

Abstract
Current interventions fail to recover injured myocardium after infarction and prompt the need for development of cardioprotective strategies. Of increasing interest is the therapeutic use of microRNAs to control gene expression through specific targeting of mRNAs. In this Review, we discuss current microRNA-based therapeutic strategies, describing the outcomes and limitations of key microRNAs with a focus on target cell types and molecular pathways. Last, we offer a perspective on the outlook of microRNA therapies for myocardial infarction, highlighting the outstanding challenges and emerging strategies.
INTRODUCTION
Heart disease remains as the leading cause of mortality worldwide, with myocardial infarction (MI) affecting more than 700,000 individuals annually in the United States alone (1). Because the adult heart lacks ability to innately repair and regenerate after injury, MI results in permanent loss of myocardial cells (2). The damaged heart undergoes pathological remodeling, leading to reduced contractile function and often heart failure (3). Standard therapies mainly focus on revascularizating the occluded artery to salvage as much of the myocardium as possible but fail to adequately recover injured myocardium. Any advances in the treatment of MI would thus have major clinical significance.
The human heart is composed of an array of different cell types including cardiomyocytes (CMs), cardiac fibroblasts (CFs), endothelial cells (ECs), and immune cells. Each cell type contributes to cardiac function in a distinct way, with intercellular communication and coordination being vital to maintaining normal organ function (Fig. 1). The pathological changes in cardiac remodeling after MI involve each of these cell types and multiple molecular mechanisms during four partially overlapping phases: the inflammatory, proliferative, maturation, and remodeling phases (3). The inflammatory phase is initiated by massive cell death in the infarct area. Although CFs degrade the extracellular matrix (ECM), changes in EC-mediated vascular permeability allow immune cells to migrate into the injured area and facilitate clearance of damaged cells. In the proliferative phase, inflammation is dampened by macrophage phenotypic switching, and reparative processes begin. Fibroblasts and ECs proliferate, deposit collagen, and establish a new microvascular network in the infarct area. In the maturation phase, ECM proteins secreted by activated fibroblasts undergo cross-linking to form a stable scar. Last, during long-term remodeling, maladaptive processes including cardiomyocyte pathological hypertrophy, remote fibrosis, and capillary rarefaction occur to contend with the stresses arising from the failing heart (Fig. 1).
Fig. 1. Summary of myocardial infarct progression with a focus on the role of each cell type.
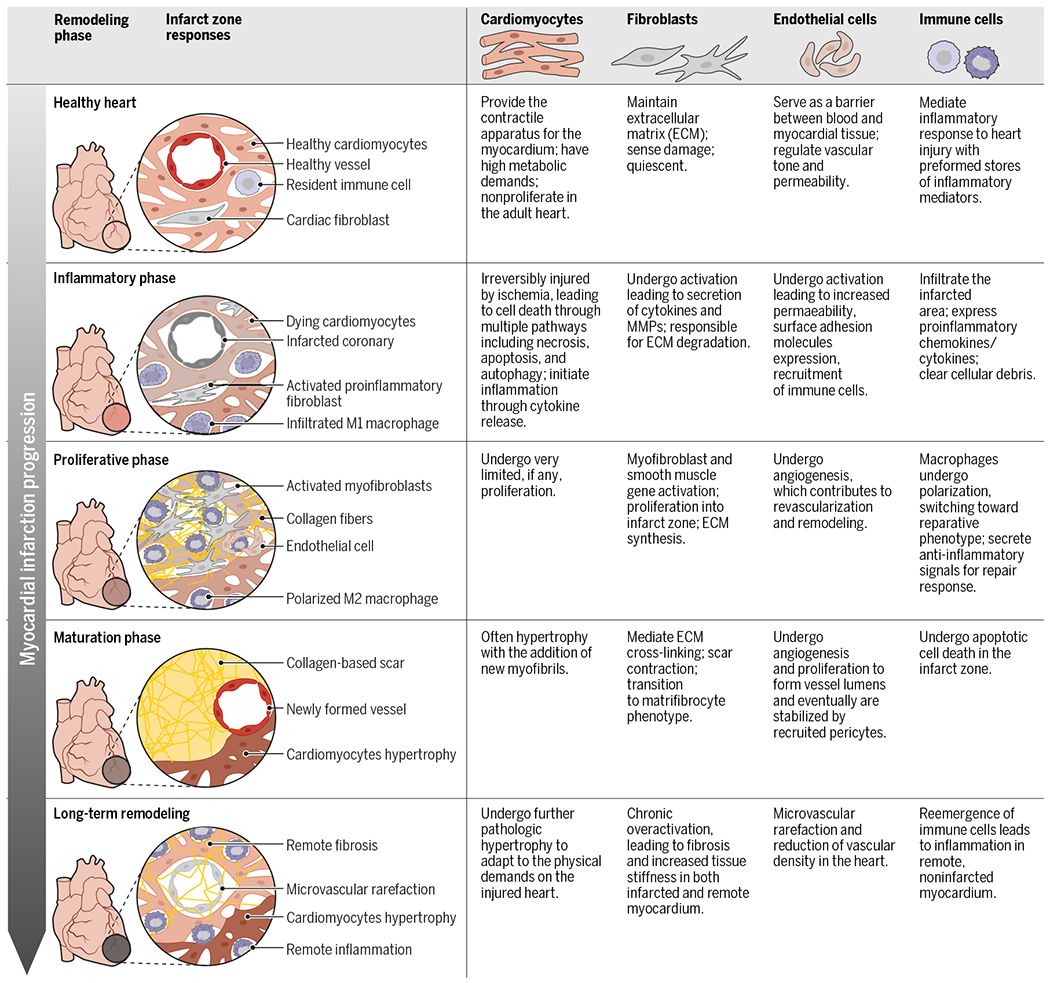
Immediately after infarction, the inflammatory phase begins and is characterized by irreversible cell death of all cell types in the infarct and the initiation of the inflammatory process through cytokine release. Next, the proliferative phase is characterized by the activation of CFs and the initiation of scar development. Macrophages also undergo polarization and switch to an anti-inflammatory phenotype during this phase. The maturation phase is characterized by cardiomyocyte hypertrophy in response to increased cardiac demand and the maturation of the scar that forms over the infarcted area. Last, long-term remodeling is characterized by pathological changes including maladaptive cardiomyocyte hypertrophy, chronic overactivation of CFs, decreased vascular density, and reemergence of an inflammatory response.
Recently, microRNAs (miRNAs) have emerged as a promising therapy for MI. miRNAs are small noncoding RNAs that regulate gene expression at the posttranscriptional level (4). Mature, single-stranded miRNAs are incorporated into miRNA-induced silencing complexes, the functional unit that targets mRNAs with near-perfect base pairing to inhibit gene expression (5). Known to interact with the majority of mammalian protein-coding genes, miRNAs are powerful mediators of a diverse spectrum of processes, both during normal heart development and physiology and during cardiovascular disease progression (6). miRNAs are implicated in every phase of MI progression; in response to ischemia, miRNAs in both mouse and human hearts have been shown to be dysregulated, contributing to the progression of many pathological processes.
For treatment of MI, miRNAs represent particularly attractive therapeutic targets due to several unique characteristics. Most important is the pleotropic ability of a single miRNA to regulate multiple pathologically disrupted biological pathways across different cell types (7). This is in stark contrast to traditional drug-based approaches that target singular molecules and pathways. Such a pleiotropic approach is especially powerful for treatment of MI because injury is not instigated by a single genetic link or biological process but rather by multiple coordinated processes. In addition, miRNAs are ideal therapeutic targets because they are small, precisely defined nucleic sequences for which mimics or antisense oligonucleotides (ASOs) can effectively and efficiently be designed with high affinity and specificity. Last, when compared to cell-based approaches, miRNA therapies may provide similar benefits without the challenges of immune rejection and poor cell engraftment.
Although miRNA therapies have potential for treating MI, a major challenge that limits their clinical advancement is a lack of understanding of the molecular mechanisms behind their therapeutic properties. Specifically, the pleotropic effects of a miRNA on each cell type and on different biological pathways within the heart must be carefully delineated to facilitate clinical translation. In this Review, we examine recently published studies describing the therapeutic manipulation of miRNAs in the treatment of MI. We describe the effect of miRNAs on each cell type and how miRNAs directly target molecular pathways to modulate cell type–specific behavior. Last, we discuss key prospects and challenges for developing miRNA therapies.
CHARACTERISTICS OF miRNA THERAPIES
Focusing on literature within the past 3 years and foundational studies from the past 10 years, we identified a total of 213 relevant studies detailing 116 unique miRNAs (Fig. 2). Therapeutic miRNA targets were identified by two methods: (i) miRNA profiling after MI, which revealed substantial dysregulation of miRNAs natively expressed in the heart; and (ii) miRNA screening in in vitro injury models, which identified potential proregenerative miRNAs not natively expressed in the heart. To understand cell type–specific effects of miRNA therapies, we organized the reviewed miRNAs by the cell types they target. For each study, we extracted a set of characteristics describing each miRNA. miRNAs were defined as beneficial if treatment with miRNA mimics resulted in improved recovery after MI and as harmful if treatment with miRNA inhibitors resulted in improved recovery after MI. We noted the model systems each study used in vitro and in vivo as well as the delivery of the miRNA therapies (miRNA formulation and the method of administration). For each miRNA studied, we detail the target pathway and the specific target molecule.
Fig. 2. Summary of miRNAs surveyed in this Review.
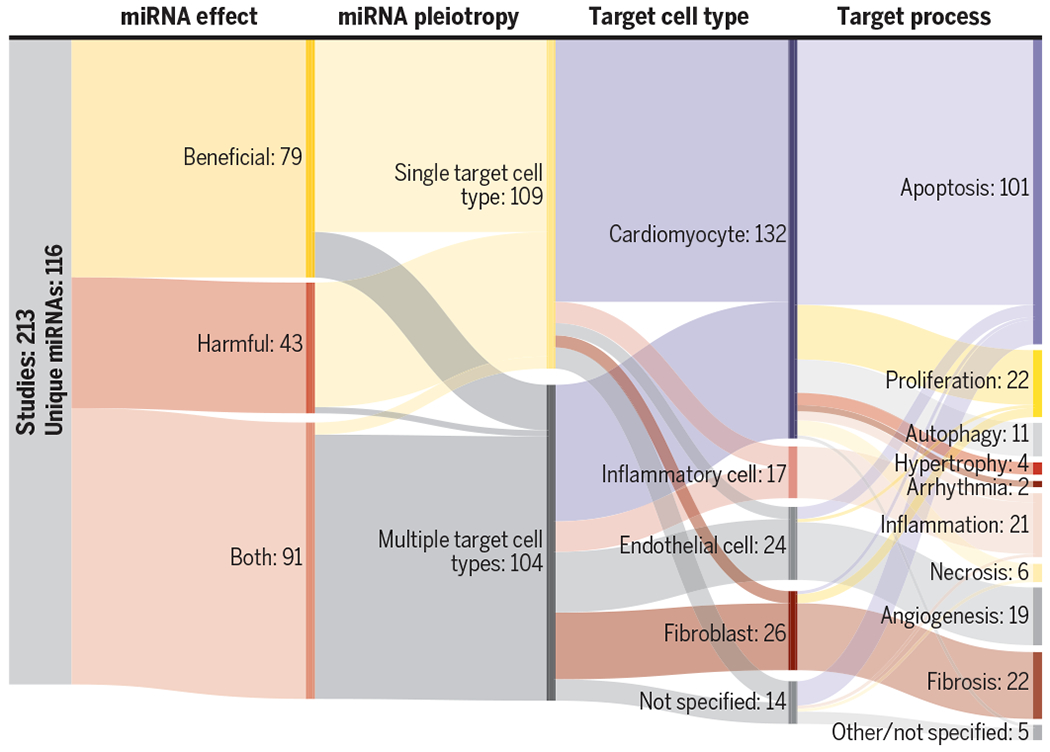
For each study, in this Review, we categorized the overall effect of the miRNA studied, whether the miRNA was shown to affect multiple cell types (pleiotropy), which cell types were studied, and the physiologic process that the miRNA was shown to target. The numbers following each division represents the number of studies that is in that category.
Using these characteristics, we assigned a clinical applicability score representing a qualitative measure of the evidence supporting the therapeutic application of each miRNA reviewed. This score is denoted by the footnotes in Table 1. “*” indicates therapeutic efficacy in in vitro cell-based models; “**” indicates therapeutic efficacy in in vivo models with target pathway identification and characterization; and “***” indicates therapeutic efficacy in multiple model systems and well-defined mechanisms of action including comprehensively validated direct targets. Table 1 presents a select list of key miRNAs for each cell type. The complete list of surveyed miRNAs is presented in table S1 and summarized in table S2.
Table 1. Select cell type–specific miRNA therapies in the treatment of MI.
Complete list of miRNAs reviewed in table S1. IRI, ischemia-reperfusion injury; AngII, angiotensin II; TAC, transverse aortic constriction; IV, intravenous; IM, intramyocardial; IP, intraperitoneal; I. Ventr, intraventricular; EV, extracellular vesicle; Transf, transfection; Lenti, lentiviral; EndMT, endothelial-to-mesenchymal transition; Mito, mitochondrial; ROS, reactive oxygen species.
| miRNA | Effect | Injury model | Delivery method | Target process | Target pathway | Target molecule | Clinical application | Reference |
|---|---|---|---|---|---|---|---|---|
| Cardiomyocytes | ||||||||
|
| ||||||||
| miR-1 | Harmful | In vivo IRI | IV | Apoptosis | PKC apoptosis | PKCε | ** | (43) |
|
| ||||||||
| miR-9 | Harmful | In vivo ischemia | IM | Proliferation | FSTL | FSTL | *** | (73) |
|
| ||||||||
| miR-22 | Harmful | In vivo ischemia | IP | Autophagy | Autophagy | PPARα | *** | (59) |
|
| ||||||||
| miR-302/367 | Beneficial | In vivo ischemia | IV | Proliferation | Hippo-YAP | MST1 | *** | (69) |
|
| ||||||||
| miR-762 | Harmful | In vivo IRI | IV | Apoptosis | ROS production | ND2 | *** | (49) |
|
| ||||||||
| miR-873 | Beneficial | In vivo IRI | IV | Necrosis | Necroptosis | RIPK1/RIPK3 | *** | (53) |
|
| ||||||||
| Let-7a | Beneficial | In vivo AngII | I. Ventr. | Hypertrophy | Undefined | CaM | *** | (80) |
| Cardiac fibroblasts | ||||||||
|
| ||||||||
| miR-21 | Harmful | In vivo ischemia | IM | Fibrosis | ERK/MAPK | SPRY1 | *** | (112) |
|
| ||||||||
| miR-21 | Harmful | In vivo TAC | IV | Apoptosis | ERK/MAPK | SPRY1 | *** | (111) |
|
| ||||||||
| miR-92a | Harmful | In vitro TGF-β | EV | Fibrosis | TGF-β | SMAD7 | * | (106) |
|
| ||||||||
| miR-101 | Beneficial | In vivo ischemia | I. Ventr. | Fibrosis | TGF-β | cFOS | *** | (96) |
|
| ||||||||
| miR-155 | Beneficial | In vivo genetic | NA | Proliferation | RAS | SOS1 | ** | (178) |
| Endothelial cells | ||||||||
|
| ||||||||
| miR-32 | Harmful | In vitro | Transf. | Proliferation | Undefined | KLF2 | * | (228) |
|
| ||||||||
| miR-155 | Harmful | In vivo ischemia | IM | Angiogenesis | AMPK | RAC1 | *** | (142) |
|
| ||||||||
| miR-324 | Beneficial | In vitro H2O2 | Transf. | Apoptosis | Mitofission | MtFR1 (indirect) | * | (135) |
|
| ||||||||
| miR-532 | Beneficial | In vivo ischemia | IM | Angiogenesis | EndMT | PRSS23 | *** | (151) |
| Inflammatory cells | ||||||||
|
| ||||||||
| miR-24 | Beneficial | In vivo ischemia | EV | Inflammatory infiltration | Intrinsic pathway | BIM | *** | (161) |
|
| ||||||||
| miR-155 | Harmful | In vivo ischemia | IV | Inflammatory activation | NF-κB | SOCS1 | *** | (175) |
|
| ||||||||
| miR-182 | Beneficial | In vivo IRI | EV | Inflammatory resolution | PI3k/AKT/mTOR | TLR4 | ** | (162) |
|
| ||||||||
| miR-224 | Beneficial | In vivo ischemia | Lenti. | Inflammatory activation | TGF-β | Undefined | * | (157) |
Indicates therapeutic efficacy in in vitro cell-based models.
Indicates therapeutic efficacy in in vitro and in vivo models with target pathway identification and characterization.
Indicates therapeutic efficacy in multiple model systems and well-defined mechanisms of action including comprehensively validated direct targets.
Most miRNAs play both beneficial and harmful roles in the progression of MI. In addition, a majority of miRNAs affect multiple cell types, thus demonstrating the important pleiotropic effects of miRNAs when used in cardiac therapy. Here, we first highlight post-MI events by cell type, briefly describing how key processes in each cell type affect their phenotype and function. After providing this contextual framework, we examine the recent advances in miRNA therapies targeting each cell type.
ROLE OF miRNAs IN CARDIOMYOCYTE RESPONSE TO MYOCARDIAL INFARCTION
Cardiomyocytes are the functional cells of the heart, responsible for generating contractile force. Because of their large energy demands, cardiomyocytes are highly susceptible to death upon loss of blood supply. Cardiomyocytes have limited proliferative ability, and those lost during MI cannot be replaced (8). Thus, two major approaches to therapeutically target cardiomyocytes after MI are prevention of cardiomyocyte cell death after ischemic stress and induction of cardiomyocyte proliferation after resolution of injury.
Cardiomyocyte response to myocardial infarction
Apoptosis and necrosis
The primary pathways of regulated cardiomyocyte cell death during MI are apoptosis and necrosis, both of which have distinct molecular mechanisms and cellular conditions. Mechanistically, apoptosis and necrosis can both be subdivided into mitochondria- and receptor-mediated pathways, known as the intrinsic and extrinsic pathways during apoptosis and as mitochondrial necrosis and necroptosis during necrosis. During MI, the intrinsic pathway is initiated when free radical generation and low adenosine triphosphate (ATP) lead to mitochondrial stress and subsequent perturbation of the B cell lymphoma 2 (BCL-2) family of proteins. Simultaneously, mitochondrial necrosis is initiated when intracellular Ca2+ overload alters the dynamics of the mitochondrial membrane, leading to a disruption in permeability. This results in a rapid decrease in ATP, which prevents the cell from carrying out necessary repair functions, leading to organelle dysfunction and plasma membrane rupture. The extrinsic pathway of apoptosis and necroptosis are both triggered during MI by release of stimuli outside the cell that activates death receptors on the cell surface such as Fas ligand activating Fas receptor (FasR) and tumor necrosis factor–α (TNFα) activating TNF receptor 1 (TNFR1) (9).
Autophagy
In response to the ischemic or metabolic stress during MI, cardiomyocytes can activate the process of autophagy, which allows them to catabolize damaged or dysfunctional macromolecular structures by lysosomal degradation to maintain cellular homeostasis. Primary players in cardiomyocyte autophagy include the mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) pathways (10). Although undoubtedly critical in cardiomyocyte response to MI, the role of autophagy in mediating the extent of myocardial injury is uncertain.
Proliferation and remodeling
Historically, adult cardiomyocytes have been described as postmitotic, but this paradigm has shifted, with studies indicating that cardiomyocytes proliferate to a small extent in the adult myocardium (11). Although limited, exogenously promoted cardiomyocyte proliferation has become a potential therapeutic strategy for heart failure. Over the past decade, several promising targets have emerged that induce cardiomyocyte proliferation, including Hippo–Yes-associated protein (YAP), homeodomain-only protein (HOPX), and follistatin-related protein 1 (FSTL) (12–14).
