator of this process. Disrupting the crucial interaction of eIF4E with its scaffold protein eIF4G or degrading eIF4E altogether are two independent routes to inhibit eIF4E function. Here we attempted to synergistically apply both these approaches. Although we did not achieve successful degradation of eIF4E, our successful targeting of eIF4E in this study with a newly identified small molecule inhibitor, i4EG-BiP, paves way for future efforts.
MATERIAL AND METHODS
Expression and purification of eIF4E
A construct of human eIF4E was expressed in transformed Escherichia coli BL21(DE3). A Δ26-eIF4E or GB1-eIF4E construct in a pET-28(+) backbone was used for crystallography and NMR studies, respectively. Bacteria were grown in Luria Broth (LB) at 37°C. Protein expression was induced by the addition of 0.1 mM isopropyl-β-D-thiogalactopyranoside at OD600 = 0.6 followed by incubation overnight at 23°C. Cells were harvested with a yield of 3 g/L wet pellet and stored at −30 °C. Bacterial pellets were resuspended by slow pipetting in lysis buffer constituting of 50 mM Tris·HCl, pH 7.5, 100 mM NaCl, 1% Triton-X, 5 mM tris(2-carboxyethyl)phosphine (TCEP), 1 cOmplete™ Protease Inhibitor Cocktail tablet, lysozyme, RNase, and DNase.
Cells were subsequently homogenized in a cell microfluidizer and the lysates were centrifuged at 38,000 × g for 1.5 h. After centrifugation the clarified lysate supernatant was first passed through a 0.45 μm cellulose acetate syringe-filter and then passed over a diethylaminoethylcellolose (DEAE) column previously equilibrated with the same lysis buffer. The DEAE flow-through was loaded on adipic–agarose-m7GDP column and after 0.5 h of binding, the column was washed with 50 mL of wash buffer (10 mM HEPES, pH 7.5, 125 mM NaCl, and 1 mM TCEP) five times followed by elution four times with 10 mL elution buffer (10 mM HEPES, pH 7.5, 125 mM NaCl,100 μM m7GTP plus 10 mM TCEP). The eluted protein concentration was assessed by Bradford assay (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions and then concentrated to a final volume of 3 mL by ultrafiltration through a 15 mL, 10-kDa-cutoff Millipore centrifugal filter. Following concentration, up to 4 mL of eluate was subjected to size-exclusion chromatography using a Superdex75 16/10 preparative column (GE Healthcare) equilibrated with 10 mM HEPES, pH 7.5, 125 mM NaCl, and 1 mM TCEP buffer. The collected pure protein fractions were concentrated to a final concentration (by ultrafiltration) to 1 mg/mL as evaluated by NanoDrop™ at 280 nm. The total yield of the aforementioned process was approximately 3-5 mg of pure protein from 1 L of culture.
Protein crystallization
Crystallization was performed using the sitting drop method and all crystals were grown in drops containing 1 μL of protein-ligand solution and 1 μL of crystallization solution containing 10–25% (vol/vol) 3.3-kDa PEG, 100 mM MES, pH 6.0, 10% (vol/vol) isopropanol. The protein-ligand solution used for crystallization was prepared by mixing eIF4E (9 mg/mL) and the small molecule (4EGI1-BP or analogs) solution (12.5 mM in DMSO) at an approximately 1:1 stoichiometry. The resulting protein–small molecule mixture was then serially diluted from 9 to 1 mg/mL and used to set up sitting drop crystal trials. Crystals began to form in 2 days and grew to full size at day 4. They were inspected and based on their size and morphology, the optimal conditions of protein concentration and PEG were determined. Crystals were harvested and quickly transferred to a cryoprotectant solution of crystallization buffer plus 10% (v/v) glycerol before flash freezing in liquid nitrogen.
X-ray diffraction data collection:
X-ray diffraction data were collected at the APS X-Ray Synchrotron Source at the Argonne National Laboratory from single protein ligand complex crystals. The data were integrated using XDS. The results indicated monoclinic space group P21 crystals with diffraction up to 1.9 Å resolution. Phases were calculated by molecular replacement using PHASER and an initial model of eIF4E (PDB ID 4TPW). Subsequently, models were manually inspected and refined in Coot followed by further rounds of phase calculations by molecular replacement in PHENIX and model structure refinement until a minimum R-free was reached. The position of the 4EGI1-BP ligand was clearly seen in the Fo-Fc map before the ligand was included in the model.
