

Abstract
In this preclinical study, we evaluated the efficacy and feasibility of creating broad human immunodeficiency virus (HIV) resistance by simultaneously disrupting the human CCR5 and CXCR4 genes, which encode cellular co-receptors required for HIV-1 infection. Using a clinically scalable system for transient ex vivo delivery of Cas9/guide RNA (gRNA) ribonucleoprotein (RNP) complexes, we demonstrated that CRISPR-mediated disruption of CCR5 and CXCR4 in T lymphocyte cells significantly reduced surface expression of the co-receptors, thereby establishing resistance to HIV-1 infection by CCR5 (R5)-tropic, CXCR4 (X4)-tropic, and dual (R5/X4)-tropic strains. Similarly, disruption of CCR5 alleles in human CD34+ hematopoietic stem and progenitor cells (HSPCs) successfully led to the differentiation of HIV-resistant macrophages. In a humanized mouse model under HIV-1 challenge, CXCR4-disrupted CD4+ T cells were enriched in the peripheral blood and spleen, indicating survival advantage because of resistance to viral infection. However, in human CD4+ T cells with both CCR5 and CXCR4 disruption, we observed poor engraftment in bone marrow, although significant changes were not observed in the lung, spleen, or peripheral blood. This study establishes a clinically scalable strategy for the dual knockout of HIV-1 co-receptors as a therapeutic strategy, while also raising caution of disrupting CXCR4, which may abate engraftment of CD4+ T cells in bone marrow.
Keywords: HIV, CRISPR, CCR5, CXCR4, gene editing, bone marrow, macrophage, T cells, adoptive T cell therapy
Graphical abstract
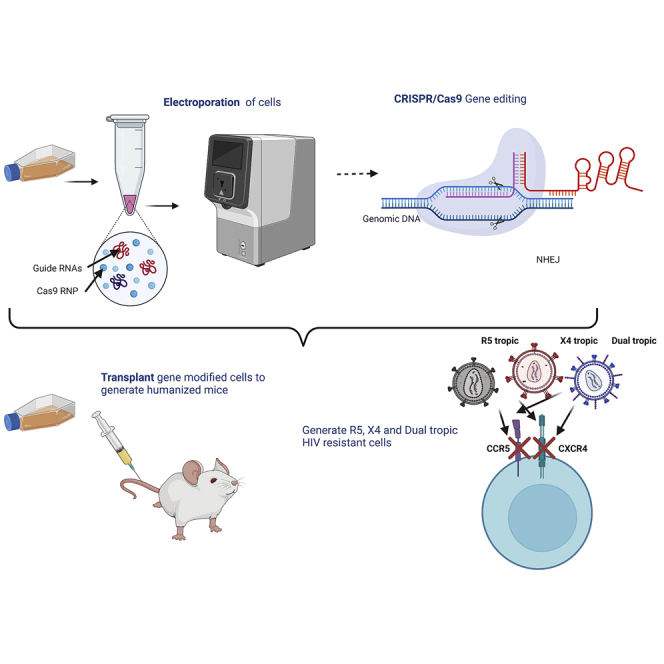
CRISPR-mediated disruption of CCR5 and CXCR4 protects cells from infection of HIV-1 R5-trpoic, X4-tropic, and dual-tropic strains, but also abates engraftment of CD4+ T cells in bone marrow. This study establishes a clinically scalable strategy for the disruption of both HIV-1 co-receptors while also raising caution.
Introduction
Human immunodeficiency virus type 1 (HIV-1), the virus that causes AIDS, currently afflicts more than 38 million people worldwide.1 Despite the effectiveness of antiretroviral therapy (ART) in controlling HIV-1 replication and infection, these drugs are unable to eradicate the virus from a patient. Complicating matters, accessibility to ART and daily compliance are challenging for millions living with HIV, and HIV-infected individuals disproportionately suffer from accelerated aging and an increased risk of age-related health complications.2 Innovative therapeutic strategies are currently being explored as potential alternatives to ART,3 including gene-editing strategies that inhibit viral infection.4
The HIV-1 replication cycle begins with the viral particle binding to the CD4 receptor and then to either the CCR5 or the CXCR4 co-receptor on the target cells. Binding then triggers fusion of the viral and host cell membranes, thereby facilitating entry into the cell, where the viral genome undergoes reverse transcription and integration into the host genome. Of the two primary co-receptors, CCR5 is the cellular co-receptor used by the majority of HIV-1 strains for binding and entry5 and is critical for primary infection via mucosal transmission.6 In some individuals, a natural 32-bp deletion in the CCR5 gene results in a truncated CCR5 protein that is not expressed on the cell surface. Approximately 1% of individuals of northern European descent carry the homozygous CCR5Δ32 allele, and although these individuals are healthy despite lacking a functional CCR5 gene, they are also highly resistant to HIV-1 infection.7,8 The first two documented functional cures of HIV-1 were with patients who received allogeneic transplantation with hematopoietic stem cells from CCR5Δ32 homozygous donors for the treatment of acute myeloid leukemia9,10 or refractory Hodgkin’s lymphoma.11 However, this general strategy has been met with mixed success, and several other patients have experienced complications because of allogeneic stem cell transplantation or relapse of underlying cancer,12,13 while others have been marked by the emergence of CXCR4 (X4)-tropic HIV-1 strains that do not use the CCR5 co-receptor.14
Numerous gene-editing tools have been used against CCR5 to inhibit R5-tropic HIV-1 infection in vitro and in vivo, including ZFN,15, 16, 17, 18 transcription activator-like effector nuclease (TALEN),19, 20, 21 and CRISPR-Cas systems.22, 23, 24 Due to the possibility of HIV resistance to CCR5 gene disruption, which occurs through natural tropism shift, it is likely necessary to disrupt CXCR4 to eradicate HIV-1 infections in most individuals. Hence ZFN25,26 and CRISPR-Cas27,28 systems have been designed edit CXCR4 for the inhibition of X4-tropic HIV-1. Moreover, a few studies have explored the simultaneous disruption of CCR5 and CXCR4 alleles using two zinc-finger nucleases (ZFNs)29 or two single guide RNAs (sgRNAs) via CRISPR-Cas9.30 Although many of these approaches are still in the preclinical stage, clinical trials primarily focused on the use of ZFN31,32 or CRISPR-Cas933 for CCR5 editing have yielded promising results in clinical safety and efficacy tests, while CXCR4 gene-editing strategies have not yet been tested clinically.