Because of the loss of functional cells after MI, cardiomyocytes undergo hypertrophy and morphological changes to adapt to the physical demands of the injured heart. Although initially compensatory, prolonged increases in functional demands eventually lead to pathological remodeling and a decrease in cardiomyocyte function. Infarcted tissue also interrupts gap junctions between cardiomyocytes, causing aberrant electrical conduction leading to arrhythmias (15).
Recent advances in miRNA therapies targeting cardiomyocytes
Most studies (132 of 213 studies) describing the therapeutic application of miRNAs to MI have cardiomyocytes as the primary target cell type. These studies have focused on identifying and using miRNAs capable of either preventing cardiomyocyte death (88 of 132 studies) or promoting cardiomyocyte proliferation (18 of 132 studies). Here, we summarize key therapeutic miRNAs and their mechanisms of action in cardiomyocytes (Fig. 3).
Fig. 3. Summary of miRNAs that target cardiomyocytes after MI.
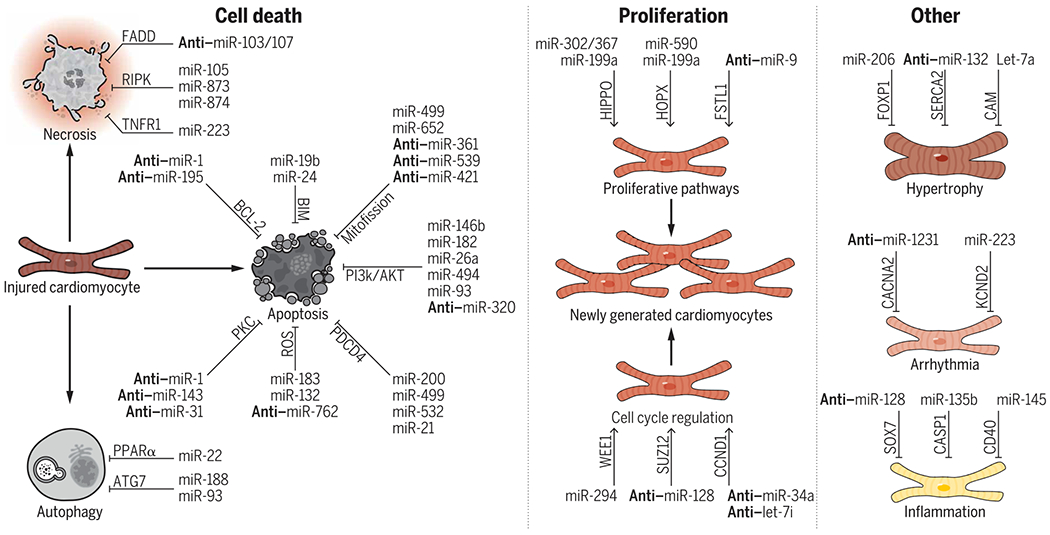
miRNAs have been shown to regulate apoptosis, necrosis, and autophagy-mediated cardiomyocyte cell death. For cardiomyocyte proliferation, miRNAs have been shown to modulate multiple proliferative pathways as well as the cell cycle directly. Last, miRNAs have also been shown to regulate important processes including hypertrophy, arrhythmia, and inflammation. Select well-characterized miRNAs, their targets (written above the arrows), and the processes that they regulate are shown (i.e., miR-19b prevents apoptosis through BIM signaling). miRNAs prefaced with “anti-” denote that the inhibition of the miRNA is therapeutic (i.e., the inhibition of miR-1 prevents apoptosis through BCL-2 signaling). For the complete list of miRNAs reviewed that affect cardiomyocytes, please see table S1.
Apoptosis
Many miRNAs have been identified that regulate components of apoptosis pathways, with beneficial miRNAs inhibiting these pathways to prevent cardiomyocyte apoptosis and harmful miRNAs inducing apoptosis after injury. In early apoptosis, mitochondrial fission leads to activation of mitochondria-mediated cardiomyocyte death. In a series of studies, Wang and colleagues (16) described five miRNAs that target components of mitochondrial fission machinery. They identified two beneficial miRNAs, miR-499 and miR-652, which prevent mitochondrial fission. miR-499 directly inhibits both the α and β isoforms of the calcineurin catalytic subunit, thereby decreasing the accumulation of dynamin-related protein 1 (DRP1), a guanosine triphosphatase required for mitochondrial fission (16). miR-652 inhibits the proapoptotic mitochondrial membrane protein MTP18 (17). In contrast, miR-361 and miR-539 were identified as harmful miRNAs that inhibit prohibitins 1 and 2, respectively, increasing mitochondrial fission and apoptosis after MI (18, 19). miR-421 was also identified as harmful after MI as it directly targets the mitochondrial Ser/Thr kinase PTEN induced kinase 1 (PINK1), which inhibits mitochondrial fission (20).
Harmful miRNAs including miR-195 and miR-1 have been shown to directly inhibit the antiapoptotic protein BCL-2 that regulates the intrinsic pathway (21, 22). Alternatively, beneficial miRNAs such as miR-19b and miR-24 have been shown to directly inhibit the proapoptotic protein Bcl-2-like protein 11 (BIM) (23, 24). In a foundational study, Qian et al. (24) demonstrated that the overexpression of miR-24 via intramyocardial injection of a miR-24 mimic attenuated ischemia-induced injury and restored cardiac function by directly reducing BIM expression. miRNAs affect other components of the intrinsic pathway: For example, miR-125b inhibits proapoptotic Bcl-2 homologous antagonist/killer 1 (BAK1), miR-17 inhibits proapoptotic apoptotic protease activating factor-1 (APAF-1), and miR-27a inhibits Bcl-2 interaction protein 3 (BNIP3) (25–27). To modulate the extrinsic pathway, miR-133b can down-regulate the death receptor FasR, and the effector caspase-3 can be inhibited by miR-1192 (28, 29). Last, miR-327 has been shown to inhibit apoptosis repressor with caspase recruitment domain (ARC), a potent repressor of the intrinsic and extrinsic signaling pathways (30).
In cardiomyocytes, multiple pathways respond to ischemic stress and injury after MI and ultimately determine whether the cell undergoes apoptosis. The PI3k/AKT pathway plays a major role in controlling cell survival and the inhibition of programmed cell death through stimulatory phosphorylation of prosurvival genes and the inhibitory phosphorylation of proapoptotic genes (31). Many miRNAs modulate the PI3k/AKT pathway in cardiomyocytes after MI. Specifically, the central AKT inhibitor phosphatase and tensin homolog (PTEN) can be directly targeted by miRNAs miR-146b, miR-182, miR-26a, miR-494, and miR-93 (32–35). Song et al. (36) demonstrated that miR-320 directly inhibits the ligand insulin growth factor 1 (IGF-1) after ischemia-reperfusion injury (IRI), thus preventing IGF receptor-mediated activation of the PI3k/AKT pathway. Restoring IGF-1 function using a lentivirus expressing miR-320 inhibitor led to a decrease in the number of apoptotic cardiomyocytes and preserved cardiac function (36). Another notable pathway regulating apoptosis centers around the molecule programmed cell death 4 (PDCD4), which is up-regulated during apoptosis and functions as a proapoptotic inhibitor of gene transcription and translation. During MI, many miRNAs directly inhibit PDCD4 and prevent apoptosis in cardiomyocytes including miR-200, miR-499, miR-532, and, importantly, miR-21, a miRNA with notable pleiotropic effects (37–42).
Additional pathways implicated in cardiomyocyte apoptosis include protein kinase C (PKC), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Notch, and reactive oxygen species (ROS) homeostasis. In PKC-mediated apoptosis, the harmful miRNAs miR-1, miR-143, and miR-31 have been shown to directly target antiapoptotic protein kinase C epsilon (PRKCE), thereby leading to increased cardiomyocyte apoptosis (43–45). In NF-κB signaling, miR-145a exerts beneficial effects by inhibiting early growth response 1 (EGR1)–mediated NF-κB activation. In Notch signaling, the harmful miRNAs miR-363 and miR-429 directly inhibit the Notch 1 receptor, preventing Notch-mediated antiapoptotic signaling (46, 47). In contrast, miR-322 targets the Notch inhibitor FBXW7, up-regulating Notch signaling and preventing apoptosis after MI (48). In the regulation of ROS, miRNAs can directly affect mitochondrial respiration. For example, the harmful miR-762 inhibits the mitochondrial nicotinamide adenine dinucleotide + hydrogen (NADH) dehydrogenase subunit ND2 leading to increased ROS production, whereas the beneficial miR-183 inhibits the mitochondrial membrane ion channels voltage-dependent anion channel 1 (VDAC1) leading to decreased ROS production (49, 50). In addition, Su et al. (51) showed that miR-132 inhibits histone deacetylase HDAC3, preventing its ability to up-regulate genes that promote ROS accumulation in cardiomyocytes.
Necrosis
Large numbers of cardiomyocytes undergo necrotic cell death after ischemic injury, providing the main stimulus for postinfarction inflammatory activation. Here, beneficial miRNAs prevent cardiomyocyte necrosis, whereas harmful miRNAs induce necrosis after injury. The central component of necroptosis activation is the assembly of the receptor-interacting protein kinase 1 (RIPK1)–RIPK3-mixed lineage kinase-like (MLKL) signaling complex. Two miRNAs, miR-105 and miR-873, prevent necroptosis through the direct down-regulation of RIPK proteins (52, 53). In addition, miRNAs can modulate the expression of death receptors responsible for initiating necroptosis. The beneficial miR-223 inhibits the receptors TNFR1 and DR6, thereby preventing necroptosis; whereas the harmful miR-103/107 inhibits the antinecrotic receptor Fas-associated death domain protein, thereby activating necroptosis (54, 55). Last, the beneficial miR-874 inhibits caspase-8 preventing its ability to activate the RIPK signaling complex (56).
Autophagy
Studies have identified miRNAs that modulate post-ischemic autophagy, sometimes with contradictory effects on cardiac function. For example, Wang et al. (57) demonstrated that miR-188 suppresses autophagy-mediated apoptosis after MI through direct inhibition of the autophagy-related gene ATG7. Similarly, Liu et al. (58) showed that miR-93 protects cardiomyocytes from apoptosis through inhibition of ATG7. In a comprehensive study, Gupta et al. (59) identified miR-22 as a potent inhibitor of cardiac autophagy through its targeting of peroxisome proliferator–activated receptor α (PPARα), a nuclear receptor known to activate autophagy. However, inhibition of miR-22 leading to the activation of autophagy after MI resulted in improved postinfarction remodeling and improved cardiac function (59). Similarly, Ucar et al. (60) demonstrated that the miR-212/132 family inhibited autophagy and that their inhibition restored the autophagic response, resulting in the rescue of heart failure in mice. Other studies have identified miRNAs that can either promote or inhibit autophagy including miR-142, miR-145, miR-221, miR-301, miR-18a, miR-199a, miR-30a, and miR-558; however, the ultimate therapeutic effects of these miRNAs seem to depend on their individual targets and not the overall activation or inactivation of autophagy (61–68).
Proliferation
Cardiac function can be restored after MI by generation of new cardiomyocytes. To this end, multiple miRNAs have been identified that can regulate cardiomyocyte proliferation. Here, beneficial miRNAs promote, whereas harmful miRNAs prevent cardiomyocyte proliferation after injury.
The Hippo-YAP pathway is a critical regulator of organ size and growth that plays an important role in cardiomyocyte proliferation (12). In a foundational study that delivered a transient proliferative stimulus using miRNA mimics, Tian et al. (69) showed that the delivery of miR-302/367 to the infarcted myocardium promoted cardiomyocyte proliferation and regeneration by targeting macrophage stimulating 1 (MST1), a central Hippo-YAP player. HOPX is a key regulator of heart development and induces cardiomyocyte proliferation when overexpressed (14). In a series of studies, the Giacca lab determined that miR-590 and miR-199a both promote cell cycle reentry in adult cardiomyocytes by targeting HOPX (70). They subsequently demonstrated that the proproliferative effect of miR-199a is also dependent on its regulation of the Hippo-Yap pathway (71). Overexpression of miR-199a using adeno-associated virus 6 in a swine model of MI resulted in unimpeded cardiomyocyte proliferation that progressively reduced the cardiac scar size but eventually led to sudden cardiac death in 70% of treated animals (72). The loss of FSTL1 has been shown to be a maladaptive response to injury, whereas its restoration resulted in increased numbers of proliferating cardiomyocytes (13). Recently, Xiao et al. (73) has demonstrated that miR-9 directly inhibits FSTL1 after MI and that the inhibition of miR-9 results in restored FSTL1, increases cardiomyocyte proliferation, and preserves cardiac function.
In parallel to targeting these canonical pathways, a set of miRNAs prevents proliferation by directly targeting cell cycle regulators such as cyclins. miR-34a and let-7i inhibit cyclin D1 and cyclin D2, respectively, suppressing their functions through the cell cycle (74, 75). In addition, miR-294 inhibits checkpoint kinase Wee1, preventing its suppression of the CDK1/cyclin B1 complex and cell cycle reentry (76). Last, miR-128 inhibits the chromatin modifier SUZ12, preventing it from activating positive cell cycle regulators cyclin E and CDK2 (77).
Other effects
Beyond cardiomyocyte cell death and proliferation, miRNAs can affect other facets of cardiac physiology. The harmful miRNAs miR-1231 and miR-223 induce arrhythmias in the heart after MI through inhibition of the ion channels CACNA2D2 and KCND2, respectively (78, 79). The beneficial miRNAs Let-7a and miR-206 prevent pathological cardiomyocyte hypertrophy by targeting calmodulin (CAM) and forkhead box protein P1 (FOXP1), respectively (80, 81). Meanwhile, the harmful miRNAs of the miR-212/132 family can induce hypertrophy through inhibition of FOXO3 and subsequent hyperactivation of NFAT signaling (60). A series of miRNAs have been shown to modulate the ability of cardiomyocytes to activate the inflammatory cascade: miR-128 targets SOX7, resulting in increased inflammation; miR-135b targets caspase-1, resulting in reduced inflammation; and miR-145 targets CD40, also resulting in reduced inflammation (82–84).
ROLE OF miRNAs IN FIBROBLAST RESPONSE TO MYOCARDIAL INFARCTION
After MI, CFs become activated and differentiated into myofibroblasts, which play an integral role in the rapid formation of a scar necessary to prevent ventricular wall rupture. However, excessive CF activation, proliferation, and ECM deposition after MI contribute to pathological cardiac fibrosis, which can exacerbate injury and lead to heart failure. Reversion of this activated phenotype with miRNAs is a key approach toward attenuating this pathological response.
Fibroblast responses to myocardial infarction
Inflammation
Immediately after MI, fibroblasts produce proinflammatory cytokines in response to cardiomyocyte death and inflammatory milieu. Interleukin-1α (IL-1α) stimulates CFs to secrete TNFα and IL-1β (85). Fibroblast signaling acts as a source of IL-1β positive feedback, attracting immune cells into the infarct zone (86). Cytokine-activated inflammatory fibroblasts modulate the secretion of proteases including matrix metalloproteinases (MMPs) that are essential for clearing the infarct of damaged matrix debris (87).
Proliferation
The process that has drawn the most focus in fibroblast repair of the heart is their differentiation into myofibroblasts and migration into the infarct zone. Transforming growth factor–β (TGF-β) is a key regulator of this phase (88). After MI, TGF-β1 is generated by macrophages producing angiotensin II (AngII). AngII has autocrine function, causing the up-regulation of TGF-β1 (89). TGF-β1 signaling involves a complex cascade of proteins including activating and inhibitory SMADs, with pleiotropic effects. TGF-β suppresses MMPs (90) while significantly increasing the production of collagens type 1 and 3, causing ECM synthesis (91). Activated myofibroblasts express α-smooth muscle actin (αSMA), allowing for scar contraction. Notably, TGF-β1 is secreted at an increased rate globally in the myocardium after injury, not just directly at the site of infarction (92), indicating widespread ramifications of MI.
Scar maturation
During the maturation phase, ECM proteins secreted by activated fibroblasts are cross-linked to form a stable scar. Over time, the myofibroblast population in the scar decreases (93) through apoptosis and transition into a recently described fibroblast phenotype termed the matrifibrocyte that is capable of maintaining the scar integrity (94). There is increasing evidence that the myofibroblast phenotype is reversible in vitro: CFs isolated from patients with heart failure revert to quiescence upon TGF-β1 inhibition (95). Therefore, the failure of myofibroblasts to deactivate has broad implications for clinical worsening of MI. Overactivation is driven by multifactorial processes, including the persistent secretion of AngII in parts of the heart remote from the site of injury (96). MiRNA therapies targeting fibroblasts seek to break the positive feedback loop of myofibroblast activation, in which the stiffening of the heart matrix induces even more TGF-β secretion.
Recent advances in miRNA therapies targeting cardiac fibroblasts
The inhibition of myofibroblast activity is a major focus of ongoing research with the identification of beneficial miRNAs that prevent and harmful miRNAs that induce fibroblast activation. Particular attention has been put on the TGF-β signaling pathway (15 of 26 studies). Here, we summarize key miRNAs and their mechanisms of action in CFs (Fig. 4A).
Fig. 4. Summary of miRNAs that target fibroblasts, endothelial cells, and immune cells.
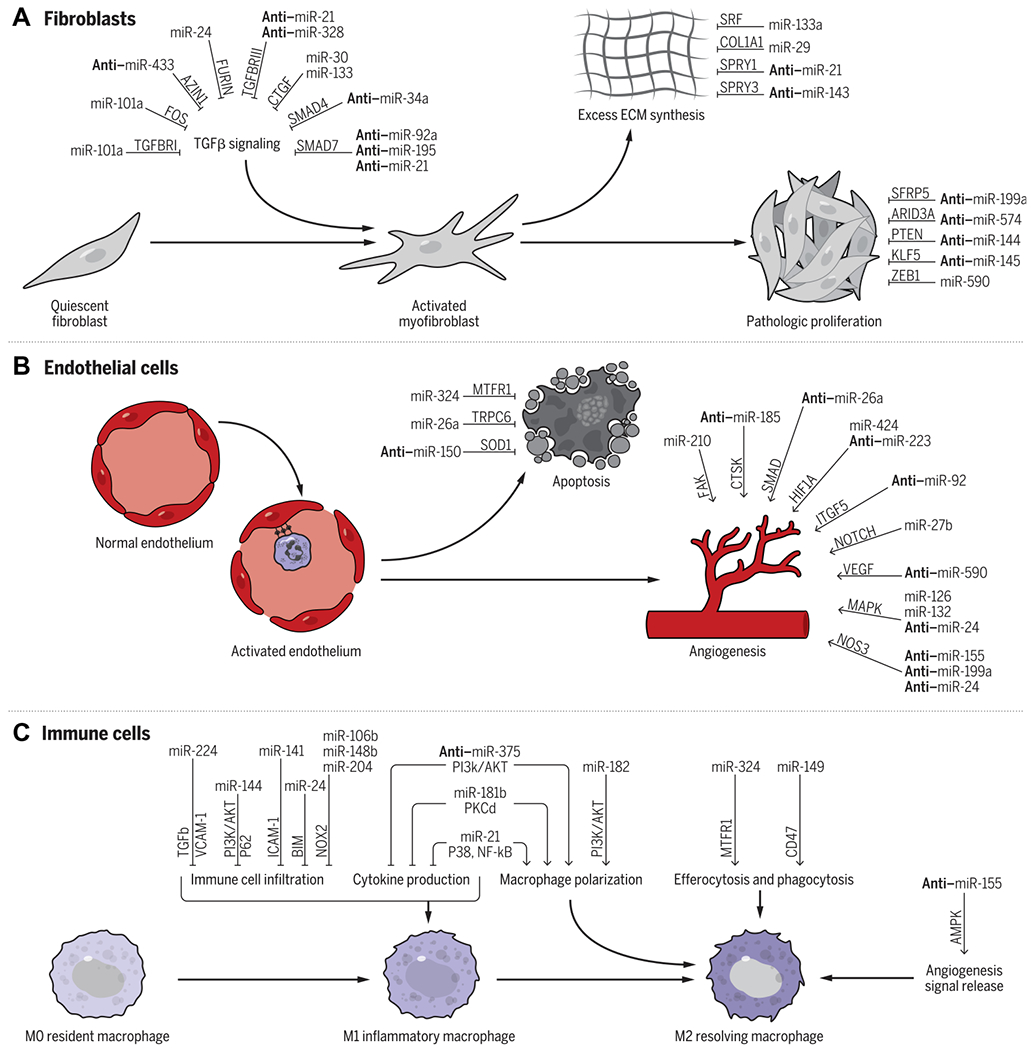
(A) In CFs, miRNAs can regulate TGFβ signaling as well as ECM synthesis and pathological proliferation. (B) In ECs, a large number of miRNAs have been shown to regulate angiogenesis, whereas others have been shown to regulate EC apoptosis. (C) In immune cells, miRNAs can regulate multiple aspects of macrophage function including immune cell infiltration, cytokine production, macrophage polarization, efferocytosis, and phagocytosis, as well as angiogenesis signal release. Select well-characterized miRNAs, their targets (written above the arrows), and the processes that they regulate are shown (i.e., miR-24 prevents TGFβ activation through FURIN signaling). miRNAs prefaced with anti–denote that the inhibition of the miRNA is therapeutic (i.e., the inhibition of miR-433 prevents TGFβ activation through AZIN1 signaling). For the complete list of miRNAs reviewed, please see table S1.