Nuclear magnetic resonance experiments
15N-1H-TROSY-HSQC spectra were recorded on a Bruker Avance III 800 MHz spectrometer with a TXO-style cryogenically cooled probe. 15N-labeled GB1-eIF4E was concentrated to a final concentration of 100 μM in a buffer composed of 50 mM sodium phosphate, pH 6.5, 50 mM potassium chloride, 2 mM DTT and 5% D2O. A reference spectrum was recorded at 298 K by addition of DMSO to a final concentration of 1.5%. NMR titration was performed by recording 15N-1H-TROSY-HSQC spectra in the presence of either i4EG-BiP or d4E-4 at 50, 100 and 300 μM concentrations (diluted from 20 mM stock solutions in DMSO). NMR experiments were processed with nmrPipe and analyzed using the ccpNMR software (version 2.4.1) (54).
Docking of d4E-4 to eIF4E
The protein structure used for docking was the structure of i4EG-BiP bound to eIF4E, with i4EG-BiP removed. The receptor structure was prepared in PDBQT format with AutoDock Tools, which is part of MGLTools (55), by assigning AutoDock atom types and merging the nonpolar hydrogen atoms. The ligand was prepared in PDBQT format with Open Babel (56), which included computation of the 3-dimensional structure. The docking box was 18 Å x 30 Å x 18 Å, and the docking exhaustiveness was set to 4. The receptor was held rigid during the docking procedure, and 10 replicates of the docking procedure were executed.
Fluorescence Polarization Experiments
For the fluorescence polarization assays, a GST-eIF4E construct was used. It was expressed in E. coli and purified using the same procedure described above for Δ26-eIF4E without the DEAE column step. The assay was used to test the activity of various compounds against eIF4E/eIF4G–peptide complex formation. For the fluorescent probe we used a purified eIF4G-peptide conjugated with fluorescein derived by peptide synthesis with the sequence KKQYDREFLLDFQFK-FITCH. Assay mixtures consisted of 150 nM eIF4G-peptide plus 0.3 μM GST-eIF4E in 100 mM Na-phosphate, pH 7.5. A 384-well black plate was used to prepare serial dilutions of 4EGI1-BP and other compounds from a 12.5 mM stock solution in DMSO at a starting ligand concentration of 500 μM. The fluorescence polarization signal was recorded using an EnVison™ plate reader.
Cell–Specific Bioluminescence Imaging (CS-BLI) cell viability assay
CS-BLI was performed according to a previously published high-throughput cell viability assay (38). Briefly, Luciferase-expressing MM.1S cells and MCF7 were cultured in T75 flasks. RPMI or DMEM media (as appropriate), supplemented with 10% FBS, and penicillin and streptomycin were used accordingly. 104 cells were seeded in 50 μL of media in each well of a 96-well, tissue-culture-treated, white flat bottom plate and left to adhere overnight in a cell-culture incubator. The next day we supplemented the well volume with 50 μL of media with serially diluted compounds. After 24 hours we added 5 μL of 1 mg/ml stock of beetle D-luciferin, let it equilibrate at 37°C for 30 min, and top-read the chemiluminescence plate using a BioTek Synergy HTX plate reader. The reads were repeated at 48 and 72 hours. Collected data were normalized, processed, and plotted with Scilab and GraphPad-Prism.
Cell Culture
HeLa cells were grown in 4.5 g/L glucose DMEM supplemented with 10% fetal bovine serum (Gibco 16000-49) and 1% penicillin/streptomycin. Cells were seeded into opaque white 96-well assay plates at a density of 5x103 cells per well. 24 h post-seeding, the medium was replaced with medium containing the drug at the specified concentrations in equal volumes of DMSO. Cells were assayed for viability 48 h later using CellTiter-Glo (Promega) and plates were read on a CLARIOstar plus (BMG Labtech) plate reader. Values were normalized to the DMSO control. A minimum of two experiments was performed with representative data shown.
