

Abstract
Gaucheromas, which are pseudotumors consisting of a cluster of Gaucher cells, are rare complications in Gaucher's disease (GD) and reported in patients treated with enzyme replacement therapy (ERT). Gaucheromas commonly develop in the lymph nodes in the mesenteric and mediastinal regions and can cause serious complications including protein-losing enteropathy. A large mesenteric Gaucheroma showed a significant reduction in size after initiation of substrate reduction therapy (SRT) with eliglustat in an adult patient with GD type 3. Combination therapy with ERT and SRT should be considered to prevent Gaucheromas in patients with GD.
Keywords: Gaucher's disease, enzyme replacement therapy, combination therapy
Introduction
Gaucher's disease (GD) is the most common lysosomal storage disease. It is an autosomal recessive disorder due to a deficiency of glucocerebrosidase which is responsible for the degradation of glucocerebroside, an abundant component of cell membranes. A deficiency of glucocerebrosidase results in the accumulation of glucocerebroside in macrophages (Gaucher cells) in the mononuclear phagocytic system throughout the body. GBA1 encodes the enzyme; however, genotype–phenotype correlations are incomplete, although a few known genotypes have been associated with certain phenotypes. Three major clinical types include type 1 nonneuronopathic, type 2 acute neuronopathic, and type 3 subacute/chronic neuronopathic. Enzyme replacement therapy (ERT) for GD was approved by the U.S. Food and Drug Administration in 1991 as the first drug for a lysosomal storage disorder. Since then ERT has been widely used as the standard management for GD type 1 and type 3. Substrate reduction therapy (SRT) with miglustat and eliglustat has been available since 2003 and 2014, respectively. These medications are specific inhibitors of glucosylceramide synthase.
Gaucheromas are rare complications in GD and have been reported in patients treated with ERT. Gaucheromas were first reported by Lim et al in 2002. 1 Since then ∼ 23 cases of GD with Gaucheromas have been reported, primarily in mesenteric and mediastinal lymph nodes. 2 We also reported previously a case of GD type 3 with an extremely rare complication of large soft tissue Gaucheromas. 3 Gaucheromas can cause serious clinical consequences and ERT does not seem to prevent Gaucheromas from developing. Combination therapy with ERT and SRT theoretically should have a combinational advantage over ERT alone in preventing accumulation of glucocerebroside in macrophages. However, Mhanni et al reported a 16-year-old patient with GD type 3 who received ERT with imiglucerase at 100 IU/kg/2 weeks for 10 years and later received combination therapy with miglustat for 4 years, but still developed severe protein-losing enteropathy due to mesenteric Gaucheromas. 4 Here, we presented an adult case of GD type 3 who developed a large mesenteric Gaucheroma in teenager, and showed a significant reduction of Gaucheroma after initiation of combination therapy with imiglucerase and eliglustat.
Clinical Report
Patient was a 24-year-old woman with GD type 3 ( GBA1 , homozygous L444P) who was born to nonconsanguineous Hispanic parents. She had been well until the age of 13 months when she had a choking episode. She was brought to a local emergency room where hepatomegaly was noted; subsequently, a diagnosis of GD was made. She has been treated with ERT with imiglucerase at 120 IU/kg every 2 weeks since the age of 16 months. She received special education due to mild intellectual disability and was also noted to have bilateral hearing impairment requiring hearing aids at 4 years of age. She had occasional bone pain for which nonsteroidal anti-inflammatory drugs were used. Necrosis of the right femoral head was diagnosed with magnetic resonance imaging (MRI) at 18 years of age. A large calcific density in the abdomen suggestive of calcified lymph nodes was found by abdominal X-ray studies at the age of 16 years, which was further studied with MRI and computed tomography confirming a cystic retroperitoneal mass with calcification at the base of the mesentery compressing the superior mesenteric vein and encasing the superior mesenteric artery. Bowel wall thickening involving the jejunum with increased mucosal enhancement as well as hypoalbuminemia (2.6 g/dL) due to protein-losing enteropathy were diagnosed at the age of 20 years. Fecal α-1 antitrypsin level was elevated at 265 mg/dL (ref: < 55). Clinical diagnosis of mesenteric Gaucheroma was made based on the clinical symptoms and imaging studies. A biopsy could not be performed due to the abundant collateral vessels surrounding the mass. In addition to ERT, SRT with eliglustat at 84 mg/d was implemented at 21 years of age since she was found to be a poor metabolizer based on cytochrome p450 2D6 activities. Follow-up imaging studies after the combination therapy of SRT and ERT for 31 months showed a significant reduction of the mesenteric mass ( Fig. 1A, 1B ). Although the small bowel thickening and ascites decreased, hypoalbuminemia (2.3 g/dL) persisted. The symptom of postprandial abdominal discomfort improved within a few months after implementation of SRT. Serum total acid phosphatase concentrations were not significantly changed after initiation of SRT: the average values over 3-year period before (6.97 U/L, n = 8) and after (5.00 U/L, n = 5) implementation of SRT being compared.
