

Abstract
Background and aims.
Despite the high clinical significance of sarcopenia in alcoholic cirrhosis, there are currently no effective therapies because the underlying mechanisms are poorly understood. We determined the mechanisms of ethanol-induced impaired phosphorylation of mechanistic target of rapamycin complex 1 (mTORC1) and AMP kinase (AMPK) with consequent dysregulated skeletal muscle protein homeostasis (balance between protein synthesis and breakdown).
Approach and results.
Differentiated murine myotubes, gastrocnemius muscle from mice with loss and gain of function of regulatory genes following ethanol treatment, and skeletal muscle from alcoholic cirrhotics were used. Ethanol increases skeletal muscle autophagy by dephosphorylating mTORC1, circumventing the classical kinase regulation by protein kinase B (Akt). Concurrently and paradoxically, ethanol exposure results in dephosphorylation and inhibition of AMPK, an activator of autophagy and inhibitor of mTORC1 signaling. However, AMPK remains inactive with ethanol exposure despite lower cellular and tissue ATP indicating a “pseudofed” state. We identified protein phosphatase 2A (PP2A) as a key mediator of ethanol-induced signaling and functional perturbations using loss and gain of function studies. Ethanol impairs binding of endogenous inhibitor of PP2A (I2-PP2A) to PP2A resulting in methylation and targeting of PP2A to cause dephosphorylation of mTORC1 and AMPK. Activity of phosphoinositide 3-kinase-γ (PI3Kγ), a negative regulator of PP2A, was decreased in response to ethanol. Ethanol-induced molecular and phenotypic perturbations in wild type mice were observed in PI3Kγ−/− mice even at baseline. Importantly, overexpressing kinase-active PI3Kγ but not the kinase-dead mutant reversed ethanol-induced molecular perturbations.
Conclusions.
Our study describes the mechanistic underpinnings for previously unrecognized ethanol-mediated dysregulation of protein homeostasis by PP2A that leads to sarcopenia with a potential for therapeutic approaches by targeting the PI3Kγ-PP2A axis.
Keywords: Signaling, protein homeostasis, ethanol, skeletal muscle, phosphoinositide-3-kinase-gamma
Introduction
Alcohol (ethanol) is a major cause of liver disease and in patients with alcoholic cirrhosis, sarcopenia or loss of skeletal muscle mass, is frequent, severe and adversely affects clinical outcomes(1–3). Sarcopenia in alcohol-related liver disease (ALD) occurs due to dysregulated protein homeostasis, an imbalance between muscle protein synthesis and proteolysis, primarily, via autophagy(1). Ethanol impairs skeletal muscle protein synthesis and activates autophagy(1, 4, 5) , but whether these perturbations occur in parallel to cause sarcopenia, and importantly, the molecular mechanisms that underlie the dysregulated protein homeostasis are currently not known. Given the high clinical significance of alcohol use disorders(2), adverse consequences of dysregulated muscle protein homeostasis resulting in sarcopenia, and potential context specificity of tissue responses to ethanol, we dissected the molecular perturbations in skeletal muscle in response to ethanol exposure in a comprehensive array of preclinical models and in alcoholic cirrhotics.
A number of regulatory signaling pathways with opposing effects on protein synthesis and autophagy maintain protein homeostasis(6–8). The convergence point for signaling responses that regulate protein homeostasis is the mechanistic target of rapamycin complex 1 (mTORC1), a kinase complex activated by phosphorylation of its mTOR component that targets downstream molecules to activate protein synthesis and inhibit autophagy during nutrient and energy sufficiency(8, 9). In contrast, AMP kinase (AMPK) is phosphorylated and activated during nutrient and energy insufficiency(10, 11), directly increasing autophagy by targeting regulatory sites on Unc like kinase 1 (ULK1)(12). AMPK also indirectly inhibits mTORC1 activity through the tuberous sclerosis complex 1/2 (TSC1/2) while simultaneously decreasing protein synthesis(12–15). These opposing effects of mTORC1 and AMPK are metabolic checkpoints that regulate protein homeostasis, depending on cellular nutrient and energy status(8, 12, 15). Activation or inactivation of mTORC1 and AMPK is determined by their phosphorylation status, which is regulated by upstream kinases and specific phosphatases(16).
Previous studies reported that ethanol alters mTORC1 or AMPK signaling and that these changes depend on the cellular and/or tissue context of ethanol exposure(17–22). In the present studies, we found that ethanol dysregulates proteostasis, with simultaneous protein synthesis impairment and increased autophagy, and with decreased phosphorylation of mTOR. However, despite reduced tissue ATP levels, a cellular energy sensor and mTORC1 inhibitor, AMPK was not activated. Previous work has suggested kinase-dependent regulation of mTORC1 and AMPK signaling(8, 10). We show that the upstream kinase for mTOR phosphorylation, protein kinase B (Akt)(23, 24), was not altered by ethanol. Instead, ethanol exposure caused simultaneous and paradoxical inactivation of mTOR and AMPK by dephosphorylation due to activation of protein phosphatase 2A (PP2A). Finally, de-repression and activation of PP2A was due to ethanol-induced inactivation (rather than activation, as has been reported to date during various stress states(25, 26)) of the gamma isoform of phosphoinositide 3-kinase (PI3Kγ), a known inhibitor of PP2A.
Materials and Methods
Antibodies:
All the antibodies used in these studies were commercially purchased except for the phospho-I2PP2A. See supplementary methods for details on the commercial antibodies.
Generation of phospho-I2PP2A antibody.
Polyclonal antibody against the phosphorylated form of I2PP2A was generated by immunizing rabbits with a synthetic peptide corresponding to residues surrounding Ser9 of human I2-PP2A (APAAKCpSKKELNC) as previously described(25). The specificity of the antibody was tested by immunoblotting in vitro phosphorylated purified I2-PP2A (phospho-I2PP2A) protein and in lysates from isoproterenol-stimulated HEK 293 cells (25).
Human Studies.
Vastus lateralis muscle from alcoholic cirrhotics and healthy patients (n=5; 4M,1F each) were obtained as previously described and clinical descriptors of these patients have been reported earlier(27). These studies were approved by the Institutional Review Board at the Cleveland Clinic, conformed to the Declaration of Helsinki, and were performed after a written informed consent was obtained from the patients.
Mouse studies.
12-week-old female and male PI3Kγ knockout (PI3Kγ−/−) and wild-type mice in a C57BL/6 background were allowed free access to a Lieber-DeCarli liquid diet containing ethanol (Dyets Inc., 710260) or isocalorically substituted maltodextrins as previously described (1). Experiments were done in female mice (n=8 each for pair-fed; n=10 each for ethanol-fed) due to their greater susceptibility to ethanol. In a subgroup (n=4 each in pair-fed and n=5 each in ethanol-fed mice) in vivo autophagy flux was quantified as previously described(28). Key experiments were also performed in male mice (n=4 each for pair-fed; n=6 each for ethanol-fed). Animals were housed in the Biological Resource Unit, at the Cleveland Clinic, with a 12 h light/dark cycle. The Cleveland Clinic Institutional Animal Care and Use Committee approved all procedures using mice. All animals received human care and the study protocols comply with the institutional guidelines.
In vitro cell culture studies and stable transfection:
C2C12 murine myoblasts were grown and differentiated as previously described(1) and exposed to 100 mM ethanol and processed as detailed in the supplementary methods. Sub-confluent myoblasts were transfected with various PI3Kγ constructs or shRNA targeting PP2A catalytic subunit and stable cells were selected using appropriate antibiotics as described in supplementary methods. All experiments were done on at least 3 biological replicates.
Protein synthesis:
Rates of protein synthesis in vitro were quantified by incorporation of puromycin, as previously reported(29). Since shRNA-PP2A myoblasts were selected on puromycin, incorporation of 3H phenylalanine was used for quantifying protein synthesis(29) as detailed in supplementary methods. In vivo protein synthesis was quantified by a flooding dose of D5-phenylalanine as described in supplementary methods.
Immunoblotting and immunoprecipitation studies:
Total proteins were extracted from flash-frozen skeletal muscle or cultured cells and resolved on gradient gel followed by densitometry quantification of the blots using ImageJ® software as described earlier(1). For immunoprecipitation studies, the lysates were incubated with protein A/G agarose beads and respective antibodies overnight at 4°C, and immunoprecipitates were washed in lysis buffer before resolution on SDS-PAGE gel (see supplementary methods).
