

Abstract
Nephrotic syndrome (NS) associated with autosomal recessive congenital ichthyosis (ARCI) is a rare association. In this article, we described a 4-year-old boy with steroid-resistant NS (SRNS) who had a history of ichthyotic skin lesions since birth. Renal biopsy revealed focal segmental glomerulosclerosis (tip variant). The skin biopsy was consistent with the findings of ichthyosis. Next-generation sequencing revealed a homozygous pathogenic variant (c.1625_1626del) in the exon 12 of the ALOX12B gene, confirming the diagnosis of ARCI2. The ALOX12B gene belongs to the lipoxygenase family and has a pivotal role in the formation of lipid layers in the epidermis. Leukotrienes have a counter-regulatory effect within the inflamed glomeruli, which influences the vascular tone and glomerular basement membrane permeability, that can be implicated in the pathogenesis of the NS. This child is currently in remission, on tacrolimus and low-dose prednisolone, with emollients and is on regular follow-up. SRNS associated with congenital ichthyosis secondary to a mutation in the ALOX12B gene has never been reported so far. The knowledge regarding this novel association will help the treating physicians in diagnosing this condition early, which will enable proper genetic counseling and prognostication of the disease to the family.
Keywords: autosomal recessive congenital ichthyosis, calcineurin inhibitors, focal segmental glomerulosclerosis, nephrotic syndrome
Introduction
Nephrotic syndrome (NS) associated with autosomal recessive congenital ichthyosis (ARCI)-2 (ARCI2, OMIM 242100) is a rare association due to mutation in the ALOX12B gene. NS is a common glomerular disease in children characterized by massive proteinuria, hypoalbuminemia, edema, and hypercholesteremia. 1 2 ARCI are inherited disorders due to the mutation in the genes responsible for skin barrier function. 3 They are a heterogeneous group of nonsyndromic ichthyosis with disorders of keratinization, clinically manifesting as abnormal scaly skin all over the body. 4 This disorder is restricted to the skin with the majority having severe cutaneous manifestations. The reported annual incidence of NS is 1.15 to 16.9 per 100,000 children. 5 The estimated prevalence of ARCI varies from one case per 139,000 to 300,000 individuals. 6 The disease frequency can be affected by the degree of consanguinity. There are three predominant ARCI clinical phenotypes, depending on the degree of severity, ranging from harlequin ichthyosis—which is the most severe form to congenital ichthyosiform erythroderma and lamellar ichthyosis. 7 The association of NS with X-linked ichthyosis (XLI) has also been reported in literature. 8 9 10 However, the association of NS and ARCI has never been reported till date to the best of our knowledge. We report a unique case of ARCI2 who had a pathogenic homozygous mutation in the ALOX12B gene and presented with steroid-resistant NS (SRNS) with a good response to calcineurin inhibitors.
Case Presentation
Case 1
A 4-year-old boy, born of the nonconsanguineous parents, presented with slowly progressive swelling of the whole body for the past 15 days and he was diagnosed with the first episode of NS (based on hypoalbuminemia, hypercholesterolemia, and nephrotic range proteinuria) and was started on prednisolone at 2 mg/kg/day; however, he did not attain remission despite 6 weeks of full dose steroids. He was considered as a case of SRNS and was started on tacrolimus (0.1 mg/kg/day) on which he attained remission at 7 weeks. He also had skin lesions since birth in the form of dark brown and scaly lesions all over the body, which were more evident over flexural aspects of the limbs.
On physical examination, the patient's height was 94 cm (−3.53 SD) and weighed 16.6 kg (−0.77 SD). There was mild pallor, no icterus/cyanosis/clubbing. Blood pressure was 114/78 mm Hg (>95th percentile). Skin examination showed large polygonal dark brown scales all over the body, which were more prominent on the axilla and flexor aspects of upper and lower limbs ( Fig. 1 ).
Fig. 1.
