

Abstract
Human neutrophil elastase (HNE) is a potent protease that plays an important physiological role in many processes but is also involved in a variety of pathologies that affect the pulmonary system. Thus, compounds able to inhibit HNE proteolytic activity could represent effective therapeutics. We present here a new series of pyrazolopyridine and pyrrolopyridine derivatives as HNE inhibitors designed as modifications of our previously synthesized indazoles and indoles in order to evaluate effects of the change in position of the nitrogen and/or the insertion of an additional nitrogen in the scaffolds on biological activity and chemical stability. We obtained potent HNE inhibitors with IC50 values in the low nanomolar range (10–50 nM), and some compounds exhibited improved chemical stability in phosphate buffer (t1/2 > 6 h). Molecular modeling studies demonstrated that inhibitory activity was strictly dependent on the formation of a Michaelis complex between the OH group of HNE Ser195 and the carbonyl carbon of the inhibitor. Moreover, in silico ADMET calculations predicted that most of the new compounds would be optimally absorbed, distributed, metabolized, and excreted. Thus, these new and potent HNE inhibitors represent novel leads for future therapeutic development.
Keywords: Human neutrophil elastase, Inhibitors, Nitrogen heterocycle, Stability, ADMET, Molecular docking
1. Introduction
Heterocyclic nuclei are considered privileged scaffolds in medicinal chemistry, and a wide variety of combinations of nitrogen, sulfur, and oxygen atoms in five- and six-membered rings have been reported. Notably, nitrogen heterocycles play a central role in drug discovery and are represented in ~85% of all biologically active chemical molecules. Furthermore, the number and the position of the nitrogen atoms in the ring influence some pharmacokinetic properties, such as solubility, hydrophilic/lipophilic characteristics, and the ability to form hydrogen bonds.1 We have identified a number of heterocyclic scaffolds that were modified based on the desired protein target in order to increase the number or the strength of the interactions with a catalytic site or receptor binding domains.2–5 For example, the identification and modification of nitrogen heterocycles has been essential in the discovery of new potent inhibitors of human neutrophil elastase (HNE), a proteolytic enzyme belonging to the serine protease family. This enzyme is involved in a variety of chronic diseases, including cardiopulmonary pathologies,6 and its regulation represents a promising therapeutic approach, as evident by the interest of many pharmaceutical companies. In fact, research in this field is quite active, and a number of HNE inhibitors are currently in various stages of clinical trials,7 such as Alvelestat developed by AstraZeneca in various formulations8, BAY85-8501 in phase II trials for bronchiectasis (Bayer HealthCare),9 and CHF6333, a novel potent inhaled HNE inhibitor by Chiesi Farmaceutici, in phase I trials for cystic fibrosis.10,11 Currently, only two HNE inhibitors are on the market. The first one, Prolastin® (purified α1-AT), is a peptide drug used in the treatment of α1-AT deficiency.12 The second is Sivelestat (ONO-5046, Elaspol® 100), a non-peptide inhibitor approved for intravenous use in Japan and South Korea for the treatment of ALI and ARDS associated with systemic inflammatory response syndrome.13
We have designed and synthesized a number of potent HNE inhibitors with various scaffolds.14–18 Among these compounds, the indazole and 7-azaindole nuclei resulted the best nitrogen bicycles, including HNE inhibitors active in the low nanomolar range.19–22 Based on these previous studies, we report here new HNE inhibitors that were developed through modification of the indazole and 7-azaindole (1H-pyrrolo[2,3-b]pyridine) nuclei in order to determine if the introduction of another nitrogen atom and/or moving nitrogen from its original position could affect biological activity (Fig. 1). The profile of selected compounds was further examined in depth with stability and molecular modeling studies. Moreover, in silico pharmacokinetic (ADMET) profiles of the most potent HNE inhibitors obtained (IC50 < 1 μM) were evaluated in order to identify candidates for future in vivo studies.
Fig. 1.
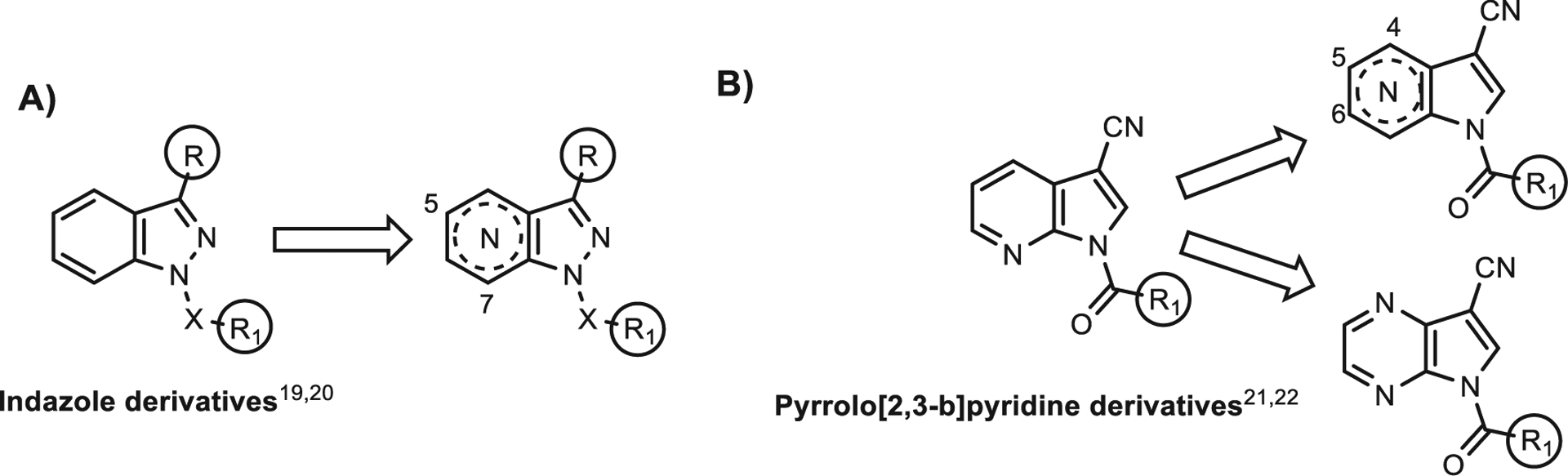
Indazole and pyrrolo[2,3-b]pyridine derivative modification.
2. Results and discussion
Two different strategies were pursued in our studies: 1) insertion of an additional nitrogen atom at position 5 or 7 of the indazole nucleus, resulting in 5- and 7-azaindazole derivatives (1H-pyrazolo[3,4-b]pyridine and 1H-pyrazolo[4,3-c]pyridine) (Fig. 1A) and insertion at position 4 of the 7-azaindole nucleus, resulting in 5H-pyrrolo[2,3-b]pyrazines (Fig. 1B); and 2) moving the nitrogen in the pyrrolopyridine scaffold from position 7 to positions 4, 5, and 6 of the pyridine ring (Fig. 1B). Finally, we synthesized some pyrazolopyrimidines containing an exocyclic amide group instead of the endocyclic amide function present in all of our previous compounds. In all new compounds, we inserted those fragments and substituents that lead to the optimal biological results based on our previous studies and those reported in the literature.
2.1. Chemistry
All compounds were synthesized as reported in Schemes 1–5, and the structures were confirmed on the basis of analytical and spectral data. To obtain the 3-substituted derivatives of the 5- and 7-azaindazoles, we followed the procedures shown in Scheme 1 and 2. Starting from the previously described compounds of type 1 (1a,b,23 1c,d,24 1e,f,25 1g26 and 1 h27), we synthesized the final compounds 2a-t (Scheme 1) following the indicated reaction conditions. Treatment with m-toluoyl chloride or cyclopropanecarbonyl chloride, triethylamine in anhydrous dichloromethane at room temperature resulted in synthesis of compounds 2a,b,l-o,r-t, while derivatives 2e-i,p,q are obtained using sodium hydride in dry tetrahydrofuran at room temperature. Compound 2c was obtained starting from intermediate 1b using benzyl bromide and potassium carbonate in dry acetonitrile at reflux, whereas 2d was obtained using benzoic acid, activating reagents (OHBt and DCC) and triethylamine in dry tetrahydrofuran.
Scheme 1. Reagents and Conditions:
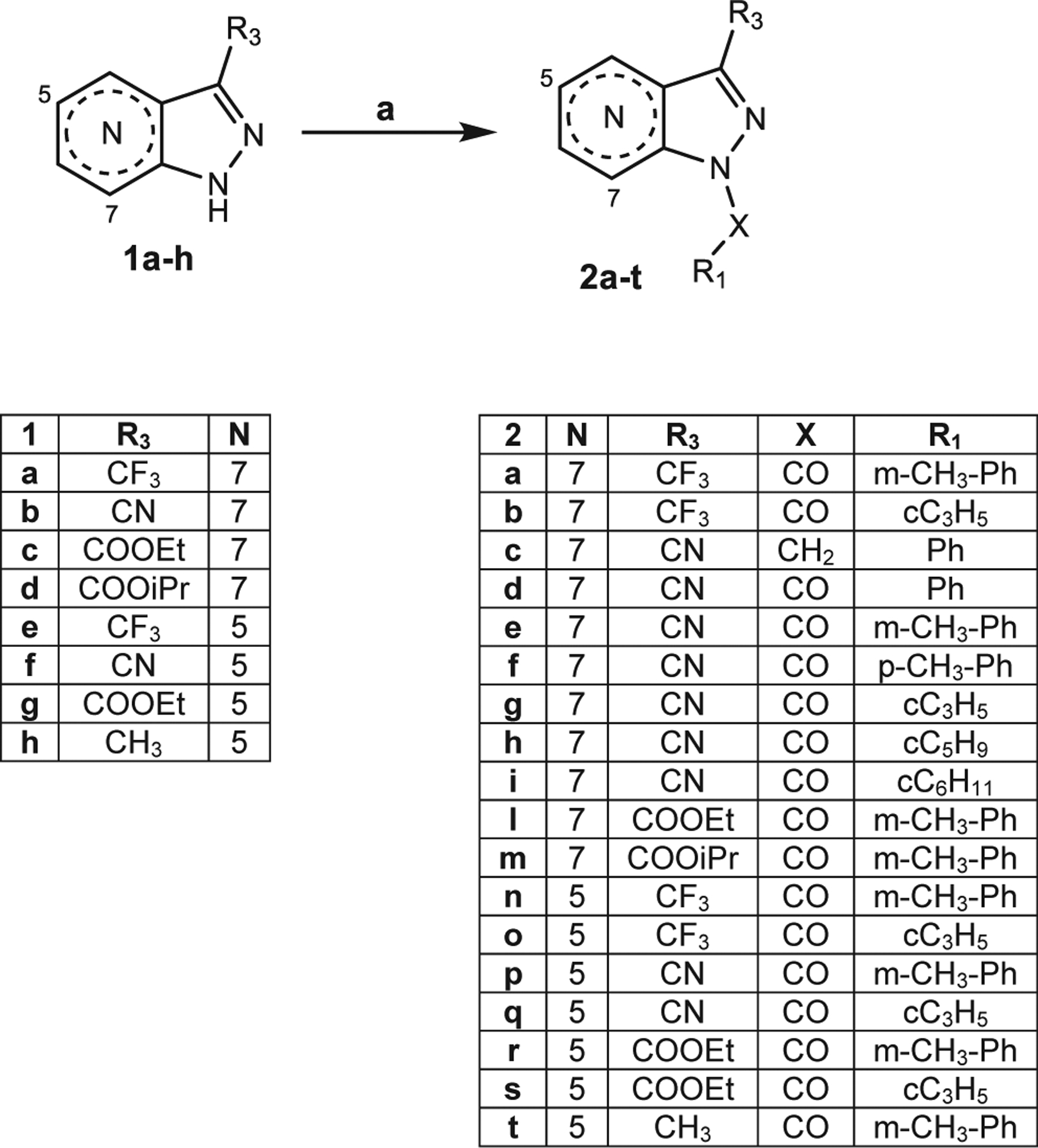
(a) for 2a,b,l-o,r-t: R-COCl, Et3N, anhydrous CH2Cl2, 0 °C, 2 h, then r.t., 2 h; for 2c: Benzyl bromide, anhydrous CH3CN, K2CO3, reflux, 4 h; for 2d: Ph-COOH, dry THF, HOBt, Et3N, DCC, 0 °C, 30′, then r.t., 48 h; for 2e-i,p,q: dry THF, NaH, 0 °C, 30′, then R-COCl, r.t., o/n.
Scheme 5. Reagents and Conditions:
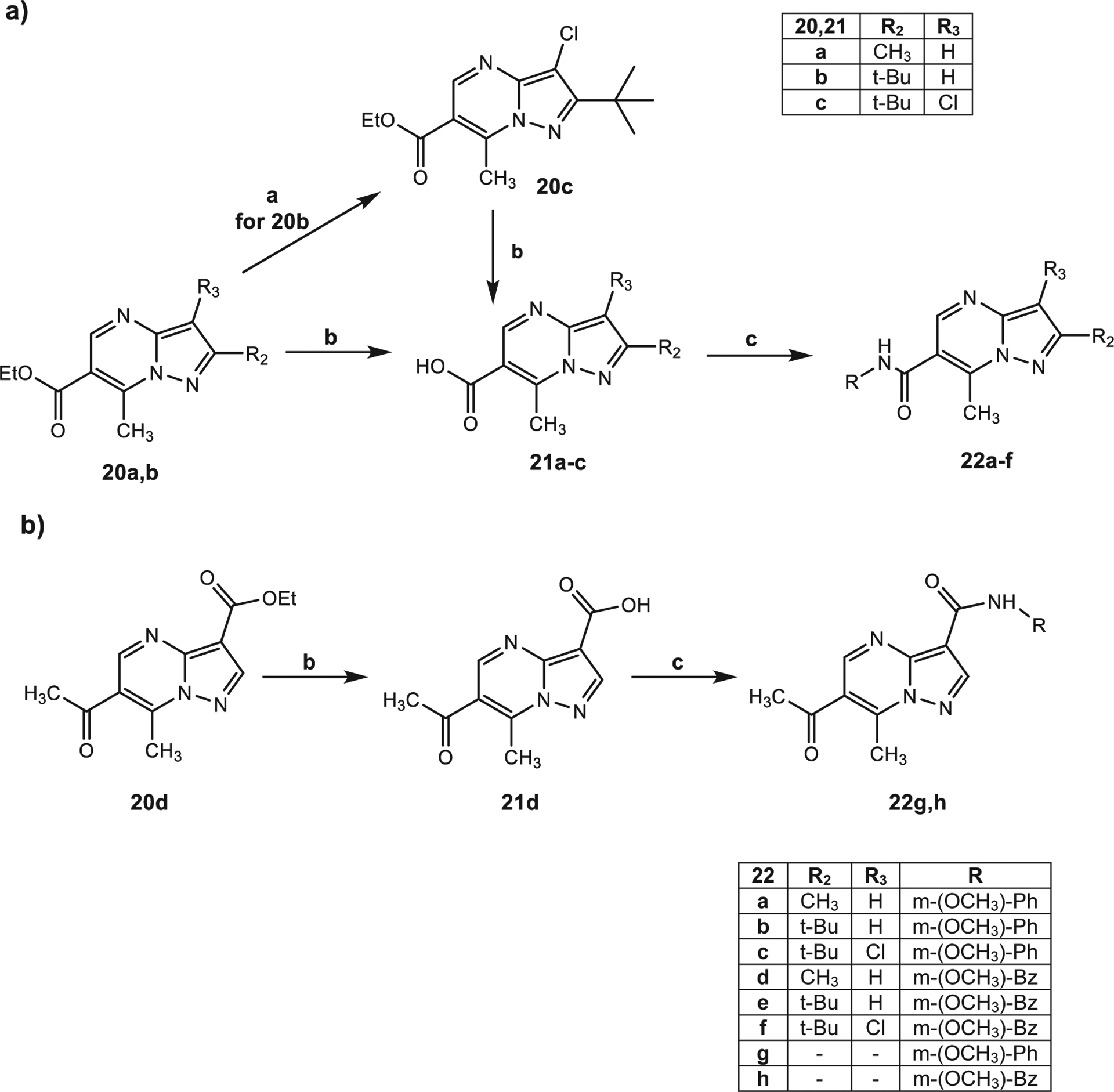
(a) NCS, CCl4, (PhCOO)2, reflux, 12 h; b) NaOH 6 N, reflux, 2 h; c) CCl3CN, PPh3, dry DCM, r.t., 4 h then appropriate amine (m-anisidine or 3-methoxybenzylamine), Et3N, r.t., 12 h.
Scheme 2. Reagents and Conditions:
(a) dry THF, NaH, 0 °C, 30′, then cC3H5-COCl, r.t., o/n; (b) HCl 37%, SnCl2·2H2O, r.t., 30′; (c) Et3N, DMF/1,4-Dioxane, R-COCl, 50 °C, 4 h; (d) CH3COCl, Et3N, anhydrous CH2Cl2, 0 °C, 2 h, then r.t., 2 h.
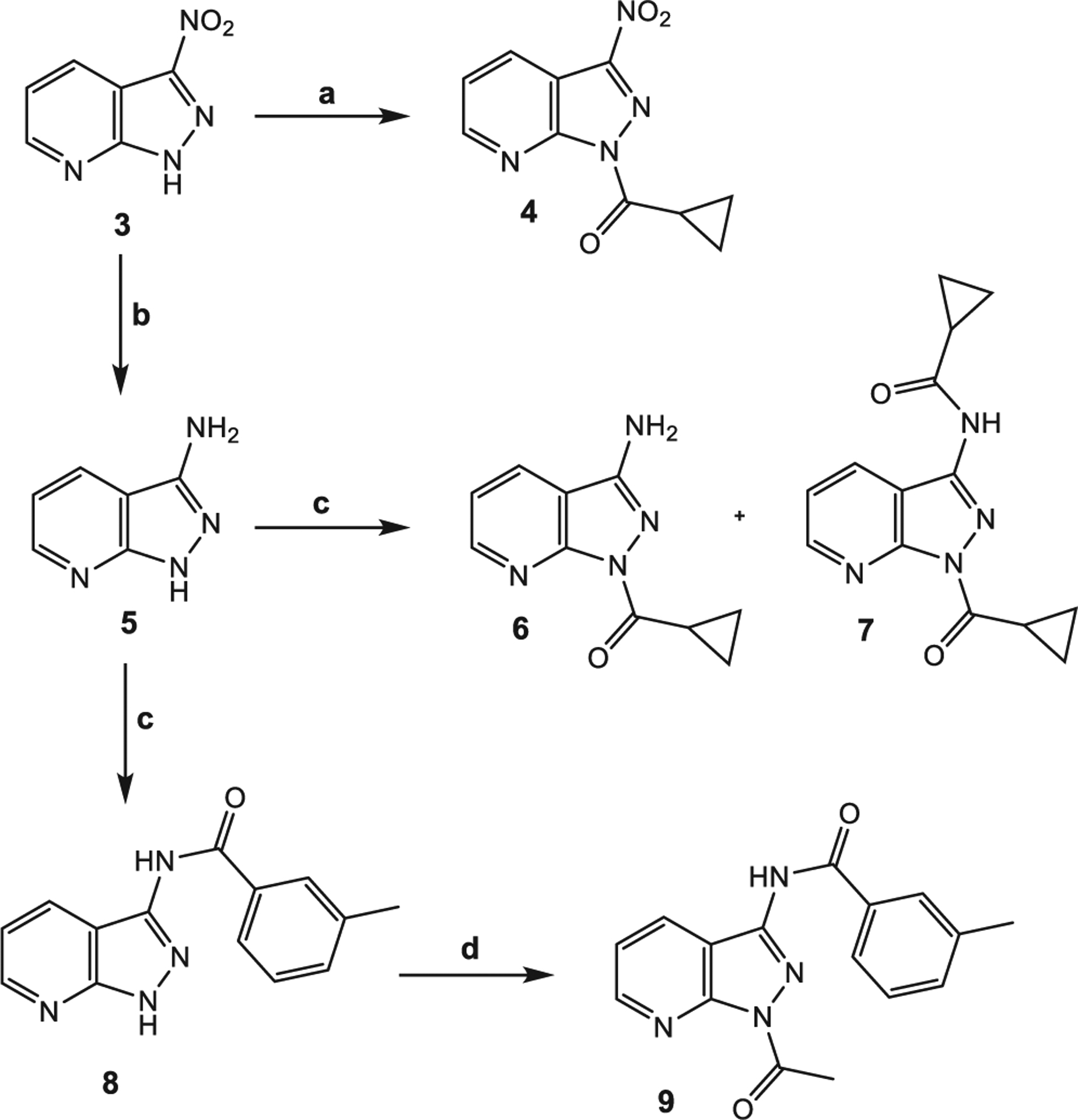
Scheme 2 shows synthesis of the pyrazolo[3,4-b]pyridine derivatives containing a nitro (4), an amino (6), or an amide group (7–9) at position 3. The 3-nitro derivative 4 was obtained through an acylation reaction starting from compound 328 and following the procedure previously reported in Scheme 1. Reduction of the nitro group in compound 3 with tin(II) chloride in acid medium, led to intermediate 528 in high yield, which was then treated with 3-methylbenzoyl chloride, triethylamine in 1,4-dioxane and DMF. According to the results reported in the literature for pyrazolo-quinolines,29 acylation selectively occurred on the amino group at position 3 under these conditions, resulting in the final compound 8. Compound 8 was further treated with acetyl chloride under the conditions shown in Scheme 1 to obtain the final derivative 9. Different results were observed when intermediate 5 was reacted with cyclopropanecarbonyl chloride, since a mixture of mono-acyl 6 and bi-acyl derivative 7 was obtained.
