

Abstract
Hamamelidaceae is an important group that represents the origin and early evolution of angiosperms. Its plants have many uses, such as timber, medical, spice, and ornamental uses. In this study, the complete chloroplast genomes of Loropetalum chinense (R. Br.) Oliver, Corylopsis glandulifera Hemsl., and Corylopsis velutina Hand.‐Mazz. were sequenced using the Illumina NovaSeq 6000 platform. The sizes of the three chloroplast genomes were 159,402 bp (C. glandulifera), 159,414 bp (C. velutina), and 159,444 bp (L. chinense), respectively. These chloroplast genomes contained typical quadripartite structures with a pair of inverted repeat (IR) regions (26,283, 26,283, and 26,257 bp), a large single‐copy (LSC) region (88,134, 88,146, and 88,160 bp), and a small single‐copy (SSC) region (18,702, 18,702, and 18,770 bp). The chloroplast genomes encoded 132–133 genes, including 85–87 protein‐coding genes, 37–38 tRNA genes, and 8 rRNA genes. The coding regions were composed of 26,797, 26,574, and 26,415 codons, respectively, most of which ended in A/U. A total of 37–43 long repeats and 175–178 simple sequence repeats (SSRs) were identified, and the SSRs contained a higher number of A + T than G + C bases. The genome comparison showed that the IR regions were more conserved than the LSC or SSC regions, while the noncoding regions contained higher variability than the gene coding regions. Phylogenetic analyses revealed that species in the same genus tended to cluster together. Chunia Hung T. Chang, Mytilaria Lecomte, and Disanthus Maxim. may have diverged early and Corylopsis Siebold & Zucc. was closely related to Loropetalum R. Br. This study provides valuable information for further species identification, evolution, and phylogenetic studies of Hamamelidaceae plants.
Keywords: chloroplast genomes, comparative analysis, Hamamelidaceae, phylogenetic relationship
In this study, the complete cp genomes of three Hamamelidaceae species were determined by Illumina high‐throughput sequencing technology, and the basic structures, conservation, and variability of these sequences were revealed. This study provides valuable information for further researches on the species identification, evolution, and phylogeny of Hamamelidaceae plants.

1. INTRODUCTION
Hamamelidaceae is an important group representing the origin and early evolution of angiosperms and is well known for its broad and scattered geographic distribution and endemics (Endress, 1993; Zhang & Lu, 1995). Hamamelidaceae fossils have been found in Upper Cretaceous‐early Tertiary strata (Manchester et al., 2009; Zhang & Lu, 1995); thus, the flora of Hamamelidaceae may have arisen earlier than the Cretaceous. This family contains 28 genera and about 120 species (Judd et al., 2007), which mainly occur in Eastern Asia, while others are distributed in the Americas, Africa, and Oceania. The plants in Hamamelidaceae are all woody, including Liquidambar L., Altingia Noronha, Exbucklandia R. W. Brown, Chunia Hung T. Chang, Mytilaria Lecomte, and Semiliquidambar Chang, which are used in the construction and furniture industries (Qin et al., 2019). Liquidambar, Altingia, Semiliquidambar, Hamamelis Gronov. ex L., Fortunearia Rehder & E. H. Wilson, and Corylopsis Siebold & Zucc. are used as medicinal materials (Kim et al., 2020; Simon et al., 2021). In addition, most of the genera have ornamental value, particularly Rhodoleia Champ. ex Hook. and Corylopsis.
The chloroplast is an important plant organelle and photosynthetic organ (Douglas, 1994). It is also a semiautonomous genetic organelle that contains independent chloroplast DNA (cpDNA), which has a length of 110–160 kb (Choi & Park, 2015). In general, cpDNA has a circular structure that includes one large single‐copy (LSC) region, one short single‐copy (SSC) region, and two inverted repeat (IR) regions, with the IR region separating the LSC and SSC regions (Ferrarini et al., 2013; Wu et al., 2014; Xue et al., 2019). The chloroplast genome is independent of the nuclear genome and corresponds to maternal inheritance with independent transcription and transport systems (Wu et al., 2020). Considering the similar structures, highly conserved sequences, and stable maternal heredity, the chloroplast genome has become an ideal resource for species identification, population genetics, phylogenetic, and genetic engineering studies (Fan et al., 2021; Nock et al., 2014). Moreover, gene mutations, rearrangements, duplications, and losses can be detected in the chloroplast genomes of the angiosperm lineages (Li et al., 2020; Luo et al., 2021). Structural changes in the genome can be used to study the taxonomic significance and phylogenetic relationships, and provide information for the development of genomic markers (Cheng et al., 2020; Watson et al., 2002). Repeat sequences are DNA sequence motifs that are repeated hundreds or thousands of times at different positions in the genome (Biscotti et al., 2015). They are ubiquitous in genomes and play important roles in evolution. Repeat sequences are mainly divided into two categories: one is tandem repeats, which mainly include some shorter repeats, such as simple sequence repeats (SSRs), and the other is interspersed repeated sequences, which are commonly known as transposons (Treangen & Salzberg, 2011). SSRs are composed of 1–6 nucleotide repeat units and are also called microsatellites, which have been widely used as molecular markers in population genetics and evolutionary biology (Bondar et al., 2019; Dashnow et al., 2015) due to their highly reproducible, codominance, multi‐allelic, and chromosome‐specific nature (Miri et al., 2014; Oliveira et al., 2006; Vieira et al., 2016). Interspersed repeated sequences account for most of the plant genomic repeats (Zhao & Ma, 2013), whereas retrotransposons play an important role in genome amplification (Ammiraju et al., 2007; Baucom et al., 2009; Paterson et al., 2009; SanMiguel et al., 2009; Schnable et al., 2009) and contribute to the expansion and contraction of the genome and the difference in the interspecific sequence (Morgante et al., 2007). The complete chloroplast genome contains all genes used to reconstruct the evolutionary history and provides more valuable and high‐quality information for evolutionary and phylogenetic analyses (Li et al., 2019). Complete chloroplast genome sequences are easily obtained due to the rapid development of large‐scale high‐throughput sequencing techniques, such as the Illumina and PacBio sequencing platforms (Huang et al., 2019; Kim et al., 2021; Lin et al., 2018; Yang et al., 2019; Ye et al., 2020).
Hamamelidaceae is a key family to study the phylogeny of angiosperms (Zhang et al., 2001). The relationships between genera in this family have been controversial for a long time (Hao & Wei, 1998; Li Bogle et al., 1999; Li Bogle et al., 1999; Li et al., 1997; Magallon, 2007; Xie et al., 2010). For example, Ye et al. (2020) reported that Hamamelis is sister to the clade that includes Parrotia C. A. Mey. and Distylium Siebold & Zucc., which is consistent with previous studies (Li, Bogle, & Donoghue, 1999; Li, Bogle, & Klein, 1999; Magallon, 2007; Shi et al., 1998; Xie et al., 2010). The results of another study showed that Parrotia subaequalis (H. T. Chang) R. M. Hao & H. T. Wei is in the Distylium genus (Chen et al., 2020), which is consistent with the result of Jiang et al. (2020). Different taxonomists have systematically divided Hamamelidaceae based on morphology, anatomy, and palynology (Bogle & Philbrick, 1980; Harms, 1930; Reinsch, 1890), but the traditional identification method based on morphological characteristics cannot be used to clearly distinguish Hamamelidaceae species (Deng et al., 1992; Endress, 1969, 1989; Zhang, 1999). In recent years, phylogenetic analyses of Hamamelidaceae species have been carried out with the rapid development of molecular technology (Li et al., 2000; Shi et al., 1998; Wen & Shi, 1999; Xiang et al., 2019; Xie et al., 2010; Zhou et al., 2019), and early studies focused on DNA fragment‐labeling techniques or phylogenetic analyses based on nuclear or chloroplast DNA fragments. However, limited nuclear or chloroplast DNA fragments do not provide sufficient phylogenetic information to effectively solve interspecific relationships (Hao & Wei, 1998; Li et al., 1997). Complete chloroplast genomes provide more valuable and higher‐quality information for evolutionary and phylogenetic analyses and reduce the sampling error inherent in studies of one or a few genes that may indicate critical evolutionary events (Cho et al., 2019). Thus, a clear phylogenetic relationship with Hamamelidaceae or the relationships between and within genera may be established based on conserved chloroplast genomes. Can the LSC, SSC, and IR regions of chloroplast genomes be used to establish a phylogenetic relationship within Hamamelidaceae?
In the present study, the complete chloroplast genomes of Loropetalum chinense, Corylopsis glandulifera, and Corylopsis velutina (Hamamelidaceae) were sequenced using Illumina technology, and their features were characterized. Our research purposes were to: (1) study the molecular structures of these three chloroplast genomes; (2) examine the variations in the repeat sequences and the SSRs in the three chloroplast genomes; (3) discover the divergence hotspot regions to provide potential molecular markers for future phylogenetic studies; and (4) establish and analyze the phylogenetic relationships of Hamamelidaceae species based on their complete chloroplast genome sequences, as well as the LSC, SSC, and IR regions. The data obtained in this study will provide valuable reference information for further studies on species identification and evolution, as well as population genetics and phylogenetic analyses of Hamamelidaceae.
2. MATERIALS AND METHODS
2.1. Plant material, DNA extraction, and sequencing
Fresh and healthy leaves of L. chinense and C. velutina were collected from the Nanjing Forestry University in Nanjing, Jiangsu, China (32°04′N, 118°48′E). Fresh and healthy leaves of C. glandulifera were collected from Mount Huang in Anhui, China (30°8′N, 118°6′E). All voucher specimens were deposited at the Herbarium of Nanjing Forestry University, Nanjing, Jiangsu, China with collection numbers 2021–20 (L. chinense), 2021–21 (C. velutina), and 2021–29 (C. glandulifera). After the quality inspection of the genomic DNA was performed, the DNA was fragmented by mechanical interruption (ultrasound). Then, the fragmented DNA was purified, end repaired, 3′ end plus A, connected to a sequencing adapter, and agarose gel electrophoresis was used to select the fragment size. The polymerase chain reaction (PCR) product was amplified to form the sequencing library. The qualified library was sequenced with the Illumina NovaSeq 6000 platform, and the sequencing read length was 150 bp. The whole genome was sequenced by Nanjing Genepioneer Biotechnologies Inc. (Nanjing, China).
2.2. Chloroplast genome assembly and annotation
Fastp v0.20.0 (https://github.com/OpenGene/fastp) was used to trim the raw reads, and the high‐quality clean data were obtained by removing the connector sequences and low‐quality reads (the filtering criteria are in the Appendix S1). Bowtie2 v2.2.4 (Langmead & Salzberg, 2012) was used to align the clean data with the chloroplast genome database built by Genepioneer Biotechnologies a in very sensitive local mode. SPAdes v3.10.1 (Bankevich et al., 2012) was used to acquire SEED sequences, and the contigs were obtained using the kmer iterative extend seed. The contig sequences were linked into scaffolds using SSPACE v2.0 (Acemel et al., 2016) and then used in Gapfiller v2.1.1 (Boetzer & Pirovano, 2012) to fill the gaps (Xiong et al., 2020) (the assembly process is in the Appendix S1). Two methods were used to annotate the chloroplast genomes to improve the accuracy of the annotation. First, protein‐coding genes were annotated using Prodigal v2.6.3 (https://www.github.com/hyattpd/Prodigal). rRNA was predicted using Hmmer v3.1b2 (Eddy, 2008), and tRNA was predicted using Aragorn v1.2.38 (Laslett & Canback, 2004). Second, the assembled sequences were compared using Blast v2.6 (McGinnis & Madden, 2004) according to the related species published at the NCBI (https://www.ncbi.nlm.nih.gov/). Then, the two annotation results were compared and manually corrected. The circular gene maps were visualized using OGDRAW v1.2 (Lohse et al., 2007). An analysis of GC content, codon usage, and relative synonymous codon usage (RSCU) values was conducted in MEGA7 (Kumar et al., 2016). The repetitive sequences and SSRs were determined using Vmatch v2.3.0 (http://www.vmatch.de/) (parameter settings: minimum length = 30 bp, hamming distance = 3) and MISA v1.0 (MIcroSAtellite identification tool, http://pgrc.ipk‐gatersleben.de/misa/misa.html) (parameters 1–8 [single base repeat 8 times or more], 2–5, 3–3, 4–3, 5–3, 6–3), respectively.
2.3. Genome comparison
Chloroplast genome sequences are often used to measure genetic diversity within a species, gene flow between species, and ancestral population size of separated sister species (Cavender et al., 2015). Therefore, it is necessary to understand the divergence of chloroplasts between species. The online comparison tool mVISTA (Mayor et al., 2000) was applied to compare the whole chloroplast genomes of L. chinense, C. glandulifera, and C. velutina to three published chloroplast genomes of Chunia bucklandioides Chang (NC_041163), Distylium tsiangii Chun ex Walker (MN711651), and Rhodoleia championii Hook. f. (NC_045276) in Shuffle‐LAGAN mode (Frazer et al., 2004) with the L. chinense annotation as the reference. Although the IR regions are the most conserved, expansion and contraction of the IR boundary are the main reasons for differences in the sizes of chloroplast genomes (Kode et al., 2005; Raubeson et al., 2007; Yao et al., 2015). Irscope (Ali et al., 2018) was used to compare and visualize the borders of the LSC, SSC, and IR regions among the six Hamamelidaceae species. The six chloroplast genome sequences were aligned using MAFFT (Katoh & Standley, 2013) under default parameters, and then DnaSP v5.10 (Librado & Rozas, 2009) was utilized to detect nucleotide diversity (Pi). Pi values were calculated with a step size of 200 bp and a sliding window of 600 bp.
