

Abstract
Technologies that precisely delete genomic sequences in a programmed fashion can be used to study function as well as potentially for gene therapy. The leading contemporary method for programmed deletion uses CRISPR-Cas9 and pairs of guide RNAs (gRNAs) to generate two nearby double-strand breaks, which is often followed by deletion of the intervening sequence during DNA repair. However, this approach can be inefficient and imprecise, with errors including small indels at the two target sites as well as unintended large deletions and more complex rearrangements. Here we describe a prime editing-based method that we term PRIME-Del, which induces a deletion using a pair of prime editing gRNAs (pegRNAs) that target opposite DNA strands, effectively programming not only the sites that are nicked but also the outcome of the repair. We demonstrate that PRIME-Del achieves markedly higher precision than CRISPR-Cas9 and gRNA pairs in programming deletions up to 10 kb. We also show that PRIME-Del can be used to couple genomic deletions with short insertions, enabling deletions whose junctions do not fall at protospacer-adjacent motif (PAM) sites. Finally, we demonstrate that lengthening the time window of expression of prime editing components can substantially enhance efficiency without compromising precision. We anticipate that PRIME-Del will be broadly useful in enabling precise, flexible programming of genomic deletions, including in-frame deletions, as well as for epitope tagging and potentially for programming rearrangements.
Introduction
The ability to precisely manipulate the genome can critically enable investigations of the function of specific genomic sequences, including genes and regulatory elements. Within the past decade, CRISPR-Cas9-based technologies have proven transformative in this regard, allowing precise targeting of a genomic locus, with a quickly expanding repertoire of editing or perturbation modalities1. Among these, the precise and unrestricted deletion of specific genomic sequences is particularly important, with critical use cases in both functional genomics and gene therapy.
Currently, the leading method for programming genomic deletions uses a pair of CRISPR guide RNAs (gRNAs) that each target a protospacer-adjacent motif (PAM) sequence, generating a pair of nearby DNA double-strand breaks (DSBs). Upon simultaneous cutting of two sites, cellular DNA damage repair factors often ligate two ends of the genome without the intervening sequence2 through non-homologous end joining (NHEJ) (Figure 1a). Although powerful, this approach has several limitations: 1) An attempt to induce a deletion, particularly a longer deletion, often results in short insertions or deletions (indels; typically less than 10-bp) near one or both DSBs, with or without the intended deletion3–5; 2) Other unintended mutations including large deletions and more complex rearrangements can frequently occur, and go undetected for technical reasons5–8; 3) DSBs are a cytotoxic insult9; and 4) The junctions of genomic deletions programmed by this method are limited by the distribution of naturally occurring PAM sites. Notwithstanding these limitations, various studies have employed this strategy to great effect, e.g. to investigate the function of genes and regulatory elements5,10,11, as well as towards gene therapy12,13. However, limited precision, DSB toxicity and the inability to program arbitrary deletions have handicapped the utility of CRISPR-Cas9-induced deletions in functional and therapeutic genomics.
Figure 1. Precise episomal deletions using PRIME-Del.
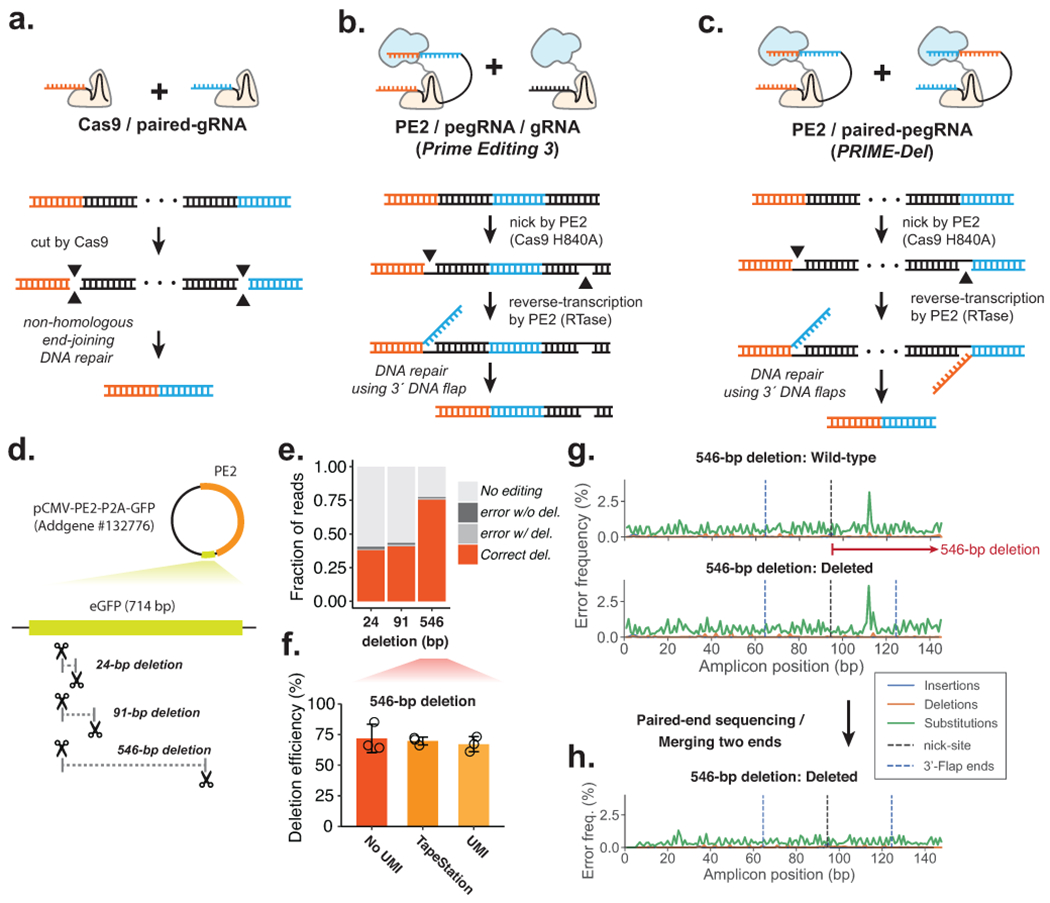
a. Schematic of Cas9/paired-gRNA deletion strategy. b. Schematic of PE3 strategy, wherein the Prime Editor-2 enzyme and gRNA complex induce a nick (denoted as a gap in the bottom DNA strand), even after the correct editing event. c. Schematic of PRIME-Del using pairs of pegRNAs that target opposite DNA strands. Each pegRNA encodes the sites to be nicked at each end of the intended deletion, as well as a 3’ flap that is complementary to the region targeted by the other pegRNA. d. Cartoon representation of deletions programmed within the episomally-encoded eGFP gene (not drawn to a scale). e. PRIME-Del-mediated deletion efficiencies and error frequencies (with or without intended deletion) were measured for 24-bp, 91-bp, and 546-bp deletion experiments in HEK293T cells (averaged over replicates; n = 5). Sequencing reads were classified as without indel modifications (“No editing”), indel errors without the intended deletion, indel errors with the intended deletion, and correct deletion without error. f. PRIME-Del-mediated deletion efficiency was measured for the 546-bp deletion experiment using three methods. Error bars represent standard deviation for three replicates. g. Insertion, deletion and substitution error frequencies across sequencing reads from 546-bp deletion experiment. Reads were aligned to reference sequence either without (top) or with (bottom) deletion. Plots are from single-end reads with collapsing of UMIs to reduce sequencing errors; also shown with additional replicates and error-class-specific scales in Supplementary Fig. 1e. Note that only one of the two 3’-DNA-flaps is covered by the sequencing read in amplicons lacking the deletion (labeled as ‘wild-type’). h. Insertion, deletion and substitution error frequencies across the amplicons from 546-bp deletion experiment after merging paired-end sequencing reads.
