

Abstract
Astrocytes protect neurons by modulating neuronal function and survival. Astrocytes support neurons in several ways. They provide energy through the astrocyte-neuron lactate shuttle, protect neurons from excitotoxicity, and internalize neuronal lipid droplets to degrade fatty acids for neuronal metabolic and synaptic support, as well as by their high capacity for glutamate uptake and the conversion of glutamate to glutamine. A recent reported astrocyte system for protection of dopamine neurons against the neurotoxic products of dopamine, such as aminochrome and other o-quinones, were generated under neuromelanin synthesis by oxidizing dopamine catechol structure. Astrocytes secrete glutathione transferase M2-2 through exosomes that transport this enzyme into dopaminergic neurons to protect these neurons against aminochrome neurotoxicity. The role of this new astrocyte protective mechanism in Parkinson´s disease is discussed.
Key Words: aminochrome; astrocytes, dopamine, dopaminergic neurons, exosomes, glutathione transferase M2-2, neuroprotection, Parkinson's disease
Introduction
The loss of dopaminergic neurons containing neuromelanin from the nigrostriatal system is one of the most important events in the generation of motor symptoms in Parkinson’s disease. The question is why these neuromelanin-loaded neurons degenerate in Parkinson’s disease and not in healthy older adults who keep these neurons intact until death. For some time it has been argued that astrocytes play a fundamental role in the protection of neurons in general. Neuromelanin synthesis involves the generation of ortho-quinones that can be neurotoxic. Therefore, the objective of this review is to analyze the protective mechanisms that prevent the loss of dopaminergic neurons loaded with neuromelanin in healthy older adults and the role of astrocytes.
Search Strategy and Selection Criteria
Studies cited in this review published from 2000 to 2020 were searched on the PubMed database using the following keywords: astrocytes neuroprotection, dopamine neurons degeneration, neuromelanin synthesis, dopamine derived ortho-quinones, aminochrome, alpha-synyclein, mechanism involved in dopaminergic neurons degeneration, neuroprotective mechanisms against aminochrome-induced neurotoxicity, endogenous neurotoxins in dopaminergic neurons.
Metabolic Coupling between Astrocytes and Neurons
Astrocyte-neuron lactate shuttle
Astrocytes protect neurons by modulating neuronal function and survival. Astrocytes support the energy requirement of neurons via the astrocyte-neuron lactate shuttle, where lactate is a precursor of glucose in gluconeogenesis (Tarczyluk et al., 2013). Lactate transporters play an important role for preservation of myelin and axon function, functional recovery following nerve injuries, motor end-plate function, and the progress of diabetic peripheral neuropathy. Brain functions and energy metabolism are also dependent on lactate interactions between neurons and glia cells (Jha et al., 2020). Recently, a study of proteins involved in the astrocyte-neuron lactate shuttle (Ldha, Atp1a2, Gys1, Ldhb, Mct1, and Pfkfb3) revealed that Pfkfb3 and Atp1a2 genes, expressing fructose-2,6-biphosphatase 3/6-phosphofructo-2-kinase and the Na+/K+ transporting ATPase, respectively, were predominantly expressed by astrocytes (Margineanu et al., 2020). The most extensively supported idea of brain energy metabolism is the lactate shuttle between the astrocyte and the neuron. However, there is evidence that astrocyte and neuronal plasma membrane receptors, such as potassium ATP-channel, and GLUT2, perform a crucial role in brain glucose metabolism (Welcome and Mastorakis, 2018). Thus, the astrocyte-neuron lactate shuttle during activation, neurotransmission, and memory consolidation is an issue of controversy (Dienelet, 2019; Figure 1).
Figure 1.
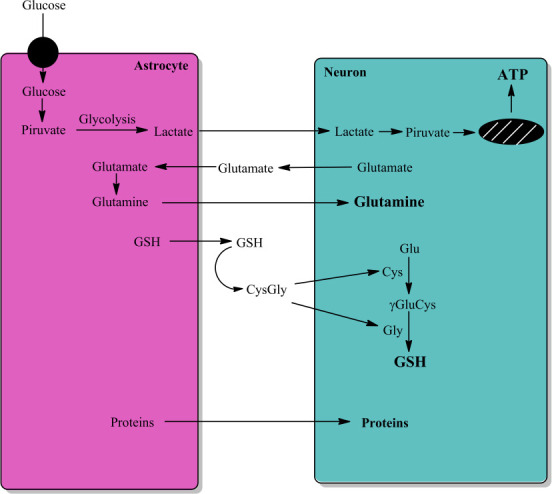
Astrocyte-neuron interactions.
The interaction between astrocytes and neurons is very important for neuron function and survival. Astrocytes convert glucose to lactate that it is transported to neuron with aim to generate energy (ATP). Astrocyte take up glutamate released by neurons to convert it to glutamine by astrocytic expressed glutamine synthetase, and neurons take up glutamine. Astrocytes secrete GSH that it is converted to Cys-Gly, which is converted to cysteine and glycine. Neurons take up both cysteine and glycine that are used in the synthesis of GSH. Astrocytes also secret a large number of proteins, including GSTM2, that neurons take up. Unpublished data. ATP: Adenosine triphosphate; Cys: L-cysteine; CysGly: L-cysteinyl-glycine; GSH: reduced tripeptide composed of L-glutamine, L-cysteine, and glycine; γGluCys: gamma-glutamyl-cysteine.
The close relationship between neurons and astrocytes can also be illustrated during an acute increase in interstitial K+ where neuronal activity couples neuron energy demand to astrocytes metabolism. An increase in K+ induces inhibition of mitochondrial respiration, activation of aerobic glycolysis, and the release of lactate. The glucose transporter GLUT1 in astrocytes is modulated by extracellular K+ that regulates the efflux and influx modes of this transporter. The regulation of astrocytes GLUT1 by K+ explains how astrocytes can preserve their glucose pool in the face of robust glycolysis stimulation. The effect of increased K+ on astrocytes results in release of lactate after activation of aerobic glycolysis, and inhibition of mitochondrial respiration (Fernández-Moncada, 2021).
