

Abstract
Diabetic kidney disease (DKD) is a highly prevalent complication of diabetes and the leading cause of end‐stage kidney disease. Inflammation is recognized as an important driver of progression of DKD. Activation of the immune response promotes a pro‐inflammatory milieu and subsequently renal fibrosis, and a progressive loss of renal function. Although the role of the innate immune system in diabetic renal disease has been well characterized, the potential contribution of the adaptive immune system remains poorly defined. Emerging evidence in experimental models of DKD indicates an increase in the number of T cells in the circulation and in the kidney cortex, that in turn triggers secretion of inflammatory mediators such as interferon‐γ and tumor necrosis factor‐α, and activation of cells in innate immune response. In human studies, the number of T cells residing in the interstitial region of the kidney correlates with the degree of albuminuria in people with type 2 diabetes. Here, we review the role of the adaptive immune system, and associated cytokines, in the development of DKD. Furthermore, the potential therapeutic benefits of targeting the adaptive immune system as a means of preventing the progression of DKD are discussed.
Keywords: Adaptive immune system, Diabetic kidney disease, Inflammation
Although the role of the innate immune system in diabetic renal disease has been well characterized, the potential contribution of the adaptive immune system remains poorly defined. Here, we review the role of the T and B cells of the adaptive immune system, and associated cytokines, in the development of diabetic kidney disease.
INTRODUCTION
Diabetic kidney disease (DKD) is a serious public health problem and is the leading cause of end‐stage kidney disease (ESKD) worldwide 1 . It is a significant complication of both type 1 diabetes mellitus and type 2 diabetes mellitus. DKD is the leading cause of type 2 diabetes, with approximately 30–50% of cases attributable to diabetes 1 , 2 , 3 . The growing incidence of type 2 diabetes contributes to the increasing incidence of DKD, which is associated with increased cardiovascular complications, morbidity and mortality 4 , 5 , 6 , 7 .
The mechanisms leading to the development and progression of renal injury in diabetes are not fully understood. Confounding factors have been reported to be associated with the pathophysiology of DKD, including absolute or relative insulin deficiency 8 , oxidative stress 9 , activation of the renin–angiotensin–aldosterone system 10 , formation of polyol and advanced glycation end‐products 11 , hemodynamic alterations 12 , and variations in deoxyribonucleic acid methylation profiles 13 . The presence of proteinuria, along with elevated levels of glycated hemoglobin, systolic blood pressure and serum uric acid, and the presence of vascular comorbidities, are indicators of a faster progression of DKD 14 . Many individuals with DKD develop albuminuria 15 . The remission of albuminuria and a slowing down of the rate of loss of renal function might occur with optimization of glucose management and blood pressure control, including the use of renin–angiotensin system blockade, sodium–glucose cotransporter 2 inhibitors and glucagon‐like peptide‐1 receptor analogs 4 , 10 , 16 . However, despite these therapeutic options, DKD remains a major public health issue, not only due to the increased risk of progression to ESKD, especially in an aging population, but also because people with DKD are at a much higher risk of developing cardiovascular disease 17 , 18 , 19 , 20 . It is therefore crucial to understand the underlying mechanisms of DKD and to target these mechanisms to prevent or treat DKD.
The risk of developing DKD is associated with both systemic and the local activation of inflammatory processes 21 . Such inflammation can be triggered by stimulated renal cells and the accumulation of immune cells from both the innate and adaptive immune responses within the injured kidney, driving remodeling of renal structure and interstitial fibrosis 22 . Macrophages are phagocytic cells of the innate immune system, and are widely recognized to hasten the progression of glomerular sclerosis in experimental DKD models and clinical trials 23 , 24 , 25 . More than 90% of leukocyte infiltration in the kidney are macrophages 23 , 26 , and strategies to selectively reduce macrophage accumulation, such as knockout of macrophage chemokines or blockade of chemokine receptors, provide significant protection in mouse DKD models 27 , 28 . Growing evidence from experimental and clinical studies show that the adaptive immune system in concert with several inflammatory cytokines might also act as a key factor in the progression of renal injury in diabetes 22 , 29 , 30 .
The adaptive immune system comprises T cells and B cells. The progression of human DKD correlates with activation of T cells in the blood and increased numbers of CD4+ T cells in the kidney 21 , 31 . CD4+ T‐cell subsets have been intensively studied in DKD, including T helper (Th) 1, Th2, Th9, Th17, Th22, T regulatory cells (Tregs) and follicular helper T cells 32 . Th1 cells, but not Th2 cells, produce large amounts of cytokines, which are associated with lower levels of creatinine clearance and increasing proteinuria in people with diabetes and nephrotic syndrome (urine protein loss >3.5 g/day), as compared with those having diabetes without nephrotic syndrome 33 . The ratios of CD4 + CD25hi Tregs/Th17 cells and CD4 + CD25hi Tregs/Th1 cells are decreased in the blood of people with type 2 diabetes with reduced immunosuppressive potentiality of CD4 + CD25hi Tregs 34 . Further evaluation showed no correlation between this ratio and glycated hemoglobin in people with type 2 diabetes. Similarly, the Tregs/Th17 balance is disturbed in diabetes, with the percentage of Th17 cells being significantly higher and level of Tregs being lower in the blood of people with type 1 diabetes compared with healthy controls, which might exacerbate diabetic microvascular complications 35 , 36 . Therefore, identifying the distinct CD4+ T‐cell subset polarization and pathways involved in the dysregulation of the immune balance in DKD is extremely necessary for prognostication and treatment purposes.
Accompanying the invasion of leukocytes (such as neutrophils and macrophages) from the innate immune system, autoreactive T cells and B cells promote the production of autoantibodies, which target type IV collagen in the glomerulus (such as anti‐glomerular basement membrane glomerulonephritis [anti‐GBM GN]), or just accumulated as immunocomplexes in the glomeruli (such as a certain type of anti‐neutrophil cytoplasmic autoantibody‐associated glomerulonephritis and immunoglobulin A nephropathy) 37 . In a murine model of Adriamycin nephropathy (chemical‐induced‐nephropathy), a model of focal segmental glomerulosclerosis, macrophages modified ex vivo by interleukin (IL)‐10/transforming growth factor (TGF)‐β were able to inhibit CD4+ T‐cell proliferation and did not promote fibrosis in inflamed kidney 38 . In another mice model of crescentic glomerulonephritis (anti‐GBM GN), T cells residing in the interstitium of the kidney showed the Th1 phenotype, and produced interferon‐γ (IFN‐γ) 39 – a key factor of stimulating macrophages into the active state, as indeed presented by a pronounced infiltration of proliferating macrophages in the kidney of rats in early studies 40 , 41 , 42 . These data highlight the importance of further study on the interaction between T cells and other immune cells, such as whether T cells precede macrophages in the development and progression of kidney disease, as well as in DKD.
PATHOGENIC ROLE OF T CELLS IN THE DEVELOPMENT OF DKD
Activation of T cells under the hyperglycemia milieu and other factors in DKD
Studies have found enhanced levels of T‐cells activation and proliferations linked to hyperglycemia 43 , 44 , 45 , 46 . A recent study found reduced capacity of T‐cells proliferation in diabetic INS C94Y transgenic pigs (a large animal model showing a permanent diabetes phenotype after birth) compared with wild‐type littermates 47 . In that study, proteomic analysis showed a high abundance of pathways associated with the immune system, signal transduction and metabolic function. Of the most regulated pathways, lipophagy is of particular interest as it involves with metabolic dysfunction of immune cells 48 , which suggests an altered metabolic phenotype of immune cells under the diabetic microenvironment.
Pathogenic role of T cells on induction of albuminuria in DKD
Several studies suggest a pathogenic role of T cells for the induction of proteinuria during the development of DKD 21 , 31 , 49 . In people with type 2 diabetes, interstitial infiltration of CD4+ T cells correlated with the degree of proteinuria 21 . Similarly, in the blood of people with type 1 diabetes, the absolute number and percentage of T cells were significantly increased in patients with non‐nephrotic proteinuria (>0.5 g and <3.5g/24 h) compared with controls (<0.5 g/24 h) 31 . Animal models of diabetes enhanced the pathogenic role of T and B cells in inducing albuminuria in DKD. In one study, Rag1−/− diabetic mice models lacking mature T and B cells showed protection against increasing albuminuria, which was characterised by slower progression of urine albumin excretion rate and albumin/creatinine ratio compared to wild‐type control mice, implying that the absence of T and B cells lessened the risk of developing albuminuria 29 . Furthermore, it also showed that a systematic inhibition of T‐cell activation by the drug, abatacept, ameliorated the presence of albuminuria, even when albuminuria was established in a streptozotocin (STZ)‐induced type 1 diabetes model 50 . Notably, subsequent data showed no change of B7‐1 (an immune‐related protein that can be expressed by podocytes and leads to podocyte destruction in DKD 51 ) after abatacept treatment in kidneys of diabetic mice compared with non‐STZ mice, neither in cultured human podocytes nor in glomeruli of people with diabtetes 50 . Therefore, the protective effect of abatacept was thought to be predominantly by inactivating systematic T cells, rather than interacting with podocytes. These findings suggest that the increase of T or B cells systematically and locally contributes to the immunopathological process of the development of proteinuria in DKD.