Cereblon engagement assay
Cells stably expressing the BRD4BD2-GFP with mCherry reporter (41) were seeded at 30-50% confluency in 384-well plates with 50 μL per well of FluoroBrite DMEM media (Thermo Fisher Scientific A18967) supplemented with 10% FBS a day before compound treatment. Compounds and 100 nM dBET6 were dispensed using a D300e Digital Dispenser (HP), normalized to 0.5% DMSO, and incubated with the cells for 5 h. The assay plate was imaged immediately using an Acumen High Content Imager (TTP Labtech) with 488 nm and 561 nm lasers in a 2 μm x 1 μm grid per well format. The resulting images were analyzed using CellProfiler (57). A series of image analysis steps (an ‘image analysis pipeline’) was constructed. First, the red and green channels were aligned and cropped to target the middle of each well (to avoid analysis of the heavily clumped cells at the edges). A background illumination function was calculated for both red and green channels of each well individually and subtracted to correct for illumination variations across the 384-well plate from various sources of error. An additional step was then applied to the green channel to suppress the analysis of large auto fluorescent artifacts and enhance the analysis of cell specific fluorescence by way of selecting for objects under a given size (30 A.U.) and with a given shape (speckles). mCherry-positive cells were then identified in the red channel by filtering for objects 8-60 pixels in diameter and by using intensity to distinguish between clumped objects. The green channel was then segmented into GFP positive and negative areas and objects were labeled as GFP positive if at least 40% of it overlapped with a GFP positive area. The fraction of GFP-positive cells/mCherry-positive cells in each well was then calculated, and the green and red images were rescaled for visualization. The values for the concentrations that led to a 50% increase in BRD4BD2-eGFP accumulation (EC50) were calculated using the nonlinear fit variable slope model (GraphPad Software).
Proteomics methods:
Global quantitative proteomics sample preparation
Kelly cells were treated with DMSO (biological triplicate) or d4E-2 at 1 μM for 5 hours and cells were harvested by centrifugation at 4 °C. Cell lysis was performed by resuspension of the cell pellet in denaturing Urea buffer (8 M Urea, 50 mM NaCl, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (EPPS) pH 8.5, Protease and Phosphatase inhibitors), followed by manual homogenization by 20 passes through a 21-guage needle. Cell lysate was clarified by centrifugation and global protein quantified using a Bradford assay (Bio-Rad). 100 μg of protein from each treatment was reduced, alkylated, digested and TMT labelled for LC-MS analysis as previously described(58). The TMT labelled sample was offline fractionated into 96 fractions by high pH reverse phase HPLC (Agilent LC1260) through an aeris peptide xb-c18 column (phenomenex) with mobile phase A containing 5% acetonitrile and 10 mM NH4HCO3 in LC-MS grade H2O, and mobile phase B containing 90% acetonitrile and 5 mM NH4HCO3 in LC-MS grade H2O (both pH 8.0). The resulting 96 fractions were recombined in a non-contiguous manner into 24 fractions and desalted using C18 solid phase extraction plates (SOLA, Thermo Fisher Scientific) followed by subsequent mass spectrometry analysis.
LC-MS data collection and analysis
Data were collected using an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) and coupled with an UltiMate 3000 RSLCnano System. Peptides were separated on an EasySpray ES803a.rev2 75 μm inner diameter microcapillary column (Thermo Fisher Scientific). Peptides were separated over a 190 min gradient of 9 - 32% acetonitrile in 1.0% formic acid with a flow rate of 300 nL/min. Quantification was performed using a MS3-based TMT method as described previously (McAlister et al., 2014), with the addition of Real-Time Search MS3 acquisition implemented between MS2 and MS3 scans. The data were acquired using a mass range of m/z 340 – 1350, 120,000 resolution, standard AGC and maximum injection time of 50 ms for the peptide measurements in the Orbitrap. Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 34%, custom AGC target and a maximum injection time of 35 ms. Real-Time Search was performed with a Swissprot human database (December 2019), searching for tryptic peptides with maximum of 1 missed cleavage, static alkylation of cysteines (57.0215 Da), static TMT labelling of lysine residues and peptide N-termini (304.2071 Da) and variable oxidation of methionine (15.9949 Da).