PP2A activity assay:
PP2A activity was performed as previously described using the malachite green assay kit (Millipore-Sigma, Burlington, MA)(25). The lysates or immunoprecipitates were washed with phosphatase assay buffer and subjected to PP2A activity assay by addition of PP2A specific threonine phospho-peptide substrate. In addition to using commercially available PP2A phospho-peptide substrate, we also generated in-house the threonine phosphopeptide used for PP2A activity assays, described in the supplementary methods.
PI3Kγ activity assay.
Clarified myotube or muscle extract was used to immunoprecipitate PI3Kγ using anti-PI3Kγ antibody and the immunoprecipitates were subjected to in vitro lipid kinase assay using phosphatidyl-inositol (PtdIns) and the labelled PtdIns were resolved using thin layer chromatography as detailed in supplementary methods and described by us earlier(25).
Statistical analyses:
All data were expressed as mean±SD unless specified. Qualitative variables were compared using the chi square test. Quantitative variables were compared by the Student’s t-test or for multiple group comparisons, analysis of variance with Bonferroni post-hoc analysis.
Results
Ethanol-treated myotubes have dysregulated protein homeostasis
Ethanol treatment results in a time-dependent reduction in puromycin incorporation, indicating decreased global protein synthesis (Fig. 1A) without altering cell viability in myotubes(Supplementary Fig. 1A). In addition to reduced protein synthesis, there was increased lipidation of LC3 and expression of Beclin1 in myotubes showing an increase in autophagy flux (Fig. 1B). In skeletal muscle from ethanol-fed compared with pair-fed mice, LC3 lipidation and Beclin 1 expression were higher and p62 lower (Fig. 1C). These observations show that ethanol inhibits protein synthesis and, concurrently, increases autophagy flux in myotubes and markers of autophagy in skeletal muscle of ethanol-fed mice.
Fig. 1. Ethanol causes simultaneous reduction in protein synthesis and increased autophagy in myotubes and mouse muscle.
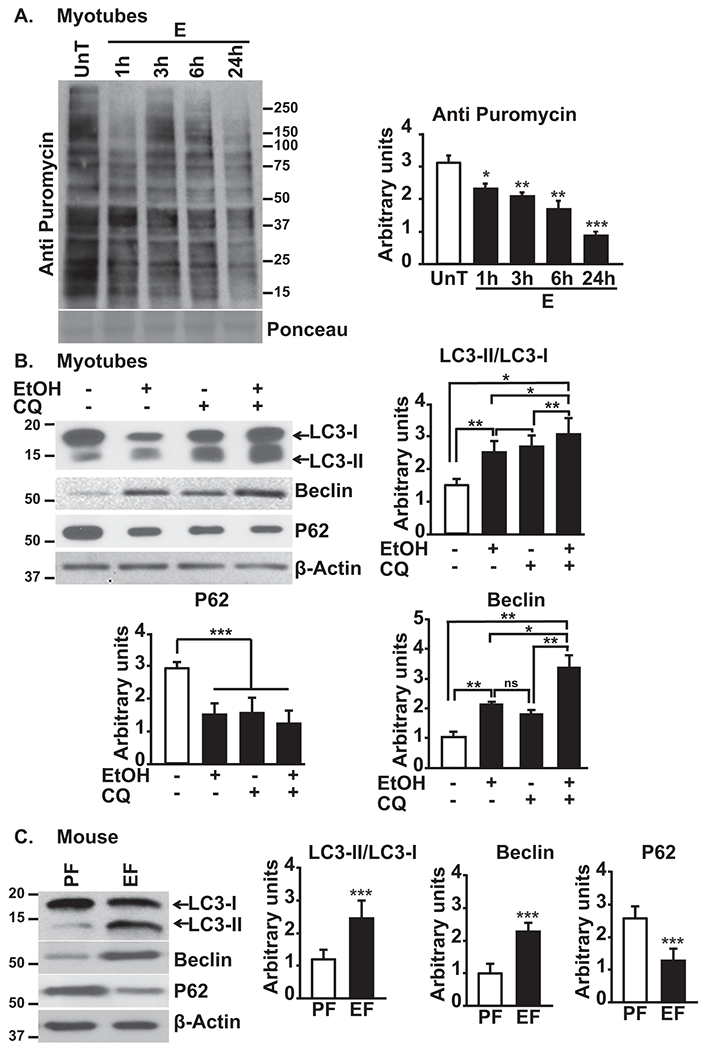
A. Representative immunoblots and densitometry (entire lane) for puromycin incorporation in C2C12 myotubes treated with 100mM ethanol for varying time points. B. Representative immunoblots and densitometry of LC3II lipidation, Beclin1 and p62 in myotubes treated with and without 100mM ethanol and/or chloroquine n=3 biological replicates). C. Representative immunoblots and densitometry of LC3 lipidation, Beclin1 and p62 in the gastrocnemius muscle of ethanol or pair-fed mice . Data: mean±SD. *p<0.05; **p<0.01; ***p<0.001 vs. untreated control myotubes (ANOVA) or pair-fed mice (unpaired Student’s ‘t’ test). E:ethanol-treated; EF:ethanol-fed mice (n=4); PF:pair-fed mice (n=6); UnT:untreated myotubes.
Ethanol treatment impairs upstream signaling components that regulate protein homeostasis.
mTORC1 is a key regulator of protein synthesis and autophagy(8, 12, 30). Consistent with decreased protein synthesis and increased autophagy markers, ethanol impairs phosphorylation of the mTOR component of mTORC1 and its downstream targets P70S6K and ribosomal S6 protein in myotubes (Fig. 2A), and in skeletal muscles from ethanol-fed mice (Fig. 2B). Intriguingly, activation of upstream canonical activator of mTOR, Akt(8, 23, 24) is unaltered (both Thr308 and Ser473) in ethanol-treated myotubes and skeletal muscles from ethanol-fed mice and alcoholic cirrhotics (Supplementary Fig. 1B).
Fig. 2. Ethanol impairs regulatory signaling in myotubes and skeletal muscle from mice and human subjects.
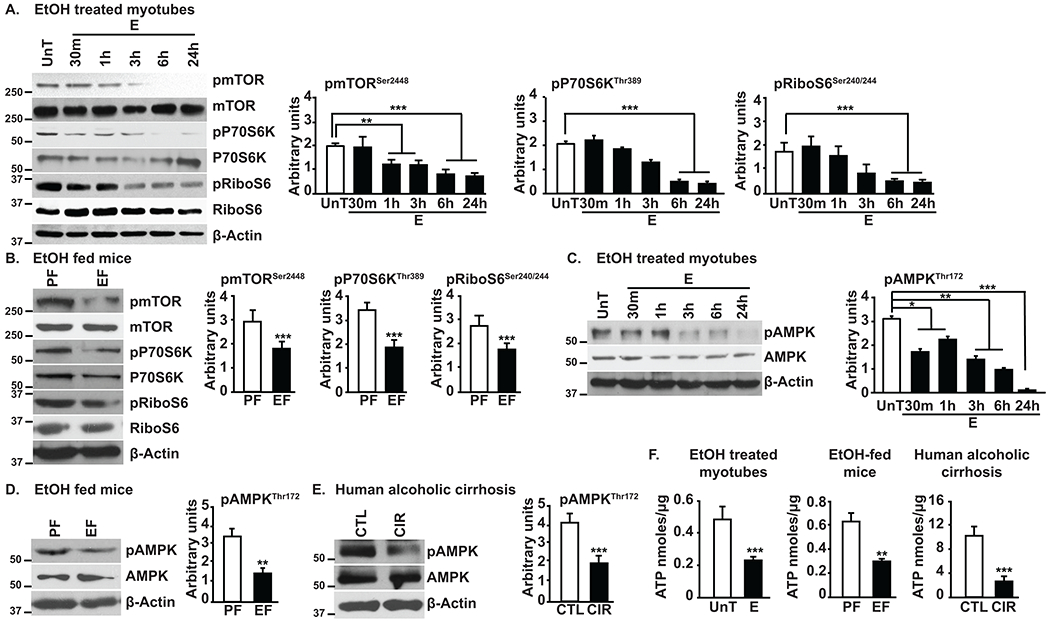
A,B. Representative immunoblots and densitometry of mTOR phosphorylation and mTORC1 signaling in ethanol-treated C2C12 myotubes and gastrocnemius muscle of ethanol or pair-fed mice. C-E. Representative immunoblots and densitometry of phosphorylated AMPK in ethanol-treated myotubes, muscle from ethanol or pair-fed mice and skeletal muscle from alcoholic cirrhosis and controls. F. ATP content in myotubes, mouse or human skeletal muscle. All data expressed as mean±SD from n=3 biological replicates of myotubes, n=4 in PF and n=6 EF mice; human cirrhotics and controls n=5 each. *p<0.05; **p<0.01; ***p<0.001 vs. untreated control myotubes (ANOVA) or pair-fed mice/control subjects (unpaired Student’s ‘t’ test). CIR:alcoholic cirrhotics; CTL:control subjects, E, EtOH:ethanol; EF:ethanol-fed mice; PF:pair-fed mice; UnT:untreated myotubes.