Scheme 3 shows the synthetic pathway used to obtain the pyrrolopyridines 13a-f. The starting material 10a-c (10a,b30 and 10c31) was treated with hydroxylamine hydrochloride and NaHCO3 at high temperature, leading to the corresponding oximes 11a-c (11c32). Dehydration with POCl3 resulted in the 3-CN derivatives 12a-c (12c32), which were finally acylated at position 1 with the appropriate acyl (aroyl) chloride and triethylamine in anhydrous dichloromethane to obtain the azaindoles 13a-f.
Scheme 3. Reagents and Conditions:
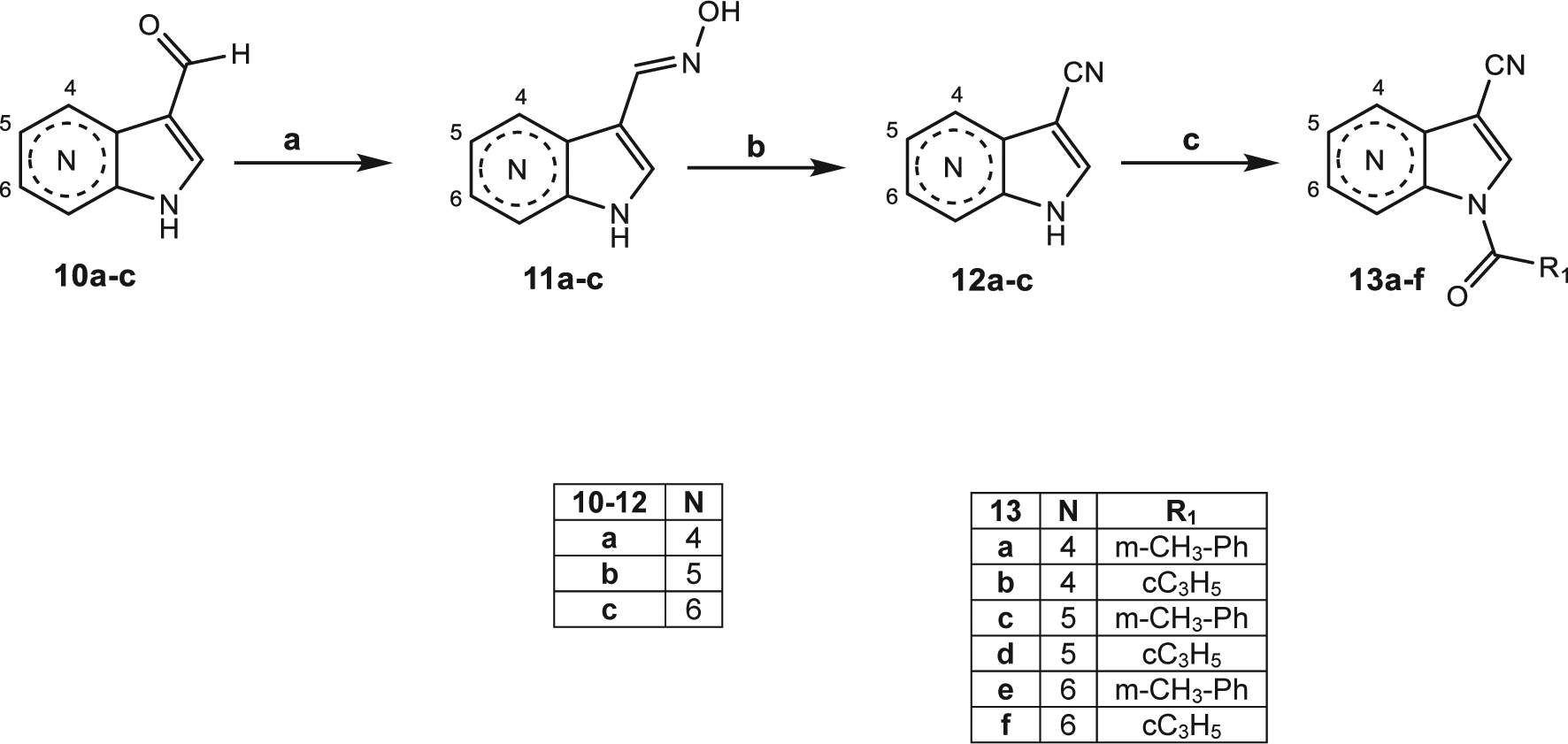
(a) NH2OH·HCl, H2O, 60 °C, 30 min; NaHCO3, reflux, 4 h; (b) POCl3, reflux, 2 h; (c) R-COCl, Et3N, anhydrous CH2Cl2, 0 °C, 2 h, then r.t., 2 h.
The synthesis of compounds with a 5H-pyrrolo[2,3-b]pyrazine scaffold (15a,b and 19a,b) is shown in Scheme 4. The final compounds 15a,b were obtained starting from intermediate 1433 and following the acylation procedures described above. To obtain compounds 19a,b, the CN group was inserted at position 3 as follows: formylation of compound 14 with HTMA and glacial acetic acid (1633), transformation into the corresponding oxime (17), and dehydration to compound 18, which was subjected to acylation. Unfortunately, compounds 15a and 19a, which contained a m-toluoyl fragment at position 1, were not chemically stable and quickly turn into the precursors 14 and 18 at room temperature, respectively.
Scheme 4. Reagents and Conditions:
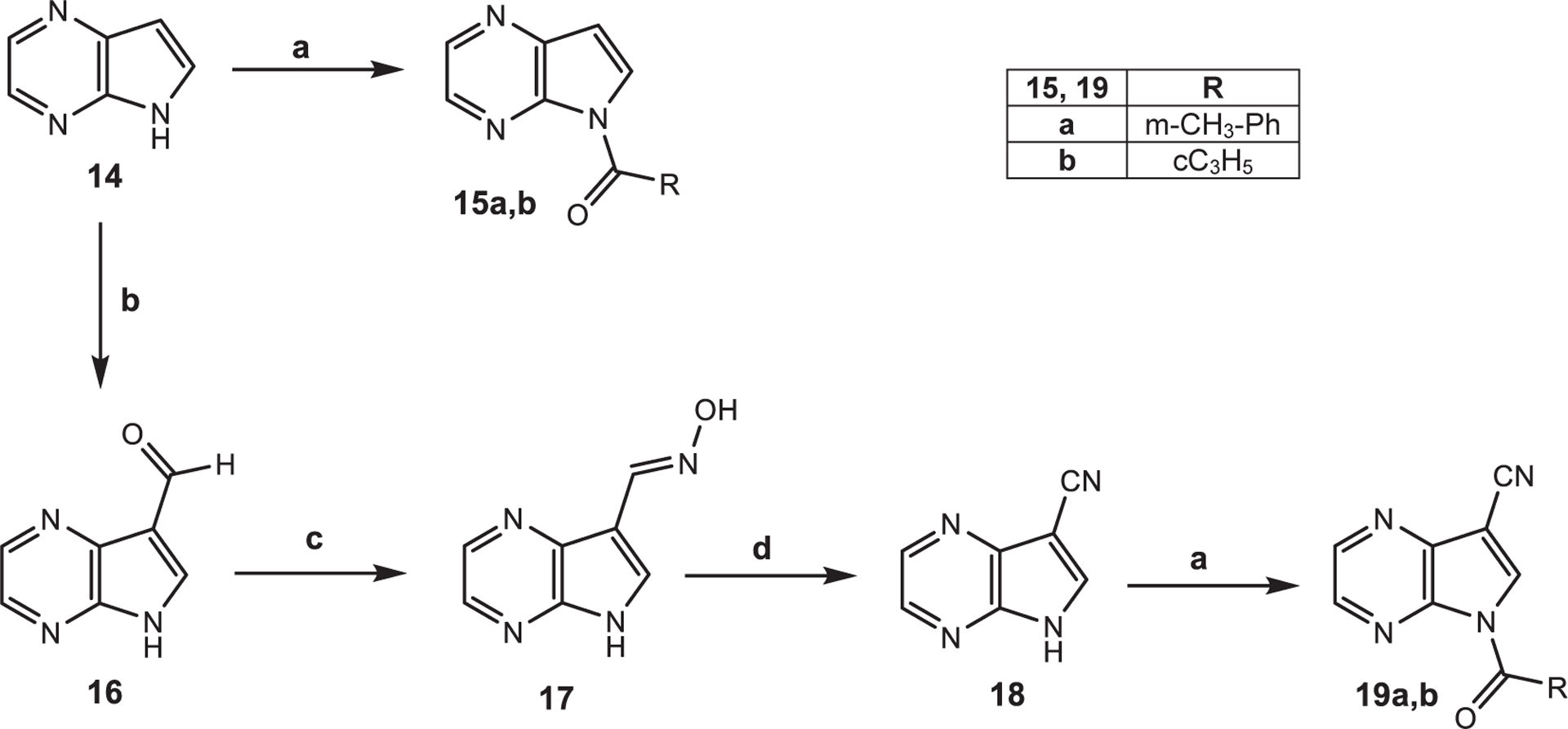
(a) R-COCl, Et3N, anhydrous CH2Cl2, 0 °C, 2 h, then r.t., 2 h; (b) HTMA, glacial CH3COOH, reflux, 2 h; (c) NH2OH·HCl, H2O, 60 °C, 30 min; NaHCO3, reflux, 4 h; (d) POCl3, reflux, 2 h.
Scheme 5 shows the steps leading to synthesis of the pyrazolopyrimidine derivatives 22a-h. Part a of Scheme 5 shows the synthesis of pyrazolopyrimidines containing the amide function at position 6, and Part b shows synthesis of the final compounds 22 g,h displaying the amide group at position 3 of the bicyclic scaffold. Starting with compounds 20a,b34 and 20d35, which were previously described, or compound 20c, which was obtained from 20b through chlorination with N-chlorosuccinimide (NCS) and benzoyl peroxide in tetrachloromethane at reflux, the 6-carbethoxy (20a-c) and 3-carbethoxy (20d) derivatives were hydrolyzed using 6 N NaOH at reflux, resulting in the corresponding acids 21a-d (21a,b,d36–38). These compounds were treated with trichloroacetonitrile, triphenylphosphine and m-anisidine or 3-methoxybenzilamine to obtain the final compounds 22a-h.
2.2. Biological evaluation and structure-activity relationship (SAR) analysis
All compounds were evaluated for their ability to inhibit HNE, and the results are presented in Tables 1–3, together with data from representative reference compounds from the previous series of N-benzoylindazoles (A20), 7-azaindoles (B21 and C21), and the drug Sivelestat. Keeping the cyano group that is present in the reference indazole A at position 3 of the 7-azaindazole compounds, we introduced various substituents at position 1 (Table 1). The introduction of a phenyl (2d) or cyclopentyl (2 h) ring resulted in retention of HNE inhibitory activity (IC50 = 140 and 160 nM, respectively), whereas introduction of a cyclohexyl ring resulted in loss of activity for compound 2i. The 7-aza analogue of compound A displaying a m-toluoyl fragment at 1-N (2e), was 47 fold less potent than A (2e: IC50 = 330 nM versus A: IC50 = 7 nM). Isomer 2f obtained by the shift of the methyl group from the meta to the para position was even less active (IC50 = 1.5 μM). The best compound of the 3-CN derivatives was 2 g, which contains a cyclopropyl group at the amide function (IC50 = 34 nM). Elimination of the amide function led to complete loss of HNE inhibitory activity (compound 2c), confirming the importance of this group in the catalytic mechanism. Replacement of the cyano group at position 3 with various groups or functions selected from our previous publications or from the literature also gave interesting results. Ester (2 l,m), amine (6), amide (7, 9) derivatives were only moderately active, with HNE inhibition in the micromolar range. In contrast, introduction of the trifluoromethyl (2a,b) or the nitro group (4) led to potent HNE inhibitors, with IC50 values comparable to or better than that of Sivelestat (2b, IC50 = 50 nM; 4, IC50 = 21 nM). Thus, the most active compounds in the 7-azaindazole series were 2b, 2 g and 4, which contain common elements, including a group cyclopropyl at the amide function and the presence of a strong electron withdrawing group at position 3 (i.e., CN, CF3, or NO2).
Table 1.
HNE inhibitory activity of compounds 2a-t, 4, 6, 7, and 9.
|
|||||
|---|---|---|---|---|---|
| Compound | N | R3 | X | R1 | IC50 (μM)a |
| 2a | 7 | CF3 | CO | m-CH3-Ph | 0.42 ± 0.12 |
| 2b | 7 | CF3 | CO | cC3H5 | 0.050 ± 0.01 |
| 2c | 7 | CN | CH2 | Ph | N.A.b |
| 2d | 7 | CN | CO | Ph | 0.16 ± 0.05 |
| 2e | 7 | CN | CO | m-CH3-Ph | 0.33 ± 0.03 |
| 2f | 7 | CN | CO | p-CH3-Ph | 1.5 ± 0.051 |
| 2g | 7 | CN | CO | cC3H5 | 0.034 ± 0.012 |
| 2h | 7 | CN | CO | cC5H9 | 0.14 ± 0.04 |
| 2i | 7 | CN | CO | cC6H11 | 1.1 ± 0.22 |
| 2l | 7 | COOEt | CO | m-CH3-Ph | 2.0 ± 0.44 |
| 2m | 7 | COOiPr | CO | m-CH3-Ph | 0.98 ± 0.31 |
| 2n | 5 | CF3 | CO | m-CH3-Ph | 0.033 ± 0.011 |
| 2o | 5 | CF3 | CO | cC3H5 | 0.087 ± 0.021 |
| 2p | 5 | CN | CO | m-CH3-Ph | 0.010 ± 0.003 |
| 2q | 5 | CN | CO | cC3H5 | 0.079 ± 0.023 |
| 2r | 5 | COOEt | CO | m-CH3-Ph | 0.016 ± 0.005 |
| 2s | 5 | COOEt | CO | cC3H5 | 0.069 ± 0.026 |
| 2t | 5 | CH3 | CO | m-CH3-Ph | 0.760 ± 0.14 |
| 4 | 7 | NO2 | CO | cC3H5 | 0.021 ± 0.002 |
| 6 | 7 | NH2 | CO | cC3H5 | 9.9 ± 1.3 |
| 7 | 7 | NHCO-cC3H5 | CO | cC3H5 | 1.5 ± 0.14 |
| 9 | 7 | NHCO-m-CH3-Ph | CO | CH3 | 25.2 ± 1.3 |
| Sivelestat | 0.050 ± 0.020 | ||||
| A 20 | – | CN | CO | m-CH3-Ph | 0.007 ± 0.0015 |
IC50 values are presented as the mean ± SD of three independent experiments.
N.A.: no inhibitory activity was found at the highest concentration of compound tested (50 μM).
Table 3.
HNE inhibitory activity of compounds 22a-h.
| ||||
| Compound | R | R2 | R3 | IC50 (μM)a |
|---|---|---|---|---|
| 22a | m-(OCH3)-Ph | CH3 | H | N.A.b |
| 22b | m-(OCH3)-Ph | t-Bu | H | N.A.b |
| 22c | m-(OCH3)-Ph | t-Bu | Cl | N.A.b |
| 22d | m-(OCH3)-Bn | CH3 | H | N.A.b |
| 22e | m-(OCH3)-Bn | t-Bu | H | N.A.b |
| 22f | m-(OCH3)-Bn | t-Bu | Cl | N.A.b |
| 22 g | m-(OCH3)-Ph | – | – | N.A.b |
| 22 h | m-(OCH3)-Bn | – | – | N.A.b |
| Sivelestat | 0.050 ± 0.020 | |||
IC50 values are presented as the mean ± SD of three independent experiments.
N.A.: no inhibitory activity was found at the highest concentration of compound tested (50 μM).
Analysis of the 5-azaindazole derivatives (2n-t) revealed a slightly different trend. First, the substitutions with CF3 or CN groups (compound 2n-q), as well as the weaker electron withdrawing ester group (2r,s), were tolerated, resulting in potent HNE inhibitors (IC50 = 10–87 nM). On the other hand, substitution with a methyl group (2 t) resulted in significant loss of activity (IC50 = 760 nM). In addition, replacement with an m-toluoyl fragment resulted in compounds with the highest activity, as is evident when comparing these pairs: 2n/2o, 2p/2q, and 2r/2s, which was different from the trend observed with the 7-azaindazoles. Compound 2p, which is the 5-aza analogue of A, is the most potent compound of this series, had comparable activity with A (IC50 = 10 nM).
Modification of the azaindoles 13a-f, which are isomers of the 7-azaindoles B and C and the pyrrolopyrazines of type 15 and 19, led to the formation of highly active derivatives (Table 2). Specifically, movement of the nitrogen in the pyridine ring from position 7 of B and C to position 4 or 5 resulted in compounds with IC50 values of 14–97 nM, with 13c being the best term of this series. In contrast, shift of the nitrogen to position 6 (compounds 13e and 13f) led to reduced activity compared to the reference compounds B and C, indicating that the nitrogen in this position does not interact well with the target. The pyrrolopyrazine 19a, which differs from the reference compound C by the introduction of a second nitrogen at position 4, retained high HNE inhibitory activity (IC50 = 56 nM), whereas elimination of the CN at position 3 (compound 15) resulted in reduced activity by two orders of magnitude.