MS3 scans were acquired in the Orbitrap with HCD collision energy set to 45%, custom AGC target, maximum injection time of 86 ms, resolution at 50,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
Proteome Discoverer 2.4 (Thermo Fisher Scientific) was used for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. The MS/MS spectra were searched against a Swissprot human database (December 2019) containing both the forward and reverse sequences. Searches were performed using a 20 ppm precursor mass tolerance, 0.6 Da fragment ion mass tolerance, tryptic peptides containing a maximum of two missed cleavages, static alkylation of cysteine (57.0215 Da), static TMT labelling of lysine residues and N-termini of peptides (304.2071 Da), and variable oxidation of methionine (15.9949 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. The peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (summed signal-to-noise across channels < 100 and precursor isolation specificity < 0.5), and the resulting data was filtered to only include proteins with a minimum of 2 unique peptides quantified. Reporter ion intensities were normalized and scaled using in-house scripts in the R framework (R Development Core Team, 2014). Statistical analysis was carried out using the limma package within the R framework(59).
eIF4E pull down and Western Blot
HEK293 and HeLa cells were grown for 24 h, harvested by centrifugation and lysed by multiple freeze-thaw cycles. 300 μL (1 μg/μL) of cell lysates, prepared in freeze-thaw lysis buffer (25mM Tris-HCl, pH 7.5,150 mM KCl, 0.1% Triton X), were treated with the indicated concentrations of 4EGI-1 and i4EG-BiP at 37 °C for 1 h. This was followed by addition of 50 μL of a 50% mixture of adipic-agarose-m7GDP beads (40) followed by incubation for 1 h at 4 °C. After washing the resin three times washing with lysis buffer, the bound proteins were resolved by SDS-PAGE, and analysed by immunoblotting. Briefly this consisted of transfer to an activated PVDF membrane, blocking, and staining with a polyclonal antibody against 4E-BP1 (Cell Signaling Technology) and monoclonal antibodies against eIF4E and eIF4G (Transduction Laboratories) in PBS-T buffer. Secondary antibodies labelled with IRDye® 800CW and IRDye 680RD were used to amplify the signal according to the manufacturer’s protocol. The immunoblot was imaged using two-color Western blot detection with an Odyssey® Imager.
Dual luciferase assay
HEK 293T cells were cultured to 70% confluence in six-well plates. Cells were transfected with 500 ng of a bicistronic reporter construct pFL-EMCV-IRES-RL, containing firefly luciferase followed by the EMCV IRES and Renilla luciferase, using polyethyleneimine (PEI) (high molecular weight, Sigma) in a 1:6 ratio using a PEI stock prepared at 1 mg/mL in DI water for transfection. The cells were treated 16 hours after transfection with the indicated concentrations of 4EGI-1 and i4EG-BiP. 3 hours after treatment, the cells were lysed in 1× passive lysis buffer (Promega), and luciferase activity was measured with a dual luciferase reporter assay system (Promega) using an EnVision™ plate reader (PerkinElmer).
Supplementary Material
Design of an eIF4E inhibitor i4EG-BiP, that binds to an internal site
High resolution crystal structure of eIF4E bound to the inhibitor i4EG-BiP
Specific inhibition of cap-dependent translation by i4EG-BiP
Design cell-permeable eIF4E-targeting PROTACs based on i4EG-BiP and 4EGI-1
Synthesis and testing of eIF4E PROTACs in biophysical and cellular assays
ACKNOWLEDGEMENT
PDF acknowledges the Chleck Foundation. HA acknowledges funding from the Claudia Adams Barr Program for Innovative Cancer Research and from NIH (GM136859). G.W. acknowledges support from NIH grant GM132079 and CA200913. We thank Dr. Kendra Leigh for her critical comments on the manuscript.
Footnotes
SUPPLEMENTARY DATA
Supplementary Data and movie are available online.
CONFLICT OF INTEREST
E. S. F. is a founder, science advisory board member and equity holder in Civetta, Jengu (board member) and Neomorph, an equity holder in C4, and a consultant to Astellas, Novartis, Deerfield, RA Capital and EcoR1. The Fischer lab receives or has received research funding from Novartis, Astellas, Ajax and Deerfield. G.W. is co-founder of PIC Therapeutics, Cellmig biolabs, Skinap therapeutics and Virtual Discovery. H.A. is an equity holder in PIC Therapeutics. The research described here is scientifically and financially independent of the efforts in any of the above-mentioned companies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession numbers 7MEU and will be released upon publication.
