

Abstract
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor and an attractive therapeutic target for cancer and other human diseases. Despite 20 years of persistent research efforts, targeting STAT3 has been very challenging. We report herein the structure-based discovery of potent small-molecule STAT3 degraders based upon the proteolysis targeting chimera (PROTAC) concept. We first designed SI-109 as a potent, small-molecule inhibitor of the STAT3 SH2 domain. Employing ligands for cereblon/cullin 4A E3 ligase and SI-109, we obtained a series of potent PROTAC STAT3 degraders, exemplified by SD-36. SD-36 induces rapid STAT3 degradation at low nanomolar concentrations in cells and fails to degrade other STAT proteins. SD-36 achieves nanomolar cell growth inhibitory activity in leukemia and lymphoma cell lines with high levels of phosphorylated STAT3. A single dose of SD-36 results in complete STAT3 protein degradation in xenograft tumor tissue and normal mouse tissues. SD-36 achieves complete and long-lasting tumor regression in the Molm-16 xenograft tumor model at well-tolerated dose-schedules. SD-36 is a potent, selective, and efficacious STAT3 degrader.
Graphical Abstract
INTRODUCTION
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family of transcription factors that transmits signals from cell surface receptors to the nucleus.1 Persistent activation of STAT3 is often associated with poor prognosis of human cancers since activated STAT3 signaling not only provides advantages of growth, survival, and metastasis to tumor cells but also suppresses the antitumor immune response.2,3 STAT3 is an attractive therapeutic target for human cancer and other human diseases.
In the canonical mechanism, STAT3 is recruited from the cytosol to the cell membrane where it interacts with different cytokine receptors through its Src homology 2 (SH2) domain. This results in phosphorylation of its Tyr705 residue (Y705) and its subsequent dimerization and translocation into the nucleus for gene transcription.1 It has been proposed that dimerization of STAT3 is required for its translocation from cytosol into the nucleus and for its transcriptional activity, and pharmacological inhibition of STAT3 SH2 domain could effectively block the dimerization and transcriptional activity of STAT3.1 Consequently, considerable efforts have been made in the past 2 decades to develop small molecular inhibitors of the STAT3 SH2 domain.3,4
Because STAT3 homodimerization is mediated by a phosphotyrosine-containing peptide from one monomer and a well-defined binding pocket in the SH2 domain of the second monomer, peptidomimetic inhibitors have been designed based upon STAT3 phosphotyrosine-containing peptides. Although previous peptidomimetic inhibitors have been reported to achieve high binding affinities to STAT3 in biochemical binding assays, they are generally not cell-permeable due to the presence of their negatively charged phosphate group and their peptidic characteristics.3,4 Nonpeptide small-molecule inhibitors of the STAT3 SH2 domain have been reported, but they often have weak to moderate binding affinities to STAT3, making it difficult to establish firmly that STAT3 is the primary cellular target.3,4 Furthermore, because STAT3 shares a high sequence homology of its SH2 domain with other STAT members, it has been challenging to design SH2 domain inhibitors for STAT3 that are highly selective over other STAT proteins. Significantly, in contrast to initial hypotheses, it has now become clear that the monomeric STAT3 protein can translocate from cytosol into the nucleus and is transcriptionally active.5 Consequently, even if a potent, cell-permeable, and highly selective inhibitor of the STAT3 SH2 domain is ultimately discovered, it can be predicted that it would only be able to partially block the gene transcriptional activity of STAT3. Accordingly, there is a clear need to develop a new therapeutic approach to targeting STAT3.
Therefore, in a therapeutic setting, specific down-regulation or knock-down of STAT3 may be much more effective than inhibition of its dimerization. To support this idea, AZD9150,6 which was designed as a STAT3 antisense oligonucleotide (ASO), has shown promising preclinical and clinical activity, despite some major limitations associated with ASO molecules.3,7
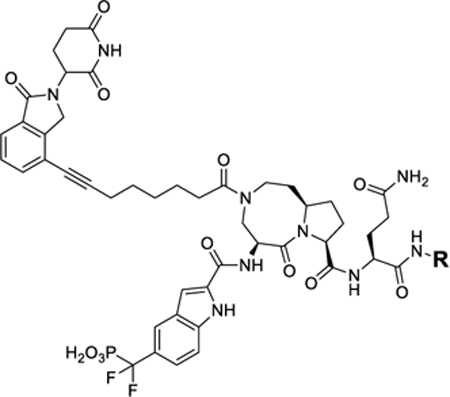
Recently, the proteolysis targeting chimeras (PROTAC) strategy has gained momentum for its promise in the discovery and development of an entirely new type of therapeutics for the treatment of human diseases.8–11 A PROTAC molecule is a bifunctional small molecule designed to recruit cellular degradation machinery to induce targeted protein degradation.8 We therefore proposed to design PROTAC degraders of STAT3 as a new strategy to effectively target STAT3. In the present study, we report our structure-based discovery of SD-36 as a potent, exceptionally selective, and highly efficacious PROTAC degrader of the STAT3 protein.
RESULTS AND DISCUSSION
Design of Small Molecule STAT3 SH2 Domain Inhibitors Containing a Novel Phosphotyrosine Mimetic.
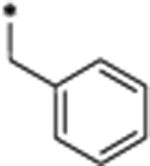
Our laboratory has previously reported the design of peptidomimetic compounds with high binding affinities to the STAT3 SH2 domain, such as CJ-887 (Figure 1),12 which binds to the human recombinant STAT3 SH2 protein with Ki = 47 nM in a fluorescence polarization (FP) assay (Figure S1 and Table S1). Despite this high binding affinity to STAT3, CJ-887 is ineffective in inhibition of STAT3 Y705 phosphorylation in cells, suggesting its poor cell permeability. We hypothesized that the poor cell-permeability of CJ-887 could be largely attributed to its doubly ionized natural phosphate group.13,14 A second major issue associated with CJ-887 is its expected susceptibility to degradation by protein tyrosine phosphatases (PTPs).13,14 To overcome these major limitations, we have performed extensive optimization of CJ-887, with the key steps outlined in Figure 1.
Figure 1.

Design of potent and cell-permeable small molecule inhibitors of the STAT3 SH2 domain: chemical structures and binding affinities of our previously reported compound CJ-887 and newly designed and synthesized inhibitors.
Cyclization of the amino group with the phenyl group in CJ-887 forms an indole (2), which converts CJ-887, a phosphotyrosine into a conformationally constrained, non-amino acid phosphate.26 The resulting compound 2 binds to STAT3 with a Ki value of 25 nM and is thus twice as potent as CJ-887 (Figure 1 and Table S1). To address the potential susceptibility of the phosphate ester bond in 2 to hydrolysis by PTPs, we replaced the -OPO3H2 group with a non-hydrolyzable -CH2-PO3H2 group,13,14 yielding compound 3. Compound 3 binds to STAT3 with a Ki value of 761 nM and is thus 30 times weaker than 2. We reasoned that the reduced binding affinity of 3 is partially attributable to the weaker acidity of the -CH2PO3H2 group compared to that of the -OPO3H2 group. To test this idea, we replaced the -CH2PO3H2 group with -CF2PO3H2, which has been used previously in the design of phosphotyrosine mimetics.13–17 Compound 4, containing the -CF2PO3H2 group binds to STAT3 with a very high affinity (Ki = 7 nM)26 and is >100 times more potent than 3 and 7 times more potent than the initial peptidomimetic 1 (CJ-887).
Our predicted binding model of compound 4 in a complex with the STAT3 SH2 domain (Figure S2) suggested that part of the eight-membered ring in 4 is exposed to the solvent environment. To facilitate the synthesis of STAT3 degraders, we replaced one of the carbon atoms in the eight-membered ring with a nitrogen atom, producing 5 with a methyl substituent,26 and 6 with an acetyl substituent. Both 5 and 6 bind to STAT3 with the same high affinity, achieving Ki values of 14 nM. Next we installed a second phenyl group at the α position of the benzyl group in 5 and 6 to restrict the conformation of the original phenyl group, yielding 7 and SI-109 (8),26 respectively. Compounds 7 and SI-109 bind to STAT3 with Ki values of 12 nM and 14 nM, respectively.
To directly compare the phosphotyrosine mimetic used in compounds 4–8 with other phosphotyrosine mimetics, we used SI-109 as the template molecule and synthesized 9, 10, and 11 (Figure 1). Compound 9 in which the -CF2PO3H2 moiety in SI-109 is replaced with a phosphate (-OPO3H2) is 2 times less potent than SI-109. Compound 10 in which the -CF2PO3H2 moiety in SI-109 is replaced with -CH2PO3H2 is 14 times less potent than SI-109. Compound 11 containing 4-phosphonodifluoromethylcinnamate,17 binds to STAT3 with Ki value of 164 nM and is >10 times less potent than SI-109. These binding data show that the combination of the bicyclic indole with the difluoromethylphosphate moiety has yielded an excellent phosphotyrosine mimetic, which is used in compounds 4–8.
Hence, optimization of 1 (CJ-887) has yielded a series of high-affinity, small-molecule ligands for the STAT3 SH2 domain, including SI-109 (8), which was employed in our subsequent design of PROTAC STAT3 degraders.
Determination of the Cocrystal Structure of SI-109 in a Complex with STAT3.
To understand the structural basis for the high binding affinity of our designed STAT3 inhibitors, we determined the cocrystal structure of SI-109 in a complex with the STAT3 SH2 domain at a resolution of 3.15 Å (PDB access code 6NUQ, Figure 2 and Supporting Information, Table S2).
Figure 2.

Cocrystal structure of SI-109 with STAT3 (PDB code 6NUQ). (A) The Fo – Fc electron density generated from an omit map contoured at 3σ shows placement of atoms of SI-109. (B) Detailed interactions of STAT3 with SI-109. SI-109 (cyan) and side chains of interacting residues of STAT3 with SI-109 are shown in stick form. Hydrogen bonds are depicted as dashed lines.
Comparison of the cocrystal structure of SI-109 in a complex with STAT3 and the STAT3 SH2 dimeric crystal structure showed that SI-109 captures all the critical interactions with STAT3 that are observed in the STAT3 dimeric complex. The difluoromethylphosphate in SI-109 forms extensive hydrogen bonding interactions with the side chains of Arg609, Ser611, and Ser613 and the backbone amide nitrogen atoms of Glu612 and Ser613. An additional hydrogen bond is formed between the pro-(S)-F atom of the difluoromethyl group with Arg609. The indole moiety in SI-109 sits in a shallow cleft formed between Pro639 and Ser636 of STAT3. The glutamine group in SI-109 forms two hydrogen bonds with the side chain of Gln644. The backbone carbonyl moiety of the glutamine group in SI-109 forms two water-mediated hydrogen bonds with the hydroxyl group of Tyr640 and the amino group of Lys658. The pro-(S)-phenyl group of the diphenylmethyl group in SI-109 interacts with Leu666, Met660, and Ile659, whereas a pro-(R)-phenyl group has no specific interactions with any residue in STAT3 and is fully exposed to solvent environment. Although no specific interactions are observed between the [8,5]-bicyclic ring system in SI-109 and the STAT3 protein, optimal hydrogen bonds are formed between the amide groups in the proximity of the [8,5]-bicyclic ring and the protein. Specifically, the amino group tethered to the eight-membered ring forms an optimal hydrogen bond with the backbone carbonyl group of Ser636 and the carbonyl group attached to the five-membered ring forms a hydrogen bond with the hydroxyl group of Tyr657 and additionally a water-mediated hydrogen bond with the backbone carbonyl group of Lys658. Hence, the bicyclic ring system functions as a scaffold which ensures that the difluoromethylphosphonic acid and the glutamine moieties in SI-109 are ideally positioned and oriented to achieve optimal interactions with STAT3.
This cocrystal structure of SI-109 in a complex with STAT3 provides the structural basis for its high binding affinity to STAT3 and facilitated our design of STAT3 degraders.
Design of PROTAC STAT3 Degraders.
VHL/cullin 2 and cereblon (CRBN)/cullin 4A E3 ligase complexes have been successfully employed in the design of PROTAC degraders for different proteins.8–11 For the VHL/cullin 2 E3 ligase complex, high-affinity peptidomimetic ligands have been discovered18–21 and have been successfully used for the design of highly potent PROTAC degraders for different proteins.8–11 For the cereblon (CRBN)/cullin 4A E3 ligase complex, ligands for cereblon, such as lenalidomide and pomalidomide, are small-molecule drugs with molecular weights between 250 and 260, which are ideal for the design of PROTAC degraders with excellent physiochemical properties. Accordingly, we have decided to employ lenalidomide and pomalidomide (Figure 3) for the design of STAT3 degraders.
Figure 3.