Podocytes play an integral role in maintaining the glomerular filtration barrier integrity 52 , 53 , where proteinuria occurs when compromised 52 , 54 . One clinical study using 37 biopsies from Pima people with type 2 diabetes showed that podocyte detachment was significantly higher in people with macroalbuminuria (1.48%) than those with normal albuminuria (0.41%) or microalbuminuria (0.37%) 55 , confirming the same positive relationship between podocyte numbers and albuminuria, as shown in people with type 1 diabetes 56 . Notably, podocytes detachment, rather than podocytes numbers, showed a positive association with albuminuria in that study. This evidence indicates that podocyte dysfunction, instead of podocyte loss, might be a prime driver of impaired permselectivity in the glomerulus. Although DKD pathogenesis theories focused on podocyte dysfunction for a long time, the underlying mechanism contributing to the initial injury of podocytes has not been fully identified 57 . A recent study used ovalbumin (OVA) as a model antigen together with transgenic OT‐I T cells bearing a T‐cell receptor specific for OVA257–264 (SIINFEKL) 58 . Using this model, SIINFEKL peptide promoted OT‐I T‐cell proliferation and secretion of diabetes associated‐inflammatory cytokines, such as IFN‐γ and IL‐17, and in turn aggravated podocyte injury and apoptosis 59 , 60 . This finding suggests that T cells and podocytes act synergistically in the profibrotic progression of DKD.
TISSUE‐RESIDENT MEMORY T CELLS IN INFLAMMATORY DISEASE: POTENTIAL TARGETS FOR THERAPY
Ontogeny of tissue‐resident memory T cells
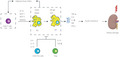
After a pathogen encounter, there is a rapid production of effector T cells from naïve T cells that migrate swiftly to lymphoid and non‐lymphoid tissues 61 . After infection resolution, antigen‐specific effector T cells might differentiate into various memory T‐cell subsets with distinct trafficking properties 62 . Effector memory T cells can circulate through lymphoid and non‐lymphoid organs, whereas central memory T cells recirculate between lymph nodes, lymph and blood by the vascular addressin L‐selectin (CD62L) and chemokine receptor CCR7 61 , 63 . In addition, a non‐recirculating T‐cell population persists in barrier tissues, including the skin, reproductive tract, respiratory tract, salivary glands and non‐barrier tissues, including the brain and kidney 63 , 64 , 65 , 66 , 67 , 68 . These cells that exist in disequilibrium from the circulation are termed tissue‐resident memory T (Trm) cells 69 , 70 , 71 (Figure 1).
Figure 1.

T‐cell migration paradigm. Memory T cells are divided into circulating and non‐circulating subsets. Central memory T (Tcm) cells migration is similar to that of the naïve T cells, and these cells predominantly reside in lymphoid tissues. Effector memory T (Tem) cells can pass through lymphoid and non‐lymphoid organs, and join the blood through lymphatic vessels. Tissue‐resident memory (Trm) cells are positioned within tissues and do not recirculate during steady‐state conditions. NLT, non‐lymphoid tissue; SLO, secondary lymphoid organs.
Prime markers of Trm cells in the human kidney
The location where the effector T cells differentiation occurs is important for shaping the phenotypic expression of Trm cells 61 . Trm cells adopt a pattern of surface molecules lacking lymph node homing molecules CCR7 and CD62L, which set it apart from both central memory T and effector memory T cells 61 , 72 . CD103+ Trm cells among CD8+ T‐cell subsets were widely detected in the small intestine, skin epidermis, sensory ganglia and brain 70 , 73 , and CD69 was widely expressed on the cell surface of Trm cells, so the co‐expression of CD103 and CD69 was commonly used as the existence of Trm cells in many studies 63 , 74 , 75 , 76 . However, the co‐expression of CD103 and CD69 as markers for Trm cells has limitations, and a comprehensive analysis focusing on the migrational properties of T cells found that T cells lacking CD103 or CD69 can also be resident within the pancreas, salivary glands and female reproductive tract (FRT) 77 . With regard to the kidney, one study showed that a subset of CD8+ T cells displayed with a Trm‐like phenotype (CD62L‐CD69+CD103+) in the kidney of mice infected with lymphocytic choriomeningitis virus Armstrong 64 . However, it is worthy to note that a substantial proportion of CD8+ T cells in the mouse kidney lack expression of CD103, in agreement with those found in healthy human kidney biopsies 78 . Additionally, CD69 is also a marker of recent T‐cell activation and is expressed on T cells at sites of chronic inflammation 79 , 80 . Therefore, these results raise caveats that we need to interpret with caution, as Trm cells are highly heterogeneous based on their locations, and keep in mind the overlap between CD103+CD69+ Trm cells and activated CD103+ cells in human kidneys.
Potential function of tissue‐resident memory T cells in inflammatory diseases
The functional properties of Trm cells in diverse tissues have been widely explored 74 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , and it is likely that the heterogeneous subsets of CD8+ Trm cells contribute to persistent autoimmune disorders, such as in psoriasis, arthritis, cutaneous lupus erythematosus, autoimmune hepatitis and β‐cell destruction in type 1 diabetes 74 , 84 , 85 , 86 , 87 . It was shown that Trm cells produced cytokines, such as IFN‐γ, that can stimulate B cells, natural killer cells and dendritic cells, and the ensuing innate immune cascade, therefore, added to the oxidative stress in different tissues 81 , 88 . However, Trm cells also play a protection role in the regulation of immune homeostasis in non‐barrier tissues (brain, liver) and barrier tissues (female reproductive tract and skin) 81 , 82 , 83 . In non‐barrier tissues, such as the brain, CD8+ Trm cells have the ability to kill OVA‐loaded targets 82 . Within the human liver, CD69+CD8+ effector memory T cells in the liver expressed a significantly higher level of the Trm‐like phenotype (CD8+CD69+CD103+), with much lower cytotoxic proteins, as compared with peripheral effector cells 83 . This diminished cytolytic activity has been posited to protect the tissue (such as the brain and liver) from immune pathology when exposed to low or non‐specific stimuli 83 , 91 . Epithelial sites, including the FRT and skin, were intensively studied 65 , 81 , 89 . It was shown in an elegant study 81 that lymphocytic choriomeningitis virus gp33 peptide‐specific CD8+ Trm cells were established locally in the FRT of P14 chimeric mice, which have a transgenic T‐cell receptor for lymphocytic choriomeningitis virus gp33‐41 epitopes 92 . On challenge with recombinant vaccinia virus expressing OVA, a significantly reduction of viral load was detected within the FRT of mice harboring locally activated CD8+ Trm cells, as compared with naïve mice. Interestingly, memory T cells were found to be far higher on the mice skin that was pre‐treated with a non‐specific inflammatory stimulus 65 and, thus, in the absence of antigen stimulation, inferring that Trm cells can be generated locally and provide protection without the existence of local antigen.
Knowledge of the functional features of Trm cells in kidney disease remains limited so far. An expansion of CD8+ Trm cells was observed in the kidneys of people with lupus nephritis or MRL/lpr mice (a model for an autoimmune disease resembling systemic lupus erythematosus), and correlated with kidney disease severity, providing the evidence that renal CD8+ Trm cells are involved in the pathogenesis of kidney injury 93 . Further studies are necessary to fill in the gap of showing discrete functional properties of Trm cells in inflammatory disease, and the beneficial effects might be able to be leveraged in specific tissue microenvironments of inflammatory disease, including DKD.
TREGSS IN DKD
Tregs make up 5–20% of the CD4+ T cells and include: (i) inducible Tregs produced in the periphery; and (ii) natural Tregs produced in the thymus 94 . It is reported that inducible Tregs implement their suppressive effect mainly by the effects of TGF‐β1 and IL‐10 95 . TGF‐β1 can effectively induce the differentiation of Trm cells in the kidney 96 . As for IL‐20, which is a pro‐inflammatory cytokine of the IL‐10 family, studies in diabetic mice showed increased IL‐20 expression in renal podocytes, and anti‐IL‐20 mAb (7E) treatment reduced mesangial cell expansion and inflammatory responses, as indicated by a lower level of inducible nitric oxide synthase, tumor necrosis factor‐α (TNF‐α) and monocyte chemotactic protein‐1 (MCP‐1) expression in renal tissue 97 . Naturally arising Tregs express FOXP3 and among them, CD4+CD25+ Tregs are the most investigated subsets in the context of autoimmune diseases 98 , 99 . CD4+CD25+FOXP3+Tregs maintain the self‐tolerance immune balance and modulate a wide range of immune responses, including activation, proliferation and effector function 98 , 100 . A recent study showed that adoptive transfer of FOXP3+ Tregs displayed an improved insulin sensitivity and a significant decline in albumin : creatinine ratio in the urine of db/db type 2 diabetic mice 101 , which implies that CD4+FOXP3+ Tregs might play a crucial part in reversing the progression of DKD. Furthermore, FOXP3+ Tregs can negatively regulate the effect of other T cells by competing for antigen‐presenting cells through CTLA‐4, whereby the administration of CTLA4‐Fc (abatacept) subcutaneously further validated the protection effect of FOXP3+ Tregs, showing reduced levels of albuminuria in high‐fat diet type 1 diabetic mice 50 . These findings suggest that the Tregs might represent a protective regulator in the development of DKD.