2.4. Phylogenetic analysis
To investigate the phylogenetic positions of L. chinense, C. glandulifera, and C. velutina within the Hamamelidaceae lineages, 28 complete chloroplast genome sequences (25 Hamamelidaceae species) were downloaded from the NCBI GenBank, along with Altingia chinensis (Champ.) Oliver ex Hance, Liquidambar formosana Hance, and Cercidiphyllum japonicum Sieb. et Zucc. as outgroups (Table S1). Maximum likelihood (ML) and Bayesian inference (BI) methods were used to perform phylogenetic analyses based on the following four datasets: (1) the complete chloroplast genome sequences; (2) LSC regions of the chloroplast genomes; (3) SSC regions of the chloroplast genomes; and (4) IR regions of the chloroplast genomes. The ML analysis (Guindon et al., 2010) was conducted using IQ‐TREE (Nguyen et al., 2015) and Ultrafast bootstrap (Minh et al., 2013), and the BI analysis was conducted using MrBayes (Ronquist et al., 2012). All datasets were aligned using MAFFT (Katoh & Standley, 2013) under default parameters. ModelFinder (Kalyaanamoorthy et al., 2017) was used to select the best‐fit model using Akaike's Information Criterion and GTR (general time‐reversible)+F+I+G4 was selected as the best substitution model for the complete chloroplast genome sequences and the LSC regions. GTR+F+G4 was selected as the best substitution model for the SSC regions and GTR+F+I was selected for the IR regions. The ML analysis was conducted with 1,000 repetitions of Ultrafast bootstrap and 1,000 bootstrap replicates of the Shimodaira/Hasegawa approximate likelihood‐ratio test (SH‐aLRT) (Guindon et al., 2010). The Markov chain Monte Carlo algorithms were run for 2,000,000 generations and sampled every 100 generations for the BI analysis. The first 25% of the generations were discarded as burn‐in. MAFFT, ModelFinder, IQ‐TREE, Ultrafast bootstrap, and MrBayes were used in PhyloSuite (Zhang, Gao, et al., 2020; Zhang, Wang, et al., 2020). The phylogenetic relationships were visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
3. RESULTS
3.1. Chloroplast genome features of the three Hamamelidaceae species
The chloroplast genomes of C. glandulifera (accession no. MZ642354), C. velutina (accession no. MZ823391), and L. chinense (accession no. MZ642355) have been submitted to GenBank at the NCBI. The genome sizes ranged from 159,402 bp (C. glandulifera) to 159,444 bp (L. chinense). These chloroplast genomes had a circular assembly and exhibited a typical quadripartite structure containing an LSC region (88,134–88,160 bp) and an SSC region (18,702–18,770 bp) separated by IR regions (26,257–26,083 bp) (Figure 1, Table 1). The overall GC contents of the three chloroplast genomes were almost identical (37.97%–38.03%) (Table 1) and the GC contents of the LSC and SSC regions were lower than those of the IR regions (Table 2).
FIGURE 1.
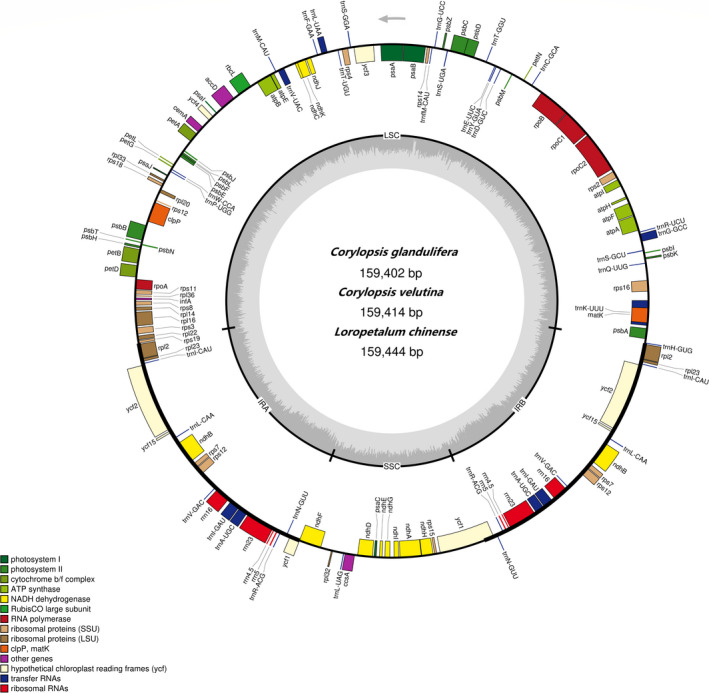
The chloroplast genome maps of Corylopsis glandulifera, Corylopsis velutina, and Loropetalum chinense. Genes on the inside of the circle are transcribed clockwise and those on the outside are transcribed counter‐clockwise. The darker gray inner circle corresponds to the GC content, whereas the lighter gray indicates the AT content. Different colors represent different functional genes
TABLE 1.
Summary of the complete chloroplast genomes of the three Hamamelidaceae species
| Genome features | Corylopsis velutina | Corylopsis glandulifera | Loropetalum chinense |
|---|---|---|---|
| Total length (bp) | 159,414 | 159,402 | 159,444 |
| LSC length (bp) | 88,146 | 88,134 | 88,160 |
| SSC length (bp) | 18,702 | 18,702 | 18,770 |
| IRa length (bp) | 26,283 | 26,283 | 26,257 |
| IRb length (bp) | 26,283 | 26,283 | 26,257 |
| Genes | 133 | 132 | 132 |
| Protein‐coding genes (CDS) | 87 | 87 | 85 |
| tRNA genes | 37 | 37 | 38 |
| rRNA genes | 8 | 8 | 8 |
| GC% | 38.03 | 38.03 | 37.97 |
TABLE 2.
Base composition of the complete chloroplast genomes of the three Hamamelidaceae species
| Species | Region | A (%) | T (U) (%) | C (%) | G (%) | AT (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| Corylopsis velutina | LSC | 31.26 | 32.60 | 18.61 | 17.53 | 63.86 | 36.14 |
| SSC | 33.65 | 33.67 | 17.11 | 15.57 | 67.32 | 32.68 | |
| IR | 28.44 | 28.44 | 21.55 | 21.55 | 56.88 | 43.10 | |
| Total | 30.61 | 31.36 | 19.40 | 18.63 | 61.97 | 38.03 | |
| Corylopsis glandulifera | LSC | 31.26 | 32.59 | 18.61 | 17.53 | 63.85 | 36.14 |
| SSC | 33.69 | 33.67 | 17.11 | 15.54 | 67.36 | 32.64 | |
| IR | 28.45 | 28.45 | 21.55 | 21.55 | 56.90 | 43.10 | |
| Total | 30.62 | 31.35 | 19.41 | 18.62 | 61.97 | 38.03 | |
| Loropetalum chinense | LSC | 31.29 | 32.65 | 18.58 | 17.49 | 63.94 | 36.07 |
| SSC | 33.62 | 33.70 | 17.19 | 15.49 | 67.32 | 32.67 | |
| IR | 28.46 | 28.46 | 21.53 | 21.53 | 56.92 | 43.06 | |
| Total | 30.63 | 31.39 | 19.39 | 18.59 | 62.02 | 37.97 |
The chloroplast genomes of C. glandulifera and L. chinense encoded 132 genes, including 87 protein‐coding genes, 37 tRNA genes, and 8 rRNA genes in C. glandulifera and 85 protein‐coding genes, 38 tRNA genes, 8 rRNA genes, and 1 pseudogene (ycf1) in L. chinense. A total of 133 distinct genes were annotated in the C. velutina chloroplast genome, including 87 protein‐coding genes, 37 tRNA genes, 8 rRNA genes, and 1 pseudogene (ycf1) (Table 1). After removing the duplicates, 80 protein‐coding genes, 30 tRNA genes, and 4 rRNA genes remained in C. glandulifera and C. velutina, while 79 protein‐coding genes, 29 tRNA genes, and 4 rRNA genes remained in L. chinense. The LSC region comprised 62 protein‐coding genes and 22 tRNA genes, while the SSC region comprised 11 protein‐coding and 1 tRNA gene of the three chloroplast genomes (Figure 1, Figures S1 and S2). Twenty genes contained introns: 17 genes (ndhA, ndhB, petB, petD, atpF, rpl16, rpl2, rps16, rpoC1, trnA‐UGC, trnG‐GCC, trnG‐UCC, trnI‐GAU, trnK‐UUU, trnL‐UAA, trnV‐UAC, and trnE‐UUC) contained 1 intron, while 3 genes (rps12, clpP, and ycf3) possessed two introns (Table 3). trnK‐UUU featured the longest intron (2,441–2,457 bp) and the shortest intron was found in trnL‐UAA (521–516 bp) (Table 4). Notably, rps12 was considered a trans‐spliced gene separated by two introns, with 1 exon in the LSC region and the other 2 in the IR regions (Figure 1).
TABLE 3.
Lists of genomic genes for Corylopsis velutina, Corylopsis glandulifera, and Loropetalum chinense
| Function |
C. velutina Genes |
C. glandulifera Genes |
L. chinense Genes |
|---|---|---|---|
| Photosystem I | psaA,psaB,psaC,psaI,psaJ | ||
| Photosystem II | psbA,psbB,psbC,psbD,psbE,psbF,psbH,psbI,psbJ,psbK,psbL,psbM,psbN,psbT,psbZ | ||
| NADH dehydrogenase | ndhA*,ndhB*(2),ndhC,ndhD,ndhE,ndhF,ndhG,ndhH,ndhI,ndhJ,ndhK | ||
| Cytochrome b/f complex | petA,petB*,petD*,petG,petL,petN | ||
| ATP synthase | atpA,atpB,atpE,atpF*,atpH,atpI | ||
| Rubisco | rbcL | ||
| Large subunit ribosomal proteins | rpl14,rpl16*,rpl2*(2),rpl20,rpl22,rpl23(2),rpl32,rpl33,rpl36 | ||
| Small subunit ribosomal proteins | rps11,rps12**(2),rps14,rps15,rps16*,rps18,rps19,rps2,rps3,rps4,rps7(2),rps8 | ||
| RNA polymerase | rpoA,rpoB,rpoC1*,rpoC2 | ||
| Ribosomal RNAs | rrn16(2),rrn23(2),rrn4.5(2),rrn5(2) | ||
| Transfer RNAs | trnA‐UGC*(2),trnC‐GCA,trnD‐GUC,trnE‐UUC,trnF‐GAA,trnG‐GCC*,trnG‐UCC,trnH‐GUG,trnI‐CAU(2),trnI‐GAU*(2),trnK‐UUU*,trnL‐CAA(2),trnL‐UAA*,trnL‐UAG,trnM‐CAU,trnN‐GUU(2),trnP‐UGG,trnQ‐UUG,trnR‐ACG(2),trnR‐UCU,trnS‐GCU,trnS‐GGA,trnS‐UGA,trnT‐GGU,trnT‐UGU,trnV‐GAC(2),trnV‐UAC*,trnW‐CCA,trnY‐GUA,trnfM‐CAU | trnA‐UGC*(2),trnC‐GCA,trnD‐GUC,trnE‐UUC,trnF‐GAA,trnG‐GCC,trnG‐UCC*,trnH‐GUG,trnI‐CAU(2),trnI‐GAU*(2),trnK‐UUU*,trnL‐CAA(2),trnL‐UAA*,trnL‐UAG,trnM‐CAU,trnN‐GUU(2),trnP‐UGG,trnQ‐UUG,trnR‐ACG(2),trnR‐UCU,trnS‐GCU,trnS‐GGA,trnS‐UGA,trnT‐GGU,trnT‐UGU,trnV‐GAC(2),trnV‐UAC*,trnW‐CCA,trnY‐GUA,trnfM‐CAU | trnA‐UGC*(2),trnC‐GCA,trnD‐GUC,trnE‐UUC,trnE‐UUC*,trnF‐GAA,trnG‐GCC*,trnG‐UCC,trnH‐GUG,trnI‐CAU(2),trnI‐GAU*(2),trnK‐UUU*,trnL‐CAA(2),trnL‐UAA*,trnL‐UAG,trnM‐CAU,trnN‐GUU(2),trnP‐UGG,trnQ‐UUG,trnR‐ACG(2),trnR‐UCU,trnS‐GCU(2),trnS‐UGA,trnT‐GGU,trnT‐UGU,trnV‐GAC(2),trnV‐UAC*,trnW‐CCA,trnY‐GUA,trnfM‐CAU |
| Other | matK,clpP**,cemA,accD,ccsA,infA | ||
| Unknown function | #ycf1,ycf1,ycf15(2),ycf2(2),ycf3**,ycf4 | ycf1(2),ycf15(2),ycf2(2),ycf3**,ycf4 | #ycf1,ycf1,ycf2(2),ycf3**,ycf4 |
*, Gene with one intron; **, Gene with two introns; #, Pseudogene; (2): Gene with two copies.
