

Abstract
Osteopathia striata with cranial sclerosis is an X-linked dominant bone dysplasia with osteosclerosis. It should be suspected in girls with macrocephaly, intellectual disability with unique facial dysmorphic features. We described the clinical and radiological profile of a patient with this rare disorder. A novel heterozygous variant was identified in the AMER1 gene which leads to premature truncation of the AMER1 protein. Facial gestalt recognition using artificial intelligence and radiographic features were used to narrow the differential diagnosis.
Keywords: AMER1, Face2Gene, osteopathia striata with cranial sclerosis
Introduction
Osteopathia striata with cranial sclerosis (OSCS) (MIM:300373) is an X-linked dominant skeletal dysplasia with osteosclerosis caused by pathogenic variants in the AMER1 gene (Xq11). It is present in females with macrocephaly, mild learning disabilities, sclerosis of the long bones and skull, longitudinal striations on the radiographs and rarely cleft palate. 1 The disorder is lethal in males and the occasionally surviving male exhibits severe congenital malformations affecting the cardiac, intestinal, and genitourinary systems. 2 The use of artificial intelligence based on deep convolutional neural networking is gaining popularity for recognition of dysmorphic syndromes. These serve as an aid to clinicians to narrow the differential diagnosis especially in genetic disorders where the phenotype is nonspecific. 3
We presented a 12-year-old girl patient with mild intellectual disability (ID) and macrocephaly, in whom an initial provisional diagnosis of AMER1 gene-related OSCS was made with the help of the Face2Gene software. This was later confirmed both radiologically and molecularly. Thus, we add this clinically identifiable rare disorder to the list of disorders with macrocephaly, short stature, and ID, as well as put forward the important role played by artificial intelligence in reaching a quicker diagnosis.
Case Presentation
A 12-year, 7-month-old girl paient was referred to the genetics department in view of mild ID and facial dysmorphism. She was the second child born to nonconsanguineous parents and had an uneventful antenatal and birth history. Development was delayed in all domains. Head control was attained at 6 months, sitting at 10 months, and walking with support at 22 months. Fine motor development also had a concordant delay with pincer grasp achieved at 2 years and drawing circles at 3 years. Cognition and language were more severely affected. She started to speak meaningful words at the age of 3 years and could state her name and gender after the age of 6 years. Our patient attended normal school but was always behind her chronological aged peers. Her parents reported behavioral issues especially noncompliance. There is also a history of nocturnal enuresis, which is present to date. There is no history of similarly affected members in the family. She has one healthy brother and each parent has one brother each with healthy children ( Fig. 1 ).
Fig. 1.
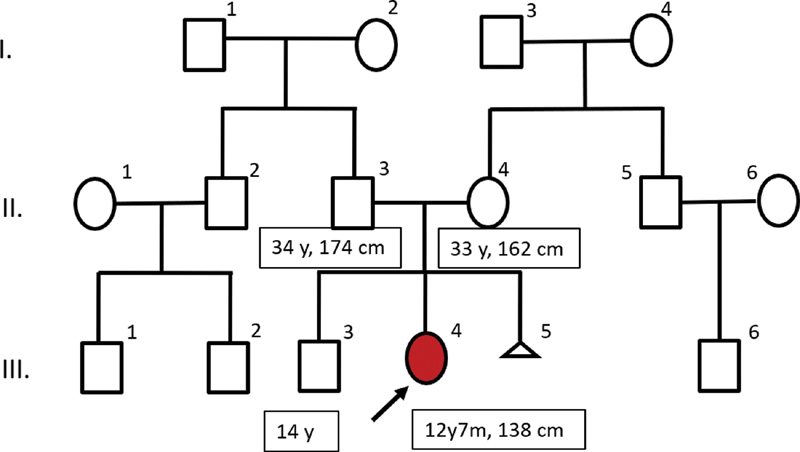
Three-generation family tree of the child (with the arrow) with osteopathia striata with cranial sclerosis. Dysmorphic facial features included a large head, frontal bossing, hypertelorism, deep-set eyes, uplifted ear lobes, low set ears, and broad nasal bridge
On examination, she was conscious, cooperative, and well oriented to time, place, and person. Anthropometry : height 138 cm (−2 standard deviation [SD]), upper segment/lower segment = 0.9, weight 34.7 kg (−0.6 SD), and head circumference (+3.2 SD) suggestive of macrocephaly. Parental head circumferences were normal. Her midparental height (MPH) is 161.5 cm (mother: 162 cm and father: 174 cm). Her predicted height is 148 cm (< − 2 SD below MPH). Dysmorphic facial features included a large head, frontal bossing, hypertelorism, deep-set eyes, uplifted ear lobes, low set ears, and broad nasal bridge ( Fig. 2A–C ). Strabismus was noted in the right eye. Bilateral rocker bottom feet with medially deviated toes were noted. Bilateral clinodactyly was present. Neurological examination revealed mild impairment of cognitive function, difficulty in calculation with normal speech and cranial nerve examination. Her tone, power, and deep tendon reflexes were normal. The rest of the systemic examination was noncontributory. At this point, several disordered were considered in the differential diagnosis, these include metabolic disorders, mucopolysaccharidosis, Primrose's syndrome, mild hypothyroidism, monogenic causes such as RASopathy, chromosomal disorders such as DiGeorge's syndrome, and skeletal dysplasias. Before deciding which genetic test to order, the following work-up was done.