An appropriate ratio between pro‐inflammatory and anti‐inflammatory factors is critical to maintain a balanced microenvironment and reduce the risk of inflammatory disease. Th17 cells can produce IL‐17, which has been shown to contribute to the pro‐inflammatory progression of diabetes 30 . In contrast, Tregs cells are known to be potent suppressors of autoimmunity, and reported to dampen Th1, Th2 and Th17 cells response, specifically 102 , 103 , 104 . In people with type 2 diabetes and/or impaired kidney function, a reduced level of peripheral Tregs and an elevated serum Th17 : Tregs ratio were seen compared with controls, and positively related to the urine albumin : creatinine ratio 104 , 105 , 106 , 107 . Collectively, more studies are required to elucidate the clinical application of Tregs as a potential cellular immunotherapy for people with DKD, such as selective treatment of low‐dose IL‐2 to modulate CD4+ Tregs.
SIGNATURE T‐CELL‐RELATED CYTOKINES IN DKD
It has been shown that systematic and local inflammation are implicated in the development of DKD, and specific cytokines are widely studied 22 , 108 , 109 , 110 , 111 , 112 , 113 , 114 , 115 , 116 , 117 . In reverse, in vitro and in vivo studies showed that short‐term hyperglycemia can persistently exacerbate the inflammation response by activating epigenetic mark, histone 3 lysine 4 monomethylation (H3K4me1) in the promoter of the nuclear factor‐κB subunit p65, which controls cytokine production from diverse immune cells 113 . This pro‐inflammatory cascade persists even in a subsequent normoglycemia environment 113 . Therefore, it is of paramount importance to identify signature cytokines and prognostic indicators for the renal injury associated with DKD.
IL‐17A
The IL‐17 family is composed of six homodimeric cytokines (IL‐17A–F), which are mainly produced by activated memory T cells 118 , 119 . IL‐17A, commonly referred to as IL‐17, is largely produced by T‐helper cells (Th17), and was originally considered to have pro‐inflammatory properties in type 1 diabetes, probably due to the influence of the inducible nitric oxide release on β‐cells 120 , 121 . Several studies reported an increased number of IL‐17‐producing cells, and production of IL‐17 in the serum, spleen and pancreas of non‐obese diabetic (NOD) mice (a model of type 1 diabetes mellitus) as diabetes progressed 121 , 122 , 123 , 124 . Similarly, a significantly higher level of IL‐17 was also shown in the serum of people with type 1 diabetes and type 2 diabetes 125 , 126 , 127 . IL‐17 deficiency was shown to play an integral role in suppressing the development of diabetes, as well as protection on the kidney, characterized by reduced albuminuria, glomerular injury and hypertrophy, macrophage accumulation, and renal fibrosis in STZ‐induced diabetic mice (a model of type 1 diabetes) 128 . Mycophenolate mofetil suppressed the proliferation of IL‐17A+CD4+ T cells (Th17 cells) during the development of DKD in STZ‐induced diabetic mice from 8 to 16 weeks, whereas the number of IFN‐γ+CD4+ Th1 cells was only decreased at an early stage, highlighting the importance of the involvement of Th17 cells compared with Th1 cells 129 . This downregulation of Th17 cells by mycophenolate mofetil was also found to be associated with albuminuria reduction 129 . Another study in line with the pathogenesis impact of IL‐17A found that structural lesions, including mesangial matrix accumulation and glomerular basement membrane thickening, can be ameliorated with neutralizing antibody against IL‐17A 30 . Together, these findings suggest the critical regulatory role of IL‐17A in immune diseases and the promising effect of IL‐17A neutralization on DKD treatment. Nevertheless, many other studies have shown controversial results. A low dose of IL‐17A protected STZ diabetic mice from kidney disease, and a more pronounced renal change, such as tubular injury, interstitial fibrosis and mesangial expansion, was observed in IL‐17A knockout STZ diabetic mice compared with wild‐type mice 130 . Furthermore, the transfer of adoptive IL‐17‐producing γδT cells in NOD mice showed that it did not aggravate diabetes, but protected NOD mice by decreasing its incidence of diabetes 131 . In human studies, there was no significant difference between the level of serum IL‐17 in people with type 2 diabetes mellitus and those without diabetes 105 , 132 . Additionally, in a cross‐sectional study of Asian and Indian populations, the level of serum IL‐17 was significantly lower in people with diabetes with or without renal lesions 133 . Regarding the possible mechanism, an elegant study showed that CD4+ Th17 cells were capable of co‐secreting IL‐10, playing regulatory properties, and stimulus with TGF‐β and IL‐6 can completely abrogate the pathogenic function of Th17 cells, despite a rise of IL‐17 in vitro and in vivo 134 . Hence, it is potentially because IL‐17‐producing Th17 cells might produce other cytokines that play a greater role in determining the pathological or tolerogenic effects, or the role of IL‐17 might be mitigated by other cytokines in different microenvironments of the disease course. Therefore, further evaluation is still required to consolidate the dose–effect of IL‐17A and subsequent inflammatory signaling pathway in DKD.
IL‐2
The pathological role of IL‐2/IL‐2 receptor (IL‐2R) in kidney injury has long been shown in early studies 135 . It was shown that serum soluble IL‐2R (sIL‐2R) levels increased in people with renal dysfunction, and a significant negative correlation was observed between sIL‐2R and creatinine clearance 136 . In vitro activated T cells showed marked upregulation of binding affinity and surface receptor number under exposure to advanced glycation end‐products (oxidative derivatives resulting from hyperglycemia), and an elevated expression of different pro‐inflammatory Th1 cytokines, such as IFN‐γ and IL‐2 135 . In patients with DKD and overt nephropathy (daily urine protein loss >3.5 g/day), a significantly higher level of serum IL‐2R was presented compared with those with normoalbuminuria (<30 mg/day) or microalbuminuria (30–300 mg/day) 33 . This evidence suggests that IL‐2R acts in concert with other Th1 cytokines, and might be an important driver in the pathological development of kidney injury in DKD 33 .
Given the fact that IL‐2 is essential for the development and normal function of Tregs, which are critical in preventing autoimmune diseases, it is feasible to make an effort to explore an efficient strategy of IL‐2, or analogs of IL‐2, treatment in people with type 1 diabetes. A recent study reported that low‐dose mouse IL‐2/CD25 (mIL‐2/CD25) prevented diabetes in NOD mice and regulated diabetes in hyperglycemic mice 137 . However, studies of low‐dose IL‐2 in chronic kidney disease in people with type 2 diabetes are scant. Studies showed that low‐dose IL‐2 selectively expanded CD4+CD25+FOXP3+ Tregs in patients with chronic kidney disease, and these Tregs hampered the production of pro‐inflammatory Th1 and Th17 cells 104 ; hence, it will be interesting to explore whether low‐dose IL‐2 can provide beneficial effect to patients with DKD.
TNF and TNF receptor
TNF was identified five decades ago 138 , 139 as the product of monocytes that induced acute and chronic systematic inflammatory responses 140 , 141 , 142 . Apart from hematopoietic cells, TNF has also been found in intrinsic renal parenchymal cells, glomerular visceral epithelial cells and mesangial cells 143 , 144 , 145 . The TNF superfamily and its receptors on the surface of T cells are of paramount significance to normal T‐cell function, as discussed elsewhere 146 , 147 .
TNF was first shown to have profound pro‐inflammatory effects in DKD in 1991 148 . Results from that study demonstrated that the incubation of peritoneal macrophages from normal rats with glomerular basement membrane from the diabetic group showed the ability to synthesize increased levels of TNF and IL‐1, compared with macrophages incubated with glomerular basement membrane from normal rats. This finding indicates that advanced glycation end‐products, which were generated on the glomerular basement membrane in the setting of diabetes, might strongly boost the production of these cytokines, and could contribute to the alteration of glomerular microcirculation, as shown by substantially increased urinary albumin excretion in diabetic mice. Indeed, TNF‐α microribonucleic acid and protein levels in the renal interstitial fluid and urine were augmented before a significant elevation of urinary albumin excretion in a STZ‐induced diabetic rodent model 149 , 150 , 151 . A positive relationship between urinary levels of TNF‐α and urinary albumin excretion was further confirmed, with a reciprocal role for albuminuria on the production of TNF‐α 150 . Conversely, TNF antagonist was reported to reduce urinary excretion of TNF, likely by preventing Na retention (which was shown to promote hypertension and cause renal hypertrophy by TGF‐β effects 152 , 153 ) in distal tubule cells through an increase of tubular Na transport and sodium‐dependent solute uptake 154 , and attenuated the loss of kidney function in a diabetic rat model 150 , 155 . Similarly, in clinical trials, many studies showed that the serum or urinary levels of TNF‐α in people with type 2 diabetes mellitus were higher than those without diabetes 156 , 157 , 158 . These changes show the possibility that TNF‐α might serve as a promising marker in predicting the risk for progressive kidney impairment in DKD.
There has been an increasing appreciation of the importance of TNF receptor (TNFR) as a predictor of the development and progression of DKD for both type 1 diabetes and type 2 diabetes 159 , 160 , 161 , 162 . TNFR type 1 and TNFR type 2 are the two main distinct receptors of TNF that existed in both a membrane‐embedded pattern and a soluble form in serum 163 . A small pilot study showed that an increase in soluble TNFR type 1 level is independently associated with an early decline in the estimated glomerular filtration rate (eGFR) 159 . For advanced kidney disease, an 8‐ to 12‐year follow‐up study has shown that circulating TNFR type 1 and TNFR type 2 levels, but not free or total TNF‐α levels, were strongly associated with the risk of progression to ESKD in type 2 diabetes patients 160 . This relationship was found to be independent of other known risk factors or markers for progression of DKD, such as albuminuria 160 . These results have challenged the value of TNF‐α levels as a DKD‐related biomarker. As such, further studies in animal models of diabetes are required to determine the factors that drive an increase in TNFR, as well as the significance and causal relationship of these changes in the pathogenesis of DKD.
