

Abstract
Background/aims:
Safe and effective therapies for COVID-19 are urgently needed. In order to meet this need, the Accelerating COVID-19 Therapeutic Interventions and Vaccines public-private partnership initiated the Therapeutics for Inpatients with COVID-19 (TICO). TICO is a multi-arm, multi-stage platform master protocol, which facilitates the rapid evaluation of the safety and efficacy of novel candidate antiviral therapeutic agents for adults hospitalized with COVID-19. Five agents have so far entered the protocol, with rapid answers already provided for three of these. Other agents are expected to enter the protocol throughout 2021. This protocol contains a number of key design and implementation features that, along with challenges faced by the protocol team, are presented and discussed.
Methods:
Three clinical trial networks, encompassing a global network of clinical sites, participated in the protocol development and implementation. TICO utilizes a multi-arm, multi-stage design with an agile and robust approach to futility and safety evaluation at 300 patients enrolled, with subsequent expansion to full sample size and an expanded target population if the agent shows an acceptable safety profile and evidence of efficacy. Rapid recruitment to multiple agents is enabled through the sharing of placebo, the confining of agent-specific information to protocol appendices, and modular consent forms. In collaboration with the Food and Drug Administration, a thorough safety data collection and DSMB schedule was developed for the study of potential therapeutic agents with limited in-human data in hospitalized patients with COVID-19.
Results:
As of August 08th 2021, five agents have entered the TICO master protocol and a total of 1909 participants have been randomized to one of these agents or matching placebo. There were a number of challenges faced by the study team that needed to be overcome in order to successfully implement TICO across a global network of sites. These included ensuring drug supply and reliable recruitment allowing for changing infection rates across the global network of sites, the need to balance the collection of data and samples without overburdening clinical staff and obtaining regulatory approvals across a global network of sites.
Conclusion:
Through a robust multi-network partnership, the TICO protocol has been successfully used across a global network of sites for rapid generation of efficacy data on multiple novel antiviral agents. The protocol design and implementation features used in this protocol, and the approaches to address challenges, will have broader applicability. Mechanisms to facilitate improved communication and harmonization among country-specific regulatory bodies are required to achieve the full potential of this approach in dealing with a global outbreak.
Keywords: SARS-CoV-2, COVID-19, Multi-arm Multi-stage, platform trials
Background/aims
There is an urgent need for novel and effective antivirals against SARS-CoV-2 to reduce the substantial morbidity and mortality seen with COVID-19. To address this need, the Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) public-private partnership1 selected three clinical trial networks, the International Network for Strategic Initiatives in Global HIV Trials (INSIGHT),2 the Cardiothoracic Surgical Trials Network3 and the Prevention and Early Treatment of Acute Lung Injury network4 to collaborate, design and implement the ACTIV-3 protocol (Therapeutics for Inpatients with COVID-19 (TICO)). Given the urgent clinical need and the large number of emerging anti-SARS-CoV-2 therapeutic agents to be tested, the protocol team opted for a multi-arm multi-stage (MAMS) platform master protocol design. MAMS platforms have a number of advantages over traditional clinical trial design. These include, the ability to share/pool placebo controls across multiple agents, and the use of intermediate efficacy, futility and safety assessments such that only the most promising agents go forward into full enrollment, and the less promising are rejected early, thus avoiding overlapping or redundant work on parallel protocols. Along with these advantages, MAMS platforms maintain scientific rigor including double blinding, randomization, placebo control, using a single database and regular reviews of interim data by an independent Data and Safety Monitoring Board (DSMB) and provide guidelines for early termination based on group sequential methods.5, 6 These features ensure the most efficient use of already stretched clinical, and regulatory resources.
While platform protocol designs have been used successfully in many different settings, including during the current pandemic (e.g. RECOVERY trial (NCT04381936), WHO SOLIDARITY trial (ISRCTN83971151) and REMAP-CAP trial (NCT02735707)), these studies have primarily investigated re-purposed agents with relatively well-established safety profiles. TICO, however, was intended to provide rapid efficacy and safety data for novel antiviral agents in hospitalized patients, and to enable downstream drug regulatory approvals if an agent shows efficacy. Facilitated by a successful multi-network partnership and U.S Food and Drug Administration (FDA) collaboration, the protocol was designed and implemented rapidly (9 weeks from first protocol meeting to first participant randomised). So far, the TICO master protocol has generated results for three novel agents, LYCoV5557 (Eli Lilly and Company), Vir-78318 (GlaxoSmithKline and Vir Biotechnology), and Brii-196/1988 (Brii Biosciences Limited). Two other agents, AZD7442 (AstraZeneca) and MP0420 (Molecular Partners) remain under study, with further agents poised to enter the protocol throughout 2021 and beyond. There were a number of key design and implementation features of the TICO master protocol that enabled the rapid recruitment and results generated by this protocol. These features are presented in the Results section of this manuscript, while the challenges faced by the study team are presented and discussed in the Challenges and summarized in Table 1.
Table 1.