Fig. 2.
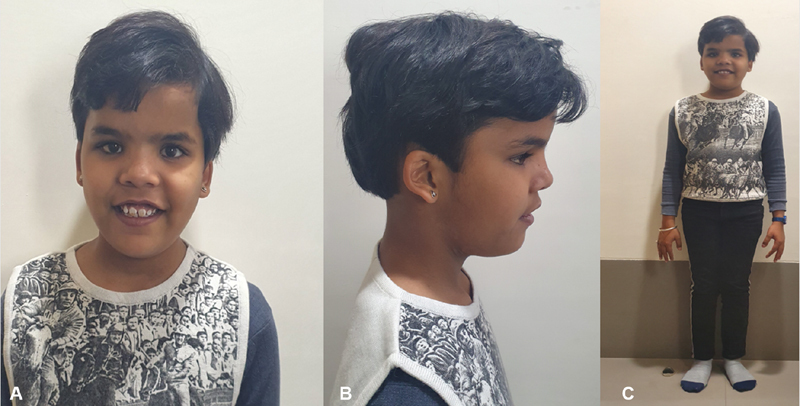
( A ) Frontal profile, ( B ) side profile of the face, and ( C ) full length photograph, showing facial dysmorphism (broad forehead, macrocephaly, hypertelorism, broad nasal bridge, low set ears, uplifted ear lobes, and large central incisors) and proportionate short stature.
Basic work-up for short stature was performed and was normal. These include fasting thyroid function test (thyroid-stimulating hormone 2.1 mU/L [1.3–3], T4 6 ng/dL [5.9–13.2]), fasting serum cortisol 12 µg/L (10–20) which was performed by radioimmunoassay and growth hormone stimulation test with clonidine (0.25 mg). X-ray of her hands revealed a bone age that was consistent with her chronological age. Formal assessment of intelligence quotient (IQ) and social quotient (SQ) by standard tests classified the patient into borderline ID (IQ 80 and SQ 79) according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria. OMIM (online Mendelian inheritance in man) search was performed and the child's facial image was entered into Face2Gene application. The top three results from our search were OSCS, KBG syndrome, and Smith–Magenis' syndrome.
In view of the differentials and the short stature, radiographs were ordered. The radiographic survey revealed severe sclerosis of the skull, sclerotic cranial base, sclerotic mastoid, occipital bossing, linear striations on the diametaphysis of femur suggestive of osteopathia striata and hypoplastic fibulae ( Fig. 3 ). Magnetic resonance imaging of the brain was performed in view of macrocephaly and ID. It revealed the presence of normal pressure hydrocephalus. Echocardiography showed a small muscular ventricular septal defect.
Fig. 3.