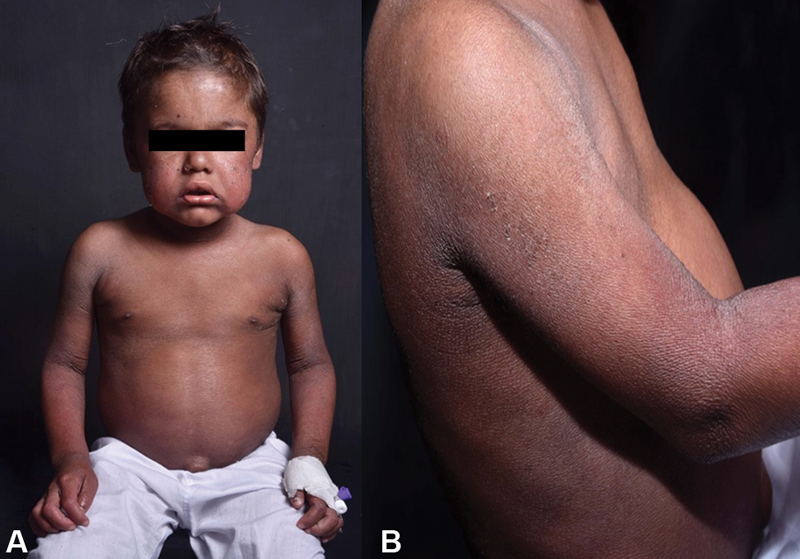
( A & B ) Skin examination showed large polygonal dark brown scales all over the body which were more prominent on the axilla and flexor aspects of upper and lower limbs.
Investigations revealed normal hemogram (hemoglobin: 13.6 g/dL; white blood cell count: 7.35 × 10 9 /L; platelets: 462 × 10 3 /L), blood urea: 49 mg/dL, serum creatinine: 0.2 mg/dL, total protein: 3.6 g/dL, serum albumin: 1.1 g/dL, total cholesterol: 425 mg/dL, 24 hour urinary protein of 59 mg/m 2 /h. Serum complement levels and antinuclear antibodies were within normal limits and viral markers for human immunodeficiency virus, hepatitis B, and C were negative. Ultrasound of kidney, ureters, and bladder was normal.
Renal Biopsy
Renal biopsy showed a maximum of 20 glomeruli per tissue section, which showed a segmental increase in mesangial matrix and hypercellularity. One of the glomeruli showed tip lesion with underlying tuft showing the presence of foam cells. Tubules showed a patchy mild acute tubular necrosis. There was no tubular atrophy seen. Interstitium and blood vessels did not show any significant pathology. Immunofluorescence was done with the panel of immunoglobulins (IgG, IgA, and IgM), complements (c1q, c3), and both light chains were negative. Electron microscopy confirmed the absence of deposits and showed the global effacement of foot processes. These findings were indicative of focal segmental glomerulosclerosis (FSGS) (tip variant; Fig. 2 ).
Fig. 2.
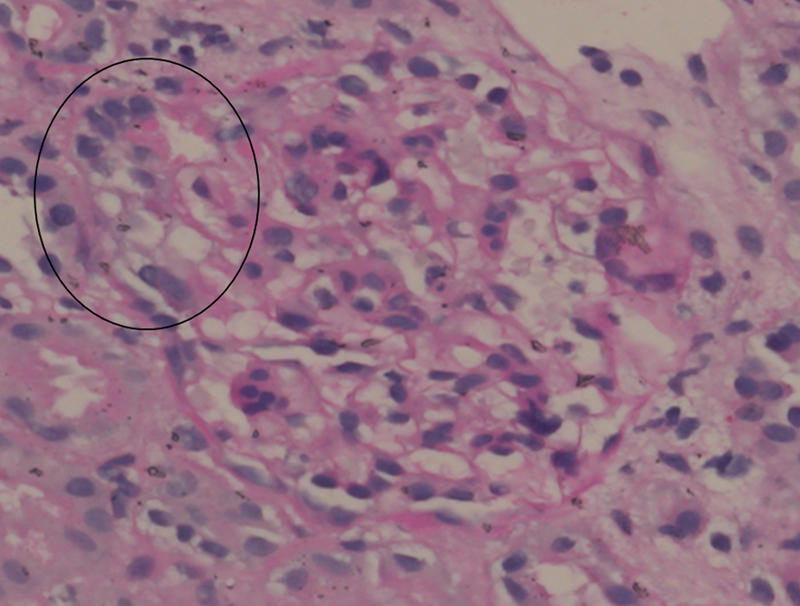
Photomicrograph shows the obliterated tip region of a glomerulus with intracapillary foam cells and overlying hypertrophied podocytes (encircled), and mild mesangial hypercellularity in rest of the glomerulus (periodic acid Schiff ×40, original magnification).