THERAPEUTIC IMPLICATIONS OF TARGETING CD8+ T CELLS IN ADIPOSE TISSUE IN DKD
The change of the microenvironment in diabetes brings about cellular alterations in adipocytes, and affects the cross‐talk between adipose tissue and other organs, including the kidney, referred to as the adipo–renal axis 164 . Plenty of evidence has shown that obesity might be a risk factor for ESKD in people with type 2 diabetes and hypertension 165 , 166 , 167 , 168 . In people with DKD and obesity who had an eGFR <40 mL/min/1.73 m2 and urine albumin excretion <30 mg/day, a short‐term intensive ketogenic diet improved their glomerular filtration rate, as well as metabolic markers, including fasting insulin, fasting glucose and insulin resistance, which shows that alteration in adipocytes might be a contributor in the regulation of kidney function 168 . A cross‐sectional study involving 200 people with diabetes found that epicardial adipose tissue was inversely associated with eGFR, and positively correlated with albuminuria 169 . These findings suggest the potential therapeutic approach of targeting adipose tissue inflammation in maintaining metabolic homeostasis in DKD, and might encourage weight loss regimens for people with impaired renal function in clinical practice.
The underlying mechanism of the aforementioned observations is not clear. Notably, T cells are involved in the inflammatory process along with macrophages within adipose tissue 170 . Nevertheless, whether the inflammation is initiated by T cells or whether it is a consequence of a response to injury has received increasing attention. A study has verified the increased presence of T cells in adipose tissue, proposing that CD8+ T cells precede adipose tissue macrophages as the immunological initiator of obesity 171 . In that study, they found that CD8+ T cells can stimulate the conversion of the anti‐inflammatory macrophages type 2 to pro‐inflammatory macrophages type 1, and systemic insulin resistance was ameliorated by depletion of CD8+ T cells and exacerbated by adoptive transfer of CD8+ T cells. This systemic insulin resistance was reported to lead to disruption of glucose uptake in human podocytes 52 . Additionally, the treatment of eliminating CD8+ T cells in diet‐induced obesity mice largely improved adipose tissue inflammation and suppressed macrophage recruitment. Although the specific molecular mechanism of the adipo–renal axis has not been shown yet, it is highly likely adipocyte‐derived factors form a complicated paracrine and endocrine pattern between local cell types in adipose tissue and other tissue, including the kidney 164 . Therefore, the inhibitor of CD8+ T cells should be placed with particular attention in the prorogation of pro‐inflammatory chaos in adipose tissue and subsequent kidney injury. Collectively, this evidence implies the significance of CD8+ T cells in the initiation and escalation of pro‐inflammatory production in obesity‐associated insulin resistance and renal diseases 52 (Figure 2).
Figure 2.

Insulin resistance and kidney damage initiated by collaboration of T cells and macrophages. CD8+ T cells act as the starter of adipose inflammation, which triggers the anti‐inflammatory macrophages type to the pro‐inflammatory type and the resultant production of cytokines. Insulin resistance occurs after this process and as a result, progressively leads to end‐organ damage, such as the kidney. IL, interleukin; IP‐10, interferon‐γ‐inducible protein 10; M1, classically activated macrophages; M2, alternatively activated macrophages; MCP‐1, monocyte chemoattractant protein 1; MCP‐3, monocyte chemoattractant protein 3; MIF, migration inhibitory factor; NF‐κB, nuclear factor‐κB; RANTES, Regulated upon Activation, Normal T cell Expressed and Secreted; TGF‐ β, transforming growth factor‐β; TNF‐α, tumor necrosis factor‐α; Treg, regulatory T cells.
B CELLS IN DKD
B cells are less explored in DKD compared with T cells 21 , 29 , with more increasing focus in their role in the pathogenesis in type 1 diabetes 172 . Type 1 diabetes is an autoimmune disorder, which is characterized by the destruction of insulin‐producing pancreatic β‐cells 172 . Although diabetogenic T cells are mainly responsible for β‐cell destruction 173 , increasing evidence has shown that B cells exercise their function through presenting islet autoantigens to diabetogenic T cells 174 , as well as the production of immunoglobulin G, facilitating the formation of an immune complex, and triggering subsequent complement activation in the blood 175 and kidney 21 , 172 , 176 , 177 . The immune complexes were reported to promote macrophage accrual 178 and ensuing inflammation in the glomerulus 179 , which further caused the release of damage‐associated molecular patterns 172 , 180 , 181 . These data might point toward a larger role of B cells by antibodies and immune complexes in the pathogenesis of DKD.
In vitro studies have shown that immunoglobulin G can promote early pro‐inflammatory activation, as well as an early upregulation of IL‐6, inciting the transition from innate to adaptive immune response through the gp130–STAT3‐dependent pathway 182 . Also, B cells can produce a wide range of cytokines shared by T helper cells, including IL‐4 and IL‐13 183 . One study proposed the role of IL‐4 and IL‐13 in nephrotic syndrome 184 . It identified that IL‐4 caused pronounced detachment of podocytes from the basement membrane in vitro, and foot process effacement and proteinuria in vivo. Furthermore, transfer of IL‐4‐deficient B cells did not induce proteinuria. For another, IL‐13 plasmid administration induced proteinuria in rats, but not in mice, probably owing to species‐specific requirements for cytokine‐induced proteinuria. Further studies are required to show the B‐cells‐induced inflammatory factors in the signaling pathway in kidney disease progression of the DKD model.
A specific subset of these B cells was recently investigated in the pathophysiological process of DKD, termed regulatory B cells (Bregs) 185 . In 27 patients with DKD, a lower number of CD19+CD24hiCD38hi Bregs were found, as well as a lower level of serum IL‐10 (an anti‐inflammatory cytokine), which was mediated through inhibition of Th1 and Th17 cells differentiation, and conversion of CD4+CD25− T cells into Tregs 186 , 187 . Additionally, a positive relationship was identified between the number of CD19+CD24hiCD38hi Bregs and eGFR, implicating a putative disease marker of CD19+CD24hiCD38hi Bregs in the progression of DKD.
FUTURE DIRECTIONS
As aforementioned, it is important to detect the sequence of events and to determine whether T cells precede macrophages in the kidney during the development of DKD. To improve clinical outcome with customized therapies, further studies are necessary for determining the T‐cell phenotype in the blood and kidneys of people with diabetes and with/without renal lesions, or DKD imposed with renal diseases. Notably, the clones of T cells present differently between the blood and kidney in people with systemic lupus erythematosus, indicating that T cells receive distinct signals in the blood and kidney. The same mechanism might be at play in DKD. Therefore, T‐cell receptor repertoire studies should be developed in kidneys, as the clone features found in the blood cannot inform us of a similar scenario in the kidney.
Trm cells have been of interest to many researchers in recent decades 79 , 188 . Although the reduced cytolytic activity of Trm cells has been observed in diverse tissue, such as the lung, brain and liver 83 , 189 , 190 , the specific function of Trm cells in the kidneys of people with diabetes has not yet been shown. For this purpose, quantitative and functional analysis of CD4+ Trm cells and CD8+ Trm cells should be considered in kidney biopsies of people with diabetes to validate if this ‘hidden cytotoxicity’ also occurs in the kidney. Importantly, two compartments of the kidney, cortex and medulla both merit further investigation to fully appreciate the T‐cell immunobiology. Furthermore, in vitro co‐culture of Trm cells with renal intrinsic cells (e.g., mesangial cell, glomerular epithelial cells and tubular cells) can be explored under normal or hypoxic conditions to mimic the fibrotic phase of the DKD, to better understand the functional interactions between Trm cells and renal intrinsic cells.
CONCLUSIONS
The pathogenesis of immune abnormalities in DKD is a process involving various factors in the context of hyperglycemia. The central role of innate immunity dominated by macrophages in DKD has widely been elucidated, whereas understanding the role of adaptive immunity is still in the early stage. Although the interaction between T cells and macrophages is documented in insulin resistance and metabolic syndrome, the direct role of T cells in contribution to DKD and the sequence of the innate and adaptive immune events in the pathogenesis of DKD remains unknown. The understanding of the role of specific immune cells from the adaptive immune response and pro‐inflammatory molecules in DKD needs to be elucidated further for new effective therapeutic options to develop.
DISCLOSURE
The authors declare no conflict of interest.
Elif Ilhan Ekinci's institution has received research funding for unrelated research projects from Eli Lilly, Novo Nordisk, Gilead, Bayer and Sanofi.
Approval of the research protocol: N/A.
Informed consent: N/A.
Approval date of registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
ACKNOWLEDGMENTS
This work was supported by a scholarship from the Chinese Scholarship Council in 2018 (No. 201806230303). We thank every co‐author for expert advice and helpful discussions. We apologize to many other works in this field that we were unable to cite here due to limited space.