Translation of gene-editing technology using disrupting co-receptors for treating HIV/AIDS demands exquisite on-target precision, ample efficiency, and delivery approaches that are scalable and clinically feasible. In the present study, we have utilized the CRISPR-Cas9 gene-editing system to disrupt CCR5, CXCR4, or both to create HIV resistance in human primary T cells in a clinically scalable system. Notably, we demonstrated that the gene-modified cells gain protection from a broad range of HIV-1 strains (R5 tropic, X4 tropic, and dual tropic) that utilize either the CCR5 or CXCR4 surface receptors, or both. Next, we evaluated the gene-modified cells in a humanized mouse model to evaluate the efficacy and feasibility of creating an HIV-resistant immune system. Although our preclinical study demonstrates that the disruption of both CCR5 and CXCR4 is feasible in a clinically scalable system and is highly effective in protecting cells from HIV infection in vivo, we observed a reduction in the bone marrow engraftment of gene-modified cells, a critical finding that has not been previously documented in similar studies.25,26,29,30
Results
CRISPR-Cas9-mediated disruption of CCR5 protects cells from HIV-1 infection
To evaluate the CRISPR-Cas9 system in creating HIV-resistant cells, we first tested the expression of both the sgRNA and human codon-optimized Streptococcus pyogenes Cas9 (spCas9) components, as well as the TagRFP reporter gene from lentiviral vectors.34 Using a sgRNA design algorithm,35 we selected unique guide sequences to target CCR5 with the CRISPR-Cas9 system. CEM.NKRCCR5+ cells (i.e., human CD4+ lymphoblast cells with retroviral vector expression of human CCR536) were transduced with the lentiviral vectors at a low multiplicity of infection (MOI, ∼0.1). A control vector was created that carried an irrelevant sgRNA sequence in addition to the spCas9 and TagRFP expression cassettes. One week after transduction, transduced cells were sorted by fluorescence-activated cell sorting (FACS) for TagRFP expression and analyzed for CCR5 surface expression by flow cytometry to assess CRISPR-mediated gene knockout. Surface expression of CCR5 was significantly reduced in the cells treated with CCR5-CRISPR (81.7% CCR5+ cells in control versus 4.3% CCR5+ cells in CCR5-CRISPR; Figure 1A). Genomic DNA was analyzed for gene editing using Surveyor Nuclease Assay, which revealed 61.2% ablation efficiency of CCR5 after FACS enrichment of TagRFP cells and 37.4% ablation in unsorted cells (Figure 1B). Gene disruption was further characterized by next generation sequencing (NGS) analysis across the CCR5 target site, which revealed significant and frequent insertions and deletions (indels) at the sgRNA target site, consistent with the imprecise DNA repair mechanism of non-homologous end joining (NHEJ) (Figure S1). Deep sequencing of the CCR5 target site revealed CRISPR-induced indels in 87.9% of the total reads (Figure 1C), ranging from single-base-pair insertions or deletions to insertions or deletions exceeding 100 bp. To investigate whether CRISPR-mediated disruption of the CCR5 gene facilitated HIV resistance, we challenged the gene-modified CEM cells with R5-tropic HIV-1BaL and observed HIV replication over a 4-week time course. HIV-1 replication was suppressed in the CCR5-CRISPR cells, with supernatant p24 antigen levels greater than 100-fold lower than the control group at 14, 17, 21, and 28 days after HIV-1BaL challenge (Figure 1D).
Figure 1.

CRISPR-Cas9 gene disruption of CCR5 in CD4+ T cells
(A) CEM CCR5+CD4+ T cells were transduced with CCR5-CRISPR vector or control vector, and TagRFP+ cells were analyzed for CCR5 surface expression by flow cytometry. (B) Detection of indels by Surveyor assay in TagRFP+ CEM T cells. (C) Deep sequencing analysis of CCR5 genome disruption in TagRFP+ CEM T cells. (D) HIV-1BaL replication in CEM CCR5+CD4+ T cells treated with CCR5-CRISPR, as measured by HIV-1 p24 antigen in supernatant. The assay was performed in triplicate, and error bars represent standard deviation.
HIV-1 resistance of CCR5 CRISPR-Cas9-modified CD34+ differentiated macrophages
R5-tropic HIV-1 strains (e.g., HIV-1BaL) are historically referred to as macrophage-tropic (M-tropic), because they are capable of infecting macrophages by utilizing the CCR5 co-receptor in addition to the CD4 receptor. Thus, we evaluated the antiviral efficacy of CRISPR-mediated gene disruption of CCR5 in primary macrophages that were derived from CD34+ hematopoietic stem and progenitor cells (HSPCs). Human CD34+ HSPCs were isolated from cord blood, transduced with the CCR5-CRISPR or control CRISPR lentiviral vectors, and sorted by FACS based on TagRFP expression (Figure 2A). The TagRFP-expressing CD34+ cells were differentiated into macrophages, as described in the Materials and methods (Figure 2B). Macrophages were then challenged with HIV-1BaL and evaluated for viral replication by p24 ELISA measurement of the supernatants over 28 days. HIV-1 replication was suppressed at all time points in macrophages treated with CCR5-CRISPR relative to the control group, with p24 antigen levels reductions exceeding 10-fold at days 3, 7, and 21, a ∼25-fold reduction in viremia at day 14, and a ∼5-fold reduction at day 28 (Figure 2C). These results demonstrate that CRISPR-mediated disruption of CCR5 in CD34+ HSPC-derived macrophages confers resistance to HIV-1 infection and replication.
Figure 2.

Hematopoietic differentiation and HIV resistance of macrophages from CRISPR-Cas9-CCR5 modified HSPCs
(A) Morphology of macrophages generated from parental and CRISPR-modified HSPCs in unsorted cells. RFP indicated cells transduced with the CCR5-CRISPR vectors in differentiated macrophages. (B) Flow cytometric analysis of macrophage-specific markers in macrophages generated from CRISPR-modified HSPCs after enrichment of TagRFP+ cells by FACS. (C) Resistance of macrophage from CCR5-CRISPR-modified HSPCs to HIV-1 infection compared with unmodified cells. The assay was performed in triplicate, and error bars represent standard deviation.
CRISPR-Cas9 gene disruption of CXCR4 confers resistance to X4-tropic HIV-1 in cell lines and primary T cells
Although CRISPR-mediated disruption of the CCR5 gene may confer resistance to R5-tropic HIV-1, it may not inhibit strains that infect T cells by utilizing the CXCR4 (X4-tropic or T-tropic) or both CXCR4 and CCR5 co-receptors (dual-tropic). Thus, we designed guide CRISPR RNA (crRNA) sequences targeting CXCR4 as an approach for inhibiting X4-tropic HIV-1. We first compared the efficacy of different sgRNAs for each target, delivered using lentiviral vectors to disrupt surface CXCR4 expression on Jurkat CD4+ T cells. Flow cytometry analysis revealed a significant decrease in surface CXCR4 expression, with 15.4% CXCR4+ cells transduced with CXCR4-CRISPR compared with 99.3% CXCR4+ cells in control-CRISPR cells (Figure 3A). These observations were corroborated with analysis of editing of genome DNA by Surveyor nuclease assay (Figure 3B) or Sanger sequencing followed by analysis using inference of CRISPR edits (ICEs) (Figure 3C), yielding 41.0% and 49.2% allelic disruption, respectively, after CXCR4-CRISPR transduction. Next, we assessed the biological effects of CXCR4 disruption on preventing replication of X4-tropic HIV-1 in human PBMCs (hu-PBMCs). Over a 16-day time course following HIV-1NL4-3 challenge, we observed a reduction of HIV replication in the CXCR4-CRISPR cells relative to the untreated control, as measured by ELISA of supernatant at the indicated time points (Figure 3D). Collectively, these experiments demonstrate the feasibility of using CRISPR-Cas9 to engineer HIV-resistant cells by targeting the CCR5 and CXCR4 host receptor genes.
Figure 3.

