

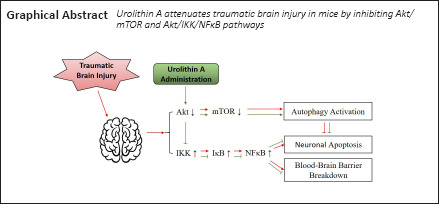
Keywords: autophagy, blood-brain barrier, cerebral edema, controlled cortical impact model, neuronal apoptosis, neuropharmacology, neuroprotection, tight junction protein, traumatic brain injury, urolithin A
Abstract
Urolithin A (UA) is a natural metabolite produced from polyphenolics in foods such as pomegranates, berries, and nuts. UA is neuroprotective against Parkinson's disease, Alzheimer's disease, and cerebral hemorrhage. However, its effect against traumatic brain injury remains unknown. In this study, we established adult C57BL/6J mouse models of traumatic brain injury by controlled cortical impact and then intraperitoneally administered UA. We found that UA greatly reduced brain edema; increased the expression of tight junction proteins in injured cortex; increased the immunopositivity of two neuronal autophagy markers, microtubule-associated protein 1A/B light chain 3A/B (LC3) and p62; downregulated protein kinase B (Akt) and mammalian target of rapamycin (mTOR), two regulators of the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR signaling pathway; decreased the phosphorylation levels of inhibitor of NFκB (IκB) kinase alpha (IKKα) and nuclear factor kappa B (NFκB), two regulators of the neuroinflammation-related Akt/IKK/NFκB signaling pathway; reduced blood-brain barrier permeability and neuronal apoptosis in injured cortex; and improved mouse neurological function. These findings suggest that UA may be a candidate drug for the treatment of traumatic brain injury, and its neuroprotective effects may be mediated by inhibition of the PI3K/Akt/mTOR and Akt/IKK/NFκB signaling pathways, thus reducing neuroinflammation and enhancing autophagy.
Introduction
Traumatic brain injury (TBI) can be divided into primary injury and secondary injury. Primary injury occurs at the moment of harm; its extent depends on the mechanism and force of impact. After primary injury, a complex cascade of events contributes to secondary injury, such as blood-brain barrier (BBB) disruption, oxidative stress, brain edema, neuronal apoptosis, impaired autophagy flux, and ionic homeostasis imbalance (Stocchetti et al., 2017; Desai and Jain, 2018), which contributes to patients’ morbidity and mortality. Here, we investigate a neuroprotective strategy to mitigate secondary injury after TBI.
Autophagy is a highly conserved intracellular degradation pathway by which cells deliver cytoplasmic organelles and proteins to lysosomes for degradation (Mizushima et al., 2008). This process recycles cellular substances and plays a vital role in maintaining cellular metabolism. Research over the past decade shows that dysregulation of autophagy is involved in various diseases, including tumors (Byun et al., 2017), neurodegenerative diseases (Menzies et al., 2015; Shao et al., 2021; Zhang et al., 2021), and neurotrauma (Sarkar et al., 2014; Wu and Lipinski, 2019). Dysregulated autophagy plays an important role in TBI secondary injury processes such as neuronal apoptosis, BBB disruption, and neuroinflammation (Chang et al., 2013; Sarkar et al., 2014; Wu et al., 2020; Mytych, 2021). Therefore, targeting of autophagy shows promise for the treatment and prevention of TBI-induced secondary injury.
Urolithin A (UA) is a natural metabolite produced from food that contains ellagitannins, such as pomegranates, berries and nuts. During digestion, ellagitannins taken from food spontaneously hydrolyze into ellagic acid, which is further converted into urolithins A, B, C, and D by gut microbiota (Espín et al., 2013; Nuñez-Sánchez et al., 2014; Kujawska and Jodynis-Liebert, 2020). Urolithins are transported into the blood circulation by intestinal epithelial cells and exert a broad spectrum of bioactivities, such as autophagy activation, anti-inflammation, and antioxidant effects (Ishimoto et al., 2011; González-Sarrías et al., 2017; Komatsu et al., 2018; Xu et al., 2018; Ahsan et al., 2019). Urolithins have a broad range of safe dosage (Heilman et al., 2017; Andreux et al., 2019) and are permeable to the BBB (Yuan et al., 2016; Kujawska et al., 2019). Recent studies showed that UA is the most potent urolithin to exert its beneficial effects in various diseases, including diabetes (Tuohetaerbaike et al., 2020; Lee et al., 2021), inflammatory bowel diseases (Singh et al., 2019), ischemic brain injury (Ahsan et al., 2019), and Alzheimer's disease (Gong et al., 2019). Andreux et al. (2019) reported that oral consumption of UA could increase biogenesis of mitochondria and improve muscle health by mitophagy enhancement in a cohort study consisting of older adults. Singh et al. (2019) found that UA treatment significantly protects tight junction proteins of the gut barrier and reduces inflammation in a rodent model of inflammatory bowel disease. Regarding central nervous system diseases, Gong et al. (2019) demonstrated that UA improved cognitive impairment in an Alzheimer's disease mouse model by targeting multiple pathological processes, such as neuronal apoptosis, amyloid-beta plaque formation, and inflammatory signaling. Another study reported that UA administration reinforced ischemia-induced autophagy and reduced ischemic brain injury in a mouse model of middle cerebral artery occlusion (Ahsan et al., 2019). However, it is still unknown whether UA is neuroprotective following TBI. On the basis of the above studies, we hypothesized that UA could protect the BBB, reduce brain edema, and preserve neuron survival by alleviating neuroinflammation and reinforcing autophagy, thus exerting neuroprotective effects against TBI-induced secondary injury. This study aimed to evaluate UA as a promising neuroprotective drug for TBI treatment and to explore its underlying mechanism of action.
Materials and Methods
Animals and experimental design
Estrogen has neuroprotective effects on TBI (Chakrabarti et al., 2016), and therefore we excluded female mice from the present study to avoid data misinterpretation. Male C57BL/6J mice aged 8–10 weeks and weighing 22–25 g were purchased from Shanghai SLAC Laboratory Animal Co., Ltd., Shanghai, China (license No. SCXK (Hu) 2017-0005). All animals used in this work were kept in individual standard cages under a 12-hour light/dark cycle with controlled temperature and humidity and full access to food and water. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, China (approval No. 2019-0180) on February 2, 2019. All experiments were designed and reported according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (Percie do Sert et al., 2020).
UA (Cat# S5312, Selleck Chemicals, Houston, TX, USA) was dissolved in DMSO (Cat# D8418, MilliporeSigma, Burlington, MA, USA) to make a 40 mg/mL stock solution, which was further diluted in 0.9% normal saline and 0.5% Tween-80 to a prescribed concentration before being used for intraperitoneal injections. Mice were randomly grouped into sham, TBI + vehicle, and TBI + UA (2.5 mg/kg) groups. Mice in the TBI + vehicle group were treated with the same volume of vehicle (0.1 mL per injection). The first dose of UA was injected immediately after controlled cortical impact (CCI) injury, and an additional dose of the same concentration and volume was given every 24 hours until sacrifice. The dosage of UA was determined by brain water content assay (described below).