TABLE 4.
Characteristics and sizes of the intron and exon genes from the three Hamamelidaceae species
| Species | Gene | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| Corylopsis velutina | trnK‐UUU | 37 | 2,441 | 37 | ||
| rps16 | 39 | 850 | 225 | |||
| trnG‐GCC | 34 | 688 | 48 | |||
| atpF | 159 | 712 | 411 | |||
| rpoC1 | 435 | 735 | 1,632 | |||
| ycf3 | 126 | 746 | 228 | 741 | 153 | |
| trnL‐UAA | 37 | 515 | 50 | |||
| trnV‐UAC | 39 | 574 | 37 | |||
| rps12 | 114 | – | 232 | 538 | 26 | |
| clpP | 69 | 635 | 291 | 812 | 228 | |
| petB | 6 | 744 | 651 | |||
| petD | 9 | 690 | 474 | |||
| rpl16 | 9 | 1,001 | 402 | |||
| rpl2 | 393 | 653 | 435 | |||
| ndhB | 777 | 682 | 756 | |||
| rps12 | 232 | – | 26 | 538 | 114 | |
| trnI‐GAU | 42 | 939 | 30 | |||
| trnA‐UGC | 38 | 842 | 35 | |||
| ndhA | 552 | 1,073 | 540 | |||
| trnA‐UGC | 38 | 842 | 35 | |||
| trnI‐GAU | 42 | 939 | 30 | |||
| ndhB | 777 | 682 | 756 | |||
| rpl2 | 393 | 653 | 435 | |||
| Corylopsis glandulifera | trnK‐UUU | 37 | 2,443 | 35 | ||
| rps16 | 39 | 851 | 225 | |||
| trnG‐UCC | 34 | 687 | 48 | |||
| atpF | 159 | 712 | 411 | |||
| rpoC1 | 435 | 735 | 1,632 | |||
| ycf3 | 126 | 746 | 228 | 741 | 153 | |
| trnL‐UAA | 37 | 516 | 50 | |||
| trnV‐UAC | 39 | 574 | 37 | |||
| rps12 | 114 | – | 232 | 538 | 26 | |
| clpP | 69 | 631 | 291 | 812 | 228 | |
| petB | 6 | 744 | 651 | |||
| petD | 9 | 690 | 474 | |||
| rpl16 | 9 | 1,001 | 402 | |||
| rpl2 | 393 | 653 | 435 | |||
| ndhB | 777 | 682 | 756 | |||
| rps12 | 232 | – | 26 | 538 | 114 | |
| trnI‐GAU | 42 | 939 | 30 | |||
| trnA‐UGC | 38 | 842 | 35 | |||
| ndhA | 552 | 1,073 | 540 | |||
| trnA‐UGC | 38 | 842 | 35 | |||
| trnI‐GAU | 42 | 939 | 30 | |||
| ndhB | 777 | 682 | 756 | |||
| rpl2 | 393 | 653 | 435 | |||
| Loropetalum chinense | trnK‐UUU | 37 | 2,457 | 35 | ||
| rps16 | 42 | 853 | 225 | |||
| trnG‐GCC | 24 | 699 | 48 | |||
| atpF | 159 | 697 | 426 | |||
| rpoC1 | 427 | 752 | 1,625 | |||
| ycf3 | 126 | 742 | 228 | 757 | 156 | |
| trnL‐UAA | 37 | 512 | 50 | |||
| trnV‐UAC | 39 | 574 | 32 | |||
| rps12 | 114 | – | 232 | 538 | 26 | |
| clpP | 69 | 644 | 291 | 836 | 228 | |
| petB | 6 | 781 | 654 | |||
| petD | 9 | 690 | 474 | |||
| rpl16 | 9 | 1,005 | 402 | |||
| rpl2 | 393 | 653 | 435 | |||
| ndhB | 777 | 682 | 756 | |||
| rps12 | 232 | – | 26 | 538 | 114 | |
| trnI‐GAU | 42 | 890 | 35 | |||
| trnA‐UGC | 38 | 842 | 35 | |||
| ndhA | 552 | 1,042 | 540 | |||
| trnA‐UGC | 38 | 842 | 35 | |||
| trnE‐UUC | 33 | 939 | 41 | |||
| trnI‐GAU | 42 | 890 | 35 | |||
| ndhB | 777 | 682 | 756 | |||
| rpl2 | 393 | 653 | 435 |
3.2. Codon usage analysis
Analyzing codon usage is essential to evaluate the evolution of the chloroplast genome (Chi et al., 2020; Sun et al., 2021). RSCU values were computed for the C. glandulifera, C. velutina, and L. chinense chloroplast genomes based on the protein‐coding sequences. Figure 2 shows the codon content of 20 amino acids and stop codons in all protein‐coding genes of the chloroplast genomes of the three species. The coding regions of C. glandulifera, C. velutina, and L. chinense were composed of 26,797, 26,574, and 26,415 codons, respectively. The most prevalent amino acid was leucine (2,811 codons in C. glandulifera, 2,781 codons in C. velutina, and 2,764 codons in L. chinense), while the rarest one was cysteine (319 codons in C. glandulifera, 317 codons in C. velutina, and 318 codons in L. chinense). Codons with no preference value were set to 1.00. Codons for arginine, leucine, and serine were the most abundant (RSCU = 6), while those for methionine and tryptophan were the least abundant (RSCU = 1) (Figure 2), indicating no codon bias for these two amino acids. In addition, nearly all the A/U‐ending codons had RSCU values >1, whereas the C/G‐ending codons had RSCU values <1 (Table S2), indicating that most of the amino acids tended to use A/U‐ending codons rather than C/G‐ending codons.
FIGURE 2.
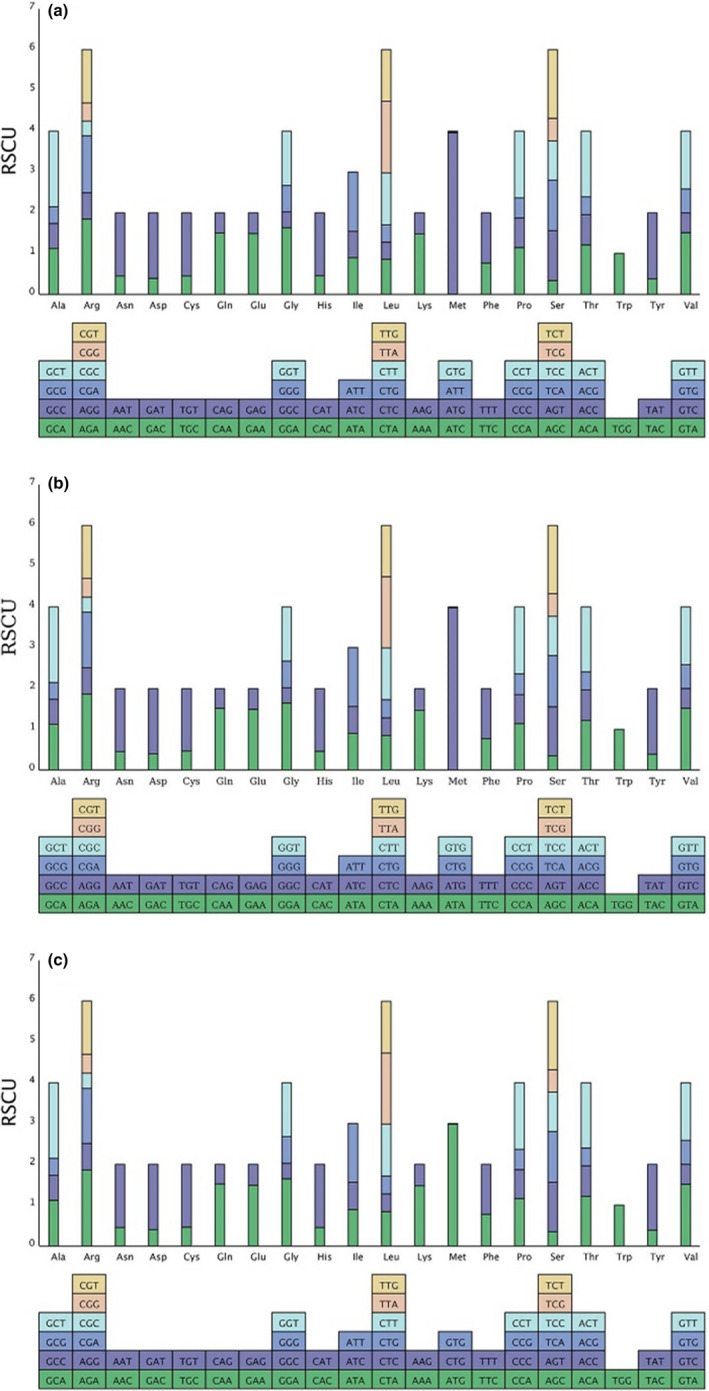
Codon content of 20 amino acids and stop codons in the protein‐coding genes of the chloroplast genomes of the three Hamamelidaceae species. (a) Loropetalum chinense; (b) Corylopsis glandulifera; (c) Corylopsis velutina
3.3. Repeat sequence analysis
Structures longer than 30 bp are known as long repeats (Asaf et al., 2018), and there are four types of long repeats, such as forward, palindromic, reverse, and complement repeats. In this study, three types of repeated sequences (forward, reverse, and palindromic) were detected in the chloroplast genomes of the three Hamamelidaceae species. In detail, there were 43 (19 forward, 22 palindromes, and 2 reverse), 42 (19 forward, 21 palindrome, and 2 reverse), and 37 (18 forward, 17 palindrome, and 2 reverse) long repeats in C. glandulifera, C. velutina, and L. chinense, respectively (Figure 3a). The lengths of the dispersed repeats were 30–35 bp (Figure 3b). Most of the long repeats were located in the ycf gene and the intergenic spacer (IGS) (Table S3). The types and content of the long repeats were similar in species from the same genus.
FIGURE 3.
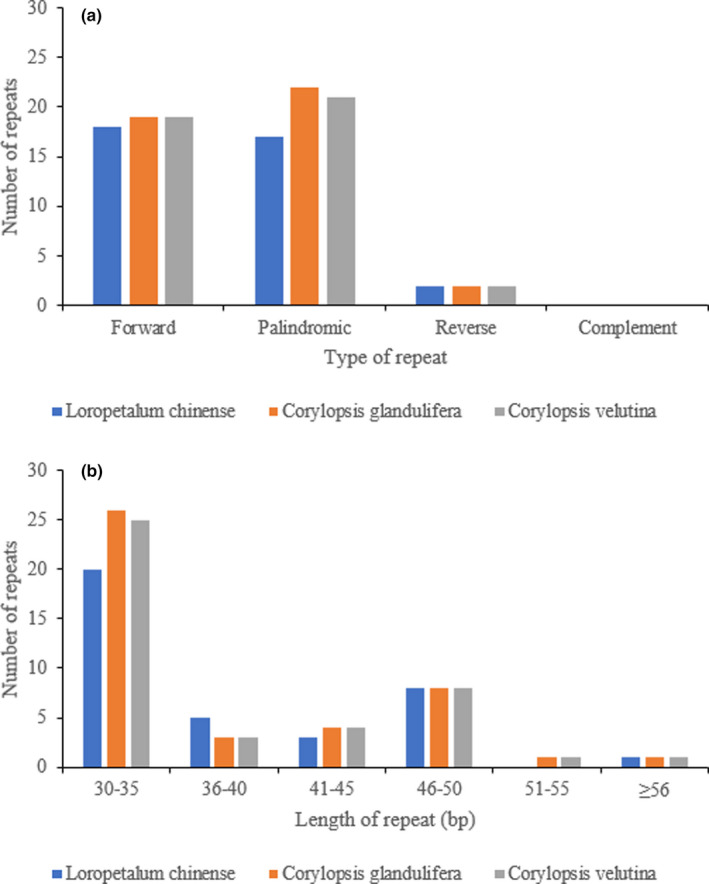
Analysis of repeated sequences in the three Hamamelidaceae chloroplast genomes. (a) Frequency of repeat types; (b) Frequency of repeat sequences by length
3.4. SSR analysis
Six types of SSRs were detected, including mononucleotides, dinucleotides, trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides with a total of 175–178 SSRs in the three species. The majority of the SSRs were located in intergenic regions. Most of the SSRs were located in the LSC regions rather than in the SSC or IR regions (Table S4). There were 143–152 mononucleotides, 9–10 dinucleotides, 58–66 trinucleotides, 4–5 tetranucleotides, 2 pentanucleotides, and 0–1 hexanucleotide (only in L. chinense). Among these SSRs, mononucleotide repeats were the most abundant, while pentanucleotide repeats numbered the least. Most mononucleotides and dinucleotides were composed of A and T (Figure 4).
FIGURE 4.