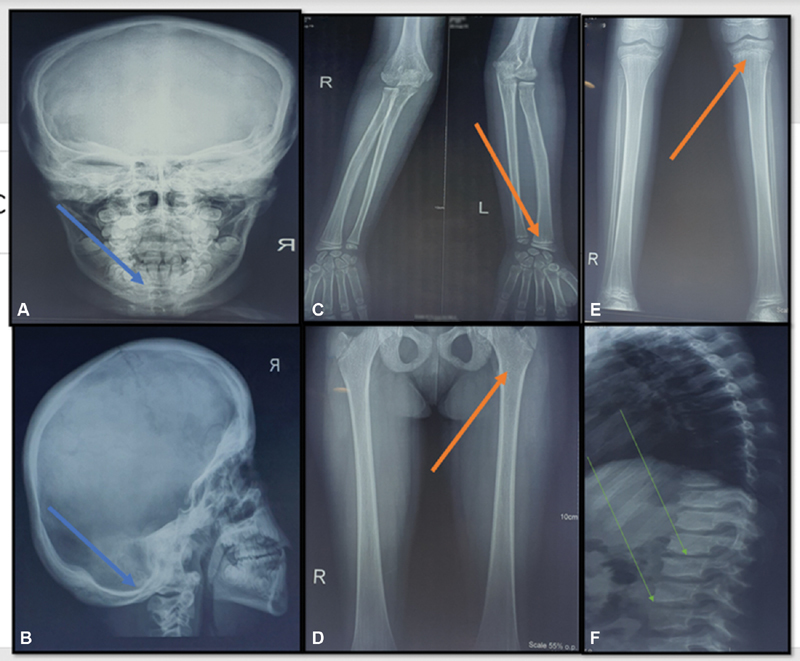
( A, B ) X-ray of skull (lateral and AP views): sclerotic mandible, cranial sclerosis (blue arrows). ( C ) X-ray of distal humerus, radius, and ulna (AP view): sclerosis of margins of diaphysis, linear striations at the distal ulna (orange arrow). ( D, E ) X-ray of knee joint, tibia, fibula, pelvis, and femur (AP view): linear striations in the shaft of femur and tibia, hypoplastic fibula (orange arrows). ( F ) X-ray of spine (lateral view): mild thoracolumbar scoliosis and irregular vertebral endplate (green arrows). AP, anteroposterior.
The radiographs, as well as the facial gestalt, were consistent with the diagnosis of OSCS. For validation of the diagnosis, targeted genetic testing using next-generation sequencing was ordered. It revealed a novel heterozygous two-base pair deletion in exon 2 of the AMER1 gene. This variant, ENST00000330258.3, c.226–227del (p.Gly76ThrfsTer25) ( Fig. 4 ) leads to premature truncation of the protein, 25 amino acids downstream of codon 76. This variant was not previously reported in the 1000 Genome or the Exome Aggregation Consortium databases and it is predicted by MutationTaster, Sorting Intolerant from Tolerant, and Protein Variation Effect Analyzer to be damaging. The reference codon is conserved across species. The variant is classified as pathogenic according to the American College of Medical Genetics classification of the variants, thus confirming the diagnosis.
Fig. 4.
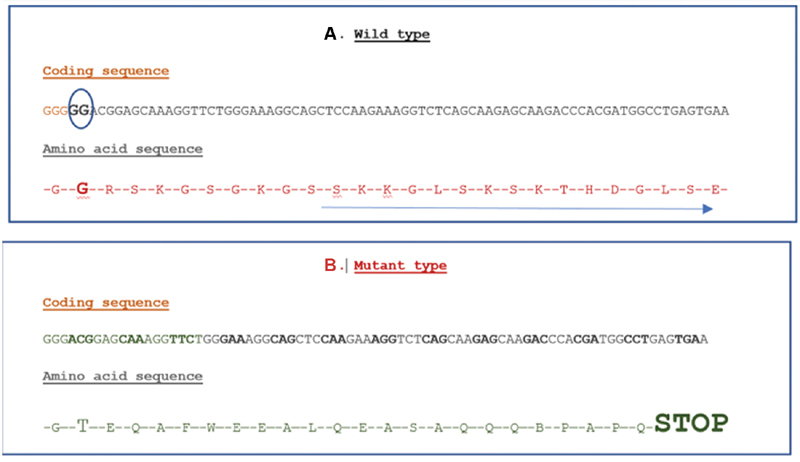
( A ) Normal coding sequence and amino acid sequence of the AMER1 gene. The blue circle around the nucleotides GG at position 226,227 denotes that these are deleted in the mutant sequence. Glycine at the 76th position of amino acid sequence is shown. ( B ) Mutant sequence with the GG being deleted, leading to a change in the reading frame and finally causing premature truncation of the protein 25 base pair downstream the 76th codon (Gly-Thr).
Discussion
OSCS is an extremely rare disorder with less than 100 patients reported worldwide. It is likely an underdiagnosed disorder due to the mild phenotypic manifestations in females. This disorder should be borne in mind by pediatricians, endocrinologists, and neurologists in the evaluation of children and adolescents with macrocephaly and ID. Further, the distinctive facial features including hypertelorism, broad nasal bridge, and abnormally formed ears are specific handles that would warrant targeted investigations for this disorder.