TGF-β signaling
Many miRNAs regulate parts of the TGF-β pathway, including its production, cleavage, and signal transduction. The classical upstream regulator of TGF-β, c-Fos, is regulated by miR-101a, with overexpression of miR-101a leading to reduced fibrosis and improved function after MI (96). Concurrently, Zhao et al. (97) identified TGF-β receptor 1 (TGFBR1) as another target of miR-101a, using a rat model to show decreases in TGF-β signal transduction. Another potential target for TGF-β modulation is AZIN1, as the knockdown of AZIN1 up-regulated the TGF-β expression, with miR-433 directly reducing AZIN1 in vivo (98). Transfection of miR-433 into CFs increased fibroblast proliferation and αSMA expression, whereas injection of a miR-433 antagomir in rats increased AZIN1 in post-MI hearts, leading to a reduction in fibrosis. Proteases cleaving latent TGF-β to its active form can also be targeted by miRNAs to reduce fibrosis. Wang et al. (99) showed that miR-24 inhibits furin, one such protease. In vitro overexpression of miR-24 increased TGF-β secretion and Smad2/3 phosphorylation, and miR-24 was found to be underexpressed in MI tissue.
Many miRNAs target the cellular receptors that bind TGF-β. Hong et al. (100) identified TGFBR1 (one of two main receptors for TGF-β) as a target of miR-22. Overexpression of miR-22 reduced AngII activation of CFs in vitro. Liang et al. (101) described a reciprocal loop by which TGF-β up-regulates miR-21, which then inhibits TGFBR3, a negative regulator of TGF-β signaling. The inhibition of TGFBRIII increases TGF-β secretion and Smad3 phosphorylation and therefore collagen secretion. Similarly, Du et al. (102) showed that miR-328 targets TGFBRIII and that an injection of anti–mir-328 can improve cardiac fibrosis post MI in mice.
After binding to the receptor, TGF-β signaling is mediated by decapentaplegic (DPP) homologs (SMAD) family. SMAD2/3 couple to the receptor and are phosphorylated by TGF-β binding, subsequently binding to SMAD4. SMAD6 and SMAD7 inhibit TGF-β signaling. miRNA mimics, which target SMAD2/3/4 and anti-miRs against those that target SMAD6/7, can prevent fibrosis. Yuan et al. (103) showed the suppression SMAD7 by miR-21, with maladaptive response in fibroblasts. miR-34a is up-regulated after MI, and in vivo inhibition of miR-34a reduced post-MI cardiac fibrosis: SMAD4 was identified as a direct target of miR-34a (104). Ischemic exosomes isolated from mouse cardiomyocytes after MI were abundant in miR-92a, which directly targets SMAD7. Overexpression of miR-92a contributed to the activation of fibroblasts (105). These post-MI cardiomyocyte exosomes contain miR-195, which also targets SMAD7 (106).
Last, the downstream effectors of TGF-β, such as connective tissue growth factor (CTGF) (107, 108), can also be targeted by miRNAs. Duisters et al. (109) initially identified both miR-133 and miR-30 as inhibitors of CTGF. Later, Chen et al. (110) delivered miR-30a using adenovirus after MI and observed increased heart function and decreased collagen deposition.
Other pathways
The harmful role of miR-21 in cardiac fibrosis was established by the Thum lab (111, 112), which showed, in a mouse model of heart failure, that miR-21 was dysregulated in fibroblasts but not cardiomyocytes. Inhibition of miR-21 increased the percentage of apoptotic fibroblasts in the failing heart. They demonstrated that miR-21 targets Sprouty1, resulting in increased prosurvival extracellular signal–regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) signaling. Delivery of anti–miR-21 in vivo attenuated fibrosis. Similarly, Cardin et al. (113) demonstrated that miR-21 is up-regulated in atria after MI and contributes to the development of fibrosis and arrhythmia in atrial fibrillation. miR-21 knockdown reduced atrial fibrosis and stabilized electrical conduction.
Yuan et al. (114) identified miR-144 as a PTEN inhibitor in miniature swine. Overexpression of miR-144 or transfection of a PTEN-targeting small-interfering RNA (siRNA) in primary human CFs in vitro increased collagen 1 and αSMA mRNA expression.
Since smooth muscle gene expression is a hallmark of differentiated myofibroblasts, miRNAs that regulate smooth muscle phenotype are potentially therapeutic. miR-143 and miR-145 are transcribed from the same cluster and play a critical role in smooth muscle differentiation (115). Li and colleagues (116) showed that miR-143 is up-regulated in human MI tissue samples, and miR-143 inhibitors reversed the effects of TGF-β stimulation on fibroblasts in vitro. miR-143 directly binds to the 3′ untranslated region of Sprouty3, activating p38, ERK, and c-Jun N-terminal kinase pathways. In another study, Wang et al. (117) showed that miR-145 is sufficient to increase the myofibroblast phenotype of CFs by targeting KLF5, which is a negative regulator of the myocardin serum response factor (SRF) pathway. miR-145 inhibition in vivo reduced αSMA expression but increased scar size, perhaps indicating a decrease in contractile function of the fibroblasts.
The miR-133a family, mir-133a-1, miR-133a-2, and miR-133b, are also key regulators of the SRF pathway. Of these, the two miR-133a genes are identical and specifically expressed in cardiac myocytes, whereas miR-133b is expressed in skeletal muscle (118). miR-133a is down-regulated in the hearts of patients with MI as well as in animal models (119). miR-133a double knockout mice display severe fibrosis and early mortality (118). Expressed specifically in cardiomyocytes, miR-133a appears to act in a paracrine manner, reducing the secretion of profibrotic cytokines. Duisters et al. (109) showed that miR-133a regulates the production of CTGF in cardiomyocytes, a downstream effector of TGF-β signaling.
ECM proteins can also be targeted as the end product of myofibroblast activation. The Olson lab demonstrated that miR-29 is decreased after MI and that it targets ECM protein mRNAs, including collagens, fibrillin, and elastins (120). Inhibition of miR-29 in vivo induced collagen mRNA expression in the heart and other organs. miR-29 was found to correlate to collagen expression after MI (121), and collagen 1 was identified as a target of miR-133a (122) in AngII-induced injury. However, an opposite observation was found with genetic knockout of the miR-29 cluster in mice, causing cardioprotection and decrease in fibrosis (123). The authors suggest that miR-29 may be more dominantly expressed in cardiomyocytes versus CFs and that whereas miR-29 may be antifibrotic in the fibroblasts, it is deleterious in cardiomyocytes, with the knockout effect dependent on the CM-specific effects.
Wingless and Int-1 (WNT) signaling is often dysregulated in fibrosis (124). miR-199a was shown to be up-regulated during fibroblast activation and to target secreted frizzled-related protein 5 in vitro. Inhibition of miR-199a reduced fibroblast migration and proliferation (125). Other studies have found that targeting fibroblast proliferation is beneficial. Cui et al. (126) showed that miR-574-5p was up-regulated in TGF-β–induced fibroblast activation. miR-574-5p targets ARID3A, which has been implicated in mediating cell cycle progression. MiR-590 targets ZEB1, an activator of transcriptional regulator CXCR4, and its overexpression inhibited migration, proliferation, and collagen secretion in CFs (127).
ROLE OF miRNAs IN ENDOTHELIAL CELL RESPONSE TO MYOCARDIAL INFARCTION
The cardiac muscle has a dense vascular network to meet the high metabolic demands of the tissue, and the continuous EC monolayer lining serves as a barrier between the blood and myocardium (128). MI severely affects these functions and induces endothelial activation, which facilitates recruitment of inflammatory cells during the inflammatory phase and mediates the repair and remodeling of the vascular network within the injured cardiac tissue via angiogenesis. Thus, miRNA therapy targeting ECs focuses mainly on regulating inflammatory recruitment and inducing angiogenesis.
Endothelial cell responses to myocardial infarction
Inflammatory recruitment
Immediately after MI, ischemia activates cardiac ECs to a proinflammatory and prothrombotic phenotype (129). The ischemic environment causes an increase in the generation of ROS and the presence of proinflammatory cytokines, such as TNFα and IL-6 (130). These conditions induce the expression of cell surface adhesion molecules, providing sites of adhesion to facilitate the recruitment and attachment of circulating leukocytes. The proinflammatory phenotype results in reduced endothelial nitric oxide (NO) production and bioavailability, impairing the ability of the endothelium to regulate vascular permeability and tone in response to external stimuli. Although endothelial activation mediates the inflammatory response within the cardiac tissue, prolonged activation and continued imbalance of ROS and NO generation can lead to permanent adverse effects, such as endothelial dysfunction and cellular apoptosis (130).
Angiogenesis
Angiogenesis, the process by which new blood vessels form from existing vasculature, is essential for cardiac repair after MI, as it is needed to restore sufficient blood flow to the injured tissue (131). Revascularization occurs at the site of the infarct as well as throughout the surrounding injured tissue via proliferation and migration of ECs. Ischemia induces expression of growth factors, which promote EC survival, migration, and proliferation, such as the vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) families (132). The hypoxic environment created during ischemia up-regulates the expression of hypoxia-inducible transcription factors (HIFs), such as HIF-1α, which activate the angiogenic process.
Recent advances in miRNA therapies targeting endothelial cells
Of the studies reviewed, relatively few focused on the EC population (24 of 213). Although some studies focus on preventing EC apoptosis or address other effects (5 of 24), most studies target the process of angiogenesis (19 of 24). Here, we summarize key miRNAs and their mechanisms of action in ECs (Fig. 4B).
Apoptosis
Apoptosis of ECs disrupts the barrier function of the endothelium and impairs its ability to effectively regulate the inflammatory response after injury. miRNA involvement in EC apoptosis has been primarily studied within the context of atherosclerotic models, which is a major cause of complications such as MI. Here, beneficial miRNAs prevent, whereas harmful ones promote EC apoptosis. The atherogenic factor oxidative low-density lipoprotein (ox-LDL) induces EC dysfunction and apoptosis and has been used both in vitro and in vivo to identify and investigate associated miRNAs. miR-26a was identified as dysregulated during atherosclerosis: Its expression was suppressed in a dose-dependent manner in human aortic ECs (HAECs) treated with ox-LDL (133). Overexpression of miR-26a targeted and repressed activity of transient receptor potential cation channel subfamily C member 6 (TRPC6), a calcium-permeable channel subunit, inhibiting apoptosis by inhibiting cytosolic calcium overload, which triggers the calcium activated apoptotic pathway.
In a separate study, ox-LDL–induced apoptosis was investigated in HAECs and human umbilical vein endothelial cells (HUVECS), and miR-150 was up-regulated during the process (134). Overexpression of miR-150 enhanced cellular apoptosis, whereas inhibition of miR-150 alleviated apoptosis. The proapoptotic miR-150 negatively regulates expression of Cu/Zn superoxide dismutase (SOD1) through the direct targeting of transcription factor ELK1. Last, miR-324 was shown to protect against hypoxia-induced apoptosis in endothelial progenitor cells (EPCs), which are active participants in the recovery process after MI, supporting both vascular endothelium repair and angiogenesis (135). Increasing expression of miR-324 down-regulates the MTFR1 gene, which, in turn, reduces mitochondrial fragmentation, stabilizes mitochondrial membrane potential, and results in decreased cellular apoptosis in hypoxic conditions.
Angiogenesis
Angiogenesis is a tightly regulated process involving multiple molecular pathways. miRNAs modulate many of the pathways involved, with beneficial miRNAs promoting and harmful miRNAs impeding the angiogenic process.
Multiple miRNAs that regulate angiogenesis modulate pathways activated by binding of VEGF (specifically VEGFA) to cell surface receptors, including HIF-1α, MAPK, and endothelial nitric oxide synthase (eNOS), which all contribute to promoting angiogenesis (132). Modulating HIF-1a is the proangiogenic miR-424, which is up-regulated in ECs under hypoxic conditions and targets Cullin 2 (136) to stabilize and up-regulate HIF-1α. In contrast, the harmful miR-223, although also up-regulated in ischemic conditions, negatively regulates HIF-1α signaling by targeting ribosomal protein S6 kinase B1 and inhibits EC migration and proliferation (137). In the MAPK pathway, the proangiogenic miR-126 and miR-132 both suppress negative regulators of MAPK activity, Spred-1 and RASA1, respectively, whereas anti-angiogenic miR-24 targets p21-activated kinase (PAK4), inhibiting MAPK activity (138–141). miR-24 also targets the eNOS signaling pathway, along with miR-199a and miR-155, all of which suppress migration, proliferation, and tube formation of ECs (142, 143). Inhibition of these miRNAs increases NO bioavailability and promotes angiogenesis in these studies. Although these miRNAs target molecules of pathways activated by the binding of VEGF, certain miRNAs target VEGF directly, such as the antiangiogenic miR-590 (144).
Several other miRNAs affect angiogenesis by modulating signaling pathways that are not VEGF associated. In two separate studies, miR-210 was shown to promote angiogenesis, with overexpression increasing EC proliferation and migration by up-regulating hepatocyte growth factor expression and targeting focal adhesion kinase (145, 146). miR-92 also showed, in two separate studies, that it targets the proangiogenic integrin α5 (ITGF5) and that its inhibition substantially increased angiogenesis and granulation tissue formation (147, 148). Because miR-26a targets SMAD1, its inhibition led to increased angiogenic activity and reduced infarct size in a murine model of MI. In contrast, miR-27b promotes vascularization by targeting notch ligand DLL4 (149). Last, miR-185 inhibits proliferation, migration, and tube formation by targeting the CatK gene (150). Inhibition of miR-185 significantly promoted these functions of angiogenesis under hypoxic conditions.
Other effects
Bayoumi et al. (151) determined that miR-532 overexpression decreased endothelial-to-mesenchymal transition (EndMT) in cardiac ECs. miR-532, which is up-regulated by β2 adrenergic receptor/β-arrestin activity, exerts this cardioprotective function by targeting protease serine 23 (PRSS23), a positive regulator of maladaptive EndMT. The metabolic state of ECs can also alter MI outcomes, as Bartman et al. (152) identified that miR-21 increased glycolytic activity in ECs in hypoxic conditions. miR-21 was up-regulated under hypoxic conditions only in ECs, not in cardiomyocytes or fibroblasts, despite being predominantly expressed in fibroblasts under normoxic conditions. Inhibition of miR-21 resulted in larger infarct sizes in a myocardial IRI murine model.
ROLE OF miRNAs IN IMMUNE CELL RESPONSE TO MYOCARDIAL INFARCTION
MI triggers an inflammatory cascade in the heart starting with an acute proinflammatory response, which is gradually replaced by an anti-inflammatory reparative phase. Therapeutic advancements generally aim to dampen the initial inflammatory response and promote the proreparative phase.
Inflammatory responses to myocardial infarction
Activation of inflammation
Upon injury, resident immune cells located in perivascular areas are activated, resulting in an acute inflammatory response. In the early inflammatory phase, the infiltration of immune cells in the infarct area and accumulation of proinflammatory chemokines/cytokines result in not only the clearance of cellular debris but also in further damage to cardiac tissue. Macrophages, among the largest resident cell populations in cardiac tissue, are predominantly responsible for the production of inflammatory cytokines post MI. They release IL-1, IL-6, and TNFα, triggering further responses from multiple cell types, both locally and remotely. The activation of this inflammatory cascade mediates remodeling throughout the myocardium.
Resolution of inflammation
Increasing evidence indicates that the initial inflammatory response elicited by MI induces resident macrophages to switch from an inflammatory M1 phenotype to a resolving M2 phenotype. M2 macrophages secrete anti-inflammatory signals critical for the repair response, allowing wound healing and scar formation, thereby limiting infarct size. Failure to activate reparative responses leads to adverse cardiac remodeling with further damage and, ultimately, heart failure.
Recent advances in miRNA therapies targeting immune cells
Of the studies surveyed, 17 of 213 focus on immune cells. Although some studies have applied miRNA therapies to either reduce the damage of the inflammatory response (10 of 17 studies) or focus on promoting the reparatory pathways (3 of 17 studies), several other effective therapies have targeted both responses, exerting the overall functional benefits as an ensemble (4 of 17 studies). In targeting immune cells, beneficial miRNAs aim to prevent infiltration of immune cells and inflammatory signal production in the infarct area and promote M2 macrophage polarization, efferocytosis, phagocytosis, and angiogenesis signal release. Here, we summarize key miRNAs and their mechanisms of action in immune cells (Fig. 4C).
Infiltration
Infiltration of immune cells such as macrophages and neutrophils into the infarct area is one of the initial inflammatory responses after MI. Many miRNAs have been shown to regulate this process, providing interesting therapeutic targets. Recently, miR-141 has been shown to suppress neutrophil/leukocyte adhesion to ECs by modulating expression of the cell adhesion associated protein intercellular adhesion molecule–1, which plays a crucial role in regulating the migration of leukocytes across the endothelium into the myocardium. In vivo, pretreating mice with intravenous miR-141 mimics before IRI successfully reduced CD11b+ myeloid cells and F4/80+ macrophages accumulation in the ischemic myocardium (153).
miR-21 knockout mice showed enhanced infiltration of CD11b+ monocytes/macrophages in myocardium, whereas miR-21 overexpression markedly inhibited it (154). However, contrasting findings in another study showed that miR-21 inhibition in cardiac allografts led to reduced infiltration of CD45+ leukocytes, but there was no difference observed for macrophage infiltration (155). Intravenous injection of miR-144 mimics resulted in reduced infiltration of macrophages in the border zone, decreased MMP-mediated inflammation, and reduced fibrosis and apoptosis. The beneficial effects of miR-144 therapy were associated with mTOR- and P62-mediated autophagy signaling with improved cardiomyocyte survival. Alterations in TGF-β signaling also demonstrated a role for miR-144 in modulating the local inflammatory response (156).