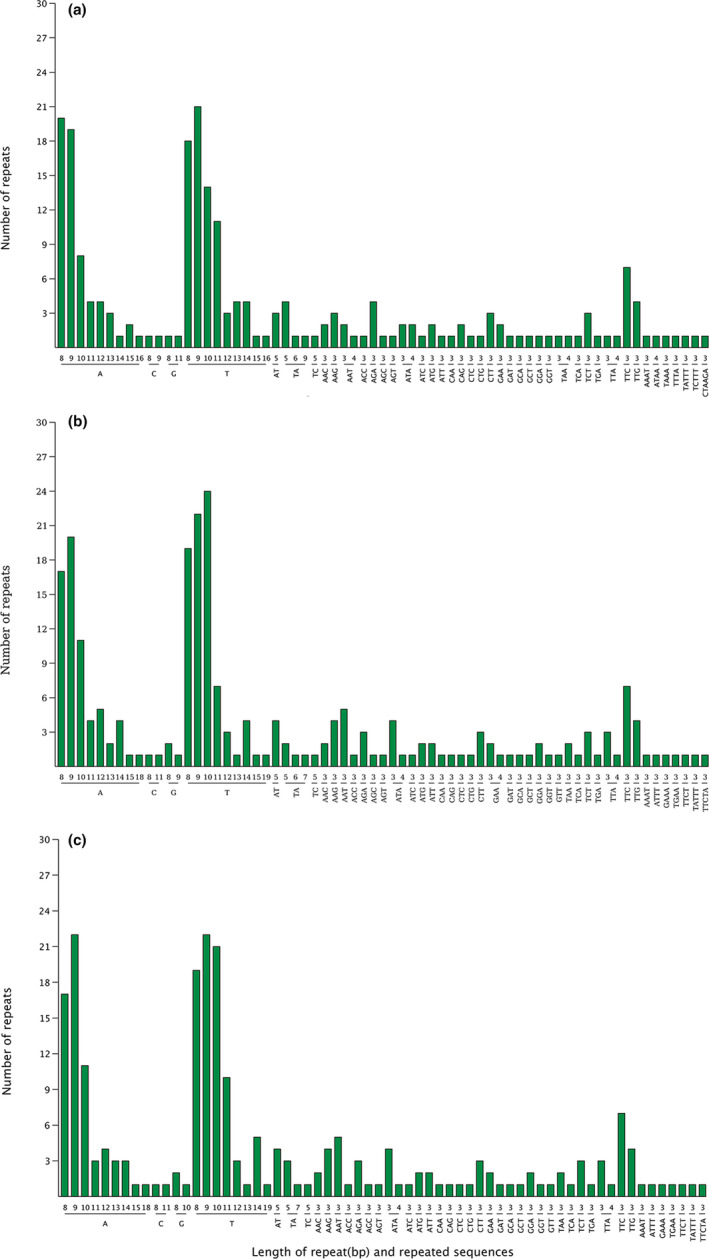
Frequency of SSRs in the different repeat class types. (a) Loropetalum chinense; (b) Corylopsis glandulifera; (c) Corylopsis velutina
3.5. Comparative genomic analysis
To investigate genomic divergence, the percentage of sequence identity was calculated for six species of Hamamelidaceae using the mVISTA program with L. chinense as the reference. The results showed that the similarity among the six species was high and the variability in the IR regions was less than that in the LSC and SSC regions. Furthermore, the chloroplast genomes were more highly variable in their noncoding regions than in their coding regions and this is consistent with the pattern found in most angiosperms (Yang et al., 2020) (Figure 5).
FIGURE 5.
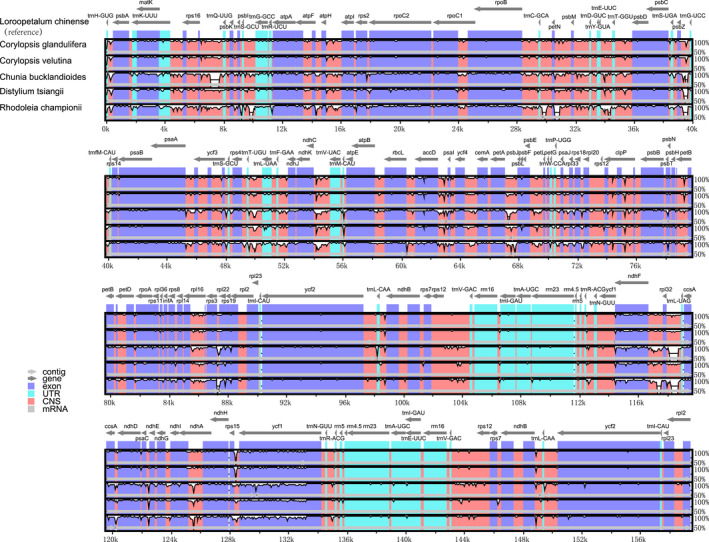
Complete chloroplast genome alignments of six Hamamelidaceae species using the mVISTA program, with the chloroplast genome of Loropetalum chinense as a reference. The horizontal axis indicates the coordinates within the chloroplast genome. The vertical scale indicates the percent identity within 50–100%. Annotated genes are displayed along the top
The chloroplast genome contains many variable nucleotides, which can be used to resolve closely related species or genera as valuable DNA barcoding (Liu et al., 2019; Xiong et al., 2020). In this study, variable loci were identified in the six species, with polymorphism information (Pi) values ranging from 0.0000 to 0.08600. According to the sliding‐window analysis, the variation in the LSC region was the greatest, followed by the SSC region, and the IR regions were the least variable (Figure 6). Seven of these loci, that is, matK‐rps16 (0.05856), rps16 (0.05844), petG‐trnW (0.08333), trnW‐trnP (0.08600), psaC (0.06344), psaC‐ndhE (0.06233), and ndhG (0.06011), showed high values (>0.055). Among them, 4 fragments were distributed in the LSC region and 3 in the SSC region (Figure 6).
FIGURE 6.
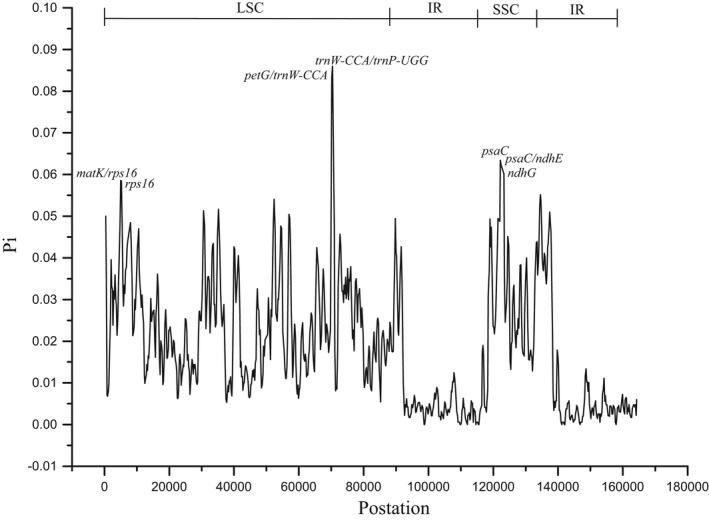
Nucleotide diversity (Pi) values among the six Hamamelidaceae species. X‐axis: the position in the genome; Y‐axis: Pi value. Pi, polymorphism information
3.6. IR contraction and expansion
Figure 7 shows the comparisons of the IR/LSC and IR/SSC boundaries among the chloroplast genomes of the six Hamamelidaceae species. The length of the chloroplast genome of Chunia bucklandioides was the longest (159,814 bp), while that of Rhodoleia championii was the shortest (159,115 bp) among the six species. The genes rps19, ndhF, ycf1, and trnH were located at the LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC boundaries, respectively. rps19 crossed the LSC/IRb boundary, with 2–6 bp within the IRb region, while trnH crossed the IRa/LSC boundary, with 6–30 bp within the IRb region, except in Chunia bucklandioides. NdhF was located away from the IRb/SSC boundary in the SSC regions of Rhodoleia championii, Distylium tsiangii, and Chunia bucklandioides but crossed the IRb/SSC boundary with 2 bp within the IRb region in C. velutina, C. glandulifera, and L. chinense. Notably, ycf1 crossed the SSC/IRa boundary, with 1,000–1,085 bp within the IRa region in all six species.
FIGURE 7.
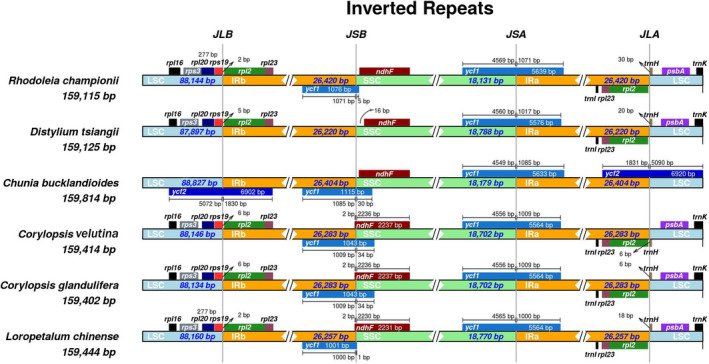
Comparison of the borders of the large single‐copy (LSC), small single‐copy (SSC), and inverted repeat (IR) regions among the six Hamamelidaceae chloroplast genomes. Genes are denoted by colored boxes. The gaps between the genes and the boundaries are indicated by the base lengths (bp)
3.7. Phylogenetic analysis
The chloroplast genome sequences observed provide essential data with which to further elucidate and understand the phylogenetic relationships among Hamamelidaceae species. The two phylogenetic analyses (ML and BI) revealed nearly identical topologies based on the complete chloroplast genomes, LSC regions, and SSC regions (the complete chloroplast genome was completely consistent with the LSC region), and all of the nodes in the phylogenetic trees had high bootstrap support values except Distylium (some Distylium species) and Sycopsis Oliv. (Sycopsis sinensis Oliver) (Figures 8, 9, 10). Hamamelidaceae species gathered on a large branch and species in the same genus were clustered together to a certain degree. The Hamamelidaceae branch was divided into two clades with Chunia and Mytilaria related to other 9 genera. Disanthus was related to other 8 genera in which Corylopsis and Loropetalum were found to be sister to other 6 genera (Sinowilsonia Hemsl., Fortunearia, Sycopsis, Distylium, Parrotia, and Hamamelis). In addition, Corylopsis and Loropetalum were sister genera to each other. However, ML and BI analyses revealed incongruent topologies based on the IR regions. Moreover, some of the nodes had very low bootstrap support values (Figure S3), indicating that the IR regions were not suitable for identification or phylogenetic analysis.
FIGURE 8.
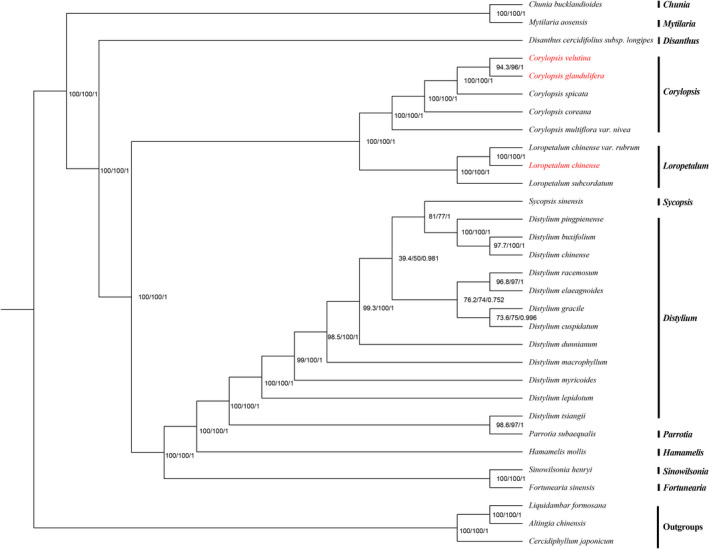
Bayesian inference (BI) and maximum likelihood (ML) phylogenetic trees were constructed using the general time‐reversible (GTR)+F+I+G4 model based on the chloroplast genomes of 31 species. Numbers are support values for ML‐SH‐Alrt, ML‐UFBoot, and BI‐PP (SH‐aLRT/UFBoot/PP). The species investigated in this study are colored in red
FIGURE 9.
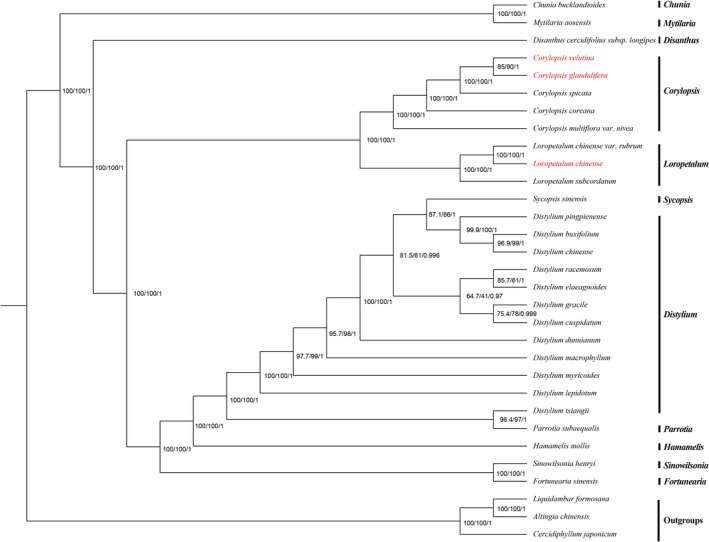
Bayesian inference (BI) and maximum likelihood (ML) phylogenetic trees were constructed using the general time‐reversible (GTR)+F+I+G4 model based on the LSC regions. Numbers on the branches are support values for ML‐SH‐Alrt, ML‐UFBoot, and BI‐PP (SH‐aLRT/UFBoot/PP). The species investigated in this study are colored in red
FIGURE 10.