CRISPR-Cas9 gene disruption of CXCR4 in Jurkat T cells
(A) Jurkat cells were transduced with CXCR4 CRISPR or control vector and analyzed for CXCR4 surface expression by flow cytometry. (B) Surveyor nuclease assay detects indels in CXCR4 CRISPR-modified Jurkat cells. (C) Sanger sequencing and ICE analysis of CRISPR-Cas9-induced genome disruption by CXCR4. (D) HIV-1NL4-3 replication in PBMCs treated with CXCR4 CRISPR, as measured by p24 in supernatant.
CXCR4 CRISPR-edited primary CD4+ T cells are protected in Hu-PBMC mice after infection with HIV-1 virus
Although lentiviral delivery of the CRISPR-Cas9 system can achieve on-target efficacy, constitutive expression of the Cas9 and sgRNA components is also associated with high frequencies of off-target editing and is thus not suitable for clinical applications.37 As an alternative delivery system, recombinant Cas9 protein may be complexed with the guide RNA (gRNA) for ex vivo delivery into cells by transient transfection or electroporation. The Cas9/gRNA ribonucleoprotein (RNP) provides burst-like kinetics that maximize the on-target efficiency, while minimizing less kinetically favorable off-target events.38 Thus, we elected to deliver the Cas9 RNP to human primary CD4+ T cells using MaxCyte STX electroporation (MaxCyte, Inc.), because a similar approach has been previously demonstrated for the preparation of ZFN-mediated gene-edited T cells at a clinical scale.39 Specifically, we utilized the Alt-R CRISPR-Cas9 system (Integrated DNA Technologies, Inc.), which consists of spCas9 recombinant protein complexed with a trans-activating crRNA (tracrRNA) and a chemically modified crRNA that is specific for CXCR4. We utilized the hu-PBMC NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) mouse model to evaluate whether knockout of CXCR4 in CD4+ T cells could protect cells in vivo from infection with X4-tropic HIV-1NL4-3 (Figure 4A). Two days after electroporation in human primary CD4+ T cells, flow cytometry analysis revealed that the subpopulation of CXCR4-negative T cells had increased from 2.3% to 20.2% in the CXCR4-CRISPR group (Figure 4B). Editing of the CXCR4 alleles was also confirmed by Surveyor assay, which revealed 46% gene disruption (Figure 4C). Mice were analyzed for engraftment at 2 weeks after transplantation and were challenged with HIV-1NL4-3 at 4 weeks after transplantation. At 2 weeks after infection (i.e., 6 weeks after transplantation), we observed an increase in CXCR4 gene disruption in T cells collected from the CXCR4-CRISPR mice, suggesting an enrichment of CXCR4-negative cells by the selective pressure of X4-tropic HIV-1 infection (Figure S2D). Notably, at the same time point, the mice engrafted with CXCR4 knockout cells exhibited ∼30-fold lower levels of plasma viremia than in the mock-treated mice (Figure 4F). Moreover, we observed significantly higher levels of total CD3+ T cells and CXCR4−CD4+ T cells in the CXCR4-CRISPR-treated mice than in controls (Figures 4D–4F), indicating that selective pressure of the virus may lead to expansion of CXCR4 knockout cells.
Figure 4.

Positive selection for CXCR4 knockout cells by HIV-1NL4-3 infection in hu-PBMCs
(A) Schematic of the timeline of building hu-PBMC mouse model and HIV infection by using mixed human primary PBMCs with CXCR4 CRISPR-modified CD4+ T cells. (B) Cell surface CXCR4 co-receptor knockout in CD4+ T cells after MaxCyte electroporation of CXCR4 guide RNAs and Cas9 RNPs. Cells were fixed in 2% formaldehyde and analyzed by flow cytometry 48 h after transfection. (C) Surveyor assay detection of the allelic disruption of CXCR4 gene in the CXCR4 CRISPR-modified cells. (D–F) Flow cytometry analysis of human CD3+ T cells in mice whole blood after transplantation by using CXCR4 CRISPR-modified or unmodified cells (control). (G) qRT-PCR was performed using plasma from hu-PBMC mice. Mice whole blood was collected by retro-orbital bleeding 2 weeks after HIV-1NL4-3 infection (6 weeks after transplantation). Data were presented by comparing two groups of mice that were transplanted by using the control and CXCR4 CRISPR-modified cells.
At 12 weeks after transplantation (i.e., 8 weeks after HIV-1NL4-3 challenge), the experiment was terminated, and the CXCR4-CRISPR modified cells were collected from the spleens of humanized mice (Figure S2A). We analyzed the gene modification level of CXCR4-CRISPR in the mice spleens by ICE analysis, which revealed 37.0% of CXCR4 alleles were disrupted (Figures S2B and S2E). Moreover, the mice engrafted with CXCR4 knockout cells exhibited significantly higher levels of CD4+ T cells in the spleen (22.5% CXCR4-CRISPR or 0.2% mock-treated) than the mice that received mock-treated cells (Figure S2C). These results indicate that CRISPR-mediated gene disruption of CXCR4 protects CD4+ T cells in vivo from infection of X4-tropic HIV-1 and virus-induced cell death.
CCR5 and CXCR4 genome disruption confers primary T cells resistant to broad HIV-1 infection
Because CRISPR-mediated disruption of CCR5 confers resistance to R5-tropic HIV-1 and disruption of CXCR4 confers resistance to X4-tropic HIV-1, it may be necessary to edit both surface receptors to create resistance to all HIV-1 infection. To test this hypothesis, we delivered Cas9 RNP complexes with CCR5 and CXCR4 gRNAs (referred hereafter as R5X4-CRISPR). After transfection of the R5X4-CRISPR system into primary CD4+ T cells, we first analyzed the knockout efficacy of CCR5 and CXCR4 receptors on the cell surface. Analysis by flow cytometry revealed that the gene-modified cells exhibited a decrease in CCR5 surface expression from 88.7% in control cells to 54.9% (Figure 5A) and from 77.1% to 26.3% in CXCR4 expression (Figure 5B). In total, the proportion of dual-positive CCR5+CXCR4+ cells decreased from 85.2% to 36.8%, while levels of dual-negative CCR5−CXCR4− cells increased from 10.6% to 49.8% (Figure S3). This demonstrates that transient delivery of CRISPR-Cas9 is effective in knocking out both of the co-receptors that are required for HIV infection in human primary CD4+ T cells.
Figure 5.

Gene disruption of CCR5 and CXCR4 induces HIV resistance in primary CD4+ T cells using MaxCyte electroporation
(A and B) Human primary CD4+ T cells were transfected with CCR5 and CXCR4 guide RNAs with Cas9 RNP by MaxCyte electroporation using the “P4” setting. Surface expression of (A) CCR5 and (B) CXCR4 co-receptors was measured by flow cytometry. Control cells were transfected with Cas9 RNP, but no guide RNA. (C) Flow cytometry analysis of CCR5 and CXCR4 expression on the surface of CD4+ T cells treated with CCR5 and CXCR4 CRISPR and then infected by using each HIV-1 virus strain (BaL, NL4-3, or 89.6) after CD3/CD28 activation. Cells were collected 5 weeks after infection, stained for surface receptors, and prepared for fixation and permeabilization for intracellular p24 staining. The bar graph represents that in the CCR5 and CXCR4 CRISPR-treated cells, percentages of CCR5−CXCR4−, CCR5+CXCR4−, CCR5−CXCR4+, and CCR5+/CXCR4+ cells were compared after different strain infection. The table graph represents that in the CCR5 and CXCR4 CRISPR-treated cells, percentages of CCR5−CXCR4−, CXCR4+CCR5−, CXCR4−CCR5+, and CXCR4+CCR5+ cells were compared after different strain infection. (D) Flow cytometer analysis of intracellular p24 in CD4+ T cells in (C). The percentages of p24 cells were compared between control group (black bars) and CCR5 and CXCR4 CRISPR-treated group (gray bars).