Lipid degradation
Another important interrelation between astrocytes and neurons concerns lipid degradation. Lipid droplets accumulate fatty acids as energy rich reservoirs. Astrocytes internalize neuronal lipid droplets to eliminate lipids and degrade fatty acids to give neuronal metabolic and synaptic support. Apolipoprotein E reduces lipid droplets internalization into astrocytes, and reduces fatty acid oxidation, resulting in lipid accumulation in astrocytes (Qi, 2021). In this way, neurotoxic fatty acids generated in excessive active neurons are transported by apolipoprotein E-positive lipid particles into astrocytes as lipid droplets, where mitochondrial β-oxidation of the fatty acids deposited in lipid droplets induce the expression of lipid detoxification genes (Ioannou et al., 2019). Astrocytes expressing apolipoprotein E4 accumulate more and smaller lipid droplets in comparison to astrocytes expressing apolipoprotein E3. Astrocytes expressing apolipoprotein E4 present a diminished uptake of palmitate, decreased oxidation of palmitate and oleate, higher dioxygen consumption during fatty acid oxidation, more accumulation of lipid droplets-derived metabolites because of incomplete oxidation, and are more sensitive to carnitine palmitoyltransferase-1 inhibition (Farmer et al., 2019).
Medium-sized side chain fatty acids generated from dietary triglycerides penetrate the blood-brain barrier and can be used as an alternative energy source or activate related signaling functions in astrocytes. Decanoic acid, a saturated medium-sized side chain fatty acid and a ligand of Gαs protein-coupled receptors, is a signaling molecule in energy metabolism in astrocytes. Decanoic acid induces the synthesis of cAMP and the release of lactate, as well as the synthesis of GABA and its release from astrocytes, modulating neighboring neurons (Lee et al., 2018).
Glutamate-glutamine shuttle
Astrocytes have higher capacity for glutamate uptake than neurons after synaptic release of glutamate from glutamatergic neurons. Astrocytic glutamine synthetase catalyzes the formation of glutamine from glutamate taken up by astrocytes. Neurons, in turn, take up glutamine synthetized in astrocytes, which can be a mechanism to replenishment glutamate lost in oxidative metabolism (Schousboe, 2020). A protective mechanism that involves astrocyte-neuron coupling has been proposed: Astrocyte activation is required for memantine protective effects against okadaic acid, a strong PP2A and PP1 inhibitor, which induces cognitive decline. Memantine enhanced the liberation of S100B protein and reduced glutamate levels in the hippocampus and in the cerebrospinal fluid (Torrez et al., 2019).
Astrocytes are functionally and metabolically coupled to neurons by the uptake and recycling of neurotransmitters, which interconnects the function and metabolism of astrocytes and neurons. Astrocytes also secrete metabolites, such as lactate, glutamate, glutamine, adenosine triphosphate, D-serine, and regulate plasticity by pre- and postsynaptic mechanisms and synaptic activity (Mayorquin et al., 2018).
Neuroprotection
Astrocytes express Kir6.1, a principal pore unit of the K-ATP channel that is not found in neurons. In the presence of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), the Kir6.1 knockout mouse exhibited a lower concentration of striatal dopamine, increased dopamine neuron degeneration in substantia nigra compacta, and increased severe motor dysfunction, in comparison with control mice. Astrocytic Kir6.1 knockout also impaired mitophagy, which resulted in the accumulation of injured mitochondria, neuro inflammation and generation of free radicals in astrocytes. These results suggest that the astrocytic Kir6.1/K-ATP channel is neuroprotective to dopaminergic neurons (Hu et al., 2019). In addition, astrocytes protect neurons from excitotoxicity by activating the G protein-coupled receptor 30, an estrogen membrane receptor (Wang et al., 2020).
A study focused on the effect of Gq and Gi/o G protein-coupled receptors on astrocytes Ca2+-based activity, on neuronal electrical activity, and gliotransmitter release, revealed that Gi/o G protein-coupled receptor activation induced cellular activation in astrocytes and cellular inhibition in neurons, while Gq G protein-coupled receptors activation induced neuronal and astrocytic activation. Transmitter liberated from glia cells enhances neurons excitability when astrocyte activation was exerted by either Gi/o or Gq protein-mediated signaling (Durkee, 2019).
Neurotransmission and neuronal activity require energy that it is produced by mitochondrial glucose oxidation and oxidative phosphorylation, which generates reactive oxygen species under normal conditions due to the leakage of dioxygen from the electron transport chain. The redox-regulated Nrf2 transcription factor activates and induces antioxidant genes transcription in astrocytes that stimulates glutathione synthesis in neighboring neurons. The enzyme fructose-2, 6-bisphosphatase-3/6-phosphofructo-2-kinase, a crucial regulator enzyme of glycolysis, is continuously degraded by the proteasomal system in neurons, which explains its low activity in neurons. Neurons stimulate glucose oxidation through the pentose-phosphate pathway to generate NADPH, which is required for reduction of oxidized glutathione (Bolaños, 2016). Co-culture of astrocytes and neurons showed that neuronal glutathione synthesis is induced by direct activation of Nrf2 in astrocytes, and that the inhibition of astrocytic Nrf2 function inhibited neuron-induced glutathione synthesis (McGann and Mandel, 2018).
Fibroblast growth factor-1 activates astrocytes to stimulate nerve growth factor secretion. Nerve growth factor induces apoptosis in motor neurons that express p75 neurotrophin receptor by inducing formation of nitric oxide (NO) and peroxynitrite. Fibroblast growth factor-1 stimulates NO production in astrocytes by increasing the expression of inducible NO synthase. However, fibroblast growth factor-1 also stimulates Nrf2 that increases glutathione synthesis in astrocytes, thereby reducing NO-dependent neurotoxicity in motor neurons (Vargas et al., 2006).
It has been reported that the glutathione antioxidant system under synaptic activity is coupled to the N-methyl-D-aspartic acid receptor. The control of neuronal oxidative stress and apoptosis is mediated by changing the synthesis, recycling, and utilization of glutathione to promote detoxification of reactive oxygen species. This mechanism is of importance in developing brain where the disruption of N-methyl-D-aspartic acid receptor-dependent transcriptional control of glutathione synthesis can lead to neurodegeneration (Baxter et al., 2015).