Skin Biopsy
Skin biopsy revealed epidermis with hyperkeratosis and orthokeratosis with focal loss of granular layer. Dermis showed mild perivascular lymphomononuclear inflammatory infiltrate with no features of vasculitis, dysplasia, or malignancy. The skin biopsy was consistent with the findings of ichthyosis ( Fig. 3 ).
Fig. 3.
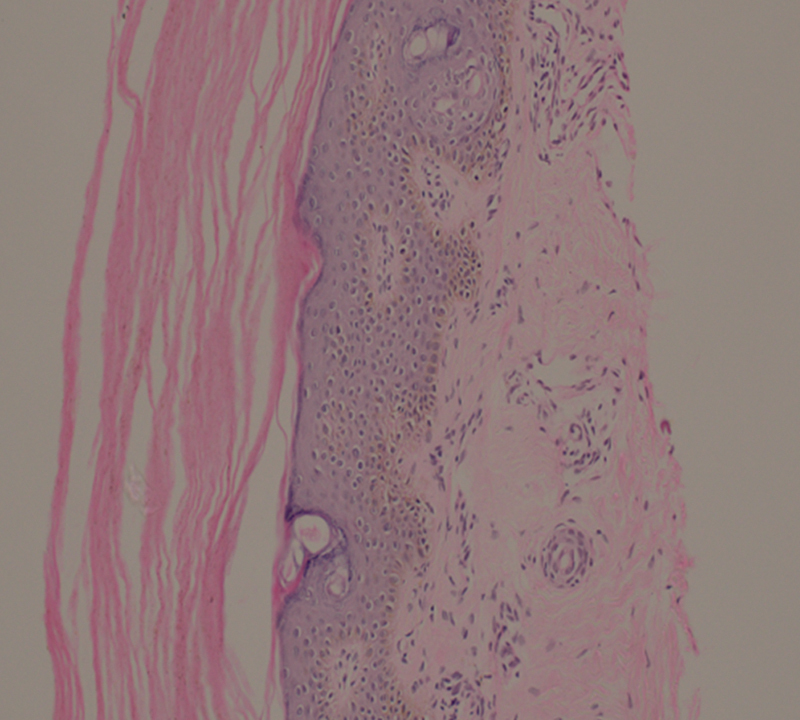
Photomicrograph of skin biopsy shows hyperkeratosis and loss of granular layer in the epidermis and perivascular mild lymphomononuclear infiltrate in absence dysplasia consistent with ichthyosis (hematoxylin and eosin ×20, original magnification).
Genetic Analysis
Genetic analysis was done using next-generation sequencing (NGS). Using a custom capture kit, DNA was used to perform targeted gene capture. On the Illumina sequencing platform, the libraries were sequenced to mean >80 to 100× coverage. Using Sentieon (v201808.07), the Genome Analysis Toolkit—best practices framework for the identification of variants in the sample—was followed. Using Sentieon aligner, the sequences obtained were aligned to the human reference genome (GRCh38.p13) and analyzed using Sentieon for removing duplicates, recalibration, and realignment of indels. To identify variants that were relevant to the clinical indication, the Sentieon haplotype caller was used. Using the VEP program against the Ensembl release 99 human gene model, gene annotation of the variants was executed. Using published variants in literature and a set of diseases databases––ClinVar, OMIM, GWAS, HGMD (v2019.4), and SwissVar, clinically relevant mutations were interpreted. Using multiple algorithms such as PolyPhen-2, SIFT, MutationTaster2, and LRT, the nonsynonymous variant effect was calculated.
NGS revealed a homozygous pathogenic variant (c.1625_1626del) in the exon 12 of the ALOX12B gene. This mutation results in a frameshift and premature truncation of the protein 13 amino acids downstream to codon 542 (p.Lys542ArgfsTer13; ENST00000647874.1). The reference region is conserved across mammals. Both the parents were the carrier for the same variant. This variant has been reported previously in ARCI by Eckl et al. 11
Final Diagnosis and Treatment
The final diagnosis of SRNS with ARCI2 was made, and the child was started on tacrolimus (0.1 mg/kg/day) and low dose-steroids (0.3 mg/kg) and he achieved remission at 7 weeks of therapy. His blood pressure was controlled on enalapril. At 2-year follow-up, he did not have any further relapses. For ichthyosis, he is on supportive care with emollients. Parents were counseled about the nature of the illness and recurrence during subsequent pregnancies.