Table 2.
HNE inhibitory activity of compounds 13a-f, 15b, and 19b.
| |||
| Compound | N | R1 | IC50 (μM)a |
|---|---|---|---|
| 13a | 4 | m-CH3-Ph | 0.089 ± 0.034 |
| 13b | 4 | cC3H5 | 0.023 ± 0.004 |
| 13c | 5 | m-CH3-Ph | 0.014 ± 0.004 |
| 13d | 5 | cC3H5 | 0.097 ± 0.01 |
| 13e | 6 | m-CH3-Ph | 0.194 ± 0.053 |
| 13f | 6 | cC3H5 | 0.183 ± 0.044 |
| 15b | – | H | 7.9 ± 2.5 |
| 19b | – | CN | 0.056 ± 0.018 |
| Sivelestat | 0.050 ± 0.020 | ||
| B 21 | 7 | m-CH3-Ph | 0.015 ± 0.004 |
| C 21 | 7 | cC3H5 | 0.087 ± 0.021 |
IC50 values are presented as the mean ± SD of three independent experiments.
The pyrazolopyrimidine derivatives 22a-h containing an exocyclic amide group instead of an endocyclic amide function were all completely inactive, indicating that this modification was not tolerated (Table 3).
The 12 most potent HNE inhibitors were evaluated for their chemical stability in aqueous buffer.39 Spontaneous hydrolysis rates and reaction orders of the inhibitors were measured in phosphate buffer at pH 7.4 and 20 °C using spectrophotometry to detect compound hydrolysis. As examples, Fig. 2 shows that the absorbance maxima at 305 nm decreased over time for compound 4, while a new peak appeared at 255 nm.
Fig. 2.
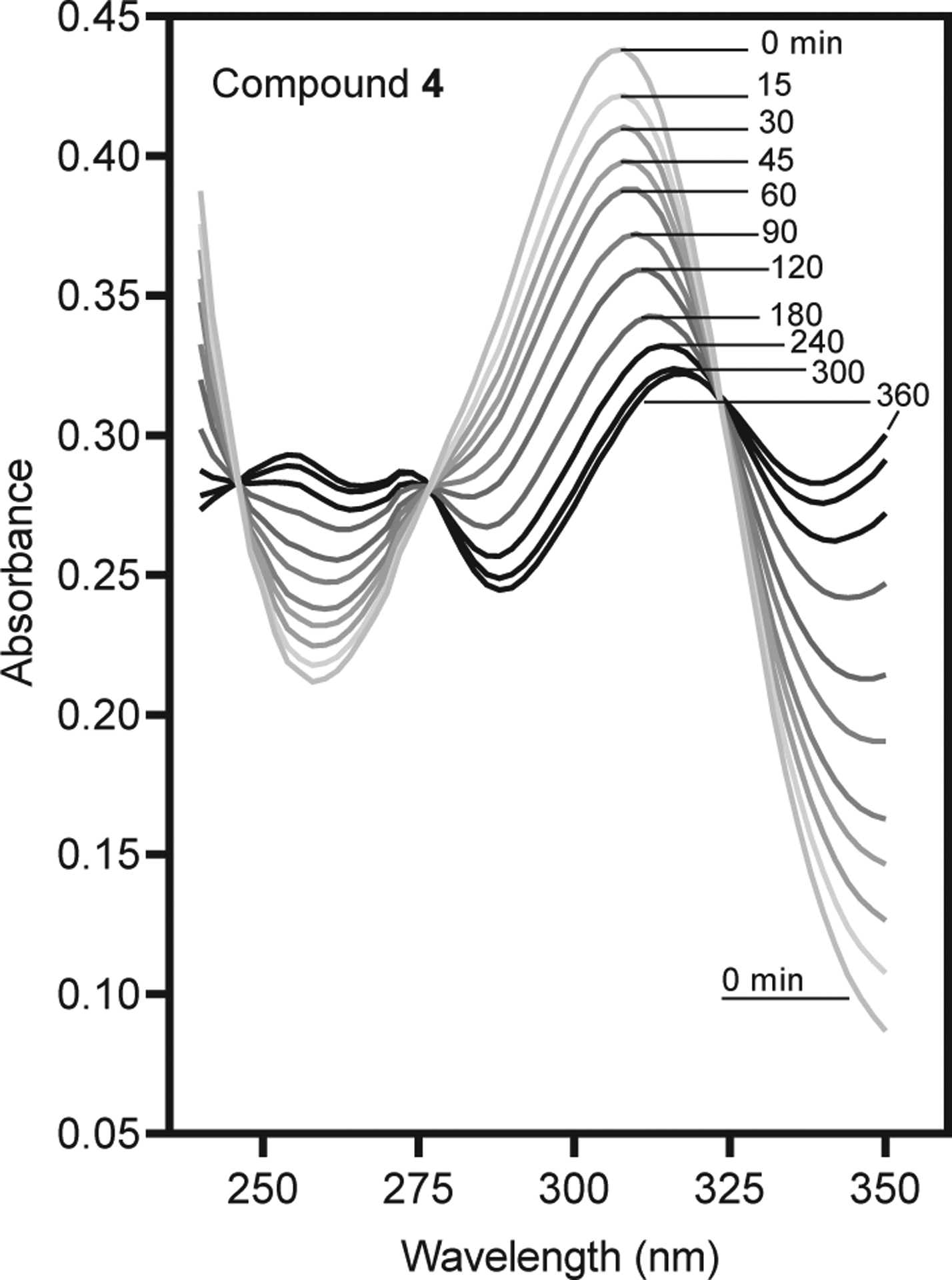
Analysis of changes in compound absorbance resulting from spontaneous hydrolysis of 4. The changes in absorbance spectra of the compounds (20 μM in 0.05 M phosphate buffer, pH 7.4, 20 °C) were monitored over time in solution.
As shown in Table 4, compounds 2b, 2o, 2s, and 13b were the most stable with t1/2 > 4 h. We found that compounds containing a cyclopropyl group at position 1 (compounds 2s and 13b) were more stable when compared to the analogs with a m-toluoyl substituent (compounds 2r and 13a, respectively). Conversely, the azaindazole derivatives (series 2 with 3 nitrogen atoms in the heterocycle) were more stable than the azaindoles and indazoles (2 nitrogen atoms in the heterocycle). Moreover, the most stable compound 2b had a CF3 at position 3 (compared to compounds 2q and 2s with CN and COOEt groups, respectively). Noticeably, acceptor substituents at position 3 exhibiting mesomeric effects with respect to the heterocyclic moiety (CN and NO2) were present in mostly unstable or moderately unstable compounds (e.g., 2p, 2q, 4, 13a, 13c, 19b). This observation is in agreement with amide hydrolysis according to the well-known acyl substitution mechanism,40 which includes a nucleophilic attack on the carbonyl carbon atom in the rate-limiting step. In addition, it should be mentioned that N-acyl derivatives of azoles are highly reactive amides, as compared to other compounds containing an amide function.41,42
Table 4.
Half-life (t1/2) for the spontaneous hydrolysis of selected derivatives. LUMO energies of the compounds calculated by DFT B3LYP/6–31 + G(d,p).
| Compound | t1/2 (min) | E(LUMO), eV |
|---|---|---|
| 2b | 880.0 ± 77.5 | −2.376 |
| 2n | 106.6 ± 2.4 | −2.547 |
| 2o | 386.1 ± 43.2 | −2.386 |
| 2p | 12.2 ± 4.0 | −2.819 |
| 2q | 79.0 ± 15.6 | −2.743 |
| 2r | 147.0 ± 12.3 | −2.536 |
| 2s | 436.8 ± 50.4 | −2.418 |
| 4 | 93.8 ± 7.6 | −3.374 |
| 13a | 76.6 ± 1.8 | −2.414 |
| 13b | 268.3 ± 29.3 | −2.242 |
| 13c | 163.3 ± 8.1 | −2.484 |
| 19b | 100.6 ± 5.2 | −2.604 |
In general, it was difficult to find a clear relationship between hydrolytic activity and electronic or steric effects of the heterocyclic moieties and substituents within the series of the investigated compounds. However, we found that the observed hydrolytic instability could be explained by the energies of the lowest unoccupied molecular orbital (LUMO) calculated for the optimized geometries of compounds 2b, 2n, 2o, 2p, 2q, 2r, 2s, 4, 13a, 13b, 13c, and 19 using density functional theory (DFT) (Table 4). The optimized structures of cyclopropyl-containing compounds, except for 13b, have Cs symmetry with heterocyclic and cyclopropane groups lying in mutually perpendicular planes. For toluoyl-substituted derivatives, the angle between the planes of the heterocycle and m-tolyl group varied between 30 and 50°. Relatively stable compounds 2b, 2o, 2s, and 13b can be distinguished from hydrolytically unstable derivatives according to their LUMO energies E(LUMO) (Fig. 1S, see Supplementary Material). The unstable compounds had systematically lower values of E(LUMO) and, hence, were more prone to reactions with nucleophiles. This conclusion agrees with the above-mentioned mechanism of the base-catalyzed hydrolysis involving an attack of the hydroxide anion on the carbonyl group. It should be noted that LUMO has a significant localization at the carbonyl carbon atom in both stable and unstable compounds (see examples in Fig. 3) and, thus, its energy can strongly affect electrophilicity of the carbonyl group. The relationship between E(LUMO) and hydrolytic stability is not quantitative, (i.e., LUMO energy is not tightly correlated with t1/2 or rate constant k‘). Clearly, reactivity is determined not just by the electronic structure of a starting compound but is also highly dependent on properties and stability of the tetrahedral intermediate formed on the addition of the hydroxide ion to the carbonyl group in the rate-limiting step.
Fig. 3.
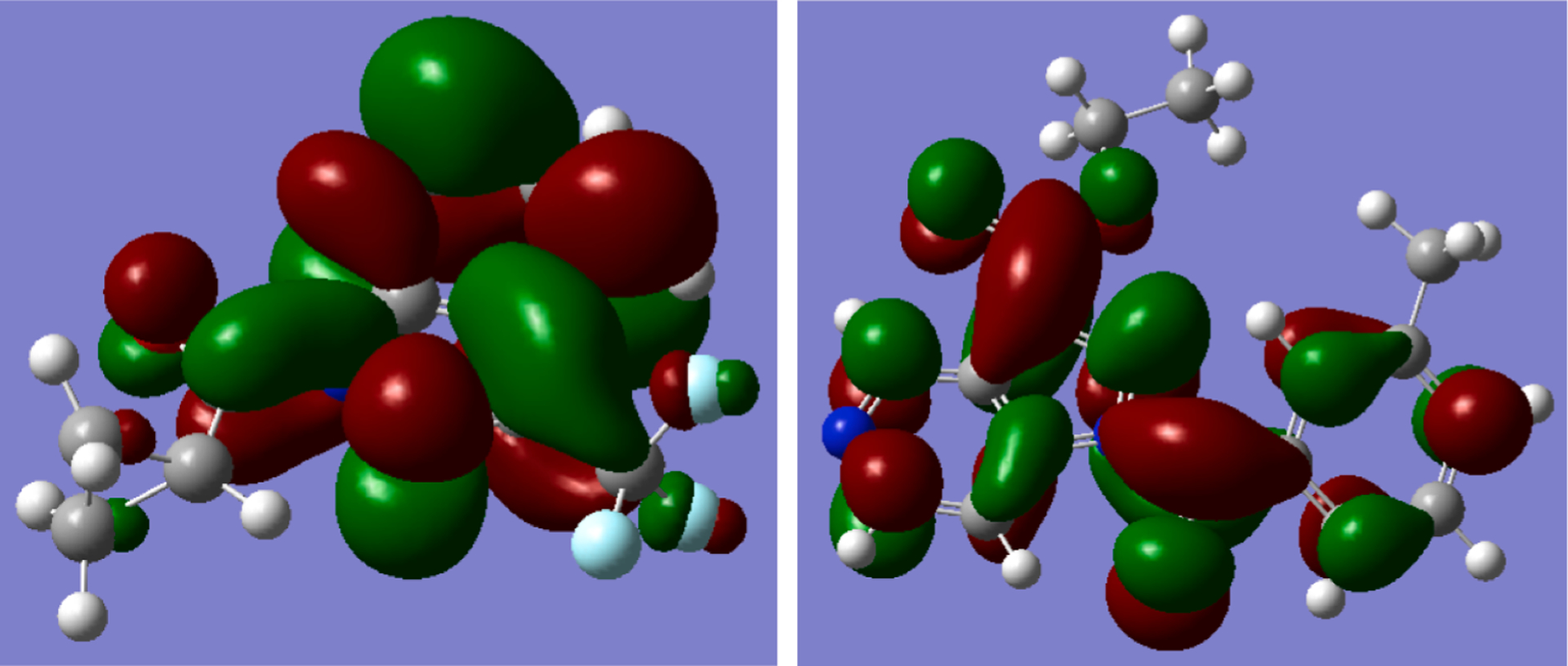
Surfaces of LUMO (isovalue 0.02) for compounds 2b (A) and 2r (B) calculated by the DFT method at B3LYP/6–31+G(d,p) level.
Classification of stable and unstable compounds according to other calculated characteristics, such as energy of the highest occupied molecular orbital (HOMO), LUMO-HOMO energy gap, or some local indices of reactivity (e.g., atomic charges, Wiberg index for the amide bond) did not lead to a separation similar to the result presented in Fig. 1S for LUMO energies.
2.3. Molecular modeling
We performed molecular modeling of selected compounds in the active site of HNE (1HNE entry of Protein Data Bank) using previously reported methods.20,21 According to the literature,43 inhibitors interact with HNE via a nucleophilic attack of the Ser195 hydroxyl oxygen atom onto a carbonyl carbon atom of a ligand, which leads to the formation of a Michaelis complex. This interaction is accompanied by proton transfer from Ser195 to Asp102. For the docking poses obtained, we have evaluated the geometric parameters d1 and α, which are important for Michaelis complex formation (Fig. 4). In addition, length of the proton transfer channel within the catalytic triad from Ser195 to Asp102 through His57 was calculated using d2 and d3 distances between the key atoms (see Methods).
Fig. 4.

Interaction of a carbonyl-containing ligand XC(O)Y with the HNE catalytic triad (Ser195, His57, and Asp102). Distance d1 and angle α determine conditions for the Michaelis complex formation. The value of α was measured as an angle between C(carbonyl)⋯O(Ser195) axis and C=O bond. Distances d2 and d3 influence on the proton transfer from Ser195 to Asp102. Length of the proton transfer channel was calculated as L = min(d2) + d3.
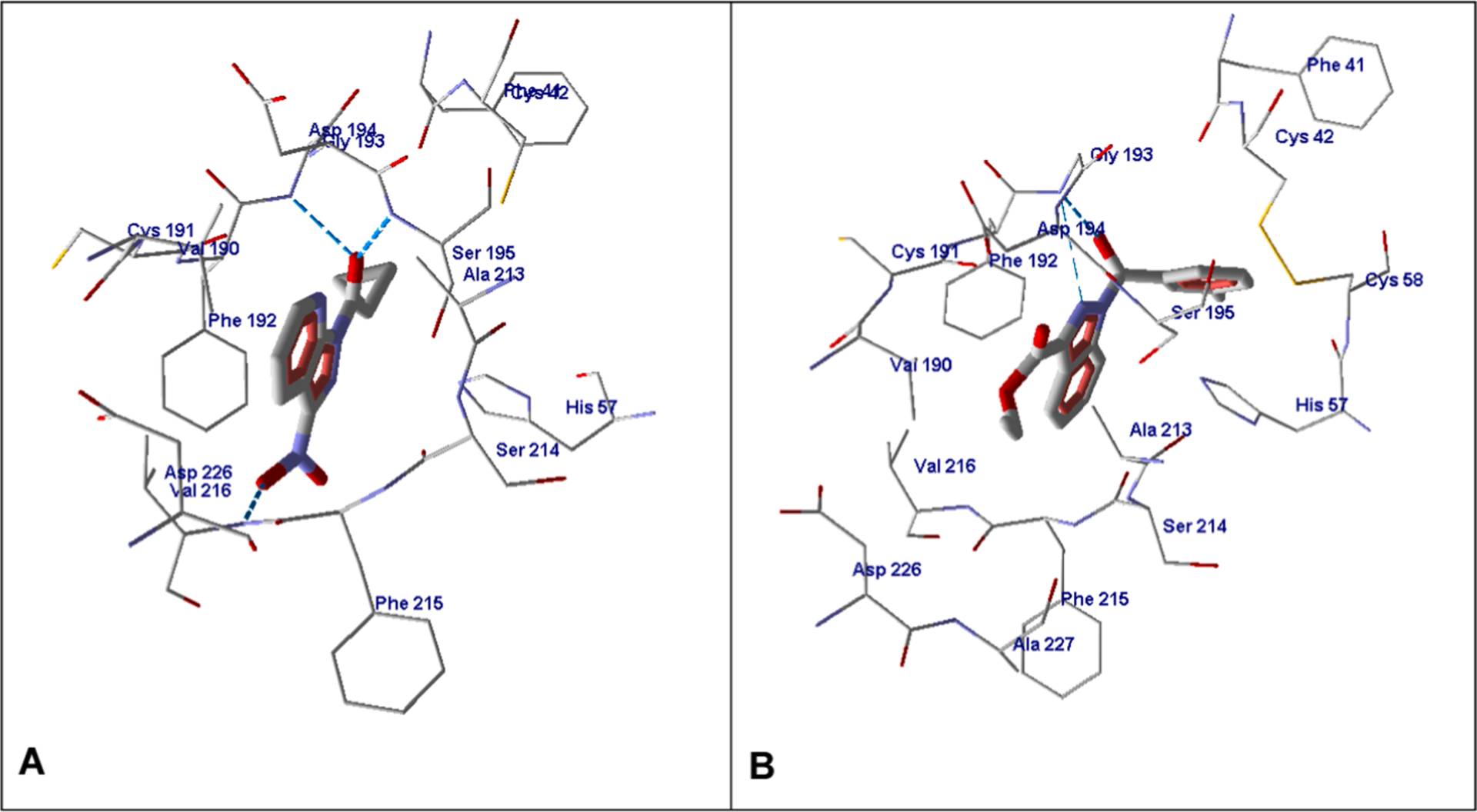
Compounds 2e (IC50 = 330 nM) and 2p (IC50 = 10 nM) contain an additional nitrogen atom at different positions of the fused aromatic ring compared to compound A.20 The docking poses of these molecules are shown in Fig. 5. Although these three molecules are isosteric, the presence of an additional nitrogen atom in the heterocycle of compounds 2e and 2p favors arrangement of their bicyclic fragments among polar amino acid residues versus compound A with an indazole heterocycle. Molecule 2e forms a H-bond with Val216 via participation of the nitrogen atom at position 7 of the heterocycle, while compound 2p is H-bonded to Ser195 with two nitrogen atoms of the five-membered ring. In addition, strong H-bonds are formed between the carbonyl oxygen of 2p and Gly193 and Ser195. Note that the nitrogen atom at position 5 of the heterocycle in 2p is located near an electropositive (blue) region of the binding site surface (Fig. 5D), which should, along with the H-bonds, stabilize the molecule in the obtained docking pose.
Fig. 5.