We next sought to determine whether CD4+ T cells with disrupted CCR5 and CXCR4 alleles would become resistant to HIV-1 infection. We challenged the R5X4-CRISPR-modified primary CD4+ T cells with HIV-1 virus that utilized the CCR5 co-receptor (HIV-1BaL), the CXCR4 co-receptor (HIV-1NL4-3), or either the CCR5 or CXCR4 co-receptors (HIV-189.6). As expected, cells that had surfaced expression of CCR5, but not of CXCR4 (CCR5+CXCR4−), were enriched 5 weeks after challenge with HIV-1NL4-3 (19.5%), but not after challenge with the other two strains that can utilize the CCR5 co-receptor (1.1% for HIV-1BaL and 2.3% for HIV-189.6) (Figure 5C). Likewise, cells with surface expression of CXCR4, but not CCR5 (CCR5−CXCR4+), were largely protected after challenge with the R5-tropic HIV-1BaL (8.8%) but disappeared after challenge with X4-tropic or dual-tropic HIV-1 strains (1.9% for HIV-1NL4-3 and 3.2% for HIV-189.6, respectively). Most notably, the CCR5−CXCR4− dual-negative subpopulation increased from 72.8% in the R5X4-CRISPR cells before HIV-1 challenge to 85.1% in the cells challenged with HIV-189.6, demonstrating an enrichment of cells that lack both CCR5 and CXCR4 co-receptors after incubation with this dual-tropic HIV-1 strain (Figure 5C). Moreover, we analyzed intracellular p24 in CD4+ T cells treated with CCR5 and CXCR4 CRISPR and then infected by using each HIV-1 virus strain after CD3/CD28 co-activation. Notably, 5 weeks after challenge with any of the three HIV-1 strains, we observed greater levels of live cells in the R5X4-treated groups (20.5% for HIV-1BaL, 23.1% for HIV-1NL4-3, and 12.3% for HIV-189.6) compared with their respective control groups (15.6% for HIV-1BaL, 3.1% for HIV-1NL4-3, and 2.9% HIV-189.6) (Figure S4). Moreover, the levels of intracellular p24 in the R5X4-CRISPR-treated cells were significantly decreased (p < 0.05) relative to each respective control group (Figure 5D).
To analyze possible off-target gene disruption after R5X4-CRISPR treatment, we examined three possible off-target sites for the CCR5 sgRNA target sequence (Table S1) and three more for the CXCR4 target sequence (Table S2), as predicted by Cas-OFFinder. Each site was analyzed using Surveyor assay, but no increases in gene disruption were observed for any of the six predicted off-target sites, whereas clear gene disruption was observed for each of the two on-target sites (Figure S5).
Poor engraftment of R5X4-CRISPR knockout CD4+ T cells in bone marrow in Hu-PBMC mice
As shown in Figure 5, knockout of both CCR5 and CXCR4 co-receptors is necessary to block infection from R5 and X4-tropic HIV-1 strains. Thus, we next evaluated the efficacy and feasibility of this approach in a preclinical in vivo model for HIV infection (Figure 6A). First, we transfected the R5X4-CRISPR RNP complex into primary CD4+ T cells by using the MaxCyte electroporation system and analyzed the knockout efficacy of CCR5 and CXCR4 receptors on the cell surface. The proportion of dual-negative CCR5−CXCR4− cells increased from 32% to 85% (Figure 6B) by flow cytometry analysis. Editing of the CCR5 and CXCR4 alleles was also confirmed by Surveyor assay, which revealed 22% and 32% gene disruption, respectively (Figure 6C).
Figure 6.

Bio-distribution of CCR5- and CXCR4-CRISPR knockout CD4+ T cells in Hu-PBMC mouse tissues
(A) Schematic of the timeline of building hu-PBMC mouse model by using mixed human primary PBMCs with CXCR4 CRISPR-modified CD4+ T cells. (B) Cell surface CCR5 and CXCR4 co-receptor knockout in CD4+ T cells after MaxCyte electroporation of CCR5 and CXCR4 guide RNAs and Cas9 RNPs. Cells were fixed in 4% formaldehyde and analyzed by flow cytometry 48 h after transfection. (C) Surveyor assay detection of CCR5 and CXCR4 allelic disruption in CD4+ T cells after MaxCyte electroporation of CCR5 and CXCR4 guide RNAs and Cas9 RNPs. (D) Eight million CRISPR-modified or unmodified CD4+ T cells with 2 million human PBMCs were transplanted into NSG mice. At the final time point, whole PBMCs, spleens, lungs, and bone marrow of all the mice from each group were harvested, and cells were analyzed by flow cytometer (n = 6, ∗p < 0.05).
At 56 days after transplantation of the gene-modified cells in NSG mice, the animals were euthanized and analyzed for engraftment in peripheral blood and tissues. From analysis of peripheral blood in the hu-PBMC mice, there were similar levels of human CD45+ lymphocytes or other surface markers, including CD3, CD4, CD8, CXCR4, and CCR5, between the dual CRISPR and control mouse groups (Figure 6D). We also evaluated engraftment in primary lymphoid tissues and lung to assess the homing and persistence of the CRISPR-modified cells. Levels of human immune (CD45+) cells, total T (CD3+) cells, CD4+ T cells, and CXCR4+ CD4+ T cells in the spleens of R5X4-CRISPR-treated mice were statistically indistinguishable from the control group (Figure 6D). Similar trends were also observed in the lung. However, in the bone marrow, the R5X4 mice had statistically significant (p < 0.05) lower levels of human CD45+ cells and CD45+CD3+ T cells, as well as similar trends of slightly lower levels of CD4+, CCR5+, and CXCR4+ T cells (Figure 6D). These results suggest that CRISPR-mediated knockout of CCR5 and CXCR4 may alter the homing, persistence, and expansion of these cells into the bone marrow and potentially other lymphoid tissues after transplantation.
Discussion
Owing to their essential roles as co-receptors for HIV entry and infection, the human CCR5 and CXCR4 chemokine receptors are major targets for gene disruption in strategies to create HIV resistance. In this study, we investigated the versatility of the CRISPR-Cas9 in simultaneously editing both CCR5 and CXCR4 receptors in human cells. We successfully disrupted CCR5 in CD4+ T cell lines (Figure 1), primary CD4+ T cells (Figure 5), and CD4+ macrophages differentiated from CD34+ HSPCs (Figure 2), which all led to R5-tropic HIV-1 resistance. Likewise, by disrupting CXCR4 in a CD4+ T cell line (Figure 3), in primary CD4+ T cells (Figure 5), and in transplanted CD4+ T cells in a humanized mouse model (Figure 4), we achieved X4-tropic HIV-1 resistance.