Recently, Liu and colleagues described ‘prime editing’, which expands the CRISPR-Cas9 genome editing toolkit in critical ways14. Prime editing utilizes a Prime Editor-2 enzyme, which is a Cas9 nickase (Cas9 H840A) fused with a reverse-transcriptase, and a 3′-extended gRNA (prime-editing gRNA or pegRNA). The Prime Editor-2 enzyme and pegRNA complex can nick one strand of the genome and attach a 3′ single-stranded DNA flap to the nicked site following the template RNA sequence in the pegRNA molecule. By including homologous sequences to the neighboring region, DNA damage repair factors can incorporate the 3′-flap sequence into the genome. The incorporation rate can be further enhanced using an additional gRNA, which makes a nick on the opposite strand, boosting DNA repair with the 3′-flap sequence but often with a decrease in precision (strategy referred to as PE3/PE3b)14 (Figure 1b). The principal advantage of prime editing lies with its encoding of both the site to be targeted and the nature of the repair within a single molecule, the pegRNA. In addition to demonstrating many other classes of precise edits, Anzalone et al. used the PE3 strategy to show that a single pegRNA/gRNA pair could be used to program deletions ranging from 5 to 80 bp achieving high efficiency (52-78%) with modest precision (on average, 11% rate of unintended indels)14. However, even the PE3 strategy could face difficulties in programming deletions larger than 100 bp, as at least in plants, observed efficiencies fall precipitously for deletions larger than 20 bp15.
We reasoned that a pair of pegRNAs could be used to specify not only the sites that are nicked but also the outcome of the repair, potentially enabling programming of deletions longer than 100 bp (Figure 1c). Here we demonstrate that this strategy, which we call PRIME-Del, induces the efficient deletion of sequences up to 10 kb in length with much higher precision than observed or expected with either the Cas9/paired-gRNA or PE3 strategies. We furthermore show that PRIME-Del can concurrently program short insertions at the deletion site. Concurrent deletion/insertion can be used to introduce in-frame deletions, to introduce epitope tags concurrently with deletions, and, more generally, to facilitate the programming of deletions unrestricted by the endogenous distribution of PAM sites. By filling these gaps, PRIME-Del expands our toolkit to investigate the biological function of genomic sequences at single nucleotide resolution.
Results & Discussion
PRIME-Del induces precise deletions in episomal DNA
We first tested the feasibility of the PRIME-Del strategy by programming deletions to an episomally encoded eGFP gene. We designed pairs of pegRNAs specifying 24-, 91- and 546-bp deletions within the eGFP coding region of the pCMV-PE2-P2A-GFP plasmid (Addgene #132776) (Figure 1d). We cloned each pair of pegRNAs into a single plasmid with separate promoters, the human U6 and H1 sequences5. We transfected HEK293T cells with eGFP-targeting paired-pegRNA and pCMV-PE2-P2A-GFP plasmids. We harvested DNA (including both genomic DNA and residual plasmid) from cells 4–5 days after transfection and PCR amplified the eGFP region. We then sequenced PCR amplicons to quantify the efficiency of the programmed deletion as well as to detect unintended edits to the targeted sequence.
We calculated deletion efficiency as the number of reads aligning to a reference sequence of the intended deletion, out of the total number of reads aligning to reference sequences either with or without the deletion. Estimated deletion efficiencies ranged from 38% (24-bp deletion) to 77% (546-bp deletion), and were consistent across replicates (note: throughout the paper, the term ‘replicate’ is used to refer to independent transfections) (Figure 1e). This result clearly indicates that the PRIME-Del strategy outlined in Fig. 1c can work. However, we were initially concerned that these were overestimates of efficiency due to the shorter, edited templates being favored by both PCR and Illumina-based sequencing, particularly for the 546-bp deletion, because it has the largest difference between amplicon sizes (766-bp vs. 220-bp for wild-type and deletion amplicons, respectively). To address this, we repeated the amplification on DNA from the 546-bp deletion experiment with a two-step PCR, first adding 15 bp unique molecular identifiers (UMIs) via linear amplification before a second, exponential phase. The addition of UMIs via linear PCR was intended to minimize PCR and sequencing biases in our estimates of deletion efficiencies16. PRIME-Del efficiency was assessed based on the sequencing data after collapsing of reads with identical UMIs, as well as on the product size distribution (Agilent TapeStation). We observed a slight decrease in deletion efficiency after duplicate removal, from 73% to 66%, comparable to the 70% efficiency measured on the TapeStation (Figure 1f). These results suggest that our initial estimates of efficiency are only modestly impacted by size-dependent biases.
For most of these sequencing data, we had only a single read extending over the intended deletion site. As such, it was difficult to distinguish unintended editing outcomes (e.g. indels at the nick sites) from PCR or sequencing errors. To address this in part, we plotted frequencies of different classes of errors (substitutions, insertions, deletions) for sequences aligning either to the unedited sequence (Figure 1g, top) or the intended deletion (Figure 1g, bottom), along the length of the sequencing read. For all replicates of the three deletion experiments (Supplementary Fig. 1), these profiles showed low rates of substitutions and indels, with nearly identical profiles and no consistent increase in the rate of any class of error at either the positions of the Prime Editor-2 enzyme nick sites or 3’ flap ends above 1%, particularly after collapsing by UMI (Fig. 1g, Supplementary Fig. 1e) or repeating sequencing with longer, paired-end sequencing reads (Figure 1h).
Simultaneous deletion and short insertion using PRIME-Del
We reasoned that because the homology sequences in the 3’-flaps program the deletion, we could potentially use PRIME-Del to concurrently introduce a short insertion at the deletion junction (Figure 2a). The desired insertion would be encoded into the pair of pegRNAs in a reverse complementary manner, just 5’ to the deletion-specifying homology sequences. With the conventional strategy for programming deletions, i.e. with Cas9 and paired gRNAs, the deletion junctions are determined by the gRNA targets, the selection of which is limited by the natural distribution of PAM sites (Figure 2b). Simultaneous deletion and short (less than 100 bps) insertion with PRIME-Del would offer at least three advantages over this conventional strategy. First, an arbitrary insertion of 1-3 bases could enable a reading frame to be maintained after editing, e.g. for deletions intended to remove a protein domain. Second, an arbitrary insertion could be used to effectively move one or both deletion junctions away from the cut-sites determined by the PAM, increasing flexibility to program deletions with base-pair precision. Third, insertion of functional sequences at the deletion junction could allow genome editing with PRIME-Del to be coupled to other experimental goals (e.g. protein tagging or insertion of a transcriptional start site).
Figure 2. Concurrent programming of deletion and insertion using PRIME-Del.