Intramyocardial injection of anti–miR-224–expressing lentivirus elevated vascular cell adhesion molecule–1 expression, regulating inflammation-associated vascular adhesion and transendothelial migration of macrophages/leukocytes, and MMP-2, an inflammatory mediator participating tissue remodeling in pathological processes (157). Other studies have shown that the overexpression of miR-106b, miR-148b, and miR-204 injection of miR-181b–enriched cardiosphere-derived exosomes (CDC-exo) or miR-24–enriched umbilical mesenchymal stem cells (MSCs) secreted exosomes (MSC-exo), and knockdown of miR-375 all suppressed CD68+ cell accumulation in the infarct border zone, leading to improved cardiac function with decreased infarct size (158–161).
Inflammatory cytokine production
The accumulation of proinflammatory cytokines plays a critical role in governing the progression and extent of tissue remodeling. The inhibition of this process by miRNA therapies successfully decreases myocardial infarct size and improves cardiac function. miR-21 mimic delivered to monocytes/macrophages in mice reduced inflammatory cytokine expression by directly inhibiting KBTBD7-mediated DAMP (damage-associated molecular patterns)–triggered inflammatory responses in macrophages (154). The delivery of miR-106b, miR-148b, and miR-204 individually encapsulated into polyketal nanoparticles in macrophages decreased Nox2 expression, inhibiting superoxide production and expression of proinflammatory genes IL-1α, IL-6, and TNF-α in vitro (158). Anti–miR-375 injection after MI in mice decreased inflammatory response by reducing the production of several proinflammatory cytokines including IL-6, interferon gamma-induced protein 10 (IP-10), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α), regulated on activation, normal T cell expressed and secreted (RANTES), and MIP1-β (160). After reperfusion in rats and swine, intramyocardial injection of miR-181b–enriched CDC-exo also attenuated the expression of proinflammatory genes by targeting PKCδ (159).
M2 macrophage polarization
Polarization of macrophages toward the anti-inflammatory M2 phenotype has been found to confer beneficial effects on cardiac repair. Intramyocardial injection of MSC-exo containing miR-182 after myocardial IRI reduced infarct size and alleviated damaged myocardial inflammation in mice. Mechanistically, miR-182 polarized macrophages toward the M2 phenotype and regulated downstream Toll-like receptor 4 (TLR4) expression, mitigating proinflammatory cascades and enhancing subsequent repair (162).
Tail vein injection of miR-21 mimic delivered to cardiac macrophages by encapsulated nanoparticles promoted macrophage switching from a proinflammatory to reparative phenotype, thus inducing the resolution of inflammation (163). The polarization state was also shifted in cardiac macrophages isolated from CDC-exo–treated rats and swine. miR-181b inhibition of PKCδ expression further demonstrates that PKCδ acts as a downstream effector of CDC-exo–mediated cardioprotection (159). Moreover, increased CD206+ cells (M2 macrophage marker) in the left ventricular infarct border zone of anti–miR-375–treated mouse hearts after MI indicate that miR-375 knockdown can trigger a macrophage switch toward the anti-inflammatory M2 phenotype (160). In vitro studies in macrophages further demonstrated that miR-375 modulated AKT signaling through phosphoinositide-dependent kinase-1 (PDK-1). PDK-1 knockdown abolished the macrophage polarization effect of miR-375, further confirming that PDK-1 is a critical mediator in miR-375–regulated macrophage phenotype shift.
Targeting efferocytosis and phagocytosis
The clearance of apoptotic cells by macrophages is thought to play a critical role in the reparative phase allowing for further recovery after MI. MI-associated transcript (MIAT), a long noncoding RNA, has been shown to regulate phagocytosis by acting as a sponge of miR-149, which directly targets antiphagocytic molecule CD47 (164). Knockdown of MIAT or miR-149 overexpression in murine macrophages reduced CD47 expression and promoted efferocytosis and phagocytosis, which consequently increased resolution of inflammation and clearance of apoptotic cells by macrophages. Similarly, transfecting EPCs derived from the peripheral blood of patients with ST-segment elevation MI with miR-324 mimic resulted in reduced apoptosis, increased proliferation, and elevated phagocytosis by targeting and inhibiting MtFr1 after peroxide-induced oxidative stress (135). The protective role of miR-324 overexpression against oxidative stress-induced EPCs injury suggests that therapies leading to correction of miRNA expression may contribute to tolerance against injury by regulating phagocytosis pathways.
Targeting angiogenesis signal release
Macrophages engage in cross-talk with other cell types and release a variety of angiogenic signals to initiate and regulate angiogenesis in ECs in response to MI. After MI, M1 macrophages secrete proinflammatory exosomes (M1-exo), which contain abundant miR-155 (142). M1-exo containing miR-155 suppressed expression of several genes, including RAC1, PAK2, Sirt1, and AMPKα2, and modulated Sirt1/AMPKα2-eNOS and Rac family small GTPase 1 (RAC1) - p21 (RAC1)–activated kinase 2 (PAK2) signaling pathways, leading to the inhibition of angiogenesis and cardiac dysfunction. The antiangiogenic function of miR-155 makes its inhibition an attractive therapeutic strategy for MI (142).
miRNAs WITH PLEIOTROPIC EFFECTS
Most miRNA studies focus on one miRNA in a singular cell type. However, the literature demonstrates that many miRNAs have pleiotropic effects on myocardial recovery after MI, spanning different pathways, processes, and cell types. Some pleiotropic miRNAs regulate many points along the same pathway, whereas others regulate different pathways. Interestingly, multiple of these miRNAs demonstrate divergent effects among cell types—promoting recovery processes in some cell types while impeding recovery processes in other cell types. Developing a clear mechanistic understanding of the pleiotropic effects of miRNAs will greatly advance their clinical translation.
miRNAs with convergent effects
Some miRNAs have convergent effects: promoting or impeding recovery processes in all cell types. For example, miR-210, miR-324, miR-494, and miR-532 all have beneficial effects on both cardiomyocytes and ECs (42, 135, 145, 146, 151, 165–169). Although miR-324 had the same target (MTFR1) and effect (prevention of apoptosis) in both cell types (135), the other three miRNAs had different targets and affected different pathways in each cell type. In separate studies, miR-24 was shown to have beneficial effects on cardiomyocytes, fibroblasts, and ECs through the prevention of apoptosis, prevention of fibrosis, and activation of angiogenesis, respectively (24, 99, 141). On the other hand, miR-92a has been shown to have harmful effects on fibroblasts and ECs through the activation of fibrosis and the prevention of angiogenesis (105, 147, 148). Because of their convergent effects on multiple cell types studied, these miRNAs may be particularly promising therapeutic targets.
miRNAs with divergent effects
Interestingly, most of the pleiotropic miRNAs identified demonstrate divergent effects: promoting recovery processes in some cell types while impeding recovery processes in other cell types. We already described miR-29, which demonstrated contradictory effects on CFs versus cardiomyocytes. Another extensively studied miRNA that falls into this category is miR-199a. miR-199a was shown to induce adult cardiomyocyte proliferation after MI (70), and subsequent studies identified multiple proproliferative pathways regulated by miR-199a in cardiomyocytes (71). Additional studies have demonstrated that miR-199a can regulate other crucial pathways in cardiomyocytes including glucose metabolism, hypertrophy, apoptosis, and autophagy with divergent effects (66, 170–173) and negatively affect angiogenesis in ECs as well as induce pathological fibrosis in fibroblasts (125, 143).
Another noteworthy miRNA is miR-21. Multiple studies have demonstrated that miR-21 prevents cardiomyocyte apoptosis after ischemic injury through direct inhibition of the proapoptotic molecule PDCD4 (38–40). In addition, treatment with miR-21 mimics has been beneficial to ECs by preventing apoptosis (152) and to immune cells by preventing inflammatory activation (152, 154, 163). However, despite consensus on the beneficial effects of miR-21 in cardiomyocytes and ECs, multiple studies have demonstrated harmful effects of miR-21 in fibroblasts, and other studies have shown contradictory effects in immune cells. The knockdown of miR-21 prevents pathological fibrosis by targeting the ERK/MAPK and TGF-β pathways (101, 111–113). In immune cells, miR-21 has been shown to both prevent macrophage infiltration and induce leukocyte infiltration (154, 155). Ultimately, despite contradictory effects, the strongest evidence for the therapeutic capabilities of miR-21 comes from a recent study by Hinkel et al. (174), which demonstrated that inhibition of miR-21 prevented myocardial dysfunction in a pig model of MI.
Multiple miRNAs have been shown to have divergent effects on fibroblasts compared to cardiomyocytes. For example, miR-22, miR-29, miR-101, and miR-155 are all beneficial to fibroblasts by preventing activation and proliferation while being harmful to cardiomyocytes by promoting apoptosis or autophagy (59, 96, 97, 100, 120, 142, 175–179). In contrast, miR-144 prevents cardiomyocyte apoptosis (beneficial) while also promoting fibroblast proliferation (harmful), by targeting the same PI3k/AKT pathway in both cell types (156). Collectively, these miRNAs demonstrate the complexity of miRNA effects across multiple cell types in the heart and suggest the difficulties with using nontargeted miRNA therapies. Thus, a cardiomyocyte-centric approach to miRNA therapy limits our understanding of pleiotropic effects.
PROSPECTS AND CHALLENGES FOR CLINICAL TRANSLATION
Promising preclinical studies have spurred interest in clinical translation of miRNA therapies. Currently, there are no miRNA therapies with U.S. Food and Drug Administration (FDA) approval; however, several early-stage biotechnology companies are focused solely on miRNAs, such as Miragen Therapeutics, Cardior Therapeutics, and Regulus Therapeutics. One phase 1 trial is currently ongoing to assess the safety of a miR-132 inhibitor (NCT04045405) in ischemic heart disease. There are several clinical trials for treatment of related pathologies, including the use of a miR-21 inhibitor for prevention of renal fibrosis (NCT03373786) and a miR-29 mimic for pulmonary fibrosis (NCT03601052).
As discussed here, mimicry of “beneficial” miRNAs or inhibition of “harmful” miRNAs both have therapeutic potential. However, the latter has benefited from past innovation in the RNA interference field, which has recently begun, achieving success in the clinic, with the first drug of its class, Patisiran, approved by the FDA in 2018. It is thus not surprising that of the current miRNA therapeutics in either phase 1 or 2 clinical trials, the majority are ASOs targeting a specific miRNA (180), although mimics are also being tested. Similar to the siRNA field, these burgeoning new therapies face challenges including considerations for therapeutic timing, optimal oligonucleotide optimization, in vivo delivery, and side effects.
Oligonucleotide modifications
Naked oligonucleotides rapidly accumulate in the kidney and liver and are quickly cleared from the circulation (181). Moreover, cleavage by serum exonucleases and degradation in the intracellular endosomal compartment reduces drug potency. To improve stability, various chemical modifications to the nucleic acid backbone have been used in the design of miRNA mimics and inhibitors.
Phosphothiorates substitute a sulfur for oxygen in the phosphate group of the nucleotide. Compared to unmodified oligonucleotides, phosphothiorates are more resistant to nucleases and thus drastically increase circulation time (182). However, compared to an unmodified phosphodiester bond, they have decreased binding affinity to their target as measured by a lower melting temperature (Tm) (183). To address these issues, second-generation designs centered around the use of 2′-O modifications of the ribose sugar and locked nucleic acid (LNA) modifications while reducing and interspersing the number of phosphothiorate modifications. 2′-O-methylation (2′OMe), first tested in the early 2000s, enhances target binding (184). Krützfeldt et al. (185) used 2′OMe oligonucleotides with spaced phosphothiorate bonds and a cholesterol group at the 3′ end to increase resistance to exonucleases while preserving nuclease activity, so termed “antagomirs” (186).
LNA modifications contain methylene linkages of the 2′O to the 4′C in the sugar backbone, locking the structure into a C3′-endo sugar conformation (186). These afford an increase in Tm while simultaneously increasing nuclease resistance (187). Given their high binding affinity, much shorter LNA oligonucleotides can be designed. This has both implications for target specificity, as shorter sequences more likely bind multiple families of miRNAs, and the related cost of synthesizing such an oligonucleotide. Recently, Obad et al. (188) reported the use of an 8-mer LNA oligo capable of powerful anti-miRNA effects.
Last, miRNA sponges have been explored as alternatives to ASOs (189). These dominant-negative inhibitors contain multiple target sites complementary to a specific miRNA seed sequence. They have several advantages over traditional oligonucleotides, including the ability to silence families of RNA sharing a common seed. However, further progress on their design and delivery is required for clinical translation, and this area of research still trails ASOs in terms of optimization (190).
Delivery vehicles
The route of administration of miRNA therapeutics greatly alters their potency. Local administration methods such as intracoronary injection (IC), hydrogel-based patches, and intramyocardial injection have been studied (table S1). The availability of percutaneous coronary intervention as the preferred method for revascularization after MI (191) makes IC injection perhaps the most effective local delivery method. Despite this, the multiple dosing of locally administered drugs to the heart is invasive and difficult in humans. In a preclinical study of MI in 135 swine, Foinquinos et al. (192) demonstrated the efficacy of IC injections of miR-132 immediately after MI. Notably, they also show that intravenous infusions were as effective as IC injections, suggesting the possibility for minimally invasive delivery of miRNA therapies.
Intravenously injected ASOs delivered without a vehicle still require large doses to be effective in vivo (185). In addition, efficient delivery to the target tissue—in this case, the heart—is essential for optimizing in vivo delivery of miRNA therapies. For this purpose, carriers such as lipid nanoparticles (LNP) and nanoparticles (193) have been explored. Traditional LNPs have been clinically validated for siRNA delivery, including Patisiran. However, their use comes at the cost of additional components and toxicities. Because these systems have been reviewed extensively (181, 194), they will not be discussed in detail here.
Extracellular vesicles, including exosomes, are plasma membrane–bound cell-secreted vesicles that transfer biologically active cargo between cells and act as the endogenous analog of LNPs. In the field of heart repair, exosomes emerged first as paracrine mediators of cell therapy. They are secreted by all cells and contain a variety of contents, including miRNAs (195). Exosomes from several cell types, including MSCs, cardiac progenitor cells, and induced pluripotent stem cell–derived cardiomyocytes (iPS-CMs), are effective in promoting cardiac repair (196–198). Several advances have been made toward clinical translation, including the development of Good Manufacturing Practice protocols for the collection of therapeutic exosomes (199) as well as toward altering their cargo. Unlike transplanted cells and synthetic LNPs, autologous exosomes do not trigger the immune system. However, barriers to their use remain, like addressing their heterogeneity and production. Eventually, highly defined engineered exosomes may represent a way to fine-tune miRNA therapy for the heart. For example, MSCs overexpressing miR-133 were more effective in cardioprotection than control vector MSCs in a rat model of MI, an effect attributed to an increase in therapeutic exosome content (200).
Tissue-specific delivery
At this time, targeted delivery of miRNA therapeutics to the heart remains an unmet need. Biodistribution studies show that LNP and exosomes are entrapped by the liver and spleen and filtered from the blood by the kidneys’ glomerular barrier due to their size (201). With only an estimated 2000 to 5000 copies of oligonucleotide required within the cell for gene knockdown, drug loading doses can be reduced if delivery can be constrained to the heart (202). The conjugation of ASOs to N-acetylgalactosamine (GalNAc) to specifically target asialoglycoprotein receptors in the liver has contributed greatly to the commercial success and translation of Patisiran (203). Identifying similar extrahepatic targets, including in the heart, has not yet been achieved and is a key focus.
To this end, homing cardiac targeting peptides and engineered exosomes have been shown to increase exosome and miRNA delivery in cardiomyocytes after IC injection (204). Chemical aptamers can alter biodistribution. Xue et al. (205) used a nanoparticle dendrimer approach to target miR-1 inhibitors to the AngII receptor 1 in post-MI hearts. Antibody-based targeting can also alter cardiac homing. Immunoglobulins are immunologically privileged proteins and are continually recycled in the circulation. Conjugation of siRNAs to an anti-CD71 Fab′ fragment allowed for durable gene silencing in cardiac tissues over a month’s time (206). Liu et al. (207) used anticardiac troponin antibodies to target liposomes containing anti–miR-1 to ischemic myocardium. However, the addition of a large macromolecule may also lead to several disadvantages. Genentech’s THIOMAB antibody-siRNA conjugates demonstrated the ability to specifically target prostate cancer in vivo but exhibited suboptimal gene silencing due to sequestering in the endocytic department (208). In addition, targeting specific receptors may have other effects besides delivering the therapeutic molecule, such as inhibition or activation of its own downstream signaling pathway, which adds complexity to this delivery modality.
Endosomal escape
After organ-specific targeting, there remains a barrier to entry at the cellular level. miR-mimics and anti-miRs are relatively large (~10,000 kDa) and highly negatively charged, preventing membrane transport. Naked nucleic acid or those bound in carriers are ubiquitously taken up by endocytosis (209). Endocytosed molecules then enter the endosomal system and eventually are degraded in the lysosome (210). To have a clinical effect, miRNA therapeutics must escape the endosomal pathway into the cytosol. Using traceable siRNAs, Gilleron et al. (211) estimated that just 1% of delivered oligonucleotides actually escape the endosomal pathway. The exact biological mechanisms of how escape occurs remain to be elucidated. Approaches to designing delivery carriers, which bypass the endosomal system via diffusion through the endosomal membrane or disrupting the compartment pH, have been explored (212). Solving the endosomal escape barrier will be critical for miRNA therapeutic development moving forward.
Side effects of miRNA therapies
Like any drug, miRNA therapeutics are not without safety concerns. For example, phosphothiorates have also been reported to react with proteins, including FGF2 (213). Although careful sequence selection can eliminate off-target RNA hybridization, many oligonucleotide modifications produce nonspecific effects, including differences in protein expression when compared to an unmodified siRNA (214).
Nucleic acids also broadly trigger the immune system, activating the TLR family (215). The use of delivery vehicles such as LNPs, which are designed to shield ASOs from the immune system, adds further reagents that may trigger an inflammatory response. A miRNA-34a mimic was halted in phase 1 (NCT02862145) studies after several patients developed severe adverse immune reactions. Development of these therapeutics can be thus unpredictable.
Abrogating the acute response to MI may also be dangerous. Therapies may be able to salvage damaged but viable cells adjacent to the infarct; however, an injured cardiomyocyte that is rescued may have impaired function, leading to arrhythmia. Similarly, interference of fibroblast function after MI has long been controversial, due to fear of cardiac rupture after failure to generate a scar. Although no studies reviewed here reported such outcomes, developing clear understandings of the mechanisms underlying miRNA therapies will go a long way toward preventing these dangerous side effects as we continue to search for more potent miRNA therapies.
The ability of miRNAs to regulate a variety of processes in different cell types leads to the additional concern of perturbations in signaling outside of the target organ. Although promoting cardiomyocyte proliferation may be desirable for cardiac recovery, altering those same pathways in other organs may result in tumorigenesis. The aforementioned miR-199a, which caused uncontrolled cardiomyocyte proliferation and subsequent arrhythmia, has also been linked to carcinogenesis and metastasis in certain cancers, such as melanoma and gastric cancer (216, 217). Similarly, inhibition of miR-34a has been shown to improve cardiac remodeling after injury (74, 218), although, at the same time, miR-34a has been studied as a tumor suppressor, and its mimicry has demonstrated anticancer effects (219, 220). Collectively, these examples further highlight a need to understand cell type–specific effects of miRNAs as well as the need for tissue-specific delivery.