In total, 126 mice were sacrificed: 36 for the brain water content assay (six groups of six rats per group), 12 for the Evans Blue (EB) extravasation assay (three groups of four rats per group), 15 for western blot analysis (three groups of five rats per group), 18 for immunofluorescence and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (three groups of six rats per group), and 45 for behavior tests (three groups of 15 rats per group). Samples were collected 72 hours after the CCI injury, except for behavior tests.
Controlled cortical impact model of traumatic brain injury
We described the TBI model used in this work in our previous study (Liu et al., 2018b). Briefly, adult male C57BL/6J mice were anesthetized intraperitoneally with ketamine (75 mg/kg, Cat# K2753, MilliporeSigma) and xylazine (10 mg/kg, Cat# X1126, MilliporeSigma), a widely used mix of anesthetics that are safe and effective (Curl, 1988). Then, mice were placed in a stereotaxic frame (Stoelting Co., Wood Dale, IL, USA) with a heating pad. First, an incision was made from the midline of the skull, and the skin and fascia were retracted using a vascular clamp. Then, a 4 mm trephine was used to perform a craniotomy over the center of the right parietal bone, between bregma and lambda and 1 mm lateral to the sagittal suture. Mice were excluded from the study if the dura was broken. Following the craniotomy, the stereotaxic frame was adjusted to ensure that the impactor tip was perpendicular to the cortical surface; then, a moderate contusion injury was made using a precision cortical impactor (PCI3000, Hatteras Instruments Inc., Cary, NC, USA) at an impact velocity of 1.5 m/s, depth of 1.5 mm, and dwell time of 100 ms (Additional Figure 1A (510.7KB, tif) ). Bleeding was stanched with sterile cotton, bone wax was placed over the craniotomy site, and the incision was closed with silk sutures. The animal remained on a heating pad until full recovery of body temperature and consciousness occurred and was then returned to its home cage. The sham group underwent the same procedure, except without the contusion injury.
Brain water content assay
We used the brain water content assay as a pilot study to determine the optimal dosage of UA, as well as to exclude neurotoxicity of the vehicle. Based on previous studies (Savi et al., 2017; Ahsan et al., 2019; Lee et al., 2021), 36 mice were randomly divided into six groups of sham, sham + vehicle, TBI + vehicle, TBI + UA (2.5 mg/kg), TBI + UA (5 mg/kg), and TBI + UA (10 mg/kg), with 6 mice in each group.
Brain water content was performed by the wet/dry weight method 72 hours after CCI. The mice were anesthetized and sacrificed, and the brains were removed immediately without intracardial perfusion. The cerebrum ipsilateral to the CCI was separated from the remaining brain and weighed with a precise analytical balance (Mettler-Toledo, Greifensee, Switzerland) to obtain the wet weight. Then, the ipsilateral cerebrum was dried in an oven (Shanghai Bluepard Instruments Co., Shanghai, China) at 60°C for 72 hours. The dry weight was obtained by weighing the brain tissue after drying. The brain water content was calculated as (wet weight – dry weight)/wet weight × 100%.
Blood-brain barrier permeability assay
BBB permeability was measured by EB dye extravasation. EB is a macromolecular dye that can only permeate a broken BBB (Kaya and Ahishali, 2011). Three days after TBI, mice were injected with 0.1 mL of EB (2% in saline, Cat# E2129, MilliporeSigma) in the jugular vein, put on a heating pad for 2 hours, and then perfused with 50 mL phosphate-buffered saline through the left ventricle to remove the EB dye from the blood circulation. The hemisphere ipsilateral to the CCI was immediately separated from the remaining brain and weighed with a precise analytical balance. After weighing, the ipsilateral hemisphere was homogenized using an ultrasonic homogenizer (Sonics & Materials Inc., Newtown, CT, USA) in 1 mL of solvent (3:1 trichloroacetic acid:ethanol). Following centrifugation (12,000 × g for 20 minutes), the solvent divided into 2 layers, and the supernatant was collected and transferred to a 96-well plate. A microplate reader (Biotek, Winooski, VT, USA) was used to detect the absorbance of the supernatant at 610 nm. Each sample was read in triplicate, and EB concentration was calculated according to the absorbance and a standard curve. Finally, the EB concentration was expressed as EB content per gram brain tissue weight.
Immunofluorescence and TUNEL assay
Mice were anesthetized, sacrificed, and perfused intracardially with 30 mL phosphate-buffered saline 72 hours after CCI. The whole brain was removed carefully from the skull, immersed in –80°C isopentane for 30 seconds, and then cut into successive coronal sections of 20 µm with a freezing microtome (Leica, Wetzlar, Germany). After attachment onto slides, the brain sections were fixed with 4% paraformaldehyde or methanol for 10 minutes, penetrated with 0.1% Triton X-100 for 10 minutes, and blocked with 10% bovine serum albumin (Cat# MB4219, Meilunbio, Dalian, China) for 60 minutes. For immunofluorescence staining, sections were incubated in primary antibodies at 4°C overnight, in secondary antibodies at room temperature for 1 hour, and in 4′,6-diamidino-2-phenylindole working solution (Cat# C1002, Beyotime Biotechnology Co., Nantong, China) for 10 minutes.
For the TUNEL assay, the sections were first stained by immunofluorescence using a neuronal nuclei antigen (NeuN) primary antibody. Then, a TUNEL assay kit (Cat# C1098, Beyotime) was used to detect apoptosis in accordance with the manufacturer's instructions. Briefly, each section was incubated with 50 µL TUNEL reaction mixture at 37°C for 30 minutes to stain nuclei of apoptotic cells red. Apoptotic neurons were defined as cells that stained positive for both NeuN and TUNEL. For each brain, three equally spaced coronal sections were selected from the injury site; we calculated the mean number of apoptotic neurons by analyzing 4–6 random microscope fields for each section of injured cortex using the 63× objective lens of a confocal microscope (Leica). For both immunofluorescence staining and the TUNEL assay, only the injured cortex was further analyzed (Additional Figure 1B (510.7KB, tif) ). Images of all sections were captured with a confocal microscope (Leica), and fluorescence intensity was quantified by LAS AF 2.8.0 software (Leica). Antibodies used in immunofluorescence are listed below: rabbit anti-zona occludens protein 1 (ZO-1; 1:100, Cat# 61-7300, RRID: AB_2533938, Thermo Fisher Scientific, Waltham, MA, USA), mouse anti-occludin (1:100, Cat# 33-1500, RRID: AB_2533101, Thermo Fisher Scientific), goat anti-CD31 (1:200, Cat# AF3628, RRID: AB_2161028, R&D Systems, Minneapolis, MN, USA), mouse anti-neuronal nuclei antigen (NeuN; 1:100, Cat# MAB377, RRID: AB_2298772, MilliporeSigma), rabbit anti-microtubule-associated protein 1 light chain 3 A/B (LC3A/B; 1:100, Cat# 12741, RRID: AB_2617131, Cell Signaling Technology, Danvers, MA, USA), rabbit anti-p62 (1:200, Cat# ab109012, RRID: AB_2810880, Abcam, Cambridge, UK), donkey anti-goat IgG–Alexa Fluor 555 (1:500, Cat# A-21432, RRID: AB_2535853, Thermo Fisher Scientific), donkey anti-rabbit IgG–Alexa Fluor 488 (1:500, Cat# A-21206, RRID: AB_2535792, Thermo Fisher Scientific), donkey anti-mouse IgG–Alexa Fluor 488 (1:500, Cat# A-21202, RRID: AB_141607, Thermo Fisher Scientific), and donkey anti-rabbit IgG–Alexa Fluor 594 (1:500, Cat# A-21207, RRID: AB_141637, Thermo Fisher Scientific).