Given unaltered Akt activation, assessment of AMPK, an energy sensor(10, 31) and negative regulator of mTOR (15, 32) was undertaken and showed reduced phosphorylation/activation of AMPK in myotubes exposed to ethanol and in muscle from ethanol-fed mice and alcoholic cirrhotics (Fig. 2C–E). AMPK was not activated despite ATP content being significantly reduced (canonical activator of AMPK) in ethanol-exposed myotubes and skeletal muscles from ethanol-fed mice and humans with alcoholic cirrhosis (Fig. 2F). Consistently, ethanol treatment decreased phosphorylation of targets of AMPK or mTORC1 kinases, Ser317,555,777 or Ser757 respectively, on downstream regulatory molecule, ULK1, that was not differentially bound to PP2A(Supplementary Fig. 2A,B). Unaltered Akt phosphorylation and reduced AMPK phosphorylation would be expected to increase mTOR phosphorylation. However, our observation of reduced mTOR phosphorylation, suggests dephosphorylation as a mechanism of ethanol-mediated signaling responses.
Akt and AMPK regulate mTORC1 signaling by phosphorylating the signaling molecule TSC2, but phosphorylation of TSC2 was not altered following ethanol exposure (Supplementary Fig. 3A,B). Immunoprecipitation studies showed that TSC2 binding to the B56 regulatory subunit of the key phosphatase PP2A was similar in ethanol- and vehicle-treated myotubes, indicating that TSC2 activation is regulated by kinases (Akt and AMPK) and not by dephosphorylation (Supplementary Fig. 3C). Consistently, ethanol-treated myotubes with or without TSC2 knockdown showed similar reduction in mTOR phosphorylation (Supplementary Fig. 3D). This finding that depletion of TSC2 does not alter mTOR response to ethanol suggests kinase-independent phosphatase-driven mechanisms of mTOR regulation and was then evaluated.
Ethanol simultaneously impairs mTORC1 and AMPK activation by PP2A-dependent dephosphorylation.
Activity of PP2A was increased in PP2A immunoprecipitates in ethanol-treated myotubes and in gastrocnemius muscle of ethanol-fed mice (Fig. 3A), showing acute activation of PP2A could underlie mTOR and AMPK dephosphorylation. Furthermore, immunoblotting for the stress-activated mitogen-activated protein kinases (MAPKs-ERK, p38 MAPK and JNK1(33)) showed differential response wherein ethanol increased p38 MAPK and JNK1 phosphorylation while simultaneously decreasing ERK phosphorylation (Supplementary Fig. 4A), showing specificity in the dephosphorylation events following ethanol exposure. Moreover, pre-treatment of myotubes with selective PP2A inhibitor, fostriecin(34), restored the phosphorylation status of mTOR and AMPK including its cognate downstream signaling molecules (Fig. 3B,C). Additionally, knockdown of the PP2A reversed the ethanol-induced reduction of mTOR phosphorylation and its downstream signaling molecules (Fig. 3D). In parallel, PP2A knockdown also reversed the ethanol-induced reduction in AMPK phosphorylation (Fig. 3E). Despite increased mTOR and AMPK phosphorylation following PP2A knockdown, there was an increase in protein synthesis with parallel reduction in autophagy markers (Fig. 3F,G) and autophagy flux (Fig. 3H).
Fig. 3. Ethanol-mediated increase in PP2A activity dephosphorylates specific signaling phosphoproteins.
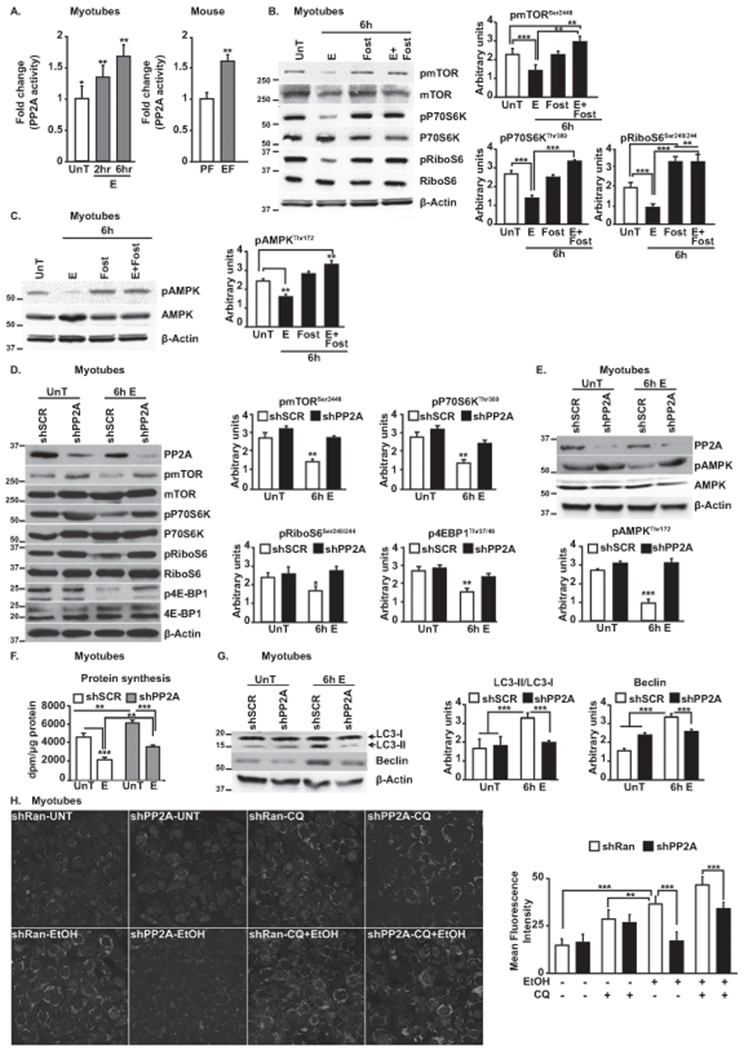
A.PP2A activity-fold change in myotubes and mouse muscle. B.Representative immunoblots and densitometry of mTOR phosphorylation and mTORC1 signaling response to fostriecin in ethanol-treated myotubes. C.Representative immunoblots and densitometry of phosphorylated AMPK with and without fostriecin. D.Representative immunoblots and densitometry of mTORC1 activation and signaling in ethanol-treated myotubes transfected with shPP2A or scrambled construct. E.Representative immunoblots and densitometry of phosphorylation of AMPK in ethanol-treated myotubes transfected with shPP2A or scrambled construct. F.Protein synthesis in ethanol-treated myotubes transfected with shPP2A. G.Representative immunoblots densitometry of autophagy markers in myotubes transfected with shPP2A or scrambled construct. H.Representative photomicrographs of myotubes transfected with GFP-LC3 treated with ethanol and lysosomal inhibitor, chloroquine. Bars graphs show number of myotubes with ≥5 punctae. Data: mean±SD from n=3 biological replicates for myotubes; quantification of punctae from at least 100 myotubes from 5 independent slides. and n=4 PF and n=6 EF mice. *p<0.05; **p<0.01; ***p<0.001 vs. untreated control myotubes (ANOVA) or pair-fed mice (unpaired Student’s ‘t’ test). E:ethanol-treated myotubes; EF:ethanol-fed mice; PF:Pair-fed mice; Scr:scrambled construct; UnT:untreated myotubes.