Challenges in protocol design and implementation
| Challenges | Implemented or proposed solutions |
|---|---|
| Ensuring drug supply across a global network of sites | • Request for waiving of relabelling requirements • Centralised drug management and distribution through two drug depots • Pragmatic registration and activation • Use of centralised pharmacies to serve multiple sites |
| Data and sample collection during a pandemic | • Reducing reporting burden through the use of protocol specified exempt events • Attempting to balance safety reporting with burden on site staff • Phone visits for discharged patients • Use of contractors for post-discharge sample collection |
| Regulatory approval and study implementation outside the U.S. | • Sharing of regulators responses across the network • Use of a master protocol design with all new agent specific changes in an appendix • Mechanism to allow communication between relevant agencies in different countries |
Methods
Protocol oversight and network integration
The National Institute of Allergy and Infectious Diseases (NIAID) serves as the overall sponsor. Sites outside the U.S. are sponsored by the University of Minnesota to accommodate the regulatory challenges posed by the European Union Global Data Protection Regulation. A Trial Oversight Committee has been established to provide oversight for the ACTIV-2 (NCT04518410), ACTIV-6 (NCT04885530) and ACTIV-3 initiatives and includes the trial co-chairs, representatives from Operation Warp Speed/Countermeasures Acceleration Group therapeutics and NIAID. Additional voting members include leaders from the National Heart, Lung and Blood institute (NHLBI), Biomedical Advanced Research and Development Authority, FDA and the National Center for Advancing Translational Sciences. The Trial Oversight Committee also has responsibility for approving agents for entry into the TICO protocol, based on recommendations from the ACTIV agent selection committee. Candidate agents are submitted for consideration for TICO through a public portal, before undergoing a systematic scientific review by the ACTIV Agent Selection Committee. The Trial Oversight Committee votes on whether an agent enters TICO and considers a number of factors, including safety, in vitro potency against the virus, potential for viral resistance to arise, target epitope and potency (if the agent is an antibody), scale-up potential and dose and route of administration. ACTIV leadership requested TICO focus initially on neutralising monoclonal antibodies, with expansion to other novel antiviral agents as these become available.
The TICO protocol team (see supplemental materials) is responsible for scientific and operational oversight. Implementation is coordinated by the INSIGHT Coordinating Centre at the University of Minnesota in collaboration with eight International Coordinating Centres (six from INSIGHT and one each representing the Cardiothoracic Surgical Trials Network and the Prevention and Early Treatment of Acute Lung Injury network). All have extensive experience managing clinical trials and work with >300 sites across North and South America, Europe, Australia, Africa and Asia. This collaboration of large diverse networks is important for three reasons. Firstly, a large global network is essential for recruitment, especially as case rates during the pandemic fluctuate regionally in unpredictable ways. Secondly, a broad range of clinical sites across multiple countries and continents results in a demographically diverse study population, allowing determination of the breadth of applicability of any beneficial treatments. Thirdly, standard approaches for operations and trial conduct naturally vary across networks, and through collaboration the most effective and efficient from each network can be elevated and disseminated as ‘best practice’ across the full collaborative network.
In order to facilitate rapid approval and implementation of the protocol across the diverse network, certain roles and responsibilities are distributed to the International Coordinating Centres, with central oversight by the INSIGHT Coordinating Centre. The INSIGHT Coordinating Centre manages drug distribution (in collaboration with PCI pharma services), central specimen storage and lab kit distribution (in collaboration with Advanced Biomedical Laboratories) and acts as the Statistical Data Management Centre. Each International Coordinating Centre is responsible for the implementation and management of clinical research sites within their networks, including registration, regulatory approval, site training, lab kit ordering, drug orders, monitoring and ensuring data quality. To further facilitate implementation, International Coordinating Centre’s often utilize in-country hubs, called Site Coordinating Centres, who have extensive experience with regulatory and other requirements unique to their network of sites. See Supplemental Table 1 for details on the International and Site Coordinating Centers as well as participating TICO sites.
Primary objective, primary endpoint and intermediate outcomes of efficacy
The TICO primary objective is to determine whether investigational agents are safe and efficacious compared with placebo when given with current standard of care (therapies strongly recommended by national/international guidelines based on high-quality evidence; including remdesivir and glucocorticoids as of August 2021). Local standard of care is also permitted and the appendix pertaining to standard of care is amended as new evidence emerges (including results from TICO itself). For more details see appendix I in the protocol (supplemental material).
The primary efficacy endpoint is time to sustained recovery through day 90, defined as when a participant is discharged from the hospital to home and remains alive and at home for at least 14 consecutive days. This patient-centered endpoint was chosen because of the extended duration of health impairment associated with COVID-19. 9–11 The longer follow-up to capture this endpoint (compared to the common 28 days12–14) was designed to provide a more comprehensive assessment of the capacity of a therapeutic agent to speed recovery from COVID-19. The operationalization of this endpoint is detailed further in supplemental materials.