J Diabetes Investig 2022; 13: 213–226
REFERENCES
- 1. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: Challenges, progress, and possibilities. Clin J Am Soc Nephrol 2017; 12: 2032–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. International Diabetes Federation . IDF Diabetes Atlas, 9th edn. Brussels, Belgium: International Diabetes Federation, 2019. [Google Scholar]
- 3. Schaubel DE, Morrison HI, Desmeules M, et al. End‐stage renal disease in Canada: prevalence projections to 2005. CMAJ 1999; 160: 1557–1563. [PMC free article] [PubMed] [Google Scholar]
- 4. American Diabetes Association AD . Standards of medical care in diabetes‐‐2014. Diabetes Care. 2014; 37: S14‐S80. [DOI] [PubMed] [Google Scholar]
- 5. Wang CCL, Hess CN, Hiatt WR, et al. Atherosclerotic cardiovascular disease and heart failure in type 2 diabetes – mechanisms, management, and clinical considerations. Circulation 2014; 133: 2459–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rawshani A, Rawshani A, Franzén S, et al. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N Engl J Med 2017; 376: 1407–1418. [DOI] [PubMed] [Google Scholar]
- 7. Harjutsalo V, Groop P‐H. Epidemiology and risk factors for diabetic kidney disease. Adv Chronic Kindey Dis 2014; 21: 260–266. [DOI] [PubMed] [Google Scholar]
- 8. Madhusudhan T, Wang H, Dong W, et al. Defective podocyte insulin signalling through p85‐XBP1 promotes ATF6‐dependent maladaptive ER‐stress response in diabetic nephropathy. Nat Commun 2015; 6: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Vriese AS, Stoenoiu MS, Elger M, et al. Diabetes‐induced microvascular dysfunction in the hydronephrotic kidney: role of nitric oxide. Kidney Int 2001; 60: 202–210. [DOI] [PubMed] [Google Scholar]
- 10. Jacobsen P, Andersen S, Rossing K, et al. Dual blockade of the renin‐angiotensin system versus maximal recommended dose of ACE inhibition in diabetic nephropathy. Kidney Int 2003; 63: 1874–1880. [DOI] [PubMed] [Google Scholar]
- 11. Tan ALY, Forbes JM, Cooper ME. AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol 2007; 27: 130–143. [DOI] [PubMed] [Google Scholar]
- 12. Hovind P, Rossing P, Tarnow L, et al. Progression of diabetic nephropathy. Kidney Int 2001; 59: 702–709. [DOI] [PubMed] [Google Scholar]
- 13. Lecamwasam A, Sexton‐Oates A, Carmody J, et al. DNA methylation profiling of genomic DNA isolated from urine in diabetic chronic kidney disease: a pilot study. PLoS One 2018; 13: 0–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Altemtam N, Russell J, El Nahas M. A study of the natural history of diabetic kidney disease (DKD). Nephrol Dial Transplant 2012; 27: 1847–1854. [DOI] [PubMed] [Google Scholar]
- 15. Tesch GH. Diabetic nephropathy – is this an immune disorder? Clin Sci 2017; 131: 2183–2199. [DOI] [PubMed] [Google Scholar]
- 16. MacIsaac RJ, Jerums G, Ekinci EI. Effects of glycaemic management on diabetic kidney disease. World J Diabetes 2017; 8: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Group. Lancet . Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS). Lancet 1998; 352: 53. [PubMed] [Google Scholar]
- 18. Rajasekeran H, Lytvyn Y, Cherney DZI. Sodium–glucose cotransporter 2 inhibition and cardiovascular risk reduction in patients with type 2 diabetes: the emerging role of natriuresis. Kidney Int 2016; 89: 524–526. [DOI] [PubMed] [Google Scholar]
- 19. Mann JFE, Ørsted DD, Brown‐Frandsen K, et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med 2017; 377: 839–848. [DOI] [PubMed] [Google Scholar]
- 20. Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375: 1834–1844. [DOI] [PubMed] [Google Scholar]
- 21. Moon J‐Y, Jeong K‐H, Lee T‐W, et al. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol 2012; 35: 164–174. [DOI] [PubMed] [Google Scholar]
- 22. Zheng Z, Zheng F. Immune cells and inflammation in diabetic nephropathy. J Diabetes Res 2016; 2016: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chow F, Ozols E, Nikolic‐Paterson DJ, et al. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int 2004; 65: 116–128. [DOI] [PubMed] [Google Scholar]
- 24. Guiteras R, Sola A, Flaquer M, et al. Exploring macrophage cell therapy on Diabetic Kidney Disease. J Cell Mol Med 2019; 23: 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen D, Ping FU, Mu W, et al. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology 2006; 11: 226–231. [DOI] [PubMed] [Google Scholar]
- 26. Li JJ, Lee SH, Kim DK, et al. Colchicine attenuates inflammatory cell infiltration and extracellular matrix accumulation in diabetic nephropathy. Am J Physiol Physiol 2009; 297: F200–F209. [DOI] [PubMed] [Google Scholar]
- 27. Chow FY, Nikolic‐Paterson DJ, Ozols E, et al. Monocyte chemoattractant protein‐1 promotes the development of diabetic renal injury in streptozotocin‐treated mice. Kidney Int 2006; 69: 73–80. [DOI] [PubMed] [Google Scholar]
- 28. Tesch GH, Pullen N, Jesson MI, et al. Combined inhibition of CCR2 and ACE provides added protection against progression of diabetic nephropathy in Nos3‐deficient mice. Am J Physiol ‐ Ren Physiol 2019; 317: F1439–F1449. [DOI] [PubMed] [Google Scholar]
- 29. Lim AKH, Ma FY, Nikolic‐Paterson DJ, et al. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia 2010; 53: 1772–1782. [DOI] [PubMed] [Google Scholar]
- 30. Lavoz C, Matus YS, Orejudo M, et al. Interleukin‐17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int 2019; 95: 1418–1432. [DOI] [PubMed] [Google Scholar]
- 31. Bending JJ, Lobo‐Yeo A, Vergani D, et al. Proteinuria and activated T‐lymphocytes in diabetic nephropathy. Diabetes 1988; 37: 507–511. [DOI] [PubMed] [Google Scholar]
- 32. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR‐T immunotherapy. Cancers 2016; 8: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu C‐C, Chen J‐S, Lu K‐C, et al. Aberrant cytokines/chemokines production correlate with proteinuria in patients with overt diabetic nephropathy. Clin Chim Acta 2010; 411: 700–704. [DOI] [PubMed] [Google Scholar]
- 34. Zeng C, Shi X, Zhang B, et al. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med 2012; 90: 175–186. [DOI] [PubMed] [Google Scholar]
- 35. Ryba‐Stanisławowska M, Skrzypkowska M, Myśliwiec M, et al. Loss of the balance between CD4+Foxp3+ regulatory T cells and CD4+IL17A+ Th17 cells in patients with type 1 diabetes. Hum Immunol 2013; 74: 701–707. [DOI] [PubMed] [Google Scholar]
- 36. Ryba M, Marek N, Hak Ł, et al. Anti‐TNF rescue CD4 +Foxp3 + regulatory T cells in patients with type 1 diabetes from effects mediated by TNF. Cytokine 2011; 55: 353–361. [DOI] [PubMed] [Google Scholar]
- 37. Holdsworth SR, Gan P‐Y, Kitching AR. Biologics for the treatment of autoimmune renal diseases. Nat Rev Nephrol 2016; 12: 217–231. [DOI] [PubMed] [Google Scholar]
- 38. Cao QI, Wang Y, Zheng D, et al. IL‐10/TGF‐β‐modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol 2010; 21: 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Q, Luan H, Wang LE, et al. Galectin‐9 ameliorates anti‐GBM glomerulonephritis by inhibiting th1 and th17 immune responses in mice. Am J Physiol ‐ Ren Physiol 2014; 306: 822–832. [DOI] [PubMed] [Google Scholar]
- 40. Lan HY, Nikolic‐Paterson DJ, Mu W, et al. Local macrophage proliferation in the progression of glomerular and tubulointerstitial injury in rat anti‐GBM glomerulonephritis. Kidney Int 1995; 48: 753–760. [DOI] [PubMed] [Google Scholar]
- 41. Lan HY, Mitsuhashi H, Ng YY, et al. Macrophage apoptosis in rat crescentic glomerulonephritis. Am J Pathol 1997; 151: 531–538. [PMC free article] [PubMed] [Google Scholar]
- 42. Flanc RS, Ma FY, Tesch GH, et al. A pathogenic role for JNK signaling in experimental anti‐GBM glomerulonephritis. Kidney Int 2007; 72: 698–708. [DOI] [PubMed] [Google Scholar]
- 43. Mahmoud F, Al‐ozairi E. Inflammatory cytokines and the risk of cardiovascular complications in type 2. Diabetes 2013; 35: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao R, Li W, Lu Y, et al. Increased peripheral proinflammatory T helper subsets contribute to cardiovascular complications in diabetic patients. Mediators Inflamm. 2014; 2014: 596967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang K, Jin F, Zhang Z, et al. Angiotensin II promotes the development of carotid atherosclerosis in type 2 diabetes patients via regulating the T cells activities: a cohort study. Med Sci Monit Int Med J Exp Clin Res 2016; 22: 4000–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gong F, Wu J, Zhou P, et al. Interleukin‐22 might act as a double‐edged sword in type 2 diabetes and coronary artery disease. Mediators Inflamm 2016; 2016: 8254797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Giese I‐M, Schilloks M‐C, Degroote RL, et al. Chronic hyperglycemia drives functional impairment of lymphocytes in diabetic INSC94Y transgenic pigs. Front Immunol 2021; 11: 607473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kounakis K, Chaniotakis M, Markaki M, et al. Emerging roles of lipophagy in health and disease. Front Cell Dev Biol 2019; 7: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moriya R, Manivel JC, Mauer M. Juxtaglomerular apparatus T‐cell infiltration affects glomerular structure in Type 1 diabetic patients. Diabetologia 2004; 47: 82–88. [DOI] [PubMed] [Google Scholar]