The metabolic coupling between astrocytes and neurons performs an important role in the prevention of oxidative stress in the brain, where astrocytes support the survival of neurons by secreting glutathione that can be taken up by neurons. This coupling between astrocytes and neurons is unidirectional, because neurons are not able to secrete glutathione to protect astrocytes. The high rate of oxidative metabolism of glucose in the brain that leaks electrons from the electron transport chain, reducing dioxygen to superoxide requires permanent secretion of glutathione by astrocytes to maintain established levels of thiols in the neurons (Wang and Cynader, 2000). However, astrocytes are not always neuroprotective. It has been suggested that reactive astrocytes can be divided in neuroprotective phenotype (A1) and neurotoxic phenotype (A2), where astrocytes A2 has been reported to be linked to Parkinson’s disease (Li et al., 2020).
Protein secretion
The metabolic coupling between neurons and astrocytes also involves astrocyte secretion of a large number of proteins such as superoxide dismutase [Cu-Zn], ceruloplasmin chain 1, antioxidant protein 2, 1-cys peroxiredoxin, apolipoprotein E chain 1, heat shock protein HSP90-alpha and beta, insulin-like growth factor binding protein 2, L-lactate dehydrogenase M and H chain, malate dehydrogenase cytoplasmic, metalloproteinase inhibitor 1 and 2 chain1, thioredoxin peroxidase 1 and 2, glutathione transferase class-mu, creatine kinase B chain, phosphoglycerate kinase 1, and carboxypeptidase H chain 1, among other proteins. Interestingly, most proteins secreted from astrocytes can also be detected in cerebrospinal fluid (Lafon-Cazal et al., 2003).
Parkinson’s Disease
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease after Alzheimer’s disease. PD has been recognized for the development of motor symptoms that are evident after 60–70% degeneration of the dopaminergic nigrostriatal neurons. However, non-motor symptoms have been observed in PD patients including olfactory dysfunction, sleep dysfunctions, and autonomic dysfunctions (Braak et al., 2003; Schrag et al., 2015; Roos et al., 2019; Wang et al., 2019; De Rui et al., 2020). Numerous mechanisms linked in the degeneration of dopamine neurons that contain neuromelanin have been reported, such as mitochondrial damage and dysfunction, protein degradation disruption of lysosomal and proteasomal systems, alpha-synuclein (SNCA) harmful oligomers formation, endoplasmic reticulum (ER) stress, neuro inflammation, and oxidative stress (Martinez-Vicente and Vila, 2013; Taylor et al, 2013; Ross et al., 2015; Zaltieri et al., 2015; Mercado et al., 2016; Moors et al., 2016; Ganguly et al., 2018; Fields et al., 2019; Jankovic et al., 2020). However, the identity of the neurotoxin that triggers these mechanisms in idiopathic PD is still not known.
A remarkable characteristic of idiopathic PD is the extremely slow rate of neurodegeneration and disease progression after diagnosis, taking years before motor symptoms develop. The possible link of exogenous neurotoxins and environmental factors in the degeneration of the nigrostriatal neurons seems to be excluded, because drug addicts that used synthetic drugs containing near pure MPTP developed a severe Parkinsonism in just 3 days (Williams, 1986). These results also discount the prion-like hypothesis based on the propagation of SNCA aggregates through Lewy bodies from different regions until the propagation affect the nigrostriatal neurons. Evidently, this propagation will not be as slow as the extremely slow rate of neurodegeneration and progression observed in PD (Volpicelli-Daley and Brundin, 2018). The particularly slow rate of the neurodegenerative process and progression in PD suggest that the degeneration of dopaminergic neurons containing neuromelanin must be induced by an endogenous neurotoxin, which triggers the degeneration of a single neuron. In this focused mode of degeneration, the accumulation of dead neurons induced by an endogenous neurotoxin will take years, which is observed in PD.
The endogenous neurotoxin that can be involved in the degeneration of dopamine neurons containing neuromelanin must be generated inside of these neurons in order to not induce a propagative and rapid degeneration. Possible endogenous neurotoxins that are generated inside dopaminergic neurons include neurotoxic oligomers of SNCA, 3,4-dihydroxyphenylacetaldehyde and o-quinones generated during dopamine oxidation to neuromelanin.
Alpha-synuclein
SNCA form fibril aggregates that generate large deposits in Lewy bodies, which are observed in postmortem tissues of PD brains. The possible role of SNCA aggregation and the deposition of fibrils in Lewy bodies in the loss of dopamine neurons containing neuromelanin is controversial because macrophages remove and degrade all the remains of dead neurons. Therefore, the Lewy bodies observed in postmortem tissue are the neurons and glia that have survived the degenerative process in PD, suggesting a protective role of Lewy bodies. Another observation supporting the idea that SNCA aggregation to fibrils is not involved in the degeneration of dopamine neurons containing neuromelanin in idiopathic PD is the absence of Lewy bodies in Parkinson’s disease associated to parkin and LRRK2 mutations (Mori et al., 1998; Giassonet al., 2006; Rajput et al., 2006; Gaig et al., 2007; Ling 2013). It has been proposed that SNCA aggregation to oligomers is involved in the neurodegeneration of the nigrostriatal neurons, since these oligomers induce mitochondrial damage and impairment, synaptic dysfunction, ER stress, oxidative stress, and autophagy dysregulation when the gene is mutated (Fields et al., 2019; Du et el., 2020). In the familial form of PD mutations of the synuclein gene, such as A53T, induces the generation of neurotoxic oligomers of SNCA, however the question is the origin of these neurotoxic oligomers in the idiopathic form of PD. Dopamine oxidation has been suggested to induce the generation of neurotoxic oligomers in idiopathic PD. A study on the role of dopamine concentration and SNCA expression in aged mice revealed that only the combination of dopamine and SNCA caused motor impairment and progressive neurodegeneration of the substantia nigra (Mor et al., 2019).
3,4-Dihydroxyphenylacetaldehyde
3,4-Dihydroxyphenylacetaldehyde is generated during dopamine degradation. Monoamine oxidase catalyzes the oxidative deamination of dopamine to 3,4-dihydroxyphenylacetaldehyde, which is converted to 3,4-dihydroxyphenylacetic acid ammonia, and hydrogen peroxide by aldehyde dehydrogenase (ADH)-1. The catechol structure of 3,4-dihydroxyphenylacetaldehyde suggests that it can be oxidize to an o-quinone by reducing dioxygen to superoxide. It has been reported 3,4-dihydroxyphenylacetaldehyde is harmful by triggering oxidative stress and SNCA aggregation (Goldstein, 2020). The catechol structure of 3,4-dihydroxyphenylacetaldehyde suggests that it can be oxidized to an o-quinone by reducing dioxygen to superoxide. It has also been reported that 3,4-dihydroxyphenylacetaldehyde can induce covalent bonds with proteins such as L-aromatic-amino-acid decarboxylase and tyrosine hydroxylase (Goldstein, 2020; Yang et al., 2020).