Case 2
Our patient’s elder brother had similar skin lesions (dark brown, polygonal scaly lesions) all over the body since birth and developed swelling of the body with decreased urine output at 6 months of age. He was admitted to a local hospital and was treated as a case of NS. He succumbed to his illness at 7 months of age secondary to infection-related issues. The genetic evaluation could not be performed in this child.
Discussion
This boy had ARCI and later developed SRNS. Genetic analysis identified a homozygous pathogenic variant in the ALOX12B gene. Concurrence of ARCI and NS has been never described in the literature, and it could be merely a chance association. However, the association of congenital ichthyosis has been previously reported and we did not find any variation in the genes associated with NS, and we considered this as a novel association. A genetic mutation has been described to be seen in up to 30% of patients with primary NS (PNS), although the exact incidence is not known. 12 More than 50 genes are currently associated with NS; among them, the three most common genes— NPHS1 , NPHS2 , and WT1 —associated with NS which encode important podocyte proteins. 13 Only four cases with NS and congenital ichthyosis have been reported so far, and three out of four had a large deletion in the STS gene. However, the association of NS and ARCI2 has never been reported in the literature.
ARCI is a nonsyndromic type of ichthyosis, and mutations in at least a dozen of genes have been described until date, including TGM , ABCA12 , NIPAL4 , CYP4F22 , ALOX12B , ALOXE3 , NIPAL4 , CERS3 , SDR9C7 , PNPLA1 , and SULT2B1 . 14 15 ALOX12B gene is located in chromosome 17p13.1, belongs to the lipoxygenase family plying a significant role in the formation of lipid layers in the epidermis. 16 Arachidonic acid is oxidized by the primary enzyme, 5-lipooxygenase (5-LO), and converted to leukotriene (LT) A 4 (LTA 4 ) in the lipoxygenase pathway. 17 LTs of the 5-LO pathway are potent mediators of inflammation, and it has been implicated to have a potential role in the early damage of glomerular structure and function in experimental glomerulonephritis. 18 Adverse effects of LT over the glomerular microcirculation manifests in the form of vasoconstriction or neutrophil infiltration. 19 Vasoactive arachidonate metabolites have a role in the pathogenesis of proteinuria as LTs have a counter-regulatory effect on the inflamed glomeruli, which influences the vascular tone and glomerular basement membrane permeability. 20 During glomerular inflammation, LTs have been detected in the urine, serum, and renal tissue. 21 22 Bakr et al 23 found a significant correlation between the expression of 5-LO and LTA 4 hydrolase and degree of proteinuria in active NS cases. Similarly, Rifai et al 24 found that proteinuria was significant in patients who express 5-LO and LTA 4 hydrolase when compared with those who had no enzyme expression in the renal tissue.
Menegatti et al 17 investigated the role of LT in the pathogenesis of glomerulonephritis with NS. In the renal tissues of patients with membranous nephropathy (MN), non-IgA mesangial glomerulonephritis, minimal change disease (MCD), and FSGS, the gene expression of 5-LO and LTA 4 hydrolase was examined. Co-expression of 5-LO and LTA 4 hydrolase was seen in four patients of FSGS and four patients of MN. The distribution and expression of 5-LO were limited to interstitial cells surrounding the peritubular capillaries, which were studied by in situ hybridization. Impaired renal function and changes in the interstitium were seen in these eight patients, which correlated with the 5-LO expression when comparing the clinical and immunohistological data. Their study findings suggested that LT might play a pivotal role in the pathogenesis of FSGS and MN.
In peripheral blood mononuclear cells of 48 children with active PNS, 27 children in remission, and 20 controls, Bakr et al 23 studied the gene expression of 5-LO and LTA 4 hydrolase. All patients in the active PNS group expressed 5-LO and LTA 4 hydrolase and none in the controls. In the remission group, 5-LO was expressed in 22.2%, while LTA 4 hydrolase was expressed in 51.9%. The degree of proteinuria and the expression of 5-LO ( r = 0.27, p = 0.03) and LTA 4 hydrolase ( r = 0.44, p = 0.001) was found to be significantly correlated among the active PNS group. They concluded that LT might have a role in the pathogenesis of PNS in children.