Fig. 1.
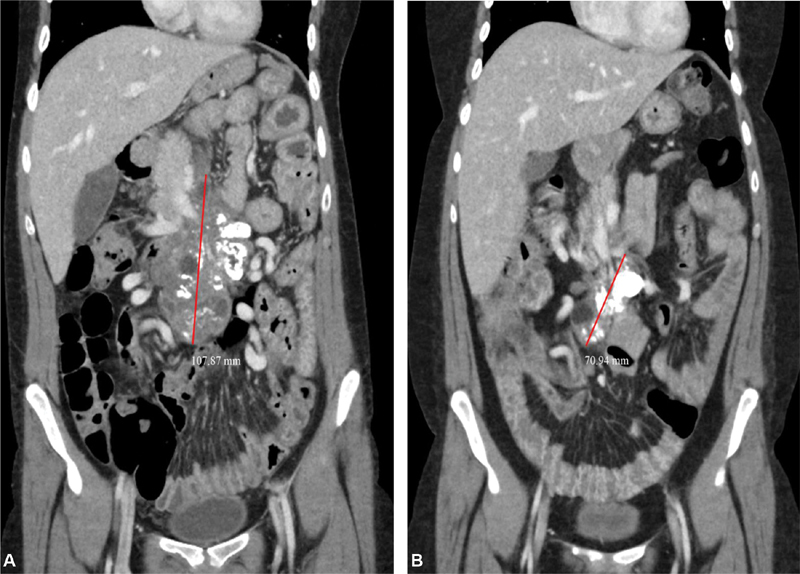
( A, B ) Sequential coronal CT scans of a large mesenteric Gaucheroma. ( A ) At 21 years of age (baseline study obtained 7 days after implementation of SRT), a large, partially calcified, mid-abdominal mass with solid and cystic components is shown. Red line marks the maximal length of the mass (dimensions: 4.9 × 7.8 × 10.8 cm). ( B ) CT scan in the same coronal plane 31 months after the initiation of combination therapy demonstrating marked reduction in size of the solid (dimensions: 5.5 × 3.7 × 7.1 cm) and cystic components of the mass. CT, computed tomography; SRT, substrate reduction therapy.
Discussion
Approximately 24 cases of Gaucheromas including the proposita have been reported. 2 3 4 5 Although Gaucheromas are rare complications in GD, they can cause serious clinical consequences due to size effect of the mass: intestinal lymphatic obstruction, lymphangiectasia, compression of mesenteric vein, postprandial abdominal discomfort, and protein-losing enteropathy leading to hypoalbuminemia, ascites, and generalized edema. 5 Table 1 summarizes the reported cases with Gaucheromas including proposita. 2 3 4 5 Approximately 65% had GD type 3. Genotyping showed that 60% of the pathogenic alleles in 24 patients were p.L444P, which is known to be associated with severe disease, often with neurologic complications. Most of the patients with Gaucheromas were diagnosed in the pediatric age range: 75% of the patients were younger than 10 years. Three exceptional cases developed Gaucheromas as teenagers or adults despite ERT implementation in early childhood. Since these three patients were treated with the higher dose of imiglucerase (100–120 IU/kg/2 weeks) rather than the standard dose of 60 IU/kg/2 weeks, it may have delayed the development of Gaucheroma. The most common primary sites were the intra-abdominal (mesenteric: 71%) and the mediastinal (29%) spaces. 2 All of the patients with intra-abdominal and mediastinal Gaucheromas were treated with ERT, which apparently did not prevent their development. Combination therapy with ERT and SRT with eliglustat was implemented in the present case resulting in a significant reduction of a mesenteric Gaucheroma and improvement of clinical symptoms associated with Gaucheroma. We also reported an adult patient with GD type 3 who developed large soft tissue Gaucheromas which were promptly reduced with combination therapy of imiglucerase and eliglustat. 6 These two cases suggest that combination therapy with imiglucerase and eliglustat may inhibit Gaucher cells from growing to develop Gaucheromas. Although Mhanni et al reported that combination therapy with SRT with miglustat and ERT was not effective in their case with calcified mesenteric lymphadenopathy, our patient presented here clearly has demonstrated the effectiveness of combination therapy with eliglustat. 5 Smid et al reported that patients with GD treated with eliglustat had a greater biochemical response than miglustat, and the markers associated with GD responded more to eliglustat than to miglustat, 7 which may explain what we observed in the proposita.