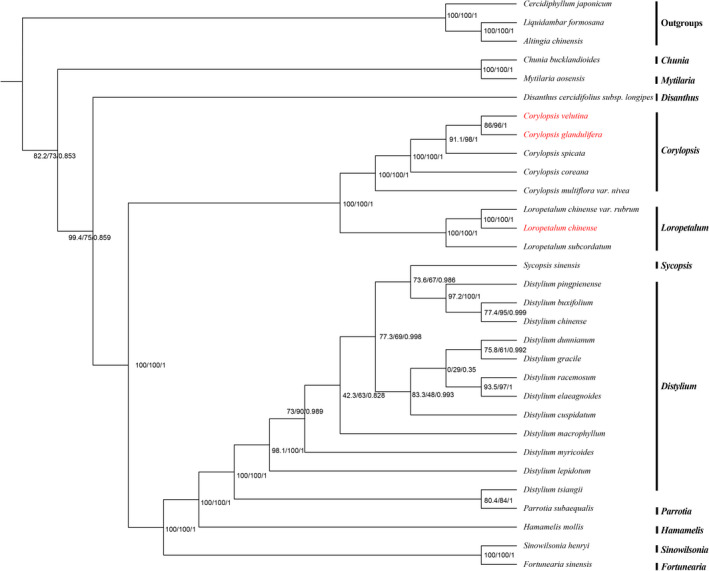
Bayesian inference (BI) and maximum likelihood (ML) phylogenetic trees were constructed using the general time‐reversible (GTR)+F+G4 model based on the SSC regions. Numbers on the branches are support values for ML‐SH‐Alrt, ML‐UFBoot, and BI‐PP (SH‐aLRT/UFBoot/PP). The species investigated in this study are colored in red
4. DISCUSSION
The chloroplast genome provides valuable information for species identification, as well as population genetics, phylogenetic, and genetic engineering studies (Daniell et al., 2016; Luo et al., 2021; Wu et al., 2021). In this study, the complete chloroplast genomes of three Hamamelidaceae species were sequenced using Illumina high‐throughput sequencing technology. The results showed that the three Hamamelidaceae species had classical chloroplast structure (He et al., 2017; Mader et al., 2018; Xu et al., 2017; Yang, Hu, et al., 2018; Yang, Zhao, et al., 2018) and the GC content was lower than the AT content. This was generally the same as seen in other angiosperm chloroplast genomes (Asaf et al., 2018; Raubeson et al., 2007). The results also showed that the GC content in the IR regions was the highest, which may be due to the presence of a large number of rRNA in the IR regions. GC skewness is considered a dominant factor in codon bias. Several studies have indicated that high AT content is the main reason for synonymous codons ending in A/U (Clegg et al., 1994; Shimda & Sugiuro, 1991), which may be related to natural selection and mutation during evolution (Liu et al., 2019). In addition, SSRs are usually composed of a higher number of A + T bases than G + C bases (Hu et al., 2017; Kuang et al., 2011; Simeone et al., 2018; Yang, Hu, et al., 2018; Yang, Zhao, et al., 2018), which is consistent with our observations, and this may also be related to the high AT content in the nucleotide composition.
The lengths of the exons and introns in genes are important information in chloroplast genomes. Genes are interrupted by introns in major groups of organisms (Fan et al., 2021). One‐intron genes vary among species, while clpP, rps12, and ycf3 are two‐intron genes (Wu et al., 2020; Zhang, Gao, et al., 2020; Zhang, Wang, et al., 2020). This finding is consistent with our observations. ClpP protease encoded by the clpP gene widely exists in mitochondria and chloroplasts of prokaryotes and eukaryotes, where it plays a vital role in regulating protein metabolism (Chen et al., 2014; Zhang et al., 2014). The rps12 gene is a trans‐spliced gene with the 5′ end located in the LSC region and duplicated 3′ ends located in the IR regions (Guo et al., 2018). In addition, ycf3 is related to photosynthesis (Boudreau et al., 1997; Naver et al., 2001). Consequently, detecting the clpP and ycf3 genes will contribute to further investigation of the chloroplasts in Hamamelidaceae.
The LSC and SSC regions are usually variable, while expansion and contraction are noted in the highly conserved IR regions (Asaf, 2017), which may be a critical factor underlying the size variation in the chloroplast genomes (Daniell et al., 2016; Kolodner et al., 1976). The difference in the size of the chloroplast genomes among the six Hamamelidaceae species compared in this study was not significant, which could be due to their similar expansion and contraction in the IR regions (such as rps19, ndhF, ycf1, and trnH located at the LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC boundaries, respectively) except C. bucklandioides. The longest chloroplast genome among the six Hamamelidaceae species was observed in C. bucklandioides, which may be associated with the size expansion of ycf2 in the IR regions. Expansion or contraction of the IR regions in these species is supposed to be related to gene retention or loss, and we suggest that gene‐loss events would have occurred during the evolution of this family and differentiation of the species.
The nucleotide diversity analysis also demonstrated that the IR regions contained fewer variable loci than the SC regions (LSC and SSC regions). Moreover, genes with Pi values > 0.055 were mainly located in the SC regions. Chloroplast genomes have a copy‐dependent repair mechanism to ensure consistency and stability of the two IR regions in sequence, which enhances the stability and conservation of the genome (Khakhlova & Bock, 2006; Perry & Wolfe, 2002). This could explain why the IR regions contain less sequence divergence than the LSC or SSC regions (Shaw et al., 2007). None of the intron‐containing genes (ndhA, ndhB, petB, petD, atpF, rpl16, rpl2, rpoC1, trnA‐UGC, trnG‐GCC, trnG‐UCC, trnI‐GAU, trnK‐UUU, trnL‐UAA, trnV‐UAC, trnE‐UUC, rps12, clpP, and ycf3) had a Pi value >0.055, except rps16, suggesting that intron‐containing genes are more highly conserved than exon‐containing genes only in the chloroplast genome. In other words, higher variability was found in exon‐containing genes, which provides more valuable information for species evolution.
The mVISTA analysis also showed that the variability in the IR regions was less than that in the LSC and SSC regions and that more variable sites were located in noncoding regions than in coding regions, which could be utilized for the development of new molecular markers for species identification and taxonomic studies in Hamamelidaceae. These variations were observed in the ndhF, accD, and ycf1 genes and intergenic regions, such as trnH‐psbA, rps16‐trnQ, atpH‐atpI, petN‐psbM, trnT‐psbD, psbZ‐trnG, ndhC‐trnV, accD‐psaI, petA‐psbJ, rps18‐rpl20, rps15‐ycf1, and trnL‐ndhB. Among these divergence hotspot regions, trnH‐psbA has already been selected as a suitable barcode for plants (Hollingsworth et al., 2009; Yang et al., 2017), as well as rbcL‐accD (Shaw et al., 2014), ndhF (Chen et al.,2021; Yang et al., 2017; Yang, Hu, et al., 2018; Yang, Zhao, et al., 2018), ycf1 (Dong et al., 2015), accD (Li et al., 2018), rps16 (Chen et al.,2021), rps16‐trnQ (Liu et al., 2016), and petA‐psbJ (Katarzyna et al., 2018; Liu et al., 2016; Wang, 2010). Further research is necessary to determine whether the remaining divergence hotspot regions could be used as candidate DNA barcodes or to assess the taxonomic evolution and phylogenetics of Hamamelidaceae.
Chloroplast genome data are valuable for analyzing species definitions because organelle‐based “barcodes” can be established for some species and then applied to reveal the phylogenetic relationships among species (Fan et al., 2021; Yang et al., 2013). Moreover, with the continuous development of next‐generation sequencing technology, particularly the application of second‐generation sequencing technology, chloroplast genome sequencing has become simpler. Thus, more studies have used complete chloroplast genome sequences to evaluate the phylogenetic relationships among angiosperms. In this study, almost all published complete chloroplast genome sequence data of Hamamelidaceae were used to construct the phylogenetic relationships by ML and BI analyses. The two phylogenetic analyses (ML and BI) revealed congruent topologies based on the complete chloroplast genomes and LSC regions, while the results of the SSC regions were slightly different in the two datasets. This may be because the LSC region accounts for a large part of the complete chloroplast genome and varies highly. However, the IR region was not suitable for identification or the phylogenetic analysis possibly because it is highly conserved. The outgroups, Altingia chinensis, Liquidambar formosana, and Cercidiphyllum japonicum, clustered into a monophyletic clade and were sister to Hamamelidaceae. According to previous molecular studies on Saxifragales, Altingiaceae (Altingia, Liquidambar), and Cercidiphyllaceae (Cercidiphyllum), Hamamelidaceae originated successively in the evolutionary history of angiosperms, and the three groups are paraphyletic (Dong et al., 2013, 2018; Soltis et al., 2013; Tarullo et al., 2021; Xiang et al., 2019). Alternatively, a different relationship of these paraphyletic groups was inferred from the morphological and molecular data, with an earlier divergence time for Cercidiphyllum than for Liquidambar (Magallon, 2007). Our results are slightly different from previous studies, possibly due to sample limitations. Nevertheless, we still support the establishment of Altingiaceae (APG IV, 2016). The results show that the Hamamelidaceae species investigated in this study were divided into two clades and the species in the same genus were clustered together to a certain degree. Among them, Chunia bucklandioides and Mytilaria laosensis (Xiang et al., 2019) were the first to diverge in Hamamelidaceae, indicating the relatively high genetic divergence between these two species and others, followed by Disanthus cercidifolius subsp. longipes, which were early‐diverging taxa in Hamamelidaceae. Interestingly, these three genera are monotypic. Corylopsis and Loropetalum formed a monophyletic group, while Fortunearia was closely related to Sinowilsonia and they are monotypic genera endemic to China (Chen et al., 2020; Jiang et al., 2020; Ye et al., 2020). The sister relationships of the three clades in Hamamelidaceae, such as Chunia + Mytilaria, Disanthus, and Corylopsis + Loropetalum, are consistent with previous reports (Bobrov et al., 2020; Tarullo et al., 2021; Xiang et al., 2019), while Distylium is not monophyletic. Moreover, some Distylium support values in the phylogenetic trees were low, particularly in the ML analysis. Although Dong et al. (2021) conducted a phylogenetic analysis on Distylium species, the support values were not high, possibly due to the close affinity within the Distylium genus and therefore the classification or circumscription would be difficult within Distylium. There are still unsolved enigmas in the phylogeny of Hamamelidaceae. This group is disjunctly distributed across Western, Southern, Eastern, and Southeast Asia; North, Central, and South America; Eastern Africa; and Northeastern Australia (Bobrov et al., 2020; Tarullo et al., 2021). The diversity in Hamamelidaceae is not fully understood, and extinct and extant new species are still being reported (Averyanov et al., 2017; Haynes et al., 2020; Huang et al., 2017). Therefore, the morphological and molecular evidence may not be complete due to sampling difficulties. Conversely, the unresolved mysteries in Hamamelidaceae may lead to more follow‐up studies. To fully understand the phylogeny of Hamamelidaceae species, studies on more genera and more genes need to be conducted in the future. Nevertheless, the phylogenetic trees constructed in this study provide a valuable resource for investigating the classification, phylogeny, and evolutionary history of Hamamelidaceae.
5. CONCLUSION
In this study, the complete chloroplast genomes of three Hamamelidaceae species were determined and the basic structures, conservation, and variability in these sequences were revealed. The IR regions were more conserved than the LSC or SSC region, while the noncoding regions contained more variability than the gene coding regions. SSRs and divergent hotspot regions could be used to develop molecular markers for population genetic and phylogenetic studies. The complete chloroplast genomes, LSC regions, and SSC regions were used to establish good phylogenetic relationships and solve the relationships between and within genera, while the IR region was not suitable for identification or phylogenetic analysis. Notably, the relationship within the genus Distylium has not been well resolved. More studies on the relationship within this genus are needed to fully understand the phylogeny of Hamamelidaceae species. The results of this study provide a valuable reference for further studies on species identification, determination of evolutionary relationships, and the development of genetic resources within Hamamelidaceae.
CONFLICT OF INTEREST
The authors declare no conflicts of interest regarding publication of this paper.