A study of postmortem tissue from substantia nigra pars compacta of PD patients showed that ADH1 expression was decreased, suggesting that 3,4-dihydroxyphenylacetaldehyde accumulates in these patients (Grünblatt et al., 2004). Another investigation also reported a decrease in ADH1 expression in Parkinson’s disease patients (Liu et al., 2014). However, we must remember that the postmortem tissue of Parkinson’s disease patients with an average age of seventy-seven years contain neurons and glia cells that have survived the degenerative process induced by PD, suggesting that low expression of ADH1 is not linked in the loss of dopamine neurons.
The results from investigations with mice deficient of ADH have been contradictory. One study that used the ADH1A1 null mouse demonstrated significantly increased dopamine levels. However, no negative effect on substantia nigra dopaminergic neurons growth and development was observed, because the expression level of dopamine transporter, tyrosine hydroxylase, or vesicular monoamine transporter-2 were unchanged (Anderson et al., 2011). Another study with mice null for both ADH1A1 and ADH2 showed different results since significant degeneration of tyrosine hydroxylase immunopositively neurons in the substantia nigra was observed, accompanied with deficits in motor performance and an increase in 3,4-dihydroxyphenylacetaldehyde and 4-hydroxynonenal (Wey et al., 2012).
Based on the observed low expression of ADH1 in postmortem samples of PD patient neurons that survived the degenerative process over many years, and the lack of effect of ADH1A1 null mouse on substantia nigra dopaminergic neurons growth and development, it seems plausible that 3,4-dihydroxyphenylacetaldehyde accumulation is not involved to the loss of dopaminergic neurons of the nigrostriatal system in PD.
Ortho-quinones generated during neuromelanin synthesis
Neuromelanin formation requires dopamine oxidation to transient o-quinones such as dopamine o-quinone (DAoQ), aminochrome (AM), and 5,6-indolequinone (5,6-IQ) in a successive oxidative event (Segura-Aguilar, 2021a). Neuromelanin synthesis is a natural and inoffensive metabolic route, since this pigment increase when the life span increase in healthy aged individuals, which have undamaged dopamine neurons containing neuromelanin in substantia nigra postmortem tissue (Cassidy et al., 2019). However, under special circumstances these o-quinones can be harmful inside dopaminergic neurons, resulting in their loss in PD.
DAoQ is unstable only at pH higher than 2.0 and therefore, the amino group of this o-quinone undergo cyclization instantly to form AM at physiological pH (0.15 s–1, Herrera et al., 2017). DAoQ generates harmful adducts with mitochondrial proteins, parkin, dopamine transporter, and tyrosine hydroxylase (Xu et al., 1998; Whitehead et al., 2001; LaVoie et al., 2005; Van Laar et al., 2009). DAoQ binds to many mitochondrial proteins when this o-quinone is formed in the presence of isolated mitochondria, but only a few proteins form adducts with DAoQ when dopamine is oxidized within a cell. Due to the high reactivity of DAoQ, the question remains whether the adducts that formed inside the cell are DAoQ or AM adducts. 5,6-IQ is also high reactive, and it polymerizes instantly to neuromelanin by autocondensation and the polymerization rate increases when AM concentration and pH rise. The neuromelanin structure is composed by 5,6-IQ units assembled by van der Waals interactions. It has been reported that 5,6-IQ reacts with glutathione and NADH and forms adducts with SNCA triggering the generation of oligomers (Bisaglia et al., 2007, 2010; Pezzella et al., 2007). 5,6-IQ forms adduct with Nurr1 and stimulates Nurr1 activity such as the transcription of genes for dopamine homeostasis (Bruning et al., 2019). AM is the less unstable o-quinone generated during dopamine oxidation to neuromelanin. This was illustrated by AM NMR spectra being stable 40 min before the appearance of 5,6-IQ NMR spectra (Bisaglia et al., 2007). AM is harmful by binding to proteins such as SNCA or when it is one-electron reduced by flavoenzymes that catalyze one-electron reductions of quinones (Herrera et al., 2017). In addition, AM has several other harmful effects in the brain; it induces mitochondrial damage, SNCA aggregates into harmful oligomers, and triggers proteasomal and lysosomal impairment. It also induces ER stress, neuro inflammation, oxidative stress, disruption of cytoskeleton architecture, and progressive neuronal dysfunction, as well as decreases synaptic dopamine release while increasing GABA release, inhibits axonal transport of monoaminergic vesicles to synaptic terminals, and induces cell shrinkage (Herrera et al., 2017; Figure 2).
Figure 2.
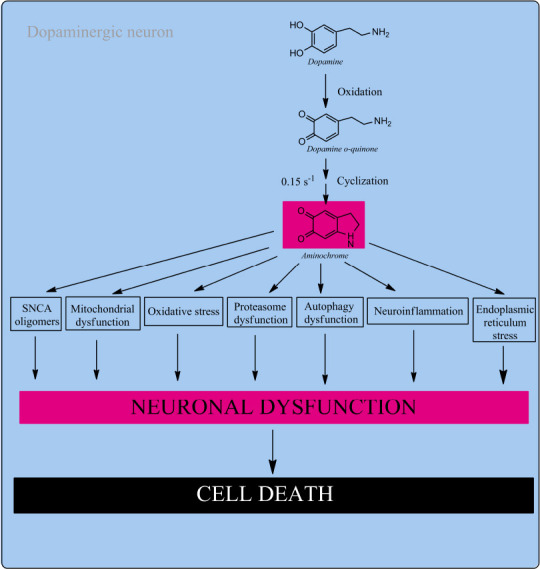
Aminochrome neurotoxicity.
Neuromelanin synthesis in dopaminergic neurons require the formation of DAoQ that is stable at pH lower than 2.0 that instantly cyclizes to AM. AM rearranges to 5,6-IQ, the precursor of neuromelanin, but when the neuroprotective capacity of DT-diaphorase is surpassed aminochrome is neurotoxic. AM is harmful by triggering the formation of SNCA harmful oligomers, mitochondrial damage, oxidative stress, proteasomal impairment, autophagy impairment, neuro inflammation and ER stress. AM induces a progressive and slow neuronal dysfunction decreasing dopamine release and increase GABA release, dramatic morphological changes such as cell shrinkage. Unpublished data. 5,6-IQ: 5,6-Indolequinone; AM: aminochrome; DAoQ: dopamine o-quinone; ER: endoplasmic reticulum; GABA: γ-aminobutyric acid; SNCA: alpha-synuclein.