Design of putative STAT3 PROTAC degraders based upon cereblon ligands and our STAT3 SH2 domain inhibitor SI-109.
Our cocrystal structure of SI-109 in a complex with the STAT3 SH2 domain shows that the amino group of the eight-membered ring in SI-109 is exposed to the solvent (Figure 2) and is a suitable site at which to tether a ligand for an E3 ligase complex in the design of PROTAC STAT3 degraders. We have designed and synthesized a series of potential STAT3 degraders linking this site in SI-109 with a cereblon ligand through a linker (Figure 3).
Determination of the Optimal Linker Length.
Our previous studies demonstrated that the length of the linker plays a key role in protein degradation potencies in our BET and MDM2 PROTAC degraders.22–25 We synthesized and evaluated a series of putative STAT3 degraders with different linker lengths (Table 1).
Table 1.
STAT3 Degraders with Various Linker Lengths
| ||
|---|---|---|
| Cmpds | Linker | DC50 (STAT3, μM) Molm-16, 4h |
|
| ||
| SI-109 | >10 | |
| 12 |
|
2.63 |
| 13 |
|
0.26 |
| SD-36 (14) |
|
0.06 |
| 15 |
|
0.09 |
| 16 |
|
0.15 |
An acute myeloid leukemia (AML) cell line, Molm-16 expresses high levels of phosphorylated STAT3 at the Tyr705 residue (pSTAT3Y705) protein and total (phosphorylated and unphosphorylated) STAT3 protein.26 We thus used the Molm-16 cell line to evaluate our putative STAT3 degraders for their ability to degrade both the total STAT3 protein and pSTAT3Y705 protein. Because PROTAC degraders can induce protein degradation rapidly, we treated the Molm-16 cells for 4 h with our putative STAT3 degraders and obtained the data shown in Table 1 and Figure S3.
In this study, we examined the degradation of both the total STAT3 protein and PstatsY705 protein for our designed STAT3 degraders and found that our STAT3 degraders induce degradation of the total STAT3 protein and pSTAT3Y705 protein with very similar potencies. Hence, we determined the DC50 value, the concentration required to reduce the protein level by 50% of the total STAT3 protein for each compound, and used this value to establish the structure–activity relationships.
Western blotting data show that the STAT3 inhibitor SI-109 has no effect on the level of total STAT3 protein and only modestly decreases the level of the pSTAT3Y705 protein at concentrations up to 1 μM. Compound 12, with a total of six non-hydrogen atoms in its linker between SI-109 and the cereblon ligand, has no effect on the level of total STAT3 protein up to 1 μM and reduces pSTAT3Y705 protein only minimally at 1 μM (Table 1 and Figure S3). However, with higher concentrations, 12 dose-dependently reduces the levels of both the total STAT3 and PstatsY705 proteins and has an DC50 value of 2.63 μM for the total STAT3 protein (Table 1 and Figure S3). Increasing the length of the linker in 12 by one carbon yielded 13, which effectively reduces the levels of both the total STAT3 and pSTATSY705 proteins and with DC50 = 0.26 μM, is 10 times more potent than 12. Increasing the linker length in 13 by one additional methylene group yielded SD-36 (14).26 SD-36 is very effective and potent in reducing the levels of the total STAT3 and pSTAT3Y705 proteins and achieves a DC50 of 0.06 μM.
Increasing the linker length by introduction of one more methylene group in SD-36 resulted in 15, which has a DC50 of 0.09 μM and is thus similarly potent as SD-36. Increasing the linker length in 15 by two methylenes yielded 16, which has DC50 = 0.15 μM and is thus 2–3 times less potent than SD-36. Our data thus established that a linker length of 8–9 atoms, as in SD-36 or 15, is optimal for these STAT3 degraders to reduce the levels of both the total STAT3 and pSTAT3Y705 proteins.
Investigation of the Linker Composition.
Our previous studies showed that the linker composition has a major effect on the ability of our MDM2 and BET PROTAC degraders to induce degradation of the target proteins.23–25 Accordingly, we investigated the linker composition using SD-36 as the template compound and obtained the data summarized in Table 2 and Figure S4.
Table 2.
STAT3 Degraders with Various Linker Compositions
| ||
|---|---|---|
| Cmpds | Linker | DC50 (STAT3, μM) Molm-16, 4h |
|
| ||
| SD-36 |
|
0.06 |
| 17 |
|
0.65 |
| 18 |
|
0.24 |
| 19 |
|
0.05 |
| 20 |
|
0.18 |
| 21 |
|
0.25 |
| 22 |
|
0.18 |
| 23 |
|
2.47 |
| 24 |
|
0.38 |
| 25 |
|
0.16 |
| 26 |
|
0.09 |
Changing the amide group in SD-36 to an amino group yielded 17, which has DC50 = 0.65 μM and is >10 times less potent than SD-36 in reducing the levels of total STAT3 and pSTAT3Y705 proteins (Table 2 and Figure S4A). Conversion of the amide group into a carbamate or urea group led to 18 and 19, respectively. While 18 is about 4 times less potent than SD-36 in inducing STAT3 degradation, 19 is as potent as SD-36. Replacing a CH2-CH2 linker moiety in SD-36 with a pyrazole ring in either side of the linker resulted in 20 and 21, respectively, and both compounds are 3–4 times less potent than SD-36. Replacement of a CH2-CH2-CH2 linker moiety in SD-36 with a piperidine ring yielded 22 or 23. Compound 22 is 3 times less potent than SD-36, and 23 is 40 times less potent than SD-36 (Figure S4B).
Because both 17 and 23 contain a linker with a positively charged group and are much less potent than SD-36, a positive charge in the linker of these degraders would appear to be detrimental to their ability to degrade STAT3 protein. To test this idea further, we changed the amino group in 23 to an amide, affording compound 24. Although 24 is still 6 times less potent than SD-36, it is more potent than 23 in reducing the levels of the total STAT3 and the PstatsY705 proteins, supporting the hypothesis. Changing the ethynyl group in SD-36 to -CH2-CH2- and -NH-CH2- led to compounds 25 and 26, respectively, both of which are 2–3 times less potent than SD-36 in reducing the levels of STAT3 and pSTAT3Y705.
Taken together, these data show that both the length and the composition of the linker play an important role in the ability of our designed STAT3 degraders to reduce the levels of STAT3 and pSTATSY705, and the optimal linkers are those used in SD-36 and compound 19.
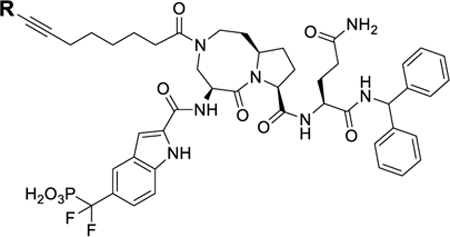
Examination of the CRBN Ligand Portion in SD-36.
We next performed modifications of the CRBN ligand portion in SD-36 (Table 3).
Table 3.
STAT3 Degraders with Modifications of the Cereblon Ligand Portion
| ||
|---|---|---|
| Code Name | R | DC50 (STAT3, μM) Molm-16, 4h |
|
| ||
| SD-36 |
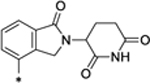
|
0.06 |
| 27 |
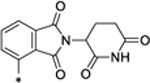
|
3.54 |
| 28 |
|
0.41 |
| 29 |
|
0.36 |
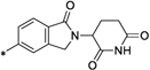
Thalidomide and lenalidomide bind to CRBN with similar modes and affinities.27 We replaced the lenalidomide moiety in SD-36 with a thalidomide moiety, producing 27. Surprisingly, 27 has DC50 = 3.54 μM and is thus >50 times less potent than SD-36 (Table 3 and Figure S5).
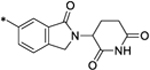
The cocrystal structure of lenalidomide in a complex with CRBN suggests two additional linking positions in the phenyl ring of lenalidomide, and accordingly, we synthesized 28 and 29. While these two compounds effectively degrade STAT3 and pSTAT3Y705, both are 5–10 times less potent than SD-36 (Table 3 and Figure S5).
Modifications of the STAT3 Ligand Portion of SD-36.
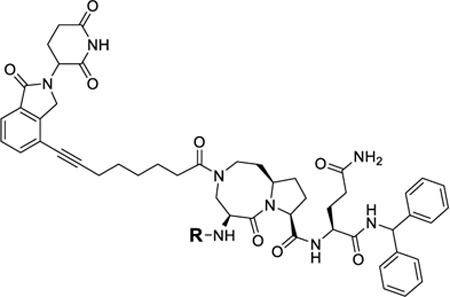
The cocrystal structure of SI-109 in a complex with STAT3 shows that one of the phenyl groups lacks specific interactions with STAT3 protein (Figure 2). We modified this part of the STAT3 ligand in SD-36 to investigate the effect on STAT3 degradation (Table 4).
Table 4.
STAT3 Degraders with Modified STAT3 Ligand Portion
| ||
|---|---|---|
| Cmpds | R | DC50 (STAT3, μM) Molm-16, 4h |
|
| ||
| SD-36 |
|
0.06 |
| 30 |
|
0.22 |
| 31 |

|
0.64 |
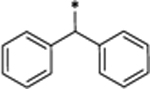
Removal of one phenyl group from SD-36 yielded compound 30, which is 4 times less potent than SD-36 in reducing STAT3 protein (Table 4 and Figure S6). Removal of both the phenyl groups from SD-36 resulted in 31, which achieve DC50 = 0.64 μM and is thus 10 times less potent than SD-36. These data indicate that the two phenyl groups in SD-36 are important for SD-36 to achieve its high potency in inducing degradation of STAT3.
Evaluation of Various Phosphotyrosine Mimetics.
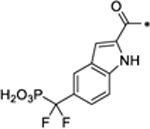
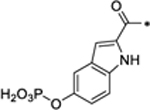
In SD-36, we employed 5-(difluoro(phosphoro)methyl)-1H-indole-2-carboxylic acid to mimic the phosphotyrosine group. To investigate the importance of this phosphotyrosine mimic in achieving potent and effective STAT3 degradation by SD-36, we synthesized 32, 33, and 34 (Table 5) with three different phosphotyrosine mimetics.15–17
Table 5.
STAT3 Degraders with Modifications of the STAT3 Ligand Portion
| ||
|---|---|---|
| Cmpds | R | DC50 (STAT3, μM) Molm-16, 4h |
|
| ||
| SD-36 |
|
0.06 |
| 32 |
|
>10 |
| 33 |
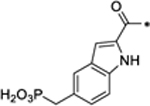
|
>10 |
| 34 |
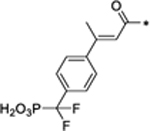
|
4.45 |
Compounds 32 and 33 foil to degrade both the total STAT3 and pSTAT3Y705 proteins at concentrations up to 10 μM (Figure S7). Compound 34 reduces the levels of both the total STAT3 and pSTAT3Y705 proteins with DC50 = 4.45 μM and is >70-times less potent than SD-36 (Table 5 and Figure S7). Hence, the phosphotyrosine mimic in SD-36 is critically important for achieving potent and effective degradation of the total STAT3 and pSTAT3Y705 proteins.
Design and Synthesis of a Control Compound for SD-36.
To facilitate our investigation of the cellular mechanism of action of our potent STAT3 degrader SD-36, we synthesized SD-36Me (Figure 4), in which a methyl group was installed to block the binding of SD-36 to cereblon.26–28
Figure 4.

Design of SD-36Me as a control compound.
SD-36Me is completely ineffective in inducing STAT3 degradation in Molm-16 cells at concentrations up to 10 μM (Figure S8), clearly suggesting that the degradation of STAT3 by SD-36 is cereblon and cullin-4A ligase-dependent, consistent with the PROTAC mechanism. We employed SD-36Me and the STAT3 inhibitor SI-109 as control compounds for further investigation of the activity, specificity, and cellular molecular mechanism of SD-36.
Evaluation of Representative Degraders in the SU-DHL-1 Lymphoma Cell Line.
The SU-DHL-1 anaplastic large cell lymphoma (ALCL) cell line has a high level of pSTAT3Y705 protein,26 and we evaluated the ability of several representative STAT3 degraders in the SU-DHL-1 cell line and obtained the data shown in Table 6 and Figure S9.
Table 6.
Degradation Potencies of STAT3 Protein and Cell Growth-Inhibitory Activities of STAT3 Degraders in the Molm-16 and SU-DHL-1 Cell Lines
| Molm-16 |
SU-DHL-1 |
|||
|---|---|---|---|---|
| compd | DC50 (STAT3, μM), 4 h | IC50 ± SD (μM), 4 days | DC50 (STAT3, μM), 16 h | IC50 ± SD (μM), 4 days |
| SI-109 (8) | >10 | 6.45 ± 4.16 | >10 | >10 |
| 12 | 2.63 | 0.95 ± 0.081 | nta | nta |
| 13 | 0.26 | 0.078 ± 0.029 | nta | nta |
| SD-36 (14) | 0.06 | 0.013 ± 0.010 | 0.028 | 0.61 ± 0.014 |
| 15 | 0.09 | 0.025 ± 0.007 | nta | nta |
| 16 | 0.15 | 0.042 ± 0.004 | nta | nta |
| 17 | 0.65 | 0.147 ± 0.027 | 0.919 | >10 |
| 18 | 0.24 | 0.128 ± 0.050 | 0.049 | 2.37 ± 0.70 |
| 19 | 0.05 | 0.091 ± 0.081 | 0.011 | 0.43 ± 0.16 |
| 20 | 0.18 | 0.241 ± 0.246 | nta | nta |
| 21 | 0.25 | 0.232 ± 0.175 | nta | nta |
| 22 | 0.18 | 0.099 ± 0.054 | nta | nta |
| 23 | 2.47 | 0.36 ± 0.056 | nta | nta |
| 24 | 0.38 | 0.067 ± 0.032 | nta | nta |
| 25 | 0.16 | 0.063 ± 0.016 | nta | nta |
| 26 | 0.09 | 0.059 ± 0.016 | nta | nta |
| 27 | 3.54 | 0.582 ± 0.654 | >10 | >10 |
| 28 | 0.41 | 0.104 ± 0.071 | nta | nta |
| 29 | 0.36 | 0.711 ± 0.262 | nta | nta |
| 30 | 0.22 | 0.210 ± 0.109 | 0.055 | 3.24 ± 0.57 |
| 31 | 0.64 | 0.226 ± 0.114 | 0.89 | >10 |
| 32 | >10 | >2 | nta | nta |
| 33 | >10 | >2 | nta | nta |
| 34 | 4.45 | 1.25 ± 0.71 | >10 | >10 |
| SD-36Me (35) | >10 | 5.21 ± 6.04 | >10 | >10 |
Not tested.
SD-36 effectively and potently reduces the levels of STAT3 and pSTAT3Y705 with DC50 of 28 nM in the SU-DHL-1 cell line with a 16 h treatment time (Table 6 and Figure S9). SI-109 and SD-36Me had no effect on the levels of STAT3 and activated pSTATSY705 at concentrations up to 10 μM in SU-DHL-1 cells.
Compound 19, which potently induces degradation of total STAT3 and pSTAT3Y705 proteins in Molm-16 cells with DC50 = 50 nM, is also potent and effective in reducing STAT3 and pSTAT3Y705 protein levels in SU-DHL-1 cells with a DC50 value of 11 nM.
Compounds 18 and 30 are 4 times less potent than SD-36 in degradation of STAT3 protein in Molm-16 cells, and both compounds are 2 times less potent than SD-36.
Compounds 17 and 31, which are >10 times less potent than SD-36 in Molm-16 cells, are 30 times less potent than SD-36 in SU-DHL-1 cells.
Compounds 27 and 34 have DC50 values of 3–5 μM in the Molm-16 cell line, but in the SU-DHL-1 cell line, both compounds are ineffective in reducing the levels of STAT3 and pSTATSY705 proteins in SU-DHL-1 cells up to 5 μM.
Evaluation of Degradation Selectivity of SD-36 on Other STAT Proteins in Cells.
There are seven STAT family members, STAT1, −2, −3, −4, −5A, −5B, and −6, which share a highly homologous SH2 domain. To investigate the degradation selectivity of SD-36, we evaluated SD-36 for its selectivity in both Molm-16 and SU-DHL-1 cells with SI-109 and SD-36Me included as controls.
As shown in Figure 5, SD-36 is very effective in reducing the level of STAT3 protein at concentrations as low as 0.1 μM but it has little or no effect on STAT1, STAT2, STAT4, STAT5A/B, and STAT6 proteins in both Molm-16 cell and SU-DHL-1 cell lines up to 10 μM. Hence, SD-36 demonstrates > 100-fold selectivity for degradation of STAT3 over other STAT members in both Molm-16 cell and SU-DHL-1 cell lines. The control compound SD-36Me has no effect on the levels of any STAT protein at 10 μM. Similarly, the STAT3 inhibitor SI-109 has no effect on the levels of any STAT protein.
Figure 5.

Western blotting analysis of STAT proteins in Molm-16 and SU-DHL-1 cells treated with SD-36 or control compounds SD-36Me and SI-109. (A) Molm-16 cells were treated with SD-36 at 0.01–10 μM or SD-36Me and SI-109 at 10 μM for 4 h. (B) SU-DHL-1 cells were treated with SD-36 at 0.01–10 μM or SD-36Me and SI-109 at 10 μM for 18 h.
Our data therefore show that SD-36 achieves an exceptional degradation selectivity for STAT3 over other STATs (Figure 5).
To gain a further insight into the degradation selectivity of SD-36 for STAT3 over other STAT proteins, we determined its binding affinities to different STAT proteins.26 Our binding data showed that while SD-36 has a high affinity for STAT3 with a Kd value of 44 nM, it also displays a good affinity to STAT1 and STAT4 with Kd values of approximately 1 μM.26 SD-36 has a much weaker affinity to other STAT proteins tested.26 Hence, although SD-36 binds to STAT1 and STAT4 with a good affinity, it fails to degrade these two STAT proteins, as well as other STAT proteins in cells. We posit that the high degradation selectivity for SD-36 is achieved through the formation of a stable and productive degradation ternary complex consisting of STAT3:SD-36:CRBN/cullin4A, as compared to much less stable and productive ternary complexes for other STAT proteins.29 Further investigations are needed to fully understand the exceptional STAT3 degradation selectivity for SD-36.
Evaluation of STAT3 Degraders for Their Growth-Inhibitory Activities.
As SD-36 is able to effectively and selectively degrade STAT3 protein in Molm-16 and SU-DHL-1 cells, we therefore tested all of our STAT3 degraders reported in this study for their ability to inhibit cell growth in the Molm-16 cell line and a number of representative STAT3 degraders in the SU-DHL-1 cell line. SI-109 and SD-36Me were included as control compounds in the cell growth experiments. The data are summarized in Table 6.
In the Molm-16 cell line, SD-36 exerts a potent growth-inhibitory activity with IC50 = 13 nM after a 4-day treatment. The control compound SD-36Me shows modest growth-inhibitory activity with IC50 = 5.21 μM and is 400 times less potent than SD-36.
There is an excellent correlation between the STAT3 degradation potencies and cell growth inhibitory activities for these STAT3 degraders in the Molm-16 cell line. All compounds that effectively and potently degrade STAT3 protein also potently inhibit cell growth in the Molm-16 cell line. Compounds such as 32 and 33, which fail to degrade STAT3 protein up to 10 μM, also display very weak activity in inhibition of cell growth.
In the SU-DHL-1 line, SD-36 effectively inhibits cell growth and achieves IC50 values of 0.61 μM, while STAT3 inhibitors SI-109 and SD-36Me have no effect on cell viability up to 10 μM. Compound 19, which effectively degrades STAT3 with DC50 = 28 nM, displays an IC50 value of 0.43 μM. Compounds 18 and 30, which achieve DC50 values of 49 nM and 55 nM, respectively, have IC50 values of 2.37 μM and 3.24 μM, respectively, in the SU-DHL-1 line. Compounds 17 and 31, which have DC50 values of 0.92 μM and 0.89 μM, respectively, have minimal activity in inhibition of cell growth with IC50 values of >10 μM. Compound 34, which fails to degrade STAT3 up to 10 μM, is also ineffective in inhibition of cell growth (IC50 > 10 μM). In general, STAT3 degraders show weaker cell growth inhibitory activity in the SU-DHL-1 line than in the Molm-16 cell line.
Hence, effective degradation of STAT3 results in robust growth inhibition in both Molm-16 and SU-DHL-1 cell lines, and there is a good correlation between STAT3 degradation and cell growth inhibition in both cell lines.
Pharmacodynamics Evaluation of SD-36 in Native and Xenograft Tumor Tissues in Mice.
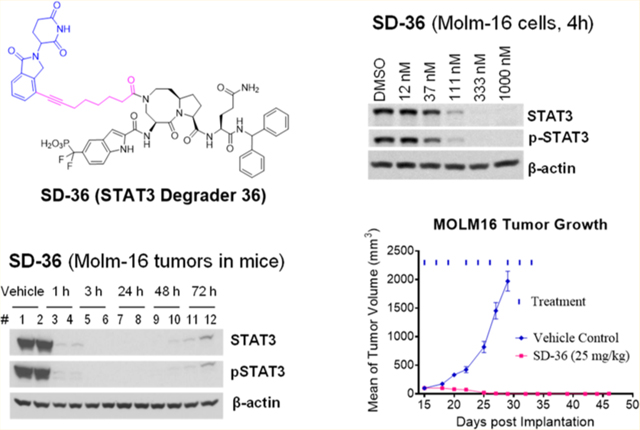
Our cellular data demonstrate that SD-36 and 19 are two potent STAT3 degraders. We performed pharmacodynamics (PD) experiments to evaluate the ability of SD-36 and 19 to degrade STAT3 protein in mice.
We first investigated SD-36 and 19 for their ability to degrade STAT3 protein in native mouse tissue. Western blotting analysis showed that a single intravenous dose of both SD-36 and 19 at 25 mg/kg is very effective in reducing STAT3 protein in native mouse spleen tissue with the effect persisting for more than 48 h, and SD-36 appears to be more effective than 19 (Figure 6A). We thus chose to further evaluate SD-36 in vivo.
Figure 6.