- 50. Herrera M, Söderberg M, Sabirsh A, et al. Inhibition of T‐cell activation by the CTLA4‐Fc Abatacept is sufficient to ameliorate proteinuric kidney disease. Am J Physiol Physiol 2017; 312: F748–F759. [DOI] [PubMed] [Google Scholar]
- 51. Fiorina P, Vergani A, Bassi R, et al. Role of podocyte B7–1 in diabetic nephropathy. J Am Soc Nephrol 2014; 25: 1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lennon R, Pons D, Sabin MA, et al. Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol Dial Transplant 2009; 24: 3288–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int 2008; 74: 22–36. [DOI] [PubMed] [Google Scholar]
- 54. White KE, Bilous RW, Cordonnier DJ, et al. Structural alterations to the podocyte are related to proteinuria in type 2 diabetic patients. Nephrol Dial Transplant 2004; 19: 1437–1440. [DOI] [PubMed] [Google Scholar]
- 55. Weil EJ, Lemley KV, Mason CC, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int 2012; 82: 1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Toyoda M, Najafian B, Kim Y, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 2007; 56: 2155–2160. [DOI] [PubMed] [Google Scholar]
- 57. Yu S‐W, Leventhal JS, Cravedi P. Totally tubular, dude: rethinking DKD pathogenesis in the wake of SGLT2i data.pdf. J Nephrol 2021; 34: 629–631. [DOI] [PubMed] [Google Scholar]
- 58. Hogquist KA, Jameson SC, Heath WR, et al. T cell receptor antagonist peptides induce positive selection. Cell 1994; 76: 17–27. [DOI] [PubMed] [Google Scholar]
- 59. Li S, Liu Y, He Y, et al. Podocytes present antigen to activate specific T cell immune responses in inflammatory renal disease. J Pathol 2020; 252: 165–177. [DOI] [PubMed] [Google Scholar]
- 60. Pritchard GH, Cross EW, Strobel M, et al. Spontaneous partial loss of the OT‐I transgene. Nat Immunol 2016; 17: 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Beura LK, Masopust D. SnapShot: resident memory T cells. Cell 2014; 157: 1488–1488.e1. [DOI] [PubMed] [Google Scholar]
- 62. Davies B, Prier JE, Jones CM, et al. Cutting edge: tissue‐resident memory T cells generated by multiple immunizations or localized deposition provide enhanced immunity. J Immunol 2017; 198: 2233–2237. [DOI] [PubMed] [Google Scholar]
- 63. Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 2015; 21: 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Casey KA, Fraser KA, Schenkel JM, et al. Antigen‐independent differentiation and maintenance of effector‐like resident memory T cells in tissues. J Immunol 2012; 188: 4866–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mackay LK, Stock AT, Ma JZ, et al. Long‐lived epithelial immunity by tissue‐resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci 2012; 109: 7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sathaliyawala T, Kubota M, Yudanin N, et al. Distribution and compartmentalization of human circulating and tissue‐resident memory T cell subsets. Immunity 2013; 38: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thome J, Yudanin N, Ohmura Y, et al. Spatial map of human t cell compartmentalization and maintenance over decades of life. Cell 2014; 159: 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gebhardt T, Mueller SN, Heath WR, et al. Peripheral tissue surveillance and residency by memory T cells. Trends Immunol 2013; 34: 27–32. [DOI] [PubMed] [Google Scholar]
- 69. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol 2013; 13: 309–320. [DOI] [PubMed] [Google Scholar]
- 70. Carbone FR, Mackay LK, Heath WR, et al. Distinct resident and recirculating memory T cell subsets in non‐lymphoid tissues. Curr Opin Immunol 2013; 25: 329–333. [DOI] [PubMed] [Google Scholar]
- 71. Korpinen E, Groop P‐H, Fagerudd JA, et al. Increased secretion of TGF‐β1 by peripheral blood mononuclear cells from patients with type 1 diabetes mellitus with diabetic nephropathy. Diabet Med 2001; 18: 121–125. [DOI] [PubMed] [Google Scholar]
- 72. Mackay LK, Kallies A. Transcriptional regulation of tissue‐resident lymphocytes. Trends Immunol. 2017; 38: 94–103. [DOI] [PubMed] [Google Scholar]
- 73. Jameson SC, Masopust D. Understanding subset diversity in T cell memory. Immunity 2018; 48: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kuric E, Seiron P, Krogvold L, et al. Demonstration of tissue resident memory CD8 T cells in insulitic lesions in adult patients with recent‐onset type 1 diabetes. Am J Pathol 2017; 187: 581–588. [DOI] [PubMed] [Google Scholar]
- 75. Radenkovic M, Uvebrant K, Skog O, et al. Characterization of resident lymphocytes in human pancreatic islets. Clin Exp Immunol 2017; 187: 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mueller SN, Mackay LK. Tissue‐resident memory T cells: local specialists in immune defence. Nat Rev Immunol 2016; 16: 79–89. [DOI] [PubMed] [Google Scholar]
- 77. Steinert EM, Schenkel JM, Fraser KA, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 2015; 161: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. van der Putten C, Remmerswaal EBM, Terpstra ML, et al. CD8 and CD4 T cell populations in human kidneys. Cells 2021; 10: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Masopust D, Soerens AG. Tissue‐resident T cells and other resident leukocytes. Annu Rev Immunol 2019; 37: 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sancho D, Gómez M, Sánchez‐Madrid F. CD69 is an immunoregulatory molecule induced following activation. Trends Immunol 2005; 26: 136–140. [DOI] [PubMed] [Google Scholar]
- 81. Schenkel JM, Fraser KA, Beura LK, et al. Resident memory CD8 t cells trigger protective innate and adaptive immune responses. Science 2014; 346: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wakim LM, Woodward‐Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci 2010; 107: 17872–17879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stelma F, De Niet A, Sinnige MJ, et al. Human intrahepatic CD69 + CD8+ T cells have a tissue resident memory T cell phenotype with reduced cytolytic capacity. Sci Rep 2017; 7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Suárez‐Fariñas M, Fuentes‐Duculan J, Lowes MA, et al. Resolved psoriasis lesions retain expression of a subset of disease‐related genes. J Investig Dermatol 2011; 131: 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Petrelli A, Van Wijk F. CD8+ T cells in human autoimmune arthritis: the unusual suspects. Nat Rev Rheumatol 2016; 12: 421–428. [DOI] [PubMed] [Google Scholar]
- 86. Gu H‐J, Song S, Roh JY, et al. Expression pattern of tissue‐resident memory T cells in cutaneous lupus erythematosus. Lupus 2021; 30: 1427–1437. [DOI] [PubMed] [Google Scholar]
- 87. You Z, Li Y, Wang Q, et al. The clinical significance of hepatic CD69+CD103+CD8+ resident‐memory T cells in autoimmune hepatitis. Hepatology 2021; 74: 847–863. [DOI] [PubMed] [Google Scholar]
- 88. Ariotti S, Hogenbirk MA, Dijkgraaf FE, et al. Skin‐resident memory CD8+ T cells trigger a state of tissue‐wide pathogen alert. Science 2014; 346: 101–105. [DOI] [PubMed] [Google Scholar]
- 89. Jiang X, Clark RA, Liu L, et al. Skin infection generates non‐migratory memory CD8 + T RM cells providing global skin immunity. Nature 2012; 483: 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gálvez‐Cancino F, López E, Menares E, et al. Vaccination‐induced skin‐resident memory CD8+T cells mediate strong protection against cutaneous melanoma. Oncoimmunology 2018; 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Smolders J, Remmerswaal EBM, Schuurman KG, et al. Characteristics of differentiated CD8+ and CD4+ T cells present in the human brain. Acta Neuropathol 2013; 126: 525–535. [DOI] [PubMed] [Google Scholar]
- 92. Pircher H, Bürki K, Lang R, et al. Tolerance induction in double specific T‐cell receptor transgenic mice varies with antigen. Nature 1989; 342: 559–561. [DOI] [PubMed] [Google Scholar]
- 93. Zhou M, Guo C, Li X, et al. JAK/STAT signaling controls the fate of CD8+CD103+ tissue‐resident memory T cell in lupus nephritis. J Autoimmun 2019; 2020: 102424. [DOI] [PubMed] [Google Scholar]
- 94. Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol 2013; 4: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gregori S, Goudy KS, Roncarolo MG. The cellular and molecular mechanisms of immuno‐suppression by human type 1 regulatory T cells. Front Immunol 2012; 3: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ma C, Mishra S, Demel EL, et al. TGF‐β controls the formation of kidney‐resident T cells via promoting effector T cell extravasation. J Immunol 2017; 198: 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hsu Y‐H, Li H‐H, Sung J‐M, et al. Interleukin‐20 targets podocytes and is upregulated in experimental murine diabetic nephropathy. Exp Mol Med 2017; 49: e310–e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self‐tolerance and autoimmune disease. Immunol Rev 2006; 212: 8–27. [DOI] [PubMed] [Google Scholar]