Western blot assay
Mice were anesthetized and sacrificed 72 hours following CCI. Brain samples of injured cortex were collected and homogenized in lysis buffer using ultrasound and then diluted in loading buffer (Cat# P0015, Beyotime) to make all samples the same protein concentration after centrifugation (Additional Figure 1C (510.7KB, tif) ). For sodium dodecyl sulfate-polyacrylamide gel electrophoresis, samples were loaded onto gels made with the Polyacrylamide Gel Fast Preparation Kit (Cat# PG110-114, Epizyme, Shanghai, China) and were subsequently transferred to polyvinylidene fluoride membranes (Cat# 10600023, Cytiva, Marlborough, MA, USA) by electroblotting. Polyvinylidene fluoride membranes were blocked with 5% nonfat milk and then incubated in primary antibodies overnight at 4°C. After washing in phosphate-buffered saline, membranes were incubated in horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Proteins were detected using the Omni-ECL Pico Light Chemiluminescence Kit (Cat# SQ202, Epizyme) and a chemiluminescent imaging system (Tanon Science and Technology Co., Shanghai, China). The relative optical density was calculated by ImageJ 1.52a software (Schneider et al., 2012). Antibodies used for western blot are listed below: rabbit anti-ZO-1 (1:1000, Cat# 61-7300, RRID: AB_2533938, Thermo Fisher Scientific), mouse anti-occludin (1:1000, Cat# 33-1500, RRID: AB_2533101, Thermo Fisher Scientific), mouse anti-β-actin (1:2000, Cat# 66009-1-Ig, RRID: AB_2687938, Proteintech, Rosemont, IL, USA), rabbit anti-cleaved caspase-3 (1:500, Cat# 9664, RRID: AB_2070042, Cell Signaling Technology), rabbit anti-caspase-3 (1:1000, Cat# 14220, RRID: AB_2798429, Cell Signaling Technology), rabbit anti-bcl-2 (1:1000, Cat# 3498, RRID: AB_1903907, Cell Signaling Technology), rabbit anti-LC3A/B (1:1000, Cat# 12741, RRID: AB_2617131, Cell Signaling Technology), rabbit anti-p62 (1:5000, Cat# ab109012, RRID: AB_2810880, Abcam), rabbit anti-protein kinase B (Akt; 1:1000, Cat# 4691, RRID: AB_915783, Cell Signaling Technology), rabbit anti-phospho-Akt (p-Akt; 1:1000, Cat# 4060, RRID: AB_2315049, Cell Signaling Technology), rabbit anti-mTOR (1:1000, Cat# 2972, RRID: AB_330978, Cell Signaling Technology), rabbit anti-phospho-mTOR (p-mTOR; 1:1000, Cat# 2971, RRID: AB_330970, Cell Signaling Technology), rabbit anti-nuclear factor kappa B (NFκB; 1:1000, Cat# 8242, RRID: AB_10859369, Cell Signaling Technology), rabbit anti-phospho-NFκB (p-NFκB; 1:1000, Cat# 3033, RRID: AB_331284, Cell Signaling Technology), mouse anti-inhibitor of NFκB (IκB) kinase alpha (IKKα; 1:1000, Cat# 11930, RRID: AB_2687618, Cell Signaling Technology), rabbit anti-phospho-IKKα/β (p-IKKα/β, 1:1000, Cat# 2697, RRID: AB_2079382, Cell Signaling Technology), anti-mouse horseradish peroxidase–linked secondary antibody (1:5000, Cat# 7076, RRID: AB_330924, Cell Signaling Technology), and anti-rabbit horseradish peroxidase–linked secondary antibody (1:5000, Cat# 7074, RRID: AB_2099233, Cell Signaling Technology).
Modified neurological severity score and rotarod test
The modified neurological severity score (mNSS) test was performed to evaluate the neurological deficits of each mouse at 1, 3, 7, and 14 days after CCI. Scoring details are as follows. Motor deficits: raising the mouse by the tail to evaluate forelimb flexion (0–3) and placing the mouse on the floor to evaluate gait (0–3). Balance deficits: placing the mouse on a balance beam to assess posture (0–6). Reflex deficits: testing pinna and corneal reflexes (0–2). A score of 0 indicates normal behavior, whereas a score of 14 indicates maximal deficit (Chen et al., 2001).
The rotarod test (Carter et al., 2001) was performed to evaluate motor coordination of the mice. Mice were trained for 72 hours (three sessions/day for 5 minutes/session) before CCI; there was at least a 15-minute interval between each session. The speed of the rod was increased from 0 to 20 rounds/minute on the first day to 0 to 30 rounds/minute on the second day and 0 to 40 rounds/minute on the third day. Mice that could not stay on the rod for at least 5 minutes were excluded from this test. On the 4th day, CCI was performed; mice were subjected to the rotarod test before CCI and at 1, 3, 7, and 14 days after CCI. The latency (time taken until the mouse fell off the rod) was recorded to evaluate the motor coordination function.
Statistical analysis
No statistical methods were used to predetermine sample sizes; however, our sample sizes are similar to those reported in a previous publication (Gong et al., 2019). No animals or data points were excluded from the analysis. The evaluator was blind to the animal groupings. All data are expressed as mean ± standard deviation (SD). All the experiments were analyzed using one-way analysis of variance followed by Bonferroni's post hoc test. P < 0.05 was considered statistically significant. IBM SPSS Statistics for Windows, version 19.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, CA, USA) were used for statistical analysis and visualization, respectively.
Results
Urolithin A reduces brain edema and protects tight junction proteins and blood-brain barrier function following traumatic brain injury
The results from the brain water content assay, a pilot study, showed that TBI significantly increased brain water content (P < 0.001, compared with sham) and was reversed by UA administration of 2.5 mg/kg (P = 0.016, compared with TBI + vehicle). Compared with TBI + UA (2.5 mg/kg), 5 and 10 mg/kg UA did not further reduce brain water content (P = 0.395). No significant difference was found between sham and sham + vehicle (P = 0.227; Figure 1A). Therefore, we used 2.5 mg/kg as the dosage for subsequent experiments to reduce unnecessary sacrifice of experimental animals.
Figure 1.

Urolithin A attenuates blood-brain barrier leakage and tight junction protein disruption 72 hours following traumatic brain injury.
(A) A pilot study. UA reduced brain water content following TBI at dosages of 2.5, 5, and 10 mg/kg (n = 6). (B) Representative images of the EB extravasation assay. EB concentration increased following TBI and reduced after UA administration. (C) Quantification of EB concentration for each group (n = 4). (D, E) Immunostaining of occludin and ZO-1 (green, Alexa Fluor 488) with CD31 (red, Alexa Fluor 555) in injured cortex following TBI. White arrows show disruption of tight junction proteins. Scale bars: 10 µm. (F) Representative western blots of occludin and ZO-1. (G) Quantification of relative protein expression (n = 5) normalized to the optical density of β-actin. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni's post hoc test). BBB: Blood-brain barrier; DAPI: 4,6-diamino-2-phenyl indole; EB: Evans blue; n.s.: no statistical significance between indicated groups; Ocln: occludin; TBI: traumatic brain injury; UA: urolithin A; Ve: vehicle; ZO-1: zonula occluden-1.