Regulation of PP2A target proteins during exposure to ethanol is mediated by the B56 regulatory subunit
Given that PP2A activity changes in response to ethanol, post-translational modifications including methylation that activates PP2A, and phosphorylation that reduces its activity(35–38), were assessed by immunoblotting. PP2A-activating methylation was increased while inhibitory phosphorylation was decreased in all 3 systems studied (Fig. 4A–C). Further, mTOR and AMPK immunoprecipitates showed increased association with the B56 regulatory subunit in ethanol-treated myotubes and skeletal muscle from ethanol-fed mice and alcoholic cirrhotic patients (Fig. 4D–F). Akt immunoprecipitates showed minimal interaction with B56 in ethanol-treated myotubes (Supplementary Fig. 4B) demonstrating the specificity of the PP2A holoenzyme interaction with target molecules.
Fig. 4. Ethanol activates PP2A via inhibitory I2PP2A and targets specific signaling molecules via the B56 regulatory subunit in myotubes and muscle tissue from mice and human subjects.
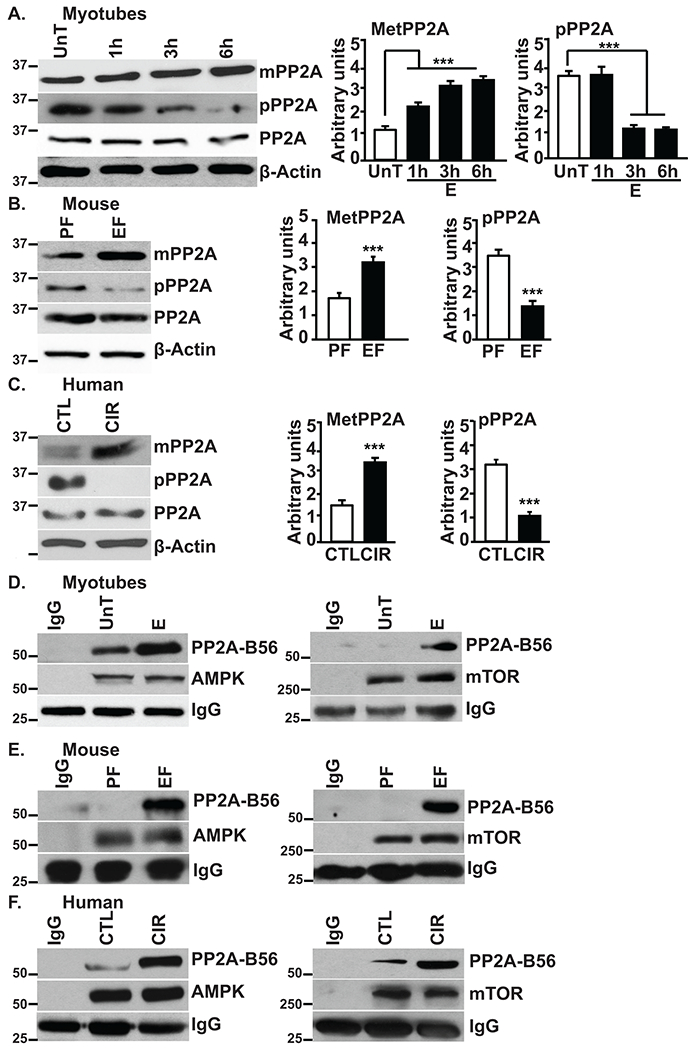
A-C.Representative immunoblots and densitometry of methylated and phosphorylated PP2A in myotubes and skeletal muscle from mice and human subjects. D-F.Immunoprecipitate of AMPK and mTOR immunoblotted for PP2A regulatory subunit, B56 in myotubes and skeletal muscle from mice and human subjects. Data: mean±SD from n=3 biological replicates for myotubes; n=4 PF and n=6 for EF mice and n=5 each for human subjects. **p<0.01; ***p<0.001 vs. untreated control myotubes (ANOVA) or pair-fed mice/control subjects (unpaired Student’s ‘t’ test). CIR:cirrhosis patients; CTL:control subjects; E:ethanol-treated myotubes; EF:ethanol-fed mice; PF:pair-fed mice; UnT:untreated myotubes.
Loss of PP2A inhibition with ethanol occurs by inhibition of the PI3Kγ-I2PP2A axis
In addition to targeting of PP2A to specific signaling proteins, PP2A catalytic activity is regulated by the binding of inhibitory proteins I1- and I2-PP2A to the catalytic subunit (39, 40). PP2A immunoprecipitates showed marked decrease in interaction with I2-PP2A with no changes in interaction with I1-PP2A in muscle from ethanol-fed mice or skeletal muscle from alcoholic cirrhotics (Supplementary Fig. 5A,B). To determine the mechanism of regulation of PP2A, I2-PP2A immunoprecipitates were assessed for co-immunoprecipitating PP2A following ethanol treatment. Ethanol exposure resulted in reduced I2-PP2A interaction with PP2A-C (PP2A catalytic subunit C)(Fig. 5A). Similarly, I2-PP2A immunoprecipitates from skeletal muscle from ethanol-fed mice showed markedly reduced interaction with PP2A-C (Fig. 5B). Since phosphorylated I2-PP2A binds and inhibits PP2A(25), the phosphorylation state of I2-PP2A was assessed using custom-made anti-phospho I2-PP2A (Ser9) antibody. Lower I2-PP2A phosphorylation was observed in the skeletal muscle of ethanol-fed mice (Fig. 5C) and alcoholic cirrhotic patients (Fig. 5D). These observations show decreased phosphorylation and binding of inhibitory I2PP2A to PP2A.
Fig. 5. Ethanol increases PP2A by inhibition of the kinase domain of PI3KY in myotubes and skeletal muscle from mice and human subjects.
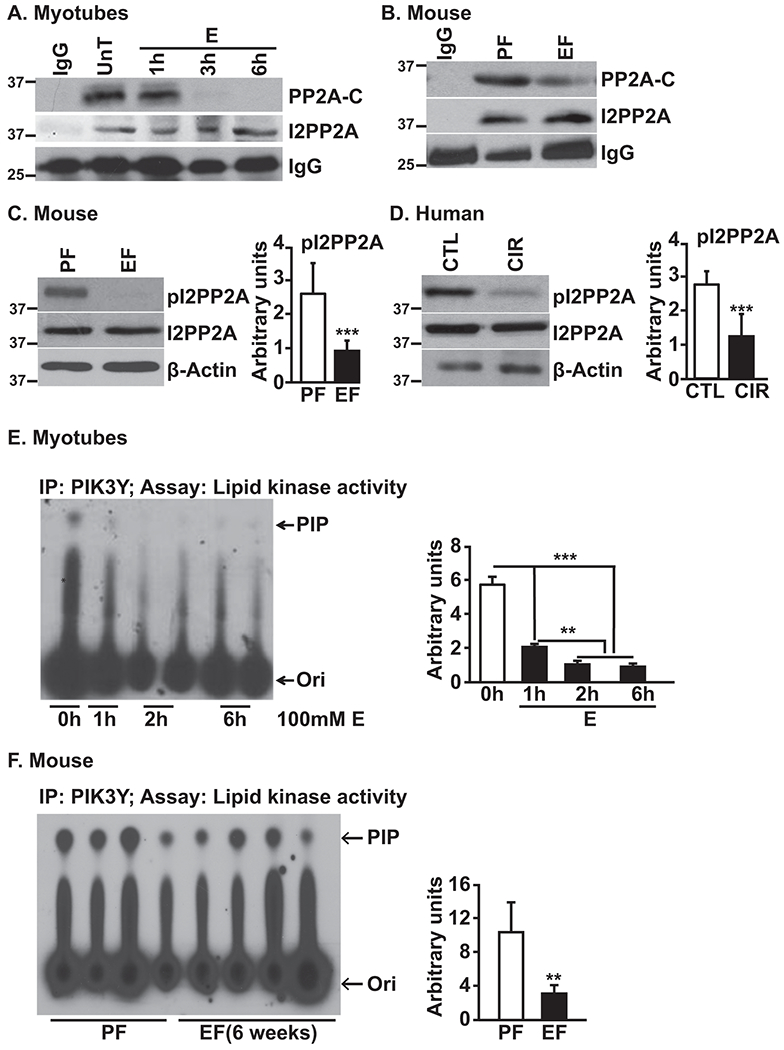
A-B.Immunoprecipitate of I2PP2A, from myotubes and gastrocnemius muscle from mice, probed for PP2A Catalytic subunit. C-D.Immunoblots of phosphorylated and total I2PP2A from skeletal muscle from mice and human subjects. E-F.Representative thin layer chromatograms and densitometry of phosphatidyl-inositol-3-phosphate as a measure of PI3KY activity in myotubes and gastrocnemius muscle from mice. Data: mean±SD from n=3 biological replicates for myotubes; n=4 PF and n=6 for EF mice and n=5 each for human subjects. *p<0.05; **p<0.01; ***p<0.001 vs. untreated control myotubes (ANOVA) or pair-fed mice/control subjects (unpaired Student’s ‘t’ test). CIR:cirrhosis patients; CTL:control subjects; E:ethanol-treated myotubes; EF:ethanol-fed mice; PF:pair-fed mice; PIP:phosphatidyl inositol phosphate; Ori:origin; UnT:untreated myotubes.