AUTHOR CONTRIBUTIONS
NingJie Wang: Conceptualization (lead); Data curation (lead); Methodology (equal); Resources (lead); Software (lead); Visualization (lead); Writing – original draft (lead). ShuiFei Chen: Conceptualization (equal); Data curation (equal); Funding acquisition (equal); Writing – original draft (equal). Lei Xie: Conceptualization (equal); Writing – review & editing (equal). Lu Wang: Methodology (equal); Resources (equal). YueYao Feng: Resources (equal). Ting Lv: Formal analysis (equal); Visualization (equal). YanMing Fang: Funding acquisition (lead); Writing – review & editing (lead). Hui Ding: Funding acquisition (equal); Writing – review & editing (equal).
Supporting information
Appendix S1
ACKNOWLEDGMENTS
This research was supported by the Major biodiversity conservation project of the Ministry of Ecology and Environment of China (ZDGC2019‐007‐04), the China Postdoctoral Science Foundation (2020M681629), and the Jiangsu Postdoctoral Research Funding Program (2021K038A). The authors would like to thank TopEdit (www.topeditsci.com) for its linguistic assistance during the preparation of this manuscript.
Wang, N. , Chen, S. , Xie, L. , Wang, L. , Feng, Y. , Lv, T. , Fang, Y. , & Ding, H. (2022). The complete chloroplast genomes of three Hamamelidaceae species: Comparative and phylogenetic analyses. Ecology and Evolution, 12, e8637. 10.1002/ece3.8637
DATA AVAILABILITY STATEMENT
The original sequencing data have been submitted to the NCBI database and received GenBank accession numbers MZ642354 (C. glandulifera), MZ823391 (C. velutina), and MZ642355 (L. chinense). The data used to support the findings of this study are included in Appendix S1.
REFERENCES
- Acemel, R. D. , Tena, J. J. , Irastorza‐Azcarate, I. , Marlétaz, F. , Gómez‐Marín, C. , de la Calle‐Mustienes, E. , Bertrand, S. , Diaz, S. G. , Aldea, D. , Aury, J.‐M. , Mangenot, S. , Holland, P. W. H. , Devos, D. P. , Maeso, I. , Escrivá, H. , & Gómez‐Skarmeta, J. L. (2016). A single three‐dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nature Genetics, 48, 336–341. 10.1038/ng.3497 [DOI] [PubMed] [Google Scholar]
- Ali, A. , Jaakko, H. , & Peter, P. (2018). IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics, 34, 3030–3031. 10.1093/bioinformatics/bty220 [DOI] [PubMed] [Google Scholar]
- Ammiraju, J. S. S. , Zuccolo, A. , Yu, Y. , Song, X. , Piegu, B. , Chevalier, F. , Walling, J. G. , Ma, J. , Talag, J. , Brar, D. S. , SanMiguel, P. J. , Jiang, N. , Jackson, S. A. , Panaud, O. , & Wing, R. A. (2007). Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. The Plant Journal, 52(2), 342–351. 10.1111/j.1365-313X.2007.03242.x [DOI] [PubMed] [Google Scholar]
- APG IV (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society, 181, 1–20. 10.1111/boj.12385 [DOI] [Google Scholar]
- Asaf, S. , Khan, A. L. , Khan, M. A. , Shahzad, R. , Lubna , Kang, S. M. , Al‐Harrasi, A. , Al‐Rawahi, A. , & Lee, I.‐J. (2018). Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinus taeda L.) with related species. PLoS One, 13(3), e0192966. 10.1371/journal.pone.0192966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaf, S. , Khan, A. L. , Khan, M. A. , Waqas, M. , Kang, S.‐M. , Yun, B.‐W. , & Lee, I.‐J. (2017). Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: Structures and comparative analysis. Scientific Reports, 7, 7556. 10.1038/s41598-017-07891-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averyanov, L. V. , Endress, P. K. , Quang, B. H. , Nguyen, K. S. , & Nguyen, D. V. (2017). Disanthus ovatifolius (Hamamelidaceae), a new species from northwestern Vietnam. Phytotaxa, 308(1), 104–110. 10.11646/phytotaxa.308.1.9 [DOI] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulikov, A. S. , Lesin, V. M. , Nikolenko, S. I. , Pham, S. , Prjibelski, A. D. , Pyshkin, A. V. , Sirotkin, A. V. , Vyahhi, N. , Tesler, G. , Alekseyev, M. A. , & Pevzner, P. A. (2012). SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19, 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucom, R. S. , Estill, J. C. , Leebens‐Mack, J. , & Bennetzen, J. L. (2009). Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Research, 19(2), 243–254. 10.1101/gr.083360.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscotti, M. A. , Olmo, E. , & Heslop‐Harrison, J. S. (2015). Repetitive DNA in eukaryotic genomes. Chromosome Research, 23(3), 415–420. 10.1007/s10577-015-9499-z [DOI] [PubMed] [Google Scholar]
- Bobrov, A. V. , Roslov, M. S. , & Romanov, M. S. (2020). Phylogenetic biogeography of Hamamelidaceae s. l. based on molecular data. Vestnik of Saint Petersburg University. Earth Sciences, 65(2), 224–244. 10.21638/spbu07.2020.201 [DOI] [Google Scholar]
- Boetzer, M. , & Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biology, 13, R56. 10.1186/gb-2012-13-6-r56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogle, A. L. , & Philbrick, C. T. (1980). A generic atlas of Hamamelidaceous pollens. Contributions from the Gray Herbarium, 210, 29–103. [Google Scholar]
- Bondar, E. I. , Putintseva, Y. A. , Oreshkova, N. V. , & Krutovsky, K. V. (2019). Siberian larch (Larix sibirica Ledeb.) chloroplast genome and development of polymorphic chloroplast markers. BMC Bioinformatics, 20, 38. 10.1186/s12859-018-2571-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau, E. , Takahashi, Y. , Lemieux, C. , Turmel, M. , & Rochaix, J. D. (1997). The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO Journal, 16(20), 6095–6104. 10.1093/emboj/16.20.6095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavender, B. J. , González, R. A. , Eaton, D. A. R. , Hipp, A. A. L. , Beulke, A. , & Manos, P. S. (2015). Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Molecular Ecology, 24, 3668–3687. 10.1111/mec.13269 [DOI] [PubMed] [Google Scholar]
- Chen, L. , Lan, L. H. , He, H. D. , Chen, Z. B. , She, S. Q. , Liu, Y. Z. , & Lü, B. (2014). Progress in the studies of ClpP: from bacteria to human mitochondria. Chinese Journal of Cell Biology, 36(6), 717–725. [Google Scholar]
- Chen, S. F. , Li, Y. , Wang, L. , Xie, L. , Ge, X. M. , Xu, Z. , Hu, Y. P. , Zhang, W. W. , Fang, M. Y. , & Ding, H. (2020). The complete chloroplast genome sequence of Distylium macrophyllum (Hamamelidaceae). Mitochondrial DNA Part B, 5(1), 701–702. 10.1080/23802359.2019.1703606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. J. , Nie, X. H. , Ai, W. S. , Meng, Y. , Xiao, Y. B. , Zhou, C. R. , Peng, Y. S. , Wang, Q. M. , & Peng, G. P. (2021). Species clustering and salt tolerance identification of bambusoideae by chloroplast DNA barcoding. Journal of Hunan Agricultural University (Natural Sciences), 47. https://kns.cnki.net/kcms/detail/43.1257.S.20210610.1045.004.html [Google Scholar]
- Cheng, Y. , Zhang, L. , Qi, J. , & Zhang, L. (2020). Complete chloroplast genome sequence of Hibiscus cannabinus and comparative analysis of the Malvaceae Family. Front Genet, 11, 227. 10.3389/fgene.2020.00227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, X. F. , Zhang, F. Q. , Dong, Q. , & Chen, S. L. (2020). Insights into comparative genomics, codon usage bias, and phylogenetic relationship of species from Biebersteiniaceae and Nitrariaceae based on complete chloroplast genomes. Plants, 9(11), 1605. 10.3390/plants9111605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, M. S. , Yang, J. Y. , Yang, T. J. , & Kim, S. C. (2019). Evolutionary comparison of the chloroplast genome in the Woody Sonchus Alliance (Asteraceae) on the Canary Islands. Genes, 10(3), 217. 10.3390/genes10030217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K. S. , & Park, S. (2015). The complete chloroplast genome sequence of Aster spathulifolius (Asteraceae); genomic features and relationship with Asteraceae. Gene, 572(2), 214–221. 10.1016/j.gene.2015.07.020 [DOI] [PubMed] [Google Scholar]
- Clegg, M. T. , Gaut, B. S. , Learn, G. H. , & Morton, B. R. (1994). Rates and patterns of chloroplast DNA evolution. Proceedings of the National Academy of Sciences of the United States of America, 91, 6795–6801. 10.1073/pnas.91.15.6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniell, H. , Lin, C. S. , Ming, Y. , & Chang, W. J. (2016). Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biology, 17(1), 134. 10.1186/s13059-016-1004-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashnow, H. , Tan, S. , Das, D. , Easteal, S. , & Oshlack, A. (2015). Genotyping microsatellites in next‐generation sequencingdata. BMC Bioinformatics, 16(S2). 10.1186/1471-2105-16-s2-a5 [DOI] [Google Scholar]
- Deng, M. B. , Wei, H. T. , & Wang, X. Q. (1992). Shaniodendron, a New Genus of Hamamelidaceae from China. Acta Phytotaxonomica Sinica, 30(1), 57–61. http://www.plantsystematics.com/CN/Y1992/V30/I1/57 [Google Scholar]
- Dong, W. , Liu, Y. , Xu, C. , Gao, Y. , Yuan, Q. , Suo, Z. , Zhang, Z. , & Sun, J. (2021). Chloroplast phylogenomic insights into the evolution of Distylium (Hamamelidaceae). BMC Genomics, 22, 293. 10.1186/s12864-021-07590-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, W. P. , Xu, C. , Cheng, T. , Lin, K. , & Zhou, S. L. (2013). Sequencing angiosperm plastid genomesmade easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biology and Evolution, 5(5), 989–997. 10.1093/gbe/evt063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, W. , Xu, C. , Li, C. , Sun, J. , Zuo, Y. , Shi, S. , Cheng, T. , Guo, J. , & Zhou, S. (2015). ycf1, the most promising plastid DNA barcode of land plants. Scientific Reports, 5, 8348. 10.1038/srep08348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, W. P. , Xu, C. , Wu, P. , Cheng, T. , Yu, J. , Zhou, S. L. , & Hong, D. Y. (2018). Resolving the systematic positions of enigmatic taxa: Manipulating the chloroplast genome data of Saxifragales. Molecular Phylogenetics and Evolution, 126, 321–330. 10.1016/j.ympev.2018.04.033 [DOI] [PubMed] [Google Scholar]
- Douglas, S. E. (1994). Chloroplast origins and evolution. In Bryant D. A. (Ed.), The Molecular biology of cyanobacteria. Advances in photosynthesis (pp. 91–118). Springer, 10.1007/978-94-011-0227-8_5 [DOI] [Google Scholar]
- Eddy, S. R. (2008). A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Computational Biology, 4(5), e1000069. 10.1371/journal.pcbi.1000069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endress, P. K. (1969). Molinadendron, eine neue Hamamelidaceae‐Gattung aus Zentralamerika. Botanische Jahrbücher Für Systematik, 89, 353–358. [Google Scholar]
- Endress, P. K. (1989). A suprageneric taxonomic classification of the Hamamelidaceae. Taxon, 38(3), 371–376. [Google Scholar]
- Endress, P. K. (1993). Hamamelidaceae. In Kubitzki K., Rohwer J. G., & Bittrich V. (Eds.), The families and genera of vascular plants (Vol. II, pp. 322–331). Spring‐Verlag. [Google Scholar]
- Fan, R. , Ma, W. , Liu, S. , & Huang, Q. (2021). Integrated analysis of three newly sequenced fern chloroplast genomes: Genome structure and comparative analysis. Ecology and Evolution, 11, 4550–4563. 10.1002/ece3.7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarini, M. , Moretto, M. , Ward, J. A. , Šurbanovski, N. , Stevanović, V. , Giongo, L. , Viola, R. , Cavalieri, D. , Velasco, R. , Cestaro, A. , & Sargent, D. J. (2013). An evaluation of the PacBio RS platform for sequencing and de novo assemblyof a chloroplast genome. BMC Genomics, 14(670). 10.1186/1471-2164-14-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer, K. A. , Pachter, L. , Poliakov, A. , Rubin, E. M. , & Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Research, 32, 273–279. 10.1093/nar/gkh458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J. F. , Lefort, V. , Anisimova, M. , Hordijk, W. , & Gascuel, O. (2010). New algorithms and methods to estimate maximum‐likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biology, 59, 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Guo, S. , Guo, L. , Zhao, W. , Xu, J. , Li, Y. , Zhang, X. , Shen, X. , Wu, M. , & Hou, X. (2018). Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii . Molecules, 23(2), 246. 10.3390/molecules23020246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, R. M. , & Wei, H. T. (1998). A new combination of Hamamelidaceae. Acta Phytotaxonomica Sinica, 36, 80. [Google Scholar]