To generate CRISPR-modified CD4+CCR5−CXCR4− T cells, we utilized a scalable system that has been used for clinical manufacturing of gene-modified cells.39 Upon treatment with the Cas9 RNP complexes with CCR5 and CXCR4 gRNAs, we observed efficient gene editing for both receptors in primary CD4+ T cells, resulting in approximately 50% CCR5−CXCR4− double-negative cells (Figure S3, bottom right panel). The gene-modified cells were resistant to broad HIV-1 infection and were selectively enriched by the selective pressure of the virus, with protection against R5-tropic, X4-tropic, and dual-tropic strains of HIV-1 (Figures 5C and 5D). In the hu-PBMC NSG mouse model, the CRISPR-modified cells were well tolerated, because the percentages of gene-modified cells were maintained over time in mice (Figures 4D and 6D). Moreover, in CXCR4-CRISPR humanized mice, X4-tropic HIV-1 resistance resulted in the selective enrichment of CD4+ T cells in spleen tissue compared with non-CRISPR mice (Figure S2C). Although CRISPR-mediated disruption of CXCR4 was successful in reducing viremia and protecting CD4+ T cells in vivo (Figure 4E), we observed that levels of R5X4-CRISPR-modified CD4+ T cells were significantly lower than unmodified controls in the bone marrow (Figure 6D).
Although gene disruption of CCR5 continues to be evaluated clinically with promising results,32 gene-editing strategies for CXCR4 have not advanced to the clinic. Moreover, unlike the naturally occurring CCR5-Δ32 homozygous mutation, homozygous CXCR4 knockouts are embryonic lethal in a murine model.40 CXCR4 is known to function as a surface receptor for cell homing, such as for the homing of HSPCs in the bone marrow,41 while the CXCR4 antagonist AMD3100 (plerixafor) is used clinically to mobilize CD34+ HSPCs from the bone marrow into the peripheral blood.42 However, it is unknown whether gene disruption of CXCR4 would abate engraftment of CD4+ T cells in bone marrow or other lymphoid organs. Previous studies have engineered ZFNs to disrupt CXCR425,26 or both CCR5 and CXCR429 in CD4+ T cells to create X4-tropic HIV-1 resistance in tissue culture and in vivo. Similar to our observations, these studies also showed decreases in HIV-1 plasma viremia and protection of the modified CD4+ T cells in hu-PBMC mouse models. However, these studies evaluated levels of CD4+ T cells and viremia only in the peripheral blood and spleen, with no analyses of the engraftment in other potential T cell niches, such as the bone marrow or lung.
In this study, we aimed to evaluate the feasibility and efficacy of targeting both co-receptors using a transient and ex vivo CRISPR-based system. Although we used SpCas9 in this study, smaller Cas9 orthologs, such as SaCas9 or Cas12a, might be more advantageous than SpCas9 for in vivo delivery in an AAV vector.43,44 Other CRISPR-Cas gene-editing systems have been developed to improve on-target precision or to edit the targeted DNA site(s) without inducing DNA double-strand breaks (DSBs).45,46 Editing without the induction of a DSB would likely lower the risk of genomic rearrangements, particularly when multiple sites are simultaneously targeted. Nevertheless, the major conclusions from this study—editing of both co-receptors creates HIV-resistant cells but may disrupt the homing and persistence of the modified cells in the bone marrow—would seem to apply to any CRISPR-based approach, as well as other gene-editing systems, including ZFNs and TALENS.
This study demonstrates the feasibility of simultaneously disrupting the CCR5 and CXCR4 co-receptors in a clinically scalable system and lays the groundwork for clinical translation. Notably, this is the first report of reduced engraftment of T cells in bone marrow following CRISPR-mediated disruption of CCR5 and CXCR4. Poor engraftment in the bone marrow may limit the duration of an adoptive T cell therapy, because the bone marrow sustains lifelong persistence of memory T cells.47 Thus, due to this potential limitation in the long-term persistence of the gene-modified cells, it is not clear that this strategy would be viable in humans. Future studies might explore the expression of other chemokine receptors that could supplant the requirement for CXCR4 or whether engraftment in human bone marrow is even necessary for the long-term persistence of the gene-modified CD4+ T cells.
Materials and methods
Cell lines and viruses
CEM.NKR CCR5+ cells (abbreviated as CEM-CCR5) and Jurkat cells are CD4+ T lymphoblastic cell lines obtained from NIH HIV Reagent Program (catalog #4376), which is cultured in RPMI 1640 media supplemented with 10% fetal bovine serum and 2 mM L-glutamine. Human embryonic kidney (HEK) 293T cells were from ATCC (catalog #CRL-3216). HIV-1 infectious virus (HIV-1BaL, catalog #510; HIV-189.6, catalog #1966) and molecular clone plasmid (HIV-1NL4-3, catalog #114) were obtained from the NIH HIV Reagent Program. These HIV-1 strains are commonly used in laboratory and animal research.
PBMCs and primary CD4+ T cells
hu-PBMCs were isolated from leukocyte reduction system chambers (i.e., buffy cones), which were obtained from four different healthy human donors at the City of Hope Amini Apheresis Center (Duarte, CA, USA). PBMCs were separated by centrifugation with Ficoll-Paque Premium (BD). Primary human CD4+ T cells were further purified and enriched by the CD4+ T cell isolation kit (Miltenyi Biotech) according to the manufacturer’s instructions and then maintained in complete RPMI medium supplemented with 10% FBS. Experiments presented in Figures 4, 5, and 6 were each performed with PBMCs from different anonymous healthy donors.
gRNA design and CRISPR-Cas9 lentiviral vector constructs
gRNA sequences for the ccr5 and cxcr4 target sites were designed using the computational tool originally described by Hsu et al.35 The pL-CRISPR-SFFV-tRFP plasmid was obtained from Addgene (Plasmid #57826) and originally deposited by the Ebert lab.34
Lentiviral vector production
Lentiviral vectors were packaged in HEK 293T cells by calcium phosphate precipitation. In brief, 15 μg of transfer plasmid was co-transfected with helper plasmids (15 μg of pCMV-Pol/Gag, 5 μg of pCMV-Rev, and 5 μg of pCMV-VSVG) into HEK 293T cells with 90–95% confluency per 10-cm dish. Viral supernatant was harvested 48 h post-transfection, concentrated by ultracentrifugation, and stored at −80°C until use. Viral titers were determined by transduction of HT1080 cells and analyzed for EGFP expression with FACS analysis.
Flow cytometry analysis
To analyze cell surface expression of CCR5 and CXCR4, we incubated cells with an allophycocyanin (APC)- or PE-CF594-conjugated mouse anti-human CCR5 (Becton Dickinson), PerCP-Cy5-conjugated mouse anti-human CXCR4 (Becton Dickinson) for 30 min at 4°C. Cells then were washed twice with FACS buffer (PBS containing 1% BSA and 0.02% NaN3) and then fixed with 2% formaldehyde. FACS analysis was performed on Fortessa (BD Biosciences, Mountain View, CA, USA). Data were analyzed by using FlowJo software. Live cells were gated prior to co-receptor expression analysis based on forward and side scatter profiles. Gating for CCR5 and CXCR4 expression was established with unstained and single-stained controls.
To isolate Tag-RFP cell populations from total CEM-CCR5 cells transduced with lentiviral vectors expressing Cas9 nuclear localization sequence (NLS) and single-guide RNAs (sgRNAs), we sorted cells using an Aria SORP cell sorter (BD Biosciences).