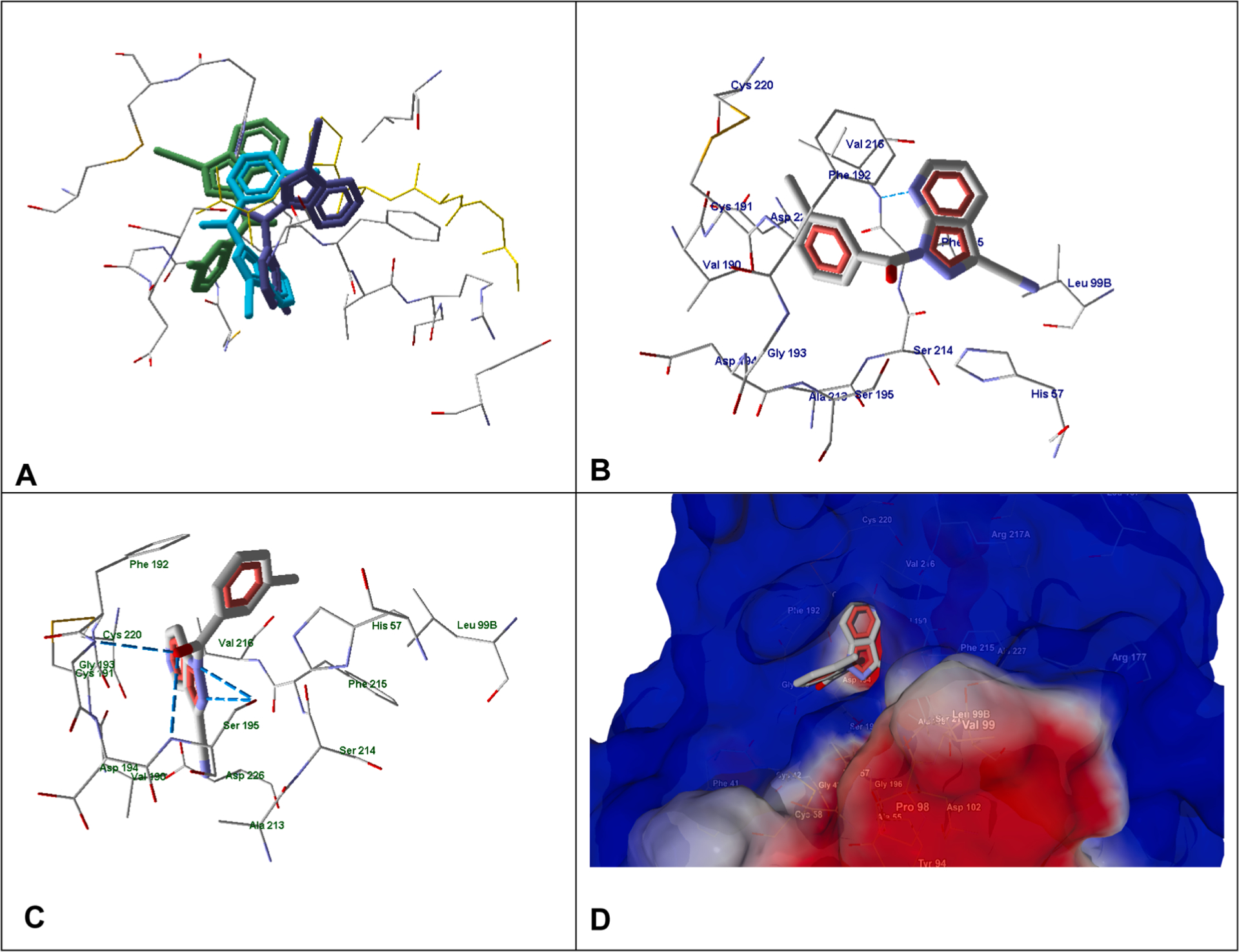
Docking poses of 2e, 2p and reference compound A. Panel A. Docking poses of 2e (violet), 2p (light blue), and reference compound A (green). Co-crystallized methoxysuccinyl-Ala-Ala-Pro-Ala chloromethyl ketone (MSACK) is shown in yellow sticks. Residues within 4 Å from the co-crystallized ligand are visible. Panel B. Docking pose of compound 2e. Panel C. Docking pose of compound 2p. For Panels B and C, residues within 4 Å of the pose are visible. Panel D. Docking pose of compound 2p. The semi-transparent surface of HNE is shown.
Compound 4 containing nitro and cyclopropyl groups is anchored in the HNE binding site due to H-bonds with Val216 (with the participation of the nitro group), Gly193, and Ser195 (with the participation of the amide oxygen atom) (Fig. 6A). Compound 2r has two potential centers for nucleophilic attack by Ser195 (i.e., carbonyl oxygen atoms of the amide and ester groups). H-bonds with Gly193 formed with participation of the amide oxygen atom and one of the nitrogen atoms in the five-membered ring play a role in anchoring 2r inside the binding site (Fig. 6B). The d1 distances O=C⋯O(Ser195) in the pose obtained for compound 2r are 2.946 and 3.817 Å for the carbonyl carbon atoms of the ester and amide groups, respectively (Table 5). From this point of view, nucleophilic attack on the ester group is more likely. In addition, the angle α in this case is 86.7°, being in the range of 80–120°, which is favorable for formation of the Michaelis complex,44–46 which is in contrast to α = 132.9° obtained for the amide C=O bond.
Fig. 6.
Docking pose of compound 4 (Panel A) and 2r (Panel B). For both panels, residues within 4 Å of the pose are visible.
Table 5.
HNE inhibitory activity of selected compounds and geometric parameters of the enzyme–inhibitor complexes predicted by molecular modelling.a
| Compound | IC50 (nM) | α(°) | d1 | d2 | d3 | L |
|---|---|---|---|---|---|---|
| Å | ||||||
| 2e | 330 | 76.3 | 3.620 | 2.196, 3.606 | 3.291 | 5.487 |
| 4 | 21 | 85.2 | 3.298 | 2.349, 3.883 | 3.294 | 5.643 |
| 2p | 10 | 89.8 | 3.001 | 1.790, 3.334 | 2.826 | 4.616 |
| 2r | 16 | 86.7 | 2.946b | 1.769, 3.340 | 2.856 | 4.625 |
| 13e | 194 | 64.9 | 3.622 | 1.857, 3.396 | 2.872 | 4.729 |
| 13a | 89 | 79.7 | 3.256 | 4.852, 5.345 | 2.478 | 7.330 |
| 13c | 14 | 140.0 | 4.324 | 1.790, 3.334 | 2.815 | 4.605 |
Geometric parameters correspond to the formation of Michaelis complex with the ester carbonyl group.
The indicated d1 value corresponds to the carbonyl carbon atom of the ester function of compound 2r. For amide carbon atom of this compound, the d1 distance equals 3.817 Å.
A comparative analysis of molecules containing one nitrogen atom in the five-membered ring (13a, 13c, 13e, and B21) shows that they have a somewhat similar orientation within the binding site (Fig. 7A). Both heterocyclic and m-methylbenzoyl groups were located approximately in the same corresponding regions of space inside the enzyme. Some features of their interactions with the HNE amino acids should be mentioned and are illustrated in Fig. 7. Compounds of this series form H-bonds with Gly193 involving participation of the amide oxygen atom (13e and 13c), or with Ser195 involving participation of the oxygen and nitrogen atoms of the amide function (13e and 13a). In addition, compound 13a is H-bonded to Val216 via the nitrogen atom at position 4 of the heterocycle. A comparison of the docking poses of compounds 13c and 2p, of which the latter has an additional nitrogen atom in the five-membered ring instead of the CH group, showed a slight difference in orientation of the molecules within the binding site (not shown). Although positions of cyano and carbonyl groups of these compounds are close in space, the heterocyclic and m-toluoyl fragments do not mutually coincide. Obviously, the difference in the arrangement of 2p and 13c docking poses is due to peculiarities of the 2p H-bonding pattern (Fig. 7, panel C).
Fig. 7.
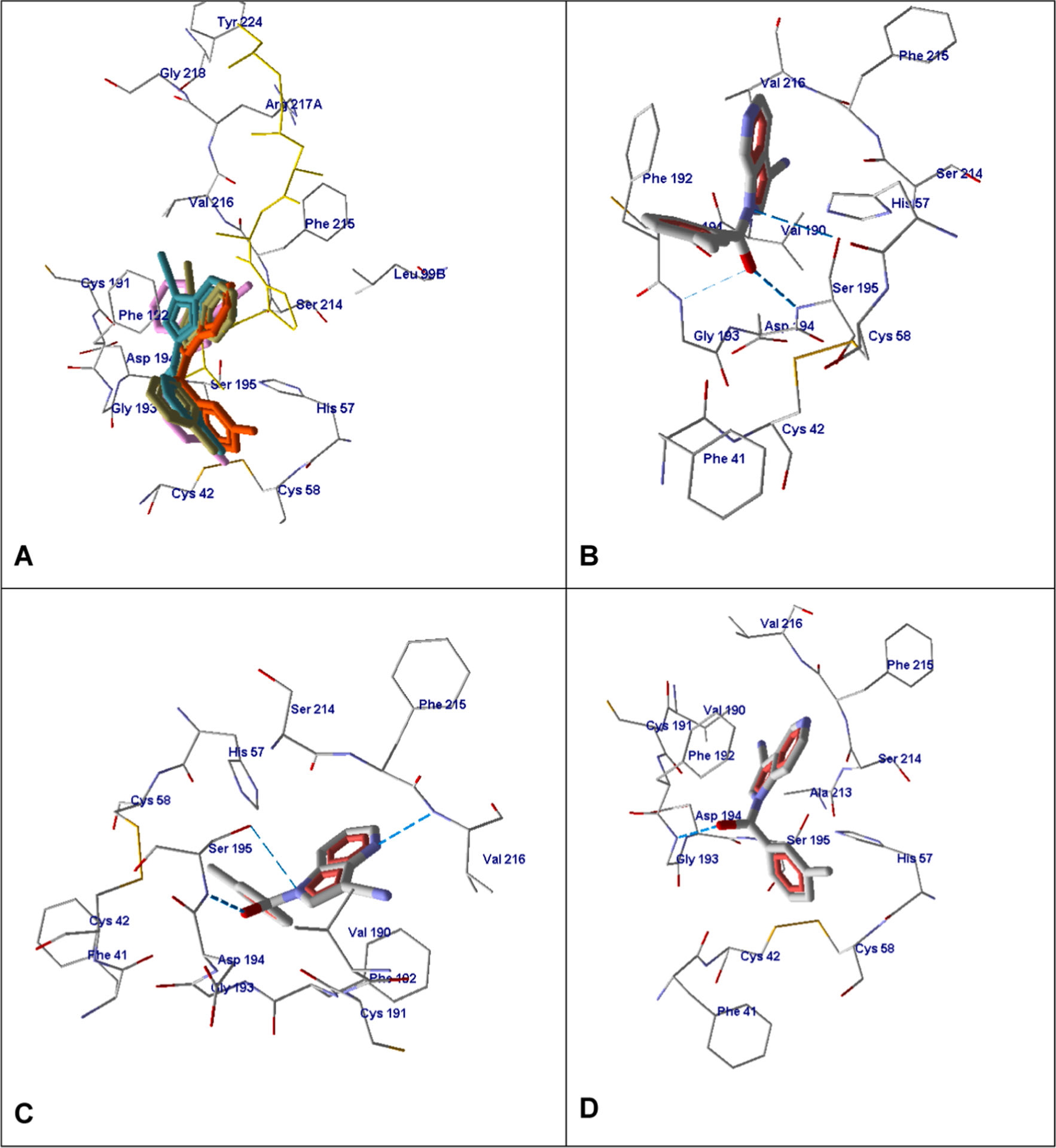
Docking poses of 13e, 13a, 13c, and reference compound B. Panel A. Docking poses of compounds B (magenta), 13e (dark green), 13a (light blue), and 13c (orange). Co-crystallized MSACK is shown in yellow sticks. Residues within 4 Å of the co-crystallized ligand are visible. Panel B. Docking pose of compound 13e. Panel C. Docking pose of compound 13a. Panel D. Docking pose of compound 13c. For Panels B-D residues within 4 Å of the pose are visible.
Table 5 shows the geometric parameters (distance d1 and angle α) for the docking poses, as well as the distances d2 and d3 between the key atoms of the catalytic triad of Ser195, His57, and Asp102 (Fig. 4), which determine the length L of the proton transfer channel. Values of α for most of the compounds evaluated were between 80 and 120° and are optimal for formation of the Michaelis complex. The exceptions were the less active compounds 2e and 13e. The pose of another compound with reduced activity (13a) is characterized by an angle α close to 80°; however, the proton transfer channel L for this molecule has a rather high value due to mutual displacements of the residues of the catalytic triad upon binding of 13c to HNE.
2.4. ADMET assessment
To facilitate selection of lead compounds and to further support the molecular modelling and in vitro evaluation, we also performed an in silico absorption, distribution, metabolism, and excretion-toxicity (ADMET) pharmacokinetics evaluation. The in silico assessment was generated through the evaluation of pharmacokinetic profiles and possible adverse side effects for compounds 2a-t, 4, 6, 7, 9, 13a-f, 15, and 19. ADMET molecular studies were conducted using SwissADME (http://swissadme.ch)47 and pkCSM (http://biosig.unimelb.edu.au/pkcsm/)48 (see Supplemental Tables 1S–10S). Most of the compounds were predicted to be orally available, with high gastrointestinal absorption and high water solubility (Tables 2S, 3S). Compounds with predicted moderate solubility (10−5 mol/L range) were 2a, 2i, 2l, 2m, 2n, and 2r. Only compounds 2o, 6 and 7 were predicted to be P-glycoprotein substrates, whereas most of the compounds were predicted to be inhibitors of the CYP1A2. Some compounds were also predicted to inhibit CYP2C19/CYP2C9, but no inhibition was predicted for CYP3A4 and CYP2D6. Otherwise, most of the compounds were predicted to be CYP3A4 substrates but should not be metabolized by CYP2D6 (Table 7S). Interestingly, none of the compounds violated the Lipinski rule of 5, nor did they violate the other drug-likeness rules (Ghose, Egan, Veber, and Muegee).49–53
The absorption and distribution calculated parameters are shown in an Egan–Egg model (Fig. 8) (Brain or IntestinaL EstimateD, BOILED-Egg). The Egan–Egg model highlights that most of the compounds were predicted to passively permeate the blood-brain barrier, whereas compounds 2g, 2q, 2s, 4, 6, 7, and 19 are only able to be passively absorbed by the gastrointestinal tract pkCSM calculated absorption properties showed > 95% intestinal absorption (aside from molecule 6 with 77.3%), due to the calculated optimal (>0.90) Caco-2 cell permeability (Table 5S). Moreover, most of the compounds are predicted to be adsorbed by the skin (exploitable for transdermal drug delivery) as shown by the Log Kp < −2.5 (see skin permeability, Table 5S).
Fig. 8.
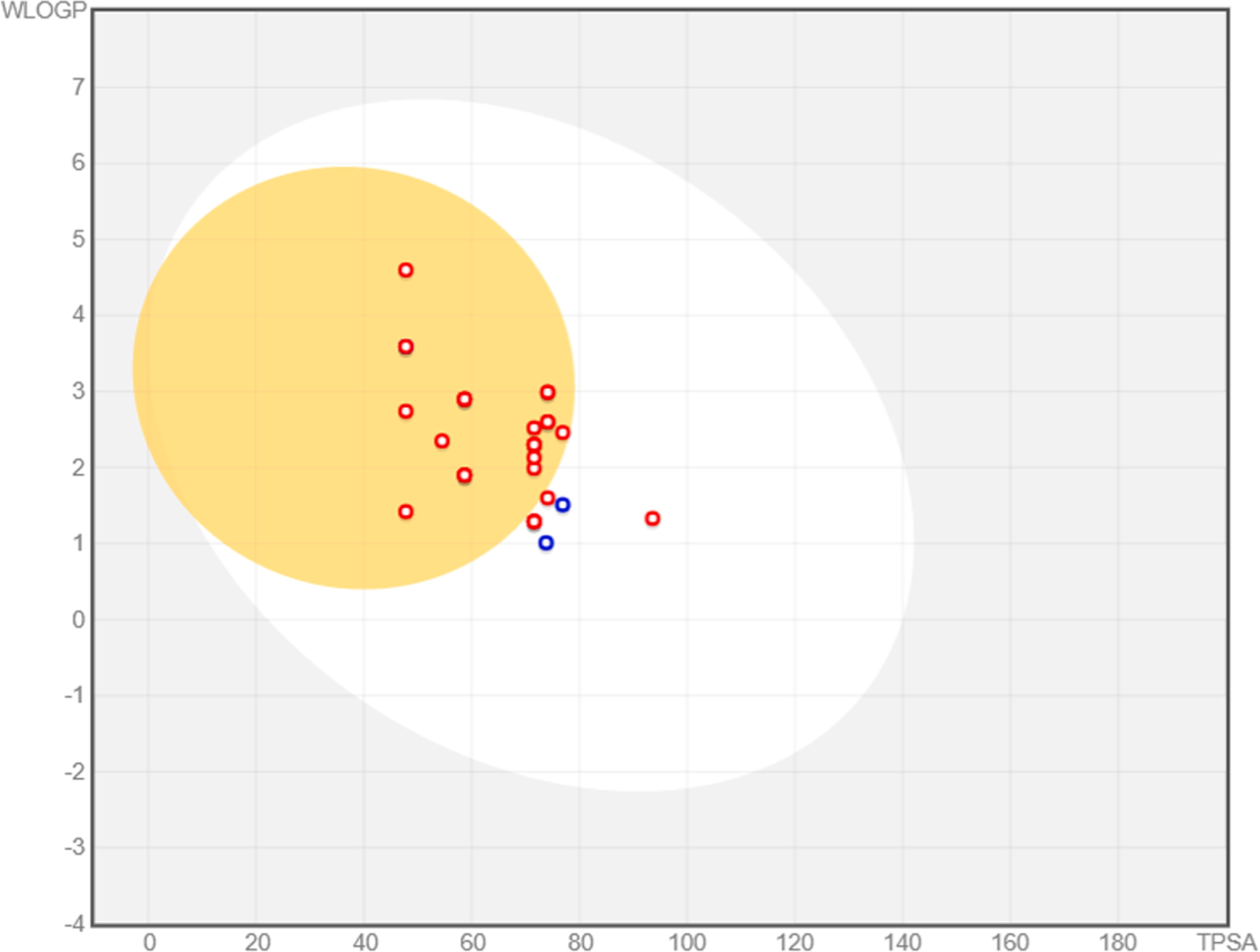
BOILED-Egg plot. Points located in the BOILED-Egg’s yellow are the compounds predicted to permeate the BBB passively, differently the ones in the white are the molecules predicted to be only passively absorbed by the gastrointestinal tract. Blue dots indicate molecules expected to be refluxed from the central nervous system (CNS) by the P-glycoprotein, whereas the red ones are not transported by the P-glycoprotein.