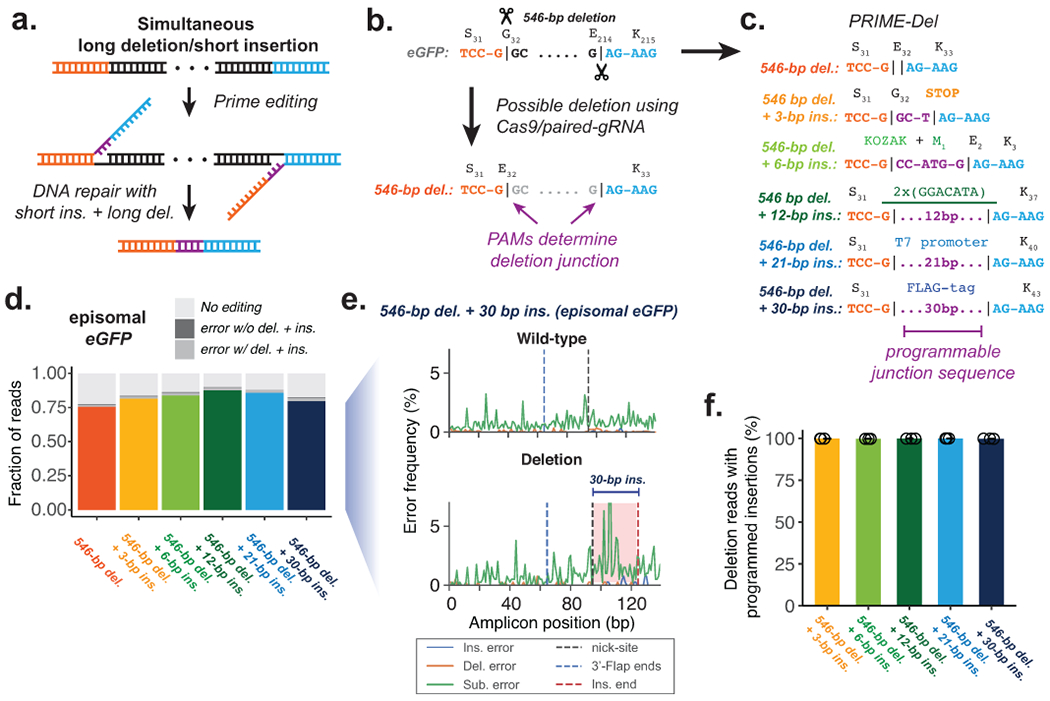
a. Schematic of strategy, with reverse complementary sequences corresponding to the intended insertion in purple. b. Conventional strategy for deletion with Cas9 and pairs of gRNAs. Potential deletion junctions are restricted by the natural distribution of PAM sites. c. Pairs of pegRNAs were designed to encode five insertions, ranging in size from 3 to 30 bp, together with a 546 bp deletion in eGFP. d. Estimated deletion efficiencies and indel error frequencies (with or without intended deletion) in using these pegRNA pairs to induce concurrent deletion and insertion in HEK293T cells (averaged over replicates; n = 3). e. Representative insertion, deletion and substitution error frequencies plotted across sequencing reads from concurrent 546-bp deletion and 30-bp insertion condition. Plots are from single-end reads without UMI correction. Note that only one of the two 3’-DNA-flaps is covered by the sequencing read in amplicons lacking the deletion (labeled as ‘wild-type’). f. The percentage of reads containing the programmed deletion that also contain the programmed insertion. Error bars represent standard deviation for at least three replicates.
To test this concept, we designed pegRNA pairs encoding five insertions ranging from 3 to 30 bp at the junction of a 546-bp programmed deletion within eGFP (Figure 2c). While our main objective was to test the effect of insertion length on deletion efficiency, we chose insertion sequences for their importance in molecular biology: The 3-bp insertion sequence generates an in-frame stop codon. The 6-bp insertion sequence includes the start codon with the surrounding Kozak consensus sequence. The 12-bp insertion sequence includes tandem repeats of m6A post-transcriptional modification consensus sequence of GGACAT17. The 21-bp insertion sequence includes T7 RNA polymerase promoter sequence. The 30-bp insertion sequence encodes for the in-frame FLAG-tag peptide sequence when translated. The estimated efficiencies for simultaneous short insertion and deletion within the episomal eGFP gene in HEK293T cells were comparable to the 546-bp deletion alone, ranging from 83% to 90% for the various programmed insertions (Figure 2d). Also, insertion, deletion and substitution error rates at deletion junctions and across programmed insertions were comparable to the background error frequencies (Figure 2e, Supplementary Figure 2a). As expected, the vast majority (>99%) of reads containing the programmed deletion also contained the insertion (Figure 2f), indicating that the full lengths of the pair of 3’-DNA flaps generated following the programmed pegRNA sequences specify the repair outcome (Figure 2a).
PRIME-Del induces precise deletions in genomic DNA
Encouraged by our initial results on editing episomal DNA, we next tested PRIME-Del on a copy of the eGFP gene integrated into the genome. We first generated the polyclonal HEK293T cells that carry the eGFP gene by lentiviral transduction, followed by flow-sorting to select GFP-positive cells (Figure 3a). We then tested the same pairs of pegRNAs encoding concurrent deletion and insertions (546-bp deletion with or without short insertions at the deletion junction) by transfecting pegRNAs and Prime Editor-2 enzyme without eGFP (pCMV-PE2; Addgene #132775) to these cells. Although editing efficiencies decreased substantially in comparison to episomal eGFP (7-17%; Figure 3b), we remained unable to detect errors that were clearly associated with editing (Figure 3c, Supplementary Figure 2b). Specifically, there was no consistent pattern of error classes above background level accumulating at the nick-site or 3’-DNA-flap incorporation sites. Also, as previously, the vast majority of reads with the 546-bp deletion also contained programmed insertions (Supplementary Figure 2c).
Figure 3. Precise genomic deletions using PRIME-Del.
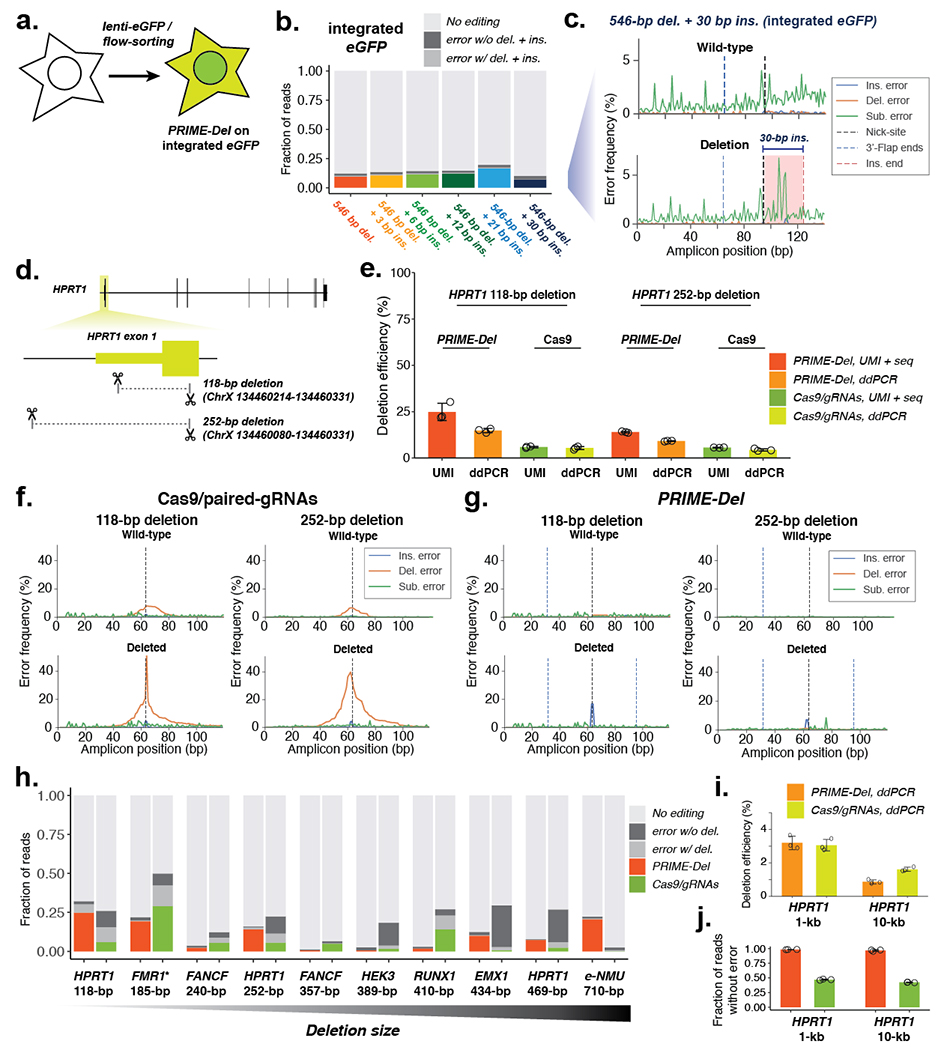
a. Schematic of generation of the eGFP-integrated HEK293T cell line. b. Estimated deletion efficiencies and error frequencies in using PRIME-Del for concurrent deletion and insertion on genomically integrated eGFP in HEK293T cells. (n = 3) c. Representative insertion, deletion and substitution error frequencies plotted across sequencing reads from concurrent 546-bp deletion and 30-bp insertion condition on genomically integrated eGFP. Plots are from single-end reads without UMI correction. d. Cartoon representation of deletions programmed within the HPRT1 gene. e. Deletion efficiencies measured for the 118-bp and 252-bp deletion using either PRIME-Del or Cas9/paired-gRNA (abbreviated to Cas9) strategies in HEK293T cells, quantified using either the unique-molecular identifier-based sequencing assay (UMI) or the droplet-digital PCR (ddPCR) assay. Error bars represent standard deviation for three replicates. f. Representative insertion, deletion and substitution error frequencies plotted across sequencing reads from 118-bp deletion (left) and 252-bp deletion (right) at HPRT exon 1, using the Cas9/paired-gRNA strategy. Different error classes are colored the same as in (c). g. Same as (f), but for PRIME-Del strategy. h. Estimated deletion efficiencies and indel error frequencies for different deletions across the genome for PRIME-Del (left) and Cas9/paired-gRNA (right) methods (averaged over replicates; n = 3). UMI-based sequencing assay was used for quantification (except the GC-rich amplicon of FMR1*, where added DMSO interfered with the UMI-addition reaction). i. Deletion efficiencies measured for 1-kb and 10-kb deletions at HPRT1 using either PRIME-Del (left) or Cas9/paired-gRNA (right) with ddPCR-based assay in HEK293T cells. Error bars represent standard deviation for three replicates. j. Fraction of reads with precise deletion measured for the 1-kb and 10-kb deletion on HPRT1 gene with either PRIME-Del (left) or Cas9/paired-gRNA (right) using sequencing of the deletion amplicons. Error bars represent standard deviation for three replicates.