Alternative preclinical models
Another challenge in miRNA therapeutic development, similar to genome-based therapies, is the lack of adequate drug-testing platforms. Genomic differences in nonprimate animal models limit their use as a predictive model of the effects of a specific miR-mimic or anti-miR. Moreover, interspecies differences in ion channels and biological pathways fail to recapitulate human biology; for example, the mouse heart beats nine times faster than the human heart (221). Current cardiovascular drug testing relies heavily on simplified models such as human embryonic kidney 293 cells and Chinese hamster ovary cells overexpressing ion channels (222). Of the studies surveyed here, most used primary rat cardiomyocytes or the rat H9C2 cell line, highlighting a need for in vitro human disease models. Although primary human cardiomyocytes would be an ideal candidate for such a model, they are in prohibitively short supply and difficult to isolate. Recently, iPS-CMs have emerged as a promising source to fill this void (223). iPS-CMs can be produced in near limitless quantity and matured to adult-like phenotypes (224), allowing for high-throughput screening of thousands of miRNA candidates. The generation of more complex, three-dimensional models of human cardiac tissues using iPS-CMs can further be used to model MI (225). Engineered cardiac tissue can also help shed light on miRNA transfer between cardiac cell types and myocyte-nonmyocyte communication, through extracellular vesicles or otherwise (226). Elucidating this biology may help further the design and development of miRNA therapeutics. How closely these models recapitulate human biology remains to be seen (227); however, they represent exciting technologies that can potentially decrease drug development cost.
Key remaining questions
There are several questions, which, when elucidated, would greatly advance the field of miRNA therapy for MI. A key question that remains is the timing of therapeutic delivery. The processes of cardiomyocyte death, fibroblast activation, and immune cell response occur acutely after ischemia onset. Because of the acute nature of cardiac injury, is it then necessary for intervention to take place rapidly after symptom onset? Or can a treatment administered in the later stages of wound healing (3 to 4 weeks) still show clinical improvement? Along a similar vein, could miRNA therapies potentially be preventative, suppressing these maladaptive processes before they begin? Very few preclinical studies have examined the timing of these deliveries and instead apply both the insult and the treatment simultaneously. Furthermore, the potential duration of clinical benefit from miRNA therapy is not well defined. Given the short-acting nature of miRNAs, it seems that repeated dosing would be required for long-lasting clinical benefit if early intervention is not possible. Should we target one cell type’s pathobiology or multiple? Is there a dominant effect of manipulating one cell type over another? The pleiotropic nature of miRNAs with convergent and divergent effects is one of its key advantages, yet it adds a degree of complexity to miRNA drug discovery unparalleled in other treatment modalities. The recent advancements in human iPS-derived in vitro models, including the ability to generate complex, three-dimensional models of human cardiac tissues, may represent a powerful and efficient strategy in answering some of these challenging questions.
CONCLUSION
Current interventions fail to recover the injured myocardium after MI, requiring the development of previously unexplored cardioprotective strategies. Here, we reviewed miRNAs as powerful regulators involved in post-MI remodeling and as the potential targets for MI therapy. Focusing on literature within the past 3 years and the foundational studies within the past 10 years, we identified a total of 213 publications describing the modulation of miRNAs in the treatment of MI, involving 116 unique miRNAs. Most of these miRNAs have been shown to play both beneficial and harmful roles in the progression of MI with divergent effects on different target cell types. To successfully translate the miRNA-based therapies of MI to the clinic, a more detailed understanding of the mechanisms and dynamics of miRNA effects on each cell type needs to be clearly defined. Clinical translation of these therapies will also require improved administration protocols, organ-specific delivery, and more predictive preclinical models.
Supplementary Material
Table S2. Summary of miRNAs reviewed.
Table S1. Complete list of miRNAs reviewed.
Funding:
We gratefully acknowledge funding support of our cardiac research by NIH [grants UH3EB025765 (G.V.-N.), P41EB027062 (G.V.-N.), HL076485 (G.V.-N.), T32GM00736 (T.N.), and F30HL145921 (B.L.)], NYSTEM [C32606GG (G.V.-N.)], NSF [grant ERC CELL-MET 16478 (G.V.-N.)], and NYSTEM [grant C32606GG (G.V.-N.)].
Footnotes
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/13/580/eabd0914/DC1
References (228–298)
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Abdulhak AB, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Jarlais DCD, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FGR, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo J-P, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, Grath JM, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KMV, Nasseri K, Norman P, O’Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Arden Pope C III, Porrini D, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara DP, Roberts T, De León FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh P-H, Yip P, Zabetian A, Zheng Z-J, Lopez AD, Murray CJL, Al Mazroa MA, Memish ZA, Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunt SA, Taking heart—cardiac transplantation past, present, and future. N. Engl. J. Med 355, 231–235 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Prabhu SD, Frangogiannis NG, The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res 119, 91–112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghildiyal M, Zamore PD, Small silencing RNAs: An expanding universe. Nat. Rev. Genet 10, 94–108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartel DP, MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Olson EN, MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci. Transl. Med 6, 239ps3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frampton AE, Castellano L, Colombo T, Giovannetti E, Krell J, Jacob J, Pellegrino L, Roca-Alonso L, Funel N, Gall TMH, De Giorgio A, Pinho FG, Fulci V, Britton DJ, Ahmad R, Habib NA, Coombes RC, Harding V, Knösel T, Stebbing J, Jiao LR, MicroRNAs cooperatively inhibit a network of tumor suppressor genes to promote pancreatic tumor growth and progression. Gastroenterology 146, 268–277.e18 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Yellon DM, Hausenloy DJ, Myocardial reperfusion injury. N. Engl. J. Med 357, 1121 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Elmore S, Apoptosis: A review of programmed cell death. Toxicol. Pathol 35, 495–516 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubli DA, Gustafsson ÅB, Cardiomyocyte health: Adapting to metabolic changes through autophagy. Trends Endocrinol. Metab 25, 156–164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu T-D, Guerquin-Kern J-L, Lechene CP, Lee RT, Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493, 433–436 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Liu S, Heallen T, Martin JF, The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol 15, 672–684 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Wei K, Serpooshan V, Hurtado C, Diez-Cuñado M, Zhao M, Maruyama S, Zhu W, Fajardo G, Noseda M, Nakamura K, Tian X, Liu Q, Wang A, Matsuura Y, Bushway P, Cai W, Savchenko A, Mahmoudi M, Schneider MD, van den Hoff MJB, Butte MJ, Yang PC, Walsh K, Zhou B, Bernstein D, Mercola M, Ruiz-Lozano P, Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 525, 479–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Zhang W, The role of HOPX in normal tissues and tumor progression. Biosci. Rep 40, BSR20191953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrow DA, Myocardial Infarction: A Companion to Braunwald’s Heart Disease (Elsevier, 2016). [Google Scholar]
- 16.Wang J-X, Jiao J-Q, Li Q, Long B, Wang K, Liu J-P, Li Y-R, Li P-F, MiR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med 17, 71–78 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Wang K, Gan T-Y, Li N, Liu C-Y, Zhou L-Y, Gao J-N, Chen C, Yan K-W, Ponnusamy M, Zhang Y-H, Li P-F, Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 24, 1111–1120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Liu C-Y, Zhang X-J, Feng C, Zhou L-Y, Zhao Y, Li P-F, MiR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell Death Differ. 22, 1058–1068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K, Long B, Zhou L-Y, Liu F, Zhou Q-Y, Liu C-Y, Fan Y-Y, Li P-F, CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat. Commun 5, 3596 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Wang K, Zhou L-Y, Wang J-X, Wang Y, Sun T, Zhao B, Yang Y-J, An T, Long B, Li N, Liu C-Y, Gong Y, Gao J-N, Dong Y-H, Zhang J, Li P-F, E2F1-dependent MIR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat. Commun 6, 7619 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Hang P, Sun C, Guo J, Zhao J, Du Z, BDNF-mediates down-regulation of microRNA-195 inhibits ischemic cardiac apoptosis in rats. Int. J. Biol. Sci 12, 979–989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang Y, Zheng J, Sun Y, Wu Z, Liu Z, Huang G, MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int. Heart J 50, 377–387 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Yang W, Han Y, Yang C, Chen Y, Zhao W, Su X, Yang K, Jin W, MicroRNA-19b-1 reverses ischaemia-induced heart failure by inhibiting cardiomyocyte apoptosis and targeting Bcl2 l11/BIM. Heart Vessels 34, 1221–1229 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D, miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J. Exp. Med 208, 549–560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayoumi AS, Park K-M, Wang Y, Teoh J-P, Aonuma T, Tang Y, Su H, Weintraub NL, Kim L-M, A carvedilol-responsive microRNA, miR-125b-5p protects the heart from acute myocardial infarction by repressing pro-apoptotic bak1 and klf13 in cardiomyocytes. J. Mol. Cell. Cardiol 114, 72–82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song S, Seo H-H, Lee S-Y, Lee CY, Lee J, Yoo K-J, Yoon C, Choi E, Hwang K-C, Lee S, MicroRNA-17-mediated down-regulation of apoptotic protease activating factor 1 attenuates apoptosome formation and subsequent apoptosis of cardiomyocytes. Biochem. Biophys. Res. Commun 465, 299–304 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Ren S, Xia J, Wei Y, Xi Y, EIF4A3-induced circ-BNIP3 aggravated hypoxia-induced injury of H9c2 cells by targeting miR-27a-3p/BNIP3. Mol. Ther. Nucleic Acids 19, 533–545 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He S-F, Zhu H-J, Han Z-Y, Wu H, Jin S-Y, Irwin MG, Zhang Y, MicroRNA-133b-5p is involved in cardioprotection of morphine preconditioning in rat cardiomyocytes by targeting fas. Can. J. Cardiol 32, 996–1007 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Tian M-M, Mi C-J, Chen K-L, Ji Y-C, Wang L, Zhang J, Cheng K, Exercise protects the heart against myocardial infarction through upregulation of miR-1192. Biochem. Biophys. Res. Commun 521, 1061–1069 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Yang J, Zhang J, Liu X-W, Yang C-J, Fan Z-X, Wang H-B, Yang Y, Zheng T, Yang J, Inhibition of microRNA-327 ameliorates ischemia/reperfusion injury-induced cardiomyocytes apoptosis through targeting apoptosis repressor with caspase recruitment domain. J. Cell. Physiol 235, 3753–3767 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C, PI3K/Akt and apoptosis: Size matters. Oncogene 22, 8983–8998 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Zhao L, Yang XR, Han X, MicroRNA-146b induces the Pi3k/Akt/NF-κB signaling pathway to reduce vascular inflammation and apoptosis in myocardial infarction by targeting PTEN. Exp. Ther. Med 17, 1171–1181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao L, Zhou Q, Wang L, Hou G, MicroRNA-182-5p protects H9c2 cardiomyocytes from hypoxia-induced apoptosis by down-regulation of PTEN. Int. J. Clin. Exp. Pathol 10, 5220–5226 (2017). [Google Scholar]
- 34.Xing X, Guo S, Zhang G, Liu Y, Bi S, Wang X, Lu Q, miR-26a-5p protects against myocardial ischemia/reperfusion injury by regulating the PTEN/PI3K/AKT signaling pathway. Braz. J. Med. Biol. Res 53, e9106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ke Z-P, Xu P, Shi Y, Gao A-M, MicroRNA-93 inhibits ischemia-reperfusion induced cardiomyocyte apoptosis by targeting PTEN. Oncotarget 7, 28796–28805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song C-L, Liu B, Diao H-Y, Shi Y-F, Zhang J-C, Li Y-X, Liu N, Yu Y-P, Wang G, Wang J-P, Li Q, Down-regulation of microRNA-320 suppresses cardiomyocyte apoptosis and protects against myocardial ischemia and reperfusion injury by targeting IGF-1. Oncotarget 7, 39740–39757 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun R, Zhang L, Long non-coding RNA MALAT1 regulates cardiomyocytes apoptosis after hypoxia/reperfusion injury via modulating miR-200a-3p/PDCD4 axis. Biomed. Pharmacother 111, 1036–1045 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Luther KM, Haar L, Guinness MM, Wang Y, Lynch TL IV, Phan A, Song Y, Shen Z, Gardner G, Kuffel G, Ren X, Zilliox MJ, Jones WK, Exosomal miR-21a-5p mediates cardioprotection by mesenchymal stem cells. J. Mol. Cell. Cardiol 119, 125–137 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Gu H, Liu Z, Li Y, Xie Y, Yao J, Zhu Y, Xu J, Dai Q, Zhong C, Zhu H, Ding S, Zhou L, Serum-derived extracellular vesicles protect against acute myocardial infarction by regulating miR-21/PDCD4 signaling pathway. Front. Physiol 9, 348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C, MicroRNA-21 protects against the H2O2-induced injury on cardiac myocytes via its target gene PDCD4. J. Mol. Cell. Cardiol 47, 5–14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Jia Z, Zhang C, Sun M, Wang W, Chen P, Ma K, Zhang Y, Li X, Zhou C, miR-499 protects cardiomyocytes from H2O2-induced apoptosis via its effects on Pdcd4 and Pacs2. RNA Biol. 11, 339–350 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma J, Zhang J, Wang Y, Long K, Wang X, Jin L, Tang Q, Zhu L, Tang G, Li X, Li M, MiR-532-5p alleviates hypoxia-induced cardiomyocyte apoptosis by targeting PDCD4. Gene 675, 36–43 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Pan Z, Sun X, Ren J, Li X, Gao X, Lu C, Zhang Y, Sun H, Wang Y, Wang H, Wang J, Xie L, Lu Y, Yang B, miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLOS ONE 7, e50515 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong H, Tao T, Chen S, Liang C, Qiu Y, Zhou Y, Zhang R, MicroRNA-143 promotes cardiac ischemia-mediated mitochondrial impairment by the inhibition of protein kinase Cepsilon. Basic Res. Cardiol 112, 60 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Men M, Yang W, Zheng H, Xue S, MiR-31 downregulation protects against cardiac ischemia/reperfusion injury by targeting protein kinase C epsilon (PKCε) directly. Cell. Physiol. Biochem 36, 179–190 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Meng X, Ji Y, Wan Z, Zhao B, Feng C, Zhao J, Li H, Song Y, Inhibition of miR-363 protects cardiomyocytes against hypoxia-induced apoptosis through regulation of Notch signaling. Biomed. Pharmacother 90, 509–516 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Xu H, Jin L, Chen Y, Li J, Downregulation of microRNA-429 protects cardiomyocytes against hypoxia-induced apoptosis by increasing Notch1 expression. Int. J. Mol. Med 37, 1677–1685 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Su X, Shen Y, Jin Y, Luo T, Kim I-M, Weintraub NL, Tang Y, MiR322 mediates cardioprotection against ischemia/reperfusion injury via FBXW7/notch pathway. J. Mol. Cell. Cardiol 133, 67–74 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan K, An T, Zhai M, Huang Y, Wang Q, Wang Y, Zhang R, Wang T, Liu J, Zhang Y, Zhang J, Wang K, Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. 10, 500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin D, Cui B, Ma J, Ren J, MiR-183-5p protects rat hearts against myocardial ischemia/reperfusion injury through targeting VDAC1. Biofactors 46, 83–93 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Su Q, Liu Y, Lv X-W, Dai R-X, Yang X-H, Kong B-H, LncRNA TUG1 mediates ischemic myocardial injury by targeting miR-132-3p/HDAC3 axis. Am. J. Physiol. Circ. Physiol 318, H332–H344 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Shin S, Choi J-W, Moon H, Lee CY, Park J-H, Lee J, Seo H-H, Han G, Lim S, Lee S, Kim SW, Hwang K-C, Simultaneous suppression of multiple programmed cell death pathways by miRNA-105 in cardiac ischemic injury. Mol. Ther. Nucleic Acids 14, 438–449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang K, Liu F, Liu C-Y, An T, Zhang J, Zhou L-Y, Wang M, Dong Y-H, Li N, Gao J-N, Zhao Y-F, Li P-F, The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 23, 1394–1405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qin D, Wang X, Li Y, Yang L, Wang R, Peng J, Essandoh K, Mu X, Peng T, Han Q, Yu K-J, Fan G-C, MicroRNA-223-5p and −3p cooperatively suppress necroptosis in ischemic/reperfused hearts. J. Biol. Chem 291, 20247–20259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J-X, Zhang X-J, Li Q, Wang K, Wang Y, Jiao J-Q, Feng C, Teng S, Zhou L-Y, Gong Y, Zhou Z-X, Liu J, Wang J-L, Li P.-f., MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting FADD. Circ. Res 117, 352–363 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Wang K, Liu F, Zhou L-Y, Ding S-L, Long B, Liu C-Y, Sun T, Fan Y-Y, Sun L, Li P-F, MiR-874 regulates myocardial necrosis by targeting caspase-8. Cell Death Dis. 4, e709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang K, Liu C-Y, Zhou L-Y, Wang J-X, Wang M, Zhao B, Zhao W-K, Xu S-J, Fan L-H, Zhang X-J, Feng C, Wang C-Q, Zhao Y-F, Li P-F, APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat. Commun 6, 6779 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Liu J, Jiang M, Deng S, Lu J, Huang H, Zhang Y, Gong P, Shen X, Ruan H, Jin M, Wang H, miR-93-5p-containing exosomes treatment attenuates acute myocardial infarction-induced myocardial damage. Mol. Ther. Nucleic Acids 11, 103–115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gupta SK, Foinquinos A, Thum S, Remke J, Zimmer K, Bauters C, de Groote P, Boon RA, de Windt LJ, Preissl S, Hein L, Batkai S, Pinet F, Thum T, Preclinical development of a microRNA-based therapy for elderly patients with myocardial infarction. J. Am. Coll. Cardiol 68, 1557–1571 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, Remke J, Caprio M, Jentzsch C, Engelhardt S, Geisendorf S, Glas C, Hofmann TG, Nessling M, Richter K, Schiffer M, Carrier L, Napp LC, Bauersachs J, Chowdhury K, Thum T, The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun 3, 1078 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su Q, Liu Y, Lv X-W, Ye Z-L, Sun Y-H, Kong B-H, Qin Z-B, Inhibition of lncRNA TUG1 upregulates miR-142-3p to ameliorate myocardial injury during ischemia and reperfusion via targeting HMGB1- and Rac1-induced autophagy. J. Mol. Cell. Cardiol 133, 12–25 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Hu S, Cao S, Tong Z, Liu J, FGF21 protects myocardial ischemia-reperfusion injury through reduction of miR-145-mediated autophagy. Am. J. Transl. Res 10, 3677–3688 (2018). [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Q, Zhou Y, Richards AM, Wang P, Up-regulation of miRNA-221 inhibits hypoxia/reoxygenation-induced autophagy through the DDIT4/mTORC1 and Tp53inp1/p62 pathways. Biochem. Biophys. Res. Commun 474, 168–174 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Li Y, Yang R, Guo B, Zhang H, Zhang H, Liu S, Li Y, Exosomal miR-301 derived from mesenchymal stem cells protects myocardial infarction by inhibiting myocardial autophagy. Biochem. Biophys. Res. Commun 514, 323–328 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Lin B, Feng D, Xu J, Cardioprotective effects of microRNA-18a on acute myocardial infarction by promoting cardiomyocyte autophagy and suppressing cellular senescence via brain derived neurotrophic factor. Cell Biosci. 