As PI3Kγ phosphorylates I2-PP2A thereby increasing its binding affinity to PP2A and inhibiting PP2A activity(25), immunoblotting was performed to assess for changes in PI3Kγ expression in response to ethanol. No appreciable changes in PI3Kγ expression was observed in ethanol-treated myotubes (Supplementary Fig. 6A) or skeletal muscles from ethanol-fed mice (Supplementary Fig. 6A). Given that PI3Kγ expression may not correlate with its activity, in vitro PI3Kγ activity was measured in PI3Kγ immunoprecipitates. Surprisingly, significant reduction in PI3Kγ activity was observed in ethanol-treated myotubes (Fig. 5E) and in muscles from ethanol-fed mice (Fig. 5F). These data show that decreased PI3Kγ could reduce I2-PP2A phosphorylation and consequent increased PP2A activity.
PI3Kγ expression reverses ethanol-mediated inhibition of mTOR and AMPK
Increased PI3Kγ activity by PI3Kγ overexpression in myotubes did not alter/inhibit PP2A activity in -untreated cells. However, PI3Kγ overexpression reduced ethanol-induced increased PP2A activity to basal levels (Fig. 6A). Immunoblotting for mTOR and signaling targets of mTORC1 in ethanol-treated myotubes showed that overexpression of PI3Kγ restored their phosphorylation status and signaling responses (Fig. 6B) while reversing ethanol-induced AMPK dephosphorylation (Fig. 6C). Functional responses included restoration of protein synthesis and autophagy flux in ethanol-treated myotubes (Fig. 6D,E) and myotube diameter (Fig. 6,F). These observations suggest that mTORC1 pathways play a dominant role overwhelming AMPK driven responses.
Fig. 6. Ethanol-induced increase in PP2A activity and its consequences are mediated via PI3KY.
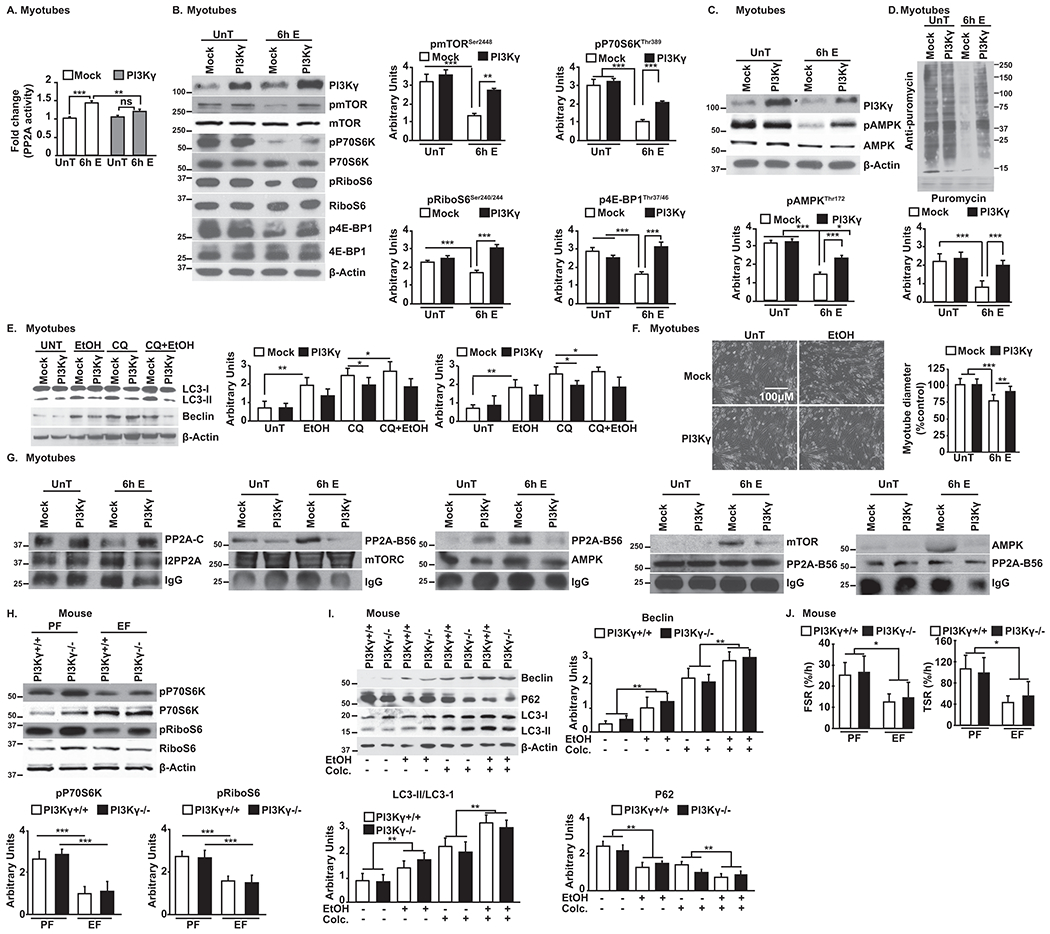
A.PP2A activity in PI3KY-overexpressing myotubes. B-F.Representative immunoblots and densitometry of mTOR phosphorylation and mTORC1 signaling, phosphorylated AMPK, protein synthesis, autophagy flux, and myotube size (representative photomicrographs and diameter) in PI3KY-overexpressing myotubes. G.Immunoprecipitate of I2PP2A immunoblotted for PP2A Catalytic subunit; immunoprecipitates of AMPK and mTOR immunoblotted for PP2A regulatory subunit, B56; immunoprecipitate of PP2A regulatory subunit, B56 immunoblotted for AMPK and mTOR. H,I. Representative immunoblots of mTORC1 signaling and autophagy flux in muscle from ethanol and pair-fed PI3KY+/+ and PI3KY−/− female mice. J. Fractional (FSR) and total (TSR) muscle protein synthesis rates in female ethanol and pair-fed PI3KY+/+ and PI3KY−/− female mice. Data: mean±SD from n=3 biological replicates for myotubes; n=4 PF and n=5 for EF mice (each with and without colchicine) and n=5 each for human subjects. *p<0.05; **p<0.01; ***p<0.001 vs. untreated control myotubes (ANOVA) or pair-fed mice (unpaired Student’s ‘t’ test). E:ethanol-treated myotubes; EF:ethanol-fed mice; PF:pair-fed mice; UnT:untreated myotubes. All immunoprecipitates from myotubes.
Given the increasing recognition of the role for kinase-independent function of PI3Kγ(26, 41, 42), kinase-dead PI3Kγ (PI3Kγ-KD) was expressed in myotubes and signaling responses with ethanol exposure were assessed. Expression of PI3Kγ-KD resulted in little change in phosphorylation of mTOR and AMPK (Supplementary Fig. 6B), ethanol treatment of cells expressing PI3Kγ-KD still increased PP2A activity (Supplementary Fig. 6C) indicating that ethanol effects are mediated via kinase-dependent function of PI3Kγ. PI3Kγ mediates its downstream signals via its lipid kinase activity(26, 42, 43) through plasma membrane recruitment. Signaling responses with ethanol exposure were not altered by overexpression of plasma membrane targeted myristoylated PI3Kγ (PI3Kγ-MT) (Supplementary Fig. 6D) suggesting that membrane localization of PI3Kγ is not critical in regulating ethanol-mediated signals.