To test PRIME-Del on native genes, we designed two pairs of pegRNAs that respectively specified 118 and 252-bp deletions within exon 1 of HPRT1 (Figure 3d). We have previously performed a scanning deletion screen across the HPRT1 locus using a Cas9/paired-gRNA strategy5. To directly compare PRIME-Del with Cas9/paired-gRNAs in programming genomic deletions, we attempted the same deletions with the same guides but substituting Prime Editor-2 enzyme with Cas9 in transfection of HEK293T cells. We quantified the resulting deletion efficiencies using two independent methods: First, we used the aforedescribed strategy of appending 15-bp unique molecular identifier (UMI) sequence via linear PCR step, before the standard PCR and sequencing readout. Resulting sequencing reads are collapsed by shared UMIs to minimize possible biases introduced in the PCR amplification and sequencing cluster generation steps. Second, we used droplet-digital PCR (ddPCR), which partitions genomic DNA into emulsion droplets before PCR amplification and fluorescence read-out of TaqMan probes within each droplet. We designed our probe to bind at the deletion junction, which would generate fluorescence signals specifically in the presence of the deletion. Our design of reporter probe aims to quantify the precise editing efficiencies, as errors introduced at the deletion junction are less likely to induce efficient binding of the probe during PCR18. Signals from deletions were normalized to the reference signal from detecting the copy-number of RPP30 gene, which has been previously characterized and often used as a standard in ddPCR assay18. At exon 1 of HPRT1, we observed comparable deletion efficiencies for the PRIME-Del and Cas9/paired-gRNA strategies in HEK293T, ranging from 5% to 30% efficiencies for 118-bp and 252-bp deletions (Figure 3e). Of note, we observed consistently lower efficiencies with the ddPCR assay compared to the UMI-based sequencing assay. While this could be due to overestimation of efficiencies by the UMI-based approach, we also note that PCR amplification of the target region may be inefficient in the ddPCR assay based on the lack of clear separation of fluorescence intensities between positive and negative droplets (Supplementary Figure 3c,d).
As is well established3–5, the Cas9/paired-gRNA strategy often resulted in errors (mostly short deletions), whether with or without the intended deletion (Figure 3f,g; Supplementary Figure 3a). Of reads lacking the intended 118-bp or 252-bp deletions, 12% or 12% also contained an unintended indel at the observable target site, respectively (these are underestimates, because they only account for one of two target sites) (Figure 3f, top). Of reads containing the intended 118-bp or 252-bp deletions, 38% or 34% also contained an unintended indel at the deletion junction, respectively (Figure 3f, bottom). Such junctional errors are an established consequence of error-prone repair by NHEJ. In contrast, unintended indels were far less common with PRIME-Del (Figure 3g; Supplementary Figure 3b). Of reads lacking the intended 118-bp or 252-bp deletions, 1.1% or 0.5% also contained an unintended short indel at the observable target site, respectively (Figure 3g, top). Of reads containing the intended 118-bp or 252-bp deletions, 12% or 2.7% also contained an unintended indel at the deletion junction, respectively (Figure 3g, bottom). The pattern of higher correct editing efficiencies for PRIME-Del over the Cas9/paired-gRNA strategy is also suggested by the ddPCR measurements, where the PRIME-Del reports a nearly 2-fold higher precisely edited population for both deletions.
For PRIME-Del, especially with the 118-bp deletion on HPRT1, the observation of an appreciable rate of insertions at the deletion junction in association with intended deletions (Figure 3g, bottom; Supplementary Figure 3b) contrasts with our earlier observations at eGFP, where these rates were consistently equivalent to background. Further investigation of the error mode revealed that these errors corresponded to long insertions (mean 47-bp +/− 12-bp; Supplementary Figure 4). The most frequent long insertion at the 118-bp deletion junction was 55-bp, a chimeric sequence between two 32-bp 3’-DNA flap sequences, overlapping at a ‘GCCCT’ sequence, suggesting its origin from the annealing of GC-rich ends of 3’-DNA flaps. Similar chimeric sequences were observed as insertions at the 252-bp deletion junction, overlapping at ‘GCCG’ within their 3’-DNA flaps. Nonetheless, even with these long insertions, 82% and 91% of all reads containing an indel matched the intended deletion exactly with PRIME-Del, but only 38% and 49% with the Cas9/paired-gRNA strategies (Figure 3h). Indel errors from the Cas9/paired-gRNA strategy are likely underestimated, because errors at only one of two Cas9 cut-sites are captured by our sequencing strategy.
The structure of the observed insertions and the lack of similar errors in applying PRIME-Del to the eGFP locus suggested that this issue might be addressable through alternative pegRNA designs. As one approach, we either shortened or lengthened the RT template portion of both pegRNAs. For 118-bp deletion that used 32-bp RT template lengths for both pegRNAs, we shortened to either 17- and 25-bp long homology arms or lengthened to 42- and 46-bp long homology arms (Supplementary Figure 5a). Both lengthening and shortening homology arms resulted in decreased deletion efficiencies (29% and 26% of the efficiencies observed with the standard designs for short and long homology arms, respectively) (Supplementary Figure 5b). However, among deleted products, lengthening the homology arms also tended to decrease the long-insertion error frequency (to 30% of the standard design), while shortening the homology arms increased the insertion error frequency (to 129% of the standard design) (Supplementary Figure 5d). Similar trends was observed with the 252-bp deletion, where shortening or lengthening homology arms decreased the deletion efficiency (Supplementary Figure 5c), while lengthening the homology arm increased precision (Supplementary Figure 5e). As a further control, substituting the sequence of the RT template to that used for programming a 546-bp deletion at eGFP failed to induce deletions for both 118-bp and 252-bp constructs targeting HPRT1 (Supplementary Figure 5b,c), fortifying the conclusion that PRIME-Del deletions are specific to DNA repair guided by the homology arm sequences.
We further applied genomic deletion using PRIME-Del at additional native loci, altogether testing 10 different deletions at 7 loci (Figure 3h). We performed all deletions in HEK293T cells, quantified deletion efficiencies and error frequencies using UMI-based sequencing assay, and directly compared PRIME-Del with the Cas9/paired-gRNA method (i.e. using the same guides but substituting in Cas9). Deletion sizes ranged from 118 bp at HPRT1 exon 1 to 710 bp at e-NMU (enhancer for NMU gene) locus. In all 10 cases, we observe substantially lower error rates with PRIME-Del compared to the Cas9/paired-gRNA method. In five out of ten cases, we observe that the precise deletion is more efficient with PRIME-Del compared to the Cas9/paired-gRNA method, suggesting that higher precision does not compromise the deletion efficiencies in general. We did not observe a strong relationship between the deletion size and efficiency in this range (118 to 710 bps) for either method.
Inversion of the sequence between two DSBs is a well-documented phenomenon when using the Cas9/paired-gRNA method3,19 (Supplementary Figure 6a). To understand the frequency of inversion events using PRIME-Del, we aligned sequencing reads to a reference that was generated by inverting the sequence between two nick-sites. Across 10 deletions in 7 loci at which we performed PRIME-Del, we observed that virtually no reads aligned to the inverted reference (Supplementary Figure 6b), while for Cas9/paired-gRNA controls, inversions were detected up in up to 2% of reads (Supplementary Figure 6b).
To evaluate the length limits of PRIME-Del, we designed two additional deletions, sized 1,064 bps (1 kb) and 10,204 bps (10 kb) at the HPRT1 locus. Since our sequencing-based assay is not well suited to detect amplicons greater than 1 kb, we used sequencing to quantify error frequencies in the deletion product alone, and ddPCR to measure the efficiency of precise deletion, again comparing Prime Editor-2 and Cas9 side-by-side. We observed that while deletion efficiencies between PRIME-Del and the Cas9/paired-gRNA method were comparable in HEK293T cells (Figure 3i), PRIME-Del achieves much higher precision, consistent with our observations while inducing shorter deletions. For the 1-kb deletion, both PRIME-Del and the Cas9/paired-gRNA method achieved nearly 3% deletion efficiency. For the 10-kb deletion, PRIME-Del and the Cas9/paired-gRNA method achieved 0.8% and 1.6% deletion efficiency, respectively. Upon sequencing amplicons derived from a PCR specific to the post-deletion junction, 98% and 97% of reads lacked indel errors at the junction with PRIME-Del for the 1-kb and 10-kb deletions, respectively, while only 47% and 42% of reads lacked indel errors with the Cas9/paired-gRNA strategy (Figure 3j).
To test whether the PRIME-Del can be “multiplexed”, we pooled plasmids encoding paired-pegRNAs programming four different but overlapping deletions (118, 252, 469 and 1064 bps) at the HPRT1 locus, and transfected HEK293T cells with these together with a plasmid encoding the Prime Editor-2 enzyme. After incubating cells for 4 days and extracting genomic DNA, we used sequencing-based quantification to estimate 5.1%, 8.5% and 2.8% efficiencies for the 118-, 252-, and 469-bp deletions, and ddPCR to estimate 2% efficiency for the 1064-bp deletion (Supplementary Figure 7). Altogether, we estimate that 18% of HPRT1 loci carry one of the four programmed deletions, which is comparable to the averaged efficiency of four deletions performed by transfecting a single construct of paired-pegRNA plasmid separately (12%). Our result suggests that PRIME-Del can be used to concurrently program multiple deletions by using pooled paired-pegRNA constructs similar to Cas9/paired-gRNA method5,10,11.
Extending the editing time window enhances prime editing and PRIME-Del efficiency
In contrast with Cas9-mediated DSBs followed by NHEJ, both prime editing and PRIME-Del have high editing precision, producing an intended edit or conserving the original editable sequence. We reasoned that if the editing efficiencies of prime editing and PRIME-Del are limited by the transient availability of PE2/pegRNA molecules in the cell, extending Prime Editor-2 enzyme and pegRNA expression through stable genomic integration or, alternatively, repetitive transfection, would boost the rates of successful editing over time, particularly if uneditable “dead ends” outcomes are not concurrently accruing.
To facilitate prolonged expression, we generated monoclonal HEK293T and K562 cell lines expressing Prime Editor-2 enzyme (termed HEK293T(PE2) and K562(PE2), respectively). Because the Prime Editor-2 enzyme gene was larger than the lentiviral vector’s typical limit, we cloned Prime Editor-2 enzyme into the piggyBAC cargo, transfected it along with the piggyBAC transposase, and identified a monoclonal cell line with active PE2. To continuously express pegRNAs in addition to Prime Editor-2 enzyme, we generated lentiviral vectors with pegRNAs and transduced them into both HEK293T(PE2) and K562(PE2) cells (Figure 4a). We tested two different deletions at HPRT1 using PRIME-Del (the aforedescribed 118-bp and 252-bp deletions at exon 1), along with standard prime editing to insert 3-bp (CTT) into the synthetic HEK3 target sequence14. In K562(PE2), we observed a steady increase of the correctly edited population over time, both for CTT-insertion using prime editing and for 118- or 252-bp deletions using PRIME-Del. The end-point prime editing efficiencies for the CTT-insertion were very high, reaching 90% of targets with correct edits by 19 days after the first transduction of pegRNA into K562(PE2) cells (Figure 4b). The rate of precise deletions using PRIME-Del also reached nearly 50% and 25% for the 118-bp and 252-bp deletions, respectively, by 19 days. In HEK293T(PE2) cells, we observed lower CTT-insertion efficiencies for the first 10 days, but eventually reaching 80-90% by day 19 (Figure 4c). Unexpectedly, we observed the near-absence of PRIME-Del-induced deletions in HEK293T(PE2) cells (Figure 4c). However, the same HEK293T(PE2) cell line showed modest increases in editing to 5 - 50% when we attempted multiple transfections of either PE2/pegRNA without additional stable integration or Prime Editor-2 enzyme alone after stable integration of piggyBAC-pegRNA, over four weeks (Supplementary Figure 8a,b). We repeated the experiment with the same HEK293T(PE2) cell line but with a different batch of pegRNA-encoding lentivirus, which resulted in better efficiencies for the 118-bp deletion over time (Supplementary Figure 8c), suggesting that PRIME-Del is more sensitive to both pegRNA and Prime Editor-2 enzyme expression level differences between transfection and lentiviral transduction than standard prime editing, presumably because the PRIME-Del requires two concurrent PE2 actions. Together, our results confirm that extended expression of prime editing or PRIME-Del components can boost efficiency.
Figure 4. Extending the editing time window enhances prime editing and PRIME-Del efficiency.
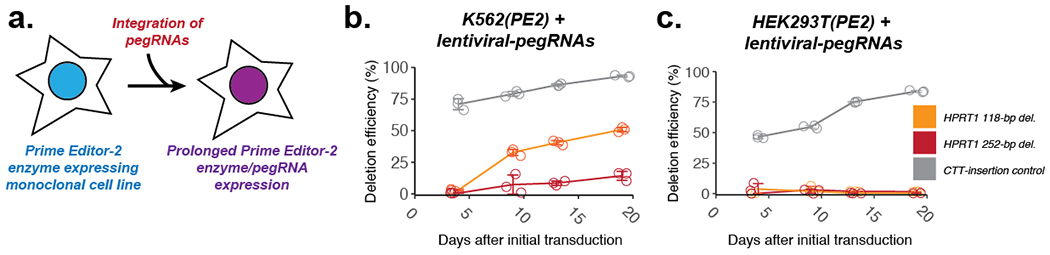
a. Schematic for stably expressing both Prime Editor-2 enzyme and pegRNAs via two-step genome integration. b-c. Editing efficiencies measured for the 118-bp and 252-bp deletions at genomic HPRT1 exon 1 using PRIME-Del (paired-pegRNA construct) or CTT-insertion using prime editing (single-pegRNA construct) in K562(PE2) cells (b) or HEK293T(PE2) cells (c), as a function of time after initial transduction of pegRNA(s). Error bars represent standard deviation for three replicates.
Potential applications of PRIME-Del
Here we introduce PRIME-Del, a paired pegRNA strategy for prime editing, and demonstrate that it achieves high precision for programming deletions, both with or without short programmed insertions. We tested deletions ranging from 20 to ~10,000-bp in length at episomal, synthetic genomic, and native genomic loci. The editing efficiency on native genes ranged from 1-30% with a single round of transient transfection in HEK293T cells, although we also observed that prolonged, high expression of prime editing or PRIME-Del components enhanced editing efficiency in K562 cells. For 12 deletions at seven genomic loci targeted with PRIME-Del, we observed high precision of editing except at HPRT1 exon 1, where long insertions were sometimes observed at the deletion junction (~5% of total reads). The GC-rich ends of 3’-DNA flap sequences of the pegRNA pairs used at HPRT1 exon 1 appear to underlie the long insertions. Optimizing pegRNA design may be able to eliminate this error mode, and we show that lengthening homology arms tends to decrease the frequency of long insertion errors. To facilitate avoidance of this particular error mode, we have developed an accompanying Python-based webtool for designing PRIME-Del paired-pegRNA sequences, which notifies the user if such sequences are present in designed pegRNA pairs.
However, even with these insertion errors, PRIME-Del consistently demonstrated higher precision than the Cas9/paired-gRNA strategy, i.e. for all 12 genomic deletions tested here, PRIME-Del resulted in fewer erroneous outcomes. For these same 12 cases, PRIME-Del exhibited markedly higher precise-deletion efficiencies for five (greater than a factor of two), comparable efficiencies for five (within a factor of two), and markedly lower efficiencies for two (less than half), compared to the Cas9/paired-gRNA method. Overall, these observations support the view that PRIME-Del achieves higher precision than the Cas9/paired-gRNA method without compromising editing efficiency.
A potential design-related limitation of PRIME-Del is that relative to the conventional Cas9/paired-gRNA strategy, it constrains the useable pairs of genomic protospacers, as they need to occur on opposing strands with the PAM sequences oriented towards one another (Figure 1c). However, the development and optimization of a near-PAMless22 prime editing enzyme23 would relax this constraint. A further limitation is that because of their longer length, cloning a pair of pegRNAs in tandem is more challenging than cloning gRNA pairs. Each pegRNA used here is 135 to 140 bp in length, such that synthesizing their unique components in tandem as a single, long oligonucleotide approaches the limits of conventional DNA synthesis technology, particularly for goals requiring array-based synthesis of paired pegRNA libraries.
Notwithstanding these limitations, PRIME-Del offers significant advantages over alternatives across several potential areas of application (Figure 5). Most straightforwardly, PRIME-Del can be used for precise programming of deletions up to 10 kb; we have yet to attempt deletions longer than 10 kb. In addition to the much lower indel error rate observed at the deletion junction compared to the Cas9/paired-gRNA strategy, inducing paired nicks is less likely to result in large, unintended deletions locally, rearrangements genome-wide (chromothripsis)24, or off-target editing7,14,25–27. These characteristics are advantageous for developing therapeutic approaches, e.g. where the PRIME-Del deletes pathogenic regions such as CGG-repeat expansions in 5’-UTR of FMR1, without undesired perturbation of nearby or distant sequences12,13.
Figure 5. Potential advantages of using PRIME-Del in various genome editing applications.
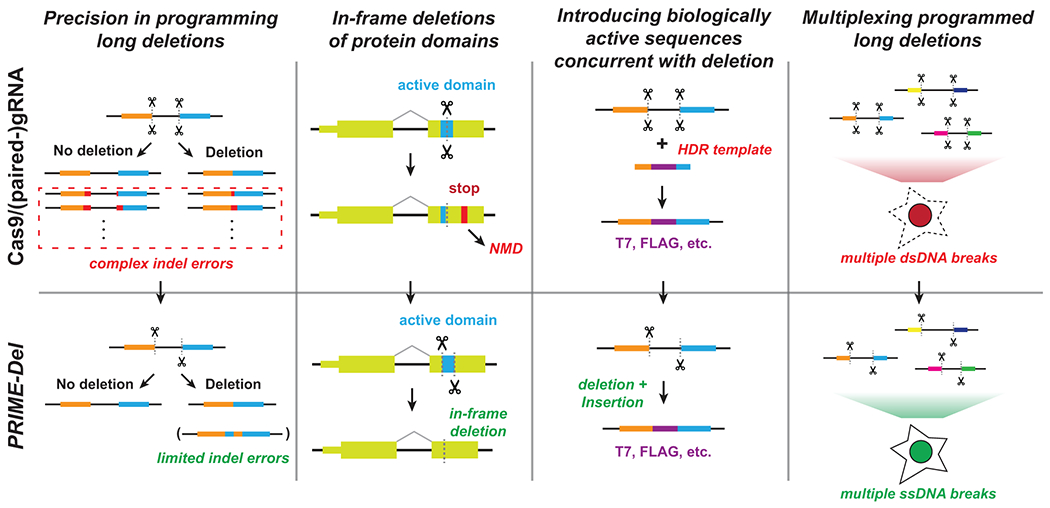
The PRIME-Del strategy can be used to program precise genomic deletions without generation of short indel errors at Cas9 target sequences. Precision deletion, combined with ability to insert a short arbitrary sequence at the deletion junction, may allow robust gene knockout of active protein domains without generating a premature in-frame stop codon, which can trigger the nonsense-mediated decay (NMD) pathway. PRIME-Del may also allow replacement of genomic regions up to 10 Kb base-pairs with arbitrary sequences such as epitope tags or RNA transcription start sites. Single-stranded breaks generated during PRIME-Del are likely to be less toxic to the cell, especially when multiple regions are edited in parallel, potentially facilitating its multiplexing.
PRIME-Del also allows simultaneous insertion of short sequences at the programmed deletion junction without substantially compromising its efficiency or precision. Inserting short sequences allows for precise deletions of protein domains while preserving the native reading frame, i.e. avoiding a premature stop codon that might otherwise elicit a complex nonsense-mediated decay (NMD) response28,29. Furthermore, inserting biologically active sequences upon deletion is likely to be advantageous in coupling PRIME-Del with technologies, i.e. by inserting epitope tags or T7 promoter sequences that can be used as molecular handles within edited genomic loci.
We also expect less toxicity via DNA damage by prime editing-based PRIME-Del than with the conventional Cas9/paired-gRNA strategy, which may facilitate multiplexing of programmed genomic deletions for frameworks such as scanDel and crisprQTL5,6. For studying the non-coding elements in transcription, efficient and precise deletions up to ~10 kb complements the current use of deactivated Cas9-tethered KRAB domain for CRISPR-interference (CRISPRi), which cannot control the range of epigenetic modifications around target regions. As such, we anticipate that PRIME-Del could be broadly applied in massively parallel functional assays to characterize native genetic elements at base-pair resolution.
Materials and Methods
pegRNA/gRNA design
For pegRNA/gRNA design, we initially used CRISPOR30 to select for 20-bp CRISPR-Cas9 spacers within a given region of interest. We avoided spacers annotated as inefficient, including U6/H1 terminator and GC-rich sequences, and generally selected spacers that had higher predicted efficiencies (Doench scores for U6 transcribed gRNAs31). The length of the RT-template portion of a pegRNA was initially set to 30-bp and extended by 1 to 2-bp if it ended in G or C14,32.
Web tool for PRIME-Del paired-pegRNA design
To facilitate PRIME-Del paired-pegRNA design, we developed a Python-based web tool that automates the design process. The software takes a FASTA-formatted sequence file as the input, identifies all possible PAM sequences within the provided region, and initially generates all potential paired pegRNA sequences to program deletions. The software can also optionally take as input scored gRNA files generated using Flashfry33, CRISPOR30 or GPP sgRNA designer30; this is highly recommended to identify effective CRISPR-Cas9 spacers. For FlashFry and CRISPOR, gRNA spacers with MIT specificity scores34 below 50 are filtered out as recommended by CRISPOR. From initially generated pegRNA pairs, the software selects relevant ones based on additional user-provided design parameters. For example, the user can define the deletion size range. The user can also define the start and end position of desired deletion, and the software will filter to pegRNA pairs present windows centered at those coordinates. pegRNAs for deletions whose junctions do not fall at PAM sites can be designed using the option ‘--precise’ (-p), which adds insertion sequences to both pegRNAs to facilitate the desired edit.
The PRIME-Del design software also enables additional design constraints to be specified. The pegRNA RT-template length (also known as the homology arm) is set to 30-bp by default, unless specified otherwise by the user. The pegRNA PBS length is set to 13-bp from the PE2 nick-site by default, unless specified otherwise by the user. The nick position relative to the PAM sequence is predicted using previously identified parameters (Lindel35), and RT-template length is adjusted accordingly if the predicted likelihood of generating a nick at a non-canonical position is greater than 25%. PegRNA sequences that include RNA polymerase III terminator sequences (more than four consecutive T’s) are filtered out. The software generates warning messages if more than 4 out of 5 bp in either 3’-DNA-flap are either G or C. Code is available at https://github.com/shendurelab/Prime-del, and interactive webpage is available at https://primedel.uc.r.appspot.com/.
pegRNA cloning
After designing pegRNA pairs, we followed the Golden-Gate cloning strategy outlined by Anzalone et al.14, assembling three dsDNA fragments and one plasmid backbone. The first dsDNA fragment contains the pegRNA-1 spacer sequence, annealed from two complementary synthetic single-strand DNA oligonucleotides (IDT) with 4-bp 5’-overhangs. The second dsDNA fragment contains the pegRNA-1 gRNA scaffold sequence, annealed from two DNA oligonucleotides with 5’-end phosphorylation at the end of 4-bp overhang. The third dsDNA fragment contains the pegRNA-1 RT template sequence and primer binding sequence (PBS), pegRNA-1 terminator sequence (six consecutive T’s), and pegRNA-2 sequence with H1 promoter sequence. This was generated by appending pegRNA-1 portion and pegRNA-2 portion to two ends of gene fragments (purchased as gBlocks from IDT) by PCR amplification. The gene fragments contained the pegRNA-1 terminator sequence, H1 promoter sequence, pegRNA-2 spacer sequence, and pegRNA-2 gRNA scaffold sequences. The forward primer included the BsmBI or BsaI restriction site, pegRNA-1 RT template sequence and PBS. The reverse primer included pegRNA-2 RT template, PBS, and BsmBI or BsaI restriction site. PCR fragments (sized between 300 and 400 bp) were purified using 1.0X AMPure (Beckman Coulter) and mixed with two other dsDNA fragments and linearized backbone vector with corresponding overhangs for Golden-Gate-based assembly mix (BsmBI or BsaI golden-gate assembly mix from New England Biolabs). For the pegRNA cloning backbone, we used either the GG-acceptor plasmid (Addgene #132777) or piggyBAC-cargo vector that carries the blasticidin-resistance gene. Each construct plasmid was transformed into Stbl Competent E. coli (NEB C3040H) for amplification and purified using a miniprep kit (Qiagen). Cloning was verified using Sanger sequencing (Genewiz).
Tissue culture, transfection, lentiviral transduction, and monoclonal line generation
HEK293T and K562 cells were purchased from ATCC. HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium with high glucose (GIBCO), supplemented with 10% fetal bovine serum (Rocky Mountain Biologicals) and 1% penicillin-streptomycin (GIBCO). K562 cells were cultured in RPMI 1640 with L-Glutamine (Gibco), supplemented with 10% fetal bovine serum (Rocky Mountain Biologicals) and 1% penicillin-streptomycin (GIBCO). HEK293T and K562 cells were grown with 5% CO2 at 37 C.
For transient transfection, about 50,000 cells were seeded to each well in a 24-well plate and cultured to 70-90% confluency. For prime editing, 375 ng of Prime Editor-2 enzyme plasmid (Addgene #132775) and 125 ng of pegRNA or paired-pegRNA plasmid were mixed and prepared with transfection reagent (Lipofectamine 3000) following the recommended protocol from the vendor. For deletion using Cas9/paired-gRNA, 375 ng of Cas9 plasmid (Addgene #52962) was used instead of Prime Editor-2 enzyme plasmid. Cells were cultured for four to five days after the initial transfection unless noted otherwise, and its genomic DNA was harvested either using DNeasy Blood and Tissue kit (Qiagen) or following cell lysis and protease protocol from Anzalone et al.14.
For lentiviral generation, about 300,000 cells were seeded to each well in a 6-well plate and cultured to 70-90% confluency. Lentiviral plasmid was transfected along with the ViraPower lentiviral expression system (ThermoFisher) following the recommended protocol from the vendor. Lentivirus was harvested following the same protocol, concentrated overnight using Peg-it Virus Precipitation Solution (SBI), and used within 1-2 days to transduce either K562 or HEK293T cells without a freeze-thaw cycle.
For transposase integration, 500 ng of cargo plasmid and 100 ng of Super piggyBAC transposase expression vector (SBI) were mixed and prepared with transfection reagent (Lipofectamine 3000) following the recommended protocol from the vendor. Prime Editor-2 enzyme-expressing single-cell clones were generated by integrating PE2 using piggyBAC transposase system, selected by marker (puromycin resistance gene), single-cell sorted into 96-well plates using flow-sort apparatus, cultured for 2-3 weeks until confluency, and screened for PE activity by transfecting CTT-inserting pegRNA alone (Addgene #132778) and sequencing the HEK3-target loci.
DNA sequencing library preparation
To quantify programmed deletion efficiency and possible errors generated by PRIME-Del, we amplified the targeted region from purified DNA (~200 to ~1000 bp in length) using two-step PCR and sequenced using Illumina sequencing platform (NextSeq or MiSeq) (Supplementary Figure 1a). Each purified DNA sample contains wild-type and edited DNA molecules, which were amplified together using the same pairs of primers through each PCR reaction. For the PCR-amplification, we designed a pair of primers for each genomic locus (amplicon) where entire amplicon sizes, with or without deletion, were greater than 200 bp to avoid potential problems in PCR-amplification, in purifying of PCR products, and in clustering onto the sequencing flow-cell.
The first PCR reaction (KAPA Robust) included 300 ng of purified genomic DNA or 2 uL of cell lysate, 0.04 to 0.4 uM of forward and reverse primers in a final reaction volume of 50 uL. We programmed the first PCR reaction to be: 1) 3 minutes at 95°C, 2) 15 seconds at 95°C, 3) 10 seconds at 65°C, 4) 45 seconds at 72°C, 25-28 cycles of repeating step 2 through 4, and 5) 1 minute at 72°C. Primers included sequencing adapters to their 3′-ends, appending them to both termini of PCR products that amplified genomic DNA. After the first PCR step, products were assessed on 6% TBE-gel and purified using 1.0X AMPure (Beckman Coulter) and added to the second PCR reaction that appended dual sample indexes and flow cell adapters. The second PCR reaction program was identical to the first PCR program except we run 5-10 cycles. Products were again purified using AMPure and assessed on the TapeStation (Agilent) before denatured for the sequencing run. For long deletions that generate amplicons sized 200 to 300 bp, we used Miseq sequencing platform at low (8 pM) input DNA concentration to minimize the short amplicons replacing the long amplicons during clustering, aiming cluster density of 300-400 k/mm2. Denatured libraries were sequenced using either Illumina NextSeq or MiSeq instruments following the vendor protocols.