The calculated values of steady state volume of distribution are relatively low for some compounds (Log VDss < −0.15, Table 6S), but most of the compounds were predicted to have a significant unbound fraction in the plasma, thus becoming available for interaction with the pharmacological target (Table 6S). The calculated values of the total clearance (Table 8S) indicate that most of the compounds were predicted to have good renal elimination (0.20–0.84 mL/min/kg), and no compounds were predicted to be substrates of the renal organic cation transporter 2. pkCSM calculated toxicity properties indicated concerns about the AMES test, the T. pyriformis toxicity and hepatoxicity. On the other hand, the compounds did not have skin sensitization properties and were not predicted to be inhibitors of hERG I and II (Table 9S). Due to the toxicology concerns indicated by pkCSM, toxicology was also evaluated with Datawarrior (v. 5.2.1 http://www.openmolecules.org/datawarrior/). In this case, all compounds were predicted to be non-mutagenic, non-tumorigenic, and not irritants. A low risk for reproductive system effects was associated with molecules 2l-n and 2r,s (Table 10S). In silico toxicity evaluations were also performed with preadmet (https://preadmet.bmdrc.kr/toxicity/) and admetsar1 (http://lmmd.ecust.edu.cn/admetsar1). The results for preadmet are presented in Table S11, while the results for admetsar1 are presented in Tables 12S–41S. Preadmet considered most of the compounds mutagenic in the same test, whereas admetsar1 considered most of the compounds as nontoxic in the same test. Despite this result, most of the compounds were considered non-carcinogenic for mouse or rat, with a medium to low risk for their hERG inhibition in both in silico evaluations. Further biological evaluation is needed due to the somewhat contrasting test results, but the ADMET general profile was good for our set of compounds, particularly the most potent compounds 2p, 2r, 4, 13b, and 13c, which had excellent ADMET profiles and shouldn’t have any particular toxicity issues. The IC50 measured potency, together with the ADMET profile will be of fundamental importance for the future selection of compounds for in vivo evaluation.
3. Experimental section
3.1. Chemistry
All melting points were determined on a Büchi apparatus (New Castle, DE) and are uncorrected. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. Merck F-254 commercial plates (Merck, Durham, NC) were used for analytical TLC to follow the course of reactions. Silica gel 60 (Merck 70–230 mesh, Merck, Durham, NC) was used for column chromatography. 1H NMR and 13C NMR spectra were recorded on an Avance 400 instrument (Bruker Biospin Version 002 with SGU, Bruker Inc., Billerica, MA). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using the solvent as an internal standard. Coupling constants (J values) are given in Hz and were calculated using TopSpin 4.0.8 software (Nicolet Instrument Corp., Madison, WI) and are rounded to the nearest 0.1 vHz. High resolution mass spectrometry (HRMS) analysis was performed with a Thermo Finnigan LTQ Orbitrap mass spectrometer equipped with an electrospray ionization source (ESI). The solvents used in HRMS were acetonitrile (Chromasolv grade) purchased from Sigma-Aldrich (Milan, Italy) and mQ water 18 MΩ cm, obtained from Millipore’s Simplicity system (Milan, Italy). The accurate mass measure was performed by introducing sample solution (1.0 μg Ml−1 in mQ water: acetonitrile 50:50) via syringe pump at 10 μL min−1, and the signal of the positive ions was acquired. The experimental conditions allowed monitoring of the protonated molecules ([M + H]+ species) with a proper dwell time to achieve 60.000 units of resolution at full width at half maximum (FWHM). Microanalyses indicated by the symbols of the elements or functions were performed with a Perkin–Elmer 260 elemental analyzer (PerkinElmer, Inc., Waltham, MA) for C, H, and N, and the results were within ± 0.4% of the theoretical values, unless otherwise stated. Reagents and starting material were commercially available.
3.1.1. General procedure for compounds (2a,b, 2l-o, and 2r-t)
To a cooled (0 °C) suspension of the appropriate substrate 1a,17 1c, d,18 1e,19 1 g,20 or 1h21 (0.32 mmol) in anhydrous CH2Cl2 (2 mL), 0.64 mmol of Et3N and 0.96 mmol of the appropriate acyl/aroyl chloride were added. The mixture was stirred at 0 °C for 2 h and then at room temperature for an additional 2 h. The solvent was evaporated, cold water was added, and the mixture was neutralized with 0.5 N NaOH. The reaction mixture was extracted with CH2Cl2 (3 × 15 mL), the solvent was dried over sodium sulfate and evaporated in vacuum. The final compound 2o was purified by crystallization with ethanol while the compounds 2a,b,l-n,r-t were purified by flash column chromatography using cyclohexane/ethyl acetate 3:1 (2a,b,n) or 1:1 (2s,t), hexane/ethyl acetate 5:2 (2l,m), dichloromethane/methanol 9:1 (2r) as eluents. Finally, all compounds were crystallized from ethanol, and melting points were performed after crystallization.
m-Tolyl-(3-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridin-1-yl) methanone (2a).
Yield = 26%; mp = 67–69 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H, m-CH3-Ph), 7.38–7.49 (m, 3H, Ar), 7.80 (d, 1H, Ar, J = 7.2 Hz), 7.85 (s, 1H, Ar), 8.26 (d, 1H, Ar, J = 8.0), 8.89 (d, 1H, Ar, J = 3.2). 13C NMR (100 MHz, CDCl3) δ 21.3 (CH3), 117.7 (C), 121.0 (CH), 122.8 (C), 128.1 (CH), 128.8 (CH), 129.7 (CH), 130.4 (C), 131.9 (CH), 134.4 (CH), 138.2 (C), 142.1 (C), 152.1 (CH), 159.1 (C), 165.0 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C15H11F3N3O = 306,0849, found 306,0847. Anal. C15H10F3N3O (C, H, N).
Cyclopropyl(3-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridin-1-yl)methanone (2b).
Yield = 23%; oil. 1H NMR (400 MHz, CDCl3) δ 1.16–1.23 (m, 2H, CH2 cC3H5), 1.38–1.44 (m, 2H, CH2 cC3H5), 3.31–3.36 (m, 1H, COCH), 7.43–7.48 (m, 1H, Ar), 8.24 (d, 1H, Ar, J = 8.0), 8.85 (d, 1H, Ar, J = 2.8). 13C NMR (100 MHz, CDCl3) δ 11.9 (CH2), 13.6 (CH), 114.3 (C), 118.8 (C), 120.9 (CH), 121.9 (C), 129.7 (CH), 151.6 (C), 152.1 (CH), 172.5 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C11H9F3N3O = 256,0692, found 256,0689. Anal. C11H8F3N3O (C, H, N).
Ethyl 1-(3-methylbenzoyl)-1H-pyrazolo[3,4-b]pyridine-3-carboxylate (2l).
Yield = 30%; mp = 185–187 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.47 (t, 3H, OCH2CH3, J = 6.8), 2.44 (s, 3H, m-CH3-Ph), 4.52 (q, 2H, OCH2CH3, J = 6.8), 7.40–7.48 (m, 3H, Ar), 7.83 (d, 1H, Ar, J = 7.2), 7.88 (s, 1H, Ar), 8.60 (d, 1H, Ar, J = 8.0), 8.83 (d, 1H, Ar, J = 4.0). 13C NMR (100 MHz, CDCl3) δ 14.3 (CH3), 21.3 (CH3), 62.0 (CH2), 108.0 (C), 121.1 (CH), 128.1 (CH), 128.9 (CH), 130.5 (C), 131.8 (CH), 132.1 (CH), 133.0 (C), 134.3 (CH), 136.7 (C), 138.1 (C), 151.4 (CH), 159.1 (C), 165.0 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C17H16N3O3 = 310,1186, found 310,1183. Anal. C17H15N3O3 (C, H, N).
Isopropyl 1-(3-methylbenzoyl)-1H-pyrazolo[3,4-b]pyridine-3-carboxylate (2m).
Yield = 28%; mp = 192–194 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.46 (d, 6H, CH(CH3)2, J = 6.0), 2.44 (s, 3H, m-CH3-Ph), 5.43–5.48 (m, 1H, CH(CH3)2), 7.40–7.50 (m, 3H, Ar), 7.85 (d, 1H, Ar, J = 7.6), 7.90 (s, 1H, Ar), 8.57 (d, 1H, Ar, J = 8.0), 8.82 (d, 1H, Ar, J = 3.2). 13C NMR (100 MHz, CDCl3) δ 21.3 (CH3), 21.9 (CH3), 70.0 (CH), 116.9 (C), 121.0 (CH), 128.3 (CH), 129.0 (CH), 131.8 (CH), 132.1 (CH), 134.2 (CH), 138.1 (C), 138.9 (C), 151.3 (CH), 153.2 (C), 160.8 (C), 166.7 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C18H18N3O3 = 324,1343, found 324,1346. Anal. C18H17N3O3 (C, H, N).
m-Tolyl(3-(trifluoromethyl)-1H-pyrazolo[4,3-c]pyridin-1-yl)methanone (2n).
Yield = 66%; mp = 80–81 °C (colorless solid). 1H NMR (400 MHz, CDCl3) δ 2.46 (s, 3H, m-CH3-Ph), 7.41–7.49 (m, 2H, Ar), 7.92 (d, 2H, Ar, J = 6.4), 8.42 (d, 1H, Ar, J = 5.6), 8.80 (d, 1H, Ar, J = 6.0), 9.29 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 21.4 (CH3), 110.4 (CH), 116.6 (C), 118.9 (C), 121.7 (C), 128.2 (CH), 128.8 (CH), 130.7 (C), 131.9 (CH), 134.5 (CH), 138.3 (C), 144.2 (CH), 145.0 (C), 148.4 (CH), 167.6 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C15H11F3N3O = 306,0849, found 306,0846. Anal. C15H10F3N3O (C, H, N).
Cyclopropyl(3-(trifluoromethyl)-1H-pyrazolo[4,3-c]pyridin-1-yl)methanone (2o).
Yield = 30%; mp = 93–94 °C (colorless solid). 1H NMR (400 MHz, CDCl3) δ 1.25–1.30 (m, 2H, CH2 cC3H5), 1.39–1.44 (m, 2H, CH2 cC3H5), 3.18–3.24 (m, 1H, COCH), 8.37 (d, 1H, Ar, J = 6.0), 8.75 (d, 1H, Ar, J = 5.6), 9.28 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 12.0 (CH2), 12.9 (CH), 110.1 (CH), 119.0 (C), 121.7 (C), 143.4 (C), 144.1 (CH), 148.3 (CH), 174.3 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C11H9F3N3O = 256,0692, found 256,0693. Anal. C11H8F3N3O (C, H, N).
Ethyl 1-(3-methylbenzoyl)-1H-pyrazolo[4,3-c]pyridine-3-carboxylate (2r).
Yield: 49%; mp = 87–89 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 1.48 (t, 3H, CH3CH2, J = 7.2), 2.44 (s, 3H, m-CH3-Ph), 4.55 (q, 2H, CH3CH2, J = 7.2), 7.39–7.46 (m, 2H, Ar), 7.94 (d, 2H, Ar, J = 7.2), 8.36 (d, 1H, Ar, J = 6.0), 8.74 (d, 1H, Ar, J = 5.6), 9.58 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 14.3 (CH3), 21.4 (CH3), 62.3 (CH2), 110.1 (CH), 120.8 (C), 128.3 (CH), 129.0 (CH), 131.1 (C), 132.1 (CH), 134.4 (CH), 138.3 (C), 141.0 (C), 145.3 (C), 146.3 (CH), 147.8 (CH), 160.8 (C), 168.0 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C17H16N3O3 = 310,1186, found 310,1182. Anal. C17H15N3O3 (C, H,N).
Ethyl 1-(cyclopropanecarbonyl)-1H-pyrazolo[4,3-c]pyridine-3-carboxylate (2s).
Yield: 44%; mp = 94–96 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.23 (m, 2H, CH2 cC3H5), 1.36 (m, 2H, CH2 cC3H5), 1.50 (t, 3H, CH2CH3, J = 7.2), 3.25–3.31 (m, 1H, CH cC3H5), 4.57 (q, 2H, CH2CH3, J = 7.2), 8.25 (d, 1H, Ar, J = 5.6), 8.67 (d, 1H, Ar, J = 5.6), 9.52 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 8.8 (CH), 12.2 (CH2), 14.1 (CH3), 60.9 (CH2), 105.3 (C), 113.7 (CH), 126.2 (C), 136.7 (C), 143.1 (CH), 148.0 (CH), 160.6 (C), 172.3 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C13H14N3O3 = 260,1030, found 260,1030. Anal. C13H13N3O3 (C,H,N).
(3-Methyl-1H-pyrazolo[4,3-c]pyridin-1-yl)(m-tolyl)methanone (2t).
Yield = 43%; mp = 104–105 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 2.45 (s, 3H, m-CH3-Ph), 2.67 (s, 3H, 3-CH3), 7.39–7.45 (m, 2H, Ar), 7.89 (d, 2H, Ar, J = 8.8), 8.38 (d, 1H, Ar, J = 6.6), 8.70 (d, 1H, Ar, J = 5.6), 9.11 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 12.4 (CH3), 21.4 (CH3), 102.9 (C), 110.3 (CH), 122.8 (C), 127.9 (CH), 128.4 (CH), 131.5 (CH), 132.4 (C), 133.5 (CH), 138.9 (C), 144.0 (CH), 144.8 (C), 147.9 (CH), 148.5 (C), 168.1 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C15H14N3O = 252,1131, found 252,1129. Anal. C15H13N3O (C, H, N).
3.1.2. 1-Benzyl-1H-pyrazolo[3,4-b]pyridine-3-carbonitrile (2c)
To a suspension of the intermediate 1b17 (0.42 mmol) in anhydrous CH3CN (2 mL), 0.50 mmol of K2CO3 and 0.84 mmol of benzyl bromide were added. The mixture was stirred at reflux for 4 h. After evaporation of the solvent, ice-cold water (20 mL) was added and the precipitate was recovered by vacuum filtration. The final compound 2c was purified by crystallization with ethanol. Yield = 40%; mp = 110–112 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 5.77 (s, 2H, CH2-Ph), 7.42–7.49 (m, 6H, Ar), 8.18 (d, 1H, Ar, J = 8.0), 8.68 (d, 1H, Ar, J = 2.8). 13C NMR (100 MHz, CDCl3) δ 52.1 (CH2), 105.0 (C), 112.9 (C), 117.1 (C), 119.6 (CH), 128.4 (CH), 128.4 (CH), 128.8 (CH), 135.3 (C), 150.6 (CH), 154.5 (C). IR = 2241 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C14H11N4 = 235,0978, found 235,0981. Anal. C14H10N4 (C, H, N).
3.1.3. 1-Benzoyl-1H-pyrazolo[3,4-b]pyridine-3-carbonitrile (2d)
To a solution of intermediate 1b17 (1.04 mmol), 2.08 mmol of Benzoic acid and 1.56 mmol of HOBt in dry THF (5 mL), 2.08 mmol of triethylamine was added. The solution was brought to 0 °C, 1.56 mmol of N,N’-dicyclohexyl carbodiimide (DCC) was added and stirred at 0 °C for about 15′. The reaction mixture was stirred at room temperature for 48 h. After evaporation of the solvent, the crude compound was diluted in CH2Cl2 and first washed with 2 M Na2CO3 solution and then with brine. The organic phase was recovered, dried over sodium sulfate and evaporated under vacuum. The final compound 2d was purified by crystallization with ethanol. Yield = 16%; mp = 208–210 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 7.55–7.65 (m, 3H, Ar), 7.68 (d, 1H, Ar, J = 6.0), 8.03 (d, 2H, Ar, J = 6.4), 8.31 (d, 1H, Ar, J = 6.8), 8.93 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 111.6 (C), 118.1 (C), 121.6 (CH), 122.3 (C), 128.4 (CH), 129.2 (CH), 131.1 (C), 131.6 (CH), 133.9 (CH), 152.7 (CH), 155.0 (C), 165.7 (C). IR = 2243 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C14H9N4O = 249,0771, found 249,0769. Anal. C14H8N4O (C, H, N).
3.1.4. General procedure for compounds (2e-i, 2p, 2q)
To a suspension of the appropriate substrate 1b17 or 1f19 (0.86 mmol) in 10 mL of anhydrous THF, 1.72 mmol of sodium hydride and 1.03 mmol of the appropriate acyl/aroyl chloride were added. The mixture was stirred at room temperature overnight. The solvent was evaporated, cold water was added, and the mixture was neutralized with 2.5 N NaOH. The precipitate was recovered by vacuum filtration to obtain the final compounds 2e-i,p,q, which were purified by crystallization with ethanol.
1-(3-Methylbenzoyl)-1H-pyrazolo[3,4-b]pyridine-3-carbonitrile (2e).
Yield = 25%; mp = 210–213 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 2.46 (s, 3H, m-CH3-Ph), 7.41–7.55 (m, 3H, Ar), 7.77–7.82 (m, 2H, Ar), 8.31 (d, 1H, Ar, J = 8.0), 8.92 (d, 1H, Ar, J = 2.8). 13C NMR (100 MHz, CDCl3) δ 21.7 (CH3), 105.0 (C), 110.5 (C), 114.2 (C), 119.6 (CH), 127.3 (CH), 128.4 (CH), 129.8 (CH), 130.4 (C), 130.7 (CH), 133.0 (C), 134.4 (CH), 138.9 (C), 150.2 (CH), 165.7 (C). IR = 2241 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C15H11N4O = 263,0927, found 263,0926. Anal. C15H10N4O (C, H, N).
1-(4-Methylbenzoyl)-1H-pyrazolo[3,4-b]pyridine-3-carbonitrile (2f).
Yield = 10%; mp = 140–143 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 2.48 (s, 3H, p-CH3-Ph), 7.37 (d, 2H, Ar, J = 8.0), 7.50–7.55 (m, 1H, Ar), 7.94 (d, 2H, Ar, J = 8.0), 8.30 (d, 1H, Ar, J = 8.0), 8.92 (d, 1H, Ar, J = 3.6). 13C NMR (100 MHz, CDCl3) δ 21.9 (CH3), 105.0 (C), 111.7 (C), 118.0 (C), 121.5 (CH), 122.0 (C), 127.9 (CH), 129.2 (CH), 132.4 (CH), 145.3 (C), 151.9 (C), 152.6 (CH), 165.6 (C). IR = 2241 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C15H11N4O = 263,0927, found 263,0927. Anal. C15H10N4O (C, H, N).
1-(Cyclopropanecarbonyl)-1H-pyrazolo[3,4-b]pyridine-3-carbonitrile (2 g).
Yield = 34%; mp = 147–149 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.24–1.29 (m, 2H, CH2 cC3H5), 1.45–1.50 (m, 2H, CH2 cC3H5), 3.32–3.37 (m, 1H, COCH), 7.48–7.53 (m, 1H, Ar), 8.27 (d, 1H, Ar, J = 8.0), 8.88 (d, 1H, Ar, J = 4.4). 13C NMR (100 MHz, CDCl3) δ 12.3 (CH2), 13.7 (CH), 105.0 (C), 110.4 (C), 114.1 (C), 121.4 (CH), 129.2 (CH), 133.0 (C), 152.5 (CH), 173.6 (C). IR = 2243 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C11H9N4O = 213,0771, found 213,0772. Anal. C11H8N4O (C, H, N).
1-(Cyclopentanecarbonyl)-1H-pyrazolo[3,4-b]pyridine-3-carbonitrile (2 h).