To investigate whether UA alleviates TBI-induced BBB disruption, Evans blue dye extravasation was performed to evaluate BBB permeability. The TBI + vehicle group exhibited more extravasation compared with the sham group, and UA administration after TBI alleviated the extravasation (Figure 1B). Quantification of the extravasation showed a significant increase in the EB concentration in brain tissue in the TBI + vehicle group compared with the sham group (P < 0.001), and UA administration after TBI significantly reduced EB concentration compared with TBI + vehicle (P = 0.005; Figure 1C), indicating that UA administration can alleviate TBI-induced BBB leakage.
Tight junction proteins are closely associated with BBB function (Obermeier et al., 2013), and therefore we investigated the changes in expression of ZO-1 and occludin in injured cortex. Immunofluorescence staining showed that tight junction proteins accompany the vascular marker CD31. Following TBI, the integrity of tight junction proteins was disrupted and multiple gaps were observed (white arrows; Figure 1D and E), but there was no disruption of CD31 integrity. The TBI + UA group exhibited fewer gaps compared with the TBI + vehicle group (Figure 1D and E). Western blot was used to assess the expression of tight junction proteins (Figure 1F and G). Consistent with immunofluorescence staining, the expression of tight junction proteins was significantly reduced after TBI (P < 0.001, compared with sham), and UA administration after TBI alleviated the reduction of tight junction proteins compared with TBI + vehicle (P < 0.001).
Urolithin A attenuates neuronal apoptosis following traumatic brain injury
To assess whether UA could attenuate neuronal apoptosis following TBI, we performed TUNEL/NeuN double immunostaining of cerebral tissue ipsilateral to the injury site (Figure 2A). Extensive neuronal apoptosis was observed in the cortex following TBI. The number of apoptotic neurons was significantly decreased in the TBI + UA group compared with that in the TBI + vehicle group (P < 0.001), indicating that UA attenuated neuronal apoptosis after TBI (Figure 2B).
Figure 2.

Urolithin A attenuates neuronal apoptosis 72 hours following traumatic brain injury.
(A) TUNEL assay (red) and costaining of NeuN (green, Alexa Fluor 488) in injured cortex. TUNEL+/NeuN+ cells are apoptotic neurons. UA administration reduced the number of apoptotic neurons in injured cortex following TBI. Scale bars: 25 µm. (B) Count of apoptotic neurons in the cortex for each group (n = 6). (C) Representative western blots of cleaved caspase-3, caspase-3, and bcl-2. (D) Quantification of relative protein expression (n = 5). Data are represented as mean ± SD. **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni's post hoc test). Cl-caspase3: Cleaved caspase3; n.s.: no statistical significance between indicated groups; NeuN: neuronal nuclei; TBI: traumatic brain injury; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling; UA: urolithin A; Ve: vehicle.
Caspase-3 and bcl-2 are key regulators of the apoptotic pathway (Franklin, 2011). Therefore, we investigated the cortical expression of cleaved caspase-3, caspase-3, and bcl-2 by western blot. Consistent with the TUNEL assay, our results showed that UA administration after TBI significantly reduced the protein expression of cleaved caspase-3 and increased the protein expression of bcl-2 compared with TBI + vehicle (P < 0.001 and P = 0.008, respectively; Figure 2C and D). Caspase-3 levels did not significantly change among the three groups (P = 0.488).
Urolithin A reinforces neuronal autophagy following traumatic brain injury
UA administration activates autophagy (Ahsan et al., 2019; Andreux et al., 2019; Lin et al., 2020). To investigate autophagy levels in cortical neurons, we used immunofluorescence to identify LC3 and p62, two autophagy markers, and the neuron marker NeuN in injured cortex (Figure 3A and B). We found that LC3 fluorescence intensity significantly increased following TBI, and the TBI + UA group had higher LC3 fluorescence intensity than the TBI + vehicle group (P < 0.001). In comparison, p62 fluorescence intensity significantly decreased following TBI, and the TBI + UA group had lower p62 fluorescence intensity than the TBI + vehicle group (P = 0.010; Figure 3C). Both LC3 and p62 immunostaining were surrounded by or merged with NeuN immunostaining in injured cortex. Consistent with our immunostaining results, western blot of injured cortex also showed increased LC3-II expression and decreased p62 expression following TBI (P < 0.001 for both LC3-II and p62, compared with sham), and the TBI + UA group had higher LC3-II expression (P = 0.006) and lower p62 expression (P = 0.003; Figure 3D and E) than the TBI + vehicle group. These results indicate that neuronal autophagy is activated following TBI, and UA administration could reinforce neuronal autophagy.
Figure 3.
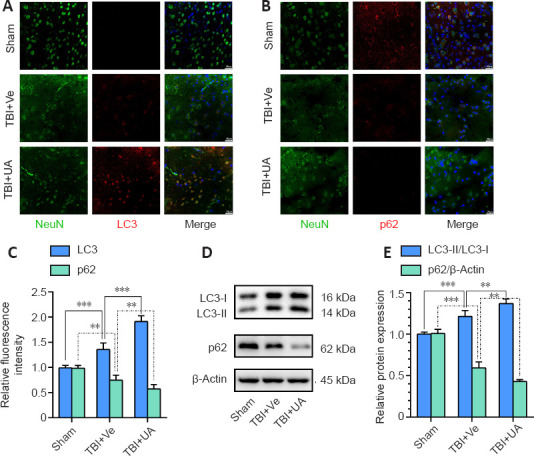
Urolithin A reinforces neuronal autophagy in injured cortex activated by traumatic brain injury.
(A, B) Representative immunostaining of LC3 (red, Alexa Fluor 594) and p62 (red, Alexa Fluor 594) with NeuN (green, Alexa Fluor 488) 72 hours following TBI in injured cortex. Fluorescence intensity of LC3 increased after TBI and was further reinforced by UA, while fluorescence intensity of p62 decreased after TBI and was further weakened by UA. Scale bars: 25 µm. (C) Quantification of relative fluorescence intensity of LC3 and p62 (n = 6). (D) Representative western blots of LC3-II, LC3-I, and p62. (E) Quantification of relative protein expression (n = 5). Data are represented as mean ± SD. **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni's post hoc test). LC3: Microtubule associated protein 1 light chain 3; NeuN: neuronal nuclei; TBI: traumatic brain injury; UA: urolithin A; Ve: vehicle.