REFERENCES
- 1.Jia Y, Polunovsky V, Bitterman PB and Wagner CR (2012) Cap-Dependent Translation Initiation Factor eIF4E: An Emerging Anticancer Drug Target: CAP-DEPENDENT TRANSLATION INITIATION EIF4E INHIBITION. Med. Res. Rev, 32, 786–814. 10.1002/med.21260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW and Sonenberg N (2004) eIF4E – from translation to transformation. Oncogene, 23, 3172–3179. 10.1038/sj.onc.1207549 [DOI] [PubMed] [Google Scholar]
- 3.Sonenberg N and Hinnebusch AG (2009) Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell, 136, 731–745. 10.1016/j.cell.2009.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M and Borden KLB (2006) eIF4E is a central node of an RNA regulon that governs cellular proliferation. Journal of Cell Biology, 175, 415–426. 10.1083/jcb.200607020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arcand M, Roby P, Bossé R, Lipari F, Padrós J, Beaudet L, Marcil A and Dahan S (2010) Single-Well Monitoring of Protein–Protein Interaction and Phosphorylation–Dephosphorylation Events. Biochemistry, 49, 3213–3215. 10.1021/bi100253p [DOI] [PubMed] [Google Scholar]
- 6.Soni A, Akcakanat A, Singh G, Luyimbazi D, Zheng Y, Kim D, Gonzalez-Angulo A and Meric-Bernstam F (2008) eIF4E knockdown decreases breast cancer cell growth without activating Akt signaling. Molecular Cancer Therapeutics, 7, 1782–1788. 10.1158/1535-7163.MCT-07-2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meric F and Hunt KK (2002) Translation Initiation in Cancer: A Novel Target for Therapy. Molecular Cancer Therapeutics, 1, 971–979. [PubMed] [Google Scholar]
- 8.Bordeleau M-E, Robert F, Gerard B, Lindqvist L, Chen SMH, Wendel H-G, Brem B, Greger H, Lowe SW, Porco JA, et al. (2008) Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J. Clin. Invest, 10.1172/JCI34753. 10.1172/JCI34753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazaris-Karatzas A, Montine KS and Sonenberg N Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5’ cap. Nature, 345, 544–547. 10.1038/345544a0 [DOI] [PubMed] [Google Scholar]
- 10.Kerekatte V, Smiley K, Hu B, Smith A, Gelder F and De Benedetti A (1995) The proto-oncogene/translation factor eIF4E: A survey of its expression in breast carcinomas. Int. J. Cancer, 64, 27–31. 10.1002/ijc.2910640107 [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Rosenwald IB, Hutzler MJ, Pihan GA, Savas L, Chen J-J and Woda BA (1999) Expression of the Eukaryotic Translation Initiation Factors 4E and 2α in Non-Hodgkin’s Lymphomas. The American Journal of Pathology, 155, 247–255. 10.1016/S0002-9440(10)65118-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathan C-AO, Liu L, Li BD, Abreo FW, Nandy I and De Benedetti A (1997) Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene, 15, 579–584. 10.1038/sj.onc.1201216 [DOI] [PubMed] [Google Scholar]
- 13.Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras A-C, Sonenberg N and Wagner G Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat Struct Biol, 4, 717–724. 10.1038/nsb0997-717. [DOI] [PubMed] [Google Scholar]
- 14.Marcotrigiano J, Gingras A-C, Sonenberg N and Burley SK Cocrystal Structure of the Messenger RNA 5′ Cap-Binding Protein (eIF4E) Bound to 7-methyl-GDP. Cell, 89, 951–961. 10.1016/s0092-8674(00)80280-9 [DOI] [PubMed] [Google Scholar]
- 15.Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G and Léger-Abraham M (2015) Molecular mechanism of the dual activity of 4EGI-1: Dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E-BP1. Proc Natl Acad Sci USA, 112,E4036–E4045. 10.1073/pnas.1512118112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grüner S, Peter D, Weber R, Wohlbold L, Chung M-Y, Weichenrieder O, Valkov E, Igreja C and Izaurralde E (2016) The Structures of eIF4E-eIF4G Complexes Reveal an Extended Interface to Regulate Translation Initiation. Molecular Cell, 64, 467–479. 10.1016/j.molcel.2016.09.020 [DOI] [PubMed] [Google Scholar]
- 17.Grüner S, Weber R, Peter D, Chung M-Y, Igreja C, Valkov E and Izaurralde E (2018) Structural motifs in eIF4G and 4E-BPs modulate their binding to eIF4E to regulate translation initiation in yeast. Nucleic Acids Research, 46, 6893–6908. 10.1093/nar/gky542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukhele S, Bah A, Lin H, Sonenberg N and Forman-Kay JD (2013) Interaction of the Eukaryotic Initiation Factor 4E with 4E-BP2 at a Dynamic Bipartite Interface. Structure, 21, 2186–2196. 10.1016/j.str.2013.08.030 [DOI] [PubMed] [Google Scholar]