Yield = 32%; mp = 123–125 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.73–1.79 (m, 2H, CH2 cC5H9), 1.80–1.85 (m, 2H, CH2 cC5H9), 1.98–2.08 (m, 2H, CH2 cC5H9), 2.10–2.16 (m, 2H, CH2 cC5H9), 4.12 (quin, 1H, COCH, J = 8.0), 7.47–7.53 (m, 1H, Ar), 8.25 (d, 1H, Ar, J = 8.0), 8.89 (d, 1H, Ar, J = 4.0). 13C NMR (100 MHz, CDCl3) δ 25.8 (CH2), 26.2 (CH2), 30.0 (CH2), 30.3 (CH2), 44.2 (CH), 111.7 (C), 118.3 (C), 121.4 (CH), 122.2 (C), 129.1 (CH), 151.0 (C), 152.6 (CH), 174.4 (C). IR = 2243 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C13H13N4O = 241,1084, found 241,108. Anal. C13H12N4O (C, H, N).
1-(Cyclohexanecarbonyl)-1H-pyrazolo[3,4-b]pyridine-3-carbonitrile (2i).
Yield = 24%; mp = 124–127 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.30–1.50 (m, 4H, 2 × CH2 cC6H11), 1.73–1.78 (m, 2H, CH2 cC6H11), 1.88 (d, 2H, CH2 cC6H11, J = 16.0), 2.05 (d, 2H, CH2 cC6H11, J = 16.0), 3.76 (t, 1H, COCH, J = 8.0), 7.47–7.52 (m, 1H, Ar), 8.25 (d, 1H, Ar, J = 8.0), 8.89 (d, 1H, Ar, J = 4.0). 13C NMR (100 MHz, CDCl3) δ 25.3 (CH2), 25.6 (CH2), 29.0 (CH2), 43.9 (CH), 111.6 (C), 118.2 (C), 121.4 (CH), 122.2 (C), 129.1 (CH), 151.0 (C), 152.6 (CH), 171.1 (C). IR = 2243 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C14H15N4O = 255,1240, found 255,1243. Anal. C14H14N4O (C, H, N).
1-(3-Methylbenzoyl)-1H-pyrazolo[4,3-c]pyridine-3-carbonitrile (2p).
Yield = 47%; mp = 192–194 °C (colorless solid). 1H NMR (400 MHz, CDCl3) δ 2.48 (s, 3H, m-CH3-Ph), 7.46 (t, 1H, Ar, J = 7.6), 7.52 (d, 1H, Ar, J = 7.2), 7.88 (d, 2H, Ar, J = 6.8), 8.45 (d, 1H, Ar, J = 6.0), 8.84 (d, 1H, Ar, J = 6.0), 9.34 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 21.4 (CH3), 110.5 (CH), 111.2 (C), 122.1 (C), 124.0 (C), 128.4 (CH), 128.8 (CH), 130.3 (C), 131.9 (CH), 134.9 (CH), 138.5 (C), 143.7 (CH), 144.2 (C), 148.8 (CH), 167.3 (C). IR = 2241 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C15H11N4O = 263,0927, found 263,0926. Anal. C15H10N4O (C, H, N).
1-(Cyclopropanecarbonyl)-1H-pyrazolo[4,3-c]pyridine-3-carbonitrile (2q).
Yield = 44%; mp = 152–154 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.30–1.35 (m, 2H, CH2 cC3H5), 1.42–1.47 (m, 2H, CH2 cC3H5), 3.14–3.20 (m, 1H, COCH), 8.41 (d, 1H, Ar, J = 6.0), 8.78 (d, 1H, Ar, J = 5.6), 9.34 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 12.4 (CH2), 12.9 (CH), 110.2 (CH), 111.2 (C), 122.2 (C), 123.7 (C), 142.6 (C), 143.7 (CH), 148.8 (CH), 173.9 (C). IR = 2243 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C11H9N4O = 213,0771, found 213,0768. Anal. C11H8N4O (C, H, N).
3.1.5. Cyclopropyl(3-nitro-1H-pyrazolo[3,4-b]pyridin-1-yl)methanone (4)
Compound 4 was obtained using the general procedure followed for compounds 2e-i, 2p, 2q, but starting from intermediate 3.22 The compound 4 was purified by crystallization with ethanol. Yield = 30%; mp = 137–139 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.28–1.33 (m, 2H, CH2 cC3H5), 1.47–1.52 (m, 2H, CH2 cC3H5), 3.29–3.35 (m, 1H, COCH), 7.58–7.63 (m, 1H, Ar), 8.68 (dd, 1H, Ar, J1 = 1.4 and J2 = 8.0), 8.90 (d, 1H, Ar, J = 4.4). 13C NMR (100 MHz, CDCl3) δ 12.6 (CH2), 13.8 (CH), 110.6 (C), 122.7 (CH), 131.2 (CH), 142.6 (C), 151.8 (C), 152.9 (CH), 172.2 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C10H9N4O3 = 233,0669, found 233,0671. Anal. C10H8N4O3 (C, H, N).
3.1.6. General procedure for compounds (6–8)
1H-pyrazolo[3,4-b]pyridin-3-amine (522) (1.27 mmol) was dissolved in 8 mL of DMF and triethylamine (12.67 mmol). The appropriate acyl/aroyl chloride (1.33 mmol) dissolved in 2.5 mL dry 1,4-dioxane was added dropwise to the reaction mixture at 10 °C, then left stirring at 50 °C for 4 h. Ice water was added to the reaction mixture, and the product was extracted with ethyl acetate (3 × 15 mL), dried over sodium sulfate, and evaporated under vacuum. The crude compounds 6–8 were purified by flash column chromatography using dicloromethane/methanol 95:5 as eluent. Finally, all compounds were crystallized from ethanol, and melting points were performed after crystallization.
(3-Amino-1H-pyrazolo[3,4-b]pyridin-1-yl)(cyclopropyl)methanone (6).
Yield = 5%; oil. 1H NMR (400 MHz, DMSO-d6) δ 0.81–0.88 (m, 2H, CH2 cC3H5), 1.19–1.25 (m, 2H, CH2 cC3H5), 3.50–3.57 (m, 1H, COCH), 6.53 (exch br s, 2H, NH2), 7.33–7.38 (m, 1H, Ar), 8.30 (d, 1H, Ar, J = 6.0), 8.57 (s, 1H, Ar). 13C NMR (100 MHz, DMSO-d6) δ 8.7 (CH), 12.2 (CH2), 106.5 (C), 114.1 (CH), 126.7 (CH), 142.9 (C), 148.0 (CH), 155.3 (C), 174.0 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C10H11N4O = 203,0927, found 203,0925. Anal. C10H10N4O (C, H, N).
N-(1-(Cyclopropanecarbonyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)cyclopropanecarboxamide (7).
Yield = 15%; mp = 222–225 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 0.93–1.00 (m, 2H, CH2 cC3H5), 1.13–1.19 (m, 4H, 2 × CH2 cC3H5), 1.33–1.39 (m, 2H, CH2 cC3H5), 1.81–1.87 (m, 1H, NHCOCH), 3.60–3.65 (m, 1H, N-COCH), 7.25–7.30 (m, 1H, Ar), 8.65 (d, 1H, Ar, J = 1.2), 8.75 (d, 1H, Ar, J = 7.6), 9.41 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 25.3 (CH2), 25.6 (CH2), 28.9 (CH2), 43.2 (CH), 111.6 (C), 118.2 (C), 121.4 (CH), 122.2 (C), 129.1 (CH), 151.0 (C), 152.6 (CH), 174.1 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C14H15N4O2 = 271,1190, found 271,1193. Anal. C14H14N4O2 (C, H, N).
3-Methyl-N-(1H-pyrazolo[3,4-b]pyridin-3-yl)benzamide (8).
Yield = 15%; mp = 205–208 °C (white powder). 1H NMR (400 MHz, DMSO-d6) δ 2.37 (s, 3H, m-CH3-Ph), 7.13–7.18 (m, 1H, Ar), 7.40–7.45 (m, 2H, Ar), 7.80–7.85 (m, 2H, Ar), 8.27 (d, 1H, Ar, J = 6.8), 8.49 (s, 1H, Ar), 10.95 (exch br s, 1H, NHCO), 13.31 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 20.9 (CH3), 91.5 (C), 114.0 (CH), 124.5 (CH), 126.7 (CH), 128.7 (CH), 131.4 (CH), 132.4 (CH), 134.1 (C), 138.5 (C), 148.0 (CH), 150.7 (C), 154.0 (C), 164.7 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C14H13N4O = 253,1084, found 253,1082. Anal. C14H12N4O (C, H, N).
3.1.7. N-(1-Acetyl-1H-pyrazolo[3,4-b]pyridin-3-yl)-3-methylbenzamide (9)
Compound 9 was obtained following the general procedure performed for compounds 2a,b, 2l-o and 2r-t but starting from intermediate 8 and using acetyl chloride as reagent. The crude product was purified by flash column chromatography using cyclohexane/ethyl acetate 1:3 as eluent. Yield = 11%; oil. 1H NMR (400 MHz, CDCl3 + D2O) δ 2.43 (s, 3H, m-CH3-Ph), 2.91 (s, 3H, NHCOCH3), 7.30–7.35 (m, 1H, Ar), 7.40–7.45 (m, 2H, Ar), 7.76 (d, 1H, Ar, J = 6.0), 7.79 (s, 1H, Ar), 8.69 (d, 1H, Ar, J = 3.2), 8.84 (d, 1H, Ar, J = 8.4). 13C NMR (100 MHz, CDCl3) δ 20.9 (CH3), 21.5 (CH3), 106.4 (C), 113.9 (CH), 124.5 (CH), 126.7 (CH), 128.7 (CH), 131.4 (CH), 132.0 (C), 132.5 (CH), 134.1 (C), 138.5 (C), 148.0 (CH), 155.4 (C), 164.7 (C), 169.2 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C16H15N4O2 = 295,1190, found 295,1184. Anal. C16H14N4O2 (C, H, N).
3.1.8. General procedure for compounds (11a,b)
A mixture of the appropriate 1H-pyrrolo[2,3-b]pyridine-3-carbal-dehyde 10a24 or 10b24 (0.78 mmol) and hydroxylamine hydrochloride (2.35 mmol) in 2 mL of water was heated at 60 °C for 30 min, and NaHCO3 (2.35 mmol) was added to the reaction mixture, which was finally left under reflux for 4 h. The reaction mixture was cooled to room temperature, the solid obtained was filtered, washed with the excess of ice-cold water and dried. The crude products were recrystallized from hexane to obtain the pure compounds.
(E)-1H-Pyrrolo[3,2-b]pyridine-3-carbaldehyde oxime (11a).
Yield = 65%; mp = 221–222 °C (colorless solid). 1H NMR (400 MHz, DMSO-d6) δ 7.14–7.19 (m, 1H, Ar), 7.81 (d, 1H, Ar, J = 8.0), 7.84 (s, 1H, CH = N-OH), 8.38 (d, 1H, Ar, J = 4.4), 8.43 (s, 1H, Ar), 11.31 (exch br s, 1H, CH = N-OH), 11.73 (exch br s, 1H, NH). ESI-HRMS (m/z) calculated for [M + H]+ ion species C8H8N3O = 162,0662, found 162,0659. Anal. C8H7N3O (C, H, N).
(E)-1H-Pyrrolo[3,2-c]pyridine-3-carbaldehyde oxime (11b).
Yield = 91%; mp = 227–228 °C (colorless solid). 1H NMR (400 MHz, DMSO-d6) δ 7.39 (d, 1H, Ar, J = 5.6), 7.86 (s, 1H, CH = N-OH), 8.21 (d, 2H, Ar, J = 7.2), 9.15 (s, 1H, Ar), 11.38 (exch br s, 1H, CH = N-OH), 11.88 (exch br s, 1H, NH). ESI-HRMS (m/z) calculated for [M + H]+ ion species C8H8N3O = 162,0662, found 162,0665. Anal. C8H7N3O (C, H, N).
3.1.9. General procedure for compounds (12a,b)
A mixture of intermediate 11a or 11b (0.92 mmol) and 4 mL of POCl3 was stirred at reflux for 2 h. After cooling, ice-cold water (20 mL) was slowly added, and the precipitate was filtered under vacuum and washed with abundant cold water to obtain the desired compounds. Finally, both compounds were crystallized from ethanol, and melting points were performed after crystallization.
1H-Pyrrolo[3,2-b]pyridine-3-carbonitrile (12a).
Yield = 86%; mp > 300 °C (white powder). 1H NMR (400 MHz, DMSO-d6) δ 7.25–7.30 (m, 1H, Ar), 7.93 (d, 1H, Ar, J = 7.2), 8.46 (d, 2H, Ar, J = 3.2), 12.36 (exch br s, 1H, NH). IR = 2240 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C8H6N3 = 144,0556, found 144,0559. Anal. C8H5N3 (C, H, N).
1H-Pyrrolo[3,2-c]pyridine-3-carbonitrile (12b).
Yield = 46%; mp = 262–263 °C dec. (white powder). 1H NMR (400 MHz, DMSO-d6) δ 7.53 (d, 1H, Ar, J = 5.2), 8.34 (d, 1H, Ar, J = 5.6), 8.37 (s, 1H, Ar), 8.92 (s, 1H, Ar), 12.51 (exch br s, 1H, NH). IR = 2240 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C8H6N3 = 144,0556, found 144,0555. Anal. C8H5N3 (C, H, N).
3.1.10. General procedure for compounds (13a-f)
Compounds 13a-f were obtained using the general procedure followed for compounds 2a,b,l-o,r-t but starting from intermediate 12a-c (12c26). The crude products were purified by crystallization from ethanol (13a-c and 13f) or by flash column chromatography using cyclohexane/ethyl acetate 1:1 (13d,e) as eluent. Finally, all compounds were crystallized from ethanol, and melting points were performed after crystallization.
1-(3-Methylbenzoyl)-1H-pyrrolo[3,2-b]pyridine-3-carbonitrile (13a).
Yield = 55%; mp = 190–191 °C (colorless solid). 1H NMR (400 MHz, CDCl3) δ 2.48 (s, 3H, m-CH3-Ph), 7.41–7.46 (m, 1H, Ar), 7.49–7.56 (m, 4H, Ar), 8.09 (s, 1H, Ar), 8.61 (d, 1H, Ar, J = 8.0), 8.74 (d, 1H, Ar, J = 4.8). 13C NMR (100 MHz, CDCl3) δ 21.4 (CH3), 94.8 (C), 112.5 (C), 121.3 (CH), 124.3 (CH), 126.7 (CH), 128.9 (C), 129.1 (CH), 130.0 (CH), 131.6 (C), 134.4 (CH), 136.8 (CH), 139.5 (C), 145.7 (C), 148.2 (CH), 167.8 (C). IR = 1726 cm−1 (CO), 2222 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C16H12N3O = 262,0975, found 262,0970. Anal. C16H11N3O (C, H, N).
1-(Cyclopropanecarbonyl)-1H-pyrrolo[3,2-b]pyridine-3-carbonitrile (13b).
Yield = 34%; mp = 134–135 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 1.22–1.27 (m, 2H, CH2 cC3H5), 1.38–1.43 (m, 2H, CH2 cC3H5), 2.24–2.29 (m, 1H, COCH), 7.37–7.42 (m, 1H, Ar), 8.47 (s, 1H, Ar), 8.67 (d, 2H, Ar, J = 8.4). 13C NMR (100 MHz, CDCl3) δ 11.2 (CH2), 13.7 (CH), 95.2 (C), 112.6 (C), 121.3 (CH), 124.6 (CH), 128.5 (C), 134.2 (CH), 145.4 (C), 147.9 (CH), 171.7 (C). IR = 1718 cm−1 (CO), 2231 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C12H10N3O = 212,0818, found 212,0819. Anal. C12H9N3O (C, H, N).
1-(3-Methylbenzoyl)-1H-pyrrolo[3,2-c]pyridine-3-carbonitrile (13c).
Yield = 40%; mp = 180–181 °C (colorless solid). 1H NMR (400 MHz, CDCl3) δ 2.48 (s, 3H, m-CH3-Ph), 7.50–7.57 (m, 4H, Ar), 7.99 (s, 1H, Ar), 8.23 (d, 1H, Ar, J = 4.8), 8.69 (d, 1H, Ar, J = 4.8), 9.16 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 21.4 (CH3), 92.1 (C), 111.2 (CH), 112.6 (C), 126.7 (CH), 129.2 (CH), 130.0 (CH), 131.5 (C), 134.7 (CH), 135.8 (CH), 139.6 (C), 139.9 (C), 142.4 (CH), 145.7 (CH), 167.6 (C). IR = 1707 cm−1 (CO), 2233 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C16H12N3O = 262,0975, found 262,0973. Anal. C16H11N3O (C, H, N).
1-(Cyclopropanecarbonyl)-1H-pyrrolo[3,2-c]pyridine-3-carbonitrile (13d).
Yield = 24%; mp = 139–140 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 1.25–1.30 (m, 2H, CH2 cC3H5), 1.41–1.46 (m, 2H, CH2 cC3H5), 2.23–2.28 (m, 1H, COCH), 8.32 (d, 2H, Ar, J = 4.8), 8.56 (s, 1H, Ar), 9.12 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 11.4 (CH2), 14.3 (CH), 92.6 (C), 111.4 (CH), 112.7 (C), 124.1 (C), 133.2 (CH), 139.2 (C), 142.5 (CH), 146.1 (CH), 171.6 (C). IR = 1710 cm−1 (CO), 2232 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C12H10N3O = 212,0818, found 212,0817. Anal. C12H9N3O (C, H, N).
1-(3-Methylbenzoyl)-1H-pyrrolo[2,3-c]pyridine-3-carbonitrile (13e).
Yield = 14%; mp = 157–159 °C (colorless solid). 1H NMR (400 MHz, CDCl3) δ 2.51 (s, 3H, m-CH3-Ph), 7.51–7.62 (m, 4H, Ar), 8.08 (s, 1H, Ar), 8.35 (d, 1H, Ar, J = 12.0), 8.71 (d, 1H, Ar, J = 5.6), 9.79 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 21.4 (CH3), 92.7 (C), 112.8 (C), 114.2 (CH), 126.7 (CH), 129.1 (CH), 130.0 (CH), 131.6 (C), 132.1 (C), 133.2 (C), 134.0 (CH), 137.1 (CH), 138.9 (CH), 139.6 (C), 144.3 (CH), 167.7 (C). IR = 1709 cm−1 (CO), 2231 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C16H12N3O = 262,0975, found 262,0972. Anal. C16H11N3O (C, H, N).
1-(Cyclopropanecarbonyl)-1H-pyrrolo[2,3-c]pyridine-3-carbonitrile (13f).
Yield = 61%; mp = 188–189 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 1.31–1.36 (m, 2H, CH2 cC3H5), 1.48–1.53 (m, 2H, CH2 cC3H5), 2.26–2.31 (m, 1H, COCH), 7.87 (d, 1H, Ar, J = 5.2), 8.54 (s, 1H, Ar), 8.64 (d, 1H, Ar, J = 5.2), 9.79 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 11.4 (CH2), 14.1 (CH), 92.9 (C), 112.9 (C), 114.0 (CH), 131.6 (C), 133.7 (C), 134.6 (CH), 139.1 (CH), 144.0 (CH), 171.2 (C). IR = 1722 cm−1 (CO), 2233 cm−1 (CN). ESI-HRMS (m/z) calculated for [M + H]+ ion species C12H10N3O = 212,0818, found 212,0820. Anal. C12H9N3O (C, H, N).