The study uses two intermediate outcomes, assessed at Day 5, as part of the initial assessment of futility (described further in the next section); the Pulmonary and Pulmonary Plus ordinal outcomes (Table 2). The Pulmonary outcome is a 7-category outcome largely based on the degree of respiratory failure, adapted from a similar outcome used in the ACTT-1 study13 and an initial World Health Organization master protocol.15 The Pulmonary Plus ordinal outcome adds extra-pulmonary conditions to the pulmonary outcome and covers a range of organ dysfunction associated with COVID-19. Three key considerations drove the intermediate outcome selection: capacity to quickly assess for potential efficacy and safety, a hypothesized high correlation with the primary endpoint of time to sustained recovery, and capacity to capture both pulmonary and non-pulmonary events among participants. Use of the primary endpoint for the initial futility and safety assessments was deemed impracticable, as it requires substantial follow-up time for ascertainment. Intermediate assessments must thus be made at much earlier time points. In unpublished analyses of ACTT-1 data and review of the literature on COVID-19 prognostication, the probability that this intermediate outcome correlated with time to recovery (essentially discharge by 28 days) was very high. Analyses of these data also suggested that day 5 would be provide good prognostication of recovery. A strong positive association between the ordinal outcomes at day 5 and the primary endpoint was observed with the first agent assessed using the TICO protocol.16 The statistical analysis plan related to the analysis of these outcomes, and other secondary outcomes, is provided in the supplemental materials.
Table 2.
Intermediate Ordinal Outcomes
| Pulmonary outcome | Pulmonary Plus outcome |
|---|---|
| 1. Can independently undertake usual activities with minimal or no symptoms | 1. Can independently undertake usual activities with minimal or no symptoms |
| 2. Symptomatic and currently unable to independently undertake usual activities but no need of supplemental oxygen (or not above premorbid requirements) | 2. Symptomatic and currently unable to independently undertake usual activities but no need of supplemental oxygen (or not above premorbid requirements) |
| 3. Supplemental oxygen (<4 liters/min, or <4 liters/min above premorbid requirements) | 3. Supplemental oxygen (<4 litres/min, or <4 litres/min above premorbid requirements) |
| 4. Supplemental oxygen (>4 liters/min, or >4 liters/min above premorbid requirements, but not high-flow oxygen) | 4. Supplemental oxygen (>4 litres/min, or >4 litres/min above premorbid requirements, but not high-flow oxygen) or any of the following: stroke (NIH Stroke Scale [NIHSS] <14), meningitis, encephalitis, myelitis, myocardial infarction, myocarditis, pericarditis, new onset CHF NYHA class III or IV or worsening to class III or IV, arterial or deep venous thromboembolic events. |
| 5. Non-invasive ventilation or high-flow oxygen | 5. Non-invasive ventilation or high-flow oxygen, or signs and symptoms of an acute stroke (NIHSS >14) |
| 6. Invasive ventilation, extracorporeal membrane oxygenation (ECMO), mechanical circulatory support, or new receipt of renal replacement therapy | 6. Invasive ventilation, ECMO, mechanical circulatory support, vasopressor therapy, or new receipt of renal replacement therapy |
| 7. Death | 7. Death |
Multi-arm, Multi-stage design of TICO
TICO is designed as a randomized, double blinded, placebo-controlled phase III MAMS platform master protocol. For any agent, at the outset of the trial, only participants without severe end-organ disease (Disease Stratum 1) are enrolled. This more restricted enrollment continues until approximately 150 participants per study arm are enrolled and followed for 5 days. At this point, the DSMB caries out a pre-specified assessment of futility, based on the two ordinal outcomes (pulmonary and pulmonary plus), assessed at Day 5. Safety of the investigational agents is assessed. For investigational agents passing this initial futility assessment, enrolment expands seamlessly, without any unblinding of data, to also include patients with end organ disease (Disease Stratum 2).
Table 3 outlines eligibility criteria related to both disease strata. The target population is narrower initially to expedite identification of early signals of safety and efficacy as patients with end organ dysfunction are unlikely to recover over 5 days and assessment of safety is more challenging. The expansion to include more severely ill participants is contingent on FDA and DSMB recommendations. If the initial futility assessment is passed, futility assessments at future interim analyses are based on the primary endpoint of sustained recovery and use pre-specified guidelines to determine early evidence of benefit, harm or futility for the investigational agent. There is no pre-specified sample size for the additional interim analyses. These are performed at subsequent full DSMB reviews. For monitoring benefit, type 1 error is controlled with the use of the Lan-DeMets spending function analogue of the O’Brien-Fleming Boundaries. Once the full sample size is reached (estimated to be 1000 participants, equally allocated to each investigational agent and placebo), a confirmatory efficacy and safety analysis takes place (Figure 1). Procedures for data collection and primary endpoint ascertainment do not change for agents that pass the initial futility assessment, and all patients recruited prior to the futility assessment are included in the final efficacy assessment. For considerations related to the sample size chosen for both the initial futility assessment and the final efficacy assessment, see supplemental materials.
Table 3.