PI3K/Akt/mTOR and Akt/IKK/NFκB pathways are involved in the neuroprotective effect of urolithin A
Many publications have reported that UA acts as an inhibitor of Akt phosphorylation, thus reinforcing autophagy via the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR signaling pathway (Komatsu et al., 2018; Xu et al., 2018; Totiger et al., 2019). Akt is also a key regulator in the Akt/IKK/NFκB signaling pathway, which regulates neuroinflammation (Ozes et al., 1999). Therefore, we speculate that the neuroprotective effects of UA may be partially achieved by inhibiting Akt phosphorylation, thus reinforcing autophagy and attenuating neuroinflammation via the PI3K/Akt/mTOR and Akt/IKK/NFκB signaling pathways, respectively (Figure 4A). Western blot analysis of injured cortex samples showed that the phosphorylation levels of Akt and mTOR decreased after TBI (P < 0.001, compared with sham) and were further decreased by UA administration after TBI (P = 0.010 and P = 0.001, for Akt and mTOR, respectively, compared with TBI + vehicle). The phosphorylation levels of IKKα and NFκB increased after TBI (P < 0.001, compared with sham) and were reduced by UA administration after TBI (P < 0.001; Figure 4B and C). These results indicate the involvement of the PI3K/Akt/mTOR and Akt/IKK/NFκB signaling pathways in the neuroprotective effects of UA to reinforce autophagy and attenuate neuroinflammation.
Figure 4.
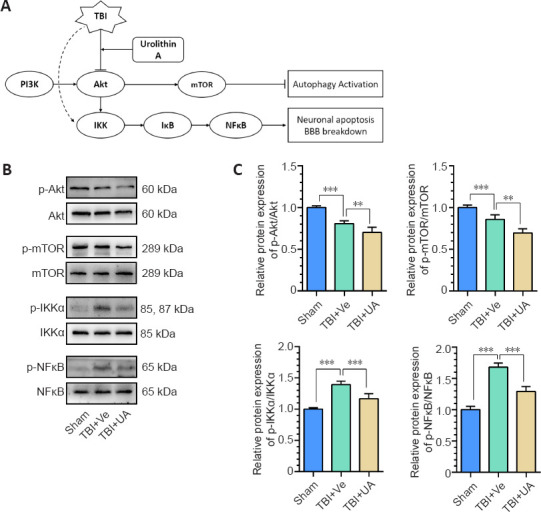
PI3K/Akt/mTOR and Akt/IKK/NFκB signaling pathways may be involved in the neuroprotective effect of urolithin A.
(A) Pattern diagram of involved pathways. UA downregulates Akt phosphorylation, thus reinforcing autophagy via the PI3K/Akt/mTOR signaling pathway and alleviates inflammation via the Akt/IKK/NFκB signaling pathway. (B) Representative western blots of p-Akt, Akt, p-mTOR, mTOR, p-IKKα, IKKα, p-NFκB, and NFκB. (C) Quantification of relative protein expression (n = 5). Data are represented as mean ± SD. **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Bonferroni's post hoc test). Akt: Protein kinase B; BBB: blood-brain barrier; BBB: blood-brain barrier; IKK: inhibitor of NFκB kinase; mTOR: mammalian target of rapamycin; NFκB: nuclear factor kappa B; p-Akt: phospho-Akt; PI3K: phosphatidylinositol 3-kinase; p-IKKα: phospho-IKKα; p-mTOR: phospho-mTOR; p-NFκB: phospho-NFκB; TBI: traumatic brain injury; TBI: traumatic brain injury; UA: urolithin A; Ve: vehicle.
Urolithin A ameliorates neurological deficits following traumatic brain injury
We evaluated the neurological deficits in mice by the mNSS score and the rotarod test. Our results showed that the mNSS score of the TBI + UA group was significantly reduced 72 hours after TBI; this reduction continued to 14 days following TBI (P = 0.658, P = 0.002, P = 0.007, and P = 0.003, for 1, 3, 7, and 14 days after TBI, respectively; Figure 5A). The rotarod test showed that the TBI + UA group had a longer latency to fall than the TBI + vehicle group at 3, 7, and 14 days following TBI (P = 0.226, P = 0.008, P < 0.001, and P < 0.001, for 1, 3, 7, and 14 days after TBI, respectively; Figure 5B).
Figure 5.
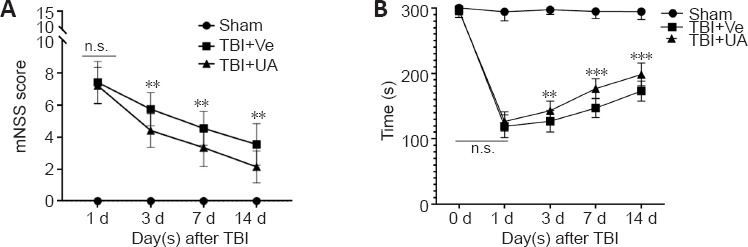
Urolithin A ameliorates neurological deficits following traumatic brain injury.
(A) mNSS score was calculate at 1, 3, 7, and 14 days following TBI (n = 15). (B) Rotarod tests were performed before TBI and 1, 3, 7, 14 days following TBI, and latencies to falls were recorded to assess motor and balance deficits (n = 15). Data are represented as mean ± SD. **P < 0.01, ***P < 0.001, vs. TBI + Ve group (one-way analysis of variance followed by Bonferroni's post hoc test). mNSS: Modified neurological severity score; n.s.: no statistical significance between indicated groups; TBI: traumatic brain injury; UA: urolithin A; Ve: vehicle.
Discussion
Currently there is no specific therapy for TBI, and all curative treatments focus on relieving TBI-induced secondary damage. UA can permeate the BBB (Yuan et al., 2016; Kujawska et al., 2019) and has neuroprotective effects against central nervous system disorders such as Alzheimer's disease (Gong et al., 2019), ischemic brain injury (Ahsan et al., 2019), and Parkinson's disease (Kujawska et al., 2019). Our present study demonstrated for the first time that UA produces neuroprotective effects against TBI at a dose of 2.5 mg/kg in an experimental TBI mouse model by protecting the BBB, attenuating brain edema, reducing neuronal apoptosis, and alleviating neurobehavioral deficits. Inhibition of the PI3K/Akt/mTOR and Akt/IKK/NFκB pathways and activation of neuronal autophagy may be involved in these effects.
As previously described, UA is a natural metabolite produced from food enriched in ellagitannins, which is further transformed by gut microbiota to urolithins (Espín et al., 2013; Nuñez-Sánchez et al., 2014; Kujawska and Jodynis-Liebert, 2020). The bioavailability of UA is highly divergent among individuals and depends on the composition of their gut microbiota (Gerhauser, 2018). Therefore, intraperitoneal injection of UA bypasses the transformation step performed by gut microbiota, thus avoiding individual variations in bioavailability. UA has a favorable safety profile: a clinical trial reported no side effects in volunteers administered with up to 2000 mg/day UA (Andreux et al., 2019). Recent studies have revealed that the pharmacological effects of UA mainly stem from its anti-inflammatory and autophagy-activating properties (Ishimoto et al., 2011; González-Sarrías et al., 2017; Komatsu et al., 2018; Xu et al., 2018; Ahsan et al., 2019), and we validated these properties in an experimental TBI mouse model.