3.1.11. General procedure for compounds (15a,b and 19a,b)
Compounds 15a,b and 19a,b were obtained using the general procedures followed for compounds 2a,b,l-o,r-t but starting from intermediate 1427 for 15a,b or 18 for 19a,b. The final compounds were purified by flash column chromatography using dichloromethane/methanol 98:2 (15a and 19a) or cyclohexane/ethyl acetate 3:1 (15b and 19b) as eluents. Finally, all compounds were crystallized from ethanol, and melting points were performed after crystallization.
(5H-Pyrrolo[2,3-b]pyrazin-5-yl)(m-tolyl)methanone (15a).
Yield: 19%; mp = 108–110 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 2.40 (s, 3H, m-CH3-Ph), 6.87 (d, 1H, Ar, J = 4.0 Hz), 7.40 (t, 1H, Ar, J = 7.6), 7.49 (d, 1H, Ar, J = 7.6), 7.55 (d, 1H, Ar, J = 7.6), 7.60 (s, 1H, Ar), 8.13 (d, 1H, Ar, J = 4.0), 8.24 (d, 1H, Ar, J = 2.4), 8.62 (d, 1H, Ar, J = 2.4). 13C NMR (100 MHz, CDCl3) δ 21.2 (CH3), 100.8 (CH), 106.7 (CH), 127.8 (CH), 128.7 (CH), 130.5 (C), 131.6 (CH), 133.5 (C), 134.3 (CH), 137.2 (CH), 138.3 (C), 138.5 (CH), 141.7 (C), 167.8 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C14H12N3O = 238,0975, found 238,0971. Anal. C14H11N3O (C,H,N).
Cyclopropyl(5H-pyrrolo[2,3-b]pyrazin-5-yl)methanone (15b).
Yield: 25%; mp = 117–119 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.21–1.29 (m, 2H, CH2 cC3H5), 1.37–1.42 (m, 2H, CH2 cC3H5), 4.00 (ept, 1H, CH cC3H5, J1 = 3.6 and J2 = 4.4), 6.85 (d, 1H, Ar, J = 4.0), 8.33 (m, 2H, Ar), 8.53 (d, 1H, Ar, J = 2.4). 13C NMR (100 MHz, CDCl3) δ 11.8 (CH2), 14.6 (CH), 106.3 (CH), 130.0 (CH), 137.4 (CH), 140.5 (CH), 141.7 (C), 143.1 (C), 172.7 (C) ESI-HRMS (m/z) calculated for [M + H]+ ion species C10H10N3O = 188,0818, found 188,0817. Anal. C10H9N3O (C,H,N).
5-(3-Methylbenzoyl)-5H-pyrrolo[2,3-b]pyrazine-7-carbonitrile(19a).
Yield: 4%; mp = 140–141 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 2.45 (s, 3H, m-CH3-Ph), 7.38–7.48 (m, 1H, Ar), 7.53–7.58 (m, 2H, Ar), 7.65 (s, 1H, Ar), 8.42 (d, 1H, Ar, J = 2.4), 8.52 (s, 1H, Ar), 8.68 (d, 1H, Ar, J = 2.4). 13C NMR (100 MHz, CDCl3) δ 20.9 (CH3), 83.6 (C), 113.6 (C), 119.4 (CH), 120.7 (C), 128.1 (CH), 129.2 (CH), 130.1 (CH), 130.5 (C), 134.8 (CH), 138.9 (C), 144.3 (CH), 145.8 (CH), 147.8 (C), 167.8 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C15H11N4O = 263,0927, found 263,0924. Anal. C15H10N4O (C,H,N).
5-(Cyclopropanecarbonyl)-5H-pyrrolo[2,3-b]pyrazine-7-carbonitrile (19b).
Yield: 20%; mp = 158–159 °C (white powder). 1H NMR (400 MHz, CDCl3) δ 1.33 (m, 2H, CH2 cC3H5), 1.46 (m, 2H, CH2 cC3H5), 3.93 (m, 1H, CH cC3H5), 8.49 (s, 1H, Ar), 8.69 (s, 1H, Ar), 8.75 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 11.4 (CH), 12.6 (CH2), 83.7 (C), 113.5 (C), 119.5 (CH), 120.7 (C), 144.2 (CH), 145.9 (CH), 147.9 (C), 172.8 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C11H9N4O = 213,0771, found 213,0774. Anal. C11H8N4O (C,H,N).
3.1.12. 5H-Pyrrolo[2,3-b]pyrazine-7-carbaldehyde oxime (17)
Compound 17 was obtained using the general procedure followed for compounds 11a,b but starting from intermediate 16.27 The desired compound was purified by crystallization from ethanol. Yield: 90%; mp = 239–240 °C dec. (colorless solid). 1H NMR (400 MHz, DMSO-d6) δ 8.12 (s, 1H, Ar), 8.27 (s, 2H, Ar), 8.43 (s, 1H, CH = N), 10.85 (exch br s, 1H, OH), 12.38 (exch br s, 1H, NH). ESI-HRMS (m/z) calculated for [M + H]+ ion species C7H7N4O = 163,0614, found 163,0617. Anal. C7H6N4O (C,H,N).
3.1.13. 5H-Pyrrolo[2,3-b]pyrazine-7-carbonitrile (18)
Compound 18 was obtained using the general procedure followed for compounds 12a,b but starting from intermediate 17. The desired compound was purified by flash column chromatography using dichloromethane/methanol 9:1 as eluent. Yield: 20%; mp > 300 °C (white powder). 1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, 1H, Ar, J = 2.4), 8.56 (d, 1H, Ar, J = 2.4), 8.75 (s, 1H,Ar), 13.20 (exch br s, 1H, NH). IR = 2227 cm−1 (CN), 3390 cm−1 (NH). ESI-HRMS (m/z) calculated for [M + H]+ ion species C7H5N4 = 145,0509, found 145,0508. Anal. C7H4N4 (C,H,N).
3.1.14. Ethyl 2-tert-butyl-3-chloro-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxylate (20c)
To a solution of the pyrazolo[1,5-a]pyrimidine 20b28 (0.50 mmol) and dichloromethane (10 mL), N-chlorosuccinimide (NCS) (100 mg) and a catalytic amount of benzoyl peroxide were added. The reaction mixture was stirred under reflux for 12 h. The solution was evaporated, and the crude product was recrystallized from diisopropyl ether. Yield = 70%; mp = 59–60 °C (white powder). 1H NMR (400 MHz, DMSO-d6) δ 1.41 (s, 9H, C-(CH3)3), 1.44 (t, 3H, OCH2CH3, J = 7.2), 3.10 (s, 3H, CH3), 4.41 (q, 2H, OCH2CH3, J = 7.2), 8.87 (s, 1H, Ar). ESI-HRMS (m/z) calculated for [M + H]+ ion species C14H18ClN3O2 = 296,1160, found 296,1155. Anal. C14H18ClN3O2 (C,H,N).
3.1.15. 2-(tert-Butyl)-3-chloro-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxylic acid (21c)
To a suspension of ethyl 2-(tert-butyl)-3-chloro-7-methylpyrazolo [1,5-a]pyrimidine-6-carboxylate 20c (1.01 mmol) in 5 mL of sodium hydroxide 40%, the minimum volume of ethanol needed to solubilize the solid was added. The reaction was kept stirring under reflux for 1 h. The mixture was cooled to 0 °C and acidified with hydrochloride acid 6 N to pH 2. The solid was filtered under vacuum and crystallized using ethanol as solvent to obtain the pure product. Yield = 96%; mp = 269–270 °C (white powder). 1H NMR (400 MHz, DMSO-d6) δ 1.45 (s, 9H, C-(CH3)3), 3.04 (s, 3H, CH3), 8.85 (s, 1H, Ar). ESI-HRMS (m/z) calculated for [M + H]+ ion species C12H15ClN3O2 = 268,0847, found 268,0845. Anal. C12H14ClN3O2 (C,H,N).
3.1.16. General procedure for compounds (22a-g)
To a solution of the appropriate acid of type 2130–32 (0.373 mmol) in 3 mL of anhydrous dichloromethane, first 1.87 mmol of trichloro acetonitrile, and then, after 5 min, 0.71 mmol of triphenylphosphine were added, and the suspension was stirred at room temperature. After 4 h, 0.373 mmol of m-anisidine or 3-methoxybenzylamine and 1.12 mmol of triethylamine were added to the mixture, which was then left for other 12 h stirring at room temperature. The solvent was evaporated, cold water was added, and the mixture was extracted with CH2Cl2 (3 × 15 mL). The organic solvent was dried over sodium sulfate and evaporated under vacuum. The final compounds 22a-g were purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 (for 22a, g,h) or 2:1 (for 22d,f), petroleum ether/ethyl acetate 5:2 (for 22b,e), dichloromethane/methanol 98:2 (for 22c) as eluents. Finally, all compounds were crystallized from ethanol, and melting points were performed after crystallization.
N-(3-Methoxyphenyl)-2,7-dimethylpyrazolo[1,5-a]pyrimidine-6-carboxamide (22a).
Yield = 19%; mp = 128–129 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 2.49 (s, 3H, CH3), 2.90 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 6.40 (s, 1H, Ar), 6.71 (dd, 1H, Ar, J1 = 2.4 and J2 = 8.8), 7.11 (d, 1H, Ar, J = 7.6), 7.23 (t, 1H, Ar, J = 8.2), 7.36 (s, 1H, Ar), 8.44 (s, 1H, Ar), 8.45 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 14.8 (CH3), 14.9 (CH3), 55.4 (CH3), 97.2 (CH), 106.0 (CH), 110.9 (CH), 112.3 (CH), 115.9 (C), 129.9 (CH), 138.8 (C), 146.3 (CH), 147.1 (C), 148.6 (C), 156.9 (C), 160.3 (C), 163.6 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C16H17N4O2 = 297,1346, found 297,1351. Anal. C16H16N4O2 (C,H,N).
2-(tert-Butyl)-N-(3-methoxyphenyl)-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide (22b).
Yield = 14%; mp = 114–115 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 1.40 (s, 9H, C-(CH3)3), 2.99 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 6.54 (s, 1H, Ar), 6.72 (dd, 1H, Ar, J1 = 2.0 and J2 = 8.2), 7.10 (d, 1H, Ar, J = 7.6), 7.25 (t, 1H, Ar, J = 8.2), 7.39 (exch br s, 1H, NH), 8.09 (s, 1H, Ar), 8.54 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 14.8 (CH3), 30.3 (CH3), 33.1 (C), 55.4 (CH3), 93.8 (CH), 106.0 (CH), 110.8 (CH), 112.3 (CH), 115.6 (C), 129.9 (CH), 138.9 (C), 145.9 (CH), 147.7 (C), 148.3 (C), 160.26 (C), 163.7 (C), 169.6 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C19H23N4O2 = 339,1815, found 339,1816. Anal. C19H22N4O2 (C,H,N).
2-(tert-Butyl)-3-chloro-N-(3-methoxyphenyl)-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide (22c).
Yield = 7%; mp = 164–166 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 1.51 (s, 9H, C-(CH3)3), 2.98 (s, 3H, CH3), 3.83 (s, 3H, OCH3), 6.74 (dd, 1H, Ar, J1 = 2.0 and J2 = 8.0), 7.10 (d, 1H, Ar, J = 7.6), 7.27 (t, 1H, Ar, J = 8.4), 7.38 (s, 1H, Ar), 7.91 (exch br s, 1H, NH), 8.59 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 14.2 (CH3), 28.5 (CH3), 34.1 (C), 55.4 (CH3), 98.2 (C), 111.0 (CH), 112.3 (CH), 129.9 (CH), 138.7 (C), 144.3 (C), 146.8 (CH), 148.1 (C), 160.3 (C), 162.7 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C19H22ClN4O2 = 373,1426, found 373,1421. Anal. C19H21ClN4O2 (C,H,N).
N-(3-Methoxybenzyl)-2,7-dimethylpyrazolo[1,5-a]pyrimidine-6-carboxamide (22d).
Yield = 6%; mp = 125–127 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 2.52 (s, 3H, CH3), 2.95 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 4.60 (d, 2H, CH2, J = 5.6), 6.46 (m, 2H, Ar + NH), 6.83 (d, 1H, Ar, J = 8.0), 6.88 (s, 1H, Ar), 6.92 (d, 1H, Ar, J = 7.6), 7.26 (m, 1H, Ar), 8.43 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 13.3 (CH3), 16.1 (CH3), 43.6 (CH2), 55.9 (CH3), 96.7 (CH), 111.0 (CH), 112.3 (CH), 119.3 (CH), 126.9 (C), 129.5 (CH), 138.3 (C), 141.7 (C), 150.8 (C), 155.8 (C), 157.9 (CH), 160.4 (C), 167.9 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C17H19N4O2 = 311,1503, found 311,1501. Anal. C17H18N4O2 (C,H,N).
2-(tert-Butyl)-N-(3-methoxybenzyl)-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide (22e).
Yield = 13%; oil. 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 9H, C-(CH3)3), 2.98 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 4.62 (d, 2H, CH2, J = 5.6), 6.38 (exch br s, 1H, NH), 6.52 (s, 1H, Ar), 6.81–6.94 (m, 3H, Ar), 7.27 (m, 1H, Ar), 8.44 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 14.9 (CH3), 30.3 (CH3), 33.2 (C), 44.3 (CH2) 55.3 (CH3), 93.8 (CH), 113.2 (CH), 113.5 (CH), 120.0 (CH), 126.8 (C), 130.0 (CH), 139.2 (C), 141.8 (C), 145.9 (CH), 147.6 (C), 150.8 (C), 153.5 (C), 160.1 (C), 165.3 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C20H25N4O2 = 353,1972, found 353,1977. Anal. C20H24N4O2 (C, H,N).
2-(tert-Butyl)-3-chloro-N-(3-methoxybenzyl)-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide (22f).
Yield = 21%; mp = 140–141 °C (brown solid). 1H NMR (400 MHz, CDCl3) δ 1.50 (s, 9H, C-(CH3)3), 2.97 (s, 3H, CH3), 3.81 (s, 3H, OCH3), 4.63 (d, 2H, CH2, J = 5.6), 6.20 (exch br s, 1H, NH), 6.85 (dd, 1H, Ar, J1 = 2.4 and J2 = 8.0), 6.90 (d, 1H, Ar, J = 2.0), 6.93 (d, 1H, Ar, J = 7.6), 7.83 (t, 1H, Ar, J = 7.8), 8.51 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 14.2 (CH3), 28.5 (CH3), 44.4 (CH2), 55.3 (CH3), 98.2 (C), 113.3 (CH), 113.6 (CH), 115.9 (C), 120.0 (CH), 130.0 (CH), 139.0 (CH), 144.5 (C), 146.9 (CH), 147.9 (C), 160.1 (C), 162.5 (C), 164.9 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C20H24ClN4O2 = 387,1582, found 387,1578. Anal. C20H23ClN4O2 (C,H,N).
6-Acetyl-N-(3-methoxyphenyl)-7-methylpyrazolo[1,5-a]pyrimidine-3-carboxamide (22g).
Yield = 7%; mp = 178–179 °C (colourless solid). 1H NMR (400 MHz, CDCl3) δ 2.76 (s, 3H, CH3), 3.21 (s, 3H, COCH3), 3.85 (s, 3H, OCH3), 6.68 (m, 1H, Ar), 7.16 (m, 1H, Ar), 7.22–7.27 (m, 1H, Ar), 7.53 (s, 1H, Ar), 8.83 (s, 1H, Ar), 9.04 (s, 1H, Ar), 9.83 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 15.5 (CH3), 29.9 (CH3), 55.3 (CH3), 90.7 (C), 105.4 (CH), 110.0 (CH), 112.0 (CH), 119.8 (C), 129.6 (CH), 138.9 (C), 142.5 (C), 145.4 (C), 148.8 (CH), 150.7 (CH), 152.4 (C), 159.9 (C), 196.6 (C) ESI-HRMS (m/z) calculated for [M + H]+ ion species C17H17N4O3 = 325,1295, found 325,1296. Anal. C17H16N4O3 (C,H,N).
6-Acetyl-N-(3-methoxybenzyl)-7-methylpyrazolo[1,5-a]pyrimidine-3-carboxamide (22h).
Yield = 5%; mp = 164–166 °C (colorless solid). 1H NMR (400 MHz, CDCl3) δ 2.71 (s, 3H, CH3), 3.20 (s, 3H, COCH3), 3.79 (s, 3H, OCH3), 4.71 (d, 2H, CH2, J = 6.0), 6.82 (dd, 1H, Ar, J1 = 2.4 and J2 = 8.4), 6.94 (m, 1H, Ar), 6.98 (d, 1H, Ar, J = 7.2), 7.26 (t, 1H, Ar, J = 7.6), 8.24 (exch br s, 1H, NH), 8.81 (s, 1H, Ar), 8.92 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 16.6 (CH3), 29.1 (CH3), 43.9 (CH2), 55.9 (CH3), 105.1 (C), 111.0 (CH), 112.3 (CH), 119.2 (CH), 119.7 (C), 129.5 (CH), 138.3 (C), 142.6 (C), 145.5 (CH), 145.8 (C), 156.2 (CH), 160.4 (C), 167.9 (C), 196.7 (C). ESI-HRMS (m/z) calculated for [M + H]+ ion species C18H19N4O3 = 339,1452, found 339,1458. Anal. C18H18N4O3 (C,H,N).
3.2. HNE inhibition assay
All compounds were dissolved in 100% DMSO at 5 mM stock concentrations. The final concentration of DMSO in the reactions was 1%, and this level of DMSO had no effect on enzyme activity. The HNE inhibition assay was performed in black flat-bottom 96-well microtiter plates. Briefly, a buffer solution containing 200 mM Tris–HCl, pH 7.5, 0.01% bovine serum albumin, and 0.05% Tween-20 and 20 mU/mL of HNE (Calbiochem) was added to wells containing different concentrations of each compound. The reaction was initiated by addition of 25 μM elastase substrate (N-methylsuccinyl-Ala-Ala-Pro-Val-7-amino-4-methylcoumarin, Calbiochem) in a final reaction volume of 100 μL/well. Kinetic measurements were obtained every 30 s for 10 min at 25 °C using a Fluoroskan Ascent FL fluorescence microplate reader (Thermo Electron, MA) with excitation and emission wavelengths of 355 and 460 nm, respectively. For all compounds tested, the concentration of inhibitor that caused 50% inhibition of the enzymatic reaction (IC50) was calculated by plotting % inhibition versus logarithm of inhibitor concentration (at least six points). The data are presented as the mean values of at least three independent experiments with relative standard deviations of < 15%.
3.3. Analysis of compound stability
Spontaneous hydrolysis of selected compounds was evaluated at 20 °C in 0.05 M phosphate buffer, pH 7.4. Kinetics of hydrolysis was monitored by measuring changes in absorbance spectra over incubation time using a SpectraMax ABS Plus spectrophotometer (Molecular Devices, Sunnyvale, CA). Absorbance (At) at the characteristic absorption maxima of each compound was measured at the indicated times until no further absorbance decreases occurred (A∞). Using these measurements, we created semilogarithmic plots of log(At − A∞) vs time, and k‘ values were determined from the slopes of these plots. Half-conversion times were calculated using t1/2 = 0.693/k‘.