- 19.Igreja C, Peter D, Weiler C and Izaurralde E (2014) 4E-BPs require non-canonical 4E-binding motifs and a lateral surface of eIF4E to repress translation. Nat Commun, 5, 4790. 10.1038/ncomms5790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peter D, Igreja C, Weber R, Wohlbold L, Weiler C, Ebertsch L, Weichenrieder O and Izaurralde E (2015) Molecular Architecture of 4E-BP Translational Inhibitors Bound to eIF4E. Molecular Cell, 57, 1074–1087. 10.1016/j.molcel.2015.01.017 [DOI] [PubMed] [Google Scholar]
- 21.Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE and Forman-Kay JD (2015) Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature, 519, 106–109. 10.1038/nature13999 [DOI] [PubMed] [Google Scholar]
- 22.Dawson JE, Bah A, Zhang Z, Vernon RM, Lin H, Chong PA, Vanama M, Sonenberg N, Gradinaru CC and Forman-Kay JD (2020) Non-cooperative 4E-BP2 folding with exchange between eIF4E-binding and binding-incompatible states tunes cap-dependent translation inhibition. Nat Commun, 11, 3146. 10.1038/s41467-020-16783-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, et al. (2007) Small-Molecule Inhibition of the Interaction between the Translation Initiation Factors eIF4E and eIF4G. Cell, 128, 257–267. 10.1016/j.cell.2006.11.046 [DOI] [PubMed] [Google Scholar]
- 24.Papadopoulos E, Jenni S, Kabha E, Takrouri KJ, Yi T, Salvi N, Luna RE, Gavathiotis E, Mahalingam P, Arthanari H, et al. (2014) Structure of the eukaryotic translation initiation factor eIF4E in complex with 4EGI-1 reveals an allosteric mechanism for dissociating eIF4G. Proceedings of the National Academy of Sciences, 111, E3187–E3195. 10.1073/pnas.1410250111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Aktas BH, wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, et al. (2012) Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget, 3. 10.18632/oncotarget.598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H and Klann E (2013) Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature, 493, 411–415. 10.1038/nature11782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMahon R, Zaborowska I and Walsh D (2011) Noncytotoxic Inhibition of Viral Infection through eIF4F-Independent Suppression of Translation by 4EGi-1. Journal of Virology, 85, 853–864. 10.1128/JVI.01873-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoeffer CA, Cowansage KK, Arnold EC, Banko JL, Moerke NJ, Rodriguez R, Schmidt EK, Klosi E, Chorev M, Lloyd RE, et al. (2011) Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proceedings of the National Academy of Sciences, 108, 3383–3388. 10.1073/pnas.1013063108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, et al. (2011) Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proceedings of the National Academy of Sciences, 108, 1046–1051. 10.1073/pnas.1011477108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A and Crews CM (2018) Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chemical Biology, 25, 78–87.e5. 10.1016/j.chembiol.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.An S and Fu L (2018) Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine, 36, 553–562. 10.1016/j.ebiom.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai AC and Crews CM (2017) Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov, 16, 101–114. 10.1038/nrd.2016.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S and Bradner JE Phthalimide conjugation as a strategy for in vivo target protein degradation. Science, 348, 1376–1381. 10.1126/science.aab1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. (2015) Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & Biology, 22, 755–763. 10.1016/j.chembiol.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ottis P and Crews CM (2017) Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol, 12, 892–898. 10.1021/acschembio.6b01068 [DOI] [PubMed] [Google Scholar]
- 36.Huang H-T, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho J-H, Ko E, Jang J, et al. (2018) A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chemical Biology, 25, 88–99.e6. 10.1016/j.chembiol.2017.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaur T, Menon A and Garner AL (2019) Synthesis of 7-benzylguanosine cap-analogue conjugates for eIF4E targeted degradation. European Journal of Medicinal Chemistry, 166, 339–350. 10.1016/j.ejmech.2019.01.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, et al. (2010) Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med, 16, 483–489. 10.1038/nm.2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019) Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature, 569, 503–508. 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edery I, Altmann M and Sonenberg N (1988) High-level synthesis in Escherichia cofi of functional cap-binding eukaryotic initiation factor eIF-4E and affhity purification using a simplified cap-analog resin. Gene, 74, 517–525. [DOI] [PubMed] [Google Scholar]