BBB damage is a common secondary injury following TBI that further leads to cerebral edema and intracerebral hemorrhage, thus worsening morbidity and mortality in TBI survivors. Singh et al. (2019) reported that UA could protect tight junction proteins and, thus, gut barrier function in patients with inflammatory bowel diseases. Similarly, in our present study, we found that UA reduces brain edema and protects BBB function by alleviating the disruption of tight junction proteins. Neuroinflammation plays a crucial role in the breakdown of the BBB, and NFκB is one of the key regulators of this inflammatory response (Sulhan et al., 2020). Following TBI, NFκB can be activated by multiple pathways regulated by molecules such as reactive oxygen species and heat shock proteins (Corps et al., 2015; Corrigan et al., 2016). Akt/IKK/NFκB is one of the upstream pathways regulating NFκB activation. Activated Akt mediates the phosphorylation of the IKK complex, which continues to degrade IκB. In a resting state, IκB is located in the cytoplasm and binds to NFκB. After degradation of IκB, the activation site of NFκB is released and phosphorylated, and NFκB transfers into the nucleus to exert its inflammatory effects (Ozes et al., 1999; Hayden and Ghosh, 2004). A growing body of evidence shows that UA inhibits Akt activation (Komatsu et al., 2018; Xu et al., 2018; Totiger et al., 2019), which is in accordance with results from this present study. Western blot confirmed the downregulation of Akt, IKKα, and NFκB phosphorylation after UA administration, which indicates that UA alleviates the inflammatory response following TBI via the Akt/IKK/NFκB pathway, thus protecting BBB function and preserving neuronal survival.
Autophagy is a process by which cells conserve and recycle their organelles and toxic proteins. Baseline autophagy occurs continuously and is further activated under environmental stresses such as starvation and cell injury (Wu and Lipinski, 2019). Autophagy includes several steps. First, organelles and proteins are sequestered by a specific double membrane structure, called the phagophore, to form autophagosomes. Then, the autophagosomes are transported and fuse with lysosomes to degrade their contents. Many proteins play a crucial role in this process. LC3-I exists in the cytoplasm; when autophagy is activated, LC3-I is lipidated to form LC3-II, which is specifically recruited to the phagophore. Ubiquitylated proteins are bound by p62, which enables them to be taken up by autophagosomes, leading to degradation of the ubiquitylated proteins, as well as p62 itself. Hence, LC3-II levels positively correlate with the number of autophagosomes, and p62 levels negatively correlate with the level of substrate degradation (Narendra et al., 2010; Noda and Inagaki, 2015). It is generally believed that autophagy activates following TBI with LC3-II levels increasing and p62 levels decreasing (Zhang and Wang, 2018). These responses match the results from our present study. However, some researchers found that LC3-II and p62 were both increased after TBI, suggesting that the number of autophagosomes increase and substrates accumulate, indicating impairment of autophagy flux (Sarkar et al., 2014; Zeng et al., 2018). This divergence may be due to the severity of TBI, the time-window assessed, or sampling. An increase of p62 was more likely to be observed after severe TBI, while a decrease of p62 was more likely to be observed after mild or moderate TBI (Wu and Lipinski, 2019).
PI3K/Akt/mTOR is one of the main pathways of autophagy regulation; in particular, the mTOR complex 1 has an intense inhibitory effect on autophagy (Akira and Takeda, 2004). In this study, we observed that the phosphorylation of Akt and mTOR decreased after UA administration, suggesting involvement of the PI3K/Akt/mTOR pathway in the autophagy reinforcement effect of UA.
Though numerous studies have reported the activation of autophagy following both experimental and clinical TBI (Zhang and Wang, 2018; Zeng et al., 2020), it remains controversial whether modulating autophagy is beneficial or detrimental to TBI-induced secondary injury. Erlich et al. (2007) first found that administration of rapamycin reduced inflammation, increased neuronal survival, and improved neurobehavioral function in an experimental TBI model. Because rapamycin can augment autophagy by inhibiting PI3K/AKT/mTOR signaling, Erlich et al. (2007) concluded that rapamycin was neuroprotective following TBI by activating autophagy. Similarly, Yin et al. (2018) reported that administration of docosahexaenoic acid improves neurological function recovery and reduces brain damage after TBI by activating autophagic flux. In addition, many neuroprotective drugs are thought to alleviate TBI-induced secondary injury by autophagy activation (Xu et al., 2014; Zhang et al., 2017; Ma et al., 2019). However, some studies suggest that autophagy plays a detrimental role in TBI. Luo et al. (2011) reported that bafilomycin A1 and 3-methyladenine, two autophagy inhibitors, improve behavioral outcomes, attenuate cell injury, and reduce lesion volume and apoptosis after TBI, suggesting that autophagy is detrimental to TBI. Wang et al. (2017) found that administration of ketamine at a subanesthetic dose could exert neuroprotective effects against TBI by suppression of autophagy, and effects of an anesthetic dose of ketamine on TBI are not clear. This divergence can be explained by the multiple targets of ketamine and the complex network of associated signaling pathways. In this study, we found that UA reinforced neuronal autophagy and decreased neuronal apoptosis, suggesting that neuronal autophagy may be involved in the neuroprotective effect of UA. This supports the view that autophagy plays a beneficial role in TBI. Following TBI, large quantities of byproducts from endoplasmic reticulum stress accumulate in neurons, which will promote neuronal apoptosis; autophagy helps to reduce endoplasmic reticulum stress (Sarkar et al., 2014; Liu et al., 2015). Inhibition of autophagy can increase the expression of receptor-interacting protein kinase 1, thus inducing necroptosis (Liu et al., 2018a). These specific mechanisms need to be explored in the future.
There are several limitations to this study. First, the present research only includes in vivo investigations, and thus it is impossible to distinguish variations in function and pathway activation in different cell types. Second, we did not use inhibitors or knock-out technology to validate the specific targets in signaling pathways. Further work on these limitations needs to be performed in the future.
To conclude, our present study found that UA exerts its neuroprotective effects against TBI by protecting against BBB disruption, reducing brain edema, decreasing neuronal apoptosis, and reinforcing neuronal autophagy, thus ameliorating TBI-induced neurological deficits. Downregulation of the PI3K/Akt/mTOR and Akt/IKK/NFκB pathways may be involved in the neuroprotective effect of UA. This study provides a solid in vivo experimental basis for UA as a promising neuroprotective drug against TBI.
Additional file:
Additional Figure 1 (510.7KB, tif) : Illustration for CCI model and sampling region of the brain.
Illustration for CCI model and sampling region of the brain.
(A) The process of CCI modeling in mice. (B) Coronal section of the brain after CCI. Shaded area indicates the region where sampled for Immunofluorescence and TUNEL assay. (C) Gross specimen of the brain after CCI. After evacuating the hemorrhage, the injured cortex in shaded area would be sampled for western blot. CCI: Controlled cortical injury; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling.
Acknowledgments:
We would like to thank Prof. Guo-Yuan Yang (Institute of Med-X, Shanghai Jiao Tong University) and Prof. Yao-Hui Tang (Institute of Med-X, Shanghai Jiao Tong University) for assistance of animal behavior research.
Footnotes
P-Reviewer: Bartkowiak-Wieczorek J; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Yu J, Song LP; T-Editor: Jia Y
Conflicts of interest: The authors declare that they have no conflict of interest.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Funding: This work was supported by the National Natural Science Foundation of China, Nos. 81974189 (to HLT), 81801236 (to QYG and LC), 82001310 (to DXY).