Pharmacodynamics study of SD-36 and 19 in native mouse spleen tissue in CD-1 mice (A) and SD-36 in the Molm-16 xenograft tumors in SCID mice (B). CD-1 and SCID mice were treated intravenously with a single dose of SD-36 or 19 as indicated. Native spleen tissue or tumor lysates were analyzed by immunoblotting.
We evaluated SD-36 for its ability to degrade STAT3 in the Molm-16 xenograft tumor tissue in mice (Figure 6B). Our Western blotting data showed that a single dose of SD-36 at 50 mg/kg is very effective in reducing the levels of both total STAT3 and pSTATSY705 proteins in xenograft tumor tissue. Both total STAT3 and pSTAT3Y705 proteins were reduced by >95% after 1 h, indicating a rapid degradation of STAT3 in vivo. Even 72 h (3 days) after dosing, SD-36 still reduced the levels of total STAT3 and pSTAT3Y705 proteins by >90% as compared to the control. Our PD data thus demonstrate that a single dose of SD-36 is highly effective in inducing near complete elimination of STAT3 protein in tumor tissue in mice with the effect persisting for >3 days.
Antitumor Activity of SD-36 in the Molm-16 Xenograft Model in Mice.
Upon the basis of its impressive PD data in the Molm-16 xenograft tumor tissue, we evaluated SD-36 for its efficacy in the Molm-16 xenograft model in mice at different doses and schedules.
Administered to mice at either 25 or 50 mg/kg three times per week for 3 weeks intravenously, SD-36 is capable of inducing complete tumor regression (Figure 7A). In fact, complete tumor regression was achieved after dosing for only 2 weeks at either 25 or 50 mg/kg and persisted for more than 2 weeks after termination of the treatment.
Figure 7.

In vivo antitumor activity of SD-36 in the MoIm-16 xenograft model in mice: (A) tumor growth; (B) percentage of animal body weight change.
Because a single dose of SD-36 at 50 mg/kg is capable of reducing STAT3 protein levels for >3 days, we evaluated its efficacy with a weekly dosing schedule. SD-36 at 50 mg/kg administered once weekly for 3 weeks is very effective in inhibition of tumor growth (Figure 7A). Furthermore, SD-36 at 100 mg/kg weekly dosing was capable of achieving complete tumor regression.26 In the efficacy experiment, SD-36 caused no weight loss or other signs of toxicity (Figure 7B).
Hence, SD-36 is highly efficacious in vivo and achieves complete and long-lasting tumor regression in the Molm-16 cancer xenograft model at well-tolerated dose-schedules.
CHEMISTRY
Scheme 1 outlines the synthesis of the protected phosphotyrosine mimetic, 5-((di-tert-butoxyphosphoryl)methyl)-1H-indole-2-carboxylic acid (41). Boc protection of 36 followed by benzylic bromination provided 38. Treatment of 38 with sodium di-tert-butyl phosphite yielded a mixture of 39 and 40 which were hydrolyzed to afford compound 41.
Scheme 1. Synthesis of 5-((Di-tert-butoxyphosphoryl)methyl)-1H-indole-2-carboxylic Acid (41)a.

aReagents and conditions: (a) NaH, Boc2O, THF, 0 °C to rt; (b) NBS, Bz2O2, CCl4, reflux, 77% over 2 steps; (c) (t-BuO)2PONa 1.0 equiv of NaH 2.5 equiv, THF, then reflux for 3 h; (d) LiOH, MeOH–THF–H2O, 60 °C.
Scheme 2 shows the construction of the 5-((diethoxyphosphoryl)difluoromethyl)-1H-indole-2-carboxylic acid (46), an α,α-difluoro derivative of 41 as previously reported.26 Compound 38 was heated in P(OEt)3 at 100 °C for 12 h furnishing the phosphate (42), which was subject to transesterification, affording the benzyl ester (43). The indole nitrogen of 40 was protected with a Cbz group, giving 44. Difluorination of the double-activated methylene of 44 provided 45, and finally, hydrogenation of 45 removed two protecting groups, furnishing the carboxylic acid (46).
Scheme 2. Synthesis of 3-((Diethoxyphosphoryl)difluoromethyl)-1H-indole-2-carboxylic Acid (46)a.

aReagents and conditions: (a) P(OEt)3, 100 °C, 84%; (b) Ti(O-i-Pr)4, BnOH, 100 °C, 83%; (c) NaH, Cbz-Cl, THF, 0 °C to rt, 88%; (d) NFBS, NaHMDS, THF, −78 °C to rt, 95%; (e) H2/Pd–C, THF, 94%.
The syntheses of compounds 2–4 are shown in Scheme 3 as previously reported.26 Amide condensation of the acid (47) with the amine (48) gave compound 49 removal of whose Boc group yielded compound 50. Condensation of 50 with corresponding protected phosphotyrosine mimetics followed by hydrolysis of the dialkyl phosphate group afforded compounds 2–4.
Scheme 3. Synthesis of Compounds 2–4a.

aReagents and conditions: (a) EDC–HCl 2.0 equiv, HOBt 2.0 equiv, EtN(i-Pr)2, DCM, rt, 3 h, 89% yield; (b) CF3CO2H 2 mL, Et3Si–H 0.1 mL, DCM 3 mL, rt, 1 h; (c) (1) protected phosphotyrosine mimetics, EDC–HCl, HOBt, EtN(i-Pr)2, DCM, rt, 3 h; (2) TFA–DCM, rt, 3 h; or TMSI, 0 °C, 1 h.
Scheme 4 shows the syntheses of compounds 5 and 7 as previously reported.26 By use of the acid (51)30 as starting material, amide condensation reaction and subsequent selective removal of Boc group yielded an amine (53) which, coupled with 46, gave 54. The common intermediate (56) was prepared by deprotection of Cbz group in 51 and introduction of a methyl group followed by hydrolysis of the tert-butyl ester. The corresponding amines were coupled to 56 and the resulting diethyl phosphates were treated with TMSI to yield compounds 5 and 7.
Scheme 4. Synthesis of Compounds 5 and 7a.

aReagents and conditions: (a) (1) HATU, EtN(i-Pr)2, DMF, rt, 1 h; (2) TFA, DCM; (b) 46, HATU, EtN(i-Pr)2, DMF, rt, 1 h; (c) H2, Pd/C, MeOH; (d) (1) HCHO, sodium triacetoxyborohydride, DCE; (2) TFA, DCM; (e) R-NH2, HATU, EtN(i-Pr)2, DMF, rt, 1 h.
The syntheses of compounds 8–11 are shown in Scheme 5. The intermediate (60) was obtained by an amide condensation reaction followed by removal of the Boc protecting group under acidic conditions. Condensation of 60 with 51 led to 61, which following removal of the Cbz and subsequent acetylation and Boc deprotection gave the common intermediate (62). Amide condensation of 62 with corresponding protected phosphotyrosine mimetics and hydrolysis of the diethyl phosphate group afforded compounds 8–11.
Scheme 5. Synthesis of Compounds 8–11a.

aReagents and conditions: (a) HATU, EtN(i-Pr)2, DMF, rt, 1 h; (b) TFA, DCM; (c) 51, HATU, EtN(i-Pr)2, DMF, rt, 1 h; (d) (1) H2, Pd/C, MeOH; (2) Ac2O, EtN(i-Pr)2, DCM; (3) TFA, DCM; (e) (1) phosphotyrosine mimetics, HATU, EtN(i-Pr)2, DMF, rt, 1 h; (2) TFA–DCM, rt, 3 h; or TMSI, 0 °C, 1 h.
The preparation of the representative STAT3 degrader SD-36 is outlined in Scheme 6 as previously reported.26 Compound 63 was obtained by removal of the Boc group of intermediate 61 followed by amidation with 46. Hydrogenation of 63 gave the amine (64) which was coupled with 68 to afford 65, which was treated with TMSI to yield SD-36. The synthesis of intermediate 68 was achieved by Sonogashira coupling of commercially available starting material 66 and oct-7-ynoic acid (67). By utilization of compound 64 as a common intermediate, the final compounds (13–29) were prepared by a method similar to that used for SD-36.
Scheme 6. Synthesis of SD-36a.

aReagents and conditions: (a) (1) TFA, DCM; (2) 46, HATU, DIEA, DMF; (b) H2, Pd/C, MeOH; (c) 68, HATU, DIEA, DMF; (d) TMSI, BSTFA, DCM; (e) Pd(PPh3)Cl2, CuI, DMF/TEA, 80 °C, 1 h.
The synthesis of compounds 30 and 31 is shown in Scheme 7. By use of compound 55 as starting material, the acid (69) was obtained by amide condensation followed deprotection of the tert-butyl ester. By employment of compound 66 as a common intermediate, compounds 30 and 31 were synthesized by condensation with corresponding amine and subsequent hydrolysis of the diethyl phosphate group.
Scheme 7. Synthesis of Compounds 30 and 31a.

aReagents and conditions: (a) (1) 68, HATU, DIEA, DMF; (2) TFA, DCM; (b) (i) R-NH2, HATU, DIEA, DMF; (2) TMSI, BSTFA, DCM.
Compounds 32–34 were prepared as shown in Scheme 8. Intermediate 70 was prepared by deprotection of the Cbz group in 61 and subsequent amidation with 68 followed by removal of the Boc group. The corresponding protected phosphotyrosine mimetics were coupled to 70 followed by TMSI-promoted hydrolysis of diethyl phosphate group to afford compounds 32–34.
Scheme 8. Synthesis of Compounds 32–34a.

aReagents and conditions: (a) (1) H2, Pd/C, MeOH; (2) 68, HATU, DIEA, DMF; (3) TFA, DCM; (b) (1) phosphotyrosine mimetics, HATU, EtN(i-Pr)2, DMF, rt, 1 h; (2) TFA–DCM, rt, 3 h; or TMSI, 0 °C, 1 h.
Scheme 9 outlines the synthesis of fluorescent labeled compound 74 as previously reported.26 Intermediate 73 was prepared by condensation of 71 and 72 with HATU as the coupling reagent, followed by subsequent removal of the Boc protecting group. Compound 74 was furnished by amide condensation reaction of compound 73 and 56 followed by TMSI-promoted hydrolysis of the diethyl phosphate group.
Scheme 9. Synthesis of Fluorescent Labeled Compound 74a.