For analysis of lymphocyte populations in mouse peripheral blood, splenocytes, lung, and bone marrow, fluorochrome-conjugated antibodies were obtained from BD Biosciences: BUV395-conjugated anti-CD45 (clone HI30), peridinin-chlorophyll protein/Cy7-conjugated anti-CD3 (clone SK7), Pacific Blue-conjugated anti-CD4 (clone RPA-T4 RUO), and fluorescein isothiocyanate-conjugated anti-CD8 (clone RPA-T8 RUO).
Intracellular HIV p24 antigen staining
To analyze cells actively infected with HIV, we stained for intracellular p24 antigen (KC57-FITC; Beckman Coulter) and permeabilized using the BD Fixation/Permeabilization Kit (catalog #554714) according to the manufacturer's instructions. Cells were fixed using 4% paraformaldehyde at 4°C for 20 min and washed twice with staining buffer (Dulbecco's PBS without Mg2+ or Ca2+, 1% heat-inactivated FCS, 0.09% (w/v) sodium azide, pH adjusted to 7.4). For permeabilization, cells were incubated in BD Perm/Wash buffer (BD Biosciences) for 15 min and stained with KC57-FITC antibody (Beckman Coulter). Because we performed intracellular p24 staining in combination with cell surface antigen staining (as in Figures 5C and 5D), we first stained with the fluorochrome-conjugated monoclonal antibodies against CCR5 and CXCR4 before proceeding to the fixation and permeabilization steps.
Analysis of insertion/deletions (indels)
To detect indels generated by CRISPR, we extracted genomic DNA from the CRISPR-modified or unmodified cells using QiAmp DNA mini Kit (Qiaqen) and assayed by Surveyor nuclease assay (Transgenomic). Six hundred base pairs of the genomic region flanking the gRNA target site was PCR amplified using primers listed in Tables S1 and S2. The PCR product was annealed to form heteroduplexes, and then the heteroduplexes were digested with 1 μL CEL1 endonuclease at 42°C for 1 h. The digested DNA was analyzed on an electrophoresis system using a 2% TBE agarose gel. The mutation frequency was quantified (ImageJ software, NIH Image-BioLab). The homoduplexes are left intact. Cas9-mediated cleavage efficiency (% indel) is calculated based on the fraction of cleaved DNA.48 Alternatively, whenever noted, Sanger sequencing of PCR-amplified genomic DNA was analyzed using ICEs (Synthego Corporation, Menlo Park, CA, USA).
HIV-1 in vitro challenge assay
To test whether CCR5 and CXCR4 gene-disrupted cells were resistant to HIV-1 infection, we infected cells with CXCR4-tropic HIV-1NL-4.3, CCR5-tropic HIV-1BaL, or dual-tropic HIV-189.6 at the MOI between 0.01 and 0.1 at 37°C, 5% CO2 for overnight. Cells were then washed twice with PBS and re-suspended in fresh complete medium. After the challenge, cells and culture supernatants were collected every 3 days and replenished with fresh medium for a total of 28 days. Levels of HIV-1 gag p24 in culture supernatants were measured by ELISA as instructed by the manufacturer (PerkinElmer).
Generation of adult HSPC-derived macrophages
Human cord blood was purchased from StemCyte (Baldwin Park, CA, USA) with approval from the City of Hope Institutional Review Board (IRB 17155). Sorted CD34+ HSPCs were cultured in Iscove's modified Dulbecco's media with 20% FBS supplemented with 2 mmol/L glutamine, 25 ng/mL stem cell factor (STEMCELL Technologies, Vancouver, BC, Canada), 30 ng/mL Flt3-L (PeproTech, Rocky Hill, NJ, USA), 30 ng/mL interleukin-3 (IL-3; Gibco), and 30 ng/mL macrophage colony-stimulating factor (PeproTech) for 10 days for guided differentiation to monocytes and were then switched to DMEM with 10% FBS supplemented with 2 mmol/L glutamine, 10 ng/mL granulocyte macrophage colony-stimulating factor (PeproTech), and 10 ng/mL macrophage colony-stimulating factor (PeproTech) for 5 days for activation into macrophages. Adherent macrophage cells were collected for HIV challenge experiments. The purity of cells was typically greater than 90% CD14+ based on FACS analysis.
Primary CD4+ T cell electroporation
The transfection of primary CD4+ T cells was performed on MaxCyte STX. A total of 2×107 primary CD4+ T cells were centrifuged and washed twice with 1× PBS, and the cells were re-suspended with 100 μL of prepared EP buffer and Cas9 NLS and chemically modified gRNA with tracrRNA complex ordered from IDT. The mixture was then transferred to the OC-100 cuvette and electro-transfected with MaxCyte STX programs. After transfection, the cells were transferred to a CD3/CD28-coated six-well plate and cultured with RPMI 1640 supplemented with 10% FBS and IL-2 (100 IU/mL).
hu-PBMC NSG mouse model
NSG mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and bred at the City of Hope Animal Resources Center according to the protocols approved by the Institutional Animal Care and Use Committee of the City of Hope (IACUC 16095). Adult NSG mice at age 8–10 weeks were transplanted with hu-PBMCs via intraperitoneal injection. Specifically, each mouse received 2.0 × 106 hu-PBMCs mixed with 8.0 × 106 CRISPR-modified or un-modified human CD4+ T cells. Cryopreserved PBMCs and human CD4+ T cells were from the same donor and were thawed and recovered before each use.
HIV-1 qRT-PCR
HIV-1 viral RNA was extracted from 20–50 μL of plasma using QIAamp Viral RNA mini kit (Qiagen). qRT-PCR was performed using a TaqMan Fast Virus 1-Step Master Mix, according to the manufacturer's instructions (Applied Biosystems, Foster City, CA, USA). The primers used were LTR-F (5′-GCCTCAATAAAGCTTGCCTTGA-3′) and LTR-R (5′-GGCGCCACTGCTAGAGATTTT-3′), along with a probe (5′-FAM/AAGTAGTGTGTGCCCGTCTGTTGTGTGACT-BHQ1-3′). Assay was performed using automated CFX96 Touch Real-Time PCR Detection System (Bio-Rad).
HIV-1 p24 ELISA
The HIV-1 p24 ELISA assay was performed according to PerkinElmer manufacturer's instructions. We prepared the p24 standard curve using the positive control with diluted standard concentrations at 12.5, 25, 50, 100, 200, and 4,000 pg/mL. We diluted each sample to three dilutions: 100×, 500×, and 2,500× dilution. Next, we labeled the plate and added 20 μL Triton X-100 to all wells except substrate blank. We then added 200 μL of standards, the negative control (sample media), and all diluted samples to appropriate wells. We sealed the plate and incubated for 2 h at 37°C. Next, we washed the plate in cell washer and added 100 μL of detector antibody to all wells, except the blank. Again, we sealed the plate and incubated for another hour at 37°C and washed. We then mixed the Streptavidin A-HRP 1:100 working dilution and added 100 μL of SA-HRP Working Dilution to all wells, except blank. We sealed the plate, incubated for 30 min at room temperature, and washed. Finally, we added 100 μL OPD Substrate Solution to all wells including blank, sealed the plate, incubated for 30 min at room temperature, and stopped the reaction by adding 100 μL of Stop Solution to all wells. Absorbance was measured at 490 nm.