In myotubes overexpressing PI3Kγ, mTOR and AMPK were co-immunoprecipitated and immunoblotted for PP2A to assess for regulation of PP2A by I2-PP2A. Ethanol treatment resulted in reduced interaction of mTOR and AMPK with PP2A (Fig. 6G), and associated PP2A activity was also lower with PI3Kγ overexpression in myotubes (Supplementary Fig. 6E). Furthermore, chronic ethanol feeding of PI3Kγ−/− and PI3Kγ+/+ mice of both sexes showed similar reduction in mTORC1 signaling (Fig. 6H, Supplementary Fig. 7A), decrease in AMPK dephosphorylation, increased autophagy flux/markers (Fig. 6I, Supplementary Fig. 7A,B), decreased fractional and total muscle protein synthesis rates (Fig. 6J, Supplementary Fig. 7C,D) and reduced skeletal muscle mass (Table 1, Supplementary Table 2 ). Furthermore, the absence of PI3Kγ did not alter PP2A activity in skeletal muscle as shown in the pair-fed PI3Kγ−/− mice (Supplementary Fig. 7E). These observations show that the biochemical phenotype observed in the absence of PI3Kγ mimics the signaling responses to ethanol-mediated inhibition of PI3Kγ in both male and female mice. These studies show the mechanistic role of PI3Kγ in ethanol-mediated sarcopenia via alterations in protein homeostasis through PP2A and lay the foundation for targeted therapies.
Table 1.
Body weight and organ weights in the PI3KY−/− and PI3KY+/+ mice
| Female mice | PI3K+/+ PF | PI3K+/+ EF | PI3K−/− PF | PI3K−/− EF |
|---|---|---|---|---|
| Number | 8 | 10 | 8 | 10 |
| INITIAL BW | 19.5±2.1 | 19.5±1.5 | 19.7±3.3 | 18.6±0.9 |
| FINAL BW | 23.5±1.9 | 23.1±1.3 | 22.6±1.9 | 21.3±1.2 |
| Δ BW | 4.0±0.8 | 3.6±1.0 | 3.0±2.2 | 2.6±0.8 |
| Avg. intake (ml/d) | ND | 23.4±3.2 | ND | 23.4±3.3 |
| MUSCLE WT | 0.23±0.01 | 0.16±0.03** | 0.23±0.03 | 0.16±0.02*** |
| HEART WT | 0.12±0.01 | 0.12±0.01 | 0.11±0.01 | 0.10±0.01 |
| LIVER WT | 1.07±0.14 | 1.12±0.17 | 1.00±0.17 | 0.96±0.10 |
| TG | 50.9±18.7 | 67.6±26.4 | 52.4±16.2 | 72.5±16.8 |
| ALT | 24.5±8.8 | 34.8±6.9* | 19.9±6.4 | 41.6±17.3* |
| AST | 60.4±34.0 | 64.9±17.1 | 61.1±28.0 | 69.5±24.2 |
| Male mice | PI3K+/+ PF | PI3K+/+ EF | PI3K−/− PF | PI3K−/− EF |
| Number | 4 | 6 | 4 | 6 |
| INITIAL BW | 28.5±0.5 | 29.0±1.3 | 26.6±1.5 | 26.9±1.2 |
| FINAL BW | 31.5±1.0 | 28.5±2.0 | 29.2±1.3 | 27.8±0.8 |
| Δ BW | 3.0±0.8 | −0.6±0.8 | 2.6±0.4 | 0.9±0.8 |
| Avg. intake (ml/d) | ND | 28.1±1.2 | ND | 27.1±1.3 |
| MUSCLE WT | 0.28±0.01 | 0.17±0.02** | 0.28±0.01 | 0.16±0.01*** |
| HEART WT | 0.16±0.03 | 0.14±0.01 | 0.16±0.02 | 0.14±0.01 |
| LIVER WT | 1.45±0.02 | 1.50±0.15 | 1.37±0.15 | 1.46±0.17 |
| TG | 43.1±6.1 | 87.5±20.8 | 45.8±23.4 | 79.5±30.6 |
| ALT | 35.2±7.2 | 42.5±15.4* | 41.1±25.9 | 43.9±14.6 |
| AST | 59.5±11.8 | 64.2±18.3 | 71.7±52.9 | 58.1±10.0 |
BW body weight in gm.; TG Hepatic triglyceride content (mg/g liver tissue); AST serum aspartate amino transferase (U/L); serum ALT alanine amino transferase (U/L). All values are mean±SD.
p<0.001
p<0.01
p<0.05 vs. pair fed
Discussion
Ethanol impairs both mTORC1 and AMPK function without alterations in the canonical upstream kinase, Akt. Instead, ethanol increased targeted dephosphorylation of specific proteins via its B56 regulatory subunit. Depletion of PP2A reversed ethanol-induced inactivation of mTORC1, with functional consequences that included increased protein synthesis and reduced autophagy. Loss and gain of function studies in vitro in myotubes and in mice showed that ethanol activates PP2A by suppression of PI3Kγ activity. These data provide direct evidence that impaired protein homeostasis in response to ethanol is due to dephosphorylation of mTOR via activation of PP2A in skeletal muscle.
Ethanol exposure leads to impaired mTORC1 function leading to reduced protein synthesis which is observed in ethanol-fed rodents and skeletal muscle from alcoholic cirrhotic patients(4, 5, 27, 44, 45). Mechanistically our studies show that among the multiple processes regulating mTORC1 complex(8, 11, 23), ethanol mediates loss in protein synthesis by driving dephosphorylation of mTOR independent of the key upstream regulator, Akt(9, 24). Our comprehensive set of data also supports the role of mTORC1 rather than mTORC2 as noted from the lack of differences in phosphorylation of Akt at both phosphorylation sites. Ethanol also increases autophagy flux in myotubes and mouse skeletal muscle as demonstrated by increased LC3-II lipidation with associated loss of mTORC1 signaling, a negative regulator of autophagy(30). These findings show that dysregulated protein homeostasis in response to ethanol occurs through simultaneous loss of protein synthesis and increased autophagy.
Consistent with a nutrient-deprived like state(11), ethanol-treated myotubes or skeletal muscles from ethanol-fed mice or alcoholic cirrhotic patients have reduced ATP content. AMPK is a cellular energy sensor(12, 13) as elevated AMP/ADP following nutrient deprivation bind to AMPK resulting in phosphorylation and activation(10, 31) restoring energy homeostasis. AMPK is known to inhibit mTORC1 through phosphorylation of negative regulator TSC2(11, 32) or direct inhibitory phosphorylation of mTOR-associated protein Raptor(15), suggesting that mTORC1 function may be subservient to activated AMPK in conditions of nutrient deprivation(11, 12, 15, 31). However, despite reduced ATP content, no activation of AMPK was observed following ethanol treatment. Therefore, in contrast to the classical inverse regulation of mTORC1 by AMPK during nutrient deprivation (11, 15), both mTORC1 and AMPK are inactivated in response to ethanol.
Contrasting with classical phosphorylation-dependent activation of mTOR, we show that ethanol impairs mTOR function through active dephosphorylation. Similarly, AMPK is also dephosphorylated, showing a unique, yet comparable mechanism that underlies dysregulated protein homeostasis in response to ethanol. Although several phosphatases including PP2A, PP1 and PP2C have been implicated in the dephosphorylation of AMPK(46), less is known about dephosphorylation as a mechanism impairing mTORC1 function.
Although previous studies have reported kinase-independent regulation of mTORC1 and AMPK, less is known about the underlying mechanisms. Our data show increased PP2A activity in skeletal muscle cells following ethanol exposure. Studies have shown that ethanol activates PP1 in the lung(22), which could potentially dephosphorylate mTOR and AMPK. However, in skeletal muscle, PP2A dephosphorylates mTOR and AMPK in response to ethanol because inhibition of PP2A by fostriecin(34) reverses ethanol-induced mTOR and AMPK phosphorylation. Furthermore, PP2A knockdown restores downstream AMPK and mTORC1 signaling (reduction in LC3-II lipidation and preservation of P70S6K and 4E-BP1 phosphorylation respectively) accounting for increased protein synthesis despite ethanol exposure. These observations show that PP2A is a key phosphatase for AMPK and mTOR in skeletal muscle in response to ethanol exposure. Studies have shown that ethanol can either activate PP2A in rat hepatocytes(17) or inhibit protein phosphatase(20) suggesting context-specific responses. However, using multiple models, we have observed that ethanol through downstream mechanisms consistently activates PP2A in skeletal muscle. Even though we have not explored the role of other phosphatases including PP1(22), our data show that the observed effects of ethanol are primarily mediated via PP2A activation.