Table 1. Summary of the reported patients with Gaucheroma including the proposita ( n = 24) .
| Type 1 | Type 3 | Unknown | |||
|---|---|---|---|---|---|
| 1. Gaucher's disease | 6 (25%) | 15 (63%) | 3 (12%) | Total 24 | |
| Yes | No | Unknown | |||
| 2. Enzyme replacement therapy a | 22 (92%) | 0 (0%) | 2 (8%) | ||
| Mesenteric | Mediastinal | Liver | Cervical/axial LN | Osseous/soft tissue | |
| 3. Primary site of Gaucheroma b | 17 (71%) | 7 (29%) | 3 (13%) | 3 (13%) | 4 (17%) |
| ≦ 5 y | 5 < – ≦ 10 y | 10 y < | Unknown | ||
| 4. Age at diagnosis of Gaucheroma | 13 (54%) | 5 (21%) | 5 (21%) | 1 (4%) | |
| L444P | R359Q | K79N | N370S | Unknown | |
| 5. Genotype of GBA c | 29 (60%) | 4 (8%) | 1 (2%) | 1 (2%) | 13 (28%) |
Abbreviation: LN, lymph nodes.
All patients except one received 60 IU/kg/2 weeks. One patient received 100 IU/kg/2 weeks for 10 years and combination therapy with miglustat for 4 years until the diagnosis of Gaucheroma was made. Dose unknown for four patients before the diagnosis of Gaucheroma.
Some patients had multiple primary sites at diagnosis.
Percentage is calculated based on the total 48 haploids ( n = 24).
Considering that most cases of Gaucheromas had childhood onset, implementation of SRT in early childhood may be necessary for early prevention. The mechanism for developing Gaucheromas in patients treated with ERT remains unclear. Boven et al reported that only 5% of Gaucher cells expressed the mannose receptors. These mannose receptors, which are expressed on the surface of macrophages, mediate cellular uptake of imiglucerase to the lysosome. This may partially explain why Gaucheromas are not responding to ERT. 8 Based on our observation, eliglustat was successfully taken up by Gaucheromas in the proposita as well as the case reported by Mahajan et al. 6 Hearing loss has been described in patients with GD, particularly common in school age children with type 3. It has been thought that Gaucher cell infiltration in the middle ear results in bilateral conductive hearing loss and accumulation of glucocerebroside in the inner hair cells gives rise to sensorineural hearing loss. 9 10 Combination therapy of ERT and eliglustat may be a consideration to prevent hearing impairment in children with GD.
SRT is based on inhibition of synthesis of glucocerebroside which is one of the basic glycosphingolipids and a precursor of most of the more complex glycosphingolipids including GM1 and GM2 gangliosides and globotriaosylceramide. Gangliosides act as distinguishing cell surface markers and involve cell-to-cell communication. Globotriaosylceramide is considered to be a critical molecule in mediating apoptosis signals. 11 Although ERT has been widely used in pediatric patients with GD, the safety of eliglustat in pediatric patients has not been well established. A phase 3 clinical trial to evaluate safety and efficacy in pediatric patients with GD has been ongoing. Since SRT was initiated in addition to ERT in our case, it is difficult to evaluate efficacy of eliglustat monotherapy on Gaucheromas. Further studies are indicated to evaluate whether combination therapy with ERT and SRT is more efficacious than SRT monotherapy for Gaucheromas.