As we mentioned earlier, neuromelanin synthesis is a normal and harmless process that requires the generation of o-quinones that can be harmful under certain circumstances. The explanation for this paradox is the existence of two enzymes that inhibit the neurotoxic effects of AM.
(a) DT-diaphorase: DT-diaphorase inhibits the harmful effects of AM by reducing it with two electrons to leukoaminochrome. One-electron transferring flavoenzymes reduce AM to leukoaminochrome o-semiquinone radical, which is very unstable and instantly autoxidizes to AM in the presence of dioxygen (Segura-Aguilar et al., 1998). One-electron reduction of AM induces a redox cycling between AM and leukoaminochrome o-semiquinone radical that reduces dioxygen to superoxide and oxidizes NADH to NAD+ in the cytosol. DT-diaphorase also prevents the generation of AM-adducts with proteins, such as a- and b-tubulin, and actin (Paris et al., 2010). DT-diaphorase is a protecting enzyme against AM neurotoxicity by preventing (i) AM-dependent cell death (Lozano et al., 2010); (ii) mitochondrial damage (Herrera et al., 2017); (iii) lysosome impairment (Herrera et al., 2017); autophagy impairment (Herrera et al., 2017); (iv) generation of harmful SNCA oligomers (Herrera et al., 2017); (v) proteasome impairment (Zafar et al., 2006); (vi) aggregation of alpha- and beta-, actin, and impairment of the cytoskeleton architecture (Herrera et al., 2017). Dopaminergic neurons and astrocytes have constitutive expression of DT-diaphorase to prevent AM harmful effects (Herrera et al., 2017).
(b) GSTM2- GSTM2 catalyzes glutathione conjugation of AM and its precursor DAoQ to 4-S-glutathionyl-5,6-dihydroxyindoline and 5-glutathionyldopamine, respectively (Herrera et al., 2017). Dioxygen, superoxide and hydrogen peroxide are not able to oxidize 4-S-glutathionyl-5,6-dihydroxyindoline. 5-Glutathionyldopamine is degraded to 5-cysteinyldopamine as the final product, found in the human neuromelanin and cerebrospinal fluid (Segura-Aguilar, 2021b). GSTM2 prevents AM-dependent (i) cell death in astrocytes; (ii) autophagy dysfunction; (iii) mitochondria damage; (iv) generation of SNCA harmful oligomers; and (v) AM-induced lysosomal dysfunction (Huenchuguala et al., 2017; Segura-aguilar, 2021b).
AM is the less unstable and most studied o-quinone during neuromelanin synthesis and the most suitable endogenous neurotoxin to be used for preclinical model of Parkinson’s disease because (i) AM is generated inside of dopamine neurons containing neuromelanin degenerated during Parkinson’s disease; (ii) there is two enzymes that prevents its neurotoxic effects, explaining why healthy aged individuals have undamaged dopamine neurons containing neuromelanin. However, AM under certain circumstances is neurotoxic when the protective enzymes capacity is surpassed; (iii) AM neurotoxicity is not expansive and affects single neurons exposed to AM neurotoxicity; and (iv) AM triggers all the reported neurodegenerative mechanisms such as mitochondrial damage, the formation of harmful oligomers of SNCA, dysfunction of proteasomal and lysosomal systems, ER stress, neuro inflammation, and free radical formation (Herrera et al., 2017; Segura-Aguilar, 2021a).
Astrocytes Protect Dopaminergic Neurons against Aminochrome Neurotoxicity
A new astrocyte neuroprotective mechanism has been reported where astrocytes secrete GSTM2 that prevents AM-dependent neurotoxicity in dopaminergic neurons. These neurons do not express GSTM2 but astrocytes constitutively express this enzyme. However, astrocytes secrete GSTM2 that can be internalized by dopaminergic neurons, thereby protecting dopaminergic neurons against the harmful effects of AM (Segura-Aguilar et al., 2021b). The secretion of GSTM2 from astrocytes is mediated by exosomal release, explaining dopamine neuron ability to internalize GSTM2 into the cytosol to mediate protection against AM (Valdes et al., 2021).
This new astrocyte protective mechanism against AM neurotoxicity in dopamine neurons during dopamine oxidation to neuromelanin would be very important to prevent loss of dopaminergic neurons in substantia nigra when DT-diaphorase protective capacity is surpassed. It is important to remember that GSH conjugate of DAoQ and AM inhibit the participation of these o-quinones in neurotoxic redox cycling reactions. 5-Glutathionyldopamine, the GSH conjugate of DAoQ, is degraded to 5-cysteinyldopamine that has been observed in cerebrospinal fluids and in human neuromelanin. This is the final product eliminated from dopaminergic neurons. 4-S-Glutathionyl-5,6-dihydroxyindoline, the GSH conjugate of AM, is resistant to biological oxidizing agents preventing its participation in neurotoxic reactions that require oxidative activation (Segura-Aguilar, 2021b; Figure 3).
Figure 3.
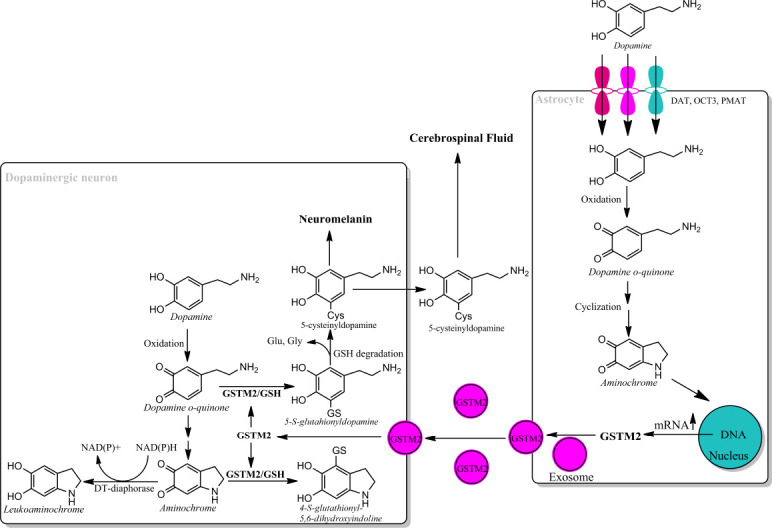
Astrocyte protects dopaminergic neurons against aminochrome neurotoxicity.