The NS in patients with XLI has been reported in the literature ( Table 1 ). NS in a case of congenital ichthyosis was first described by McGrae 25 in a 24-year-old female with keratitis, ichthyosis, and deafness (KID) syndrome. At the age of 9 years, she developed the NS. She was born of a nonconsanguineous marriage with two normal siblings and no similar illness in the family. She has scaly and red skin lesions since birth which progressed to erythematous keratoderma in early adolescence. Sensorineural hearing loss is congenital or present in early infancy in KID. They did not perform genetic analysis or renal biopsy.
Table 1. Summary of patients with congenital ichthyosis and nephrotic syndrome.
| Cases | Age/Sex | Age of onset of NS | Involved gene | Type of mutation | Renal biopsy | Response to treatment |
|---|---|---|---|---|---|---|
| 1 | 24 years/F | 9 years | NA | NA | NA | NA |
| 2 | 8 years/M | 6 years | STS | Deletion of exons 1–10 | MCD | No response to steroids, cyclophosphamide and cyclospsorine |
| 3 | 10 years/M | 10 years | STS | Deletion of exons 1–10 | NA | Responded to prednisolone |
| 4 | 4.5 years/M | 4.5 years | STS | Deletion of exons 1–10 | MCD | Responded to cyclosporine |
| 5 | 7 months/M | 6 months | NA | NA | NA | Died |
| 6* | 4 years/M | 4 years | ALOX12B | Frameshift mutation in exon 12 (c.1625_1626delAA) | FSGS | Responded to prednisolone and tacrolimus |
Abbreviations: ALOX12B, arachidonate 12-lipoxygenase, 12R type; F, female; FSGS, focal segmental glomerulosclerosis; M, male; MCD, minimal change disease; NA, not available; NS, nephrotic syndrome; STS, steroid sulfatase. ∗ Index child.
Matsukura et al 8 reported SRNS associated with XLI in an 8-year-old boy. This boy had congenital ichthyosis and NS developed at 6 years of age. Renal biopsy was suggestive of MCD. Genetic analysis showed a large deletion in the STS gene. The child did not respond to all immunosuppressive therapy given (i.e., steroids, cyclophosphamide, and cyclosporine) and he, later on, progressed to end-stage renal disease and renal transplantation was done.
Krishnamurthy et al 9 reported a 10-year-old boy with XLI, Kallmann syndrome, and unilateral left renal agenesis with NS. The onset of NS was at 10 years of age, and genetic evaluation revealed a large deletion of the STS gene. He was born from a nonconsanguineous parents, but had a significant family history of ichthyotic skin lesions in other family members. With oral prednisolone alone, his NS went into remission.
Mishra et al 10 reported male boy with NS associated with XLI due to steroid sulfatase deficiency. He had ichthyotic skin lesions since birth and developed NS at the age of 4.5 years. He was born from the third-degree consanguineous parents with a similar history of ichthyotic skin lesions in the family. His renal biopsy showed MCD and he achieved remission with cyclosporine. The deletion of the STS gene was revealed in genetic analysis.
The association of XLI with NS in all these three patients cannot be attributed to chance association alone. STS gene plays a pivotal role in the regulation of skin barrier permeability and desquamation since hydrolyses cholesterol sulfates to their unconjugated forms, cholesterol, and sulfate. Cholesterol sulfate accumulates in the stratum corneum which leads to hyperkeratosis, formation of scales, and impaired skin permeability in the absence of STS activity. Pathogenesis of XLI has been proposed to be secondary to cholesterol sulfate-induced transglutaminase 1 (TGM1) dysfunction. 26 The expression and activity of TGM1 in the keratinocytes are known. Hiiragi et al 27 conducted a study to identify the role of TGM1 in other cell types and found that large amounts of TGM1 were expressed in the epithelial tissues of the liver, kidney, and lungs. TGM1 was also concentrated around the cadherin-based adherens junction in the liver and kidney, and at the intercellular junctions of simple epithelial cells, transglutaminase cross-linking activity was seen to be concentrated, which suggest the role of TGM1 in the maintenance of epithelial cells structural integrity, particularly at the cadherin-based adherens junction. Reiser et al 28 had described the slit diaphragm of the glomerular capillary wall to a modified adherens junction. Therefore, TGM1 dysfunction in XLI patients can disrupt the normal function of slit diaphragm leading to proteinuria, which can explain the association of NS in patients with XLI.