aReagents and conditions: (a) (1) HATU, DIEA, DMF; (2) TFA, DCM; (b) (1) HATU, DIEA, DMF; (2) TMSI, BSTFA, DCM.
CONCLUSION
In this paper, we report our structure-based discovery of potent and selective PROTAC STAT3 degraders with SD-36 being the best compound. We first designed and obtained SI-109 as a potent inhibitor of the STAT3 SH2 domain, starting from our previously reported peptidomimetic STAT3 inhibitor. A key element in the successful discovery of SI-109 was the design of 5-(difluoro(phosphoro)methyl)-1H-indole-2-carboxylic acid as a novel phosphotyrosine mimetic. Determination of the cocrystal structure of SI-109 in a complex with STAT3 protein provided a structural basis for the design of STAT3 degraders.
Employing SI-109 and ligands for cereblon/cullin 4A, we designed, synthesized, and tested a series of PROTAC STAT3 degraders. Our efforts have yielded a series of potent STAT3 degraders, with SD-36 being the best. SD-36 induces degradation of STAT3 protein in cells and demonstrates >100-fold selectivity over other STAT proteins.
SD-36 potently inhibits cell growth in the Molm-16 leukemia and SU-DHL-1 lymphoma cell lines. A single dose of SD-36 is capable of achieving complete degradation of STAT3 in native mouse spleen tissue and in the Molm-16 xenograft tumor tissue with the degradation effect persisting for >2–3 days. SD-36 is capable of achieving complete and persistent tumor regression in the Molm-16 xenograft model in mice at well tolerated dose-schedules. To our knowledge, SD-36 is a first, potent, selective, and highly efficacious degrader of STAT3 protein reported. Because SD-36 effectively and selectively degrades STAT3 in cells and in tissues, it can be employed as a powerful tool to study the role(s) of STAT3 in different biological processes. Furthermore, SD-36 warrants extensive investigation as a new class of anticancer therapy for the treatment of human cancers and other human diseases. Our present study demonstrates that the PROTAC technology can be successfully employed to target those traditionally difficult-to-drug or undruggable protein targets.
EXPERIMENTAL SECTION
General Information.
All commercial reagents and solvents were used as supplied without further purification with the following exceptions: THF was freshly distilled from sodium wire. The reactions were performed under an N2 atmosphere in anhydrous solvents. The final products were purified by reverse phase HPLC (RP-HPLC) with solvent A (0.1% of TFA in water) and solvent B (0.1% of TFA in CH3CN) as eluents with a flow rate of 60 mL/min. The purity of compounds was determined by Waters ACQUITY UPLC, and all the final compounds were >95% pure. Proton nuclear magnetic resonance (1H NMR), carbon nuclear magnetic resonance (13C NMR), and phosphorus nuclear magnetic resonance (31P NMR) spectroscopies were performed on Bruker Advance 300/400 NMR spectrometers, and chemical shifts are reported in parts per million (ppm) relative to an internal standard. MS analysis was carried out with a Thermo-Scientific LCQ Fleet mass spectrometer or a Waters ultraperformance liquid chromatography (UPLC)–mass spectrometer.
2-((3S,6S)-3-((6S)-5-Amino-1-(benzylamino)-1,5-dioxopentan-2-ylcarbamoyl)-5-oxodecahydropyrrolo[1,2-a]azocin-6-ylcarbamoyl)-1H-indol-5-yl Dihydrogen Phosphate (2).
The acid (47, 66 mg, 0.2 mmol), HOBt (56 mg, 0.4 mmol), and EDC-HCl (80 mg, 0.4 mmol) were placed in round-bottomed flask. EtN(i-Pr)2 (0.2 mL, 1.0 mmol) and DCM were added via syringe, and the reaction mixture was stirred at rt for 5 min. Compound 48 was added as a solution in DCM. The reaction was stirred at rt for 3 h before being quenched with saturated NaHCO3. The aqueous layer was extracted with DCM, and the combined organic layers were washed with brine and dried over anhydrous sodium sulfate. The solvent was removed in vacuo and the residue was purified by flash column chromatography to yield 49 in 89% yield (0.14 g). 1H NMR (300 MHz, MeOD-d4): 8.52 (s, 1H), 8.40–8.20 (m, 2H), 7.50–7.00 (m, 20 H), 4.70–4.45 (m, 1H), 4.45–4.20 (m, 5H), 2.70–2.40 (m, 2H), 2.30–1.00 (m, 14H), 1.44 (s, 9H). 13C NMR (75 MHz, MeOD-d4): 174.60, 174.53, 173.96, 173.56, 157.57, 146.21, 139.92, 130.25, 129.69, 128.91, 128.68,128.38, 127.93, 80.80, 71.74, 63.17, 61.63, 54.28, 52.14,44.21, 37.46, 35.80, 33.60, 29.14, 28.95, 26.95, 23.53. ESI-MS calculated for C47H56N5O6 [M + H]+ = 786.42. Obtained: 786.92
The amine 49 (78 mg, 0.1 mmol) was dissolved in DCM (5 mL). Et3Si-H (0.1 mL) and CF3CO2H (2 mL) were added via syringe. The reaction was stirred at rt for 1 h. The volatile components were removed on a rotary evaporator. The residue compound (50) was used in the next step without further purification. Compound 50 (crude, 0.1 mmol) and 5-((di-tert-butoxyphosphoryl)oxy)-1H-indole-2-carboxylic acid31 (55 mg, 0.15 mmol) were coupled using peptide coupling conditions (3.0 equiv of HOBt, 3.0 equiv of EDC-HCl, 20 equiv of EtN(i-Pr)2, and DCM as solvent). After being purified by flash column chromatography, the obtained product was dissolved in DCM (5 mL) in a round bottomed flask equipped with a magnetic stirring bar. CF3CO2H (2 mL) and Et3Si-H (0.1 mL) were added to the reaction mixture, which was then stirred for 3 h. Solvent was removed in vacuo and the product was purified by reverse phase semipreparative HPLC to give compound 2 (25 mg, 36% yield). 1H NMR (300 MHz, MeOD-d4): 7.47 (s, 1H), 7.42 (d, J = 8.92 Hz, 1H), 7.38–7.20 (m, 5H), 7.17 (s, 1H), 7.13 (dd, J = 8.92, 1.67 Hz, 1H), 5.14–5.04 (m, 1H), 4.50–4.32 (m, 5H), 2.50–1.50 (m, 16H). 31P NMR (121 MHz, MeOD-d4): 4.4 (s). ESI-MS calculated for C32H40N6O9P [M + H]+ = 683.26. Found: 683.20. Reverse phase analytical HPLC: >99% purity.
(2-((3S,6S)-3-((S)-5-Amino-1-(benzylamino)-1,5-dioxopentan-2-ylcarbamoyl)-5-oxodecahydropyrrolo[1,2-a]azocin-6-ylcarbamoyl)-1H-indol-5-yl)methylphosphonic Acid (3).
NaH (2.2 g, 53 mmol, 2.0 equiv, 60% in mineral oil) and THF (300 mL) were mixed in a round bottomed flask equipped with a magnetic stirring bar. The suspension was cooled with an ice/water bath before addition of ethyl 5-methyl-1H-indole-2-carboxylate (36) (5.0 g, 26 mmol, 1.0 equiv) over 15 min. The solution was stirred at this temperature for 30 min, and the color of the solution turned red. Boc2O (8.1 g, 37 mmol, 1.4 equiv) was added to the solution in one portion. The reaction mixture was stirred at rt for another 24 h before quenching with ice–water. The aqueous layer was extracted with EtOAc (200 mL × 2), and the combined organic layers were washed with brine (50 mL × 2), dried over anhydrous sodium sulfate, and concentrated on a rotary evaporator. The residual crude product (37) was used directly for the next step without further purification. The crude 37, (PhCO)2O2 (242 mg, 1.0 mmol, 0.04 equiv), NBS (4,62 g, 26.0 mmol, 1.0 equiv), and anhydrous CCl4 (150 mL) were added to a round bottomed flask equipped with a magnetic stirring bar. The reaction mixture was heated at reflux for 12 h. The precipitate was filtered off, and the solvent was removed on a rotary evaporator. The residual crude product was purified by flash column chromatography to afford the desired benzylic bromide (38) as colorless oil (7.6 g, 77% yield). 1H NMR (300 MHz, CDCl3): 8.05 (d, J = 8.66 Hz, 1H), 7.61 9d, J = 1.39 Hz, 1H), 7.44 (dd, J = 8.66, 1.81 Hz, 1H), 7.06 (d, J = 0.65 Hz, 1H), 5.29 (s, 2H), 4.38 (q, J = 7.14 Hz, 2H), 1.62 (s, 9H), 1.40 (t, J = 7.14 Hz, 3H). 13C NMR (75 MHz, CDCl3): 161.8, 149.2, 146.9, 137.6, 133.0, 131.8, 128.0, 127.9, 122.7, 115.5, 114.5, 85.3, 85.0, 61.7, 34.1, 28.0, 14.4. ESI-MS calculated for C17H2179BrNO4 [M + H]+ = 382.07. Found: 382.42. C17H2181BrNO4 [M + H]+: 384.06. Found: 384.08
NaH 0.22 g (60% in mineral oil, 5.5 mmol, 2.5 equiv) and anhydrous THF (10 mL) were added to a round-bottom flask equipped with a magnetic stirring bar. (t-BuO)2POH (427 mg, 2.2 mmol, 1.0 equiv) was added dropwise as a THF solution, and the reaction mixture was stirred for 0.5 h at rt. The bromide (38) was then added as a THF solution, and the mixture was heated at 80 °C for 3 h before quenching with NH4Cl saturated solution at 0 °C. The aqueous layer was extracted with EtOAc (3 × 30 mL). The organic layers were combined and dried over anhydrous Na2SO4. The solvent was removed in vacuo and the product was purified by flash column chromatography to give a mixture of 39 and 40 (0.68 g, 1.4 mmol, 63% yield, 39:40 = 75:25, inseparable by flash column chromatography). The data of the major isomer (39) are shown below. 1H NMR (300 MHz, CDCl3): 7.99 (d, J = 8.60 Hz, 1H), 7.51–7.49 (m, 1H), 7.36–7.30 (m, 1H), 7.05 (s, 1H), 4.38 (q, J = 7.16 Hz, 2H), 3.12 (d, JP–H = 21.08 Hz, 2H), 1.63 (s, 9H), 1.41 (s, 18H), 1.41–1.39 (m, 3H). 13C NMR (75 MHz, CDCl3): 161.8, 149.2, 136.6 (d, JP–C = 2.9 Hz), 130.9, 128.9 (d, JP–H = 5.9 Hz), 128.51 (d, JP–H = 9.6 Hz), 127.6 (d, JP–C = 2.8 Hz), 123.0 (d, JP–C = 7.2 Hz), 114.5, 114.4 (d, JP–C = 2.4 Hz), 84.5, 82.0 (d, JP–C = 9.0 Hz), 61.3, 37.6 (d, JP–C = 142.9 Hz), 30.33 (d, JP–C = 3.9 Hz), 27.8, 14.2. 31P NMR (121 MHz, CDCl3): 18.10. ESI-MS calculated for C25H38NO7P [M + Na]+ = 518.23. Found: 518.24
The acid 41 was synthesized from 39 and 40 (90 mg, 2.0 equiv) using LiOH–H2O (16 mg, 4.0 equiv) as base and THF/H2O/MeOH (3 mL:3 mL:3 mL) as solvents. The reaction was heated at 60 °C. Upon completion of hydrolysis determined by TLC (~2 h), the reaction mixture was acidified to pH = 2 with 0.1 N HCl. The aqueous layer was extracted with EtOAc (3 × 30 mL), and the organic layers were combined and dried over anhydrous Na2SO4. The solvent was removed in vacuo, and the crude product was coupled with 41 without further purification.
The freshly prepared amine 50 (0.05 mmol) and 41 (0.2 mmol) were coupled using peptide coupling conditions (6.0 equiv HOBt, 6.0 equiv of EDC-HCl, 40 equiv of EtN(i-Pr)2, and DCM as solvent). The reaction was stirred at rt for 3 h before being quenched with saturated NaHCO3. The aqueous layer was extracted with DCM, and the combined organic layers were washed with brine and dried over anhydrous sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography. The obtained product was dissolved in DCM (5 mL), and CF3CO2H (3 mL) and Et3Si-H (0.1 mL) were added via syringe. The reaction mixture was stirred at rt for 3 h. The volatile components were removed on a rotary evaporator and the desired product was purified by reverse phase semipreparative HPLC to give compound 3 (20 mg, 58% yield). 1H NMR (300 MHz, MeOD-d4): 7.57 (s, 1H), 7.39 (d, J = 8.52 Hz, 1H), 7.36–7.24 (m, 5H), 7.23 (d, J = 8.52 Hz, 1H), 7.15 (s, 1H), 5.14–5.02 (m, 1H), 4.52–4.06 (m, 5H), 3.20 (d, JP–H = 21.05 Hz, 2H), 2.50–1.50 (m, 16H). 31P NMR (121 MHz, MeOD-d4): 25.3 (s). ESI-MS calculated for C33H42N6O8P [M + H]+ = 681.28. Found: 681.28. Reverse phase analytical HPLC: 98% purity.
(2-((3S,6S)-3-((S)-5-Amino-1-(benzylamino)-1,5-dioxopentan-2-ylcarbamoyl)-5-oxodecahydropyrrolo[1,2-a]azocin-6-ylcarbamoyl)-1H-indol-5-yl)difluoromethylphosphonic Acid (4).
Compound 38 (3 g, 7.9 mmol, 1.0 equiv) and (EtO)3P (1.72 mL, 10.0 mmol, 1.2 equiv) were placed in a round bottomed flask equipped with a magnetic stirring bar. The reaction mixture was heated at 100 °C for 12 h and then was loaded directly onto a silica gel column and purified by flash column chromatography to afford the desired phosphate (42) as a colorless oil (2.9 g, 84%). 1H NMR (300 MHz, CDCl3): 8.02 (d, J = 8.62 Hz, 1H), 7.53 (s, 1H), 7.35 (d, J = 8.63 Hz, 1H), 7.05 (s, 1H), 4.38 (q, J = 7.13 Hz, 2H), 4.07–3.92 (m, 4H), 3.23 (d, JP–H = 21.24 Hz, 2H), 1.63 (s, 9H), 1.39 (t, J = 7.13 Hz, 3H), 1.23 (t, J = 7.06 Hz, 6H). 13C NMR (75 MHz, CDCl3): 161.7, 149.1, 136.7 (d, JP–C = 2.88 Hz), 131.1, 128.5 (d, JP–C = 5.88 Hz), 127.7 (d, JP–C = 2.81), 126.5 (d, JP–C = 9.12 Hz), 122.9 (d, JP–C = 7.15 Hz), 114.8 (d, JP–C = 2.50 Hz), 114.3, 84.5, 62.0 (d, JP–C = 6.79 Hz), 61.3, 33.3 (d, JP–C = 128.4), 27.7, 16.3 (d, JP–C = 5.96 Hz), 14.1. 31P NMR (121 M Hz, CDCI3): 26.3 (s). ESI-MS calculated for C21H31NO7P [M + H]+ = 440.18. Found: 440.67
Compound 42 (2.9 g, 6.6 mmol, 1.0 equiv), BnOH (14 mL, 132 mmol, 20 equiv), and Ti(Oi-Pr)4 (0.32 mL, 1.6 mmol, 0.25 equiv) were placed in a round bottomed flask equipped with a magnetic stirring bar. The reaction mixture was heated at 100 °C for 12 h. The reaction mixture was cooled down to 35 °C and quenched with 1 N HCl (20 mL). The aqueous layer was extracted with EtOAc (200 mL × 2), and the combined organic extracts were washed with brine (50 mL × 2), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residual crude product was purified by flash column chromatography to afford the desired benzyl carboxylate (43) as a colorless oil (2.25 g, 83% yield). 80% purity (determined by 31P NMR): 10% ethyl carboxylate, 10% unknown. 1H NMR (300 MHz, MeOD-d4): 7.65 (s, 1H), 7.60–7.38 (m, 6H), 7.31 (dt, J = 8.57, 1.72 Hz, 1H), 7.24 (s, 1H), 5.43 (s, 2H), 4.15–4.00 (m, 4H), 3.35 (d, JP–H = 21.03 Hz, 2H), 1.30 (t, J = 7.06 Hz, 6H). 13C NMR (75 MHz, MeOD-d4): 163.0, 138.2 (d, JP–C = 2.19 Hz), 137.6, 129.6, 129.3, 129.2, 129.1, 128.8 (d, JP–C = 2.76 Hz), 128.2 (d, JP–C = 5.33 Hz), 124.3 (d, JP–C = 7.95 Hz), 124.1 (d, JP–C = 9.42 Hz), 113.4 (d, JP–C = 2.37 Hz), 109.3, 67.4, 63.6 (d, JP–C = 6.96 Hz), 33.6 (d, JP–C = 138.3 Hz), 16.7 (d, JP–C = 5.92 Hz). 31P NMR (121 M Hz, MeOD-d4): 28.3 (s), 26.4 (s). ESI-MS calculated for C21H25NO5P [M + H]+ = 402.15. Found: 402.50