Astrocytes take up dopamine after neurotransmission through dopamine transporter (DAT), organic cation transporter-3 (OCT3) and plasma membrane transporter (PMAT) into cytosol where dopamine autoxidizes at physiological pH to DAoQ. DAoQ is very unstable and instantly cyclizes to AM at rate of 0.15 s–1. AM induces an increase in mRNA expression of GSTM2 and exosomes mediate intercellular transport GSTM2 from astrocytes into dopamine neurons. GSTM2 conjugates DAoQ with GSH to 5-glutathionyldopamine that is degraded to 5-cysteinyldopamine, which will be deposited in the neuromelanin or be excreted into cerebrospinal fluid. GSTM2 also catalyze GSH conjugation of AM to 4-S-glutathionyl-5,6-dihydroxyindoline, which it is not able to oxidize in the presence of dioxygen, hydrogen peroxide and superoxide, and it is unable to participate in neurotoxic redox cycling reactions. Unpublished data. AM: Aminochrome; DAoQ: dopamine o-quinone; GSH: glutathione; GSTM2: glutathione transferase M2-2.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Funding: This work was supported by ANID-FONDECYT 1170033 (to JSA), ANID-STINT-CONICYT CS2018-7940 (to JSA, IN, JI, MV), Swedish Research Council grant 2015-04222 to BM.
References
- 1.Anderson DW, Schray RC, Duester G, Schneider JS. Functional significance of aldehyde dehydrogenase ALDH1A1 to the nigrostriatal dopamine system. Brain Res. 2011;1408:81–87. doi: 10.1016/j.brainres.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baxter PS, Bell KF, Hasel P, Kaindl AM, Fricker M, Thomson D, Cregan SP, Gillingwater TH, Hardingham GE. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat Commun. 2015;6:6761. doi: 10.1038/ncomms7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisaglia M, Mammi S, Bubacco L. Kinetic and structural analysis of the early oxidation products of dopamine: analysis of the interactions with alpha-synuclein. J Biol Chem. 2007;282:15597–15605. doi: 10.1074/jbc.M610893200. [DOI] [PubMed] [Google Scholar]
- 4.Bisaglia M, Soriano ME, Arduini I, Mammi S, Bubacco L. Molecular characterization of dopamine-derived quinones reactivity toward NADH and glutathione: implications for mitochondrial dysfunction in Parkinson disease. Biochim Biophys Acta. 2010;1802:699–706. doi: 10.1016/j.bbadis.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Bolaños JP. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J Neurochem 139 Suppl. 2016;2:115–125. doi: 10.1111/jnc.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 7.Bruning JM, Wang Y, Oltrabella F, Tian B, Kholodar SA, Liu H, Bhattacharya P, Guo S, Holton JM, Fletterick RJ, Jacobson MP, England PM. Covalent modification and regulation of the nuclear receptor Nurr1 by a dopamine metabolite. Cell Chem Biol. 2019;26:674–685. doi: 10.1016/j.chembiol.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassidy CM, Zucca FA, Girgis RR, Baker SC, Weinstein JJ, Sharp ME, Bellei C, Valmadre A, Vanegas N, Kegeles LS, Brucato G, Kang UJ, Sulzer D, Zecca L, Abi-Dargham A, Horga G. Neuromelanin-sensitive MRI as a noninvasive proxy measure of dopamine function in the human brain. Proc Natl Acad Sci U S A. 2019;116:5108–5117. doi: 10.1073/pnas.1807983116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Rui M, Inelmen EM, Trevisan C, Pigozzo S, Manzato E, Sergi G. Parkinson's disease and the non-motor symptoms: hyposmia, weight loss, osteosarcopenia. Aging Clin Exp Res. 2020 doi: 10.1007/s40520-020-01470-x. 10.1007/s40520-020-01470-x. [DOI] [PubMed] [Google Scholar]
- 10.Dienel GA. Brain glucose metabolism: integration of energetics with function. Physiol Rev. 2019;99:949–1045. doi: 10.1152/physrev.00062.2017. [DOI] [PubMed] [Google Scholar]
- 11.Du XY, Xie XX, Liu RT. The role of α-synuclein oligomers in Parkinson’s disease. Int J Mol Sci. 2020;21:8645. doi: 10.3390/ijms21228645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durkee CA, Covelo A, Lines J, Kofuji P, Aguilar J, Araque A. Gi/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia. 2019;67:1076–1093. doi: 10.1002/glia.23589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farmer BC, Kluemper J, Johnson LA. Apolipoprotein E4 alters astrocyte fatty acid metabolism and lipid droplet formation. Cells. 2019;8:182. doi: 10.3390/cells8020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernández-Moncada I, Robles-Maldonado D, Castro P, Alegría K, Epp R, Ruminot I, Barros LF. Bidirectional astrocytic GLUT1 activation by elevated extracellular K. Glia. 2021;69:1012–1021. doi: 10.1002/glia.23944. [DOI] [PubMed] [Google Scholar]
- 15.Fields CR, Bengoa-Vergniory N, Wade-Martins R. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front Mol Neurosci. 2019;12:299. doi: 10.3389/fnmol.2019.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Follmer C, Coelho-Cerqueira E, Yatabe-Franco DY, Araujo GD, Pinheiro AS, Domont GB, Eliezer D. Oligomerization and membrane-binding properties of covalent adducts formed by the interaction of a-synuclein with the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL) J Biol Chem. 2015;290:27660–27679. doi: 10.1074/jbc.M115.686584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E. G2019S LEUCINE RICH REPEAT KINASE-2 mutation causing Parkinson's disease without Lewy bodies. J Neurol Neurosurg Psychiatry. 2007;78:626–628. doi: 10.1136/jnnp.2006.107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganguly U, Chakrabarti SS, Kaur U, Mukherjee A, Chakrabarti S. Alpha-synuclein proteotoxicity and Parkinson’s disease: search for neuroprotective therapy. Curr Neuropharmacol. 2018;16:1086–1097. doi: 10.2174/1570159X15666171129100944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein DS. The “Sick-but-not-Dead” phenomenon applied to catecholamine deficiency in neurodegenerative diseases. Semin Neurol. 2020;40:502–514. doi: 10.1055/s-0040-1713874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grünblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome heat shock protein iron and oxidative stress regulated proteins cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm (Vienna) 2004;111:1543–1573. doi: 10.1007/s00702-004-0212-1. [DOI] [PubMed] [Google Scholar]