References
- 1.Ahsan A, Zheng YR, Wu XL, Tang WD, Liu MR, Ma SJ, Jiang L, Hu WW, Zhang XN, Chen Z. Urolithin A-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo. CNS Neurosci Ther. 2019;25:976–986. doi: 10.1111/cns.13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, Auwerx J, Singh A, Rinsch C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1:595–603. doi: 10.1038/s42255-019-0073-4. [DOI] [PubMed] [Google Scholar]
- 4.Byun S, Lee E, Lee KW. Therapeutic implications of autophagy inducers in immunological disorders, infection, and cancer. Int J Mol Sci. 2017;18:1959. doi: 10.3390/ijms18091959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter RJ, Morton J, Dunnett SB. Motor coordination and balance in rodents. Curr Protoc Neurosci Chapter. 2001;8:8–12. doi: 10.1002/0471142301.ns0812s15. [DOI] [PubMed] [Google Scholar]
- 6.Chakrabarti M, Das A, Samantaray S, Smith JA, Banik NL, Haque A, Ray SK. Molecular mechanisms of estrogen for neuroprotection in spinal cord injury and traumatic brain injury. Rev Neurosci. 2016;27:271–281. doi: 10.1515/revneuro-2015-0032. [DOI] [PubMed] [Google Scholar]
- 7.Chang CP, Su YC, Hu CW, Lei HY. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2013;20:515–523. doi: 10.1038/cdd.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–2688. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- 9.Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72:355–362. doi: 10.1001/jamaneurol.2014.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corrigan F, Mander KA, Leonard AV, Vink R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation. 2016;13:264. doi: 10.1186/s12974-016-0738-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curl JL. Ketamine-xylazine anaesthesia in the Djungarian hamster (Phodopus sungorus) Lab Anim. 1988;22:309–312. doi: 10.1258/002367788780746269. [DOI] [PubMed] [Google Scholar]
- 12.Desai M, Jain A. Neuroprotection in traumatic brain injury. J Neurosurg Sci. 2018;62:563–573. doi: 10.23736/S0390-5616.18.04476-4. [DOI] [PubMed] [Google Scholar]
- 13.Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Espín JC, Larrosa M, García-Conesa MT, Tomás-Barberán F. Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: the evidence so far. Evid Based Complement Alternat Med 2013. 2013 doi: 10.1155/2013/270418. 270418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franklin JL. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid Redox Signal. 2011;14:1437–1448. doi: 10.1089/ars.2010.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerhauser C. Impact of dietary gut microbial metabolites on the epigenome. Philos Trans R Soc Lond B Biol Sci. 2018;373 doi: 10.1098/rstb.2017.0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong Z, Huang J, Xu B, Ou Z, Zhang L, Lin X, Ye X, Kong X, Long D, Sun X, He X, Xu L, Li Q, Xuan A. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J Neuroinflammation. 2019;16:62. doi: 10.1186/s12974-019-1450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.González-Sarrías A, Núñez-Sánchez M, Tomás-Barberán FA, Espín JC. Neuroprotective effects of bioavailable polyphenol-derived metabolites against oxidative stress-induced cytotoxicity in human neuroblastoma SH-SY5Y cells. J Agric Food Chem. 2017;65:752–758. doi: 10.1021/acs.jafc.6b04538. [DOI] [PubMed] [Google Scholar]
- 19.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 20.Heilman J, Andreux P, Tran N, Rinsch C, Blanco-Bose W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem Toxicol. 2017;108:289–297. doi: 10.1016/j.fct.2017.07.050. [DOI] [PubMed] [Google Scholar]
- 21.Ishimoto H, Shibata M, Myojin Y, Ito H, Sugimoto Y, Tai A, Hatano T. In vivo anti-inflammatory and antioxidant properties of ellagitannin metabolite urolithin A. Bioorg Med Chem Lett. 2011;21:5901–5904. doi: 10.1016/j.bmcl.2011.07.086. [DOI] [PubMed] [Google Scholar]
- 22.Kaya M, Ahishali B. Assessment of permeability in barrier type of endothelium in brain using tracers: Evans blue, sodium fluorescein, and horseradish peroxidase. Methods Mol Biol. 2011;763:369–382. doi: 10.1007/978-1-61779-191-8_25. [DOI] [PubMed] [Google Scholar]
- 23.Komatsu W, Kishi H, Yagasaki K, Ohhira S. Urolithin A attenuates pro-inflammatory mediator production by suppressing PI3-K/Akt/NF-κB and JNK/AP-1 signaling pathways in lipopolysaccharide-stimulated RAW264 macrophages: Possible involvement of NADPH oxidase-derived reactive oxygen species. Eur J Pharmacol. 2018;833:411–424. doi: 10.1016/j.ejphar.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 24.Kujawska M, Jodynis-Liebert J. Potential of the ellagic acid-derived gut microbiota metabolite - Urolithin A in gastrointestinal protection. World J Gastroenterol. 2020;26:3170–3181. doi: 10.3748/wjg.v26.i23.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kujawska M, Jourdes M, Kurpik M, Szulc M, Szaefer H, Chmielarz P, Kreiner G, Krajka-Kuźniak V, Mikołajczak P, Teissedre PL, Jodynis-Liebert J. Neuroprotective effects of pomegranate juice against parkinson's disease and presence of ellagitannins-derived metabolite-urolithin A-in the brain. Int J Mol Sci. 2019;21:202. doi: 10.3390/ijms21010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HJ, Jung YH, Choi GE, Kim JS, Chae CW, Lim JR, Kim SY, Yoon JH, Cho JH, Lee SJ, Han HJ. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021;28:184–202. doi: 10.1038/s41418-020-0593-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin J, Zhuge J, Zheng X, Wu Y, Zhang Z, Xu T, Meftah Z, Xu H, Wu Y, Tian N, Gao W, Zhou Y, Zhang X, Wang X. Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway. Free Radic Biol Med. 2020;150:109–119. doi: 10.1016/j.freeradbiomed.2020.02.024. [DOI] [PubMed] [Google Scholar]
- 28.Liu S, Sarkar C, Dinizo M, Faden AI, Koh EY, Lipinski MM, Wu J. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015;6:e1582. doi: 10.1038/cddis.2014.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S, Li Y, Choi HMC, Sarkar C, Koh EY, Wu J, Lipinski MM. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018a;9:476. doi: 10.1038/s41419-018-0469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu YL, Yuan F, Yang DX, Xu ZM, Jing Y, Yang GY, Geng Z, Xia WL, Tian HL. Adjudin attenuates cerebral edema and improves neurological function in mice with experimental traumatic brain injury. J Neurotrauma. 2018b;35:2850–2860. doi: 10.1089/neu.2017.5397. [DOI] [PubMed] [Google Scholar]
- 31.Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ, Dai DK, Shen YW, Xu HF, Ni H, Wan L, Qin ZH, Tao LY, Zhao ZQ. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63. doi: 10.1016/j.neuroscience.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 32.Ma J, Ni H, Rui Q, Liu H, Jiang F, Gao R, Gao Y, Li D, Chen G. Potential roles of NIX/BNIP3L pathway in rat traumatic brain injury. Cell Transplant. 2019;28:585–595. doi: 10.1177/0963689719840353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 34.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mytych J. Klotho and neurons: mutual crosstalk between autophagy, endoplasmic reticulum, and inflammatory response. Neural Regen Res. 2021;16:1542–1543. doi: 10.4103/1673-5374.303014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noda NN, Inagaki F. Mechanisms of Autophagy. Annu Rev Biophys. 2015;44:101–122. doi: 10.1146/annurev-biophys-060414-034248. [DOI] [PubMed] [Google Scholar]