NaH (0.6 g, 15 mmol, 3.0 equiv, 60% in mineral oil) and THF (100 mL) were placed in a round bottomed flask equipped with a magnetic stirring bar. The suspension was cooled with ice/water bath before addition of compound 43 (2.25 g in THF, 5.5 mmol, 1.0 equiv) over 5 min. The solution was stirred at this temperature for 10 min before addition of Cbz-Cl (1.12 mL, 8 mmol, 1.5 equiv) via a syringe. The reaction mixture was stirred at rt for another 12 h before quenching with ice–water. The aqueous layer was extracted with EtOAc (200 mL × 2), and the combined organic extracts were washed with brine (50 mL × 2), dried over anhydrous sodium sulfate, and concentrated in vacuo. The residual crude product was purified by flash column chromatography to afford the desired 41 as a colorless oil (2.6 g, 88% yield). 1H NMR (300 MHz, CDCl3): 8.00 (d, J = 8.63 Hz, 1H), 7.52 (s, 1H), 7.46–7.26 (m, 11H), 7.11 (s, 1H), 5.33 (s, 2H), 5.20 (s, 2H), 4.10–3.90 (m, 4H), 3.22 (d, JP–H = 21.30 Hz, 2H), 1.21 (t, J = 7.05 Hz, 6H). 13C NMR (75 MHz, CDCl3): 161.3, 150.5, 136.6 (d, JP–C = 2.97 Hz), 135.3, 134.4, 130.6, 129.0 (d, JP–C = 5.88 Hz) 128.7, 128.6, 128.6, 128.5, 128.3, 128.2, 127.8 (d, JP–C = 2.82 Hz), 127.0 (d, JP–C = 9.10 Hz), 123.1 (d, JP–C = 7.08 Hz), 115.6, 115.0 (d, JP–C = 2.25 Hz), 69.5, 67.1, 62.1 (d, JP–C = 6.78 Hz), 33.4 (d, JP–C = 138.49 Hz), 16.3 (d, JP–C = 5.87 Hz). 31P NMR (121 M Hz, CDCI3): 26.3 (s). ESI-MS calculated for C29H30NO7P [M + Na]+ = 558.17. Found: 558.08
Compound 44 (9.17 g, 17 mmol, 1.0 equiv), (PhSO2)2NF (NFSB, 16 g, 51 mmol, 3.0 equiv), and THF (300 mL) were added to a round bottomed flask equipped with a magnetic stirring bar. The reaction mixture was cooled to −78 °C with the aid of an ethanol/dry ice bath. To this solution, NaHMDS (51 mL, 1.0 M in THF, 3.0 equiv) was added over 10 min. The reaction mixture was stirred at this temperature for 2 h before warming up to rt over 3–4 h. The reaction was quenched with saturated NH4Cl aqueous solution (100 mL). The aqueous layer was extracted with EtOAc (200 mL × 2), and the combined organic extracts were washed with brine (50 mL × 2), dried over anhydrous sodium sulfate, and concentrated in vacuo. The residual crude product was purified by flash column chromatography to afford the desired product (45) as a colorless oil (9.6 g, 95% yield). 1H NMR (300 MHz, CDCl3): 8.13 (d, J = 8.70 Hz, 1H), 7.88 (s, 1H), 7.65 (d, J = 8.90 Hz, 1H), 7.50–7.28 (m, 10H), 7.17 (s, 1H), 5.33 (s, 2H), 5.20 (s, 2H), 4.30–4.00 (m, 4H), 1.27 (t, J = 6.85 Hz, 6H). 13C NMR (75 MHz, CDCl3): 161.2, 150.3, 138.6, 135.2, 134.2, 131.5, 129.0, 128.8, 128.7, 128.6, 128.5, 128.4, 128.4–127.6 (m), 127.4, 125.2–124.4 (m), 121.0–120.6 (m), 120.5–119.5 (m), 115.5, 115.2, 70.0, 67.3, 64.9 (d, JP–C = 6.76 Hz), 16.3 (d, JP–C = 5.49 Hz). 31P NMR (121 M Hz, CDCl3): 6.3 (t, JP–F = 117 Hz). ESI-MS calculated for C29H29F2NO7P [M + H]+ = 572.17. Found: 572.25
Compound 45 (l g, 1.7 mmol, 1.0 equiv) and THF (300 mL) were placed in a round bottomed flask equipped with a magnetic stirring bar. The oxygen was removed with the aid of a vacuum line and a nitrogen balloon, and then 10% Pd/C (0.1 g, 0.1 mmol, 0.05 equiv) was added to the reaction mixture. The reaction was stirred at rt for 12 h under an H2 atmosphere (1 atm H2 balloon). The Pd/C was removed by filtration, and the solvent was removed in vacuo. The residual crude product was purified by flash column chromatography to afford the desired compound (46) as a pale green solid (0.56 g, 94% yield). Higher purity can be achieved by recrystallization from CHCl3. 1H NMR (300 MHz, MeOD-d4): 11.6 (s, 1H), 7.94 (s, 1H), 7.58 (d, J = 8.75 Hz, 1H), 7.48 (d, J = 8.75 Hz, 1H), 7.27 (s, 1H), 4.30–4.05 (m, 4H), 1.30 (td, J = 7.04 Hz, JP–H = 0.49 Hz, 6H). 13C NMR (75 MHz, MeOD-d4): 164.5, 139.7, 131.2, 128.1, 126.0–124.0 (m), 123.4–123.0 (m), 122.4–122.0 (m), 119.0–118.1 (m), 113.5, 109.6, 66.3 (d, JP–C = 7.09 Hz), 16.6 (d, JP–C = 5.34 Hz). 31P NMR (121 M Hz, MeOD-d4): 6.6 (t, JP–F = 123 Hz). ESI-MS calculated for C14H17F2NO5P [M + H]+ = 348.08. Found: 348.42.
The freshly prepared amine (50) (0.2 mmol) and the acid (46) (0.2 mmol) were coupled using peptide coupling conditions (3.0 equiv of HOBt. 3.0 equiv of EDC–HCI, and 4.0 equiv of EtN(i-Pr)2, DCM as solvent). The reaction was stirred at rt for 3 h before being quenched with saturated NaHCO3. The aqueous layer was extracted with DCM, and the combined organic layers were washed with brine and dried over anhydrous sodium sulfate. The solvent was removed in vacuo, and the residue was purified by flash column chromatography. The obtained product was dissolved in DCM (5 mL) in a round bottomed flask equipped with a magnetic stirring bar, and TMSI (0.7 mmol, 0.1 mL) was added at 0 °C. The reaction mixture was stirred for 2 h at 0 °C. Volatile components were removed on a rotary evaporator, and a mixture of MeCN–H2O–AcOH (20 mL, 8:1:1) was added. The mixture was stirred at rt for 1 h. The volatile components were removed on a rotary evaporator and the desired product was purified by reverse phase semipreparative HPLC to give compound 4 (38 mg, 26% yield). 1H NMR (300 MHz, MeOD-d4): 7.89 (s, 1H), 7.50 (d, J = 8.74 Hz, 1H), 7.45 (d, J = 8.74 Hz, 1H), 7.34–7.16 (m, 6H), 5.10–5.00 (m, 1H), 4.48–4.30 (m, 5H), 2.50–1.50 (m, 16H). 31P NMR (121 MHz, MeOD-d4): 5.6 (t, JP–F = 119 Hz). ESI-MS calculated for C33H40F2N6O8P [M + H]+ = 717.26. Found: 717.33. Reverse phase analytical HPLC: >95%.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-methyl-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Add (5).
HATU (418 mg, 1.1 mmol, 1.1 equiv) was added to a solution of 51 (462 mg, 1 mmol, 1 equiv), tert-butyl l-glutaminate 52 (202 mg, 1 mmol, 1 equiv), and DIEA (0.52 mL, 3 mmol, 3 equiv) in DMF (10 mL), and the resulting mixture was stirred at rt for 1 h. The solution was diluted with EtOAc and washed with H2O, saturated sodium bicarbonate aqueous solution, and brine and dried over sodium sulfate. After removal of the solvent in vacuo, the residue was dissolved in DCM (50 mL) and TFA (2.5 mL) was added. The resulting reaction mixture was stirred for 1 day, and the solvent was removed under vacuum. The residual was purified by HPLC to afford 53 (279 mg 51% over two steps). 1H NMR (400 MHz, MeOD) δ 7.44–7.33 (m, 5H), 5.33–5.14 (m, 2H), 4.53 (t, J = 8.6 Hz, 1H), 4.45–4.29 (m, 2H), 4.18–4.11 (m, 2H), 3.83–3.63 (m, 1H), 3.55–3.36 (m, 2H), 2.38 (tt, J = 7.3, 6.0 Hz, 3H), 2.28–2.03 (m, 3H), 2.01–1.72 (m, 4H), 1.50–1.44 m, 9H). UPLC–MS (ESI-MS) m/z: calculated for C27H40N5O7+ 546.29, found [M + H]+ 546.47.
HATU (153 g, 0.40 mmol, 1.1 equiv) was added to a solution of compounds 53 (200 mg, 0.37 mmol, 1 equiv), 46 (127 mg, 0.37 mmol, 1 equiv), and DIEA (0.19 mL, 1.1 mmoL, 3 equiv) in DMF (5 mL). The resulting mixture was stirred at rt for 1 h and diluted with EtOAc, washed with H2O, saturated sodium bicarbonate aqueous solution, and brine, and dried over sodium sulfate. After removal of the solvent in vacuo, the residue was purified by flash chromatography on silica gel to afford 54 (237 mg, 74%). 1H NMR (400 MHz, MeOD) δ 7.90 (s, 1H), 7.61–7.18 (m, 8H), 5.29–5.06 (m, 3H), 4.49 (t, J = 8.5 Hz, 1H), 4.39–4.30 (m, 2H), 4.26–4.08 (m, 4H), 4.013.44 (m, 4H), 2.52–1.62 (m, 10H), 1.50–1.43 (m, 9H), 1.29 (t, J = 7.1 Hz, 6H). UPLC–MS (ESI-MS) m/z: calculated for C37H46F2N6O11P+ [M – CH2=C(CH3)2 + H]+ 819.29, found [M – CH2=C(CH3)2 + H]+ 819.59.
10% Pd–C (50 mg) was added to a solution of compound 54 (150 mg, 0.17 mmol) in MeOH (10 mL). The solution was stirred under 1 atm of H2 at rt for 3 h before filtering through Celite and being concentrated. The resulting amine was purified by HPLC to afford compound 55 (103 mg, 81%). 1H NMR (400 MHz, MeOD) δ 7.91 (s, 1H), 7.56 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.33 (d, J = 0.6 Hz, 1H), 5.64 (dd, J = 12.1, 5.6 Hz, 1H), 4.84–4.67 (m, 2H), 4.39 (dd, J = 9.2, 5.3 Hz, 1H), 4.29–4.04 (m, 4H), 3.79–3.50 (m, 3H), 3.43 (t, J = 12.4 Hz, 1H), 2.53–2.41 (m, 1H), 2.37 (t, J = 7.4 Hz, 2H), 2.28–1.80 (m, 7H), 1.48 (s, 9H), 1.36–1.23 (m, 6H). UPLC–MS (ESI-MS) m/z: calculated for C33H48F2N6O9P+ 741.32, found [M + H]+ 741.44.
Compound 55 (103 g, 0.14 mmol, 1.0 equiv), HCHO (37%, 0.06 mL, 0.70 mmol, 5.0 equiv), DCE (5 mL), and NaBH(OAc)3 (59 mg, 0.28 mmol, 2.0 equiv) were added to a 25 mL round bottomed flask equipped with a magnetic stirring bar. The solution was stirred rt for 2 h until LC–MS detected the reaction was complete. Water (5 mL) was added to quench the reaction, and the reaction mixture was then extracted with DCM (10 mL × 3) and dried with anhydrous sodium sulfate. The mixture was filtered, and the solvent was removed in vacuo. The residual crude product was dissolved in DCM (5 mL), and TFA (2 mL) was added. The resulting mixture was stirred at rt until UPLC–MS detected it was finished. Most of the organic solvent was removed by evaporation, then the residue was purified by HPLC to afford 56 (60 mg, 64% yield over two steps). 1H NMR (400 MHz, MeOD) δ 7.93 (s, 1H), 7.59 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 7.34 (d, J = 0.6 Hz, 1H), 5.63 (dd, J = 11.9, 5.3 Hz, 1H), 4.84 (t, J = 8.9 Hz, 1H), 4.75–4.71 (m, 1H), 4.56 (dd, J = 9.9, 4.3 Hz, 1H), 4.30–4.09 (m, 4H), 4.01 (t, J = 12.7 Hz, 1H), 3.80 (dd, J = 12.1, 5.4 Hz, 1H), 3.60 (d, J = 14.3 Hz, 1H), 3.43 (t, J = 12.0 Hz, 1H), 3.13 (s, 3H), 2.56–2.49 (m, 1H), 2.45–2.16 (m, 5H), 2.14–2.02 (m, 1H), 2.00–1.82 (m, 3H), 1.31 (t, J = 7.1 Hz, 6H). UPLC–MS (ESI-MS) m/z: calculated for C30H42F2N6O9P+ 699.27, found [M + H]+ 699.05.
HATU (21 mg, 0.055 mmol, 1.1 equiv) was added to a solution of the acid 56 (35 mg, 0.05 mmol, 1 equiv), benzylamine (5.4 mg, 0.05 mmol, 1 equiv), and DIEA (46 μL, 0.15 mmoL, 3 equiv) in DMF (3 mL). The resulting mixture was stirred at rt for 10 min and purified by HPLC. The product obtained (32 mg, 0.04 mmol, 1 equiv) was dissolved in DCM (4 mL) and cooled to 0 °C before adding CF3CON(TMS)2 (63 mg, 0.24 mmol, 6.0 equiv) and 1 M TMS-I in DCM (0.16 mL, 0.16 mmol, 4.0 equiv). The reaction mixture was stirred at 0 °C for 10 min, and the solvent was removed in vacuo at 0 °C. The residue was dissolved in a mixed solvent of CH3CN (l mL), water (2 mL), and TFA (0.1 mL) and purified by HPLC to yield compound 5 (24 mg, 65% over two steps). 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.82 (s, 1H), 7.45–7.39 (m, 2H), 7.31–7.23 (m, 2H), 7.19 (dd, J = 6.0, 3.6 Hz, 4H), 5.50–5.28 (m, 1H), 4.67–4.48 (m, 2H), 4.25 (s, 2H), 4.22–4.18 (m, 1H), 3.82–3.73 (m, 1H), 3.62–3.51 (m, 1H), 3.44–3.37 (m, 1H), 3.24 (t, J = 12.2 Hz, 1H), 2.88 (s, 3H), 2.38–2.30 (m, 1H), 2.22 (t, J = 7.6 Hz, 2H), 2.14–1.92 (m, 3H), 1.87–1.63 (m, 4H). UPLC–MS (ESI-MS) m/z: calculated for C33H41F2N7O8P+ 732.27, found [M + H]+ 732.44.
((2-(((5S,8S,10aR)-3-Acetyl-8-(((S)-5-amino-1-(benzylamino)-1,5-dioxopentan-2-yl)carbamoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (6).
Compound 6 was synthesized by employing the similar method as for compound 5. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.82 (s, 1H), 7.47–7.43 (m, 1H), 7.39–7.37 (m, 1H), 7.28–7.20 (m, 2H), 7.18–7.15 (m, 4H), 5.06–4.91 (m, 1H), 4.37–4.17 (m, 5H), 3.87–3.70 (m, 1H), 3.66–3.29 (m, 3H), 2.28–2.10 (m, 3H), 2.09–1.91 (m, 5H), 1.87–1.59 (m, 5H). UPLC–MS (ESI-MS) m/z: calculated for C34H41F2N7O9P+ 760.27, found [M + H]+ 760.38.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-methyl-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (7).
Compound 7 was prepared from 56 by a procedure similar to that used for compound 5. NMR(400 MHz, CD3CN:D2O = 1:1) δ 7.91 (s, 1H), 7.62–7.43 (m, 2H), 7.42–7.14 (m, 11H), 6.10 (s, 1H), 5.48–5.46 (m, 1H), 4.76–4.57 (m, 2H), 4.42–4.39 (m, 1H), 3.92–3.84 (m, 1H), 3.67–3.65 (m, 1H), 3.49–3.46 (m, 1H), 3.36–3.30 (m, 1H), 2.96 (s, 3H), 2.51–2.23 (m, 3H), 2.17–2.04 (m, 3H), 1.92–1.87 (m, 2H), 1.80–1.72 (m, 2H). UPLC–MS (ESI-MS) m/z: calculated for C39H45F2N7O8P+ 808.30, found [M + H]+ 808.46.
((2-(((5S,8S,10aR)-3-Acetyl-8-(((S)-5-amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (SI-109, 8).
HATU (8.5 g, 22.3 mmol, 1.1 equiv) was added to a solution of the Boc-Gln-OH 57 (5.0 g, 20.3 mmol, 1 equiv), aminodiphenylmethane hydrochloride 58 (4.5 g, 20.3 mmol, 1 equiv), and DIEA (10.6 mL, 60.9 mmol, 3 equiv) in DMF (60 mL), and the resulting mixture was stirred at rt for 1 h. The solution was diluted with EtOAc and washed with H2O, saturated sodium bicarbonate aqueous solution, and brine and dried over sodium sulfate. After removal of the solvent in vacuo, the residue was purified by flash chromatography on silica gel to afford 59 (7.3 g 87%). 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 6.5 Hz, 1H), 7.35–7.23 (m, 10H), 6.36 (s, 1H), 6.21 (d, J = 8.2 Hz, 1H), 5.89 (d, J = 5.8 Hz, 1H), 5.74 (s, 1H), 4.24 (s, 1H), 2.33–2.26 (m, 1H), 2.21–2.11 (m, 1H), 2.09–2.00 (m, 1H), 1.92–1.87 (m, 1H), 1.43 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 175.42, 171.02, 156.26, 141.50, 141.32, 128.63, 127.44, 80.10, 56.92, 53.75, 31.91, 29.10, 28.31.
TFA (5 mL) was added slowly to a solution of 59 (3 g) in DCM (50 mL), and the resulting reaction solution was stirred at rt for 6 h and then evaporated. The residue, consisting of 60 was used directly in the next step without further purification.
HATU (1.59 g, 4.2 mmol, 1.1 equiv) was added to a solution of 51 (1.75 g, 3.8 mmol, 1 equiv), 60 (1.61 g, 3.8 mmol, 1 equiv), and DIEA (1.98 mL, 11.4 mmol, 3 equiv) in DMF (15 mL), and the resulting mixture was stirred at rt for 1 h. The solution was diluted with EtOAc and washed with H2O, saturated sodium bicarbonate aqueous solution and brine and dried over sodium sulfate. After removal of the solvent in vacuo, the residue was purified by flash chromatography on silica gel to afford 61 (2.4 g, 84%). 1H NMR (400 MHz, MeOD) δ 7.46–7.39 (m, 2H), 7.38–7.19 (m, 13H), 6.16–6.15 (m, 1H), 5.20–5.18 (m, 2H), 4.74–4.57 (m, 1H), 4.56–4.37 (m, 2H), 4.25–4.23 (m, 1H), 3.87–3.35 (m, 4H), 2.55–2.28 (m, 2H), 2.27–1.58 (m, 8H), 1.46 (s, 9H). UPLC–MS (ESI-MS) m/z: calculated for C41H51N6O8P+ 755.38, found [M + H]+ 755.52.