9, 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Z, Song Y, Liu L, Hou N, An X, Zhan D, Li Y, Zhou L, Li P, Yu L, Xia J, Zhang Y, Wang J, Yang X, MiR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 24, 1205–1213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang Y, Li Y, Chen X, Cheng X, Liao Y, Yu X, Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J. Mol. Med 94, 711–724 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Guo X, Wu X, Han Y, Tian E, Cheng J, LncRNA MALAT1 protects cardiomyocytes from isoproterenol-induced apoptosis through sponging miR-558 to enhance ULK1-mediated protective autophagy. J. Cell. Physiol 234, 10842–10854 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Tian Y, Liu Y, Wang T, Zhou N, Kong J, Chen L, Snitow M, Morley M, Li D, Petrenko N, Zhou S, Lu M, Gao E, Koch WJ, Stewart KM, Morrisey EE, A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med 7, 279ra38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eulalio A, Mano M, Ferro MD, Zentilin L, Sinagra G, Zacchigna S, Giacca M, Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376–381 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Torrini C, Cubero RJ, Dirkx E, Braga L, Ali H, Prosdocimo G, Gutierrez MI, Collesi C, Licastro D, Zentilin L, Mano M, Zacchigna S, Vendruscolo M, Marsili M, Samal A, Giacca M, Common regulatory pathways mediate activity of microRNAs inducing cardiomyocyte proliferation. Cell Rep. 27, 2759–2771.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandonà L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M, MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 569, 418–422 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xiao Y, Zhang Y, Chen Y, Li J, Zhang Z, Sun Y, Shen H, Zhao Z, Huang Z, Zhang W, Chen W, Shen Z, Inhibition of microRNA-9-5p protects against cardiac remodeling following myocardial infarction in mice. Hum. Gene Ther 30, 286–301 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Yang Y, Cheng H-W, Qiu Y, Dupee D, Noonan M, Lin Y-D, Fisch S, Unno K, Sereti K-I, Liao R, MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ. Res 117, 450–459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu Y, Jin G, Li B, Chen Y, Zhong L, Chen G, Chen X, Zhong J, Liao W, Liao Y, Wang Y, Bin J, Suppression of miRNA let-7i-5p promotes cardiomyocyte proliferation and repairs heart function post injury by targetting CCND2 and E2F2. Clin. Sci 133, 425–441 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Borden A, Kurian J, Nickoloff E, Yang Y, Troupes CD, Ibetti J, Lucchese AM, Gao E, Mohsin S, Koch WJ, Houser SR, Kishore R, Khan M, Transient introduction of miR-294 in the heart promotes cardiomyocyte cell cycle reentry after injury. Circ. Res 125, 14–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang W, Feng Y, Liang J, Yu H, Wang C, Wang B, Wang M, Jiang L, Meng W, Cai W, Medvedovic M, Chen J, Paul C, Davidson WS, Sadayappan S, Stambrook PJ, Yu X-Y, Wang Y, Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat. Commun 9, 700 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang J, Wu L, Li Z, Fu G, miR-1231 exacerbates arrhythmia by targeting calciumchannel gene CACNA2D2 in myocardial infarction. Am. J. Transl. Res 9, 1822–1833 (2017). [PMC free article] [PubMed] [Google Scholar]
- 79.Liu X, Zhang Y, Du W, Liang H, He H, Zhang L, Pan Z, Li X, Xu C, Zhou Y, Wang L, Qian M, Liu T, Yin H, Lu Y, Yang B, Shan H, MiR-223-3p as a novel microRNA regulator of expression of voltage-gated K+ Channel Kv4.2 in acute myocardial infarction. Cell. Physiol. Biochem 39, 102–114 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Zhou X, Sun F, Luo S, Zhao W, Yang T, Zhang G, Gao M, Lu R, Shu Y, Mu W, Zhuang Y, Ding F, Xu C, Lu Y, Let-7a is an antihypertrophic regulator in the heart via targeting calmodulin. Int. J. Biol. Sci 13, 22–31 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang Y, Del Re DP, Nakano N, Sciarretta S, Zhai P, Park J, Sayed D, Shirakabe A, Matsushima S, Park Y, Tian B, Abdellatif M, Sadoshima J, MIR-206 mediates YAP-induced cardiac hypertrophy and survival. Circ. Res 117, 891–904 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang J, Hu F, Fu X, Jiang Z, Zhang W, Chen K, MiR-128/SOX7 alleviates myocardial ischemia injury by regulating IL-33/sST2 in acute myocardial infarction. Biol. Chem 400, 533–544 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Li A, Yu Y, Ding X, Qin Y, Jiang Y, Wang X, Liu G, Chen X, Yue E, Sun X, Zahra SM, Yan Y, Ren L, Wang S, Chai L, Bai Y, Yang B, MiR-135b protects cardiomyocytes from infarction through restraining the NLRP3/caspase-1/IL-1β pathway. Int. J. Cardiol 307, 137–145 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Yuan M, Zhang L, You F, Zhou J, Ma Y, Yang F, Tao L, MiR-145-5p regulates hypoxia-induced inflammatory response and apoptosis in cardiomyocytes by targeting CD40. Mol. Cell. Biochem 431, 123–131 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Turner NA, Das A, Warburton P, O’Regan DJ, Ball SG, Porter KE, Interleukin-1α stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am. J. Physiol. Hear. Circ. Physiol 297, H1117–H1127 (2009). [DOI] [PubMed] [Google Scholar]
- 86.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U, Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123, 594–604 (2011). [DOI] [PubMed] [Google Scholar]
- 87.Lindsey ML, Zamilpa R, Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc. Ther 30, 31–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lijnen PJ, Petrov VV, Fagard RH, Induction of cardiac fibrosis by transforming growth factor-β1. Mol. Genet. Metab 71, 418–435 (2000). [DOI] [PubMed] [Google Scholar]
- 89.Sun Y, Zhang JQ, Zhang J, Ramires FJA, Angiotensin II, transforming growth factor-β1 and repair in the infarcted heart. J. Mol. Cell. Cardiol 30, 1559–1569 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Ye H, Cai P-C, Zhou Q, Ma W-L, Transforming growth factor-β1 suppresses the up-regulation of matrix metalloproteinase-2 by lung fibroblasts in response to tumor necrosis factor-α. Wound Repair Regen. 19, 392–399 (2011). [DOI] [PubMed] [Google Scholar]
- 91.Li A-H, Liu PP, Villarreal FJ, Garcia RA, Dynamic changes in myocardial matrix and relevance to disease: Translational perspectives. Circ. Res 114, 916–927 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Nagaraju CK, Dries E, Popovic N, Singh AA, Haemers P, Roderick HL, Claus P, Sipido KR, Driesen RB, Global fibroblast activation throughout the left ventricle but localized fibrosis after myocardial infarction. Sci. Rep 7, 10801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frangogiannis NG, Michael LH, Entman ML, Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc. Res 48, 89–100 (2000). [DOI] [PubMed] [Google Scholar]
- 94.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD, Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest 128, 2127–2143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagaraju CK, Robinson EL, Abdesselem M, Trenson S, Dries E, Gilbert G, Janssens S, Van Cleemput J, Rega F, Meyns B, Roderick HL, Driesen RB, Sipido KR, Myofibroblast phenotype and reversibility of fibrosis in patients with end-stage heart failure. J. Am. Coll. Cardiol 73, 2267–2282 (2019). [DOI] [PubMed] [Google Scholar]
- 96.Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, Feng S, Xie L, Lu C, Yuan Y, Zhang Y, Wang Y, Lu Y, Yang B, miR-101 inhibited post-infarct cardiac fibrosis and improved left ventricular compliance via FOS/TGFβ1 pathway. Circulation 126, 840–850 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Zhao X, Wang K, Liao Y, Zeng Q, Li Y, Hu F, Liu Y, Meng K, Qian C, Zhang Q, Guan H, Feng K, Zhou Y, Du Y, Chen Z, MicroRNA-101a inhibits cardiac fibrosis induced by hypoxia via targeting TGF RI on cardiac fibroblasts. Cell. Physiol. Biochem 35, 213–226 (2015). [DOI] [PubMed] [Google Scholar]
- 98.Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H, Xu J, Che L, Chen X, Sluijter JPG, Das S, Cretoiu D, Xu B, Zhong J, Xiao J, Li X, Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics 6, 2068–2083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang J, Huang W, Xu R, Nie Y, Cao X, Meng J, Xu X, Hu S, Zheng Z, MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med 16, 2150–2160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hong Y, Cao H, Wang Q, Ye J, Sui L, Feng J, Cai X, Song H, Zhang X, Chen X, MiR-22 may suppress fibrogenesis by targeting TGFβR I in cardiac fibroblasts. Cell. Physiol. Biochem 40, 1345–1353 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Liang H, Zhang C, Ban T, Liu Y, Mei L, Piao X, Zhao D, Lu Y, Chu W, Yang B, A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int. J. Biochem. Cell Biol 44, 2152–2160 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Du W, Liang H, Gao X, Li X, Zhang Y, Pan Z, Li C, Wang Y, Liu Y, Yuan W, Ma N, Chu W, Shan H, Lu Y, MicroRNA-328, a potential anti-fibrotic target in cardiac interstitial fibrosis. Cell. Physiol. Biochem 39, 827–836 (2016). [DOI] [PubMed] [Google Scholar]
- 103.Yuan J, Chen H, Ge D, Xu Y, Xu H, Yang Y, Gu M, Zhou Y, Zhu J, Ge T, Chen Q, Gao Y, Wang Y, Li X, Zhao Y, Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell. Physiol. Biochem 42, 2207–2219 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Huang Y, Qi Y, Du J-Q, Zhang D.-f., MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin. Ther. Targets 18, 1355–1365 (2014). [DOI] [PubMed] [Google Scholar]
- 105.Wang X, Morelli MB, Matarese A, Sardu C, Santulli G, Cardiomyocyte-derived exosomal microRNA-92a mediates post-ischemic myofibroblast activation both in vitro and ex vivo. ESC Hear. Fail 7, 285–289 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morelli MB, Shu J, Sardu C, Matarese A, Santulli G, Cardiosomal microRNAs are essential in post-infarction myofibroblast phenoconversion. Int. J. Mol. Sci 21, 201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dorn LE, Petrosino JM, Wright P, Accornero F, CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell. Cardiol 121, 205–211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lipson KE, Wong C, Teng Y, Spong S, CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 5, S24 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE, miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res 104, 170–178 (2009). [DOI] [PubMed] [Google Scholar]
- 110.Chen L, Ji Q, Zhu H, Ren Y, Fan Z, Tian N, miR-30a attenuates cardiac fibrosis in rats with myocardial infarction by inhibiting CTGF. Exp. Ther. Med 15, 4318–4324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JTR, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S, MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456, 980–984 (2008). [DOI] [PubMed] [Google Scholar]
- 112.Adam O, Löhfelm B, Thum T, Gupta SK, Puhl SL, Schäfers HJ, Böhm M, Laufs U, Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res. Cardiol 107, 278 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Cardin S, Guasch E, Luo X, Naud P, Quang KL, Shi YF, Tardif J-C, Comtois P, Nattel S, Role for MicroRNA-21 in atrial profibrillatory fibrotic remodeling associated with experimental postinfarction heart failure. Circ. Arrhythm. Electrophysiol 5, 1027–1035 (2012). [DOI] [PubMed] [Google Scholar]
- 114.Yuan X, Pan J, Wen L, Gong B, Li J, Gao H, Tan W, Liang S, Zhang H, Wang X, MiR-144-3p enhances cardiac fibrosis after myocardial infarction by targeting PTEN. Front. Cell Dev. Biol 7, 249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T, Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Invest 119, 2634–2647 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li C, Li J, Xue K, Zhang J, Wang C, Zhang Q, Chen X, Gao C, Yu X, Sun L, MicroRNA-143-3p promotes human cardiac fibrosis via targeting sprouty3 after myocardial infarction. J. Mol. Cell. Cardiol 129, 281–292 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Wang Y-S, Li S-H, Guo J, Mihic A, Wu J, Sun L, Davis K, Weisel RD, Li R-K, Role of miR-145 in cardiac myofibroblast differentiation. J. Mol. Cell. Cardiol 66, 94–105 (2014). [DOI] [PubMed] [Google Scholar]
- 118.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN, microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 22, 3242–3254 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boštjančič E, Zidar N, Štajer D, Glavač D, MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology 115, 163–169 (2010). [DOI] [PubMed] [Google Scholar]
- 120.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN, Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A 105, 13027–13032 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Port JD, Walker LA, Polk J, Nunley K, Buttrick PM, Sucharov CC, Temporal expression of miRNAs and mRNAs in a mouse model of myocardial infarction. Physiol. Genomics 43, 1087–1095 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Castoldi G, Di Gioia CRT, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini G, Condorelli G, Stella A, MiR-133a regulates collagen 1A1: Potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J. Cell. Physiol 227, 850–856 (2012). [DOI] [PubMed] [Google Scholar]
- 123.Sassi Y, Avramopoulos P, Ramanujam D, Grüter L, Werfel S, Giosele S, Brunner A-D, Esfandyari D, Papadopoulou AS, De Strooper B, Hübner N, Kumarswamy R, Thum T, Yin X, Mayr M, Laggerbauer B, Engelhardt S, Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat. Commun 8, 1614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xiang FL, Fang M, Yutzey KE, Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun 8, 712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen M-H, Liu J-C, Liu Y, Hu Y-C, Cai X-F, Yin D-C, MicroRNA-199a regulates myocardial fibrosis in rats by targeting SFRP5. Eur. Rev. Med. Pharmacol. Sci 23, 3976–3983 (2019). [DOI] [PubMed] [Google Scholar]
- 126.Cui J, Qi S, Liao R, Su D, Wang Y, Xue S, MiR-574–5p promotes the differentiation of human cardiac fibroblasts via regulating ARID3A. Biochem. Biophys. Res. Commun 521, 427–433 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Yuan X, Pan J, Wen L, Gong B, Li J, Gao H, Tan W, Liang S, Zhang H, Wang X, MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J. Cell. Mol. Med 24, 227–237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brutsaert DL, Cardiac endothelial-myocardial signaling: Its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev 83, 59–115 (2003). [DOI] [PubMed] [Google Scholar]
- 129.Falque H, Bochaton T, Bernelin H, Paccalet A, Da Silva CC, Baetz D, Bonnefoy-Cudraz E, Mewton N, Ovize M, Endothelial activation and infarct size at the acute phase of myocardial infarction. Arch. Cardiovasc. Dis. Suppl 10, 178 (2018). [Google Scholar]
- 130.Hernández-Reséndiz S, Muñoz-Vega M, Contreras WE, Crespo-Avilan GE, Rodriguez-Montesinos J, Arias-Carrión O, Pérez-Méndez O, Boisvert WA, Preissner KT, Cabrera-Fuentes HA, Responses of endothelial cells towards ischemic conditioning following acute myocardial infarction. Cond. Med 1, 247–258 (2018). [PMC free article] [PubMed] [Google Scholar]
- 131.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM, Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, Spinale FG, Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol 290, H232–H239 (2006). [DOI] [PubMed] [Google Scholar]
- 132.Fallah A, Sadeghinia A, Kahroba H, Samadi A, Heidari HR, Bradaran B, Zeinali S, Molavi O, Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. Biomed. Pharmacother 110, 775–785 (2019). [DOI] [PubMed] [Google Scholar]
- 133.Zhang Y, Qin W, Zhang L, Wu X, Du N, Hu Y, Li X, Shen N, Xiao D, Zhang H, Li Z, Zhang Y, Yang H, Gao F, Du Z, Xu C, Yang B, MicroRNA-26a prevents endothelial cell apoptosis by directly targeting TRPC6 in the setting of atherosclerosis. Sci. Rep 5, 9401 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Qin B, Shu Y, Xiao L, Lu T, Lin Y, Yang H, Lu Z, MicroRNA-150 targets ELK1 and modulates the apoptosis induced by ox-LDL in endothelial cells. Mol. Cell. Biochem 429, 45–58 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Chen P, Zhong J, Ye J, He Y, Liang Z, Cheng Y, Zheng J, Chen H, Chen C, miR-324-5p protects against oxidative stress-induced endothelial progenitor cell injury by targeting Mtfr1. J. Cell. Physiol 234, 22082–22092 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, Chandrashekhar YS, Hall JL, Roy S, Zeng Y, Ramakrishnan S, Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J. Clin. Invest 120, 4141–4154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dai G-H, Ma P-Z, Song X-B, Liu N, Zhang T, Wu B, MicroRNA-223-3p inhibits the angiogenesis of ischemic cardiac microvascular endothelial cells via affecting RPS6KB1/hif-1a signal pathway. PLOS ONE 9, e108468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JTG, Sohn-Lee C, Loyer X, Soutschek J, Brand T, Tuschl T, Heineke J, Martin U, Schulte-Merker S, Ertl G, Engelhardt S, Bauersachs J, Thum T, MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 124, 720–730 (2011). [DOI] [PubMed] [Google Scholar]
- 139.Wang S, Aurora AB, Johnson BA, Qi X, Anally JM, Hill JA, Richardson JA, Bassel-Duby R, Olson EN, The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 15, 261–271 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ma T, Chen Y, Chen Y, Meng Q, Sun J, Shao L, Yu Y, Huang H, Hu Y, Yang Z, Yang J, Shen Z, MicroRNA-132, delivered by mesenchymal stem cell-derived exosomes, promote angiogenesis in myocardial infarction. Stem Cells Int. 2018, 3290372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Meloni M, Marchetti M, Garner K, Littlejohns B, Sala-Newby G, Xenophontos N, Floris I, Suleiman M-S, Madeddu P, Caporali A, Emanueli C, Local inhibition of microRNA-24 improves reparative angiogenesis and left ventricle remodeling and function in mice with myocardial infarction. Mol. Ther 21, 1390–1402 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu S, Chen J, Shi J, Zhou W, Wang L, Fang W, Zhong Y, Chen X, Chen Y, Sabri A, Liu S, M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res. Cardiol 115, 22 (2020). [DOI] [PubMed] [Google Scholar]