Off-target analysis
Cas-OFFinder was employed to find potential OTSs with limitation of three-base mismatched sequences. From the resulting off-targets, OTSs only in gene-coding regions were selected and Surveyor nuclease assayed (Surveyor Mutation Detection Kit; Transgenomics).
For analysis of translocations between CCR5 and CXCR4 loci at the sites of DSBs, we performed PCR for 40 cycles using primers for a forward CCR5 primer and a reverse CXCR4 primer, or a reverse CCR5 primer and a forward CXCR4 primer, or a forward CCR5 primer and a forward CXCR4 primer, or a reverse CCR5 primer and a reverse CXCR4 primer. No PCR amplicons were detected (data not shown). Primer sequences are the same as used for Surveyor assay (Tables S1 and S2).
Deep sequencing and analysis
Target loci were amplified by the specific primers. Before sequencing on an Illumina HiSeq 2500 platform, the amplicons were purified, end repaired, and connected with sequencing primer. For the sequences gained by sequencing, low-quality and joint pollution data were removed to obtain reliable target sequences (clean reads) for subsequent analysis. The corresponding Read1 and Read2 (sequences gained from the 5′ and 3′ ends, respectively) were spliced. Analysis of indels was performed using the CRISPResso tool.49
Statistics and illustrations
All in vitro experiments were performed in biological triplicate, and in vivo experiments were performed with the number of animals indicated in the figure captions. Statistical significance was determined with the Student's t test. Statistical analysis was performed on GraphPad Prism software. Pre-drawn icons in illustrations were used from BioRender.
Acknowledgments
We thank the following City of Hope core facilities: Analytic Cytometry Core (ACC), Integrative Genomics and Bioinformatics Core (IGBC), and Center for Comparative Medicine (CCM) and its breeding core. The following reagents were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Jurkat cells, CEM.NKR CCR5+ cells from Dr. Alexandra Trkola; HIV-1Ba-L from Dr. Suzanne Gartner, Dr. Mikulas Popovic, and Dr. Robert Gallo; HIV-189.6 virus from Dr. Ronald Collman; and HIV-1NL4-3 Infectious Molecular Clone (pNL4-3) from Dr. Malcolm Martin. Cord blood from an anonymous donor was purchased from StemCyte. We thank members of the Burnett lab for discussions and suggestions. This research was supported by a grant from the California HIV/AIDS Research Program (CHRP) in Basic Biomedical Sciences (ID13-BRI-540) to J.C.B. and laboratory start-up funds to J.C.B.
Author contributions
S.L. and J.C.B. conceived and designed the experiments. S.L. performed the experiments. L.H. assisted with the in vivo experiments. S.L. and J.C.B. analyzed the data. S.L. and J.C.B. wrote the paper. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.01.012.
Supplemental information
References
- 1.GBD 2015 HIV Collaborators Estimates of global, regional, and national incidence, prevalence, and mortality of HIV, 1980-2015: the Global Burden of Disease Study 2015. Lancet HIV. 2016;3:e361–e387. doi: 10.1016/S2352-3018(16)30087-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deeks S.G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011;62:141–155. doi: 10.1146/annurev-med-042909-093756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deeks S.G., Lewin S.R., Ross A.L., Ananworanich J., Benkirane M., Cannon P., Chomont N., Douek D., Lifson J.D., Lo Y.R., et al. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nat. Med. 2016;22:839–850. doi: 10.1038/nm.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li S., Burnett J.C. In: Micro and Nano Technologies, Biomedical Applications of Functionalized Nanomaterials. Sarmento B., Neves J., editors. Elsevier; 2018. Chapter 18 - Biomolecular Therapeutics for HIV; pp. 541–567. [DOI] [Google Scholar]
- 5.Moyle G.J., Wildfire A., Mandalia S., Mayer H., Goodrich J., Whitcomb J., Gazzard B.G. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J. Infect. Dis. 2005;191:866–872. doi: 10.1086/428096. [DOI] [PubMed] [Google Scholar]
- 6.Zaitseva M., Blauvelt A., Lee S., Lapham C.K., Klaus-Kovtun V., Mostowski H., Manischewitz J., Golding H. Expression and function of CCR5 and CXCR4 on human Langerhans cells and macrophages: implications for HIV primary infection. Nat. Med. 1997;3:1369–1375. doi: 10.1038/nm1297-1369. [DOI] [PubMed] [Google Scholar]
- 7.Liu R., Paxton W.A., Choe S., Ceradini D., Martin S.R., Horuk R., MacDonald M.E., Stuhlmann H., Koup R.A., Landau N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 8.Paxton W.A., Martin S.R., Tse D., O'Brien T.R., Skurnick J., VanDevanter N.L., Padian N., Braun J.F., Kotler D.P., Wolinsky S.M., Koup R.A. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposure. Nat. Med. 1996;2:412–417. doi: 10.1038/nm0496-412. [DOI] [PubMed] [Google Scholar]
- 9.Allers K., Hutter G., Hofmann J., Loddenkemper C., Rieger K., Thiel E., Schneider T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- 10.Hutter G., Nowak D., Mossner M., Ganepola S., Mussig A., Allers K., Schneider T., Hofmann J., Kucherer C., Blau O., et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 11.Gupta R.K., Peppa D., Hill A.L., Galvez C., Salgado M., Pace M., McCoy L.E., Griffith S.A., Thornhill J., Alrubayyi A., et al. Evidence for HIV-1 cure after CCR5Delta32/Delta32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV. 2020;7:e340–e347. doi: 10.1016/S2352-3018(20)30069-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hütter G. More on shift of HIV tropism in stem-cell transplantation with CCR5 delta32/delta32 mutation. N. Engl. J. Med. 2014;371:2437–2438. doi: 10.1056/NEJMc1412279. [DOI] [PubMed] [Google Scholar]
- 13.Petz L.D., Burnett J.C., Li H., Li S., Tonai R., Bakalinskaya M., Shpall E.J., Armitage S., Kurtzberg J., Regan D.M., et al. Progress toward curing HIV infection with hematopoietic cell transplantation. Stem Cells Cloning. 2015;8:109–116. doi: 10.2147/sccaa.s56050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kordelas L., Verheyen J., Beelen D.W., Horn P.A., Heinold A., Kaiser R., Trenschel R., Schadendorf D., Dittmer U., Esser S. Shift of HIV tropism in stem-cell transplantation with CCR5 Delta32 mutation. N. Engl. J. Med. 2014;371:880–882. doi: 10.1056/NEJMc1405805. [DOI] [PubMed] [Google Scholar]
- 15.Li L., Krymskaya L., Wang J., Henley J., Rao A., Cao L.F., Tran C.A., Torres-Coronado M., Gardner A., Gonzalez N., et al. Genomic editing of the HIV-1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases. Mol. Ther. 2013;21:1259–1269. doi: 10.1038/mt.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez E.E., Wang J., Miller J.C., Jouvenot Y., Kim K.A., Liu O., Wang N., Lee G., Bartsevich V.V., Lee Y.L., et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi G., Choi J.G., Bharaj P., Abraham S., Dang Y., Kafri T., Alozie O., Manjunath M.N., Shankar P. CCR5 gene editing of resting CD4(+) T cells by transient ZFN expression from HIV envelope pseudotyped nonintegrating lentivirus confers HIV-1 resistance in humanized mice. Mol. Ther. Nucleic Acids. 2014;3:e198. doi: 10.1038/mtna.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W., Ye C., Liu J., Zhang D., Kimata J.T., Zhou P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE. 2014;9:e115987. doi: 10.1371/journal.pone.0115987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye L., Wang J., Beyer A.I., Teque F., Cradick T.J., Qi Z., Chang J.C., Bao G., Muench M.O., Yu J., et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Delta32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. U S A. 2014;111:9591–9596. doi: 10.1073/pnas.1407473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin L., Deng Y., He N., Wang L., Weng M. Polyethylenimine-mediated CCR5 gene knockout using transcription activator-like effector nucleases. J. Biomed. Nanotechnol. 2018;14:546–552. doi: 10.1166/jbn.2018.2545. [DOI] [PubMed] [Google Scholar]
- 21.Yu A.Q., Ding Y., Lu Z.Y., Hao Y.Z., Teng Z.P., Yan S.R., Li D.S., Zeng Y. TALENs-mediated homozygous CCR5Delta32 mutations endow CD4+ U87 cells with resistance against HIV1 infection. Mol. Med. Rep. 2018;17:243–249. doi: 10.3892/mmr.2017.7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao Q., Chen S., Wang Q., Liu Z., Liu S., Deng H., Hou W., Wu D., Xiong Y., Li J., Guo D. CCR5 editing by Staphylococcus aureus Cas9 in human primary CD4(+) T cells and hematopoietic stem/progenitor cells promotes HIV-1 resistance and CD4(+) T cell enrichment in humanized mice. Retrovirology. 2019;16:15. doi: 10.1186/s12977-019-0477-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang H., Minder P., Park M.A., Mesquitta W.T., Torbett B.E., Slukvin I.I. CCR5 disruption in induced pluripotent stem cells using CRISPR/Cas9 provides selective resistance of immune cells to CCR5-tropic HIV-1 virus. Mol. Ther. Nucleic Acids. 2015;4:e268. doi: 10.1038/mtna.2015.42. [DOI] [PubMed] [Google Scholar]
- 24.Xu L., Yang H., Gao Y., Chen Z., Xie L., Liu Y., Liu Y., Wang X., Li H., Lai W., et al. CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol. Ther. 2017;25:1782–1789. doi: 10.1016/j.ymthe.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilen C.B., Wang J., Tilton J.C., Miller J.C., Kim K.A., Rebar E.J., Sherrill-Mix S.A., Patro S.C., Secreto A.J., Jordan A.P., et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011;7:e1002020. doi: 10.1371/journal.ppat.1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan J., Wang J., Crain K., Fearns C., Kim K.A., Hua K.L., Gregory P.D., Holmes M.C., Torbett B.E. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4+ T cell resistance and enrichment. Mol. Ther. 2012;20:849–859. doi: 10.1038/mt.2011.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q., Chen S., Xiao Q., Liu Z., Liu S., Hou P., Zhou L., Hou W., Ho W., Li C., et al. Genome modification of CXCR4 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection. Retrovirology. 2017;14:51. doi: 10.1186/s12977-017-0375-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou P., Chen S., Wang S., Yu X., Chen Y., Jiang M., Zhuang K., Ho W., Hou W., Huang J., Guo D. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015;5:15577. doi: 10.1038/srep15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Didigu C.A., Wilen C.B., Wang J., Duong J., Secreto A.J., Danet-Desnoyers G.A., Riley J.L., Gregory P.D., June C.H., Holmes M.C., Doms R.W. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood. 2014;123:61–69. doi: 10.1182/blood-2013-08-521229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu S., Yao Y., Xiao H., Li J., Liu Q., Yang Y., Adah D., Lu J., Zhao S., Qin L., Chen X. Simultaneous knockout of CXCR4 and CCR5 genes in CD4+ T cells via CRISPR/Cas9 confers resistance to both X4- and R5-tropic human immunodeficiency virus type 1 infection. Hum. Gene Ther. 2018;29:51–67. doi: 10.1089/hum.2017.032. [DOI] [PubMed] [Google Scholar]
- 31.Tebas P., Stein D., Tang W.W., Frank I., Wang S.Q., Lee G., Spratt S.K., Surosky R.T., Giedlin M.A., Nichol G., et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tebas P., Jadlowsky J.K., Shaw P.A., Tian L., Esparza E., Brennan A.L., Kim S., Naing S.Y., Richardson M.W., Vogel A.N., et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Invest. 2021;131:e144486. doi: 10.1172/JCI144486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L., Wang J., Liu Y., Xie L., Su B., Mou D., Wang L., Liu T., Wang X., Zhang B., et al. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N. Engl. J. Med. 2019;381:1240–1247. doi: 10.1056/NEJMoa1817426. [DOI] [PubMed] [Google Scholar]
- 34.Heckl D., Kowalczyk M.S., Yudovich D., Belizaire R., Puram R.V., McConkey M.E., Thielke A., Aster J.C., Regev A., Ebert B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014;32:941–946. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trkola A., Matthews J., Gordon C., Ketas T., Moore J.P. A cell line-based neutralization assay for primary human immunodeficiency virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J. Virol. 1999;73:8966–8974. doi: 10.1128/jvi.73.11.8966-8974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tycko J., Myer V.E., Hsu P.D. Methods for optimizing CRISPR-cas9 genome editing specificity. Mol. Cell. 2016;63:355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beane J.D., Lee G., Zheng Z., Mendel M., Abate-Daga D., Bharathan M., Black M., Gandhi N., Yu Z., Chandran S., et al. Clinical scale zinc finger nuclease-mediated gene editing of PD-1 in tumor infiltrating lymphocytes for the treatment of metastatic melanoma. Mol. Ther. 2015;23:1380–1390. doi: 10.1038/mt.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou Y.R., Kottmann A.H., Kuroda M., Taniuchi I., Littman D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 41.Sharma M., Afrin F., Satija N., Tripathi R.P., Gangenahalli G.U. Stromal-derived factor-1/CXCR4 signaling: indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev. 2011;20:933–946. doi: 10.1089/scd.2010.0263. [DOI] [PubMed] [Google Scholar]
- 42.Liles W.C., Broxmeyer H.E., Rodger E., Wood B., Hubel K., Cooper S., Hangoc G., Bridger G.J., Henson G.W., Calandra G., Dale D.C. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102:2728–2730. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 43.Ye L., Wang J., Tan Y., Beyer A.I., Xie F., Muench M.O., Kan Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and beta-thalassemia. Proc. Natl. Acad. Sci. U S A. 2016;113:10661–10665. doi: 10.1073/pnas.1612075113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M., McCaw Z.R., Aryee M.J., Joung J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y.B., Komor A.C., Levy J.M., Packer M.S., Zhao K.T., Liu D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.You L., Tong R., Li M., Liu Y., Xue J., Lu Y. Advancements and obstacles of CRISPR-cas9 technology in translational research. Mol. Ther. Methods Clin. Dev. 2019;13:359–370. doi: 10.1016/j.omtm.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Rosa F. Two niches in the bone marrow: a hypothesis on life-long T cell memory. Trends Immunol. 2016;37:503–512. doi: 10.1016/j.it.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 48.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinello L., Canver M.C., Hoban M.D., Orkin S.H., Kohn D.B., Bauer D.E., Yuan G.C. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016;34:695–697. doi: 10.1038/nbt.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.