10% Pd–C (150 mg) was added to a solution of compound 61 (0.5 g, 0.66 mmol) in MeOH (30 mL). The solution was stirred under 1 atm of H2 at rt for 2 h before filtration through Celite and being concentrated. The resulting amine was dissolved in DCM (30 mL), and Ac2O (135 mg, 1.3 mmoL, 2 equiv) and DIEA (0.35 mL, 2.0 mmol, 3 equiv) were added. The resulting reaction mixture was stirred for 30 min and was then evaporated. The residue was treated with TFA (3 mL) in DCM (30 mL) at rt, and the resulting reaction solution was stirred at rt for 6 h and then evaporated. The residue was purified by HPLC to afford compound 62 (264 mg, 71% over three steps). 1H NMR (400 MHz, MeOD) δ 7.40–7.21 (m, 10H), 6.17 (s, 1H), 4.66–4.36 (m, 3H), 4.30–3.95 (m, 2H), 3.83–3.54 (m, 3H), 2.46–1.75 (m, 13H).
HATU (22 mg, 0.06 mmol, 1.1 equiv) was added to a solution of compound 62 (30 mg, 0.05 mmol, 1 equiv), 46 (19 mg, 0.05 mmol, 1 equiv), and DIEA (28 μL, 0.16 mmoL, 3 equiv) in DMF (2 mL). The resulting mixture was stirred at rt for 1 h and diluted with EtOAc, then washed with H2O, saturated sodium bicarbonate aqueous solution and brine, and dried over sodium sulfate. After removal of the solvent in vacuo, the residue was purified by flash chromatography on silica gel. The obtained product was dissolved in DCM (2 mL) and cooled to 0 °C before adding CF3CON(TMS)2 (82 mg, 0.32 mmol, 6.0 equiv) and 1 M of TMS-I in DCM (0.21 mL, 0.21 mmol, 4.0 equiv). The reaction mixture was stirred at 0 °C for 10 min, and the solvent was removed under vacuum at 0 °C. The residue was dissolved in a mixed solvent of CH3CN (l mL), water (2 mL), and TFA (0.1 mL) and purified by HPLC to yield SI-109 (8, 28 mg, 63% over two steps). 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.90 (s, 1H), 7.56–7.52 (m, 1H), 7.48–7.46 (m, 1H), 7.35–7.21 (m, 11H), 6.07–6.06 (m, 1H), 5.11–5.02 (m, 1H), 4.42–4.23 (m, 3H), 3.95–3.80 (m, 1H), 3.77–3.53 (m, 2H), 3.49–3.37 (m, 1H), 2.32–2.26 (m, 2H), 2.19–2.14 (m, 4H), 2.11–2.01 (m, 2H), 1.96–1.58 (m, 5H). UPLC–MS (ESI-MS) m/z: calculated for C40H45F2N7O9P+ 836.30, found [M + H]+ 836.43.
2-(((5S,8S,10aR)-3-Acetyl-8-(((S)-5-amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl Dihydrogen Phosphate (9).
Compound 9 was prepared from 62 by a similar procedure as was used for compound 2. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.39–7.35 (m, 2H), 7.27–7.17 (m, 10H), 7.06–7.03 (m, 2H), 5.99–5.98 (m, 1H), 5.03–4.94 (m, 1H), 4.20–4.23 (m, 3H), 3.88–3.46 (m, 3H), 3.43–3.23 (m, 1H), 2.24–2.19 (m, 2H), 2.16–1.94 (m, 6H), 1.89–1.56 (m, 5H). UPLC–MS (ESI-MS) m/z: calculated for C39H45N7O10P+ 802.30, found [M + H]+ 802.41.
((2-(((5S,8S,10aR)-3-Acetyl-8-(((S)-5-amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl) carbamoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1 H-indol-5-yl)methyl)phosphonic Acid (10).
Compound 10 was prepared from 62 by a similar procedure as was used for compound 3. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.48 (s, 1H), 7.38–7.36 (m, 1H), 7.33–7.09 (m, 11H), 7.06–7.02 (m, 1H), 6.00–5.99 (m, 1H), 5.07–4.92 (m, 1H), 4.40–4.22 (m, 3H), 3.80–3.59 (m, 3H), 3.41–3.29 (m, 1H), 3.13 (d, J = 21.1 Hz, 2H), 2.25–2.20 (m, 2H), 2.17–1.96 (m, 6H), 1.89–1.54 (m, 5H). UPLC–MS (ESI-MS) m/z: calculated for C40H47N7O9P + 800.32, found [M + H]+ 800.45.
((4-((E)-4-(((5S,8S,10aR)-3-Acetyl-8-(((S)-5-amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)amino)-4-oxobut-2-en-2-yl)phenyl)difluoromethyl)phosphonic Acid (11).
Compound 11 was prepared from 62 and the previously reported protected phosphotyrosine mimetic, (E)-3-(4-((diethoxyphosphoryl)-difluoromethyl)phenyl)but-2-enoic acid,32 by a similar procedure to that used for compound 8. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.56–7.40 (m, 4H), 7.28–7.07 (m, 10H), 6.19–6.17 (m, 1H), 5.94 (s, 1H), 4.94–4.71 (m, 1H), 4.29–4.17 (m, 3H), 3.78–3.03 (m, 4H), 2.32 (s, 3H), 2.19–2.16 (m, 2H), 2.08–1.88 (m, 6H), 1.85–1.48 (m, 5H). UPLC–MS (ESI-MS) m/z: calculated for C41H48F2N609P+ 837.32, found [M + H]+ 837.57.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(6-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)hex-5-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)-difluoromethyl)phosphonic Acid (12).
Compound 12 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.99–7.79 (m, 1H), 7.78–7.43 (m, 4H), 7.42–6.93 (m, 12H), 6.09–5.98 (m, 1H), 5.11–4.89 (m, 2H), 4.50–4.24 (m, 5H), 3.91–3.15 (m, 4H), 2.86–2.47 (m, 6H), 2.42–1.99 (m, 8H), 1.93–1.45 (m, 6H). UPLC–MS (ESI-MS) m/z: calculated for C57H60F2N9O12P2+ 565.71, found [M + H]2+ 565.96.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(7-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)hept-6-ynoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (13).
Compound 13 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.99–7.81 (m, 1H), 7.76–7.43 (m, 4H), 7.43–7.00 (m, 12H), 6.06–6.03 (m, 1H), 5.19–4.88 (m, 2H), 4.45–4.18 (m, 5H), 3.95–3.65 (m, 3H), 3.37–3.28 (m, 1H), 3.02–1.99 (m, 14H), 1.93–1.51 (m, 8H). UPLC–MS (ESI-MS) m/z: calculated for C58H61F2N9O12P+ 1144.41, found [M + H]+ 1144.45.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)-difluoromethyl)phosphonic Acid (SD-36, 14).
TFA (3 mL) was added slowly to a solution of 61 (2g, 2.65 mmol) in DCM (30 mL) at rt, and the resulting solution was stirred at rt for 6 h and then evaporated. The crude product was directly used in the next step without purification. HATU (1.1 g, 2.91 mmol, 1.1 equiv) was added to a solution of the crude product of the previous step (2.65 mmol, 1 equiv), 46 (0.92 g, 2.65 mmol, 1 equiv), and DIEA (1.39 mL, 7.95 mmoL, 3 equiv) in DMF (15 mL). The resulting mixture was stirred at rt for 1 h and diluted with EtOAc and washed with H2O, saturated sodium bicarbonate aqueous solution, and brine and dried over sodium sulfate. After removal of the solvent in vacuo, the residue was purified by flash chromatography on silica gel to afford compound 63. 10% Pd–C (200 mg) was added to a solution of the obtained 63 in MeOH (60 mL). The solution was stirred under 1 atm of H2 at rt for 3 h before being filtered through Celite and concentrated. The resulting amine was purified by HPLC to afford compound 64 (1.73 g, 77% over three steps). 1H NMR (400 MHz, MeOD) δ 7.92 (s, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.38–7.31 (m, 5H), 7.30–7.24 (m, 6H), 6.19 (s, 1H), 5.61 (dd, J = 12.1, 5.6 Hz, 1H), 4.78 (t, J = 8.9 Hz, 1H), 4.70–4.66 (m, 1H), 4.59 (dd, J = 9.5, 5.0 Hz, 1H), 4.29–4.05 (m, 4H), 3.72–3.61 (m, 2H), 3.54 (t, J = 12.6 Hz, 1H), 3.42 (t, J = 12.4 Hz, 1H), 2.45–2.27 (m, 3H), 2.24–2.1 (m, 3H), 2.00–1.70 (m, 4H), 1.29 (td, J = 7.1, 3.2 Hz, 6H). UPLC–MS (ESI-MS) m/z: calculated for C42H51F2N7O8P+ 850.35, found [M + H]+ 850.37.
Trimethylamine (10 mL) was added to a mixture of compound 66 (1.3 g, 4.0 mmol, 1 equiv), oct-7-ynoic acid 67 (0.56 g, 4.0 mmol, 1 equiv), CuI (154 mg, 0.8 mmol, 0.2 equiv), and Pd(PPh3)2Cl2 (282 mg, 0.4 mmol, 0.1 equiv) in DMF (10 mL). The resulting mixture was purged and refilled with argon three times and then stirred at 70–80 °C for 3 h under argon. The reaction mixture was then cooled to rt and evaporated to remove the solvent. The residue was purified by HPLC to yield compound 68 (1.18 g, 76%). 1H NMR (400 MHz, DMSO) δ 11.99 (s, 1H), 10.99 (s, 1H), 7.77–7.68 (m, 1H), 7.68–7.59 (m, 1H), 7.52 (t, J = 7.6 Hz, 1H), 5.15 (dd, J = 13.3, 5.0 Hz, 1H), 4.46 (d, J = 17.7 Hz, 1H), 4.33 (d, J = 17.7 Hz, 1H), 2.97–2.88 (m, 1H), 2.63–2.59 (m, 1H), 2.53–2.47 (m, 3H), 2.24 (t, J = 7.2 Hz, 2H), 2.13–1.94 (m, 1H), 1.78–1.27 (m, 6H). 13C NMR (101 MHz, DMSO) δ 174.90, 173.32, 171.45, 168.14, 144.23, 134.52, 132.46, 129.05, 123.05, 119.32, 96.73, 76.91, 52.14, 47.47, 34.07, 31.68, 28.32, 28.25, 24.50, 22.83, 19.14. UPLC–MS (ESI-MS) m/z: calculated for C21H23N2O5+ 383.16, found [M + H]+ 383.28.
HATU (295 mg, 0.77 mmol, 1.1 equiv) was added to a solution of 64 (600 mg, 0.71 mmol, 1 equiv), 68 (270 g, 0.71 mmol, 1 equiv), and DIEA (0.37 mL, 2.12 mmol, 3 equiv) in DMF (10 mL), and the resulting mixture was stirred at rt for 1 h. The solution was diluted with EtOAc and washed with H2O, saturated sodium bicarbonate aqueous solution, and brine and dried over sodium sulfate. After removal of the solvent in vacuo, the residue was purified by flash chromatography on silica gel to afford 65 (702 mg 82%). 1H NMR (400 MHz, MeOD:CDCl3 = 1:1) δ 7.90 (s, 1H), 7.76–7.66 (m, 1H), 7.57–7.51 (m, 2H), 7.47–7.35 (m, 2H), 7.33–7.19 (m, 11H), 6.27–6.6 (m, 1H), 5.33–4.87 (m, 2H), 4.55–4.37 (m, 4H), 4.26–3.75 (m, 7H), 3.71–3.26 (m, 2H), 2.90–2.71 (m, 2H), 2.67–1.85 (m, 14H), 1.83–1.47 (m, 8H), 1.29 (t, J = 7.1 Hz, 6H).
Compound 65 (500 mg, 0.41 mmol, 1.0 equiv) and DCM (40 mL) were placed in a round bottomed flask. The solution was cooled to 0 °C before adding CF3CON(TMS)2 (635 mg, 2.5 mmol, 6.0 equiv) and a 1 M solution of TMS-I in DCM (1.65 mL, 1.65 mmol, 4.0 equiv). The reaction mixture was stirred at 0 °C for 10 min, and then the solvent was removed under vacuum at 0 °C. The residue was dissolved in a mixture of CH3CN (5 mL), H2O (5 mL), and TFA (0.3 mL) and purified by HPLC to yield SD-36 (14, 420 mg, 88%). 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ δ 7.90–7.85 (m, 1H), 7.73–7.59 (m, 1H), 7.56–7.36 (m, 4H), 7.35–7.05 (m, 11H), 6.06–6.03 (m, 1H), 5.12–4.87 (m, 2H), 4.48–4.19 (m, 5H), 3.98–3.49 (m, 3H), 3.36–3.24 (m, 1H), 2.88–2.64 (m, 2H), 2.61–2.23 (m, 7H), 2.21–1.99 (m, 4H), 1.94–1.35 (m, 11H). UPLC–MS (ESI-MS) m/z: calculated for C59H63F2N9O12P+ 1158.43, found [M + H]+ 1158.60.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(9-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)non-8-ynoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (15).
Compound 15 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.99–7.87 (m, 1H), 7.73–7.41 (m, 5H), 7.40–6.99 (m, 11H), 6.07–6.04 (m, 1H), 5.12–4.91 (m, 2H), 4.46–4.21 (m, 5H), 3.98–3.53 (m, 3H), 3.34–3.26 (m, 1H), 2.88–2.68 (m, 2H), 2.57–2.06 (m, 10H), 1.90–1.21 (m, 14H). UPLC–MS (ESI-MS) m/z: calculated for C60H66F2N9O12P2+ 586.73, found [M + 2H]2+ 586.95.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(11-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl) undec-10-ynoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (16).
Compound 16 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.89–7.88 (m, 1H), 7.727.67 (m, 1H), 7.60–7.39 (m, 4H), 7.37–7.13 (m, 11H), 6.06–6.03 (m, 1H), 5.17–4.86 (m, 2H), 4.47–4.19 (m, 5H), 3.95–3.53 (m, 3H), 3.38–3.27 (m, 1H), 2.90–2.67 (m, 2H), 2.57–2.01 (m, 11H), 1.93–1.20 (m, 17H). UPLC–MS (ESI-MS) m/z: calculated for C62H70F2N9O12P2+ 600.74, found [M + 2H]2+ 600.88.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oct-7-yn-1-yl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (17).
Compound 17 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.86 (s, 1H), 7.67–7.64 (m, 1H), 7.48–7.37 (m, 4H), 7.29–7.14 (m, 11H), 6.01 (s, 1H), 5.45 (br, 1H), 5.02–4.96 (m, 1H), 4.68–4.53 (m, 2H), 4.32–4.21 (m, 3H), 3.73–3.70 (m, 1H), 3.54–3.49 (m, 2H), 3.29 (t, J = 1.2 Hz, 1H), 3.14–3.08 (m, 2H), 2.82–2.64 (m, 2H), 2.38–2.15 (m, 6H), 2.14–1.95 (m, 3H), 1.93–1.23 (m, 13H). UPLC–MS calculated for C59H64F2N8O11PS [M + H]+: 1144.45, found 1144.50.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(((6-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)hex-5-yn-1-yl)oxy)carbonyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (18).
Compound 18 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.90 (s, 1H), 7.72 (d, J = 7.5 Hz, 1H), 7.65–7.39 (m, 4H), 7.38–6.99 (m, 11H), 6.05–6.03 (m, 1H), 5.12–4.90 (m, 2H), 4.46–4.16 (m, 6H), 4.07–3.97 (m, 1H), 3.68–3.31 (m, 4H), 2.86–2.69 (m, 2H), 2.56–1.98 (m, 10H), 1.90–1.43 (m, 8H). UPLC–MS (ESI-MS) m/z: calculated for C588H63F2N9013P2+ 580.71, found [M + H]2+ 581.11
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-((6-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)hex-5-yn-1-yl)carbamoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (19).
Compound 19 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.84 (d, J = 4.8 Hz, 1H), 7.63–7.55 (m, 1H), 7.53–7.42 (m, 3H), 7.41–7.01 (m, 12H), 6.07 (s, 1H), 5.04–4.92 (m, 1H), 4.78 (d, J = 8.1 Hz, 1H), 4.45–4.32 (m, 4H), 4.09–4.04 (m, 1H), 3.87 (d, J = 13.5 Hz, 2H), 3.43–3.22 (m, 2H), 3.15–3.09 (m, 2H), 2.88–2.63 (m, 2H), 2.51 (t, J = 6.4 Hz, 2H), 2.39–2.16 (m, 4H), 2.12–2.02 (m, 4H), 1.96–1.90 (m, 1H), 1.83–1.43 (m, 7H). UPLC–MS (ESI-MS) m/z: calculated for C58H62F2N10O12P+ 1159.42, found [M + H]+ 1159.44.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(1-(5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pent-4-yn-1-yl)-1H-pyrazole-4-carbonyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)-carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (20).
Compound 20 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 8.14–8.03 (m, 1H), 7.86–7.60 (m, 3H), 7.51–7.10 (m, 15H), 6.00–5.99 (m, 1H), 5.15–4.95 (m, 2H), 4.27–4.18 (m, 7H), 3.82–3.48 (m, 3H), 2.74–2.66 (m, 2H), 2.38–1.95 (m, 11H), 1.92–1.54 (m, 5H). UPLC–MS calculated for C60H61F2N11O12P [M + H]+: 1196.42, found 1196.28.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(4-(4-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ethynyl)-1H-pyrazol-1-yl)butanoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (21).
Compound 21 was prepared from 64 by a procedure similar to that used for SD-36. NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.89–7.78 (m, 2H), 7.71–7.60 (m, 2H), 7.50–7.34 (m, 4H), 7.27–7.13 (m, 11H), 6.01–5.96 (m, 1H), 5.05–4.88 (m, 2H), 4.37–4.21 (m, 7H), 3.75–3.29 (m, 4H), 2.82–2.70 (m, 2H), 2.65–1.94 (m, 11H), 1.93–1.65 (m, 5H). UPLC–MS calculated for C60H61F2N11O12P [M + H]+: 1196.42, found 1196.44.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(4-(4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)but-3-yn-1-yl)piperidine-1-carbon-yl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)-carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (22).