- Harms, H. (1930). Hamamelidaceae. In Engler A., & Prantl K. (Eds.), Die Naturlichen Pflanzenfamilien (Vol. 18a, pp. 303–345). Verlag von Wilhelm Engelmann. [Google Scholar]
- Haynes, J. E. , Phillips, W. D. , Krings, A. , Lynch, N. P. , & Ranney, T. G. (2020). Revision of Fothergilla (Hamamelidaceae), including resurrection of F. parvifolia and a new species, F. milleri . Phytokeys, 144, 57–80. 10.3897/phytokeys.144.49589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, L. , Qian, J. , Li, X. , Sun, Z. , Xu, X. , & Chen, S. (2017). Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules, 22(2), 49. 10.3390/molecules22020249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth, P. M. , Forrest, L. L. , Spouge, J. L. , Hajibabaei, M. , Ratnasingham, S. , van der Bank, M. , Chase, M. W. , Cowan, R. S. , Erickson, D. L. , Fazekas, A. J. , Graham, S. W. , James, K. E. , Kim, K.‐J. , Kress, W. J. , Schneider, H. , van AlphenStahl, J. , Barrett, S. C. , van den Berg, C. , Bogarin, D. , … Little, D. P. (2009). A DNA barcode for land plants. Proceedings of the National Academy of Sciences of the United States of America, 106(31), 12794–12797. 10.1073/pnas.0905845106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y. , Woeste, K. E. , & Zhao, P. (2017). Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Frontiers in Plant Science, 7, 1939–1955. 10.3389/fpls.2016.01955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Shi, G. L. , Su, T. , & Zhou, Z. K. (2017). Miocene Exbucklandia (Hamamelidaceae) from Yunnan, China and its biogeographic and palaeoecologic implications. Review of Palaeobotany and Palynology, 244, 96–106. 10.1016/j.revpalbo.2016.10.004 [DOI] [Google Scholar]
- Huang, Y. , Yin, Q. , Van, T. D. , Meng, K. , Chen, S. , Liao, B. , & Fan, Q. (2019). Development and characterization of genomic microsatellite markers in the tree species, Rhodoleia championii, R. parvipetala, and R. forrestii (Hamamelidaceae). Molecular Biology Reports, 46, 6547–6556. 10.1007/s11033-019-05106-w [DOI] [PubMed] [Google Scholar]
- Jiang, M. , Wang, J. F. , & Zhang, H. J. (2020). Characterization and phylogenetic analysis of the complete chloroplast genome sequence of Disanthus cercidifolius subsp. longipes (Hamamelidaceae), a rare and endangered wild plant species in China. Mitochondrial DNA Part B, 5(2), 1206–1207. 10.1080/23802359.2020.1731372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd, W. S. , Campbell, C. S. , Kellogg, E. A. , Stevens, P. F. , & Donoghue, M. J. (2007). Plant systematics: A phylogenetic approach. (Vol. 2, pp. 343–344). Sinauer Associates. 10.1111/j.1096-0031.2008.00212.x [DOI] [Google Scholar]
- Kalyaanamoorthy, S. , Minh, B. Q. , Wong, T. K. F. , von Haeseler, A. , & Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nature Methods, 14, 587–589. 10.1038/nmeth.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katarzyna, K. , Marcin, N. , Kamil, M. , Ewelina, K. , & Jakub, S. (2018). Plastid super‐barcodes as a tool for species discrimination in feather grasses (Poaceae: Stipa). Scientific Reports, 8, 1924. 10.1038/s41598-018-20399-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakhlova, O. , & Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. The Plant Journal, 46, 85–94. 10.1111/j.1365-313X.2006.02673.x [DOI] [PubMed] [Google Scholar]
- Kim, M. K. , Lee, W. K. , Choi, Y. R. , Kim, J. , Kang, I. , & Kang, J. (2021). The complete chloroplast genome sequence of three medicinal species; Curcuma longa, Curcuma wenyujin, and Curcuma phaeocaulis (Zingiberaceae). Mitochondrial DNA Part B, 6(4), 1363–1364. 10.1080/23802359.2020.1768917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. , Boo, H. O. , Ahn, T. H. , & Bae, C. S. (2020). Protective effects of Erythronium japonicum and Corylopsis coreana Uyeki extracts against 1,3‐dichloro‐2‐propanol‐induced hepatotoxicity in rats. Applied Microscopy, 50(1), 29. 10.1186/s42649-020-00049-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode, V. , Mudd, E. A. , Iamtham, S. , & Day, A. (2005). The tobacco plastid accD gene is essential and is required for leafdevelopment. The Plant Journal, 44, 237–244. 10.1111/j.1365-313X.2005.02533.x [DOI] [PubMed] [Google Scholar]
- Kolodner, R. , Tewari, K. K. , & Warner, R. C. (1976). Physical studieson the size and structure of the covalently closed circularchloroplast DNA from higher plants. Biochimica Et Biophysica Acta (BBA)—Nucleic Acids and Protein Synthesis, 447(2), 144–155. [DOI] [PubMed] [Google Scholar]
- Kuang, D.‐Y. , Hong, W. , Wang, Y.‐L. , Gao, L.‐M. , Zhang, S.‐Z. , & Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome, 54(8), 663–673. 10.1139/g11-026 [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology & Evolution, 33(7), 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett, D. , & Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Research, 32, 11–16. 10.1093/nar/gkh152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. , Zhao, Y. , Xu, Z. , Yang, G. , Peng, J. , & Peng, X. (2020). Initial characterization of the chloroplast genome of Vicia sepium, an important wild resource plant, and related inferences about its evolution. Front Genet, 11, 73. 10.3389/fgene.2020.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. M. , Zhao, C. Y. , & Liu, X. F. (2019). Complete chloroplast genome sequences of Kaempferia Galanga and Kaempferia Elegans: Molecular structures and comparative analysis. Molecules, 24(3), 474. 10.3390/molecules24030474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. H. , Bogle, A. L. , & Donoghue, M. J. (1999). Phylogenetic relationships in the Hamamelidoideae inferred from sequences of trn non‐coding regions of chloroplast DNA. Harvard Papers in Botany, 4, 343–356. https://www.jstor.org/stable/41761318 [Google Scholar]
- Li, J. H. , Bogle, A. L. , & Klein, A. S. (1999). Phylogenetic relationships of the Hamamelidaceae inferred from sequences of internal transcribed spacers (ITS) of nuclear ribosomal DNA. American Journal of Botany, 86, 1027–1037. 10.2307/2656620 [DOI] [PubMed] [Google Scholar]
- Li, J. H. , Bogle, A. L. , Klein, A. S. , & Donoghue, M. J. (2000). Phylogeny and biogeography of Hamamelis (Hamamelidaceae). Harvard Papers in Botany, 5, 171–178. https://www.jstor.org/stable/41761602 [Google Scholar]
- Li, J. H. , Bogle, A. L. , Klein, A. S. , & Pan, K. Y. (1997). Close relationship between Shaniodendron and Parrotia (Hamamelidaceae), evidence from ITS sequences of nuclear ribosomal DNA. Acta Phytotaxonomica Sinica, 35, 481–493. [Google Scholar]
- Li, J. , Su, Y. , & Wang, T. (2018). The repeat sequences and elevated substitution rates of the chloroplast accD gene in Cupressophytes. Frontiers in Plant Science, 9, 533. 10.3389/fpls.2018.00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- Lin, M. , Qi, X. , Chen, J. , Sun, L. , Zhong, Y. , Fang, J. , & Hu, C. (2018). The complete chloroplast genome sequence of Actinidia arguta using the PacBio RSII platform. PLoS One, 13, e0197393. 10.1371/journal.pone.0197393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Chang, E. M. , Liu, J. F. , Huang, Y. N. , Wang, Y. , Yao, N. , & Jiang, Z. P. (2019). Complete chloroplast genome sequence and phylogenetic analysis of Quercus bawanglingensis Huang, Li et Xing, a vulnerable oak tree in China. Forests, 10(7), 587. 10.3390/f10070587 [DOI] [Google Scholar]
- Liu, Y. C. , Lin, B. Y. , Lin, J. Y. , Wu, W. L. , & Chang, C. C. (2016). Evaluation of chloroplast DNA markers for intraspecific identification of Phalaenopsis equestris cultivars. Scientia Horticulturae, 203, 86–94. 10.1016/j.scienta.2016.03.021 [DOI] [Google Scholar]
- Lohse, M. , Drechsel, O. , & Bock, R. (2007). Organellar Genome DRAW (OGDRAW): A tool for the easy generation ofhigh‐quality custom graphical maps of plastid and mitochondrial genomes. Current Genetics, 52, 267–274. 10.1007/s00294-007-0161-y [DOI] [PubMed] [Google Scholar]
- Luo, C. , Huang, W. , Sun, H. , Yer, H. , Li, X. , Li, Y. , Yan, B. O. , Wang, Q. , Wen, Y. , Huang, M. , & Huang, H. (2021). Comparative chloroplast genome analysisof Impatiens species (Balsaminaceae) in the karst area of China: Insights into genomeevolution and phylogenomic implications. BMC Genomics, 22, 571. 10.1186/s12864-021-07807-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader, M. , Pakull, B. , Blanc‐Jolivet, C. , Paulini‐Drewes, M. , Bouda, Z. , Degen, B. , Small, I. , & Kersten, B. (2018). Complete chloroplast genome sequences of four Meliaceae species and comparative analyses. International Journal of Molecular Sciences, 19(3), 701. 10.3390/ijms19030701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magallon, S. (2007). From Fossils to Molecules: Phylogeny and the core eudicot floral groundplan in Hamamelidoideae (Hamamelidaceae, Saxifragales). Systematic Botany, 32(2), 317–347. 10.1600/036364407781179617 [DOI] [Google Scholar]
- Manchester, S. R. , Chen, Z. D. , Lu, A. M. , & Uemura, K. (2009). Eastern Asian endemic seed plant genera and their paleogeographical history throughout the Northern Hemisphere. Journal of Systematics and Evolution, 47, 1–42. 10.1111/j.1759-6831.2009.00001.x [DOI] [Google Scholar]
- Mayor, C. , Brudno, M. , Schwartz, J. R. , Poliakov, A. , Rubin, E. M. , Frazer, K. A. , Pachter, L. S. , & Dubchak, I. (2000). VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics, 16, 1046–1047. 10.1093/bioinformatics/16.11.1046 [DOI] [PubMed] [Google Scholar]
- McGinnis, S. , & Madden, T. L. (2004). BLAST: At the core of a powerful and diverse set ofsequence analysis tools. Nucleic Acids Research, 32, 20–25. 10.1093/nar/gkh435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh, B. Q. , Nguyen, M. A. , & von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Molecular Biology and Evolution, 30, 1188–1195. 10.1093/molbev/mst024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miri, S. M. , Mousavi, A. , Naghavi, M. R. , & Naserian Khiabani, B. (2014). Molecular analysis of Musa mutants resistant to salinity using microsatellite markers. Trakia Journal of Sciences, 12, 114–120. [Google Scholar]
- Morgante, M. , De Paoli, E. , & Radovic, S. (2007). Transposable elements and the plant pan‐genomes. Current Opinion in Plant Biology, 10(2), 149–155. 10.1016/j.pbi.2007.02.001 [DOI] [PubMed] [Google Scholar]
- Naver, H. , Boudreau, E. , & Rochaix, J. D. (2001). Functional studies of Ycf3: Its role in assembly of photosystem I and interactions with some of its subunits. The Plant Cell, 13, 2731–2745. 10.1105/tpc.010253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L. T. , Schmidt, H. A. , von Haeseler, A. , & Minh, B. Q. (2015). IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nock, C. J. , Baten, A. , & King, G. J. (2014). Complete chloroplast genome of Macadamia integrifolia confirms the position of the Gondwanan early‐ diverging eudicot family Proteaceae. BMC Genomics, 15, S13. 10.1186/1471-2164-15-S9-S13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira, E. J. , Pádua, J. G. , Zucchi, M. I. , Vencovsky, R. , & Vieira, M. L. C. (2006). Origin, evolution and genome distribution of microsatellites. Genetics and Molecular Biology, 29, 294–307. 10.1590/S1415-47572006000200018 [DOI] [Google Scholar]
- Paterson, A. H. , Bowers, J. E. , Bruggmann, R. , Dubchak, I. , Grimwood, J. , Gundlach, H. , Haberer, G. , Hellsten, U. , Mitros, T. , Poliakov, A. , Schmutz, J. , Spannagl, M. , Tang, H. , Wang, X. , Wicker, T. , Bharti, A. K. , Chapman, J. , Feltus, F. A. , Gowik, U. , … Rokhsar, D. S. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature, 457(7229), 551–556. 10.1038/nature07723 [DOI] [PubMed] [Google Scholar]
- Perry, A. S. , & Wolfe, K. H. (2002). Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. Journal of Molecular Evolution, 55, 501–508. 10.1007/PL00020998 [DOI] [PubMed] [Google Scholar]
- Qin, G. M. , Hao, J. , Yang, J. C. , Li, R. S. , & Yin, G. T. (2019). Branch occlusion and discoloration under the natural pruning of Mytilaria laosensis . Forests, 10, 892. 10.3390/f10100892 [DOI] [Google Scholar]