3.4. Molecular modeling
Initial 3D structures of compounds 2e, 4, 2p, 2r, 13a, 13c, and 13e were built with ChemOffice Professional (Perkin Elmer, Waltham, MA) and refined by molecular mechanics with MM2 force field.54 For the docking computations, Molegro Virtual Docker (MVD) software, version 6.0 (CLC Bio, Copenhagen, Denmark), was used as described previously.20 The structure of HNE co-crystallized with a peptide chloromethyl ketone inhibitor (1HNE entry of the Protein Data Bank) determined by X-ray diffraction55 was used for the docking studies. The co-crystallized peptide ligand and water molecules were removed from the 1HNE structure. A search for docking poses was performed within a spherical area of 10 Å radius centered at the atom in the five-membered ring of the co-crystallized inhibitor. Side chains of 42 residues closest to the center of the sphere20 were set flexible, including the residues surrounding HNE binding site: His57, Cys58, Leu99B, Val190, Cys191, Phe192, Gly193, Asp194, Ser195, Ala213, Ser214, Phe215, Val216. Fifteen docking runs were performed for each compound with full flexibility of a ligand around all rotatable bonds and with flexibility of the above-mentioned side chains of the enzyme amino acid residues. Docking poses of each compound were checked for the ability to form a Michaelis complex between the carbonyl moiety in a ligand and the Ser195 hydroxyl group. For this purpose, the important geometric parameters were determined: d1 [distance O(Ser195)⋯C between the Ser195 hydroxyl oxygen atom and the ligand carbonyl carbon atom closest to O(Ser195)] and α [angle O(Ser195)⋯C=O, where C=O is the carbonyl group of a ligand closest to O(Ser195)]50 (Fig. 4). The conditions for proton transfer within the key catalytic triad from Ser195 to Asp102 via His57 were also evaluated by the calculation of distances d2 between the NH hydrogen in His57 and carboxyl oxygen atoms in Asp102, as described in our earlier paper.20 The distance between the OH proton in Ser195 and the pyridine-type nitrogen in His57 is also important for proton transfer. However, the OH group of the Ser195 is easily rotatable about the C–O bond. Hence, we measured distance d3 between the oxygen atom of Ser195 side chain and the basic nitrogen atom in His57 residue. The effective length of the proton transfer channel was determined as L = min (d2) + d3.
3.5. DFT calculations
Single-molecule DFT calculations were performed with the Gaussian 09w program, Revision-D.01 (Gaussian, Inc., Wallingford CT) for compounds 2b, 2n, 2o, 2p, 2q, 2r, 2s, 4, 13a, 13b, 13c, and 19 in gas phase. The hybrid B3LYP functional56,57 and 6–31+G(d,p) basis set58 were used with D3BJ dispersion correction59 applied. Vibrational frequency analysis was done for all the optimized geometries in order to ensure attaining energy minima for the molecules.
4. Conclusions
We identified new potent HNE inhibitors with different nitrogen heterocyclic scaffolds that were designed as modifications of effective compounds previously synthesized by our research group.20,21 The biological results indicated that the change of position of the nitrogen, as well as the insertion of an additional nitrogen atom in the nuclei of reference compounds A, B, and C did not negatively impact biological activity, since most of the new products exhibited potent HNE inhibitory activity with IC50 values in the nanomolar range. The most potent compounds were in the series of 5/7-azaindazoles (2p, 2r, and 4 with IC50 = 10, 16, and 21 nM, respectively) and 4/5-azaindoles (13b, 13c with IC50 = 23 and 14 nM, respectively). Analysis of chemical stability indicated that the new inhibitors generally had a half-life (t1/2) > 1 h and for compounds 2b, 2o, 2s, and 13b, t1/2 was > 4 h. The new azaindazole derivatives of type 2, were stable than the previously described indazoles (i.e., reference compound A, which had a t1/2 of 21.7 min)20 with the only exception being 2p (t1/2 = 12.2 min). These results suggest that this new scaffold improves chemical stability against spontaneous hydrolysis; however, chemical stability also seems to be related to the substituent at position 3 of the scaffold. In fact, both 2p and A, which differ only in a nitrogen at position 5 of the scaffold, present a CN group at position 3, which could be responsible for this increased hydrolysis.
The relatively stable compounds 2b, 2o, 2s, and 13b were well separated from hydrolytically unstable derivatives according to their LUMO energies E(LUMO). Indeed, the unstable compounds had overall lower values of E(LUMO) and, hence, were more prone to reactions with nucleophilic species. Molecular modeling studies confirmed the importance of the Michaelis complex formation for the interaction with the enzyme pocket, and we found that highly active HNE inhibitors were characterized by geometries favorable for acyl-inhibitor complex formation and by relatively short lengths of the proton transfer channel via the catalytic triad. Finally, in silico ADMET calculations predicted that most of the new compounds would be optimally absorbed, distributed, metabolized and excreted. Particularly, compounds 2p, 2r, 4, 13b, and 13c had excellent predicted ADMET profiles and no noted toxicity problems. Moreover, many of our compounds seem to be well adsorbed by the skin, opening to a new potential and easier route of administration for in vivo studies of these new HNE inhibitors.
Supplementary Material
Acknowledgements
This research was supported in part by National Institutes of Health IDeA Program Grants GM110732, GM115371, and GM103474; USDA National Institute of Food and Agriculture Hatch project 1009546; the Montana State University Agricultural Experiment Station; the Tomsk Polytechnic University Competitiveness Enhancement Program (project CEP-SAMT-208/2020); and the Program of Increasing the Competitiveness of TSU (project No. 8.2.10.2018).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2020.115836.
References
- 1.Jampilek J Heterocycles in medicinal chemistry. Molecules. 2019;24:3839. 10.3390/molecules24213839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giovannoni MP, Ciciani G, Cilibrizzi A, et al. Further studies on pyrazolo[10,50:1,6] pyrimido[4,5-d]pyridazin-4(3H)-ones as potent and selective human A1 adenosine receptor antagonists. Eur J Med Chem. 2015;89:32–41. 10.1016/j.ejmech.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 3.Biagini P, Biancalani C, Graziano A, et al. Functionalized pyrazoles and pyrazolo[3,4-d]pyridazinones: Synthesis and evaluation of their phosphodiesterase 4 inhibitory activity. Bioorg Med Chem. 2010;18:3506–3517. 10.1016/j.bmc.2010.03.066. [DOI] [PubMed] [Google Scholar]
- 4.Vergelli C, Schepetkin IA, Ciciani G, et al. 2-Arylacetamido-4-phenylamino-5-substituted pyridazinones as formyl peptide receptors agonists. Bioorg Med Chem. 2016;24:2530–2543. 10.1016/j.bmc.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrini G, Ciciani G, Ciattini S, et al. Pyrazolo[1,5-A]quinazoline scaffold as 5-deaza analogue of pyrazolo[5,1-c][1,2,4]benzotriazine system: Synthesis of new derivatives, biological activity on GABAA receptor subtype and molecular dynamic study. J Enzyme Inhib Med Chem. 2016;31:195–204. 10.3109/14756366.2015.1014475. [DOI] [PubMed] [Google Scholar]
- 6.Von Nussbaum F, Li VMJ. Neutrophil elastase inhibitors for the treatment of (cardio) pulmonary diseases: into clinical testing with pre-adaptive pharmacophores. Bioorg Med Chem. 2015;25:4370–4381. 10.1016/j.bmcl.2015.08.049. [DOI] [PubMed] [Google Scholar]
- 7.Crocetti L, Quinn MT, Schepetkin IA, Giovannoni MP. A patenting perspective on human neutrophil elastase (HNE) inhibitors (2014–2018) and their therapeutic applications. Expert Opin Ther Pat. 2019;29:555–578. 10.1080/13543776.2019.1630379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clinicaltrials.gov. NCT study code NCT03636347, NCT03679598, NCT02669251, (2020).Suns. [Google Scholar]
- 9.Von Nussbaum F, Li VM-J, Allerheiligen S, et al. Freezing the bioactive conformation to boost potency: the identification of BAY 85–8501, a selective and potent inhibitor of human neutrophil elastase for pulmonary diseases. ChemMedChem. 2015;10: 1163–1173. 10.1002/cmdc.201500131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carnini C, Brogin G, Patacchini R, et al. CHF6333: pharmacological and pharmacokinetic characterization of a novel potent inhaled inhibitor of neutrophil elastase. Am J Respir Crit Care Med. 2017;195:A4420. [Google Scholar]
- 11.Clinicaltrials.gov. NCT study code NCT04010799; 2020.
- 12.Bayer Corp: Prolastin. Company World Wide Web Site. Available from: http://edoc.com/prolastin; 2002.
- 13.Iwata K, Doi A, Ohji G, et al. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern Med. 2010;49: 2423–2432. 10.2169/internalmedicine.49.4010. [DOI] [PubMed] [Google Scholar]
- 14.Giovannoni MP, Schepetkin IA, Crocetti L, et al. Cinnoline derivatives as human neutrophil elastase inhibitors. J Enzyme Inhib Med Chem. 2016;31:628–639. 10.3109/14756366.2015.1057718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crocetti L, Bartolucci G, Cilibrizzi A, et al. Synthesis and analytical characterization of new thiazol-2-(3H)-ones as human neutrophil elastase (HNE) inhibitors. Chem Cent J. 2017;11:127. 10.1186/s13065-017-0358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giovannoni MP, Schepetkin IA, Quinn MT, et al. Synthesis, biological evaluation, and molecular modelling studies of potent human neutrophil elastase (HNE) inhibitors. J Enzyme Inhib Med Chem. 2018;33:1108–1124. 10.1080/14756366.2018.1480615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giovannoni MP, Crocetti L, Cantini N, et al. New 3-unsubstituted Isoxazolones as Potent Human Neutrophil Elastase Inhibitors: Synthesis and Molecular Dynamic Simulation. Drug Dev Res. 2020;81:338–349. 10.1002/ddr.21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crocetti L, Schepetkin IA, Ciciani G, et al. Synthesis and pharmacological evaluation of indole derivatives as Deaza analogues of potent human neutrophil Elastase inhibitors. Drug Dev Res. 2016;77:285–299. 10.1002/ddr.21323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crocetti L, Giovannoni MP, Schepetkin IA, et al. Design, synthesis and evaluation of N-Benzoylindazole derivatives and analogues as inhibitors of human neutrophil elastase. Bioorg Med Chem. 2011;19:4460–4472. 10.1016/j.bmc.2011.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crocetti L, Schepetkin IA, Cilibrizzi A, et al. Optimization of N-benzoylindazole derivatives as inhibitors of human neutrophil Elastase. J Med Chem. 2013;56: 6259–6272. 10.1021/jm400742j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crocetti L, Giovannoni MP, Schepetkin IA, et al. 1H-pyrrolo[2,3-b]pyridine: A new scaffold for human neutrophil elastase (HNE) inhibitors. Bioorg Med Chem. 2018;26: 5583–5595. 10.1016/j.bmc.2018.09.034. [DOI] [PubMed] [Google Scholar]
- 22.Giovannoni MP, Cantini N, Crocetti L, et al. Further modifications of 1H-pyrrolo[2,3-b]pyridine derivatives as inhibitors of human neutrophil elastase. Drug Dev Res. 2019;80:617–628. 10.1002/ddr.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schirok H, Griebenow N, Füstner C, Dilmac AM. Use of 3-(trifluoromethyl)-1H-pyrazolo-[3,4-b]pyridine as a versatile building block. Tetrahedron. 2005;71: 5597–5601. 10.1016/j.tet.2015.06.050. [DOI] [Google Scholar]
- 24.Aranov A, Lauffer DJ, Li P, Tomlinson RC. Compositions Useful As Inhibitors of Protein Kinases. PCT WO 2005/028475 A2; 2005.
- 25.Kawase M, Koyanagi J, Saito S. Site-selective trifluoroacetylation of dimethylamino-substituted pyridines and its use as a building block for trifluoromethyl-containing heterocycles. Chem Pharm Bull. 1999;47:718–719. 10.1248/cpb.47.718. [DOI] [Google Scholar]
- 26.Hert J, Hunziker D, Kuehne H, et al. Preparation of phenoxymethyl-heterocyclic compounds as autotaxin (ATX) inhibitors. PCT Int Appl. WO 2017037146 A1 20170309, 2017. [Google Scholar]
- 27.Tucker TJ, Sisko JT, Tynebor RM, et al. Discovery of 3-{5-[(6-amino-1H-pyrazolo [3,4-b]pyridine-3-yl)methoxy]-2-chlorophenoxy}-5-chlorobenzonitrile (MK-4965): a potent, orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitor with improved potency against key mutant viruses. J Med Chem. 2008;51:6503–6511. 10.1021/jm800856c. [DOI] [PubMed] [Google Scholar]
- 28.Lynch BM, Misbahul AI, Teo HC, Pedrotti F. Pyrazolo[3,4-blpyridines: Synthesis, reactions, and nuclear magnetic resonance spectral. Can J Chem. 1988;66:420–428. 10.1139/v88-074. [DOI] [Google Scholar]
- 29.Lapa GB, Bekker OP, Mirchink EP, Danilenko VN, Preobrazhenskaya MN. Regioselective acylation of congeners of 3-amino-1H-pyrazolo[3,4-b]quinolines, their activity on bacterial serine/threonine protein kinases and in vitro antibacterial (including antimycobacterial) activity. J Enzyme Inhib Med Chem. 2013;28: 1089–1091. 10.3109/14756366.2012.716056. [DOI] [PubMed] [Google Scholar]
- 30.Bradbury RH, Hales NJ, Rabow AA. Bicyclic derivatives for use in the treatment of androgen receptor associated conditions and their preparation. PTC Int Appl. WO 2009/081197, 2009. [Google Scholar]
- 31.Nakano H, Saito N, Parker L, et al. Rational Evolution of a Novel Type of Potent and Selective Proviral Integration Site in Moloney Murine Leukemia Virus Kinase 1 (PIM1) Inhibitor from a Screening-Hit Compound. J Med Chem. 2012;55:5151–5164. 10.1021/jm3001289. [DOI] [PubMed] [Google Scholar]
- 32.Clark BAJ, Parrick J, West PJ, Kelly AK. Diazaindenes (azaindoles). Part III. Reactions of Vilsmeier reagents leading to 3-formyl-1,6-diazaindenes and -1,4-diazabenz[f]indene. J Chem Soc C.. 1970;3:498–501. 10.1039/J39700000498. [DOI] [Google Scholar]
- 33.Clark BAJ, Parrick J, Dorgan RJJ. Formation of certain substituted 5H-pyrrolo[2,3-b] pyrazines by thermal cyclization of pyrazinylhydrazones and a route to 5H-pyrazino [2,3-b]indole; a synthesis of 5H-pyrrolo[2,3-b]pyrazine and some of its properties. J Chem Soc, Perkin Trans. 1976;1(13):1361–1363. 10.1039/P19760001361. [DOI] [Google Scholar]
- 34.Bruni F, Selleri S, Costanzo A, Guerrini G, Casilli ML, Giusti L. Reactivity of 7-(2-dimethylaminovinyl)pyrazolo[1,5-a]pyrimidines: synthesis of pyrazolo[1,5-a]pyrido [3,4-e]pyrimidine derivatives as potential benzodiazepine receptor ligands. 2. J Heterocycl Chem. 1995;32:291–298. 10.1002/jhet.5570310516. [DOI] [Google Scholar]
- 35.Bruni F, Chimichi S, Cosimelli B, Costanzo A, Guerrini G, Selleri S. A new entry to pyrazolo[1,5-a]pyrido[3,4-e]pyrimidine derivatives. Heterocycles. 1990;31: 1141–1149. 10.3987/COM-90-5408. [DOI] [Google Scholar]
- 36.Bartroli J, Turmo E, Alguero M, et al. New Azole Antifungals. 2. Synthesis and Antifungal Activity of Heterocyclecarboxamide Derivatives of 3-Amino-2-aryl-1-azolyl-2-butanol. J Med Chem. 1998;41:1855–1868. 10.1021/jm970726e. [DOI] [PubMed] [Google Scholar]
- 37.Kato N, Oka M, Murase T, et al. Discovery and pharmacological characterization of N-[2-({2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl}amino)-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide hydrochloride (anagliptin hydrochloride salt) as a potent and selective DPP-IV inhibitor. Bioorg Med Chem. 2011;19:7221–7227. 10.1016/j.bmc.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 38.Goldfarb DS. Method using lifespan-altering compounds for altering the lifespan of eukaryotic organisms, and screening for such compounds. U.S. Pat Appl Publ US 20090163545 A1 20090625 2009. [Google Scholar]
- 39.Schepetkin IA, Khlebnikov AI, Quinn MT. N-benzoylpyrazoles are novel small-molecule inhibitors of human neutrophil elastase. J Med Chem. 2007;50:4928–4938. 10.1021/jm070600+. [DOI] [PubMed] [Google Scholar]
- 40.Soderberg T Organic Chemistry with a Biological Emphasis Volume II. University of Minnesota Morris Digital Well: Chemistry Publications; 2019. [Google Scholar]
- 41.Takahashi Y, Ikeda H, Kanase Y, et al. Elucidation of the E-Amide Preference of N-Acyl Azoles. J Org. Chem 2017;82:11370–11382. 10.1021/acs.joc.7b01759. [DOI] [PubMed] [Google Scholar]
- 42.Staab HA. New Methods of Preparative Organic Chemistry IV. Syntheses Using Heterocyclic Amides (Azolides). Angew Chem Int Ed Engl. 1962;1:351–367. 10.1002/anie.196203511. [DOI] [Google Scholar]
- 43.Groutas WC, Kuang R, Venkataraman R, Epp JB, Ruan S, Prakash O. Structure-based design of a general class of mechanism-based inhibitors of the serine proteinases employing a novel amino acid-derived heterocyclic scaffold. Biochemistry. 1997;36: 4739–4750. 10.1021/bi9628937. [DOI] [PubMed] [Google Scholar]
- 44.Burgi HB, Dunitz JD, Lehn JM, Wipff G. Stereochemistry of reaction paths at carbonyl centers. Tetrahedron. 1974;30:1563–1572. 10.1016/S0040-4020(01)90678-7. [DOI] [Google Scholar]
- 45.Vergely I, Laugaa P, Reboud-Ravaux M. Interaction of human leukocyte elastase with a N-aryl azetidinone suicide substrate: conformational analyses based on the mechanism of action of serine proteinases. J Mol Graphics. 1996;14:158–167. 10.1016/S0263-7855(96)00057-4. [DOI] [PubMed] [Google Scholar]
- 46.Peters MB, Merz KM. Semiempirical comparative binding energy analysis (SE-COMBINE) of a series of trypsin inhibitors. J Chem Theory Comput. 2006;2:383–399. 10.1021/ct050284j. [DOI] [PubMed] [Google Scholar]
- 47.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem. 2015;58:4066–4072. 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 1997;23:3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 50.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68. 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 51.Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43:3867–3877. 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 52.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KM, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 53.Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem. 2001;4:1841–1846. 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- 54.Vanommeslaeghe K, Guvench O, MacKerell AD Jr. Molecular Mechanics. Curr Pharm Des. 2014;20:3281–3292. 10.2174/13816128113199990600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navia MA, McKeever BM, Springer JP, et al. Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84 Å resolution. Proc Natl Acad Sci USA. 1989;86:7–11. 10.1073/pnas.86.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Becke AD. A new mixing of Hartree-Fock and local density-functional theories. J Chem Phys. 1993;98:5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- 57.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem. 1994;98:11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- 58.Foresman JB, Frisch Æ. Exploring chemistry with electronic structure methods. 3rd edition. Gaussian Inc; 2015. [Google Scholar]
- 59.Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J Comp Chem. 2011;32:1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.