- 143.Joris V, Gomez EL, Menchi L, Lobysheva I, Di Mauro V, Esfahani H, Condorelli G, Balligand J-L, Catalucci D, Dessy C, MicroRNA-199a-3p and MicroRNA-199a-5p take part to a redundant network of regulation of the NOS (NO Synthase)/NO pathway in the endothelium. Arterioscler. Thromb. Vasc. Biol 38, 2345–2357 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Zhao X, Wei X, Wang X, Qi G, Long non-coding RNA NORAD regulates angiogenesis of human umbilical vein endothelial cells via miR-590-3p under hypoxic conditions. Mol. Med. Rep 21, 2560–2570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fan Z-G, Qu X-L, Chu P, Gao Y-L, Gao X-F, Chen S-L, Tian N-L, MicroRNA-210 promotes angiogenesis in acute myocardial infarction. Mol. Med. Rep 17, 5658–5665 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang N, Chen C, Yang D, Liao Q, Luo H, Wang X, Zhou F, Yang X, Yang J, Zeng C, Wang WE, Mesenchymal stem cells-derived extracellular vesicles, via miR-210, improve infarcted cardiac function by promotion of angiogenesis. Biochim. Biophys. Acta - Mol. Basis Dis 1863, 2085–2092 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Gallant-Behm CL, Piper J, Dickinson BA, Dalby CM, Pestano LA, Jackson AL, A synthetic microRNA-92a inhibitor (MRG-110) accelerates angiogenesis and wound healing in diabetic and nondiabetic wounds. Wound Repair Regen. 26, 311–323 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S, MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in Mice. Science 324, 1710–1713 (2009). [DOI] [PubMed] [Google Scholar]
- 149.Veliceasa D, Biyashev D, Qin G, Misener S, Mackie AR, Kishore R, Volpert OV, Therapeutic manipulation of angiogenesis with miR-27b. Vasc. Cell 7, 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li C-C, Qiu X-T, Sun Q, Zhou J-P, Yang H-J, Wu W-Z, He L-F, Tang C-E, Zhang G-G, Bai Y-P, Endogenous reduction of miR-185 accelerates cardiac function recovery in mice following myocardial infarction via targeting of cathepsin K. J. Cell. Mol. Med 23, 1164–1173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bayoumi AS, Teoh J-P, Aonuma T, Yuan Z, Ruan X, Tang Y, Su H, Weintraub NL, Kim I-M, MicroRNA-532 protects the heart in acute myocardial infarction, and represses prss23, a positive regulator of endothelial-to-mesenchymal transition. Cardiovasc. Res 113, 1603–1614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bartman CM, Oyama Y, Brodsky K, Khailova L, Walker L, Koeppen M, Eckle T, Wang M, Intense light-elicited upregulation of miR-21 facilitates glycolysis and cardioprotection through Per2-dependent mechanisms. PLOS ONE 12, e0176243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu RR, Li J, Gong JY, Kuang F, Liu JY, Zhang YS, Ma QL, Song CJ, Truax AD, Gao F, Yang K, Jin BQ, Chen LH, MicroRNA-141 regulates the expression level of ICAM-1 on endothelium to decrease myocardial ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol 309, H1303–H1313 (2015). [DOI] [PubMed] [Google Scholar]
- 154.Yang L, Wang B, Zhou Q, Wang Y, Liu X, Liu Z, Zhan Z, MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 9, 769 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gupta SK, Itagaki R, Zheng X, Batkai S, Thum S, Ahmad F, Van Aelst LN, Sharma A, Piccoli M-T, Weinberger F, Fiedler J, Heuser M, Heymans S, Falk CS, Förster R, Schrepfer S, Thum T, miR-21 promotes fibrosis in an acute cardiac allograft transplantation model. Cardiovasc. Res 110, 215–226 (2016). [DOI] [PubMed] [Google Scholar]
- 156.Li J, Cai SXX, He Q, Zhang H, Friedberg D, Wang F, Redington AN, Intravenous miR-144 reduces left ventricular remodeling after myocardial infarction. Basic Res. Cardiol 113, 36 (2018). [DOI] [PubMed] [Google Scholar]
- 157.Xu H-M, Sui F-H, Sun M-H, Guo G-L, Downregulated microRNA-224 aggravates vulnerable atherosclerotic plaques and vascular remodeling in acute coronary syndrome through activation of the TGF-β/Smad pathway. J. Cell. Physiol 234, 2537–2551 (2019). [DOI] [PubMed] [Google Scholar]
- 158.Yang J, Brown ME, Zhang H, Martinez M, Zhao Z, Bhutani S, Yin S, Trac D, Xi JJ, Davis ME, High-throughput screening identifies microRNAs that target Nox2 and improve function after acute myocardial infarction. Am. J. Physiol. Hear. Circ. Physiol 312, H1002–H1012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, Berman BP, Marbán E, Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation 136, 200–214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Garikipati VNS, Verma SK, Jolardarashi D, Cheng Z, Ibetti J, Cimini M, Tang Y, Khan M, Yue Y, Benedict C, Nickoloff E, Truongcao MM, Gao E, Krishnamurthy P, Goukassian DA, Koch WJ, Kishore R, Therapeutic inhibition of miR-375 attenuates post-myocardial infarction inflammatory response and left ventricular dysfunction via PDK-1-AKT signalling axis. Cardiovasc. Res 113, 938–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shao L, Zhang Y, Pan X, Liu B, Liang C, Zhang Y, Wang Y, Yan B, Xie W, Sun Y, Shen Z, Yu X-Y, Li Y, Knockout of beta-2 microglobulin enhances cardiac repair by modulating exosome imprinting and inhibiting stem cell-induced immune rejection. Cell. Mol. Life Sci 77, 937–952 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhao J, Li X, Hu J, Chen F, Qiao S, Sun X, Gao L, Xie J, Xu B, Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc. Res 115, 1205–1216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bejerano T, Etzion S, Elyagon S, Etzion Y, Cohen S, Nanoparticle delivery of miRNA-21 mimic to cardiac macrophages improves myocardial remodeling after myocardial infarction. Nano Lett. 18, 5885–5891 (2018). [DOI] [PubMed] [Google Scholar]
- 164.Ye ZM, Yang S, Xia Y.-p., Hu R.-t., Chen S, Li B.-w., Chen S.-l., Luo X.-y., Mao L, Li Y, Jin H, Qin C, Hu B, LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 10, 138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Arif M, Pandey R, Alam P, Jiang S, Sadayappan S, Paul A, Ahmed RPH, MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J. Mol. Med 95, 1369–1385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, Robbins RC, Wu JC, MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 122, S124–S131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Huang L, Guo B, Liu S, Miao C, Li Y, Inhibition of the LncRNA Gpr19 attenuates ischemia-reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR-324-5p/Mtfr1 axis. IUBMB Life 72, 373–383 (2020). [DOI] [PubMed] [Google Scholar]
- 168.Wang X, Zhang X, Ren X-P, Chen J, Liu H, Yang J, Medvedovic M, Hu Z, Fan G-C, MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation 122, 1308–1318 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Su Q, Lv X-W, Sun Y-H, Ye Z-L, Kong B-H, Qin Z-B, MicroRNA-494 inhibits the LRG1 expression to induce proliferation and migration of VECs in rats following myocardial infarction. Mol. Ther. Nucleic Acids 18, 110–122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 170.el Azzouzi H, Leptidis S, Dirkx E, Hoeks J, van Bree B, Brand K, McClellan EA, Poels E, Sluimer JC, van den Hoogenhof MMG, Armand AS, Yin X, Langley S, Bourajjaj M, Olieslagers S, Krishnan J, Vooijs M, Kurihara H, Stubbs A, Pinto YM, Krek W, Mayr M, Martins PADC, Schrauwen P, De Windt LJ, The hypoxia-inducible microRNA cluster miR-199a~214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab. 18, 341–354 (2013). [DOI] [PubMed] [Google Scholar]
- 171.Li Z, Liu L, Hou N, Song Y, An X, Zhang Y, Yang X, Wang J, miR-199-sponge transgenic mice develop physiological cardiac hypertrophy. Cardiovasc. Res 110, 258–267 (2016). [DOI] [PubMed] [Google Scholar]
- 172.Park K-M, Teoh J-P, Wang Y, Broskova Z, Bayoumi AS, Tang Y, Su H, Weintraub NL, Kim I.-m., Carvedilol-responsive microRNAs, miR-199a-3p and −214 protect cardiomyocytes from simulated ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol 311, H371–H383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chen L, Wang F-Y, Zeng Z-Y, Cui L, Shen J, Song X-W, Li P, Zhao X-X, Qin Y-W, MicroRNA-199a acts as a potential suppressor of cardiomyocyte autophagy through targeting Hspa5. Oncotarget 8, 63825–63834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hinkel R, Ramanujam D, Kaczmarek V, Howe A, Klett K, Beck C, Dueck A, Thum T, Laugwitz KL, Maegdefessel L, Weber C, Kupatt C, Engelhardt S, AntimiR-21 prevents myocardial dysfunction in a pig model of ischemia/reperfusion injury. J. Am. Coll. Cardiol 75, 1788–1800 (2020). [DOI] [PubMed] [Google Scholar]
- 175.Hu J, Huang C-X, Rao P-P, Zhou J-P, Wang X, Tang L, Liu M-X, Zhang G-G, Inhibition of microRNA-155 attenuates sympathetic neural remodeling following myocardial infarction via reducing M1 macrophage polarization and inflammatory responses in mice. Eur. J. Pharmacol 851, 122–132 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Ye Y, Hu Z, Lin Y, Zhang C, Perez-Polo JR, Downregulation of microRNA-29 by antisense inhibitors and a PPAR-γ agonist protects against myocardial ischaemia–reperfusion injury. Cardiovasc. Res 87, 535–544 (2010). [DOI] [PubMed] [Google Scholar]
- 177.Guo Z-X, Zhou F-Z, Song W, Yu L-L, Yan W-J, Yin L-H, Sang H, Zhang H-Y, Suppression of microRNA-101 attenuates hypoxia-induced myocardial H9c2 cell injury by targeting DIMT1-Sp1/survivin pathway. Eur. Rev. Med. Pharmacol. Sci 22, 6965–6976 (2018). [DOI] [PubMed] [Google Scholar]
- 178.Wang C, Zhang C, Liu L, Xi A, Chen B, Li Y, Du J, Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol. Ther 25, 192–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Guo J, Liu H-B, Sun C, Yan X-Q, Hu J, Yu J, Yuan Y, Du Z-M, MicroRNA-155 promotes myocardial infarction-induced apoptosis by targeting RNA-binding protein QKI. Oxid. Med. Cell. Longev 2019, 4579806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hanna J, Hossain GS, Kocerha J, The potential for microRNA therapeutics and clinical research. Front. Genet 10, 478 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Juliano RL, The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 44, 6518–6548 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Eckstein F, Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 24, 374–387 (2014). [DOI] [PubMed] [Google Scholar]
- 183.Lennox KA, Behlke MA, Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 18, 1111–1120 (2011). [DOI] [PubMed] [Google Scholar]
- 184.Meister G, Landthaler M, Dorsett Y, Tuschl T, Sequence-specific inhibition of microRNA-and siRNA-induced RNA silencing. RNA 10, 544–550 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M, Silencing of microRNAs in vivo with “antagomirs”. Nature 438, 685–689 (2005). [DOI] [PubMed] [Google Scholar]
- 186.Vickers TA, Wyatt JR, Burckin T, Bennett CF, Freier SM, Fully modified 2′ MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res. 29, 1293–1299 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Elmén J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, Hedtjärn M, Hansen JB, Hansen HF, Straarup EM, Cullagh KM, Kearney P, Kauppinen S, Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 36, 1153–1162 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, Hansen HF, Koch T, Pappin D, Hannon GJ, Kauppinen S, Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet 43, 371–380 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ebert MS, Neilson JR, Sharp PA, MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4, 721–726 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ebert MS, Sharp PA, MicroRNA sponges: Progress and possibilities. RNA 16, 2043–2050 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Keeley EC, Boura JA, Grines CL, Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: A quantitative review of 23 randomised trials. Lancet 361, 13–20 (2003). [DOI] [PubMed] [Google Scholar]
- 192.Foinquinos A, Batkai S, Genschel C, Viereck J, Rump S, Gyöngyösi M, Traxler D, Riesenhuber M, Spannbauer A, Lukovic D, Weber N, Zlabinger K, Hašimbegović E, Winkler J, Fiedler J, Dangwal S, Fischer M, de la Roche J, Wojciechowski D, Kraft T, Garamvölgyi R, Neitzel S, Chatterjee S, Yin X, Bär C, Mayr M, Xiao K, Thum T, Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat. Commun 11, 633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dowdy SF, Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol 35, 222–229 (2017). [DOI] [PubMed] [Google Scholar]
- 194.Kulkarni JA, Darjuan MM, Mercer JE, Chen S, van der Meel R, Thewalt JL, Tam YYC, Cullis PR, On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and sirna. ACS Nano 12, 4787–4795 (2018). [DOI] [PubMed] [Google Scholar]
- 195.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO, Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 9, 654–659 (2007). [DOI] [PubMed] [Google Scholar]
- 196.Bian S, Zhang L, Duan L, Wang X, Min Y, Yu H, Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J. Mol. Med. (Berl) 92, 387–397 (2014). [DOI] [PubMed] [Google Scholar]
- 197.Liu B, Lee BW, Nakanishi K, Villasante A, Williamson R, Metz J, Kim J, Kanai M, Bi L, Brown K, Di Paolo G, Homma S, Sims PA, Topkara VK, Vunjak-Novakovic G, Cardiac recovery via extended cell-free delivery of extracellular vesicles secreted by cardiomyocytes derived from induced pluripotent stem cells. Nat. Biomed. Eng 2, 293–303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gallet R, Dawkins J, Valle J, Simsolo E, de Couto G, Middleton R, Tseliou E, Luthringer D, Kreke M, Smith RR, Marbán L, Ghaleh B, Marbán E, Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur. Heart J 38, 201–211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pachler K, Lener T, Streif D, Dunai ZA, Desgeorges A, Feichtner M, Öller M, Schallmoser K, Rohde E, Gimona M, A good manufacturing practice–grade standard protocol for exclusively human mesenchymal stromal cell–derived extracellular vesicles. Cytotherapy 19, 458–472 (2017). [DOI] [PubMed] [Google Scholar]
- 200.Chen Y, Zhao Y, Chen W, Xie L, Zhao Z-A, Yang J, Chen Y, Lei W, Shen Z, MicroRNA-133 overexpression promotes the therapeutic efficacy of mesenchymal stem cells on acute myocardial infarction. Stem Cell Res. Ther 8, 268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Morishita M, Takahashi Y, Nishikawa M, Takakura Y, Pharmacokinetics of exosomes—an important factor for elucidating the biological roles of exosomes and for the development of exosome-based therapeutics. J. Pharm. Sci 106, 2265–2269 (2017). [DOI] [PubMed] [Google Scholar]
- 202.Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J, Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol 33, 870–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Springer AD, Dowdy SF, GalNAc-siRNA conjugates: Leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 28, 109–118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mentkowski KI, Lang JK, Exosomes engineered to express a cardiomyocyte binding peptide demonstrate improved cardiac retention in vivo. Sci. Rep 9, 10041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Xue X, Shi X, Dong H, You S, Cao H, Wang K, Wen Y, Shi D, He B, Li Y, Delivery of microRNA-1 inhibitor by dendrimer-based nanovector: An early targeting therapy for myocardial infarction in mice. Nanomedicine 14, 619–631 (2018). [DOI] [PubMed] [Google Scholar]
- 206.Sugo T, Terada M, Oikawa T, Miyata K, Nishimura S, Kenjo E, Ogasawara-Shimizu M, Makita Y, Imaichi S, Murata S, Otake K, Kikuchi K, Teratani M, Masuda Y, Kamei T, Takagahara S, Ikeda S, Ohtaki T, Matsumoto H, Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 237, 1–13 (2016). [DOI] [PubMed] [Google Scholar]
- 207.Liu M, Li M, Sun S, Li B, Du D, Sun J, Cao F, Li H, Jia F, Wang T, Chang N, Yu H, Wang Q, Peng H, The use of antibody modified liposomes loaded with AMO-1 to deliver oligonucleotides to ischemic myocardium for arrhythmia therapy. Biomaterials 35, 3697–3707 (2014). [DOI] [PubMed] [Google Scholar]
- 208.Cuellar TL, Barnes D, Nelson C, Tanguay J, Yu S-F, Wen X, Scales SJ, Gesch J, Davis D, van Brabant Smith A, Leake D, Vandlen R, Siebel CW, Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 43, 1189–1203 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Juliano RL, Ming X, Carver K, Laing B, Cellular uptake and intracellular trafficking of oligonucleotides: Implications for oligonucleotide pharmacology. Nucleic Acid Ther. 24, 101–113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.LÖnn P, Kacsinta AD, Cui XS, Hamil AS, Kaulich M, Gogoi K, Dowdy SF, Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci. Rep 6, 32301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, StÖter M, Epstein-Barash H, Zhang L, Koteliansky V, Fitzgerald K, Fava E, Bickle M, Kalaidzidis Y, Akinc A, Maier M, Zerial M, Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol 31, 638–646 (2013). [DOI] [PubMed] [Google Scholar]
- 212.Dominska M, Dykxhoorn DM, Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci 123, 1183–1189 (2010). [DOI] [PubMed] [Google Scholar]
- 213.Stessl M, Noe CR, Winkler J, in From Nucleic Acids Sequences to Molecular Medicine, Erdmann VA, Barciszewski J, Eds. (Springer Berlin Heidelberg, 2012), pp. 67–83. [Google Scholar]
- 214.Winkler J, Stessl M, Amartey J, Noe CR, Off-target effects related to the phosphorothioate modification of nucleic acids. ChemMedChem 5, 1344–1352 (2010). [DOI] [PubMed] [Google Scholar]
- 215.Sioud M, Recent advances in small interfering RNA sensing by the immune system. N. Biotechnol 27, 236–242 (2010). [DOI] [PubMed] [Google Scholar]
- 216.Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, Tavazoie SF, Convergent multi-miRNA targeting of ApoE grives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 151, 1068–1082 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wang Z, Ma X, Cai Q, Wang X, Yu B, Cai Q, Liu B, Zhu Z, Li C, MiR-199a-3p promotes gastric cancer progression by targeting ZHX1. FEBS Lett. 588, 4504–4512 (2014). [DOI] [PubMed] [Google Scholar]
- 218.Bernardo BC, Gao X-M, Winbanks CE, Boey EJH, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du X-J, Lin RCY, McMullen JR, Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. U.S.A 109, 17615–17620 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kim NH, Kim HS, Kim N-G, Lee I, Choi H-S, Li X-Y, Kang SE, Cha SY, Ryu JK, Na JM, Park C, Kim K, Lee S, Gumbiner BM, Yook JI, Weiss SJ, p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal 4, ra71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhang L, Liao Y, Tang L, MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res 38, 53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mathur A, Loskill P, Shao K, Huebsch N, Hong SG, Marcus SG, Marks N, Mandegar M, Conklin BR, Lee LP, Healy KE, Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep 5, 8883 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Chi KR, Revolution dawning in cardiotoxicity testing. Nat. Rev. Drug Discov 12, 565–567 (2013). [DOI] [PubMed] [Google Scholar]