- 22.Herrera A, Muñoz P, Steinbusch HWM, Segura-Aguilar J. Are dopamine oxidation metabolites involved in the loss of dopaminergic neurons in the nigrostriatal system in Parkinson’s disease. ACS Chem Neurosci. 2017;8:702–711. doi: 10.1021/acschemneuro.7b00034. [DOI] [PubMed] [Google Scholar]
- 23.Hu ZL, Sun T, Lu M, Ding JH, Du RH, Hu G. Kir6.1/K-ATP channel on astrocytes protects against dopaminergic neurodegeneration in the MPTP mouse model of Parkinson's disease via promoting mitophagy. Brain Behav Immun. 2019;81:509–522. doi: 10.1016/j.bbi.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 24.Huenchuguala S, Muñoz P, Segura-Aguilar J. The importance of mitophagy in maintaining mitochondrial function in U373MG cells. Bafilomycin A1 restores aminochrome-induced mitochondrial damage. ACS Chem Neurosci. 2017;8:2247–2253. doi: 10.1021/acschemneuro.7b00152. [DOI] [PubMed] [Google Scholar]
- 25.Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, Pasolli HA, Xu CS, Pang S, Matthies D, Hess HF, Lippincott-Schwartz J, Liu Z. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell. 2019;177:1522–1535. doi: 10.1016/j.cell.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Jankovic J, Tan EK. Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91:795–808. doi: 10.1136/jnnp-2019-322338. [DOI] [PubMed] [Google Scholar]
- 27.Jha MK, Morrison BM. Lactate transporters mediate glia-neuron metabolic crosstalk in homeostasis and disease. Front Cell Neurosci. 2020;14:589582. doi: 10.3389/fncel.2020.589582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lafon-Cazal M, Adjali O, Galéotti N, Poncet J, Jouin P, Homburger V, Bockaert J, Marin P. Proteomic analysis of astrocytic secretion in the mouse. Comparison with the cerebrospinal fluid proteome. J Biol Chem. 2003;278:24438–24448. doi: 10.1074/jbc.M211980200. [DOI] [PubMed] [Google Scholar]
- 29.LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- 30.Lee N, Sa M, Hong YR, Lee CJ, Koo J. Fatty acid increases cAMP-dependent lactate and MAO-B-dependent GABA production in mouse astrocytes by activating a Gαs protein-coupled receptor. Exp Neurobiol. 2018;27:365–376. doi: 10.5607/en.2018.27.5.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li T, Liu T, Chen X, Li L, Feng M, Zhang Y, Wan L, Zhang C, Yao W. Microglia induce the transformation of A1/A2 reactive astrocytes via the CXCR7/PI3K/Akt pathway in chronic post-surgical pain. J Neuroinflammation. 2020;17:211. doi: 10.1186/s12974-020-01891-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ling H, Kara E, Bandopadhyay R, Hardy J, Holton J, Xiromerisiou G, Lees A, Houlden H, Revesz T. TDP-43 pathology in a patient carrying G2019S leucine rich repeat kinase-2 mutation and a novel p.Q124E MAPT. Neurobiol Aging. 2013;34:2889. doi: 10.1016/j.neurobiolaging.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu G, Yu J, Ding J, Xie C, Sun L, Rudenko I, Zheng W, Sastry N, Luo J, Rudow G, Troncoso JC, Cai H. Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J Clin Invest. 2014;124:3032–3046. doi: 10.1172/JCI72176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margineanu MB, Sherwin E, Golubeva A, Peterson V, Hoban A, Fiumelli H, Rea K, Cryan JF, Magistretti PJ. Gut microbiota modulates expression of genes involved in the astrocyte-neuron lactate shuttle in the hippocampus. Eur Neuropsychopharmacol. 2020;41:152–159. doi: 10.1016/j.euroneuro.2020.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Martinez-Vicente M, Vila M. Alpha-synuclein and protein degradation pathways in Parkinson’s disease: a pathological feed-back loop. Exp Neurol. 2013;13:88–85. doi: 10.1016/j.expneurol.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Mayorquin LC, Rodriguez AV, Sutachan JJ, Albarracín SL. Connexin-mediated functional and metabolic coupling between astrocytes and neurons. Front Mol Neurosci. 2018;11:118. doi: 10.3389/fnmol.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGann JC, Mandel G. Neuronal activity induces glutathione metabolism gene expression in astrocytes. Glia. 2018;66:2024–2039. doi: 10.1002/glia.23455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mercado G, Castillo V, Soto P, Sidhu A. ER stress and Parkinson disease: pathological inputs that converge into the secretory pathway. Brain Res. 2016;16:30260–30268. doi: 10.1016/j.brainres.2016.04.042. [DOI] [PubMed] [Google Scholar]
- 39.Moors T, Paciotti S, Chiasserini D, Calabresi P, Parnetti L, Beccari T, van de Berg WD. Lysosomal dysfunction and α-synuclein aggregation in Parkinson’s disease. Diagnostic Links Mov Disord. 2016;6:791–801. doi: 10.1002/mds.26562. [DOI] [PubMed] [Google Scholar]
- 40.Mor DE, Daniels MJ, Ischiropoulos H. The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov Disord. 2019;34:167–179. doi: 10.1002/mds.27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. 1998;51:890. doi: 10.1212/wnl.51.3.890. [DOI] [PubMed] [Google Scholar]
- 42.Muñoz P, Cardenas S, Huenchuguala S, Briceño A, Couve E, Paris I, Segura-Aguilar J. DT-diaphorase prevents aminochrome-induced alpha-synuclein oligomer formation and neurotoxicity. Toxicol Sci. 2015;145:37–47. doi: 10.1093/toxsci/kfv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paris I, Muñoz P, Huenchuguala S, Couve E, Sanders LH, Greenamyre JT, Caviedes P, Segura-Aguilar J. Autophagy protects against aminochrome-induced cell death in substantia nigra-derived cell line. Toxicol Sci. 2011;121:376–388. doi: 10.1093/toxsci/kfr060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pezzella A, Crescenzi O, Natangelo A, Panzella L, Napolitano A, Navaratnam S, Edge R, Land EJ, Barone V, d’Ischia M. Chemical pulse radiolysis and density functional studies of a new labile 5,6-indolequinone and its semiquinone. J Org Chem. 2007;72:1595–1603. doi: 10.1021/jo0615807. [DOI] [PubMed] [Google Scholar]
- 45.Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021;34:108572. doi: 10.1016/j.celrep.2020.108572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Leucine rich repeat kinase-2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 47.Roos DS, Twisk JWR, Raijmakers PGHM, Doty RL, Berendse HW. Hyposmia as a marker of (non-) motor disease severity in Parkinson’s disease. J Neural Transm (Vienna) 2019;126:1471–1478. doi: 10.1007/s00702-019-02074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ross JM, Olson L, Coppotelli G. Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin. Int J Mol Sci. 2015;16:19458–76. doi: 10.3390/ijms160819458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schousboe A. Astrocytic metabolism focusing on glutamate homeostasis: a short review dedicated to vittorio gallo. Neurochem Res. 2020;45:522–525. doi: 10.1007/s11064-019-02888-0. [DOI] [PubMed] [Google Scholar]
- 50.Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14:57–64. doi: 10.1016/S1474-4422(14)70287-X. [DOI] [PubMed] [Google Scholar]
- 51.Segura-Aguilar J. In: Clinical studies and therapies in Parkinson's disease: translations from preclinical models (Segura-Aguilar J, ed) Cambridge: Elsevier; 2021a. Dopamine oxidation to neuromelanin and neurotoxic metabolites; pp. 213–223. [Google Scholar]
- 52.Segura-Aguilar J. In: Clinical studies and therapies in Parkinson's disease: translations from preclinical models (Segura-Aguilar J, ed) Cambridge: Elsevier; 2021b. Neuroprotective mechanisms against dopamine oxidationdependent neurotoxicity; pp. 229–237. [Google Scholar]
- 53.Tarczyluk MA, Nagel DA, O’Neil JD, Parri HR, Tse EH, Coleman MD, Hill EJ. Functional astrocyte-neuron lactate shuttle in a human stem cell-derived neuronal network. J Cereb Blood Flow Metab. 2013;33:1386–1393. doi: 10.1038/jcbfm.2013.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor JM, Main BS, Crack PJ. Neuroinflammation and oxidative stress: Co-conspirators in the pathology of Parkinson’s disease. Neurochem Int. 2013;62:803–819. doi: 10.1016/j.neuint.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 55.Torrez VR, Zimmer ER, Kalinine E, Haas CB, Zenki KC, Muller AP, Souza DO, Portela LV. Memantine mediates astrocytic activity in response to excitotoxicity induced by PP2A inhibition. Neurosci Lett. 2019;696:179–183. doi: 10.1016/j.neulet.2018.12.034. [DOI] [PubMed] [Google Scholar]
- 56.Valdes R, Armijo A, Muñoz P, Hultenby K, Hagg A, Inzunza J, Nalvarte I, Varshney M, Mannervik B, Segura-Aguilar J. Cellular trafficking of glutathione transferase M2-2 between U373MG and SHSY-S7 cells is mediated by exosomes. Neurotox Res. 2021;39:182–190. doi: 10.1007/s12640-020-00327-5. [DOI] [PubMed] [Google Scholar]
- 57.Van Laar VS, Mishizen AJ, Cascio M, Hastings TG. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol Dis. 2009;34:487–500. doi: 10.1016/j.nbd.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem. 2006;97:687–696. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- 59.Volpicelli-Daley L, Brundin P. Prion-like propagation of pathology in Parkinson disease. Handb Clin Neurol. 2018;153:321–335. doi: 10.1016/B978-0-444-63945-5.00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang XY, Han YY, Li G, Zhang B. Association between autonomic dysfunction and olfactory dysfunction in Parkinson’s disease in southern Chinese. BMC Neurol. 2019;19:17. doi: 10.1186/s12883-019-1243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Welcome MO, Mastorakis NE. Emerging concepts in brain glucose metabolic functions: from glucose sensing to how the sweet taste of glucose regulates its own metabolism in astrocytes and neurons. Neuromolecular Med. 2018;20:281–300. doi: 10.1007/s12017-018-8503-0. [DOI] [PubMed] [Google Scholar]
- 63.Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One. 2012;7:e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitehead RE, Ferrer JV, Javitch JA, Justice JB. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J Neurochem. 2001;76:1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x. [DOI] [PubMed] [Google Scholar]
- 65.Williams A. MPTP toxicity: clinical features. J Neural Transm Suppl. 1986;20:5–9. [PubMed] [Google Scholar]
- 66.Xiong R, Siegel D, Ross D. Quinone-induced protein handling changes: implications for major protein handling systems in quinone-mediated toxicity. Toxicol Appl Pharmacol. 2014;280:285–295. doi: 10.1016/j.taap.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu Y, Stokes AH, Roskoski R Jr, Vrana KE. Dopamine in the presence of tyrosinase covalently modifies and inactivates tyrosine hydroxylase. J Neurosci Res. 1998;54:691–697. doi: 10.1002/(SICI)1097-4547(19981201)54:5<691::AID-JNR14>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 68.Yang F, Li Y, Pan H, Wu K, Lu Y, Shi F. A novel LC-MS/MS method for quantification of unstable endogenous 3,4-dihydroxyphenylacetaldehyde in rat brain after chemical derivatization. J Pharm Biomed Anal. 2020;2020:113822. doi: 10.1016/j.jpba.2020.113822. [DOI] [PubMed] [Google Scholar]
- 69.Zafar KS, Inayat-Hussain SH, Siegel D, Bao A, Shieh B, Ross D. Overexpression of NQO1 protects human SK-N-MC neuroblastoma cells against dopamine-induced cell death. Toxicol Lett. 2006a;166:261–267. doi: 10.1016/j.toxlet.2006.07.340. [DOI] [PubMed] [Google Scholar]
- 70.Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A. Mitochondrial dysfunction and α-synuclein synaptic pathology in Parkinson’s disease: who’s on first. Parkinsons Dis. 2015;2015:108029. doi: 10.1155/2015/108029. [DOI] [PMC free article] [PubMed] [Google Scholar]