Compound 22 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.84 (s, 1H), 7.70–7.68 (m, 1H), 7.55–7.41 (m, 4H), 7.29–7.13 (m, 11H), 6.02 (s, 1H), 5.14–5.02 (m, 2H), 4.44–4.13 (m, 6H), 3.64–3.52 (m, 4H), 3.27–3.15 (m, 2H), 2.83–2.61 (m, 4H), 2.45–2.02 (m, 9H), 1.92–1.49 (m, 9H), 1.20–1.05 (m, 2H). UPLC–MS calculated for C61H66F2N10O12P [M + H]+: 1199.46, found 1199.48.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(3-(4-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ethynyl)piperidin-1-yl)propanoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (23).
Compound 23 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.88–7.85 (m, 1H), 7.74–7.70 (m, 1H), 7.64–7.57 (m, 1H), 7.50–7.42 (m, 3H), 7.29–7.13 (m, 11H), 6.01–5.97 (m, 1H), 5.20–5.00 (m, 2H), 4.43–4.18 (m, 5H), 3.76–3.32 (m, 8H), 3.21–2.72 (m, 7H), 2.39–2.04 (m, 9H), 1.85–1.65 (m, 7H). UPLC–MS calculated for C61H66F2N10O12P [M + H]+: 1199.46, found 1199.68.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(3-(4-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)ethynyl)piperidin-1-yl)-3-oxopropanoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)-carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (24).
Compound 24 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.87–7.67 (m, 2H), 7.51–7.38 (m, 3H), 7.30–7.13 (m, 12H), 6.03–5.97 (m, 1H), 5.21–5.01 (m, 2H), 4.35–4.19 (m, 5H), 3.85–3.62 (m, 5H), 3.36–3.32 (m, 4H), 2.92–2.65 (m, 4H), 2.35–2.04 (m, 9H), 1.88–1.69 (m, 7H). UPLC–MS calculated for C61H64F2N10O13P [M + H]+: 1213.44, found 1213.56.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)octanoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl)-carbamoyl)-1H-indol-5-yl)-difluoromethyl)phosphonic Acid (25).
Compound 25 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.90–7.88 (m, 1H), 7.69–7.41 (m, 4H), 7.41–7.07 (m, 12H), 6.07–6.03 (m, 1H), 5.13–4.90 (m, 2H), 4.51–4.18 (m, 5H), 3.99–3.55 (m, 3H), 3.36–3.27 (m, 1H), 3.01–2.02 (m, 13H), 1.91–1.09 (m, 15H). UPLC–MS (ESI-MS) m/z: calculated for C59H67F2N9O12P+ 1162.46, found [M + H]+ 1162.44.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(7-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)heptanoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (26).
Compound 26 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) 7.89–7.87 (m, 1H), 7.61–7.41 (m, 4H), 7.38–7.08 (m, 12H), 6.06–6.03 (m, 1H), 5.13–4.90 (m, 2H), 4.50–4.18 (m, 5H), 3.97–3.50 (m, 3H), 3.41–3.33 (m, 1H), 3.26–3.30 (m, 2H), 2.90–2.65 (m, 2H), 2.56–2.00 (m, 9H), 1.92–1.84 (m, 2H), 1.73–1.55 (m, 7H), 1.42–1.21 (m, 4H). UPLC–MS (ESI-MS) m/z: calculated for C57H60F2N9O12P2+ 565.71, found [M + H]2+ 582.65
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (27).
Compound 27 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.89–7.85 (m, 1H), 7.76–7.65 (m, 3H), 7.57–7.41 (m, 2H), 7.38–7.08 (m, 11H), 6.06–6.03 (m, 1H), 5.13–4.84 (m, 2H), 4.35–4.21 (m, 3H), 3.3.95–3.65 (m, 3H), 3.38–3.26 (m, 1H), 2.87–2.68 (m, 2H), 2.65–2.36 (m, 5H), 2.33–2.00 (m, 6H), 1.96–1.36 (m, 11H). UPLC–MS (ESI-MS) m/z: calculated for C59H61F2N9O13P+ 1172.41, found [M + H]+ 1172.29
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl) carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (28).
Compound 28 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.93–7.83 (m, 1H), 7.65–7.54 (m, 1H), 7.47–7.10 (m, 15H), 6.03–5.99 (m, 1H), 5.00–4.90 (m, 2H), 4.30–4.18 (m, 6H), 3.91–3.63 (m, 3H), 3.29–3.26 (m, 1H), 2.76–2.57 (m, 3H), 2.45–1.94 (m, 10H), 1.93–1.44 (m, 10H). UPLC–MS calculated for C59H64F2N9O12P2+: 579.72, found 580.24
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-3-oxoisoindolin-5-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl) carbamoyl)-1H-indo 1–5-yl)difluoromethyl)phosphonic Acid (29).
Compound 29 was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.95–7.82 (m, 1H), 7.68–7.61 (m, 1H), 7.53–7.09 (m, 15H), 6.03–5.99 (m, 1H), 4.99–4.90 (m, 2H), 4.31–4.15 (m, 6H), 3.93–3.64 (m, 3H), 3.29–3.28 (m, 1H), 2.78–2.69 (m, 2H), 2.57–2.13 (m, 7H), 2.03–1.94 (m, 4H), 1.92 – 1.47 (m, 10H). UPLC–MS calculated for C59H63F2N9O12P [M + H]+: 1158.43, found 1158.45.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl)carbamoyl) −1H-indol-5-yl)difluoromethyl)phosphonic Acid (30).
Compound 69 was prepared from 55 by a procedure similar to that used for compound 56. 1H NMR (400 MHz, MeOD) δ 7.91 (s, 1H), 7.76–7.67 (m, 1H), 7.64–7.53 (m, 2H), 7.51–7.40 (m, 2H), 7.33–7.24 (m, 1H), 5.27–5.07 (m, 2H), 4.59–4.40 (m, 4H), 4.35–4.25 (m, 1H), 4.25–4.09 (m, 4H), 4.02–3.99 (m, 1H), 3.82–3.54 (m, 3H), 2.95–2.47 (m, 7H), 2.45–1.85 (m, 10H), 1.83–1.49 (m, 7H), 1.30 (t, J = 7.1 Hz, 6H). UPLC–MS (ESI-MS) m/z: calculated for C50H60F2N8O13P+ 1049.40, found [M + H]+ 1049.53.
Compound 30 was prepared from 69 by a procedure similar to that used for compound 5. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.98–7.87 (m, 1H), 7.79–7.45 (m, 4H), 7.44–7.35 (m, 1H), 7.35–6.92 (m, 6H), 5.12–4.93 (m, 2H), 4.46–4.21 (m, 6H), 3.94–3.28 (m, 5H), 2.81–2.73 (m, 2H), 2.63–1.99 (m, 12H), 1.92–1.38 (m, 10H). UPLC–MS (ESI-MS) m/z: calculated for C53H59F2N9O12P+ 1082.40, found [M + H]+ 1082.42.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(methylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (31).
Compound 31 was prepared from 69 by a procedure similar to that used for compound 5. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.97–7.82 (m, 1H), 7.81–6.96 (m, 6H), 5.16–4.93 (m, 2H), 4.49–4.18 (m, 5H), 3.95–3.18 (m, 4H), 3.00–1.99 (m, 14H), 1.95–1.08 (m, 13H). UPLC–MS (ESI-MS) m/z: calculated for C47H55F2N9O12P+ 1006.37, found [M + H]+ 1006.49.
2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl Dihydrogen Phosphate (32).
Compound 70 was prepared from 61 by a procedure similar to that used for compound 62. 1H NMR (400 MHz, MeOD) δ 7.74 (d, J = 7.6 Hz, 1H), 7.63–7.56 (m, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.36–7.22 (m, 10H), 6.16 (s, 1H), 5.26–5.11 (m, 1H), 4.65–4.34 (m, 5H), 4.20–4.04 (m, 2H), 3.88–3.40 (m, 3H), 2.93–2.74 (m, 2H), 2.67–2.23 (m, 8H), 2.19–1.52 (m, 14H).
Compound 32 was prepared from 70 by a procedure similar to that used for compound 2. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.66–7.54 (m, 1H), 7.52–7.09 (m, 14H), 7.07–6.92 (m, 2H), 6.00–5.96 (m, 1H), 5.05–4.82 (m, 2H), 4.47–4.22 (m, 5H), 3.89–3.46 (m, 3H), 3.26–3.19 (m, 1H), 2.76–2.62 (m, 2H), 2.57–1.97 (m, 12H), 1.85–1.35 (m, 10H). UPLC–MS (ESI-MS) m/z: calculated for C58H64N9O13P2+ 562.72, found [M + 2H]2+ 563.17.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo-[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)methyl)-phosphonic Acid (33).
Compound 33 was prepared from 70 by a procedure similar to that used for compound 3. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.67–7.54 (m, 1H), 7.54–6.82 (m, 16H), 6.01–5.96 (m, 1H), 5.05–4.82 (m, 2H), 4.41–4.22 (m, 5H), 3.88–3.53 (m, 3H), 3.27–3.07 (m, 3H), 2.77–2.66 (m, 2H), 2.56–1.94 (m, 12H), 1.87–1.35 (m, 10H). UPLC–MS (ESI-MS) m/z: calculated for C59H66N9O12P2+ 561.73, found [M + 2H]2+ 561.97.
((4-((E)-4-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)amino)-4-oxobut-2-en-2-yl)phenyl)difluoromethyl)phosphonic Acid (34).
Compound 34 was prepared from 70 by a procedure similar to that used for compound 4. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.66–7.56 (m, 1H), 7.52–7.29 (m, 6H), 7.28–7.02 (m, 10H), 6.19 (s, 1H), 5.97–5.93 (m, 1H), 5.00–4.72 (m, 2H), 4.45–4.17 (m, 5H), 4.1–14.00 (m, 1H), 3.85–3.30 (m, 3H), 3.20–3.05 (m, 1H), 2.77–2.61 (m, 2H), 2.52–1.91 (m, 14H), 1.87–1.30 (m, 11H). UPLC–MS (ESI-MS) m/z: calculated for C60H66F2N8O12P + 1159.45, found [M + H]+ 1159.64.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(benzhydrylamino)-1,5-dioxopentan-2-yl)carbamoyl)-3-(8-(2-(1-methyl-2,6-dioxopi-peridin-3-yl)-1-oxoisoindolin-4-yl)oct-7-ynoyl)-6-oxodecahydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (35, SD-36Me).
SD-36Me was prepared from 64 by a procedure similar to that used for SD-36. 1H NMR (400 MHz, CD3CN:D2O = 1:1) δ 7.88 (d, J = 16.1 Hz, 1H), 7.72–7.59 (m, 1H), 7.56–7.36 (m, 4H), 7.35–7.06 (m, 11H), 6.06–6.03 (m, 1H), 5.12–4.87 (m, 2H), 4.45–4.20 (m, 5H), 3.98–3.50 (m, 3H), 3.37–3.22 (m, 1H), 3.08–2.94 (m, 3H), 2.88–2.73 (m, 2H), 2.61–2.37 (m, 4H), 2.34–1.99 (m, 7H), 1.95–1.81 (m, 2H), 1.79–1.40 (m, 9H). UPLC–MS (ESI-MS) m/z: calculated for C60H65F2N9O12P+ 1172.45, found [M + H]+ 1172.45.
((2-(((5S,8S,10aR)-8-(((S)-5-Amino-1-(((S)-2-((8-(3′,6′-dihydroxy-3-oxo-35H5-spiro[isobenzofuran-1,9′-xanthene]-5-carboxamido)octyl)amino)-2-oxo-1-phenylethyl)amino)-1,5-dioxopentan-2-yl)carbamoyl)-3-methyl-6-oxodeca-hydropyrrolo[1,2-a][1,5]diazocin-5-yl)carbamoyl)-1H-indol-5-yl)difluoromethyl)phosphonic Acid (74).
HATU (42 mg, 0.11 mmol, 1.1 equiv) was added to a solution of 71 (50 mg, 0.1 mmol, 1 equiv), (S)-2-((tert-butoxycarbonyl)amino)-2-phenylacetic acid 72 (25 mg, 0.1 mmol, 1 equiv), and DIEA (0.05 mL, 0.3 mmoL, 3 equiv) in DMF (3 mL). The resulting mixture was stirred at rt for 1 h, then diluted with EtOAc, and washed with H2O, saturated sodium bicarbonate aqueous solution, and brine, and dried over sodium sulfate. After removal of the solvent under vacuum, the residue was dissolved in DCM (5 mL), and TFA (1 mL) was added. The resulting mixture was stirred at rt for 3 h. After removal of the organic solvent, the residual was purified by HPLC to afford compound 73 (32 mg, 50% over two steps). HATU (21 g, 0.055 mmol, 1.1 equiv) was added to a solution of 73 (32 mg, 0.05 mmol, 1 equiv), 56 (35 mg, 0.05 mmol, 1 equiv), and DIEA (0.025 mL, 0.15 mmoL, 3 equiv) in DMF (3 mL). The resulting mixture was stirred at rt for 1 h and directly purified by HPLC. Compound 74 was achieved using a similar method as for SD-36 from 65, and the yield was 65% over two steps. 1H NMR (400 MHz, CD3CN) δ 8.30 (s, 1H), 8.06 (dd, J = 8.0, 1.5 Hz, 1H), 7.82 (s, 1H), 7.45–7.39 (m, 2H), 7.34–7.08 (m, 7H), 6.75–6.72 (m, 2H), 6.65–6.63 (m, 2H), 6.55–6.53 (m, 2H), 5.37 (s, 1H), 5.24 (s, 1H), 4.63–4.51 (m, 2H), 4.34–4.28 (m, 1H), 3.793.55 (m, 2H), 3.45–3.19 (m, 4H), 3.13–3.06 (m, 1H), 2.98–2.91 (m, 1H), 2.87 (s, 3H), 2.32–2.2l(m, 3H), 2.11–1.93 (m, 3H), 1.86–1.65 (m, 4H), 1.56–1.39 (m, 2H), 1.33–1.01 (m, 10H). UPLC–MS (ESI-MS) m/z: calculated for C63H70F2N9O15P2+ 630.73, found [M + 2H]2+ 630.92.
Expression and Purification of STAT3.
STAT3 (residues 127–688) was cloned into a pHIS-SUMO vector, transformed into Rosetta DE3 cells, and grown in LB media overnight at 20 °C. HIS-SUMO-STAT3 was purified from the cleared cell lysate using Ni-NTA resin (Qiagen), then loaded onto a Superdex 200 column (GE Healthcare) in buffer containing 50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% NP-40, 1 mM DTT, and 10% glycerol For crystallography, the HIS-SUMO tag was cleaved overnight with SUMO protease. The cleaved protein was then applied to a Ni-NTA column, concentrated, and loaded onto a Superdex 200 column pre-equilibrated with 50 mM Tris, pH 8.5, 150 mM NaCl, and 1 mM DTT.
Fluorescence Polarization (FP) Assay.
FP assay was performed as described previously.12 To determine the dissociation constant (Kd) for the interaction between STAT3 SH2 domain binder 74 and STAT3, 5 nM 74 was incubated with serially diluted STAT3 proteins in FP assay buffer (50 mM NaCI, 10 mM Hepes, pH 7.5, 1 mM EDTA pH 8.0, 0.01% Triton X-100, 2 mM DTT). FP was measured 1 h after incubation on a Tecan Infinite microplate reader. Kd values were determined from the binding isotherm derived from curves of mP vs protein concentrations. For the competitive binding assay, STAT3 recombinant protein was first incubated with tracer 74 for 5 min, then added to the serially diluted compounds for 1 h. IC50 values of 74 displacement were calculated by nonlinear regression analysis using GraphPad Prism software. The Ki values of competitive inhibitors were calculated as described previously.33
Molecular Modeling.
The binding pose of compound 4 with STAT3 protein was modeled based on the crystal structure of STAT3 (pY705) with DNA (PDB code 1BG134). First, chain A of STAT3 (pY705) was extracted from the crystal structure and protons were added using the “protonate 3D” module in MOE35 by setting the pH value at 7.0. Then, compound 4 was geometrically optimized using MOE. The GOLD program (version 5.2)36 was used for the docking calculations. In the docking simulation, the center of the binding site in STAT3 was set at V637 and the radius at 13 Å. Additional parameters in the genetic algorithm were the same as in our previous report.37 GoldScore was used as the fitness function to rank the docked poses. The top ranked pose was considered as the binding model and displayed in Figure S2. The figures were prepared using PyMOL.
Western Blot Analysis.
Western blotting was performed as described previously.22 Cells were lysed in cell lysis buffer (Cell Signaling Technology, no. 9803), separated by SDS–PAGE NuPAGE gel (Thermo Fisher Scientific), and transferred to a PVDF membrane (Millipore). PVDF membranes were first blocked for 1 h using 5% blotting-grade blocker (no. 1706404, Bio-Rad) in Tris-buffered saline with Tween 20 (TBST, Pierce) and then were incubated with the primary and subsequently their corresponding secondary antibodies. SuperSignal West Pico Plus chemiluminescent substrate (Thermo Fisher Scientific) was used for signal development.
Cell Growth Assay.
Cell viability analysis was performed as described previously.22 Cells grown in 384-well white plates (Coming Costar) were incubated with serially diluted compounds for 4 days. Cell viability was determined using the CellTiter-Glo luminescent cell viability assay (Promega) following the manufacturer’s instructions.
Crystallization and Structure Determination of STAT3 Bound to SI-109.