- 38.Nuñez-Sánchez MA, García-Villalba R, Monedero-Saiz T, García-Talavera NV, Gómez-Sánchez MB, Sánchez-Álvarez C, García-Albert AM, Rodríguez-Gil FJ, Ruiz-Marín M, Pastor-Quirante FA, Martínez-Díaz F, Yáñez-Gascón MJ, González-Sarrías A, Tomás-Barberán FA, Espín JC. Targeted metabolic profiling of pomegranate polyphenols and urolithins in plasma, urine and colon tissues from colorectal cancer patients. Mol Nutr Food Res. 2014;58:1199–1211. doi: 10.1002/mnfr.201300931. [DOI] [PubMed] [Google Scholar]
- 39.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 41.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, Pearl EJ, et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020;18:e3000410. doi: 10.1371/journal.pbio.3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarkar C, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, Lipinski MM. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10:2208–2222. doi: 10.4161/15548627.2014.981787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savi M, Bocchi L, Mena P, Dall’Asta M, Crozier A, Brighenti F, Stilli D, Del Rio D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2017;16:80. doi: 10.1186/s12933-017-0561-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shao ZQ, Dou SS, Zhu JG, Wang HQ, Wang CM, Cheng BH, Bai B. Apelin-13 inhibits apoptosis and excessive autophagy in cerebral ischemia/reperfusion injury. Neural Regen Res. 2021;16:1044–1051. doi: 10.4103/1673-5374.300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, Hiwale AA, Saiyed T, Patel P, Vijay-Kumar M, Langille MGI, Douglas GM, Cheng X, Rouchka EC, Waigel SJ, Dryden GW, Alatassi H, Zhang HG, Haribabu B, Vemula PK, et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun. 2019;10:89. doi: 10.1038/s41467-018-07859-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stocchetti N, Carbonara M, Citerio G, Ercole A, Skrifvars MB, Smielewski P, Zoerle T, Menon DK. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16:452–464. doi: 10.1016/S1474-4422(17)30118-7. [DOI] [PubMed] [Google Scholar]
- 48.Sulhan S, Lyon KA, Shapiro LA, Huang JH. Neuroinflammation and blood-brain barrier disruption following traumatic brain injury: pathophysiology and potential therapeutic targets. J Neurosci Res. 2020;98:19–28. doi: 10.1002/jnr.24331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Totiger TM, Srinivasan S, Jala VR, Lamichhane P, Dosch AR, Gaidarski AA, 3rd , Joshi C, Rangappa S, Castellanos J, Vemula PK, Chen X, Kwon D, Kashikar N, VanSaun M, Merchant NB, Nagathihalli NS. Urolithin A, a novel natural compound to target PI3K/AKT/mTOR pathway in pancreatic cancer. Mol Cancer Ther. 2019;18:301–311. doi: 10.1158/1535-7163.MCT-18-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tuohetaerbaike B, Zhang Y, Tian Y, Zhang NN, Kang J, Mao X, Zhang Y, Li X. Pancreas protective effects of Urolithin A on type 2 diabetic mice induced by high fat and streptozotocin via regulating autophagy and AKT/mTOR signaling pathway. J Ethnopharmacol. 2020;250:112479. doi: 10.1016/j.jep.2019.112479. [DOI] [PubMed] [Google Scholar]
- 51.Wang CQ, Ye Y, Chen F, Han WC, Sun JM, Lu X, Guo R, Cao K, Zheng MJ, Liao LC. Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience. 2017;343:30–38. doi: 10.1016/j.neuroscience.2016.11.029. [DOI] [PubMed] [Google Scholar]
- 52.Wu F, Xu K, Xu K, Teng C, Zhang M, Xia L, Zhang K, Liu L, Chen Z, Xiao J, Wu Y, Zhang H, Chen D. Dl-3n-butylphthalide improves traumatic brain injury recovery via inhibiting autophagy-induced blood-brain barrier disruption and cell apoptosis. J Cell Mol Med. 2020;24:1220–1232. doi: 10.1111/jcmm.14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu J, Lipinski MM. Autophagy in neurotrauma: good, bad, or dysregulated. Cells. 2019;8:693. doi: 10.3390/cells8070693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, Wang H, Lu X, Ding K, Zhang L, He J, Wei W, Wu Y. Posttraumatic administration of luteolin protects mice from traumatic brain injury: implication of autophagy and inflammation. Brain Res. 2014;1582:237–246. doi: 10.1016/j.brainres.2014.07.042. [DOI] [PubMed] [Google Scholar]
- 55.Xu J, Yuan C, Wang G, Luo J, Ma H, Xu L, Mu Y, Li Y, Seeram NP, Huang X, Li L. Urolithins attenuate LPS-induced neuroinflammation in BV2 microglia via MAPK, Akt, and NF-κB signaling pathways. J Agric Food Chem. 2018;66:571–580. doi: 10.1021/acs.jafc.7b03285. [DOI] [PubMed] [Google Scholar]
- 56.Yin Y, Li E, Sun G, Yan HQ, Foley LM, Andrzejczuk LA, Attarwala IY, Hitchens TK, Kiselyov K, Dixon CE, Sun D. Effects of DHA on hippocampal autophagy and lysosome function after traumatic brain injury. Mol Neurobiol. 2018;55:2454–2470. doi: 10.1007/s12035-017-0504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan T, Ma H, Liu W, Niesen DB, Shah N, Crews R, Rose KN, Vattem DA, Seeram NP. Pomegranate's neuroprotective effects against Alzheimer's disease are mediated by urolithins, its ellagitannin-gut microbial derived metabolites. ACS Chem Neurosci. 2016;7:26–33. doi: 10.1021/acschemneuro.5b00260. [DOI] [PubMed] [Google Scholar]
- 58.Zeng XJ, Li P, Ning YL, Zhao Y, Peng Y, Yang N, Zhao ZA, Chen JF, Zhou YG. Impaired autophagic flux is associated with the severity of trauma and the role of A(2A)R in brain cells after traumatic brain injury. Cell Death Dis. 2018;9:252. doi: 10.1038/s41419-018-0316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeng Z, Zhang Y, Jiang W, He L, Qu H. Modulation of autophagy in traumatic brain injury. J Cell Physiol. 2020;235:1973–1985. doi: 10.1002/jcp.29173. [DOI] [PubMed] [Google Scholar]
- 60.Zhang L, Wang H. Autophagy in traumatic brain injury: a new target for therapeutic intervention. Front Mol Neurosci. 2018;11:190. doi: 10.3389/fnmol.2018.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang L, Wang H, Fan Y, Gao Y, Li X, Hu Z, Ding K, Wang Y, Wang X. Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci Rep. 2017;7:46763. doi: 10.1038/srep46763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Wang J, Gao JZ, Zhang XN, Dou KX, Shi WD, Xie AM. P2X4 receptor participates in autophagy regulation in Parkinson's disease. Neural Regen Res. 2021;16:2505–2511. doi: 10.4103/1673-5374.313053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Illustration for CCI model and sampling region of the brain.
(A) The process of CCI modeling in mice. (B) Coronal section of the brain after CCI. Shaded area indicates the region where sampled for Immunofluorescence and TUNEL assay. (C) Gross specimen of the brain after CCI. After evacuating the hemorrhage, the injured cortex in shaded area would be sampled for western blot. CCI: Controlled cortical injury; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling.