

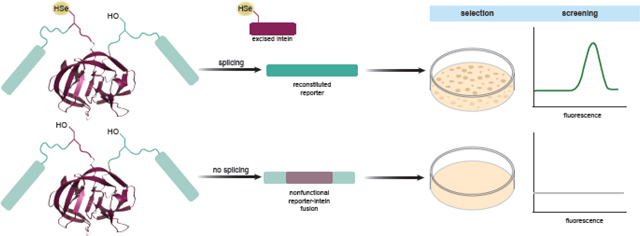
Abstract
The presence of selenocysteine in a protein confers many unique properties that make the production of recombinant selenoproteins desirable. Targeted incorporation of Sec into a protein of choice is possible by exploiting elongation factor Tu-dependent reassignment of UAG codons, a strategy that has been continuously improved by a variety of means. Improving selenoprotein yield by directed evolution requires selection and screening markers that are titratable, have a high dynamic range, enable high-throughput screening, and can discriminate against nonspecific UAG decoding. Current screening techniques are limited to a handful of reporters where a cysteine (Cys) or Sec residue normally affords activity. Unfortunately, these existing Cys/Sec-dependent reporters lack the dynamic range of more ubiquitous reporters or suffer from other limitations. Here we present a versatile strategy to adapt established reporters for specific Sec incorporation. Inteins are intervening polypeptides that splice themselves from the precursor protein in an autocatalytic splicing reaction. Using an intein that relies exclusively on Sec for splicing, we show that this intein cassette can be placed in-frame within selection and screening markers, affording reporter activity only upon successful intein splicing. Furthermore, because functional splicing can only occur when a catalytic Sec is present, the amount of synthesized reporter directly measures UAG-directed Sec incorporation. Importantly, we show that results obtained with intein-containing reporters are comparable to the Sec incorporation levels determined by mass spectrometry of isolated recombinant selenoproteins. This result validates the use of these intein-containing reporters to screen for evolved components of a translation system yielding increased selenoprotein amounts.
Keywords: genetic code expansion, tRNA, selection method, screening method, protein splicing
Graphical Abstract
Introduction
Selenium is an essential micronutrient in humans that exerts its health effects mainly through selenocysteine (Sec), the 21st amino acid.1,2 Chemically, Sec is equivalent to cysteine (Cys) with a selenium atom in place of the sulfur atom. However, the selenium atom provides Sec with unique properties.3,4 At physiological pH, Sec is considerably more nucleophilic than Cys due to its lower pKa (5.2 vs 8.5), thus existing primarily in its ionized form while Cys is primarily found in its reduced form. In addition, Sec has been shown to be more resistant than Cys to inactivation by a variety of oxidizing reagents5 and forms a significantly stronger diselenide bond compared to its corresponding disulfide bond.6 Inserting Sec into recombinant proteins has chemical advantages,7 but it can also be used as a tool for investigating reaction mechanisms, e.g., of redox enzymes,8 broadening its applications. Due to its high reactivity Sec is important in chemical protein synthesis,9,10 modulation of protein folding,11,12 and protein stability.6
Sec incorporation follows a pathway that is different from other amino acids, starting with synthesis on its cognate tRNA (tRNASec) (Figure 1A). In bacteria, tRNASec (encoded by the selC gene) is serylated by seryl-tRNA synthetase (SerRS) and then serine (Ser) is converted to Sec by selenocysteine synthase SelA. Sec-tRNASec is brought to the ribosome to decode an in-frame UGA codon by a specialized elongation factor, SelB, which interacts with a stem-loop structure (SECIS element) present in the Sec-encoding mRNA. This interaction and the specific distance between the UGA codon and the SECIS element create an appropriate context for natural UGA recoding and Sec insertion.13 Consequently, expression of archaeal and eukaryotic selenoproteins in which the SECIS element is located in the 3’-untranslated region may not be possible in Escherichia coli. However, changing the anticodon of E. coli tRNASec and keeping the SECIS present led to excellent yields for Sec-containing TrxR1 in the recoded C321.ΔA strain.14 Similarly, without additional mutations in their mRNA, the production of non-natural recombinant selenoproteins with Sec site-specifically inserted would not be feasible.
Figure 1.

(a) Selenocysteine is formed on tRNASec using a combination of seryl-tRNA synthetase (SerRS) and selenocysteine synthase (SelA). The charged tRNA decodes a UGA codon through recognition of a Selenocysteine insertion sequence (SECIS) element by a specialized elongation factor (SelB). (b) In elongation factor Tu (EF-Tu) dependent translation, allo-tRNASec is recognized by SerRS and SelA, but is brought to the ribosome by EF-Tu to decode a UAG codon. Insertion of Ser is also possible (dashed line) due to recognition of Ser-allo-tRNASec by EF-Tu.
Alternate systems have been developed that utilize tRNASec variants to insert Sec in response to UAG codons in E. coli (Figure 1B).15–18 These tRNASec variants are recognized by SerRS and SelA in a manner analogous to canonical tRNASec (Figure 1A) but are brought to the ribosome by EF-Tu. This removes the restriction placed by the SECIS element to dictate insertion of Sec at specific positions and allows the insertion of Sec anywhere in the protein. An important caveat with this technology is that EF-Tu is capable of binding Ser-tRNASec, an intermediate in the pathway of Sec-tRNASec synthesis (Figure 1B).
As stop-codon read-through at a permissive site in GFP might occur via either Ser-tRNASec or Sec-tRNASec decoding, the amount of reporter activity correlates only with the extent of UAG read-through, and not insertion of Sec. For this reason, traditional genetic reporters employed to monitor incorporation of noncanonical amino acids at UAG codons are not suitable as reporters of Sec insertion. Accurate measurements of Sec insertion have thus far predominantly relied on the isolation and mass spectrometric analysis of the target protein for confirmation of Sec insertion.19,20
Current Sec-specific reporters are limited to a small number of proteins where a Sec residue provides enzymatic activity. One common qualitative Sec-specific assay utilizes the ability of E. coli selenoenzyme formate dehydrogenase (FDHH) to reduce benzyl viologen and generate a purple color.21 However, this assay is done under anaerobic conditions and requires an E. coli strain that carries deletions of the endogenous fdhF gene (encoding FDHH) or components of the endogenous Sec insertion machinery.15–18 Analogous examination of Sec insertion through recombinant selenoprotein activity could be possible with, for example, human selenoprotein GPx1, but would be more laborious.15,16 To extend the repertoire of Sec-specific reporters, Cys-dependent proteins have been harnessed, provided that Cys replacement by Sec has significant effect on activity. Recently, a fluorescent Sec reporter22 was constructed from the phycobiliprotein (smURFP)23 whose catalytic Cys is required for chromophore ligation. Appropriate intracellular heme levels are favorable for good fluorescence. The seleno-smURFP design replaces the catalytic Cys residue with Sec and provides a simple fluorescent method to distinguish between Sec and Ser.22
Selection markers used for Sec-specific insertion include UAG-containing variants of thymidylate synthase (ThyA)15, human O6-alkylguanine-DNA alkyltransferase (hAGT),24 and β-lactamase.18,22 These existing Sec-dependent reporters are gene specific, and rely on Sec incorporation for activity. They are limited in their dynamic range and suffer from toxicity, poor distinction from Ser, or the need to use anaerobic conditions.
Here, we present a versatile strategy to engineer established reporters to become Sec-dependent. This strategy is based on inserting an intein cassette in-frame within the reporter gene to disrupt protein activity, requiring intein splicing for functional capacity. Replacement of the catalytic Cys of this intein with Sec allows the production of an active reporter that directly measures UAG-directed Sec incorporation. The results below show that a DnaB mini-intein variant25 was a suitable cassette to engineer such Sec-dependent reporters.
Results
Monitoring Sec incorporation via intein splicing – general strategy
To harness the power of general reporters such as superfolder green fluorescent protein (sfGFP) or antibiotic resistance genes for specific Sec insertion, we decided to use a Cys-dependent intein inserted in-frame within the reporter gene. Inteins may employ both Cys and Ser in their catalysis. The canonical splicing reaction requires Ser or Cys at the N-terminus of the intein (position 1, Figure S1) and Cys, Ser, or Thr at the N-terminus of the C-extein (position +1, Figure S1). The side chain in position 1 is necessary to start the splicing reaction by participating in the amide-thioester rearrangement that leads to the formation of the (thio)ester intermediate. The side chain in position +1 is necessary to execute the nucleophilic attack on the newly formed (thio)ester bond, facilitating the cyclization of the conserved Asn and subsequent intein release (Figure S1).26 Inteins have naturally evolved to work specifically within proteins they inhabit, resulting in drastically altered splicing activity when inserted into artificial protein targets.25,27,28 Thus, we sought intein sequences whose catalytic activity is (i) independent of extein context and (ii) employ a catalytic Cys but not Ser at position 1. The Synechocystis sp PCC6803 DnaB intein was shortened to form a DnaB mini-intein consisting of only the essential amino acids required for protein splicing.29 The protein was evolved to retain wildtype splicing activity when inserted into different sequences, with the most active and general variant identified as the M86 variant.25 This protein efficiently splices only when Cys is present at position 1 of M86, but is inactive with Ser in this position (Figure 2). This feature is not common with all inteins, but one that is useful when investigating Sec incorporation (Ser misincorporation is possible when Sec conversion by SelA is inefficient (Figure 1B)). Due to similar chemical reactivity of Cys and Sec, reassigning position 1 to Sec should also lead to protein splicing; thus, we generated a system that can distinguish between Ser and Sec at this crucial position (Figure 2). Following this approach, we can insert this DnaB mini-intein cassette into any gene and develop Sec-specific reporters that guide us to generate an enhanced selenoprotein translation system.
Figure 2.

Schematic of the KanR-M86 reporter showing Cys (C), Ser (S), or Sec (U) at position 1 (bolded in red) and Ser (S) at position +1 (bolded and underlined). Successful splicing will result in complete removal of the intein and reconstitution of the functional KanR protein. The M86 DnaB mini-intein self-splices out of the KanR protein efficiently with Cys at position 1 (panel A), leading to growth comparable to the KanR protein lacking the intein (KanR (no intein)). Replacement of Cys with Ser (panel B) leads to inactive M86 that is unable to splice out to generate an active protein, while replacement with Sec (panel C) leads to efficient splicing. A representative image for 3 replicates is shown.
Creation of a Sec-specific selection marker
The Sec-specific selection systems used to date include thymidylate synthase, O6-alkylguanine-DNA alkyltransferase, and β-lactamase enzymes.15,18,22,24 Therefore we chose to work with a different reporter for selection, aminoglycoside-3’-phospho-transferase-I, the product of the kanamycin resistance (kanR) gene. This enzyme was already used to monitor intein splicing in vivo via a catalytic Cys residue at position 1.25 To test the capability of this reporter to probe for Sec incorporation, we adapted the M86 DnaB mini-intein to encode Sec at position 1 and investigated insertion of the mini-intein at two different junctions (pAB_a01 and pAB_a02 plasmids; Figures 3A and B). Due to the possibility of Ser contamination in our Sec incorporation systems (Figure 1B), we required this reporter to be active with Sec and not Ser.
Figure 3.

(a) Design of the plasmids used to test the Sec incorporation by different tRNA variants using the kanamycin resistance (kanR) gene. (b) The M86 intein (highlighted in purple) was inserted at two different positions (a01 and a02)25 in the kanR gene that would disrupt protein function (PDB ID: 4EJ7).35 (c) The M86 intein was tested for efficiency in splicing at positions a01 and a02 with Cys or Sec at position 1 and growth was compared to the wildtype (WT) KanR protein without the intein Position 1 was also mutated to Ser or Gly to confirm absence of splicing. Initial spots are at OD600=2.0 with 10-fold dilutions across the plate. Each plate is at a different concentration of kanamycin (0–20 μg/mL). (d) pAB_a02 was chosen to test the efficiency of Sec incorporation in B-95.ΔAΔfabRΔselABC. A pSecUAG_tRNA plasmid lacking allo-tRNAUTu2D (ΔUTu2D) was used as a negative control. Each plate is at a different concentration of kanamycin (0–150 μg/mL). Areas of importance to compare are boxed in the same yellow line pattern. Representative images for 3 replicates are shown.
For the initial experiment with Sec-mediated splicing, allo-tRNAUTu2D was used;16 appropriate controls for Cys, Ser, and glycine (Gly) intein variants had this tRNA deleted. When M86 was inserted into the a01 site, Sec allowed growth up to 5 μg/mL kanamycin (kan), much less than the Cys containing reporter (20 μg/mL kan). However, when inserted into the a02 site of the KanR protein, Sec and Cys containing KanR-M86 proteins spliced equally well, allowing growth at wildtype (WT) levels (intein-free KanR protein, 20 μg/mL kan) (Figure 3C). Importantly, growth of pAB_a02 containing Ser or Gly (negative control) was equivalent (barely growing on 2.5 μg/mL kan), while pAB_a01 containing Ser grew well to 10−1 dilution at the same kan concentration (more details on cell dilution are given in the legend to Figure 3). Considering the lower growth with Ser and higher activity with Sec, pAB_a02 was chosen for the remainder of the experiments.
After choosing a plasmid system specific for the presence of Sec (pAB_a02), we used it as a tool to determine which of our currently used cell lines is preferable for incorporation of Sec. Unfortunately, ME6 (E. coli ΔselABC ΔfdhF)16 was found to be a poor cell line in this assay, with the cells barely growing in the absence of kan (Figure S2A). In contrast, allo-tRNAUTu2D facilitated growth up to 150 μg/mL kan in B-95.ΔAΔfabR and B-95.ΔAΔfabRΔselABC strains,30 comparable to the WT KanR protein and Cys containing pAB_a02 (Figures 3D and S2B). B-95.ΔAΔfabRΔselABC was further found to have less background growth (Gly present in position 1 allowed weak growth at 5 μg/mL kan in B-95.ΔAΔfabR) and therefore was chosen for the remainder of the experiments.
To show that incorporation of Sec is required for intein splicing, B-95.ΔAΔfabRΔselABC cells expressing the UAG variant of pAB_a02 and allo-tRNAUTu2D were grown in either the presence or absence of 10 μM sodium selenite. Cells grown in the absence of sodium selenite could not grow on plates containing 20 μg/mL kan while the addition of sodium selenite allowed growth (Figure S3A). Growth of cells up to 5 μg/mL kan in the absence of sodium selenite is likely due to the presence of a selenium source in the growth media.
Furthermore, we demonstrate that the KanR-M86 system can detect minor differences in tRNA expression. B-95.ΔAΔfabRΔselABC cells grown under different arabinose concentrations (to induce allo-tRNAUTu2D) showed that increasing tRNA levels allowed growth on plates with higher concentrations of kanamycin (Figure S3B). Cells grown in the presence of 0.001% arabinose could not grow at 50 μg/mL kan while 0.1% arabinose allowed growth up to 150 μg/mL kan.
Adaptation of a GFP reporter for specific Sec incorporation
To show the versatility of the intein-based design for engineering another Sec-specific reporter, we applied it to the commonly used fluorescent screening reporter, sfGFP. As discussed above, seleno-smURFP22 is a highly selective for Sec; yet very low fluorescence is still possible with the Ser variant. However, with the intein-based design, we can completely exclude Ser incorporation, as this amino acid prevents protein splicing.
To design the sfGFP-M86 reporter, we tested M86 splicing with four sfGFP-M86 fusion proteins (Figures 4A and B). Intein insertion positions were strategically chosen to interrupt a β-sheet and disrupt the fluorescent properties of the protein, requiring intein splicing for fluorescence. In all cases the M86 sequence was introduced preceding a naturally occurring Ser, except for pB_03, where a natural Arg was mutated to Ser (Ser is found in the WT GFP sequence). In this way the Ser residues of sfGFP occupy position +1 which is crucial for splicing (Figure S1). To mimic standard recombinant protein expression, we adopted an inducible plasmid system (pB_sfGFP) (Figures 4A and B). We tested the efficiency of M86 splicing at the four positions using a catalytic Cys at position 1 (Figure 4C). Compared to WT sfGFP (without intein), the best performing construct (pB_04) showed approximately 30% fluorescent intensity. The remaining constructs (pB_01, pB_02, pB_03) showed lower, mutually comparable yields of about 10% fluorescent intensity of WT sfGFP. As with the KanR-M86 assay, we tested pB_04 in three different E. coli strains: ME6, B-95.ΔAΔfabR, and B-95.ΔAΔfabRΔselABC. Since the fluorescence was comparable for the latter two strains, B-95.ΔAΔfabRΔselABC was chosen as it lacks the endogenous Sec machinery and is the same strain that was used for the KanR-M86 assays.
Figure 4.

(a) Design of plasmid used to encode sfGFP. This plasmid, in combination with the pSecUAG_tRNA plasmid, was used to test Sec incorporation by different tRNA variants. (b) The M86 intein (highlighted in purple) was inserted at four different positions in the sfGFP gene that would disrupt protein function (PDB ID: 2B3P).36 (c) Splicing efficiency of M86 intein was tested at all four positions in three cell lines (ME6, B-95.ΔAΔfabR, and B-95.ΔAΔfabRΔselABC) with Cys (C) at position 1 compared to the wildtype (WT) sfGFP protein without the intein. (d) Sensitivity of pB_04 to Ser (S) and Gly (G) was tested and compared to Cys (C) and Sec (U) with allo-tRNAUTu2D. A pSecUAG_tRNA plasmid lacking allo-tRNAUTU2D (ΔtRNA) was used as a negative control. Values shown are the average and standard deviation of four biological replicates.
Next, we tested the performance of pB_04 as a Sec-specific reporter (Figure 4D). We introduced a UAG codon in place of the catalytic Cys (position 1) of M86 and co-transformed this construct with the pSecUAG_tRNA plasmid (containing the necessary Sec incorporation machinery and allo-tRNAUTu2D). Under the conditions necessary for optimal Sec incorporation, Sec-dependent splicing (U) and sfGFP fluorescence was ~30 % of the Cys-containing construct (C) (Figure 4D). Importantly, pB_04 variants containing Ser (S) or Gly (G) showed no fluorescence when tested. This confirms the usefulness of sfGFP-M86 as a suitable reporter for Sec-specific insertion.
Application of the Sec reporters using known tRNASec variants to compare Sec incorporation
Current EF-Tu dependent Sec insertion systems are still lacking with respect to protein yield. In one case E. coli Hyd-1 with a single Sec residue produced yields of only 6% that of the Cys-containing Hyd-1.8 Therefore, further optimizations of EF-Tu dependent Sec insertion systems are necessary. To emphasize the ability of these intein-based reporters to screen for a better Sec-incorporation system, we employed already optimized tRNASec variants and their respective machineries. For 13-branch systems, tRNASecUX and tRNAUTux contained the compatible E. coli SelA enzyme (pSecAUG_tRNA plasmids),17,18 while the 12-branch allo-tRNA-derived variants (allo-tRNAUTu1, allo-tRNAUTu1D, allo-tRNAUTu2, and allo-tRNAUTu2D) contained the Aeromonas salmonicida SelA and SelD enzymes (Figure S4).16 Figure 5A shows that the KanR-M86 reporter is able to distinguish small changes in Sec incorporation efficiency over a wide range of kan concentrations. We observed allo-tRNAUTu1, allo-tRNAUTu1D, and allo-tRNAUTu2D to grow well at high concentrations of kan (100–150 μg/mL) (Figure 5A). Allo-tRNAUTu1D was found to grow at multiple dilutions (up to 10−4) at 150 μg/mL kan while allo-tRNAUTu1 and allo-tRNAUTu2D only survived when undiluted (more details on cell dilution are given in the legend to Figure 5). On the other hand, tRNASecUx, tRNAUTuX and tRNAUTu2 barely supported growth at 2.5–5 μg/mL kan.
Figure 5.

(a) KanR-M86 assay using pAB_a02 to test efficiency of six tRNASec variants. Initial spots are at OD600=2.0 with 10-fold dilutions across the plate. Each plate contains a different concentration of kanamycin (0–150 μg/mL). (b) sfGFP-M86 assay using pB_04 to test efficiency of the same six tRNASec variants. Values shown are the average and standard deviation of 8 biological replicates. (c) Benzyl viologen assay using E. coli FDHH (140UAG) to test the efficiency of six tRNASec variants. Initial spots are at OD600=5.0 with 2-fold dilutions across the plate. Plates shown are representatives of greater than 3 replicates.
Comparable results are obtained with the sfGFP-M86 reporter. Maximum fluorescence was obtained using allo-tRNAUTu1D and allo-tRNAUTu2D, with allo-tRNAUTu1 hovering around 50% that of allo-tRNAUTu2D (Figure 5B). On the other hand, relative activity of tRNASecUx, tRNAUTuX, and allo-tRNAUTu2 was between 25–30%, around the level of background fluorescence (ΔtRNA plasmid).
These results are in an agreement with Sec incorporation levels as monitored by the benzyl viologen assay (Figure 5C). The E. coli fdhF gene, encoding FDHH, produces active protein only when position 140 contains a Sec residue. Active protein is verified by reduction of benzyl viologen under anaerobic conditions to produce an observable purple color. Using an inducible expression system (similar to the pB_sfGFP reporter), we tested all the tRNAs investigated with the intein-based reporters. Allo-tRNAUTu2D allowed the strongest purple color to be developed at dilutions up to 2−3 while strains harboring allo-tRNAUTu1 and allo-tRNAUTu1D both had a weaker color, although equivalent to one another. Parallel to what was observed in the previous assays, under these conditions no accumulation of purple color was observed for tRNASecUx, tRNAUTuX, and allo-tRNAUTu2.
Sec incorporation measured by mass spectrometry matches results from intein-based reporters
To verify that the results obtained through in vivo assays using the intein-based reporters correlated with selenoprotein expression levels, we expressed and purified two selenoproteins, E. coli Grx1 and human GPx1, using the EF-Tu dependent Sec incorporation technology (Table 1 and Figure 1B). Recombinant selenoprotein expression levels were tested employing various tRNASec variants and their respective machineries. Using the same expression and purification protocol for both proteins, we were able to compare the trends of the protein yields observed between the tRNAs.
Table 1.
Table of values for Grx1 and GPx1 production with various tRNAs for Sec incorporation.
| tRNA | Grx1 | GPx1 | ||||
|---|---|---|---|---|---|---|
| Yield (mg)* | Sec incorporation (%) | Activity (U/mg)** | Yield (mg)* | Sec incorporation (%) | Activity (U/mg)** | |
| SecUx | 0.42 | 13.6 | 9 ± 2 | 0.40 | 6.8 | 0.08 ± 0.01 |
| UTuX | 0.15 | 18.8 | 4 ± 2 | 0.18 | 0 | 0.28 ± 0.03 |
| UTu1 | 3.8 | 19.1 | 35 ± 4 | 0.18 | 44.9 | 21 ± 4 |
| UTu1D | 1.6 | 33.7 | 53 ± 4 | 0.62 | 73.4 | 29 ± 2 |
| UTu2 | 0.30 | 19.0 | 9 ± 2 | 0.21 | ND | 1.2 ± 0.3 |
| UTu2D | 0.37 | 31.8 | 25 ± 3 | 0.26 | 11.5 | 5 ± 1 |
| Pos Control | ND | 0 | 130 ± 10 | ND | 100 | 32 ± 3 |
| Neg Control | ND | 0 | 2 ± 1 | ND | 0 | 0.07 ± 0.01 |
ND: not determined
Protein yield is from a 1 L culture
1 U is defined as the amount of enzyme that catalyzes conversion of 1 μmol of substrate per minute at 25°C ± SE.
The trend of protein yields was consistent between Grx1 and GPx1, with the exception of allo-tRNAUTu1 (Table 1). Allo-tRNAUTu1D facilitated the highest yield while tRNAUTuX resulted in the lowest (in descending order allo-tRNAUTu1D > tRNASecUx > allo-tRNAUTu2D > allo-tRNAUTu2 > tRNAUTuX). Allo-tRNAUTu1 was found to have the highest yield in Grx1 (twice that of allo-tRNAUTu1D) but the lowest in GPx1 (equivalent to tRNAUTuX).
The more important question was whether the recombinant selenoproteins were active and how much Sec was incorporated. Both Grx1 and GPx1 produced similar trends for enzymatic activity with allo-tRNAUTu1D having the highest activity and tRNAUTuX having the lowest (in descending order allo-tRNAUTu1D > allo-tRNAUTu1 > allo-tRNAUTu2D > allo-tRNAUTu2 > tRNASecUx > tRNAUTuX) (Table 1). To confirm that the enzymatic activity correlated to the amount of Sec inserted, intact mass spectrometry (MS) of the proteins was done. Deconvolution of the MS spectra found that the percentage of Sec in the proteins correlated with its observed activity; higher percentage of Sec leads to increased activity (Table 1, Figures 6 and 7). For Grx1, tRNAUTu1 Sec incorporation was low compared to its activity. This discrepancy can be explained by the difficulty in distinguishing the overlapping peaks due to Grx1_Q11 with one oxidation and Grx1_U11. It should be noted that Grx1 is naturally not a selenoprotein and its Cys homolog (positive control) is more active than when Sec is present. GPx1 is a natural selenoprotein and replacement of the active site Sec for Cys deems it inactive.31 The amount of Sec incorporation varied between the proteins though the trend remained the same. tRNAs which had low Sec incorporation in the intein-based assays were found to have a higher percentage of Sec incorporated in Grx1 compared to GPx1. However, for tRNAs with high Sec incorporation in the intein-based assays (allo-tRNAUTu1 and allo-tRNAUTu1D), GPx1 had a higher amount of Sec (44.9% and 73.4%, respectively) compared to Grx1 (19.1% and 33.7%, respectively).
Figure 6.

(a) Intact mass analysis of Grx1 to determine the amount of selenocysteine (Sec, U) incorporation for each tRNA tested with the intein-based reporters. Peaks corresponding to Sec incorporation are highlighted in red while serine (S) and glutamine (Q) incorporation are in black. The table below the spectra provides details on the identified peaks. (b) Grx1 activity assay for each tRNASec variant was visualized as the consumption of NADPH over time at concentrations up to 65 nM of a tRNASec variant. Data points are an average of three independent measurements ± SD.
Figure 7.
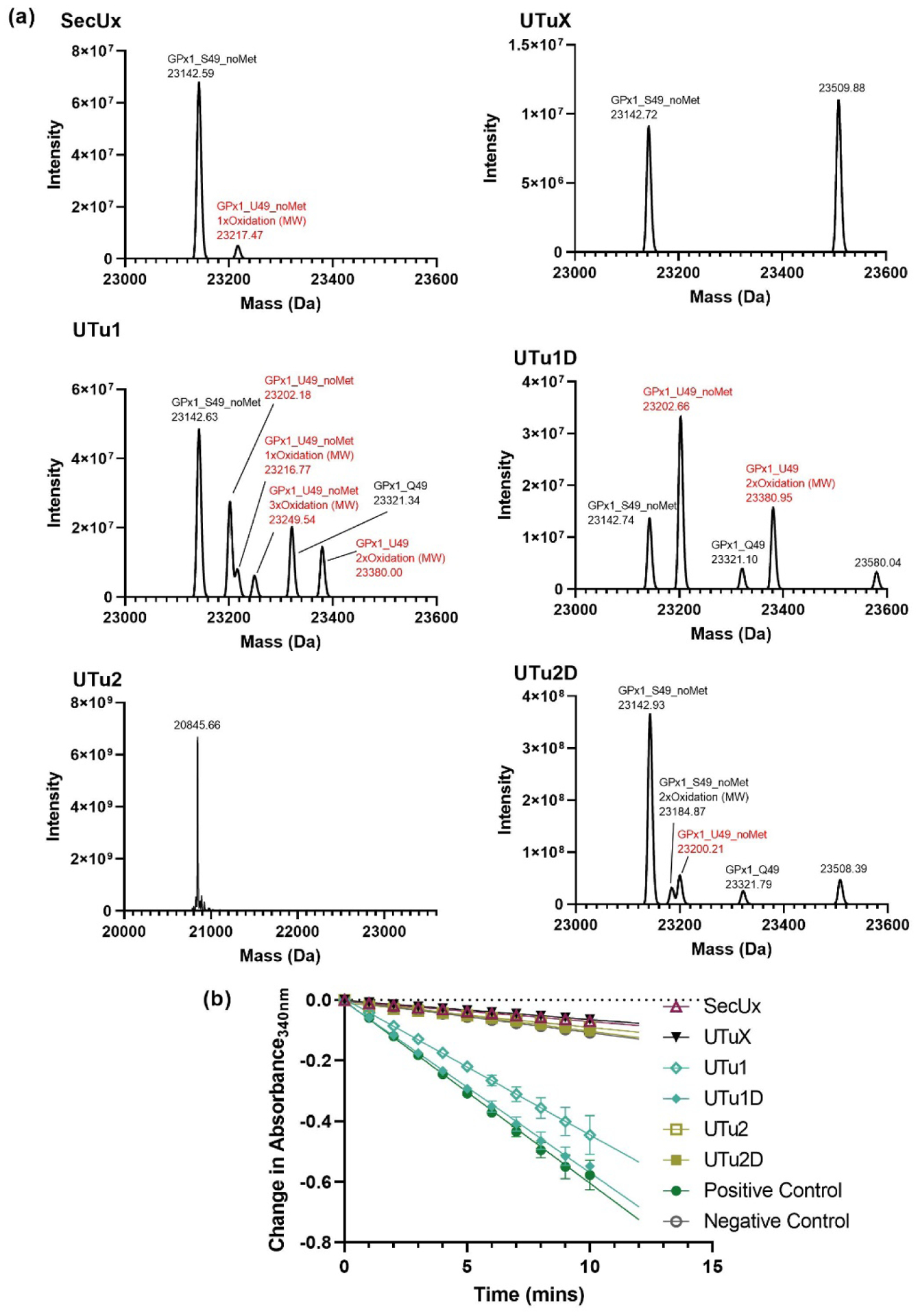
(a) Intact mass analysis of GPx1 to determine the amount of selenocysteine (Sec, U) incorporation for each tRNA tested with the intein-based reporters. Peaks corresponding to Sec incorporation are highlighted in red while serine (S) and glutamine (Q) incorporation are in black. No peaks were observed at the correct size for allo-tRNAUTu2. The truncated product detected in the intact mass analysis of GPx1 expressed in the presence of allo-tRNAUTu2 was observed in all spectra (not shown). (b) GPx1 activity assay for each tRNASec variant was visualized as the consumption of NADPH over time. Data points are an average of three independent measurements ± SD.
The MS and enzymatic activity of these recombinant selenoproteins agrees with most of the results obtained from the intein-based reporters. From the sfGFP-M86 and benzyl viologen assay, allo-tRNAUTu2D stands out as a promising candidate for Sec incorporation. However, it did not produce reasonable Sec incorporation in either Grx1 or GPx1. Instead, allo-tRNAUTu1D performed well in both intein-based reporters and the benzyl viologen assay, affording the highest Sec incorporation in both recombinant selenoproteins.
Discussion
The many desirable applications of selenoproteins drive the desire to improve the current methodologies for site-specific in vivo Sec incorporation. Two top aims, increased selenoprotein yields and consistently high levels of Sec incorporation require Sec-specific selection tools and screening reporters with high dynamic range. The currently accepted method to confirm Sec incorporation is through expressing FDHH and testing its activity via benzyl viologen reduction.21 This very sensitive method detects even the smallest amount of Sec incorporation, but requires work in an anaerobic chamber. A β-lactamase reporter used to screen a library of tRNASec variants required up to 1000 μg/mL ampicillin18, much higher than the recommended working concentration, to screen for a tRNA species with low Sec incorporation (tRNASecUx) (Table 1). Recently seleno-smURFP has provided a quantitative fluorescent reporter that has been used to improve genetically recoded E. coli strains for selenoprotein production.22 However, this reporter requires special optimization of the bacterial strain, and is poor at discriminating against Ser incorporation.
The potential use of inteins as Sec-specific reporters was already demonstrated by our group and others: Mycobacterium xenopi GyrA intein is active when the catalytic Cys is replaced by Sec, but not Ser.16,24,32 However, the GyrA intein activity was induced in vitro, in the presence of high concentrations of dithiothreitol. For the purposes of an in vivo Sec-reporter assay, we sought an intein that would be (i) catalytically active once Sec is introduced, (ii) inactive when the catalytic residue is replaced by Ser, and (iii) tolerant towards the surrounding extein sequences. The first two conditions had to be satisfied to monitor specific Sec insertion, as well as Ser misincorporation. The third condition was critical to generate a Sec-specific cassette that could be shuttled between different reporters.
The DnaB mini-intein M86 showed remarkable sensitivity to Ser insertion and is catalytically active in the presence of Sec (Figure 2). Furthermore, it had been previously evolved to perform as a general intein, with the ability to splice different proteins at multiple insertion sites.25 Since splicing activity is not equal at each insertion site, this allows the reporter to be easily tuned to extend its dynamic range. Starting with a Sec system that is not very efficient we optimized the insertion site to achieve the highest signal while maintaining sensitivity for Sec incorporation (a02 for the KanR protein and 04 for sfGFP; Figures 3C–D, 4C–D, and S2).
With these two different reporter systems, we were able to test the activity of various tRNAs previously used for Sec incorporation. Comparing the various Sec-tRNAs, allo-tRNAUTu1, allo-tRNAUTu1D, and allo-tRNAUTu2D produced greater selenoprotein amounts than tRNASecUx, tRNAUTuX, and allo-tRNAUTu2 (Table 1 and Figures 6 and 7). The top three performing tRNAs, allo-tRNAUTu1D, allo-tRNAUTu1 and allo-tRNAUTu2D enabled cells to develop significant kan resistance and fluorescence. The slightly different results between the performance of these tRNAs in the two assays can be attributed to context-dependent tRNA suitability, i.e. the mRNA-tRNA interaction, and differences in the way each reporter is expressed; KanR-M86 uses a constitutive promoter, while sfGFP-M86 expression is induced. The latter system allows a more stringent control over expression of the reporter. With addition of arabinose at the beginning of the culture, cells are first allowed to produce large quantities of tRNASec, which are subsequently serylated by endogenous SerRS, before converted to Sec by the plasmid borne copy of SelA. In this way, the pool of acylated suppressor tRNA is created prior to expression of the reporter. In the case of KanR-M86, the constitutively expressed mRNA can be translated at an earlier time point, potentially using more of the Ser-tRNASec intermediate. This may mask the subtle differences in the efficiency of Ser-to-Sec conversion between different tRNAs. Specifically, both tRNAUTu1D and tRNAUTu2D have been altered in their D-arm to have a higher affinity to A. salmonicida SelA than their previous variants (allo-tRNAUTu1 and allo-tRNAUTu2, respectively) (Figures 5B and S4).16 It is likely that this improved conversion of the reaction intermediate is more apparent in the sfGFP-M86 assay due to the controlled expression of the reporter.
Protein expression of Grx1 and GPx1 confirmed that allo-tRNAUTu1D produced the highest yield with the most Sec incorporated. The yield of Sec incorporated using tRNAUTu1 was protein dependent; significantly higher than allo-tRNAUTu2D in GPx1 compared to Grx1 (Table 1, Figures 6 and 7). This was also observed in the two intein assays, with allo-tRNAUTu1 performing better in the KanR-M86 compared to the sfGFP-M86 assay (Figures 5A and B). However, this was not the case with allo-tRNAUTu2D. allo-tRNAUTu2D exhibited low to modest Sec incorporation in GPx1 and Grx1 compared to allo-tRNAUTu1 and allo-tRNAUTu1D (Table 1, Figures 6 and 7) while displaying the highest Sec incorporation in the sfGFP-M86 assay (Figure 5B) and benzyl viologen assay (Figure 5C). This is likely due the dependence of allo-tRNAUTu2D on the sequences surrounding the recoded UAG codon. Thus, application of both intein-based reporters is preferred to identity the best performing tRNA variants (or Sec incorporation system) before expression of the desired selenoprotein.
This strategy of designing intein reporters is not limited to the KanR protein or sfGFP, but applicable to any gene of interest. In addition, the assay sensitivity can be altered by choosing a different intein insertion position in the protein. We tested out positions a01 and a02 for the KanR-M86 reporter as they had the highest splicing activity (Figure 3C). However, as the Sec incorporation system becomes more efficient, a reasonable concentration of kan may not be sufficient for selection. Therefore, by inserting the intein at a different position,25 the kan concentration may be reduced for appropriate selection. The ability to choose the gene and tune the sensitivity of the reporter makes it a versatile tool for synthetic biologists to engineer efficient machinery for Sec incorporation.
Although our studies were performed in E. coli, the intein reporter system is not limited to bacteria. Using the same strategy as described above, an appropriate reporter may be designed for use in eukaryotes, specifically in mammalian cells. There are currently 25 identified selenoproteins in humans, with many of their functions still unknown.1 Human proteins can be difficult to produce in E. coli and will lack the proper posttranslational modifications, among other problems,33 hindering our study of selenoproteins. Designing a recombinant Sec system in mammalian cells would allow overexpression of the proteins with the proper modifications for protein activity and folding. As the mammalian Sec incorporation system is quite complicated (i.e. more components compared to the bacterial system and unidentified factors in the pathway), a simple fluorescent intein-based reporter that relies solely on the incorporation of Sec without affecting endogenous machinery, should help in the development of a recombinant Sec system.
Materials and methods
Cloning and mutagenesis
KanR-M86 expression plasmids.
Plasmids encoding aminoglycoside-3’-phosphotransferase-I, the kanamycin resistance (kanR) gene with the M86 DnaB mini-intein variant inserted after Ser154 and Ser164 (pAB_a01 and pAB_a02, respectively; Table S1) were kindly provided by Henning D. Mootz.25 Position 1 of the intein (Cys, TGC) was mutated to encode Ser (TCG), Gly (GGT in pAB_a01, GGC in pAB_a02), or Sec (TAG) in both plasmids. All PCRs were performed with PfuUltra II Fusion HS DNA Polymerase (Agilent). PCR products were gel purified (Macherey-Nagel) and ligated together using NEBuilder® HiFi DNA Assembly Master Mix (NEB). Products were transformed into DH5α competent cells and sequencing of the plasmids confirmed successful mutagenesis (Quintara Biosciences).
A control plasmid with wild-type kanR lacking the intein (pAB_kan) was generated by amplifying pAB_a02 around the intein. The PCR products were gel purified (Macherey-Nagel) and the linear products were 5’-end phosphorylated and ligated in the same reaction using T4 PNK (NEB) and T4 DNA ligase (NEB), respectively. Products were transformed into DH5α competent cells and sequencing of the plasmids confirmed successful removal of M86 (Quintara Biosciences).
Sec system expression plasmids.
Construction of all plasmids encoding the Sec expression system used in this study have been previously reported.16 The pSecUAG-ADT series was used with allo-tRNAUTu1, allo-tRNAUTu1D, or allo-tRNAUTu2 and the pSecUAG-Evol series was used for allo-tRNAUTu2D. For tRNASecUx and tRNAUTuX, plasmids with the same backbone as the pSecUAG-Evol and ADT series were used but with the E. coli Sec machinery (pSecAUG_tRNA plasmids). The kanR gene was replaced with specR to allow co-transformation with the KanR-M86 constructs. A control plasmid was generated by deleting allo-tRNAUTu2D in the same manner as described above.
sfGFP-M86 expression plasmids.
E. coli codon-optimized sfGFP with a C-terminal His6-tag was amplified and inserted into pET-15b in place of the N-terminal His6-tag, thrombin site, and multiple cloning site. The inserted sfGFP gene along with the T7 promoter, T7 terminator, and lacI sequences was amplified and moved to the high-copy number plasmid pBAD, where it replaced the araBAD promoter. The M86 DnaB mini-intein was inserted after residues 27, 29, 71, and 204 (Table S1 and Figure 4B) and site-directed mutagenesis was performed to mutate position 1 (Cys, TGC) of the intein to encode Ser (AGC), Gly (GGC), or Sec (TAG) (pB_sfGFP series).
E. coli Grx1 and human GPx1 expression plasmids.
Construction of pET-Grx1(11UAG/14Ser), pET-Grx1(11Cys/14Ser), pET-GPx1(49UAG), and pET-GPx1(49Cys) has been previously described.16 pET-Grx1(11Ser/14Ser) was constructed through site-directed mutagenesis following manufacturer’s instructions to change TAG to AGC.
E. coli FDHH expression plasmids.
E. coli fdhF gene was cloned into pTRC99A with a C-terminal His6-tag. Position 140 (TGA) encodes Sec and was mutated to TAG through site-directed mutagenesis.16
Kanamycin resistance assay
Plasmids encoding the Sec expression system and KanR-M86 variant were transformed into electrocompetent E. coli cells (ME6, B-95.ΔAΔfabR, or B-95.ΔAΔfabRΔselABC)16,30, plated on LB agar plates (50 μg/mL ampicillin, 50 μg/mL spectinomycin), and incubated at 37°C overnight. Single colonies were grown in 5 mL LB cultures (50 μg/mL amp, 50 μg/mL spec, 0.1% arabinose, 10 μM sodium selenite (Na2SeO3)) at 37°C overnight. A dilution to OD600 2.0 and 10-fold serial dilutions were prepared. From each dilution, 5 μL was spotted onto LB agar plates (50 μg/mL amp, 50 μg/mL spec, 0.1% arabinose, 10 μM Na2SeO3) containing 0–150 μg/mL kan. Plates were left at 37°C overnight and imaged the next day on the ChemiDoc™ MP Imager (Bio-Rad). For streaked cells, an inoculating loop was dipped directly into the preculture and streaked onto a LB agar plate (50 μg/mL amp, 50 μg/mL spec, 0.1% arabinose, 10 μM Na2SeO3,50 μg/mL kan). For growths observing the effect of Na2SeO3 and arabinose on Sec incorporation, the required Na2SeO3 and arabinose concentrations were implemented starting at the preculture stage with the same starting colony used for each set of experiments.
GFP fluorescence assay
Plasmids encoding the Sec expression system and sfGFP-M86 variant were transformed into electrocompetent E. coli cells (ME6, B-95.ΔAΔfabR, or B-95.ΔAΔfabRΔselABC)16,30, plated on LB agar plates (50 μg/mL amp, 50 μg/mL spec), and incubated at 37°C overnight. Single colonies were grown in 0.5 mL LB (50 μg/mL amp, 50 μg/mL spec, 1% glucose, 0.1% arabinose, 10 μM Na2SeO3) at 37°C for 8 hours before transferred to a 96-well black plate with clear bottoms. Cultures were mixed 1:1 with fresh media (to a final volume of 150 μL) with and without the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) to induce sfGFP expression. Fluorescence (Ex. 485 nm, Em. 528 nm) and OD600 readings were taken every 15 minutes for 24 hours at 37°C on the Synergy HTX Plate Reader (BioTek). Each experiment was performed with a minimum of four biological replicates. The fluorescence readings were divided by the cell densities (OD600) at 16 hours and were graphed using GraphPad Prism 9.
In vivo FDHH activity assay
Plasmids encoding the Sec expression system and fdhF were transformed into electrocompetent B-95.ΔAΔfabRΔselABC cells, plated on LB agar plates (50 μg/mL amp, 50 μg/mL spec), and incubated at 37°C overnight. Single colonies were grown in 7 mL LB cultures (50 μg/mL amp, 50 μg/mL spec, 0.1% arabinose, 10 μM Na2SeO3) at 37°C overnight. A dilution of OD600 5.0 and 2-fold serial dilutions were prepared. From each dilution, 5 μL was spotted onto LB agar plates (50 μg/mL amp, 50 μg/mL spec, 0.1% arabinose, 10 μM Na2SeO3, 50 mM sodium formate (NaHCOO), 1 μM sodium molybdate (Na2MoO4), 10 μM IPTG). Plates were placed inside an anaerobic jar (90% N2, 5% H2, 5% CO2) and incubated at 37°C for 24 hours. A thin layer of LB agar (0.75% agar, 1 mg/mL benzyl viologen di-chloride, 250 mM NaHCOO, 25 mM monopotassium phosphate (KH2PO4) [pH 7.0]) was poured over the plates in an anaerobic chamber and imaged after 24 hours.
Expression and purification of E. coli Grx1
Expression and purification of C-terminally tagged E. coli Grx1 was performed as previously described with minor changes.15 Briefly, plasmids encoding the Sec expression system and Grx1 variant were transformed into electrocompetent B-95.ΔAΔfabRΔselABC, plated on LB agar plates (50 μg/mL amp, 50 μg/mL spec), and incubated at 37°C overnight. Grx1 (11Cys/14Ser) and Grx1 (11Ser/14Ser) were co-transformed with the Sec expression plasmid system void of tRNAUTu2D. Single colonies were grown in 10 mL LB (50 μg/mL amp, 50 μg/mL spec) at 37°C overnight and used to inoculate 1 L LB (50 μg/mL amp, 50 μg/mL spec, 0.1% arabinose, 10 μM Na2SeO3). Cultures were grown at 37°C until OD600 1.2, at which point the temperature was lowered to 20°C and protein expression was induced with 100 μM IPTG for 20 hours.
All purification steps were performed under anaerobic conditions (90% N2, 5% H2, 5% CO2) in an anaerobic tent (Coy Laboratories). Each pellet was resuspended in 18 mL lysis buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10% glycerol, 30 mM imidazole) and 2 mL BugBuster® 10X Extraction Reagent (EMD Millipore) along with 2 mM β-mercaptoethanol and 0.05 mg/mL lysozyme. The resuspended cells were incubated at room temperature for 20 minutes, with occasional mixing, and the lysate was centrifuged at 38 000 rpm for 45 minutes at 4°C. The supernatant was loaded onto 2 mL nickel resin (Ni-NTA Agarose, Qiagen) pre-equilibrated with lysis buffer. The beads were washed with 150 mL lysis buffer and eluted in 1 mL fractions with elution buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10% glycerol, 230 mM imidazole). Protein elution was monitored using the Bio-Rad Protein Assay Dye. Elutions were concentrated and buffer exchanged to the storage buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10% glycerol) using Amicon® Ultra Centrifugal Filters (Merck Millipore) and stored at −80°C.
Grx1 glutathione oxidoreductase activity assay
The assay was performed as previously described.15 Briefly, 35 μL 20 mM β-hydroxyethylene disulfide (HED, final is 0.7 mM) was preincubated with 950 μL buffer (1 mM reduced glutathione, 100 mM Tri-HCl [pH 8.0], 2 mM EDTA, 1 mg/mL bovine serum albumin (BSA), 0.4 mM nicotinamide adenine dinucleotide phosphate reduced (NADPH), 6 μg/mL glutathione reductase) for 2 minutes at room temperature. The reaction was initiated with the addition of 10, 25, or 50 nM Grx1 variant (in triplicate) and the consumption of NADPH was measured at 340 nm every 30 seconds for 3 minutes. The change in absorbance over time was calculated from 1–2 minutes and averaged between the three measurements, followed by buffer subtraction. This was adjusted to a change in nmol of NADPH using the extinction coefficient (ε = 0.00622 μM−1cm−1) and then plotted against the concentration of Grx1 used. The slope and error of this line were used to calculate the activity of the enzyme (in μmol of NADPH/minute/mg protein) using the molecular weight of Grx1 (10 804 Da).
Expression and purification of human GPx1
Expression and purification of human GPx1 was performed as described above for E. coli Grx1.
Glutathione peroxidase activity assay with GPx1 variants
GPx1 peroxidase activity was assessed in a 96-well plate using a Glutathione Peroxidase Assay Kit (Cayman Chemical). GPx1 samples diluted in GPX Sample Buffer (50 mM Tris-HCl [pH 7.6], 5 mM ethylenediaminetetraacetic acid (EDTA), 1 mg/mL BSA) were analyzed according to the manufacturer’s protocol. Depending on the sample, 0.1–3 μg of protein was used to achieve a decrease in absorbance between 0.02 and 0.135 per minute. Consumption of NADPH was measured at 340 nm every minute for 10 minutes on the Synergy HT Plate Reader (BioTek). A positive control of bovine erythrocyte GPx and a blank for background subtraction were included. Calculations for GPx1 activity were performed as in the manufacturer’s protocol and expressed per mg of enzyme used in the reaction. All reactions were performed in triplicate.
Intact mass spectrometry of GPx1 Sec variants
GPx1 and Grx1 Sec variants were sent to Bioinformatics Solutions, Inc (Canada) for intact mass analysis to quantify Sec incorporation. Protein samples were reduced with DTT34 before LC-MS was performed on a Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer, equipped with a heated electrospray ionization source (H-ESI) in positive ion mode with a Thermo Fisher Ultimate 3000 RSLC nano HPLC System. On H-ESI source, sheath gas was set to 2 arbitrary units (arb), and auxiliary gas was set to 6.7 arb. The vaporizer temp was at 200°C. The sample was analyzed on a MAbPac RP, 4μM, 3.0×50 mm analytical column (ThermoFisher, San Jose, CA), held at 70°C. Protein was eluted at a rate of 500 μL/min for a 10 min gradient. 0–7 mins :10% – 70% acetonitrile + 0.1% formic acid; 7–8.2 mins: 95% acetonitrile + 0.1% formic acid, 8.2–10 mins: 20% acetonitrile + 0.1% formic acid. MS spectra were acquired using full scans at 7500 resolution in the orbitrap within a range of 700–2200 m/z. The maximum injection time was limited to 50 ms, with an AGC target of 4e6. Ten micro scans were employed, and the RF lens was set to 50%. 15 V of insource CID was applied.
Thermo BioPharma Finder™ 4.1 (ThermoFisher Scientific) was used for intact mass deconvolution and peak identification.
Supplementary Material
Highlights.
a DnaB mini-intein can selectively utilize selenocysteine (Sec) for splicing
a transferable DnaB cassette that monitors UAG-directed Sec insertion is developed
traditional reporters can be disrupted with a DnaB cassette to detect Sec insertion
Sec-dependent intein-fusion reporters monitor Sec insertion with high accuracy
Acknowledgements
We thank Henning D. Mootz (Münster, Germany) for inspired advice and the gift of M86 DnaB constructs. We are grateful to Jonathan Fischer, Jeffery Tharp, Oscar Vargas-Rodriguez, and Kyle Hoffman (Bioinformatics Solutions, Inc) for experimental advice and mass spectrometry support.
Funding
This work was supported by grants the National Institute of General Medical Sciences (R35GM122560 to D.S.), and, for the genetic studies, the Department of Energy Office of Basic Energy Sciences (DE-FG0298ER2031 to D.S.). Christina Z. Chung holds a Postdoctoral Fellowship from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Schmidt RL, Simonović M, (2012). Synthesis and decoding of selenocysteine and human health. Croat Med J, 53, 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang D, Lee J, Wu C, Guo X, Lee BJ, Chun JS, Kim JH, (2020). The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp Mol Med, 52, 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reich HJ, Hondal RJ, (2016). Why nature chose selenium. ACS Chem Biol, 11, 821–841. [DOI] [PubMed] [Google Scholar]
- 4.Maroney MJ, Hondal RJ, (2018). Selenium versus sulfur: Reversibility of chemical reactions and resistance to permanent oxidation in proteins and nucleic acids. Free Radic Biol Med, 127, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snider GW, Ruggles E, Khan N, Hondal RJ, (2013). Selenocysteine confers resistance to inactivation by oxidation in thioredoxin reductase: comparison of selenium and sulfur enzymes. Biochemistry, 52, 5472–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arai K, Takei T, Okumura M, Watanabe S, Amagai Y, Asahina Y, Moroder L, Hojo H, Inaba K, Iwaoka M, (2017). Preparation of selenoinsulin as a long-lasting insulin analogue. Angew Chem Int Ed Engl, 56, 5522–5526. [DOI] [PubMed] [Google Scholar]
- 7.Metanis N, Hilvert D, (2014). Natural and synthetic selenoproteins. Curr Opin Chem Biol, 22, 27–34. [DOI] [PubMed] [Google Scholar]
- 8.Evans RM, Krahn N, Murphy BJ, Lee H, Armstrong FA, Söll D, (2021). Selective cysteine-to-selenocysteine changes in a [NiFe]-hydrogenase confirm a special position for catalysis and oxygen tolerance. Proc Natl Acad Sci U S A, 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giesler RJ, Erickson PW, Kay MS, (2020). Enhancing native chemical ligation for challenging chemical protein syntheses. Curr Opin Chem Biol, 58, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Chen Q, Rozovsky S, (2018). Selenocysteine-mediated expressed protein ligation of SELENOM. Methods Mol Biol, 1661, 265–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pegoraro S, Fiori S, Rudolph-Bohner S, Watanabe TX, Moroder L, (1998). Isomorphous replacement of cystine with selenocystine in endothelin: oxidative refolding, biological and conformational properties of [Sec3,Sec11,Nle7]-endothelin-1. J Mol Biol, 284, 779–792. [DOI] [PubMed] [Google Scholar]
- 12.Metanis N, Hilvert D, (2015). Harnessing selenocysteine reactivity for oxidative protein folding. Chem Sci, 6, 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labunskyy VM, Hatfield DL, Gladyshev VN, (2014). Selenoproteins: molecular pathways and physiological roles. Physiol. Rev, 94, 739–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng Q, Arner ES, (2017). Selenocysteine insertion at a predefined UAG codon in a release factor 1 (RF1)-depleted Escherichia coli host strain bypasses species barriers in recombinant selenoprotein translation. J Biol Chem, 292, 5476–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aldag C, Bröcker MJ, Hohn MJ, Prat L, Hammond G, Plummer A, Söll D, (2013). Rewiring translation for elongation factor Tu-dependent selenocysteine incorporation. Angew Chem Int Ed Engl, 52, 1441–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukai T, Sevostyanova A, Suzuki T, Fu X, Söll D, (2018). A facile method for producing selenocysteine-containing proteins. Angew Chem Int Ed Engl, 57, 7215–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller C, Bröcker MJ, Prat L, Ip K, Chirathivat N, Feiock A, Veszprémi M, Söll D, (2015). A synthetic tRNA for EF-Tu mediated selenocysteine incorporation in vivo and in vitro. FEBS Lett, 589, 2194–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thyer R, Robotham SA, Brodbelt JS, Ellington AD, (2015). Evolving tRNA(Sec) for efficient canonical incorporation of selenocysteine. J Am Chem Soc, 137, 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tharp JM, Ad O, Amikura K, Ward FR, Garcia EM, Cate JHD, Schepartz A, Söll D, (2020). Initiation of protein synthesis with non-canonical amino acids in vivo. Angew Chem Int Ed Engl, 59, 3122–3126. [DOI] [PubMed] [Google Scholar]
- 20.Kwok HS, Vargas-Rodriguez O, Melnikov SV, Söll D, (2019). Engineered aminoacyl-tRNA synthetases with improved selectivity toward noncanonical amino acids. ACS Chem Biol, 14, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mandrand-Berthelot M-A, Wee MYK, Haddock BA, (1978). An improved method for the identification and characterization of mutants of Escherichia coli deficient in formate dehydrogenase activity. FEMS Microbiol. Lett, 4, 37–40. [Google Scholar]
- 22.Thyer R, Shroff R, Klein DR, d’Oelsnitz S, Cotham VC, Byrom M, Brodbelt JS, Ellington AD, (2018). Custom selenoprotein production enabled by laboratory evolution of recoded bacterial strains. Nat Biotechnol, 36, 624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez EA, Tran GN, Gross LA, Crisp JL, Shu X, Lin JY, Tsien RY, (2016). A far-red fluorescent protein evolved from a cyanobacterial phycobiliprotein. Nat Methods, 13, 763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haruna K, Alkazemi MH, Liu Y, Söll D, Englert M, (2014). Engineering the elongation factor Tu for efficient selenoprotein synthesis. Nucleic Acids Res, 42, 9976–9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Appleby-Tagoe JH, Thiel IV, Wang Y, Wang Y, Mootz HD, Liu XQ, (2011). Highly efficient and more general cis- and trans-splicing inteins through sequential directed evolution. J Biol Chem, 286, 34440–34447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Ventura B, Mootz HD, (2019). Switchable inteins for conditional protein splicing. Biol Chem, 400, 467–475. [DOI] [PubMed] [Google Scholar]
- 27.Friedel K, Popp MA, Matern JCJ, Gazdag EM, Thiel IV, Volkmann G, Blankenfeldt W, Mootz HD, (2019). A functional interplay between intein and extein sequences in protein splicing compensates for the essential block B histidine. Chem Sci, 10, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amitai G, Callahan BP, Stanger MJ, Belfort G, Belfort M, (2009). Modulation of intein activity by its neighboring extein substrates. Proc Natl Acad Sci U S A, 106, 11005–11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu H, Xu MQ, Liu XQ, (1998). Protein trans-splicing and functional mini-inteins of a cyanobacterial DnaB intein. Biochim Biophys Acta, 1387, 422–432. [DOI] [PubMed] [Google Scholar]
- 30.Mukai T, Hoshi H, Ohtake K, Takahashi M, Yamaguchi A, Hayashi A, Yokoyama S, Sakamoto K, (2015). Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci Rep, 5, 9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocher C, Lalanne JL, Chaudiere J, (1992). Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase. Eur J Biochem, 205, 955–960. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Zheng F, Cheng R, Li S, Rozovsky S, Wang Q, Wang L, (2018). Site-specific incorporation of selenocysteine using an expanded genetic code and palladium-mediated chemical deprotection. J Am Chem Soc, 140, 8807–8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosano GL, Ceccarelli EA, (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol, 5, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen RE, Winther JR, (2009). An introduction to methods for analyzing thiols and disulfides: Reactions, reagents, and practical considerations. Anal Biochem, 394, 147–158. [DOI] [PubMed] [Google Scholar]
- 35.Stogios PJ, Spanogiannopoulos P, Evdokimova E, Egorova O, Shakya T, Todorovic N, Capretta A, Wright GD, Savchenko A, (2013). Structure-guided optimization of protein kinase inhibitors reverses aminoglycoside antibiotic resistance. Biochem J, 454, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS, (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol, 24, 79–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.