- 99. Dousdampanis P, Trigka K, Mouzaki A. Tregs and kidney: From diabetic nephropathy to renal transplantation. World J Transplant 2016; 6: 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Arce‐Sillas A, Álvarez‐Luquín DD, Tamaya‐Domínguez B, et al. Regulatory T cells: molecular actions on effector cells in immune regulation. J Immunol Res 2016; 2016: 1720827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Eller K, Kirsch A, Wolf AM, et al. Potential role of regulatory T cells in reversing obesity‐linked insulin resistance and diabetic nephropathy. Diabetes 2011; 60: 2954–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kornete M, Mason ES, Piccirillo CA. Immune regulation in T1D and T2D: Prospective role of Foxp3+ treg cells in disease pathogenesis and treatment. Front Endocrinol 2013; 4: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lindley S, Dayan CM, Bishop A, et al. Defective suppressor function in CD4+CD25+T‐cells from patients with type 1 diabetes. Diabetes 2005; 54: 92–99. [DOI] [PubMed] [Google Scholar]
- 104. Li Y, Liu X, Wang W, et al. Low‐dose IL‐2 expands CD4+ regulatory T cells with a suppressive function in vitro via the STAT5‐dependent pathway in patients with chronic kidney diseases. Ren Fail 2018; 40: 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang C, Xiao C, Wang P, et al. The alteration of Th1/Th2/Th17/Treg paradigm in patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Hum Immunol 2014; 75: 289–296. [DOI] [PubMed] [Google Scholar]
- 106. Abouzeid S, Sherif N. Role of alteration in Treg/Th17 cells’ balance in nephropathic patients with Type 2 diabetes mellitus. Electron Physician 2015; 7: 1613–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zhu X, Li S, Zhang Q, et al. Correlation of increased Th17/Treg cell ratio with endoplasmic reticulum stress in chronic kidney disease. Med 2018; 97: e10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wang SN, Lapage J, Hirschberg R. Role of glomerular ultrafiltration of growth factors in progressive interstitial fibrosis in diabetic nephropathy. Kidney Int 2000; 57: 1002–1014. [DOI] [PubMed] [Google Scholar]
- 109. Yamamoto T, Nakamura T, Noble NA, et al. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci 1993; 90: 1814–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Navarro‐González JF, Mora‐Fernández C, de Fuentes MM, et al. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol 2011; 7: 327–340. [DOI] [PubMed] [Google Scholar]
- 111. Wada T, Furuichi K, Sakai N, et al. Up‐regulation of monocyte chemoattractant protein‐1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int 2000; 58: 1492–1499. [DOI] [PubMed] [Google Scholar]
- 112. Hofmann MA, Schiekofer S, Isermann B, et al. Peripheral blood mononuclear cells isolated from patients with diabetic nephropathy show increased activation of the oxidative‐stress sensitive transcription factor NF‐κB. Diabetologia 1999; 42: 222–232. [DOI] [PubMed] [Google Scholar]
- 113. El‐Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008; 205: 2409–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Shruthi S, Mohan V, Amutha A, et al. Increased serum levels of novel T cell cytokines IL‐33, IL‐9 and IL‐17 in subjects with type‐1 diabetes. Cytokine 2016; 86: 6–9. [DOI] [PubMed] [Google Scholar]
- 115. Donate‐Correa J, Martín‐Núñez E, Muros‐de‐Fuentes M, et al. Inflammatory cytokines in diabetic nephropathy. J Diabetes Res 2015; 2015: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci 2013; 124: 139–152. [DOI] [PubMed] [Google Scholar]
- 117. Xia C, Rao X, Zhong J. Role of T lymphocytes in type 2 diabetes and diabetes‐associated inflammation. J Diabetes Res 2017; 2017: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Moseley TA, Haudenschild DR, Rose L, et al. Interleukin‐17 family and IL‐17 receptors. Cytokine Growth Factor Rev 2003; 14: 155–174. [DOI] [PubMed] [Google Scholar]
- 119. Jin W, Dong C. IL‐17 cytokines in immunity and inflammation. Emerg Microbes Infect 2013; 2: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Miljkovic D, Cvetkovic I, Momcilovic M, et al. Interleukin‐17 stimulates inducible nitric oxide synthase‐dependent toxicity in mouse beta cells. Cell Mol Life Sci 2005; 62: 2658–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Emamaullee JA, Davis J, Merani S, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes 2009; 58: 1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Jain R, Tartar DM, Gregg RK, et al. Innocuous IFNγ induced by adjuvant‐free antigen restores normoglycemia in NOD mice through inhibition of IL‐17 production. J Exp Med 2008; 205: 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhang J, Huang Z, Sun R, et al. IFN‐γ induced by IL‐12 administration prevents diabetes by inhibiting pathogenic IL‐17 production in NOD mice. J Autoimmun 2012; 38: 20–28. [DOI] [PubMed] [Google Scholar]
- 124. Kuriya G, Uchida T, Akazawa S, et al. Double deficiency in IL‐17 and IFN‐γ signalling significantly suppresses the development of diabetes in the NOD mouse. Diabetologia 2013; 56: 1773–1780. [DOI] [PubMed] [Google Scholar]
- 125. Honkanen J, Nieminen JK, Gao R, et al. IL‐17 immunity in human type 1 diabetes. J Immunol 2010; 185: 1959–1967. [DOI] [PubMed] [Google Scholar]
- 126. Marwaha AK, Crome SQ, Panagiotopoulos C, et al. Cutting edge: increased IL‐17–secreting T cells in children with new‐onset type 1 diabetes. J Immunol 2010; 185: 3814–3818. [DOI] [PubMed] [Google Scholar]
- 127. Alina‐Emanuela G, Gadalean F, Vlad A, et al. Pro‐inflammatory cytokines IL‐6 and IL‐17 display a particular molecular pattern in association with dysregulated miRNAs in patients with type 2 diabetes mellitus in the early stages of diabetic kidney disease. Nephrol Dial Transplant 2021; 36: i371–i380. [Google Scholar]
- 128. Ma J, Li YJ, Chen X, et al. Interleukin 17A promotes diabetic kidney injury. Sci Rep 2019; 9: 2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kim S‐M, Lee S‐H, Lee A, et al. Targeting T helper 17 by mycophenolate mofetil attenuates diabetic nephropathy progression. Transl Res 2015; 166: 375–383. [DOI] [PubMed] [Google Scholar]
- 130. Mohamed R, Jayakumar C, Chen F, et al. Low‐dose IL‐17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J Am Soc Nephrol 2016; 27: 745–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Han G, Wang R, Chen G, et al. Interleukin‐17‐producing γδ+ T cells protect NOD mice from type 1 diabetes through a mechanism involving transforming growth factor‐β. Immunology 2010; 129: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Arababadi MK, Nosratabadi R, Hassanshahi G, et al. Nephropathic complication of type‐2 diabetes is following pattern of autoimmune diseases? Diabetes Res Clin Pract 2010; 87: 33–37. [DOI] [PubMed] [Google Scholar]
- 133. Vasanthakumar R, Mohan V, Anand G, et al. Serum IL‐9, IL‐17, and TGF‐b levels in subjects with diabetic kidney disease (CURES‐134). Cytokine 2015; 72: 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. McGeachy MJ, Bak‐Jensen KS, Chen YI, et al. TGF‐β and IL‐6 drive the production of IL‐17 and IL‐10 by T cells and restrain TH‐17 cell‐mediated pathology. Nat Immunol 2007; 8: 1390–1397. [DOI] [PubMed] [Google Scholar]
- 135. Imani F, Horii Y, Suthanthiran M, et al. Advanced glycosylation endproduct‐specific receptors on human and rat T‐ lymphocytes mediate synthesis of interferon gamma: role in tissue remodeling. J Exp Med 1993; 178: 2165–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chen HS, Wu MS, Yen TS, et al. Soluble interleukin‐2 receptor in patients with glomerular diseases. Postgrad Med J 1995; 71: 617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ward NC, Lui JB, Hernandez R, et al. Persistent IL‐2 receptor signaling by IL‐2/CD25 fusion protein controls diabetes in NOD mice by multiple mechanisms. Diabetes 2020; 69: 2400–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Carswell EA, Old LJ, Kassel RL, et al. An endotoxin‐induced serum factor that causes necrosis of tumors (activated macrophage). Immunology 1975; 72: 3666–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Granger GA, Shacks SJ, Williams TW, et al. Lymphocyte in vitro cytotoxicity: Specific release of lymphotoxin‐like materials from tuberculin‐sensitive lymphoid cells. Nature 1969; 221: 1155–1157. [DOI] [PubMed] [Google Scholar]
- 140. Chu WM. Tumor necrosis factor. Cancer Lett 2013; 328: 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Awad AS, You H, Gao T, et al. Macrophage‐derived tumor necrosis factor‐α mediates diabetic renal injury. Kidney Int 2015; 88: 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Chen P, Stärkel P, Turner JR, et al. Dysbiosis‐induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015; 61: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Nakamura A, Johns EJ, Imaizumi A, et al. Role of angiotensin II‐induced cAMP in mesangial TNF‐α production. Cytokine 2002; 19: 47–51. [DOI] [PubMed] [Google Scholar]
- 144. Timoshanko JR, Sedgwick JD, Holdsworth SR, et al. Intrinsic renal cells are the major source of tumor necrosis factor contributing to renal injury in murine crescentic glomerulonephritis. J Am Soc Nephrol 2003; 14: 1785–1793. [DOI] [PubMed] [Google Scholar]
- 145. Neale TJ, Rüger BM, Macaulay H, et al. Tumor necrosis factor‐alpha is expressed by glomerular visceral epithelial cells in human membranous nephropathy. Am J Pathol 1995; 146: 1444–1454. [PMC free article] [PubMed] [Google Scholar]
- 146. Watts TH. Tnf/tnfr family members in costimulation of T cell responses. Annu Rev Immunol 2005; 23: 23–68. [DOI] [PubMed] [Google Scholar]
- 147. Croft M. The role of TNF superfamily members in T‐cell function and diseases. Nat Rev Immunol 2009; 9: 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hasegawa G, Nakano K, Sawada M, et al. Possible role of tumor necrosis factor and interleukin‐1 in the development of diabetic nephropathy. Kidney Int 1991; 40: 1007–1012. [DOI] [PubMed] [Google Scholar]
- 149. Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF‐α increase prior to the rise in albuminuria in diabetic rats. Kidney Int 2003; 64: 1208–1213. [DOI] [PubMed] [Google Scholar]
- 150. Navarro JJ, Milena FF, Mora C, et al. Tumor necrosis factor‐α gene expression in diabetic nephropathy: relationship with urinary albumin excretion and effect of angiotensin‐converting enzyme inhibition. Kidney Int 2005; 68: S98–102. [DOI] [PubMed] [Google Scholar]
- 151. DiPetrillo K, Coutermarsh B, Gesek FA. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol ‐ Ren Physiol 2003; 284: 113–121. [DOI] [PubMed] [Google Scholar]
- 152. Baumgartl H, Sigl G, Banholzer P, et al. On the prognosis of IDDM patients with large kidneys. Nephrol Dial Transplant 1998; 13: 630–634. [DOI] [PubMed] [Google Scholar]
- 153. Kleinman KS, Fine LG. Prognostic implications of renal hypertrophy in diabetes mellitus. Diabetes Metab Rev 1988; 4: 179–189. [DOI] [PubMed] [Google Scholar]
- 154. Ramia NF, Kreydiyyeh SI. TNF‐α modulates the Na+/ K+ ATPase and the Na+K+2Cl− symporter in LLC‐PK1 cells. Eur J Clin Invest 2009; 39: 280–288. [DOI] [PubMed] [Google Scholar]
- 155. Malik S, Suchal K, Khan SI, et al. Apigenin ameliorates streptozotocin‐induced diabetic nephropathy in rats via MAPK‐NF‐κB‐TNF‐α and TGF‐β1‐MAPK‐fibronectin pathways. Am J Physiol ‐ Ren Physiol 2017; 313: F414–F422. [DOI] [PubMed] [Google Scholar]
- 156. Moriwaki Y, Yamamoto T, Shibutani Y, et al. Elevated levels of interleukin‐18 and tumor necrosis factor‐α in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism 2003; 52: 605–608. [DOI] [PubMed] [Google Scholar]
- 157. Lampropoulou IT, Stangou Μ, Sarafidis P, et al. TNF‐α pathway and T‐cell immunity are activated early during the development of diabetic nephropathy in Type II Diabetes Mellitus. Clin Immunol 2020; 215: 108423. [DOI] [PubMed] [Google Scholar]
- 158. Lampropoulou IT, Stangou M, Papagianni A, et al. TNF‐ α and microalbuminuria in patients with type 2 diabetes mellitus. J Diabetes Res 2014; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. MacIsaac RJ, Farag M, Obeyesekere V, et al. Changes in soluble tumor necrosis factor receptor type 1 levels and early renal function decline in patients with diabetes. J Diabetes Investig 2019; 10: 1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Niewczas MA, Gohda T, Skupien J, et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol 2012; 23: 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Gohda T, Niewczas MA, Ficociello LH, et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol 2012; 2012: 207–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Pavkov ME, Nelson RG, Knowler WC, et al. Elevation of circulating TNF receptors 1 and 2 increases the risk of end‐stage renal disease in American Indians with type 2 diabetes. Kidney Int 2015; 87: 812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001; 104: 487–501. [DOI] [PubMed] [Google Scholar]
- 164. Zhu Q, Scherer PE. Immunologic and endocrine functions of adipose tissue: implications for kidney disease. Nat Rev Nephrol 2018; 14: 105–120. [DOI] [PubMed] [Google Scholar]
- 165. Kramer H, Luke A, Bidani A, et al. Obesity and prevalent and incident CKD: the hypertension detection and follow‐up program. Am J Kidney Dis 2005; 46: 587–594. [DOI] [PubMed] [Google Scholar]
- 166. Maric‐Bilkan C. Obesity and diabetic kidney disease. Med Clin North Am 2013; 97: 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Felizardo RJF. Obesity in kidney disease: a heavyweight opponent. World J Nephrol 2014; 3: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Friedman AN, Chambers M, Kamendulis LM, et al. Short‐term changes after a weight reduction intervention in advanced diabetic nephropathy. Clin J Am Soc Nephrol 2013; 8: 1892–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Akbas EM, Demirtas L, Ozcicek A, et al. Association of epicardial adipose tissue, neutrophil‐to‐lymphocyte ratio and platelet‐to‐lymphocyte ratio with diabetic nephropathy. Int J Clin Exp Med 2014; 7: 1794–1801. [PMC free article] [PubMed] [Google Scholar]
- 170. Morcos M, Sayed AAR, Bierhaus A, et al. Activation of tubular epithelial cells in diabetic nephropathy. Diabetes 2002; 51: 3532–3544. [DOI] [PubMed] [Google Scholar]
- 171. Nishimura S, Manabe I, Nagasaki M, et al. CD8+effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009; 15: 914–920. [DOI] [PubMed] [Google Scholar]
- 172. Smith MJ, Simmons KM, Cambier JC. B cells in type 1 diabetes mellitus and diabetic kidney disease. Nat Rev Nephrol 2017; 13: 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Burrack AL, Martinov T, Fife BT. T cell‐mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol 2017; 8: 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Serreze DV, Fleming SA, Chapman HD, et al. B lymphocytes are critical antigen‐presenting cells for the initiation of T cell‐mediated autoimmune diabetes in nonobese diabetic mice. J Immunol 1998; 161: 3912–3918. [PubMed] [Google Scholar]
- 175. Nicoloff G, Blazhev A, Petrova C, et al. Circulating immune complexes among diabetic children. Clin Dev Immunol 2004; 11: 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Xiao X, Ma B, Dong B, et al. Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. J Autoimmun 2009; 32: 85–93. [DOI] [PubMed] [Google Scholar]
- 177. Ainsworth SK, Hirsch HZ, Brackett NC, et al. Diabetic glomerulonephropathy: histopathologic, immunofluorescent, and ultrastructural studies of 16 cases. Hum Pathol 1982; 13: 470–478. [DOI] [PubMed] [Google Scholar]
- 178. Imig JD, Ryan MJ. Immune and inflammatory role in renal disease. Compr Physiol 2013; 3: 957–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Duran‐Salgado MB, Rubio‐Guerra AF. Diabetic nephropathy and inflammation. World J Diabetes 2014; 5: 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Huang S‐W, Haedt LH, Rich S, et al. Prevalence of antibodies to nucleic acids in insulin‐dependent diabetics and their relatives. Diabetes 1981; 30: 873–874. [DOI] [PubMed] [Google Scholar]
- 181. Giardina E, Triolo G, Accardo‐Palumbo A, et al. Anti‐single‐stranded DNA antibody in the sera of patients with type 2 diabetes mellitus. Relation to vascular complications. Acta Diabetol 1997; 34: 39–41. [DOI] [PubMed] [Google Scholar]
- 182. Ronda N, Cravedi P, Benozzi L, et al. Early proinflammatory activation of renal tubular cells by normal and pathologic IgG. Nephron ‐ Exp Nephrol 2005; 100: 77–85. [DOI] [PubMed] [Google Scholar]
- 183. Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol 2000; 1: 475–482. [DOI] [PubMed] [Google Scholar]
- 184. Kim AHJJ, Chung J‐J, Akilesh S, et al. B cell–derived IL‐4 acts on podocytes to induce proteinuria and foot process effacement. JCI Insight 2017; 2: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Li T, Yu Z, Qu Z, et al. Decreased number of CD19+CD24hiCD38hi regulatory B cells in Diabetic nephropathy. Mol Immunol 2019; 112: 233–239. [DOI] [PubMed] [Google Scholar]
- 186. Mizoguchi A, Mizoguchi E, Takedatsu H, et al. Chronic intestinal inflammatory condition generates IL‐10‐producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002; 16: 219–230. [DOI] [PubMed] [Google Scholar]
- 187. Flores‐Borja F, Bosma A, Ng D, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med 2013; 5: 173ra23. [DOI] [PubMed] [Google Scholar]
- 188. Turner J‐E, Becker M, Mittrücker H‐W, et al. Tissue‐resident lymphocytes in the kidney. J Am Soc Nephrol 2018; 29: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Piet B, de Bree GJ, Smids‐Dierdorp BS, et al. CD8+ T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J Clin Investig 2011; 121: 2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Smolders J, Heutinck KM, Fransen NL, et al. Tissue‐resident memory T cells populate the human brain. Nat Commun 2018; 9: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]