- 41.Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, et al. (2018) Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol, 14, 706–714. 10.1038/s41589-018-0055-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Wispelaere M, Du G, Donovan KA, Zhang T, Eleuteri NA, Yuan JC, Kalabathula J, Nowak RP, Fischer ES, Gray NS, et al. (2019) Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat Commun, 10, 3468. 10.1038/s41467-019-11429-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeppesen F and Ilium P (1972) Concentration of Ampicillin in Antral Mucosa Following Administration of Ampicillin Sodium and Pivampicillin. Acta Oto-Laryngologica, 73, 428–432. 10.3109/00016487209138962 [DOI] [PubMed] [Google Scholar]
- 44.Patnaik A, Rowinsky EK, Villalona MA, Hammond LA, Britten CD, Siu LL, Goetz A, Felton SA, Burton S, Valone FH, et al. (2002) A Phase I Study of Pivaloyloxymethyl Butyrate, a Prodrug of the Differentiating Agent Butyric Acid, in Patients with Advanced Solid Malignancies. Clin Cancer Res, 8, 2142–2148. [PubMed] [Google Scholar]
- 45.Nowak RP, Fischer ES, Gray NS, Zhang T and He Z Heterobifunctional compounds with improved specificity for the bromodomain of brd4. (U.S. Patent No. WO2019079701A1), WIPO (PCT), https://www.wipo.int/pct/en/
- 46.Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, Tan L, Shi K, Yue H, Liu X, et al. (2020) Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell, 183, 1714–1731.e10. 10.1016/j.cell.2020.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mori T, Ito T, Liu S, Ando H, Sakamoto S, Yamaguchi Y, Tokunaga E, Shibata N, Handa H and Hakoshima T (2018) Structural basis of thalidomide enantiomer binding to cereblon. Sci Rep, 8, 1294. 10.1038/s41598-018-19202-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borissenko L and Groll M (2007) 20S Proteasome and Its Inhibitors: Crystallographic Knowledge for Drug Development. Chem. Rev, 107, 687–717. 10.1021/cr0502504 [DOI] [PubMed] [Google Scholar]
- 49.Alhossary A, Handoko SD, Mu Y and Kwoh C-K (2015) Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics, 31, 2214–2216. 10.1093/bioinformatics/btv082 [DOI] [PubMed] [Google Scholar]
- 50.Trott O and Olson AJ (2009) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem, 10.1002/jcc.21334. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, Huang F, Wang J, Wang P, Lv W, Hong L, Li S and Zhou J (2015) The synergistic inhibition of breast cancer proliferation by combined treatment with 4EGI-1 and MK2206. Cell Cycle, 14, 232–242. 10.4161/15384101.2014.977096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu M, Zhang C, Li X-J, Liu Q and Wanggou S (2016) Anti-Cancer Effect of Cap-Translation Inhibitor 4EGI-1 in Human Glioma U87 Cells: Involvement of Mitochondrial Dysfunction and ER Stress. Cell Physiol Biochem, 40, 1013–1028. 10.1159/000453158 [DOI] [PubMed] [Google Scholar]
- 53.Murata T and Shimotohno K (2006) Ubiquitination and Proteasome-dependent Degradation of Human Eukaryotic Translation Initiation Factor 4E. J. Biol. Chem, 281, 20788–20800. 10.1074/jbc.M600563200 [DOI] [PubMed] [Google Scholar]
- 54.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J and Laue ED (2005) The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins, 59, 687–696. 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 55.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS and Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem, 30, 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T and Hutchison GR (2011) Open Babel: An open chemical toolbox. J Cheminform, 3, 33. 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology, 7, R100. 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL and Fischer ES (2018) Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife, 7, e38430. 10.7554/eLife.38430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W and Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research, 43, e47–e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession numbers 7MEU and will be released upon publication.