- Raubeson, L. A. , Peery, R. , Chumley, T. W. , Dziubek, C. , Fourcade, H. M. , Boore, J. L. , & Jansen, R. K. (2007). Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nupharadvena and Ranunculus macranthus . BMC Genomics, 8, 174. 10.1186/1471-2164-8-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinsch, A. (1890). Uber di e anatomischen verhaltnisse der Hamamelidaceae mit Rucksicht auf ihre systematische Gruppierung. Engler's Bot Jahrb, 2, 347–395. [Google Scholar]
- Ronquist, F. , Teslenko, M. , van der Mark, P. , Ayres, D. L. , Darling, A. , Höhna, S. , Larget, B. , Liu, L. , Suchard, M. A. , & Huelsenbeck, J. P. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61(3), 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanMiguel, P. , Gaut, B. S. , Tikhonov, A. , Nakajima, Y. , & Bennetzen, J. L. (2009). The paleontology of intergene retrotransposons of maize. Nature Genetics, 20(1), 43–45. 10.1038/1695 [DOI] [PubMed] [Google Scholar]
- Schnable, P. S. , Ware, D. , Fulton, R. S. , Stein, J. C. , Wei, F. , Pasternak, S. , Liang, C. , Zhang, J. , Fulton, L. , Graves, T. A. , Minx, P. , Reily, A. D. , Courtney, L. , Kruchowski, S. S. , Tomlinson, C. , Strong, C. , Delehaunty, K. , Fronick, C. , Courtney, B. , … Wilson, R. K. (2009). The B73 maize genome: complexity, diversity, and dynamics. Ssience, 326(5956), 1112–1115. 10.1126/science.1178534 [DOI] [PubMed] [Google Scholar]
- Shaw, J. , Lickey, E. B. , Schilling, E. E. , & Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. American Journal of Botany, 94, 275–288. 10.3732/ajb.94.3.275 [DOI] [PubMed] [Google Scholar]
- Shaw, J. , Shafer, H. L. , Leonard, O. R. , Kovach, M. J. , Schorr, M. , & Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. American Journal of Botany, 101, 1987–2004. 10.3732/ajb.1400398 [DOI] [PubMed] [Google Scholar]
- Shi, S. H. , Chang, H. T. , Chen, Y. Q. , Qu, L. H. , & Wen, J. (1998). Phylogeny of the Hamamelidaceae based on the ITS sequences of nuclear ribosomal DNA. Biochemical Systematics and Ecology, 26, 55–69. 10.1016/S0305-1978(97)00075-6 [DOI] [Google Scholar]
- Shimda, H. , & Sugiuro, M. (1991). Fine structural features of the chloroplast genome: Comparison of the sequenced chloroplast genomes. Nucleic Acids Research, 19, 983–995. 10.1093/nar/19.5.983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone, M. C. , Cardoni, S. , Piredda, R. , Imperatori, F. , Avishai, M. , Grimm, G. W. , & Denk, T. (2018). Comparative systematics and phylogeography of Quercus Section Cerris in western Eurasia: Inferences from plastid andnuclear DNA variation. PeerJ, 6, e5793. 10.7717/peerj.5793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, S. , Leon, D. , Dietmar, R. K. , Florian, C. S. , & Meinhard, S. (2021). The Microbiome of the Medicinal Plants Achillea millefolium L. and Hamamelis virginiana L. Frontiers in Microbiology, 12, 696398. 10.3389/fmicb.2021.696398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis, D. E. , Mort, M. E. , Latvis, M. , Mavrodiev, E. V. , O'Meara, B. C. , Soltis, P. S. , Burleigh, J. G. , & de Casas, R. R. (2013). Phylogenetic relationships and character evolution analysis of Saxifragales using a supermatrix approach. American Journal of Botany, 100, 916–929. 10.3732/ajb.1300044 [DOI] [PubMed] [Google Scholar]
- Sun, Y. X. , Lin, J. , Ai, Q. , Lü, Z. L. , Liu, C. J. , & Yang, L. M. (2021). Analysis of codon usage bias in the chloroplast genome of Asarum heterotropoides Fr. Schmidt var. mandshuricum (Maxim.) Kitag. In Molecular Plant Breeding. https://kns.cnki.net/kcms/detail/46.1068.S.20210811.1341.012.html [Google Scholar]
- Tarullo, C. , Rose, J. P. , Sytsma, K. J. , & Drew, B. T. (2021). Using a supermatrix approach to explore phylogenetic relationships, divergence times, and historical biogeography of Saxifragales. Turkish Journal of Botany, 45, 440–456. 10.3906/bot-2106-41 [DOI] [Google Scholar]
- Treangen, T. J. , & Salzberg, S. L. (2011). Repetitive DNA and next‐generation sequencing: Computational challenges and solutions. Nature Reviews Genetics, 13(1), 36–46. 10.1038/nrg3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, M. L. , Santini, L. , Diniz, A. L. , & Munhoz, C. (2016). Microsatellite markers: What they mean and why they are so useful. Genetics and Molecular Biology, 39, 312–328. 10.1590/1678-4685-GMB-2016-0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. P. (2010). Study of DNA barcoding in identifying species of Picea in different geographical regions. Cheng Du University of Technology. [Google Scholar]
- Watson, L. , Bates, P. , Evans, T. , Unwin, M. , & Estes, J. (2002). Molecular phylogeny of subtribe Artemisiinae (Asteraceae), including Artemisia and its allied and segregate genera. BMC Evolutionary Biology, 2(1), 17. 10.1186/1471-2148-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, J. , & Shi, S. H. (1999). A phylogenetic and biogeographic study of Hamamelis (Hamamelidaceae), an eastern Asian and eastern North American disjunct genus. Biochemical Systematics and Ecology, 27, 55–66. 10.1016/S0305-1978(98)00067-2 [DOI] [Google Scholar]
- Wu, L. , Nie, L. , Wang, Q. , Xu, Z. , Wang, Y. U. , He, C. , Song, J. , & Yao, H. (2021). Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Scientific Reports, 11, 14643. 10.1038/s41598-021-94137-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L. , Nie, L. , Xu, Z. , Li, P. , Wang, Y. , He, C. , Song, J. , & Yao, H. (2020). Comparative and phylogenetic analysis of the complete chloroplast genomes of three Paeonia Section Moutan Species (Paeoniaceae). Frontiers in Genetics, 11, 980. 10.3389/fgene.2020.00980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z. , Gui, S. , Guan, Z. , Pan, L. , Wang, S. , Ke, W. , Liang, D. , & Ding, Y. (2014). A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: Insight into the plastid evolution of basal eudicots. BMC Plant Biology, 14, 289. 10.1186/s12870-014-0289-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, X. G. , Xiang, K. L. , Ortiz, R. D. C. , Jabbour, F. , & Wang, W. (2019). Integrating palaeontological and molecular data uncovers multiple ancient and recent dispersals in the pantropical Hamamelidaceae. Journal of Biogeography, 00, 1–10. 10.1111/jbi.13690 [DOI] [Google Scholar]
- Xie, L. , Yi, T.‐S. , Li, R. , Li, D.‐Z. , & Wen, J. (2010). Evolution and biogeographic diversification of the witch‐hazel genus (Hamamelis L., Hamamelidaceae) in the Northern Hemisphere. Molecular Phylogenetics and Evolution, 56, 675–689. 10.1016/j.ympev.2010.02.018 [DOI] [PubMed] [Google Scholar]
- Xiong, Y. , Xiong, Y. , He, J. , Yu, Q. , Zhao, J. , Lei, X. , Dong, Z. , Yang, J. , Peng, Y. , Zhang, X. , & Ma, X. (2020). The Complete chloroplast genome of two importantannual clover species, Trifolium alexandrinum and T. resupinatum: genome structure, comparative analyses and phylogenetic relationships with relatives in Leguminosae. Plants, 9(4), 478. 10.3390/plants9040478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C. , Dong, W. , Li, W. , Lu, Y. , Xie, X. , Jin, X. , Shi, J. , He, K. , & Suo, Z. (2017). Comparative analysisof six Lagerstroemia complete chloroplast genomes. Frontiers in Plant Science, 8, 15. 10.3389/fpls.2017.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, S. , Shi, T. , Luo, W. , Ni, X. , Shahid, I. , Ni, Z. , Huang, X. , Yao, D. , Shen, Z. , & Gao, Z. (2019). Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina . Horticulture Research, 6, 89. 10.1038/s41438-019-0171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. H. , Liu, X. , Cui, Y. X. , Nie, L. P. , Lin, Y. L. , Wei, X. P. , Wang, Y. , & Yao, H. (2020). Molecular structure and phylogenetic analyses of the complete chloroplast genomes of three original species of Pyrrosiae Folium . Chinese Journal of Natural Medicines, 18(8), 573–581. 10.1016/S1875-5364(20)30069-8 [DOI] [PubMed] [Google Scholar]
- Yang, J. , Vázquez, L. , Chen, X. , Li, H. , Zhang, H. , Liu, Z. , & Zhao, G. (2017). Development of chloroplast and nuclear DNA markers for Chinese oaks (Quercus subgenus Quercus) and assessment of their utility as DNA barcodes. Frontiers in Plant Science, 8, 816. 10.3389/fpls.2017.00816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J.‐B. , Yang, S.‐X. , Li, H.‐T. , Yang, J. , & Li, D.‐Z. (2013). Comparative chloroplast genomes of Camellia species. PLoS One, 8(8), e73053. 10.1371/journal.pone.0073053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Hu, Y. , Ren, T. , Sun, J. , & Zhao, G. (2018). Remarkably conserved plastid genomes of Quercus group Cerris in China: Comparative and phylogenetic analyses. Nordic Journal of Botany, 36, e01921. 10.1111/njb.01921 [DOI] [Google Scholar]
- Yang, Z. , Wang, G. , Ma, Q. , Ma, W. , Liang, L. , & Zhao, T. (2019). The complete chloroplast genomes of three Betulaceae species: Implications for molecular phylogeny and historical biogeography. PeerJ, 7, e6320. 10.7717/peerj.6320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Zhao, T. , Ma, Q. , Liang, L. , & Wang, G. (2018). Comparative genomics and phylogenetic analysis revealed the chloroplast genome variation and interspecific relationships of Corylus (Betulaceae) species. Frontiers in Plant Science, 9, 927. 10.3389/fpls.2018.00927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X. , Tang, P. , Li, Z. , Li, D. , Liu, Y. , & Huang, H. (2015). The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS One, 10, e129347. 10.1371/journal.pone.0129347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, X. Z. , Zhang, M. Z. , Jiang, Y. T. , Fan, H. H. , Chen, S. P. , & Liu, B. (2020). The complete chloroplast genome of Altingia chinensis (Hamamelidaceae). Mitochondrial DNA Part B, 5(2), 1808–1809. 10.1080/23802359.2020.1750989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , Yang, L. Q. , Bian, F. , Li, N. N. , Fan, Z. X. , Gong, Y. C. , Pu, Y. Y. , Ding, H. F. , & Peng, Z. Y. (2014). Cloning and sequence analysis of Clp protease gene (TaClpP) from wheat (Triticum aestivum L.). Shandong. Agricultural Sciences, 46(6), 1–5. 10.14083/j.issn.1001-4942.2014.06.001 [DOI] [Google Scholar]
- Zhang, D. , Gao, F. , Jakovlić, I. , Zou, H. , Zhang, J. , Li, W. X. , & Wang, G. T. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Molecular Ecology Resources, 20(1), 348–355. 10.1111/1755-0998.13096 [DOI] [PubMed] [Google Scholar]
- Zhang, D. , Wang, J. , Xu, L. , Xing, Y. , Zhang, T. , Li, S. , Yang, Y. , Bao, G. , Ao, W. , & Kang, T. (2020). Characteristic and phylogenetic analysis of the complete chloroplast genomes of three medicinal plants of Schisandraceae. BioMed Research International, 10.1155/2020/3536761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Shi, S. H. , Huang, Y. L. , & Zhang, H. D. (2001). ITS sequences of Hamamelidoideae and their systematic significance. Journal of Wuhan Botanical Research, 19(6), 445–448. http://www.whzwxyj.cn/CN/Y2001/V19/I6/445 [Google Scholar]
- Zhang, Z. Y. (1999). Notes on the modern classification systems of the Hamamelidaceae. Acta Botanica Yunnanica, 21(1), 1–10. [Google Scholar]
- Zhang, Z. Y. , & Lu, A. M. (1995). Hamamelidaceae: Geographic distribution, fossil history and origin. Acta Phytotaxonomy Sinica, 33, 313–339. [Google Scholar]
- Zhao, M. , & Ma, J. (2013). Co‐evolution of plant LTR‐retrotransposons and their host genomes. Protein Cell, 4(7), 493–501. 10.1007/s13238-013-3037-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q. , Chen, Y. , Dai, J. , Wang, F. , Wu, W. , Fan, Q. , Zhou, R. , & Ng, W. L. (2019). The chloroplast genome of Chunia bucklandioides (Hamamelidaceae): A rare tree endemic to Hainan, China. Conservation Genetics Resources, 11, 427–429. 10.1007/s12686-018-1042-z [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
The original sequencing data have been submitted to the NCBI database and received GenBank accession numbers MZ642354 (C. glandulifera), MZ823391 (C. velutina), and MZ642355 (L. chinense). The data used to support the findings of this study are included in Appendix S1.