Conclusion
To conclude, the association of congenital ichthyosis and NS is rarely described in the literature. The previously reported cases were associated with a large deletion in the STS gene. SRNS associated with congenital ichthyosis secondary to a mutation in the ALOX12B gene has never been reported so far. The knowledge regarding this novel association will help the treating physicians in diagnosing this condition early, which will enable proper genetic counselling and prognostication of the disease to the family.
Funding Statement
Funding None.
Conflict of Interest None declared.
Note
Informed consent was obtained from the parents of the child. This article does not contain any studies with human participants or animals performed by any of the authors.
Authors' Contributions
L.D. collected clinical data, designed the study, reviewed the literature, and wrote the manuscript. A.K. performed and analyzed the genetic analysis. R.N. examined and reviewed the renal biopsy. S.C. collected clinical data. S.H. supervised overall concept design and critical review of the article for important intellectual content and final approval of the manuscript. I.K.S. supervised overall concept design, critical review of the article for important intellectual content, and final approval of the manuscript. K.T. supervised overall concept design, critical review of the article for important intellectual content, and final approval of the manuscript. All the authors revised and approved the final version of the manuscript.
References
- 1.Sharawat I K, Dawman L, Kumkhaniya M V, Devpura K, Mehta A. Bone mineral density and its influencing factors in children with idiopathic nephrotic syndrome: a prospective observational study. Trop Doct. 2019;49(04):292–298. doi: 10.1177/0049475519868055. [DOI] [PubMed] [Google Scholar]
- 2.Dawman L, Mehta A, Sharawat I K, Yadav R. Risk factors for steroid dependency in children with idiopathic nephrotic syndrome in India. Indian J Pediatr. 2016;83(03):261. doi: 10.1007/s12098-015-1819-y. [DOI] [PubMed] [Google Scholar]
- 3.Cortés H, Del Prado-Audelo M L, Urbán-Morlán Z. Pharmacological treatments for cutaneous manifestations of inherited ichthyoses. Arch Dermatol Res. 2020;312(04):237–248. doi: 10.1007/s00403-019-01994-x. [DOI] [PubMed] [Google Scholar]
- 4.Vahlquist A, Fischer J, Törmä H. Inherited nonsyndromic ichthyoses: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2018;19(01):51–66. doi: 10.1007/s40257-017-0313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khullar S, Banh T, Vasilevska-Ristovska J. Impact of steroids and steroid-sparing agents on quality of life in children with nephrotic syndrome. Pediatr Nephrol. 2021;36(01):93–102. doi: 10.1007/s00467-020-04684-3. [DOI] [PubMed] [Google Scholar]
- 6.González-Del Carmen M, Montaño S, Reyes-Hernández O D. High prevalence of autosomal recessive congenital ichthyosis in a Mexican population caused by a new mutation in the TGM1 gene: epidemiological evidence of a founder effect. Int J Dermatol. 2020;59(08):969–977. doi: 10.1111/ijd.14952. [DOI] [PubMed] [Google Scholar]
- 7.Takeichi T, Akiyama M. Inherited ichthyosis: non-syndromic forms. J Dermatol. 2016;43(03):242–251. doi: 10.1111/1346-8138.13243. [DOI] [PubMed] [Google Scholar]
- 8.Matsukura H, Fuchizawa T, Ohtsuki A. End-stage renal failure in a child with X-linked ichthyosis. Pediatr Nephrol. 2003;18(03):297–300. doi: 10.1007/s00467-002-1042-8. [DOI] [PubMed] [Google Scholar]
- 9.Krishnamurthy S, Kapoor S, Yadav S. Nephrotic syndrome with X-linked ichthyosis, Kallmann Syndrome and unilateral renal agenesis. Indian Pediatr. 2007;44(04):301–303. [PubMed] [Google Scholar]
- 10.Mishra K, Batra V V, Basu S, Rath B, Saxena R. Steroid-resistant nephrotic syndrome associated with steroid sulfatase deficiency-x-linked recessive ichthyosis: a case report and review of literature. Eur J Pediatr. 2012;171(05):847–850. doi: 10.1007/s00431-012-1712-x. [DOI] [PubMed] [Google Scholar]