1.2-fold molar excess of SI-109 was incubated with STAT3 (6.25 mg/mL) for 3 h at 4 °C, and the crystals were grown by sitting drop vapor diffusion against well solution containing 10% PEG 3350 and 100–150 mM ammonium nitrate at 4 °C. Prior to flash freezing in liquid nitrogen, crystals were cryoprotected in mother liquor containing 20% ethylene glycol. Data were collected at the Life Sciences Collaborative Access Team beamline 21-ID-D at the Advanced Photon Source, Argonne National Laboratory. HKL200038 was used to process data, and the structures were solved by molecular replacement using Phaser39 with PDB code 3CWG as a search model. The structures were iteratively refined with Buster40 and fit with COOT.41 Coordinates and geometric restraints for the compounds were generated in GRADE.42 The complexes crystallized in space group P41212 with one molecule in the asymmetric unit. Residues 136–687 were modeled with the exception of the following missing loop regions: 181–191, 372–378, and 419–429. Prior to deposition, the structures were validated with Molprobity43 and the PDB validation server (wwPDB validation server).
In Vivo PK/PD and Efficacy.
All the in vivo studies were carried out under the animal protocol PR000007499, which has been approved by the Institutional Animal Care & Use Committee (IACUC) of the University of Michigan and is in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. For PD studies in native mouse tissues, CD-1 female mice were treated with SD-36 or 19 or vehicle control (10% PEG400 + 90% PBS) as indicated in the figures. Mice were euthanized at the indicated time points in the figures, and mouse organs were harvested for Western blotting analysis. For efficacy and PD studies, CB.17 SCID mice were injected subcutaneously with 3 × 106 Molm-16 cells in 5 mg/mL Matrigel (Coming) for tumor growth. When the tumors reached an average volume of 150 mm3, mice were randomly assigned to different experimental groups for treatment. Drugs or vehicle control (10% PEG400 + 90% PBS) were given at the indicated dose schedules in the figures. Tumor sizes and animal weights were measured 2–3 times per week. Tumor volumes were calculated as tumor volume (mm3) = (length × width2)/2. For PD studies, mice were euthanized at the indicated time points in the figures, and tumor tissues or mouse organs were harvested for Western blotting analysis.
Supplementary Material
ACKNOWLEDGMENTS
This study is supported in part by a research contract from Oncopia Therapeutics Inc. S.W. and J.S. are grateful for the support of this work from Grant NIH P30 CA046592, the University of Michigan Comprehensive Cancer Center (now the Rogel Cancer Center) Core grant from the National Cancer Institute (NCI), NIH. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
ABBREVIATIONS USED
- STAT3
signal transducer and activator of transcription 3
- PROTAC
proteolysis targeting chimera
- SH2
Src homology 2
- ASO
antisense oligonucleotide
- FP
fluorescence polarization
- PTP
protein tyrosine phosphatase
- CRBN
cereblon
- MDM2
human murine double minute 2
- BET
bromodomain and extra-terminal
- AML
acute myeloid leukemia
- ALCL
anaplastic large cell lymphoma
- PD
pharmacodynamics
- DIEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- HATU
1-[bis(dimethylamino)methylene]-1H-l,2,3-triazolo[4,5-b]-pyridinium 3-oxide hexafluorophosphate
- TFA
trifluoroacetic acid
- TMSI
iodotrimethylsilane
- Pd/C
palladium on carbon
- NBS
N-bromosuccinimide
- THF
tetrahydrofuran
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- HOBt
1-hydroxybenzotriazole hydrate
- BSTFA
N,O-bis (trimethylsilyl)trifluoroacetamide
Footnotes
The authors declare the following competing financial interest(s): The University of Michigan has filed patent applications on SD-36 and its analogues, for which S. Wang, H. Zhou, R Xu, L. Bai, D. McEachem, C.-Y. Yang, and J. Stuckey are co-inventors. These patents have been licensed by Oncopia Therapeutics Inc., of which S. Wang is a co-founder, a paid consultant, and owns stock The University of Michigan also owns stock in Oncopia. This study is supported in part by a research contract from Oncopia.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01530.
Figures of FP binding experiments, computational model, and Western blotting analysis of total STAT3 and pSTAT3Y705 proteins; tables of binding affinities and crystallography data collection and refinement statistics; 1H NMR and UPLC–MS spectra for representative compounds (PDF)
Molecular formula strings and some data (CSV)
Structure representation (PDB)
Accession Codes
Coordinates for cocrystal structure SI-109 in complex with STAT3 have been deposited in the PDB under accession code 6NUQ and will be released upon publication.
REFERENCES
- (1).Levy DE; Darnell JE STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3 (9), 651–662. [DOI] [PubMed] [Google Scholar]
- (2).Yu H; Jove R The stats of cancer - new molecular targets come of age. Nat. Rev. Cancer 2004, 4 (2), 97–105. [DOI] [PubMed] [Google Scholar]
- (3).Johnson DE; O’Keefe RA; Grandis JR Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15 (4); 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Beebe JD,; Liu JY; Zhang JT Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther. 2018, 191, 74–91. [DOI] [PubMed] [Google Scholar]
- (5).Yang J,; Stark GR Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18 (4), 443–451. [DOI] [PubMed] [Google Scholar]
- (6).Hong D; Kurzrock R; Kim Y; Woessner R; Younes A; Nemunaitis J; Fowler N; Zhou TY; Schmidt J; Jo MJ; Lee SJ; Yamashita M; Hughes SG; Fayad L; Piha-Paul S; Nadella MVP; Mohseni M; Lawson D; Reimer C; Blakey DC; Xiao XK; Hsu J; Revenko A; Monia BP; MacLeod AR AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7 (314), 314ral85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rinaldi C; Wood MJ A Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14 (1), 9–21. [DOI] [PubMed] [Google Scholar]
- (8).Toure M,; Crews CM Small-Molecule PROTACS: new approaches to protein degradation. Angew. Chem., Int. Ed. 2016, 55 (6), 1966–1973. [DOI] [PubMed] [Google Scholar]
- (9).Lai AC,; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16 (2), 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Paiva SL,; Crews CM Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol 2019, 50, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bondeson DP,; Crews CM Targeted protein degradation by small molecules. Annu. Rev. Pharmacol Toxicol 2017, 57, 107–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chen J,; Bai L; Bernard D; Nikolovska-Coleska Z; Gomez C; Zhang J; Yi H; Wang S Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med. Chem. Lett. 2010, I (2), 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Burke TR; Yao ZJ,; Liu DG,; Voigt J,; Gao Y Phosphoryltyrosyl mimetics in the design of peptide-based signal transduction inhibitors. Biopolymers 2001, 60 (l), 32–44. [DOI] [PubMed] [Google Scholar]
- (14).Burke TR; Lee K Phosphotyrosyl mimetics in the development of signal transduction inhibitors. Acc. Chem. Res. 2003, 36 (6), 426–433. [DOI] [PubMed] [Google Scholar]
- (15).Mandal PK,; Liao WSL,; McMurray JS Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of stat3. Org. Lett. 2009, 11 (15), 3394–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mandal PK,; Iimbrick D,; Coleman DR; Dyer GA; Ren ZY,; Birtwistle JS,; Xiong CY,; Chen XM,; Briggs JM,; McMurray JS Conformationally constrained peptidomimetic inhibitors of signal transducer and activator of transcription 3: evaluation and molecular modeling. J. Med. Chem. 2009, 52 (8), 2429–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mandal PK; Gao FQ; Lu Z; Ren ZY; Ramesh R; Birtwisde JS; Kaluarachchi KK; Chen XM; Bast RC; Liao WS; McMurray JS Potent and selective phosphopeptide mimetic prodrugs targeted to the src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J. Med. Chem. 2011, 54 (10), 3549–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Buckley DL; Van Molle I; Gareiss PC; Tae HS; Michel J; Noblin DJ; Jorgensen WL; Ciulli A; Crews CM Targeting the von hippel-lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1 alpha interaction. J. Am. Chem. Soc. 2012, 134 (10), 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Galdeano C; Gadd MS; Soares P; Scaffidi S; Van Molle I; Birced I; Hewitt S; Dias DM; Ciulli A Structure-guided design and optimization of small molecules targeting the protein protein interaction between the von hippel- lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57 (20), 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Soares P; Gadd MS; Frost J; Galdeano C; Ellis L; Epemolu O; Rocha S; Read KD; Ciulli A Group-based optimization of potent and cell-active inhibitors of the von hippel-lindau (VHL) E3 ubiquitin ligase: structure-activity relationships leading to the chemical probe (2S,4R)-l-((S)-2-(l-cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyppyrroIidine-2-carboxamide (VH298). J. Med. Chem. 2018, 61 (2), 599–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lucas X; Van Molle I; Ciulli A Surface probing by fragment-based screening and computational methods identifies ligandable pockets on the von hippel-lindau (VHL) E3 ubiquitin ligase. J. Med. Chem. 2018, 61 (16), 7387–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bai LC; Zhou B; Yang CY; Ji J; McEachem D; Przybranowski S; Jiang H; Hu JT; Xu FM; Zhao YJ; Liu L; Femandez-Salas E; Xu J; Dou YL; Wen B; Sun DX; Meagher J; Stuckey J; Hayes DF; Li SQ; Ellis MJ; Wang SM Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017, 77 (9), 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhou B; Hu JT; Xu FM; Chen Z; Bai LC; Femandez-Salas E; Lin M; Liu L; Yang CY; Zhao YJ; McEachem D; Przybranowski S; Wen B; Sun DX; Wang SM Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2018, 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Qin C; Hu Y; Zhou B; Fernandez-Salas E; Yang CY; Liu L; McEachem D; Przybranowski S; Wang M; Stuckey J; Meagher J; Bai LC; Chen Z; Lin M; Yang JL; Ziazadeh DN; Xu FM; Hu JT; Xiang WG; Huang LY; Li S; Wen B; Sun DX; Wang SM Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J. Med. Chem. 2018, 61 (15), 6685–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li YB; Yang JL; Aguilar A; McEachem D; Przybranowski S; Liu L; Yang CY; Wang M; Han X; Wang SM Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 2019, 62 (2), 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bai L; Zhou H; Xu R; Zhao Y; Chinnaswamy K; McEachem D; Chen J; Yang CY; Liu Z; Wang M; Liu L; Jiang H; Wen B; Kumar P; Meagher JL; Sun D; Stuckey JA; Wang S A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression In vivo. Cancer Cell 2019, 36 (5), 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fischer ES; Bohm K; Lydeard JR; Yang HD; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM; Tichkule RB; Schebesta M; Forrester WC; Schirle M; Hassiepen U; Ottl J; Hild M; Beckwith REJ; Harper JW; Jenkins JL; Thoma NH Structure of the DDBI-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512 (7512), 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lu J; Qian YM; Altieri M; Dong HQ; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol 2015, 22 (6), 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Bondeson DP; Smith BE; Burslem GM; Buhimschi A; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol 2018, 25 (1), 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Peng YF; Sun HY; Wang SM Design and synthesis of a 1,5-diazabicyclo [6,3,0] dodecane amino acid derivative as a novel dipeptide reverse-turn mimetic. Tetrahedron Lett. 2006, 47 (27), 4769–4770. [Google Scholar]
- (31).Mandal PK; Morlacchi P; Knight JM; link TM; Lee GR; Nurieva R; Singh D; Dhanik A; Kavraki L; Corry DB; Ladbury JE; McMurray JS Targeting the src homology 2 (SH2) domain of signal transducer and activator of transcription 6 (STAT6) with cell-permeable, phosphatase-stable phosphopeptide mimics potently inhibits tyr641 phosphorylation and transcriptional activity. J. Med. Chem. 2015, 58 (22), 8970–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McMurray JS; Mandal PK; Liao WS; Ren Z; Chen X; Rajaopal R; Robertson F Preparation of Phosphopeptide Inhibitors of STAT3. WO 2010118309, 2010.
- (33).Cer RZ; Mudunuri U; Stephens R; Lebeda FJ IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and bgand binding. Nucleic Acids Res. 2009, 37 (Web Server), W441–W445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Becker S; Groner B; Muller CW Three-dimensional structure of the Stat3 beta homodimer bound to DNA. Nature 1998, 394 (6689), 145–151. [DOI] [PubMed] [Google Scholar]
- (35).Vilar S; Cozza G; Moro S Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8 (18), 1555–1572. [DOI] [PubMed] [Google Scholar]
- (36).Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and validation of a genetic algorithm for flexible docking. J. Mol Biol 1997, 267 (3), 727–748. [DOI] [PubMed] [Google Scholar]
- (37).Zhou HB; Zhou WH; Zhou B; Liu L; Chem TR; Chinnaswamy K; Lu JF; Bernard D; Yang CY; Li SS; Wang M; Stuckey J,; Sun Y; Wang SM High-affinity peptidomimetic inhibitors of the DCN1-UBC12 protein protein interaction. J. Med. Chem. 2018, 61 (5), 1934–1950. [DOI] [PubMed] [Google Scholar]
- (38).Otwinowski Z; Minor W [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (39).McCoy AJ,; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 (Part 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bricogne G,; Blanc E,; Brandi M,; Flensburg,; Keller P,; Paciorek W,; Roversi P,; Sharif A; Smart OS,; Vonrhein C,; Womack TO BUSTER, version, 2.10.3; Global Phasing Ltd.: Cambridge, U.K, 2017. [Google Scholar]
- (41).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol Crystallogr. 2010, 66 (Part 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Smart OS; Womack TO; Sharif A; Flensburg C; Keller P; Paciorek W; Vonrhein C; Bricogne G GRADE, version 1.2.13; Global Phasing Ltd.: Cambridge, U.K, 2017. [Google Scholar]
- (43).Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66 (Part 1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.