For appending 15-bp unique molecular identifiers (UMI), we performed the first PCR reaction in two-steps: First, genomic DNA was linearly amplified in the presence of 0.04 to 0.4 uM of single forward primer in two PCR cycles using KAPA Robust polymerase. We programmed the UMI-appending linear PCR reaction to be: 1) 3 minutes and 15 seconds at 95°C, 2) 1 minute at 65°C, 3) 2 minutes at 72°C, 5 cycles of repeating step 2 and 3, 4) 15 seconds at 95°C, 5) 1 minute at 65°C, 6) 2 minutes at 72°C, and another 5 cycles of repeating step 5 and 6. This reaction was cleaned up using 1.5X AMPure, and subject to the second PCR with forward and reverse primers. In this case, the forward primer anneals to the upstream of UMI sequence and is not specific to the genomic loci. After PCR amplification, products were cleaned up and added to another PCR reaction that appended dual sample indexes and flow cell adapters, similar to other samples.
Sequencing data processing and analysis
We designed the sequencing layout to cover at least 50-bp away from the deletion junction in each direction (Supplementary Figure 1a). In case of the paired-end sequencing, PEAR36 was used to merge the paired-end reads with default parameters and ‘-e’ flag to disable the empirical base frequencies. When 15-bp UMI was present in the sequencing reads, we used a custom Python script to find all reads that share the same UMI, and collapsed into a single read with the most frequent sequence. The resulting sequencing reads were aligned to two reference sequences (with or without deletion) generally using the CRISPResso2 software37. Default alignment parameters were used in CRISPResso2, with the gap-open penalty of −20, the gap-extension penalty of −2, and the gap incentive value of 1 for inserting indels at the cut/nick sites. The minimum homology score for a read alignment was explored between 50 and 95 for different amplicon length. Custom python and R scripts were used to analyze the alignment results from CRISPResso2.
Alignment was done using two reference sequences (wild-type and deletion) of same sequence length, generating two sets of reads with respective reference sequences. Deletion efficiencies were calculated as the fraction of total number of reads aligning to the reference sequence with deletion over the total number of reads aligning to either references. Genome editing has three types of error modes: substitution, insertion, and deletion. Each error frequency was plotted across two reference sequences, highlighting in each such plot the Cas9(H840A) nick-site and the 3’-DNA flap incorporation sites.
Droplet digital PCR (ddPCR) assay
We designed ddPCR probes following the recommended parameters by Bio-Rad Laboratories. We purchased pre-mixed reference probes and primers for the RPP30 gene from Bio-Rad Laboratories. Probes and PCR primers were purchased from Integrated DNA Technologies (IDT). Probes were modified with FAM on their 5’-ends and included double quenchers (IDT PrimeTime qPCR probes). Probe sequences were specifically designed to cover the deletion junction for detecting precise deletion products34. For detecting each deletion, we prepared a 20X primer mix composed of 18 uM forward-primer, 18 uM reverse-primer, and 5 uM FAM-labeled probe in 50 mM Tris-HCl buffer (pH 8.0 at room temperature). 25 uL of ddPCR reaction mixes were composed of 12.5 uL of 2X Supermix for Probes (no dUTP) (Bio-Rad Laboratories), 1.25 uL of 20X HEX-modified RPP30 reference mix (Bio-Rad Laboratories), 1.25 uL of 20X FAM-modified primer mix, 0.5 uL of cell lysate containing genomic DNA, and 9.5 uL of DNAse-free water. We added 20 uL of ddPCR reaction mix to 70 uL of Droplet generation oil for probes and used QX200 Droplet generator (Bio-Rad Laboratories) to generate droplets. Droplets were transferred to ddPCR 96-well plates (Bio-Rad Laboratories) and run on 96-well thermocyclers (Eppendorf) with the following program: 1) 10 minutes at 95°C, 2) 30 seconds at 94°C, 3) 1 minute at 50°C, 41 cycles of repeating step 2 and 3, 4) 10 minutes on 98°C, and 5) cooled down to 4°C before loading to QX200 Droplet reader. Temperature ramps were limited to 1°C per second on all steps on thermocyclers. We used QX200 Droplet reader and Bio-Rad QuantaSoft Pro software to visualize and analyze ddPCR experiments. The deletion efficiencies were taken from the ratio of FAM+ (precise-deletion) over HEX+ (RPP30 reference for genomic DNA loading) events.
Supplementary Material
Acknowledgements
We thank former and present members of the Shendure lab including Yi Yin, Jacob Tomes, Silvia Domcke, Alexander Boulgakov, Diego Calderon, Jase Gehring, Sanjay Srivatsan, and Lea Starita for helpful discussions. We thank the David Liu laboratory at Harvard University/Howard Hughes Medical Institute for sharing the prime editing plasmids. We thank Jase Gehring, Joshua Cuperus and the Stanley Fields laboratory at the Department of Genome Sciences, University of Washington, for their help with using a droplet-digital PCR instrument. This work was supported by the National Human Genome Research Institute grant 5UM1HG009408-04. J.C. is a Howard Hughes Medical Institute Fellow of the Damon Runyon Cancer Research Foundation (DRG-2403-20). J.S. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests
The University of Washington has filed a patent application based on this work, in which J.C., W.C., and J.S. are listed as inventors.
Code availability statement
Source code for PRIME-Del is available at https://github.com/shendurelab/Prime-del. An interactive webpage for designing pegRNAs for PRIME-Del is available at https://primedel.uc.r.appspot.com/.
Data availability statement
Raw sequencing data have been uploaded on Sequencing Read Archive (SRA) and made available to the public with associated BioProject ID PRJNA692623.
References
- 1.Knott GJ & Doudna JA CRISPR-Cas guides the future of genetic engineering. Science vol. 361 866–869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canver MC et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem 289, 21312–21324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrne SM, Ortiz L, Mali P, Aach J & Church GM Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res. 43, e21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gasperini M et al. CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet 101, 192–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gasperini M et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 1516 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Kosicki M, Tomberg K & Bradley A Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol 36, 765–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuccaro MV et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell (2020) doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- 9.Mehta A & Haber JE Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol 6, a016428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diao Y et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 14, 629–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu S et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol 34, 1279–1286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khosravi MA et al. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol 854, 398–405 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Dastidar S et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 46, 8275–8298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin Q et al. Prime genome editing in rice and wheat. Nat. Biotechnol 38, 582–585 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Kivioja T et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 9, 72–74 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Dominissini D et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Watry HL et al. Rapid, precise quantification of large DNA excisions and inversions by ddPCR. Sci. Rep 10, 14896 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandal PK et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15, 643–652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verkerk AJ et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Tippens ND et al. Transcription imparts architecture, function and logic to enhancer units. Nat. Genet 52, 1067–1075 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kweon J et al. Engineered prime editors with PAM flexibility. Mol. Ther (2021) doi: 10.1016/j.ymthe.2021.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leibowitz ML et al. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nature Genetics (2021) doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schene IF et al. Prime editing for functional repair in patient-derived disease models. 2020.06.09.139782 (2020) doi: 10.1101/2020.06.09.139782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owens DDG et al. Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic Acids Res. 47, 7402–7417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DY et al. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Research (2020) doi: 10.1093/nar/gkaa764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Brolosy MA et al. Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Z et al. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Concordet J-P & Haeussler M CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim HK et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol (2020) doi: 10.1038/s41587-020-0677-y. [DOI] [PubMed] [Google Scholar]
- 33.McKenna A & Shendure J FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 16, 74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen W et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Research vol. 47 7989–8003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Kobert K, Flouri T & Stamatakis A PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data have been uploaded on Sequencing Read Archive (SRA) and made available to the public with associated BioProject ID PRJNA692623.