Non-agent specific inclusion and exclusion criteria from the TICO master protocol
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| 1. Age > 18 years 2. Informed consent by the patient or the patient’s legally authorized representative 3. SARS-CoV-2 infection, documented by PCR or other nucleic acid test (NAT) within 3 days prior to randomization OR documented by NAT more than 3 days prior to randomization AND progressive disease suggestive of ongoing SARS-CoV-2 infection per the responsible investigator; 4. Duration of symptoms attributable to COVID-19 < 12 days per the responsible investigator; 5. Requiring admission for inpatient hospital acute medical care for clinical manifestations of COVID-19, per the responsible investigator, and NOT for purely public health or quarantine purposes. |
1. Prior receipt of any SARS-CoV-2 hyperimmune intravenous immunoglobulin, convalescent plasma from a person who recovered from COVID-19 or SARS-CoV-2 neutralising monoclonal antibody at any time prior to hospitalization 2. In the opinion of the responsible investigator, any condition for which, participation would not be in the best interest of the participant or that could limit protocol-specified assessments; 3. Expected inability to participate in study procedures 4. Women of child-bearing potential who are not already pregnant at study entry and who are unwilling to abstain from sexual intercourse with men or practice appropriate contraception through Day 90 of the study 5.Men who are unwilling to abstain from sexual intercourse with women of child-bearing potential or who are unwilling to use barrier contraception through Day 90 of the study. 6. [Prior to the inclusion of disease stratum 2] Presence at enrolment of any of the following: a) stroke b) meningitis c) encephalitis d) myelitis e) myocardial infarction f) myocarditis g) pericarditis h) symptomatic congestive heart failure (NYHA class III-IV) i) arterial or deep venous thrombosis or pulmonary embolism 7. [Prior to the inclusion of disease stratum 2] Current or imminent requirement for any of the following: a) invasive mechanical ventilation b) ECMO c) mechanical circulatory support d) vasopressor therapy e) commencement of renal replacement therapy at this admission (i.e. not patients on chronic renal replacement therapy). |
Figure 1.
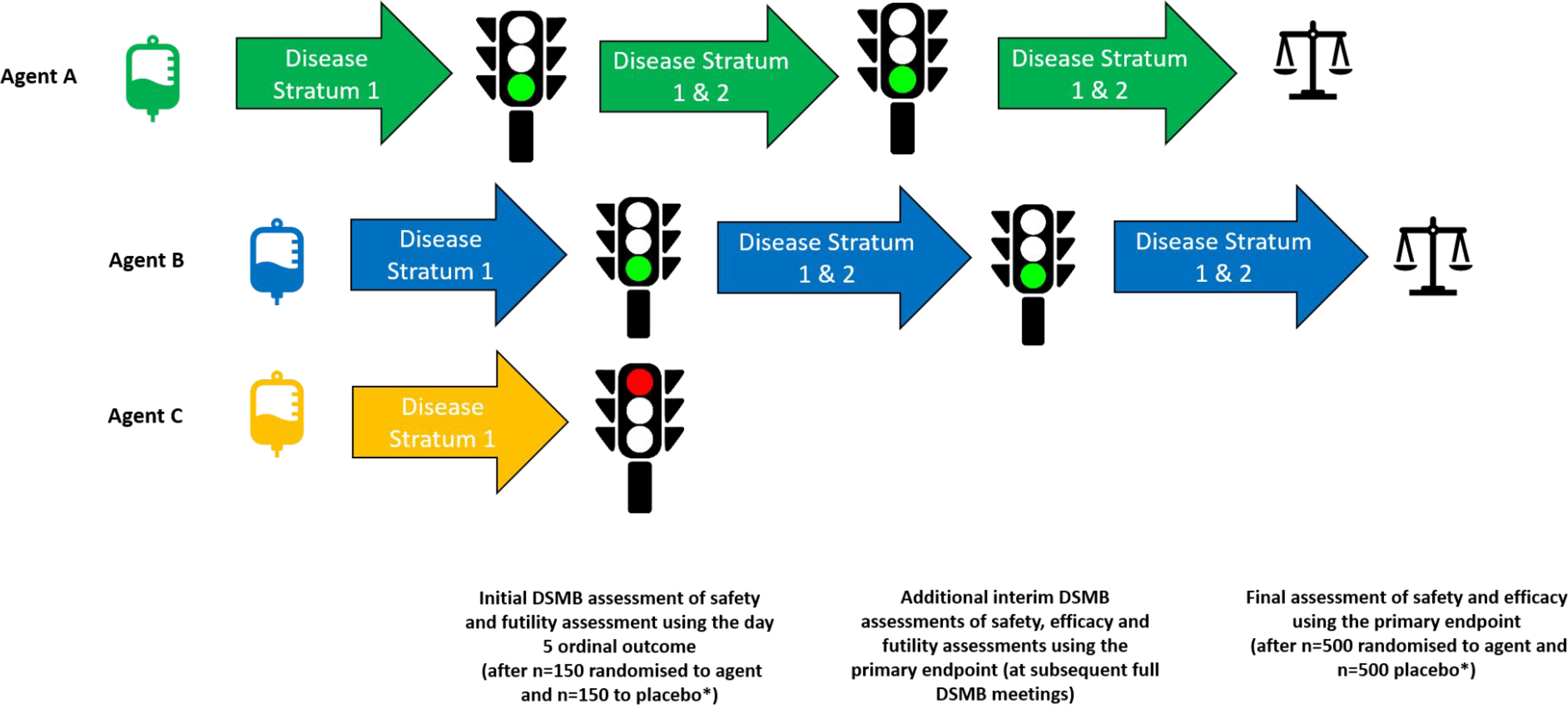
Agent entry and progression through the TICO study. The TICO study allows for multiple agents to be studied concurrently and for agents to enter the study at different time-points. In the theoretical scenario presented in this figure, Agent A is the only agent that is available for randomisation at the beginning of the study. Later, Agent B and Agent C enter the study, and new participants are able to be randomized to all three agents (and placebo). Agent A completes recruitment in Disease Stratum 1 and, after the initial futility assessment by the independent DSMB (using the day 5 ordinal outcome), the agent is approved to also include those in Disease Stratum 2 (i.e. those with end organ disease, including requirements for invasive mechanical ventilation or ECMO ). Agent B and Agent C, entering the study at the same time, but after Agent A, both progress to the initial futility assessment. However, only Agent B receives DSMB approval to proceed and randomisation to Agent C ceases. Agent A and Agent B continue to recruit in both Disease Stratum 1 and 2 and undergo additional interim safety, efficacy and futility assessments (using the primary endpoint) at subsequent full DSMB meetings before undergoing a final review of safety and efficacy (using the primary endpoint) when recruitment is complete (graphically represented by the image of scales). * As outlined in Use of Shared Placebo in TICO section of this manuscript, the placebo group may be shared across multiple agents (not graphically represented)
Use of shared placebo in TICO