We show that ethanol increases PP2A activity and dephosphorylation of critical signaling molecules, contrasting with the paradigm that PP2A activity is a passive homeostasis maintaining process(47). There is evidence that PP2A activity can be acutely regulated(48) by methylation of the PP2A catalytic subunit that increases activity(38) or tyrosine phosphorylation that inhibits PP2A activity(35). Also, increased association of PP2A with target proteins via the PP2A regulatory B-subunit(49) leads to dephosphorylation of target proteins, while inhibition of PP2A activity occurs by binding of endogenous inhibitors of PP2A(25, 39). Consistent with acute regulation following exposure to ethanol, there was a significant increase in methylated PP2A with concomitant decrease in phospho-PP2A. This suggests that the phosphorylation-methylation axis(25, 37, 39) is responsible for ethanol-mediated activation of PP2A because inhibition of PI3Kγ by ethanol releases the PP2A catalytic subunit from its regulatory control with methylation causing increased PP2A activity. Increased interaction of the PP2A regulatory B-subunit with AMPK and mTOR showed augmented targeting of PP2A causes dephosphorylation of regulatory molecules. Simultaneously, we also observed reduced I2-PP2A interaction with PP2A following ethanol treatment, supporting elevated PP2A activity. Thus, ethanol increases PP2A activity by engaging multiple pathways providing the mechanistic underpinnings for previous reports of ethanol activating PP2A in the heart(19) and hepatocytes(17, 21) with inhibition of AMPK and increased phosphorylation of riboS6, an mTORC1 target(21).
Given that PI3Kγ can mediate acute effects, our findings of reduced phospho-I2PP2A following ethanol exposure suggests that PI3Kγ-dependent mechanisms could be the major regulator of PP2A activity. Our findings that ethanol-fed PI3Kγ−/− mice have elevated PP2A activity and LC3-II lipidation with colchicine (suggesting increased autophagy flux) support this inference. PI3Kγ is a key upstream regulator of Akt(41, 43) and despite reduced PI3Kγ activity, Akt activation was unaltered in response to ethanol, showing that loss of mTOR phosphorylation was due to dephosphorylation by activation of PP2A through inhibition of PI3Kγ. Consistently, overexpression of wild-type PI3Kγ in myotubes reduced PP2A activity and reversed phosphorylation of proteins involved in protein synthesis and in AMPK. Furthermore, expression of wild-type PI3Kγ leads to increased association of I2-PP2A with PP2A and reduction in mTOR- and AMPK-associated PP2A activity. Corroborating previous studies(39), our findings suggest that increased I2-PP2A binding to PP2A may reduce the interaction with the regulatory B-subunit and subsequent interaction with target proteins. Based on our current findings and that of others(48), we believe that the acute regulation of PP2A by I2-PP2A could be the key mechanism present in skeletal muscle. In this context, ethanol inhibits PI3Kγ activity resulting in activation of PP2A that mediates dephosphorylation of target proteins and contrasting with classical kinase activation, ethanol activates PP2A with dysregulated protein homeostasis. We also did not observe any sex differences in mice phenotype or signaling responses to ethanol that was consistent with previous reports in human patients with ALD.
Our studies show that exposure to ethanol results in decreased ATP content, mTORC1 signaling, and protein synthesis, while autophagy is increased, findings characteristic of a cellular starvation state. Paradoxically, these perturbations are associated with no activation of AMPK, characteristic of a fed state. We call this unique phenotype of simultaneous inactivation of mTOR and AMPK by ethanol a “pseudofed state” because the functional responses are those of impaired mTORC1 signaling observed in a fasted state. This “pseudofed state” is phenotypically opposite to previous reports of a “pseudofasted state” wherein AMPK and mTOR are activated simultaneously in response to olanzapine(50). We also noted that ethanol-activated PP2A dephosphorylates AMPK, overriding the classical regulation despite energy surfeit/deficiency, and that PP2A activation is target specific. These findings highlight the formerly underappreciated idea that acute regulation of the PI3Kγ-PP2A axis mediate functional and phenotypic outcomes by inhibiting protein synthesis and increasing autophagy in response to ethanol (Fig. 7).
Fig. 7.
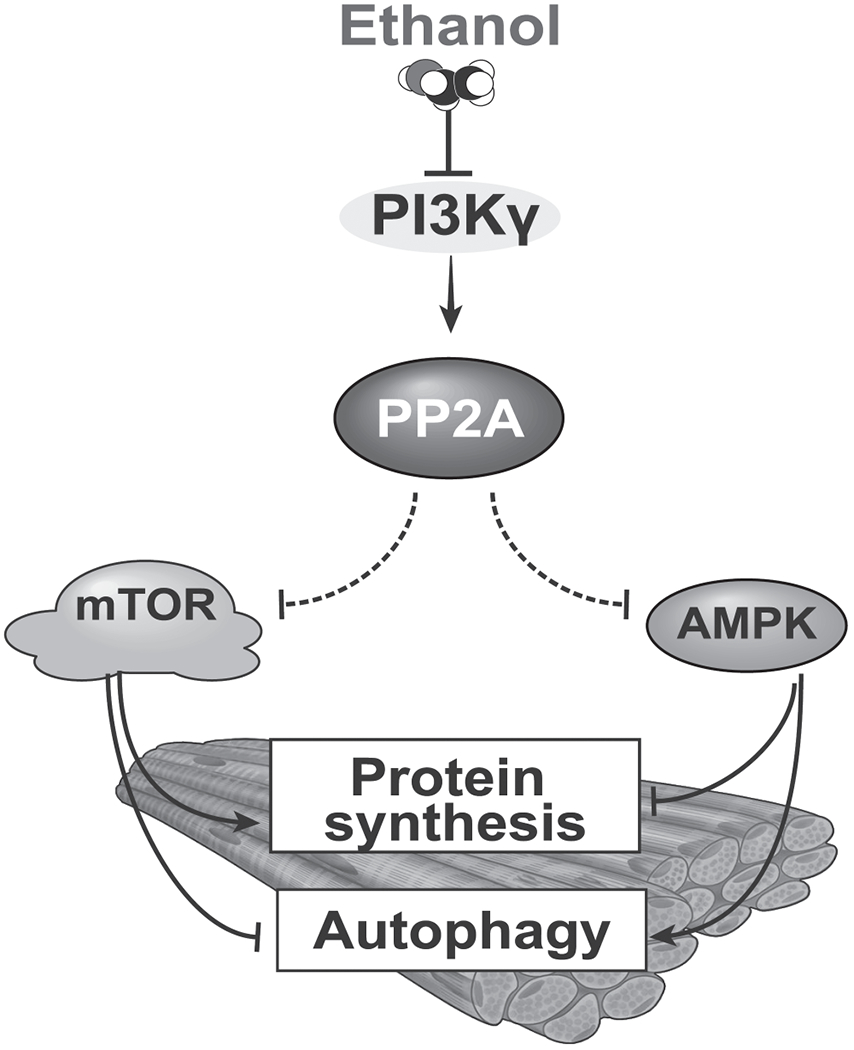
Schematic model of ethanol mediated PI3KY-PP2A axis with phosphatase dependent regulation of regulatory kinases.
The clinical significance of these data are related to the high frequency, greater severity and more rapid rate of progression of sarcopenia in ALD(3). Identifying the mechanistic basis for molecular and functional perturbations allows for development of novel targeted therapeutic approaches in these patients.
Supplementary Material
Acknowledgements
Phospho I2 PP2A were generated by the hybridoma core and the substrate for PP2A was generated by the biotechnology core at the Cleveland Clinic.
Ms. Lakshmi Veera assisted with proofreading and organizing the data.