Disorders with macrocephaly (without overgrowth) and mild ID which could be considered in the differential diagnosis of this condition include mucopolysaccharidosis, RASopathies, Primrose's syndrome, and metabolic disorders such as glutaric acidemia type1. Endocrinal abnormalities, metabolic derangements, as well as certain chromosomal disorders can also present with a similar phenotype. The radiological survey offers an important first-line investigation and the presence of osteopathia, 4 dysostosis multiplex, pectus carinatum/kyphoscoliotic spine, epiphyseal abnormalities gives a clue to the diagnosis of OSCS, MPS, RASopathy, and Primrose's syndrome, respectively. Since the radiographs did not reveal any abnormalities related to the aforementioned syndromes, these syndromes were excluded. Findings consistent with osteopathia striata helped confirm the diagnosis.
In the current era of next-generation sequencing, variants of uncertain significance pose a challenge to making accurate genetic diagnoses. Deep phenotyping is an invaluable tool to aide variant filtration and is facilitated by clinical examination and investigations, as well as artificial intelligence tools that are now available for beside use. As most of the disorders discussed earlier have a unique facial gestalt, software such as Face2Gene help shortening the diagnostic odyssey. It is a deep learning algorithm-based syndrome-specific tool based on syndrome gestalts. This proprietary technology converts a patient's photo into deidentified mathematical facial descriptors. The patient's facial descriptors are compared with syndrome gestalts to quantify similarity (gestalt scores) resulting in a prioritized list of syndromes with similar morphology.
Most of the pathogenic variants described in the AMER1 gene are nonsense variants or deletions. In spite of the truncated protein, there exists phenotypic heterogeneity even in females. In contrast to the patient by Ward et al, 5 our patient did not give any history of pain and contractures. Other manifestations include dental abnormalities, hearing loss, cranial nerve involvement, and neuromuscular involvement. While previous studies suggested, at least in some cases, a nonrandom X-inactivation pattern to explain this phenomenon, Jenkins et al 1 demonstrated that in 19 AMER1 mutation-bearing heterozygous females, X-inactivation ratios were not skewed. The location of the variants in the different domains likely determines the phenotypic outcome in an individual.
Finally, we reiterate that although a clinical diagnosis of this condition can be reached, it is of utmost importance to obtain a genetic diagnosis, since the case of X-linked disorder diagnosis is helpful in cascade screening and provision of prenatal diagnosis. In light of newer emerging therapies, a definitive diagnosis is essential for procurement of treatment.
Conclusion
In conclusion, we described a patient with OSCS and described the distinctive radiological findings which are an important clue to the disorder. We also described a significant role for AI-driven facial recognition software in aiding the clinician to reach a quicker diagnosis.
Acknowledgments
We would like to acknowledge the patient and her family. We would also like to extend our gratitude toward MedGenome laboratories for genetic testing, and Dr. Aditi, Fellow in Developmental Pediatrics, for assisting in developmental assessment of the patient. Our sincere thanks to Dr. Renu Saxena, Head, Molecular Genetics and Dr. Ratna D. Puri, Head, Institute of Medical Genetics & Genomics for logistic and academic support.
Footnotes
Conflict of Interest None declared.
References
- 1.Jenkins Z A, van Kogelenberg M, Morgan T. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet. 2009;41(01):95–100. doi: 10.1038/ng.270. [DOI] [PubMed] [Google Scholar]
- 2.Rott H D, Krieg P, Rütschle H, Kraus C. Multiple malformations in a male and maternal osteopathia strata with cranial sclerosis (OSCS) Genet Couns. 2003;14(03):281–288. [PubMed] [Google Scholar]
- 3.Bijarnia-Mahay S, Arora V. Next generation clinical practice - it's man versus artificial intelligence! Indian Pediatr. 2019;56(12):1007–1008. [PubMed] [Google Scholar]
- 4.Zicari A M, Tarani L, Perotti D. WTX R353X mutation in a family with osteopathia striata and cranial sclerosis (OS-CS): case report and literature review of the disease clinical, genetic and radiological features. Ital J Pediatr. 2012;38:27. doi: 10.1186/1824-7288-38-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward L M, Rauch F, Travers R. Osteopathia striata with cranial sclerosis: clinical, radiological, and bone histological findings in an adolescent girl. Am J Med Genet A. 2004;129A(01):8–12. doi: 10.1002/ajmg.a.30107. [DOI] [PubMed] [Google Scholar]