In TICO, each randomised participant could potentially receive any of the active agents for which they are eligible. The placebo group is then “pooled” so those randomized to the placebo of one agent will also serve as controls for other agents to which the person could have been allocated. Thus, the probability of being allocated to any one investigational agent is the same as being allocated to placebo. The more concurrent agents under study at any given time therefore increases the probability of a participant being randomized to an active agent while also reducing the total number of placebo participants required for an experimental agent to reach crucial milestones. For example, the first agent to be studied using this protocol, LYCoV555 entered the protocol by itself and 314 participants were recruited over a 10-week period prior to the futility assessment. The second two agents, the Vir-7831 and Brii-196/198, began concurrently and were able to share placebo. At the time of the futility assessment for these agents, ~11 weeks after first patient recruited, 168 participants had available day-5 data for Vir-7831, 166 for Brii-196/198 and 173 for placebo. If placebo were not shared, another 100 participants would have been needed for the futility assessment, costing time and resources, for the same result.
Separate appendices for investigational agents and modular consent forms
Key to the success of TICO was the ability for multiple agents to be studied concurrently and for new agents to enter the protocol seamlessly. To facilitate this, the master protocol itself contained all relevant information and study procedures applicable to the broad conduct of the trial. Agent-specific information (including unique eligibility criteria, if any) is provided in individual appendices (Supplemental Table 2). Thus, the entry of a new agent simply involves review of a new appendix by regulatory bodies and ethical boards, and the master protocol remains intact. This approach coupled with a modular information statement and consent form, with additional information sheets on individual drugs, and their side-effect profile, minimizes duplication for regulatory and site staff. To accommodate instances where an individual cannot or should not be randomized to one or several of the agents (e.g. if agent specific eligibility criteria excludes them or an agent is unavailable due to pending regulatory approval, supply-chain or storage issues), two further key features were added. First, a randomization application was developed that factors in potential differences in both availability of study product and eligibility criteria between agents (see supplemental materials). Second the use of modular consents, as described above, easily allows investigators to inform participants which agents they may receive and then present the appropriate drug information.
Safety data collection and DSMB schedule for the study of novel agents
Many of the agents to be studied in TICO have limited in-human safety data. To ensure patient safety and adequate capture of data for future emergency use authorisations and/or new drug application, the FDA reviewed and provided feedback on the protocol and DSMB schedule. As guided by the FDA, the specific safety collection (Supplemental Table 3) includes infusion-related reactions, targeted day 5 laboratory results (centrally graded) along with frequent assessments of adverse events, serious adverse events, and unexpected problems while hospitalised and post-discharge. For the first agent, participants were followed for 90 days. For next three agents, follow-up was extended to 18 months, due to longer half-lives for the new agents. The data collection beyond 90 days is restricted to death and hospitalizations, which was judged to provide sufficient information for safety monitoring without overburdening site staff. To review these safety data, and ensure safety of participants throughout the protocol, the DSMB conducts regular meetings while an agent is under study including a very early review (after 20–30 participants have day-5 data), at the initial futility assessment at around 150 participants in a given experimental arm, and subsequent futility assessments (for more details see page 9 of the supplemental statistical analysis plan). The DSMB also receives weekly safety reports and can choose to convene additional meetings should concerning safety signals emerge.