Financial support statement:
Funded in part by: R21 AR 071046 (SD, GD), R21 AA022742 (SD); RO1 GM119174 (SD); RO1 DK113196 (SD); P50 AA024333 (LN, SD); UO1 AA021890 (LN, SD); R56HL141744 (SD); UO1 DK061732; T32 DK083251 (NW); and the American College of Gastroenterology Clinical Research Award (NW).\
Footnotes
Conflict of interest statement: The authors have no conflicts to report.
References
- 1.Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, Naga Prasad SV, Dasarathy S. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy 2014;10:677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guirguis J, Chhatwal J, Dasarathy J, Rivas J, McMichael D, Nagy LE, McCullough AJ, et al. Clinical impact of alcohol-related cirrhosis in the next decade: estimates based on current epidemiological trends in the United States. Alcohol Clin Exp Res 2015;39:2085–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch N, Dasarathy J, Runkana A, Penumatsa R, Bellar A, Reen J, Rotroff D, et al. Continued muscle loss increases mortality in cirrhosis: Impact of aetiology of liver disease. Liver Int 2020;40:1178–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steiner JL, Lang CH. Dysregulation of skeletal muscle protein metabolism by alcohol. Am J Physiol Endocrinol Metab 2015;308:E699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Preedy VR, Peters TJ. The effect of chronic ethanol ingestion on protein metabolism in type-I- and type-II-fibre-rich skeletal muscles of the rat. Biochem J 1988;254:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz-Villanueva JF, Diaz-Molina R, Garcia-Gonzalez V. Protein Folding and Mechanisms of Proteostasis. Int J Mol Sci 2015;16:17193–17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 2013;36:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 1999;344 Pt 2:427–431. [PMC free article] [PubMed] [Google Scholar]
- 10.Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol 2015;33:1–7. [DOI] [PubMed] [Google Scholar]
- 11.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 2006;281:34870–34879. [DOI] [PubMed] [Google Scholar]
- 14.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem 2004;279:32771–32779. [DOI] [PubMed] [Google Scholar]
- 15.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008;30:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter T Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 1995;80:225–236. [DOI] [PubMed] [Google Scholar]
- 17.Liangpunsakul S, Sozio MS, Shin E, Zhao Z, Xu Y, Ross RA, Zeng Y, et al. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am J Physiol Gastrointest Liver Physiol 2010;298:G1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004;127:1798–1808. [DOI] [PubMed] [Google Scholar]
- 19.Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. J Am Coll Cardiol 2009;54:2187–2196. [DOI] [PubMed] [Google Scholar]
- 20.Higashi K, Hoshino M, Nomura T, Saso K, Ito M, Hoek JB. Interaction of protein phosphatases and ethanol on phospholipase C-mediated intracellular signal transduction processes in rat hepatocytes: role of protein kinase A. Alcohol Clin Exp Res 1996;20:320A–324A. [PubMed] [Google Scholar]
- 21.Noh BK, Lee JK, Jun HJ, Lee JH, Jia Y, Hoang MH, Kim JW, et al. Restoration of autophagy by puerarin in ethanol-treated hepatocytes via the activation of AMP-activated protein kinase. Biochem Biophys Res Commun 2011;414:361–366. [DOI] [PubMed] [Google Scholar]
- 22.Price ME, Pavlik JA, Sisson JH, Wyatt TA. Inhibition of protein phosphatase 1 reverses alcohol-induced ciliary dysfunction. Am J Physiol Lung Cell Mol Physiol 2015;308:L577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 2000;60:3504–3513. [PubMed] [Google Scholar]
- 24.Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC Jr. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci U S A 1998;95:7772–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasudevan NT, Mohan ML, Gupta MK, Hussain AK, Naga Prasad SV. Inhibition of protein phosphatase 2A activity by PI3Kgamma regulates beta-adrenergic receptor function. Mol Cell 2011;41:636–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohan ML, Naga Prasad SV. Scaffolding Function of PI3Kgamma Emerges from Enzyme’s Shadow. J Mol Biol 2017;429:763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsien C, Davuluri G, Singh D, Allawy A, Ten Have GA, Thapaliya S, Schulze JM, et al. Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology 2015;61:2018–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu J, Tsien C, Thapalaya S, Narayanan A, Weihl CC, Ching JK, Eghtesad B, et al. Hyperammonemia-mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am J Physiol Endocrinol Metab 2012;303:E983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davuluri G, Krokowski D, Guan BJ, Kumar A, Thapaliya S, Singh D, Hatzoglou M, et al. Metabolic adaptation of skeletal muscle to hyperammonemia drives the beneficial effects of l-leucine in cirrhosis. J Hepatol 2016;65:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009;20:1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross FA, Jensen TE, Hardie DG. Differential regulation by AMP and ADP of AMPK complexes containing different gamma subunit isoforms. Biochem J 2016;473:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577–590. [DOI] [PubMed] [Google Scholar]
- 33.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001;81:807–869. [DOI] [PubMed] [Google Scholar]
- 34.Walsh AH, Cheng A, Honkanen RE. Fostriecin, an antitumor antibiotic with inhibitory activity against serine/threonine protein phosphatases types 1 (PP1) and 2A (PP2A), is highly selective for PP2A. FEBS Lett 1997;416:230–234. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992;257:1261–1264. [DOI] [PubMed] [Google Scholar]
- 36.Lee J, Stock J. Protein phosphatase 2A catalytic subunit is methyl-esterified at its carboxyl terminus by a novel methyltransferase. J Biol Chem 1993;268:19192–19195. [PubMed] [Google Scholar]
- 37.Wu J, Tolstykh T, Lee J, Boyd K, Stock JB, Broach JR. Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. EMBO J 2000;19:5672–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie H, Clarke S. Methyl esterification of C-terminal leucine residues in cytosolic 36-kDa polypeptides of bovine brain. A novel eucaryotic protein carboxyl methylation reaction. J Biol Chem 1993;268:13364–13371. [PubMed] [Google Scholar]
- 39.Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 1995;34:1988–1996. [DOI] [PubMed] [Google Scholar]
- 40.Li M, Makkinje A, Damuni Z. Molecular identification of I1PP2A, a novel potent heat-stable inhibitor protein of protein phosphatase 2A. Biochemistry 1996;35:6998–7002. [DOI] [PubMed] [Google Scholar]
- 41.Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004;118:375–387. [DOI] [PubMed] [Google Scholar]
- 42.Mohan ML, Chatterjee A, Ganapathy S, Mukherjee S, Srikanthan S, Jolly GP, Anand RS, et al. Noncanonical regulation of insulin-mediated ERK activation by phosphoinositide 3-kinase gamma. Mol Biol Cell 2017;28:3112–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stoyanova S, Bulgarelli-Leva G, Kirsch C, Hanck T, Klinger R, Wetzker R, Wymann MP. Lipid kinase and protein kinase activities of G-protein-coupled phosphoinositide 3-kinase gamma: structure-activity analysis and interactions with wortmannin. Biochem J 1997;324 (Pt 2):489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lang CH, Wu D, Frost RA, Jefferson LS, Kimball SR, Vary TC. Inhibition of muscle protein synthesis by alcohol is associated with modulation of eIF2B and eIF4E. Am J Physiol 1999;277:E268–276. [DOI] [PubMed] [Google Scholar]
- 45.Hong-Brown LQ, Frost RA, Lang CH. Alcohol impairs protein synthesis and degradation in cultured skeletal muscle cells. Alcohol Clin Exp Res 2001;25:1373–1382. [PubMed] [Google Scholar]
- 46.Joseph BK, Liu HY, Francisco J, Pandya D, Donigan M, Gallo-Ebert C, Giordano C, et al. Inhibition of AMP Kinase by the Protein Phosphatase 2A Heterotrimer, PP2APpp2r2d. J Biol Chem 2015;290:10588–10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohan ML, Vasudevan NT, Gupta MK, Martelli EE, Naga Prasad SV. G-protein coupled receptor resensitization-appreciating the balancing act of receptor function. Curr Mol Pharmacol 2012. [PMC free article] [PubMed] [Google Scholar]
- 48.Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J 2016;283:1004–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem 1996;271:22081–22089. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt RH, Jokinen JD, Massey VL, Falkner KC, Shi X, Yin X, Zhang X, et al. Olanzapine activates hepatic mammalian target of rapamycin: new mechanistic insight into metabolic dysregulation with atypical antipsychotic drugs. J Pharmacol Exp Ther 2013;347:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.