In conclusion, an adult case of GD type 3 with a mesenteric Gaucheroma is presented. Although combination therapy with ERT and SRT with miglustat did not show clear effects on calcified mesenteric Gaucheromas in a case reported by Mhanni et al, 5 our case as well as the case reported by Mahajan et al 6 showed an encouraging effectiveness of combination therapy with eliglustat. Since Gaucheromas can cause serious clinical consequences, prevention of the development of Gaucheromas is important. Most patients with GD who developed Gaucheromas were treated with ERT alone; therefore, combination therapy with ERT and eliglustat should be considered in early childhood.
Acknowledgment
The authors thank the patient and the family for allowing us to submit this case report.
Funding Statement
Funding None.
Conflict of Interest None declared.
Note
Written informed consent was obtained from the patient for publication of this case report.
Ethical Approval
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Declaration of Helsinki 1975. Informed consent was obtained from the patients (and/or the parents) reported in this case report and the patient agreed with the content and with submitting this case report for consideration for publication in this journal.
The report of single case without identifiable information in the case report not requiring Institutional Review Board approval or ethics approval from the University of Southern California.
References
- 1.Lim A KP, Vellodi A, McHugh K. Mesenteric mass in a young girl--an unusual site for Gaucher's disease. Pediatr Radiol. 2002;32(09):674–676. doi: 10.1007/s00247-002-0761-0. [DOI] [PubMed] [Google Scholar]
- 2.Tseng S Y, Niu D M, Chu T H. Very rare condition of multiple Gaucheroma: a case report and review of the literature. Mol Genet Metab Rep. 2019;20:100473. doi: 10.1016/j.ymgmr.2019.100473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahajan N, Brynes R, Yano S. Large soft-tissue masses in an adult patient with Gaucher disease. J Inherit Metab Dis. 2016;39(06):887–888. doi: 10.1007/s10545-016-9966-5. [DOI] [PubMed] [Google Scholar]
- 4.Ye Z X, Gao X, Qu Q, Ye X, He X D. Gaucher disease with mesenteric lymphadenopathy: a case with 13-year follow-up. Chin Med J (Engl) 2016;129(20):2502–2503. doi: 10.4103/0366-6999.191825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mhanni A A, Kozenko M, Hartley J N, Deneau M, El-Matary W, Rockman-Greenberg C. Successful therapy for protein-losing enteropathy caused by chronic neuronopathic Gaucher disease. Mol Genet Metab Rep. 2015;6:13–15. doi: 10.1016/j.ymgmr.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahajan N, Warren M, Yano S. Reduction of large soft-tissue Gaucheromas with substrate reduction therapy. J Inherit Metab Dis. 2020;43(02):375–376. doi: 10.1002/jimd.12188. [DOI] [PubMed] [Google Scholar]
- 7.Smid B E, Ferraz M J, Verhoek M. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J Rare Dis. 2016;11:28. doi: 10.1186/s13023-016-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boven L A, van Meurs M, Boot R G. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am J Clin Pathol. 2004;122(03):359–369. doi: 10.1309/BG5V-A8JR-DQH1-M7HN. [DOI] [PubMed] [Google Scholar]
- 9.Endo S, Mizuta K, Yamatodani T. A case of improved hearing with cochlear implantation in Gaucher disease type 1. Auris Nasus Larynx. 2018;45(03):603–607. doi: 10.1016/j.anl.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Khan A, Stimpson P, Karmolinski A, Patel N. Middle-ear involvement in type I Gaucher's disease - a unique case. J Laryngol Otol. 2013;127(12):1226–1229. doi: 10.1017/S0022215113002521. [DOI] [PubMed] [Google Scholar]
- 11.Kojima Y, Fukumoto S, Furukawa K. Molecular cloning of globotriaosylceramide/CD77 synthase, a glycosyltransferase that initiates the synthesis of globo series glycosphingolipids. J Biol Chem. 2000;275(20):15152–15156. doi: 10.1074/jbc.M909620199. [DOI] [PubMed] [Google Scholar]