Results
As of 8th of August 2021, the TICO protocol has been amended three times. As outlined earlier in this manuscript, each protocol version adds a new agent or agents. V1.0 included the Lilly neutralizing monoclonal antibody Ly-CoV555. V2.0 of the protocol included the GSK/Vir neutralizing monoclonal antibody Vir-7831 and the Brii Bioscience neutralizing monoclonal antibodies Brii-196/198; randomization started after randomization to LY-CoV555 was complete. V3 of the protocol added the AstraZeneca neutralising monoclonal antibody AZD7442. V4 of the protocol added the Molecular Partners DARPin® molecule MP0420.
In total, 1909 participants were randomised into the TICO protocol as of 8 August 2021 (326 to the LyCoV555 (or its placebo), 361 to Brii-196/198, 367 to Vir-7831, 980 to AZD7442 and 125 to MP0420). Note that for agents studied at the same time, placebo was shared across more than one agent, which is why the total randomized is smaller than the sum of the participants used to study each individual agent. Of the agents that have entered the protocol so far, Ly-CoV555, Vir-7831 and Brii-196/198 did not pass the initial futility assessment and were discontinued, while AZD7442 and MP0420 remain under study. For a timeline of agent entry into TICO and key events in the study, see Figure 2.
Figure 2.
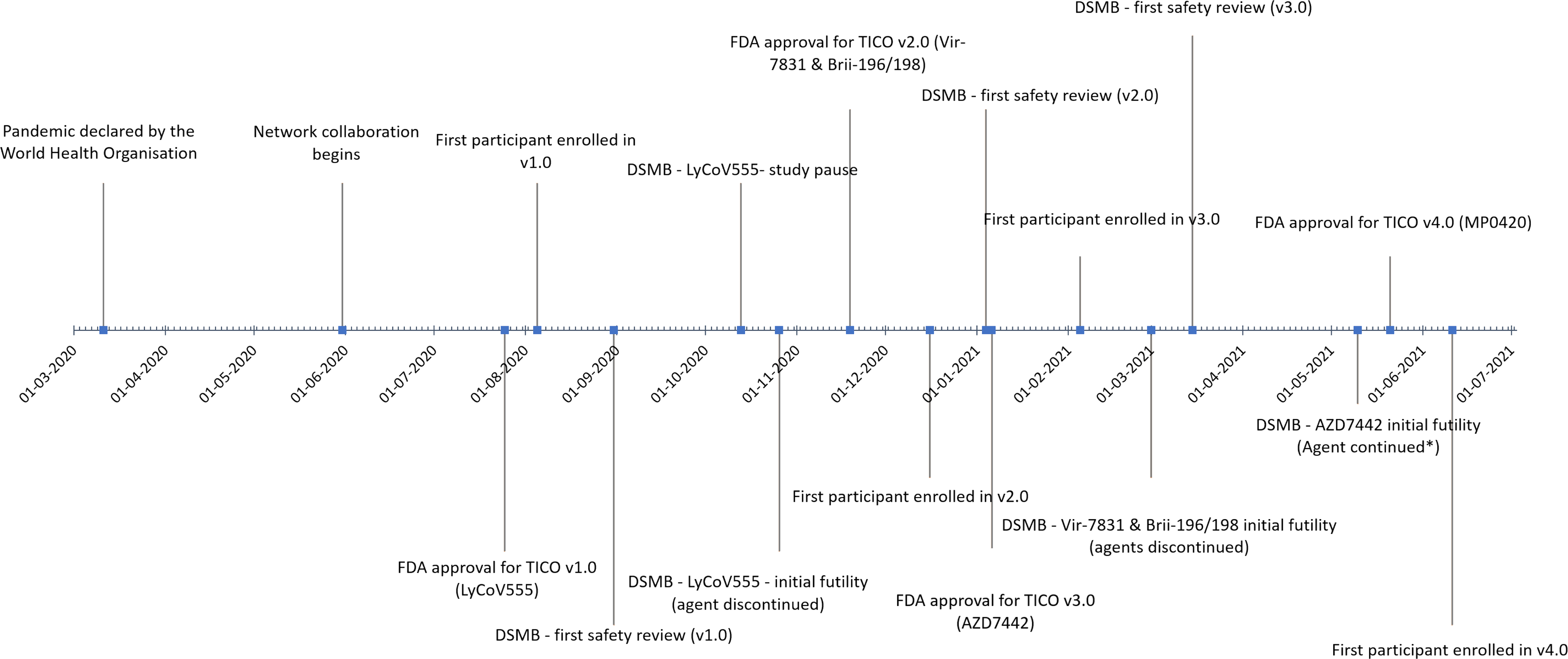
TICO Timeline and Milestones. Key milestones for the TICO protocol, including agent specific information (FDA approval, first participant enrolled, first safety review and initial futility assessment by the independent DSMB) are presented.