- 11.Eckl K-M, de Juanes S, Kurtenbach J. Molecular analysis of 250 patients with autosomal recessive congenital ichthyosis: evidence for mutation hotspots in ALOXE3 and allelic heterogeneity in ALOX12B. J Invest Dermatol. 2009;129(06):1421–1428. doi: 10.1038/jid.2008.409. [DOI] [PubMed] [Google Scholar]
- 12.SRNS Study Group . Sadowski C E, Lovric S, Ashraf S. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26(06):1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bierzynska A, McCarthy H J, Soderquest K. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91(04):937–947. doi: 10.1016/j.kint.2016.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Oji V, Tadini G, Akiyama M. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol. 2010;63(04):607–641. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Heinz L, Kim G-J, Marrakchi S. Mutations in SULT2B1 cause autosomal-recessive congenital ichthyosis in humans. Am J Hum Genet. 2017;100(06):926–939. doi: 10.1016/j.ajhg.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jobard F, Lefèvre C, Karaduman A. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet. 2002;11(01):107–113. doi: 10.1093/hmg/11.1.107. [DOI] [PubMed] [Google Scholar]
- 17.Menegatti E, Roccatello D, Fadden K. Gene expression of 5-lipoxygenase and LTA4 hydrolase in renal tissue of nephrotic syndrome patients. Clin Exp Immunol. 1999;116(02):347–353. doi: 10.1046/j.1365-2249.1999.00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badr K F. Glomerulonephritis: roles for lipoxygenase pathways in pathophysiology and therapy. Curr Opin Nephrol Hypertens. 1997;6(02):111–118. [PubMed] [Google Scholar]
- 19.Badr K F. Five-lipoxygenase products in glomerular immune injury. J Am Soc Nephrol. 1992;3(04):907–915. doi: 10.1681/ASN.V34907. [DOI] [PubMed] [Google Scholar]
- 20.O'Meara Y M, Brady H R. Lipoxins, leukocyte recruitment and the resolution phase of acute glomerulonephritis. Kidney Int Suppl. 1997;58:S56–S61. [PubMed] [Google Scholar]
- 21.Lefkowith J B, Pippin J, Nagamatsu T, Lee V. Urinary eicosanoids and the assessment of glomerular inflammation. J Am Soc Nephrol. 1992;2(11):1560–1567. doi: 10.1681/ASN.V2111560. [DOI] [PubMed] [Google Scholar]
- 22.Yared A, Albrightson-Winslow C, Griswold D, Takahashi K, Fogo A, Badr K F. Functional significance of leukotriene B4 in normal and glomerulonephritic kidneys. J Am Soc Nephrol. 1991;2(01):45–56. doi: 10.1681/ASN.V2145. [DOI] [PubMed] [Google Scholar]
- 23.Bakr A, Hawas S, Slem S, Moniem A A, Ghatab T, Tawfik M. 5-Lipoxygenase and leukotriene A4 hydrolase expression in primary nephrotic syndrome. Pediatr Nephrol. 2004;19(04):396–399. doi: 10.1007/s00467-003-1399-3. [DOI] [PubMed] [Google Scholar]
- 24.Rifai A, Sakai H, Yagame M. Expression of 5-lipoxygenase and 5-lipoxygenase activation protein in glomerulonephritis. Kidney Int Suppl. 1993;39:S95–S99. [PubMed] [Google Scholar]
- 25.McGrae J D., Jr Keratitis, ichthyosis, and deafness (KID) syndrome with adult onset of keratitis component. Int J Dermatol. 1990;29(02):145–146. doi: 10.1111/j.1365-4362.1990.tb04090.x. [DOI] [PubMed] [Google Scholar]
- 26.Nemes Z, Demény M, Marekov L N, Fésüs L, Steinert P M. Cholesterol 3-sulfate interferes with cornified envelope assembly by diverting transglutaminase 1 activity from the formation of cross-links and esters to the hydrolysis of glutamine. J Biol Chem. 2000;275(04):2636–2646. doi: 10.1074/jbc.275.4.2636. [DOI] [PubMed] [Google Scholar]
- 27.Hiiragi T, Sasaki H, Nagafuchi A. Transglutaminase type 1 and its cross-linking activity are concentrated at adherens junctions in simple epithelial cells. J Biol Chem. 1999;274(48):34148–34154. doi: 10.1074/jbc.274.48.34148. [DOI] [PubMed] [Google Scholar]
- 28.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11(01):1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]