- 223.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC, Chemically defined generation of human cardiomyocytes. Nat. Methods 11, 855–860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song LJ, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G, Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Huebsch N, Loskill P, Deveshwar N, Spencer CI, Judge LM, Mandegar MA, Fox CB, Mohamed TMA, Ma Z, Mathur A, Sheehan AM, Truong A, Saxton M, Yoo J, Srivastava D, Desai TA, So P-L, Healy KE, Conklin BR, Miniaturized iPS-cell-derived cardiac muscles for physiologically relevant drug response analyses. Sci. Rep 6, 24726 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pellman J, Zhang J, Sheikh F, Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: Mechanisms and model systems. J. Mol. Cell. Cardiol 94, 22–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gintant G, Burridge P, Gepstein L, Harding S, Herron T, Hong C, Jalife J, Wu JC, Use of human induced pluripotent stem cell-derived cardiomyocytes in preclinical cancer drug cardiotoxicity testing: A scientific statement from the american heart association. Circ. Res 125, e75–e92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dai Y, Yan T, Gao Y, Silence of miR-32-5p promotes endothelial cell viability by targeting KLF2 and serves as a diagnostic biomarker of acute myocardial infarction. Diagn. Pathol 15, 19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Roca-Alonso L, Castellano L, Mills A, Dabrowska AF, Sikkel MB, Pellegrino L, Jacob J, Frampton AE, Krell J, Coombes RC, Harding SE, Lyon AR, Stebbing J, Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in β-adrenergic signaling and enhances apoptosis. Cell Death Dis. 6, e1754 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R, Sadek HA, Olson EN, MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Invest 122, 1222–1232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Hao Y, Yuan H, Yu H, Downregulation of miR-483-5p decreases hypoxia-induced injury in human cardiomyocytes by targeting MAPK3. Cell. Mol. Biol. Lett 25, 20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 232.Ren X-P, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan G-C, MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 119, 2357–2366 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shen Y, Shen Z, Miao L, Xin X, Lin S, Zhu Y, Guo W, Zhu YZ, miRNA-30 family inhibition protects against cardiac ischemic injury by regulating cystathionine-γ-lyase expression. Antioxid. Redox Signal 22, 224–240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Li X, Zeng Z, Li Q, Xu Q, Xie J, Hao H, Luo G, Liao W, Bin J, Huang X, Liao Y, Inhibition of microRNA-497 ameliorates anoxia/reoxygenation injury in cardiomyocytes by suppressing cell apoptosis and enhancing autophagy. Oncotarget 6, 18829–18844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wu G, Tan J, Li J, Sun X, Du L, Tao S, miRNA-145-5p induces apoptosis after ischemia-reperfusion by targeting dual specificity phosphatase 6. J. Cell. Physiol 234, 16281–16289 (2019). [DOI] [PubMed] [Google Scholar]
- 236.Liu Y, Qian X-M, He Q-C, Weng J-K, MiR-421 inhibition protects H9c2 cells against hypoxia/reoxygenation-induced oxidative stress and apoptosis by targeting Sirt3. Perfusion 35, 255–262 (2019). [DOI] [PubMed] [Google Scholar]
- 237.Long B, Wang K, Li N, Murtaza I, Xiao J-Y, Fan Y-Y, Liu C-Y, Li W-H, Cheng Z, Li PF, MiR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic. Biol. Med 65, 371–379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wang X, Ha T, Zou J, Ren D, Liu L, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C, MicroRNA-125b protects against myocardial ischaemia/reperfusion injury via targeting p53-mediated apoptotic signalling and TRAF6. Cardiovasc. Res 102, 385–395 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Pan J, Alimujiang M, Chen Q, Shi H, Luo X, Exosomes derived from miR-146a-modified adipose-derived stem cells attenuate acute myocardial infarction–induced myocardial damage via downregulation of early growth response factor 1. J. Cell. Biochem 120, 4433–4443 (2019). [DOI] [PubMed] [Google Scholar]
- 240.Wang X, Ha T, Liu L, Zou J, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C, Increased expression of microRNA-146a decreases myocardial ischaemia/reperfusion injury. Cardiovasc. Res 97, 432–442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zhang S, Zhang R, Wu F, Li X, MicroRNA-208a regulates H9c2 cells simulated ischemia-reperfusion myocardial injury via targeting CHD9 through Notch/NF-kappa B signal pathways. Int. Heart. J 59, 580–588 (2018). [DOI] [PubMed] [Google Scholar]
- 242.Huang W, Tian S-S, Hang P-Z, Sun C, Guo J, Du Z-M, Combination of microRNA-21 and microRNA-146a attenuates cardiac dysfunction and apoptosis during acute myocardial infarction in mice. Mol. Ther. Nucleic Acids 5, e296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Huang L, Yang L, Ding Y, Jiang X, Xia Z, You Z, Human umbilical cord mesenchymal stem cells-derived exosomes transfers microRNA-19a to protect cardiomyocytes from acute myocardial infarction by targeting SOX6. Cell Cycle 19, 339–353 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ma J, Chen Z, Ma Y, Xia Y, Hu K, Zhou Y, Chen A, Qian J, Ge J, MicroRNA-19a attenuates hypoxia-induced cardiomyocyte apoptosis by downregulating NHE-1 expression and decreasing calcium overload. J. Cell. Biochem 121, 1747–1758 (2020). [DOI] [PubMed] [Google Scholar]
- 245.Yang Q, Yang K, Li A, microRNA-21 protects against ischemia-reperfusion and hypoxia-reperfusion- induced cardiocyte apoptosis via the phosphatase and tensin homolog/Akt- dependent mechanism. Mol. Med. Rep 9, 2213–2220 (2014). [DOI] [PubMed] [Google Scholar]
- 246.Jin Y, Ni S, miR-496 remedies hypoxia reoxygenation–induced H9c2 cardiomyocyte apoptosis via Hook3-targeted PI3k/Akt/mTOR signaling pathway activation. J. Cell. Biochem 121, 698–712 (2020). [DOI] [PubMed] [Google Scholar]
- 247.Shi Y, Han Y, Niu L, Li J, Chen Y, MiR-499 inhibited hypoxia/reoxygenation induced cardiomyocytes injury by targeting SOX6. Biotechnol. Lett 41, 837–847 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Li Q, Xie J, Li R, Shi J, Sun J, Gu R, Ding L, Wang L, Xu B, Overexpression of microRNA-99a attenuates heart remodelling and improves cardiac performance after myocardial infarction. J. Cell. Mol. Med 18, 919–928 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gong L-C, Xu H-M, Guo G-L, Zhang T, Shi J-W, Chang C, Long non-coding RNA H19 protects H9c2 cells against hypoxia-induced injury by targeting MicroRNA-139. Cell. Physiol. Biochem 44, 857–869 (2017). [DOI] [PubMed] [Google Scholar]
- 250.Shi K, Sun H, Zhang H, Xie D, Yu B, MiR-34a-5p aggravates hypoxia-induced apoptosis by targeting ZEB1 in cardiomyocytes. Biol. Chem 400, 227–236 (2019). [DOI] [PubMed] [Google Scholar]
- 251.Liu C, Tang M, Zhang X, Li J, Cao G, Knockdown of miR-665 protects against cardiomyocyte ischemia/reperfusion injury-induced ROS accumulation and apoptosis through the activation of Pak1/Akt signaling in myocardial infarction. Int. Heart J 61, 347–354 (2020). [DOI] [PubMed] [Google Scholar]
- 252.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M, Downregulation of MiR-199a derepresses hypoxia-inducible factor-1α and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res 104, 879–886 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhang J-Y, Yang Z, Fang K, Shi Z-L, Ren D-H, Sun J, Long noncoding RNA ILF3-AS1 regulates myocardial infarction via the miR-212-3p/SIRT1 axis and PI3K/Akt signaling pathway. Eur. Rev. Med. Pharmacol. Sci 24, 2647–2658 (2020). [DOI] [PubMed] [Google Scholar]
- 254.Wang X, Yuan B, Cheng B, Liu Y, Zhang B, Wang X, Lin X, Yang B, Gong G, Crocin alleviates myocardial ischemia/reperfusion-induced endoplasmic reticulum stress via regulation of miR-34a/Sirt1/Nrf2 pathway. Shock 51, 123–130 (2019). [DOI] [PubMed] [Google Scholar]
- 255.Zhu L-P, Tian T, Wang J-Y, He J-N, Chen T, Pan M, Xu L, Zhang H-X, Qiu X-T, Li C-C, Wang K-K, Shen H, Zhang G-G, Bai Y-P, Hypoxia-elicited mesenchymal stem cell-derived exosomes facilitates cardiac repair through miR-125b-mediated prevention of cell death in myocardial infarction. Theranostics 8, 6163–6177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhou T, Qin G, Yang L, Xiang D, Li S, LncRNA XIST regulates myocardial infarction by targeting miR-130a-3p. J. Cell. Physiol 234, 8659–8667 (2019). [DOI] [PubMed] [Google Scholar]
- 257.Zhao Z, Du S, Shen S, Wang L, microRNA-132 inhibits cardiomyocyte apoptosis and myocardial remodeling in myocardial infarction by targeting IL-1α. J. Cell. Physiol 235, 2710–2721 (2020). [DOI] [PubMed] [Google Scholar]
- 258.Pan Y-L, Han Z-Y, He S-F, Yang W, Cheng J, Zhang Y, Chen Z-W, MiR-133b-5p contributes to hypoxic preconditioning-mediated cardioprotection by inhibiting the activation of caspase-8 and caspase-3 in cardiomyocytes. Mol. Med. Rep 17, 7097–7104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Yang S, Li H, Chen L, MicroRNA-140 attenuates myocardial ischemia-reperfusion injury through suppressing mitochondria-mediated apoptosis by targeting YES1. J. Cell. Biochem 120, 3813–3821 (2019). [DOI] [PubMed] [Google Scholar]
- 260.Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, Hare JM, Olson EN, van Rooij E, Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res 110, 71–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Tang Y, Wang Y, Park K-M, Hu Q, Teoh J-P, Broskova Z, Ranganathan P, Jayakumar C, Li J, Su H, Tang Y, Ramesh G, Kim I-M, MicroRNA-150 protects the mouse heart from ischaemic injury by regulating cell death. Cardiovasc. Res 106, 387–397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Li Y, Li Q, Zhang O, Guan X, Xue Y, Li S, Zhuang X, Zhou B, Miao G, miR-202-5p protects rat against myocardial ischemia reperfusion injury by downregulating the expression of Trpv2 to attenuate the Ca2+ overload in cardiomyocytes. J. Cell. Biochem 120, 13680–13693 (2019). [DOI] [PubMed] [Google Scholar]
- 263.Qin Y, Yu Y, Dong H, Bian X, Guo X, Dong S, MicroRNA 21 inhibits left ventricular remodeling in the early phase of rat model with ischemia-reperfusion injury by suppressing cell apoptosis. Int. J. Med. Sci 9, 413–423 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Tréguer K, Carmona G, Bonauer A, Horrevoets AJG, Didier N, Girmatsion Z, Biliczki P, Ehrlich JR, Katus HA, Müller OJ, Potente M, Zeiher AM, Hermeking H, Dimmeler S, MicroRNA-34a regulates cardiac ageing and function. Nature 495, 107–110 (2013). [DOI] [PubMed] [Google Scholar]
- 265.Sheikh MSA, Overexpression of miR-375 protects cardiomyocyte injury following hypoxic-reoxygenation injury. Oxid. Med. Cell. Longev 2020, 7164069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Li R, Geng H-H, Xiao J, Qin X.-t., Wang F, Xing J.-h., Xia Y.-f., Mao Y, Liang J.-w., Ji X.-p., MIR-7a/b attenuates post-myocardial infarction remodeling and protects H9c2 cardiomyoblast against hypoxia-induced apoptosis involving Sp1 and PARP-1. Sci. Rep 6, 29082 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhai C-L, Tang G-M, Qian G, Hu H-L, Wang S-J, Yin D, Zhang S, MicroRNA-98 attenuates cardiac ischemia-reperfusion injury through inhibiting DAPK1 expression. IUBMB Life 71, 166–176 (2019). [DOI] [PubMed] [Google Scholar]
- 268.Bian B, Yu X-F, Wang G-Q, Teng T-M, Role of miRNA-1 in regulating connexin 43 in ischemia-reperfusion heart injury: A rat model. Cardiovasc. Pathol 27, 37–42 (2017). [DOI] [PubMed] [Google Scholar]
- 269.Ge L, Cai Y, Ying F, Liu H, Zhang D, He Y, Pang L, Yan D, Xu A, Ma H, Xia Z, MiR-181c-5p exacerbates hypoxia/reoxygenation-induced cardiomyocyte apoptosis via targeting PTPN4. Oxid. Med. Cell. Longev 2019, 1957920 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Yuan X, Chen C, Wang L, He X, Inhibition of microRNA-182 reduces hypoxia/re-oxygenation-induced HL-1 cardiomyocyte apoptosis by targeting the nuclear respiratory factor-1/mitochondrial transcription factor A (NRF-1/mtTFA) pathway (2017); www.ijcep.com/.
- 271.Liu D-W, Zhang Y-N, Hu H-J, Zhang P-Q, Cui W, Downregulation of microRNA-199a-5p attenuates hypoxia/reoxygenation-induced cytotoxicity in cardiomyocytes by targeting the HIF-1α-GSK3β-mPTP axis. Mol. Med. Rep 19, 5335–5344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Wang Y, Jiang Y, Sun X, Shen X, Wang H, Dong C, Lu B, Yan Y, Lu Y, Fasae MB, Liu B, Bai Y, Downregulation of miR-200a protects cardiomyocyte against apoptosis. Biomed. Pharmacother 123, 109303 (2020). [DOI] [PubMed] [Google Scholar]
- 273.Song Y-S, Joo H-W, Park I-H, Shen G-Y, Lee Y, Shin JH, Kim H, Kim K-S, Bone marrow mesenchymal stem cell-derived vascular endothelial growth factor attenuates cardiac apoptosis via regulation of cardiac miRNA-23a and miRNA-92a in a rat model of myocardial infarction. PLOS ONE 12, e0179972 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ruiz-Velasco A, Zi M, Hille SS, Azam T, Kaur N, Jiang J, Nguyen B, Sekeres K, Binder P, Collins L, Pu F, Xiao H, Guan K, Frey N, Cartwright EJ, Müller OJ, Wang X, Liu W, Targeting mir128-3p alleviates myocardial insulin resistance and prevents ischemia-induced heart failure. eLife 9, e54298 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Khan M, Nickoloff E, Abramova T, Johnson J, Verma SK, Krishnamurthy P, Mackie AR, Vaughan E, Garikipati VNS, Benedict C, Ramirez V, Lambers E, Ito A, Gao E, Misener S, Luongo T, Elrod J, Qin G, Houser SR, Koch WJ, Kishore R, Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res 117, 52–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Deng S, Zhao Q, Zhen L, Zhang C, Liu C, Wang G, Zhang L, Bao L, Lu Y, Meng L, Lü J, Yu P, Lin X, Zhang Y, Chen Y-H, Fan H, Cho WC, Liu Z, Yu Z, Neonatal heart-enriched miR-708 promotes proliferation and stress resistance of cardiomyocytes in rodents. Theranostics 7, 1953–1965 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Chen J, Huang Z-P, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang D-Z, Mir-17-92 induce cardiomyocyte proliferation in postnatal and adult hearts, 2013. Circ. Res 112, 1557–1566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Yang M, Kong D-Y, Chen J-C, Inhibition of miR-148b ameliorates myocardial ischemia/reperfusion injury via regulation of Wnt/β-catenin signaling pathway. J. Cell. Physiol 234, 17757–17766 (2019). [DOI] [PubMed] [Google Scholar]
- 279.Pandey R, Velasquez S, Durrani S, Jiang M, Neiman M, Crocker JS, Benoit JB, Rubinstein J, Paul A, Ahmed RPH, MicroRNA-1825 induces proliferation of adult cardiomyocytes and promotes cardiac regeneration post ischemic injury. Am. J. Transl. Res 9, 3120–3137 (2017). [PMC free article] [PubMed] [Google Scholar]
- 280.Gao F, Kataoka M, Liu N, Liang T, Huang Z-P, Gu F, Ding J, Liu J, Zhang F, Ma Q, Wang Y, Zhang M, Hu X, Kyselovic J, Hu X, Pu WT, Wang J, Chen J, Wang D-Z, Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat. Commun 10, 1802 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Aguirre A, Montserrat N, Zachiggna S, Nivet E, Hishida T, Krause MN, Kurian L, Ocampo A, Vazquez-Ferrer E, Rodriguez-Esteban C, Kumar S, Moresco JJ, Yates III JR, Campistol JM, Sancho-Martinez I, Giacca M, Belmonte JCI, In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 15, 589–604 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Icli B, Wara AKM, Moslehi J, Sun X, Plovie E, Cahill M, Marchini JF, Schissler A, Padera RF, Shi J, Cheng H-W, Raghuram S, Arany Z, Liao R, Croce K, Rae CM, Feinberg MW, MicroRNA-26a regulates pathological and physiological angiogenesis by targeting BMP/SMAD1 signaling. Circ. Res 113, 1231–1241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ribeiro-Rodrigues TM, Laundos TL, Pereira-Carvalho R, Batista-Almeida D, Pereira R, Coelho-Santos V, Silva AP, Fernandes R, Zuzarte M, Enguita FJ, Costa MC, Pinto-do-Ó P, Pinto MT, Gouveia P, Ferreira L, Mason JC, Pereira P, Kwak BR, Nascimento DS, Girão H, Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis. Cardiovasc. Res 113, 1338–1350 (2017). [DOI] [PubMed] [Google Scholar]
- 284.Peng Y, Chao F, Cai Y, Teng W, Qiu C, MiR-126 inhibits the proliferation of myocardial fibroblasts by regulating EGFL7-mediated EGFR signal pathway. Int. J. Clin. Exp. Med 10, 6158–6166 (2017). [Google Scholar]
- 285.Wei Z, Qiao S, Zhao J, Yihai L, Qiaoling L, Zhonghai W, Qing D, Lina K, Biao X, miRNA-181a over-expression in mesenchymal stem cell-derived exosomes influenced inflammatory response after myocardial ischemia-reperfusion injury. Life Sci. 232, 116632 (2019). [DOI] [PubMed] [Google Scholar]
- 286.Liu Z, Ye P, Wang S, Wu J, Sun Y, Zhang A, Ren L, Cheng C, Huang X, Wang K, Deng P, Wu C, Yue Z, Xia J, MicroRNA-150 protects the heart from injury by inhibiting monocyte accumulation in a mouse model of acute myocardial infarction. Circ. Cardiovasc. Genet 8, 11–20 (2015). [DOI] [PubMed] [Google Scholar]
- 287.Song Y, Zhang C, Zhang J, Jiao Z, Dong N, Wang G, Wang Z, Wang L, Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics 9, 2346–2360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Ge Z-W, Zhu X-L, Wang B-C, Hu J-L, Sun J-J, Wang S, Chen X-J, Meng S-P, Liu L, Cheng Z-Y, MicroRNA-26b relieves inflammatory response and myocardial remodeling of mice with myocardial infarction by suppression of MAPK pathway through binding to PTGS2. Int. J. Cardiol 280, 152–159 (2019). [DOI] [PubMed] [Google Scholar]
- 289.Lu S, Lu Y, MiR-26a inhibits myocardial cell apoptosis in rats with acute myocardial infarction through GSK-3β pathway. Eur. Rev. Med. Pharmacol. Sci 24, 2659–2666 (2020). [DOI] [PubMed] [Google Scholar]
- 290.Wu H-Y, Wu J-L, Ni Z-L, Overexpression of microRNA-202-3p protects against myocardial ischemia-reperfusion injury through activation of TGF-β1/Smads signaling pathway by targeting TRPM6. Cell Cycle 18, 621–637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 291.Jiang T, You H, You D, Zhang L, Ding M, Yang B, A miR-1275 mimic protects myocardiocyte apoptosis by regulating the Wnt/NF-κB pathway in a rat model of myocardial ischemia–reperfusion-induced myocardial injury. Mol. Cell. Biochem 466, 129–137 (2020). [DOI] [PubMed] [Google Scholar]
- 292.Li Y, Zhou J, Zhang O, Wu X, Guan X, Xue Y, Li S, Zhuang X, Zhou B, Miao G, Zhang L, Bone marrow mesenchymal stem cells-derived exosomal microRNA-185 represses ventricular remolding of mice with myocardial infarction by inhibiting SOCS2. Int. Immunopharmacol 80, 106156 (2020). [DOI] [PubMed] [Google Scholar]
- 293.Yang J, Fan Z, Yang J, Ding J, Yang C, Chen L, MicroRNA-22 attenuates myocardial ischemia-reperfusion injury via an anti-inflammatory mechanism in rats. Exp. Ther. Med 12, 3249–3255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Tan H, Qi J, Fan B-Y, Zhang J, Su F-F, Wang H-T, MicroRNA-24-3p attenuates myocardial ischemia/reperfusion injury by suppressing RIPK1 expression in mice. Cell. Physiol. Biochem 51, 46–62 (2018). [DOI] [PubMed] [Google Scholar]
- 295.Zhou F-Q, Zhao X-F, Liu F-Y, Wang S-S, Hu H-L, Fang Y, MiR-101a attenuates myocardial cell apoptosis in rats with acute myocardial infarction via targeting TGF-β/JNK signaling pathway. Eur. Rev. Med. Pharmacol. Sci 23, 4432–4438 (2019). [DOI] [PubMed] [Google Scholar]
- 296.Yu B-T, Yu N, Wang Y, Zhang H, Wan K, Sun X, Zhang C-S, Role of miR-133a in regulating TGF-β1 signaling pathway in myocardial fibrosis after acute myocardial infarction in rats. Eur. Rev. Med. Pharmacol. Sci 23, 8588–8597 (2019). [DOI] [PubMed] [Google Scholar]
- 297.Hinkel R, Penzkofer D, Zühlke S, Fischer A, Husada W, Xu Q-F, Baloch E, van Rooij E, Zeiher AM, Kupatt C, Dimmeler S, Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation 128, 1066–1075 (2013). [DOI] [PubMed] [Google Scholar]
- 298.Dong J, Zhang Z, Huang H, Mo P, Cheng C, Liu J, Huang W, Tian C, Zhang C, Li J, MiR-10a rejuvenates aged human mesenchymal stem cells and improves heart function after myocardial infarction through KLF4. Stem Cell Res. Ther 9, 151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Summary of miRNAs reviewed.
Table S1. Complete list of miRNAs reviewed.