Challenges
Ensuring drug supply across a global network of sites
A major challenge faced was ensuring timely drug supply across a global network of sites to match the dynamic infection rates across geographical areas. A number of strategies were implemented to overcome this challenge. First, regulatory bodies were asked to waive the requirements to relabel study drugs, including translation into local languages, according to the local regulatory requirements. Secondly, drug distribution was centralized to two drug depots (one in the U.S. and one in the UK which later moved to Ireland), and the INSIGHT Coordinating Centre and International Coordinating Centres closely monitored drug supply at individual sites through a central drug management database. This allowed the protocol team to monitor drug supply closely and send additional product to sites in need. Despite this, drug shipment to non-US and non-European study sites were occasionally hampered by freight availability. Thirdly, in an attempt to best utilize the global network of sites and respond to the changing nature of global infection rates TICO registers all sites proactively, when all appropriate regulatory, registration and training documentation is in place, but only activates a site and ships study product when there is evidence or expectation of local disease activity. Finally, as infection rates and recruitment capabilities vary even across the same country/city, clinical sites are encouraged to select a pharmacy that can serve multiple clinical sites within a close geographical area, as opposed to a more traditional one-site one-pharmacy model (see Pharmacy Options in Supplemental materials). This one-pharmacy, multiple-sites model has resulted in efficient drug-distribution and reduced waste. Notable successes of this model were at Centre of Excellence for Health Immunity and Infections, Rigshospitalet, Denmark (one-pharmacy, 10 sites), Hospital Universitari Germans trias I Pujol, Spain (one-pharmacy, six sites), Hospital Universitario La Paz, Spain (one-pharmacy,three sites), Evangelismos Hospital, Greece (one pharmacy, four sites), Medical Research Council, Uganda Virus Research Institute, London School of Hygiene & Tropical Medicine Uganda Research Unit, Uganda (one pharmacy, six sites), Duke University United States (one pharmacy, three sites), University of California San Francisco United States (one pharmacy, two sites) and Cleveland Clinic Foundation United States (one pharmacy, three sites).
Real-time data and sample collection during a pandemic
Detailed and well-standardized data collection during and after hospitalization (in particular regular assessments of the primary endpoint post discharge) is essential for the regular safety and clinical efficacy assessments, as well as any future emergency use authorisations or new drug applications. Further, collection of baseline and follow-up biological samples (including plasma, serum and nasopharyngeal swabs) were essential for protocol defined safety and laboratory assessments (e.g. viral load measurements) and future research (details on timing of sample collection is detailed in section 9 of the protocol). Due to local surges in case numbers during the pandemic, however, extensive data and sample collection carried the danger of overwhelming the research staff at affected clinical sites, with health care worker infections exacerbating the situation. Additionally, stringent infection control measures posed challenges for patient review and sample collection, particularly post-discharge.
To reduce the burden on site staff, data collection was carefully calibrated. For example, clinical events that were already captured as part of the ordinal outcomes or other secondary objectives were exempt from additional serious adverse event reporting (unless deemed related to an investigational agent). These “protocol specified exempt events” were defined in the protocol. Further, after day-7, adverse events of any grade were collected as a snapshot on day 14 and day 28 only, while grade 3 and 4 adverse events were collected retrospectively on day 14 and day 28). Longer term follow-up (after 90 days) was limited to vital status and hospitalisation only, which, as described above, was a balance between capturing key outcomes without overwhelming the clinical sites. Finally, some of the post-discharge study assessments were preformed over the phone, and, in some settings, contractors were hired to visit participants at home for post-discharge sample collection.
Regulatory approval and study implementation outside the U.S.
Due to the involvement of the US FDA, and a central ethics review by Advarra®, study implementation was rapid within the US. However, regulatory approval and study implementation outside of the US occurred slower and was a major challenge for the protocol team. For example, in the LYCoV555 sub-study, only Denmark, Spain, UK and Singapore received approval by both ethics and medicines agencies by the time of the futility assessment, and only Denmark and Singapore opened in time to recruit. There were three main reasons for these delays. Firstly, submission of the protocol to countries outside the US required approval by both the FDA and Advarra® before the submission process could even begin. Secondly, due to the huge increase in COVID-19 related projects, many countries were facing a backlog of COVID-19 clinical trials applications and fast-track systems developed during the early phase of the pandemic became overwhelmed. Thirdly, regulatory agencies were reviewing data on novel antiviral agents for the first time and this necessitates careful review. Often, responses to these reviews required input from the pharmaceutical companies (specifically around pre-clinical data included in the agent’s submission data). All these factors led to delays in approvals.
A number of strategies were implemented to speed up regulatory reviews, including sharing of responses across International Coordinating Centres to more swiftly deal with common questions and the use of Site Coordinating Centres to better coordinate submissions in specific countries. Future versions of the protocol may proceed more swiftly as regulatory agencies only need to review the additional appendix with no major changes to the master protocol. However, global recruitment into large platform trials has the potential to substantially speed up the development of new treatments in a pandemic and ways to improve global implementation should be prioritized moving forward. One such improvement would be a formal mechanism that allows sharing of reviews between regulatory agencies (particularly between the FDA and other agencies). This way, the regulatory agency for each new participating country would have the benefit of communicating with other regulatory bodies and reviewing prior approvals and additional requested data. The intent would be to avoid repeated questions, give more confidence to the reviewing agency and generally speed up reviews.
Conclusion
The TICO master protocol responds to the urgent need to accelerate the development of safe, efficacious, novel antivirals for hospitalized COVID-19 patients. Through a successful collaboration of clinical trial networks, TICO has been rapidly and successfully designed and implemented globally. TICO is an efficient, flexible, rigorous MAMS platform master protocol that allows for concurrent safety and efficacy evaluation of multiple novel antiviral agents, with agents able to enter at different times. The use of an early futility assessment allows for the rapid selection of only the most promising agents for full evaluation using a clinically relevant primary endpoint, and therefore quickly removing agents from the trial that fail to demonstrate potential efficacy. Crucially, the thorough safety data collection and frequent DSMB reviews allow speed and safety to co-exist. To the best of the authors knowledge, TICO is one of only two protocols (along with the ACTIV-2 protocol; NCT04518410) to utilize a MAMS design during a pandemic. The success of TICO shows the broad applicability of MAMS designs, which have previously only been used in cancer trials (e.g. STAMPEDE 5), and the unique combination of features in TICO may inform future clinical trial design in other disease areas where there is a great need to quickly concentrate resources on the most promising therapeutic agents. Finally, the challenges faced by the study team, and in particular the difficulties obtaining regulatory approval across a global network of sites, need to be addressed in order to improve the ability to rapidly respond on a global level.
Supplementary Material
Acknowledgements
The authors would like to thank the large number individuals across all the contributing trials networks, government agencies, local clinical sites, laboratory staff, pharmacists, logistics personnel, institutional review boards and regulatory bodies who contributed to the design and implementation of the TICO protocol.
Funding
TICO is funded primarily through Operation Warp Speed (now Countermeasure Acceleration Group) as a sub-contract through Leidos Biomedical Research Inc.. Additional funding was provided by the NIH (including NHLBI, NIAID), U.S. Department of Veterans Affairs, as well as the governments of Denmark (National Research Foundation; grant no 126), Australia (National Health and Medical Research Council), U.K. (Medical Research Council, MRC_UU_12023/23), and Singapore (Singapore National Medical Research Council; COVID19RF-0005).
References
- 1.Collins FS and Stoffels P. Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV): An Unprecedented Partnership for Unprecedented Times. JAMA 2020; 323: 2455–2457. [DOI] [PubMed] [Google Scholar]
- 2.INSIGHT clinical trials network, https://insight.ccbr.umn.edu/.
- 3.CTSN Cardiothoracic Surgical Trials Network, http://www.ctsurgerynet.org/.
- 4.PETAL Network. Prevention & Early Treatment of Acute Lung Injury, https://petalnet.org/.
- 5.Sydes MR, Parmar MKB, James ND, et al. Issues in applying multi-arm multi-stage methodology to a clinical trial in prostate cancer: the MRC STAMPEDE trial. Trials 2009; 10: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Royston P, Parmar MK and Qian W. Novel designs for multi-arm clinical trials with survival outcomes with an application in ovarian cancer. Stat Med 2003; 22: 2239–2256. [DOI] [PubMed] [Google Scholar]
- 7.Lundgren JD, Grund B, Barkauskas CE, et al. A Neutralizing Monoclonal Antibody for Hospitalized Patients with Covid-19. N Engl J Med 2021; 384: 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.National Institutes of Health. NIH-Sponsored ACTIV-3 Clinical Trial Closes Enrollment into Two Sub-Studies, https://www.nih.gov/news-events/news-releases/nih-sponsored-activ-3-clinical-trial-closes-enrollment-into-two-sub-studies (2021, accessed 18 March 2021).
- 9.Mitrani RD, Dabas N and Goldberger JJ. COVID-19 cardiac injury: Implications for long-term surveillance and outcomes in survivors. Heart Rhythm 2020; 17(11): 1984–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.CDC COVID-19 Resonse Team. Preliminary Estimates of the Prevalence of Selected Underlying Health Conditions Among Patients with Coronavirus Disease 2019 - United States, February 12-March 28, 2020. MMWR Morb Mortal Wkly Rep 2020; 69: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung T, Chan A, Chan EW, et al. Short- and Potential Long-term Adverse Health Outcomes of COVID-19: A Rapid Review. Emerg Microbes Infect 2020; 9(1): 2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The RECOVERY Collaborative Group, Horby P, Lim WS, et al. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N Engl J Med 2021; 348(8): 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the Treatment of Covid-19 — Final Report. N Engl J Med 2020; 383: 1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dodd LE, Follmann D, Wang J, et al. Endpoints for randomized controlled clinical trials for COVID-19 treatments. Clin Trials 2020; 17: 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Health Organization. COVID-19 Therapeutic Trial Synopsis, https://www.who.int/publications/i/item/covid-19-therapeutic-trial-synopsis (2020).
- 16.ClinicalTrials.gov. A Study of LY3819253 (LY-CoV555) in Participants Hospitalized for COVID-19, https://ClinicalTrials.gov/show/NCT04411628.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.