

Abstract
Anatomical studies and stimulation studies in the decerebrate animal have suggested that the muscle atonia of rapid eye movement (REM) sleep is mediated by a projection from cholinoceptive glutamatergic neurons in the pons to the nucleus magnocellularis (NMC) of medulla. This model suggests that glutamate release in NMC should be enhanced in REM sleep. In the present study, glutamate release across the sleep–wake cycle in NMC was measured by in vivo microdialysis. We found that glutamate release in NMC was significantly higher (p=0.0252) during REM sleep than during wakefulness (W). Glutamate release during REM sleep was not elevated either in nucleus paramedianus (NPM) or in the pontine inhibitory area (PIA) regions where cholinergic stimulation suppresses muscle tone. Acetylcholine (ACh) microinjection into PIA enhanced glutamate release in NMC. These results support the hypothesis that a glutamatergic pathway from PIA to NMC is responsible for the suppression of muscle tone in REM sleep.
Keywords: REM sleep, Atonia, Glutamate, Nucleus magnocellularis of medulla, Nucleus paramedianus of medulla, Pontine inhibitory area, In vivo microdialysis
Muscle atonia is present throughout rapid eye movement (REM) sleep. Lesion studies have implicated the pontine inhibitory area (PIA) [6,8] and medial medulla [7,25] in the generation of muscle atonia.
Magoun [22] first demonstrated that stimulation of the medial medulla of the decerebrate animal produced a non-reciprocal suppression of muscle tone. Further work in our laboratory demonstrated that electrical stimulation of pontine and midbrain regions could also produce non-reciprocal inhibition of muscle tone [16–18]. The pontine inhibitory region has been shown to be cholinoceptive [2,5,9,23,26]. Work in our laboratory has shown that this region is also responsive to non-NMDA glutamate agonists and to corticotropin releasing factor [19,20]. In addition, we have determined that the medullary atonia region can be subdivided into a rostral region, corresponding to nucleus magnocellularis (NMC) and caudal region, corresponding to nucleus paramedianus (NPM). The NMC, like the PIA, produces atonia if non-NMDA glutamate agonists or corticotropin releasing factor are applied. However, unlike the PIA, NMC does not produce atonia when cholinergic agonists are microinjected [19]. The NPM, on the other hand, does produce atonia when cholinergic receptors are activated, but not when glutamate agonists are applied [19].
Dialysis studies have been undertaken to determine the nature of transmitter release in these regions during REM sleep in the intact animal. Kodama et al. [12] showed that REM sleep was accompanied by an increased release of acetylcholine (ACh) in the PIA, confirming the hypothesis that enhanced ACh release mediated REM sleep atonia. In further work, we found a similar increase in ACh release in NPM, but not the glutamate sensitive NMC in REM sleep [11]. In the current study, we examined glutamate release in PIA, NPM and NMC in natural REM sleep with the in vivo microdialysis technique.
Six adult cats weighing 3.0–4.5 kg were anesthetized by sodium pentobarbital (35 mg/kg i.p.) and chronically implanted with standard electrodes for recording cortical electroencephalograms (EEG), ponto–geniculo–occipital (PGO) waves, eye movement potentials (EOG) and nuchal electromyograms (EMG). Guide cannulae (23G) for microdialysis probes were also stereotaxically implanted above the centers of target regions, PIA (A: −1.5, L: 2.25, D: −4.0), NMC (A: −9.5, L: 1.0, D: −10.0) and NPM (A: −13.5, L: 1.0, D: −9.5).
Following recovery from surgery, at least 12 h prior to the collection of dialysates, a microdialysis probe was inserted, through the guide cannula into PIA, NMC or NPM. Brain microdialysis and sleep monitoring Were simultaneously performed for 48 consecutive hours. We used a coaxial type microdialysis probe, made of polyamide-coated quartz glass, covered by semipermeable membrane (o.d. 220 μm, and 2 mm in length; supplied by EICOM, Kyoto). Ringer solution (Na+ 145 mM, K+ 2.7 mM, Mg2+ 1.0 mM, Ca2+ 1.2 mM, Cl− 150 mM) was perfused at a flow rate of 2 μl/min. Dialysate sampling was timed to each sleep–waking state under EEG monitoring. Each sample was collected for 5 min and stored frozen below −80°C, until analysis.
Acute decerebrate studies were performed on two cats to investigate the change of glutamate release in NMC produced by ACh microinjection in PIA. ACh microinjections (100 μg/0.5 μl) were made through a 26-gauge Hamilton 1 μl microsyringe over a period of 1 min. Dialysates were collected for 5-min intervals during the pre-stimulation, stimulation and post-stimulation with ACh microinjection.
The perfusate (10 μl) was reacted with o-phtaldialde-hyde reagent (8 μl), consisting of 500 μl of o-phthaldialdehyde in ethanol (54 mg/ml), 20 μl of mercap-thethanol (Nacalai Tesque), and 9.48 ml sodium carbonate buffer (0.1 M, pH 9.5 with HCl). Following a 2-min reaction time at room temperature (25°C), 15 μl of the derivatized sample mixture was injected into the HPLC system. Amino acid derivatives were then separated on a C18 column (30°C) using an isocratic mobile phase of 70% sodium phosphate buffer (0.1 M, pH 5.5) and 30% methanol containing 5 mg EDTA–2Na. Electrochemical detection was accomplished using a glassy-carbon electrode (700 mV; EICOM). The concentration of amino acid in the perfusate was quantified with a Labchart-180 (SIC, Japan) using an external amino acid standard. The detection limit for glutamate was 50 femtomol (fmol).
After the experiment, the animals were given a lethal dose of pentobarbital (80 mg/kg, i.p.) and perfused through the heart with 1000 ml of saline solution followed by 10% formalin in 0.1 M phosphate buffer (pH 7.2). The brain was then REMoved and cut into sagittal sections at 50 μm thickness and stained by Cresyl violet.
A total of 378 dialysate samples were obtained from the three targeted areas (Fig. 1). Forty-one sets of dialysates during each stage of slow wave sleep (SWS), REM sleep and wakefulness (W) from both NMC and NPM (41 dialysates×sleep stages), and 44 sets of dialysates from PIA (44×dialysates). The mean (±S.E.M.) amount of glutamate content in dialysate during SWS in NMC, NPM and PIA was 217.5±21.9 (fmol/min sample), 239.6±23.5 and 164.3±18.1, respectively. The differences of these group means were statistically significant [p=0.0375, F=3.372, F(123, 2), ANOVA].
Fig. 1.
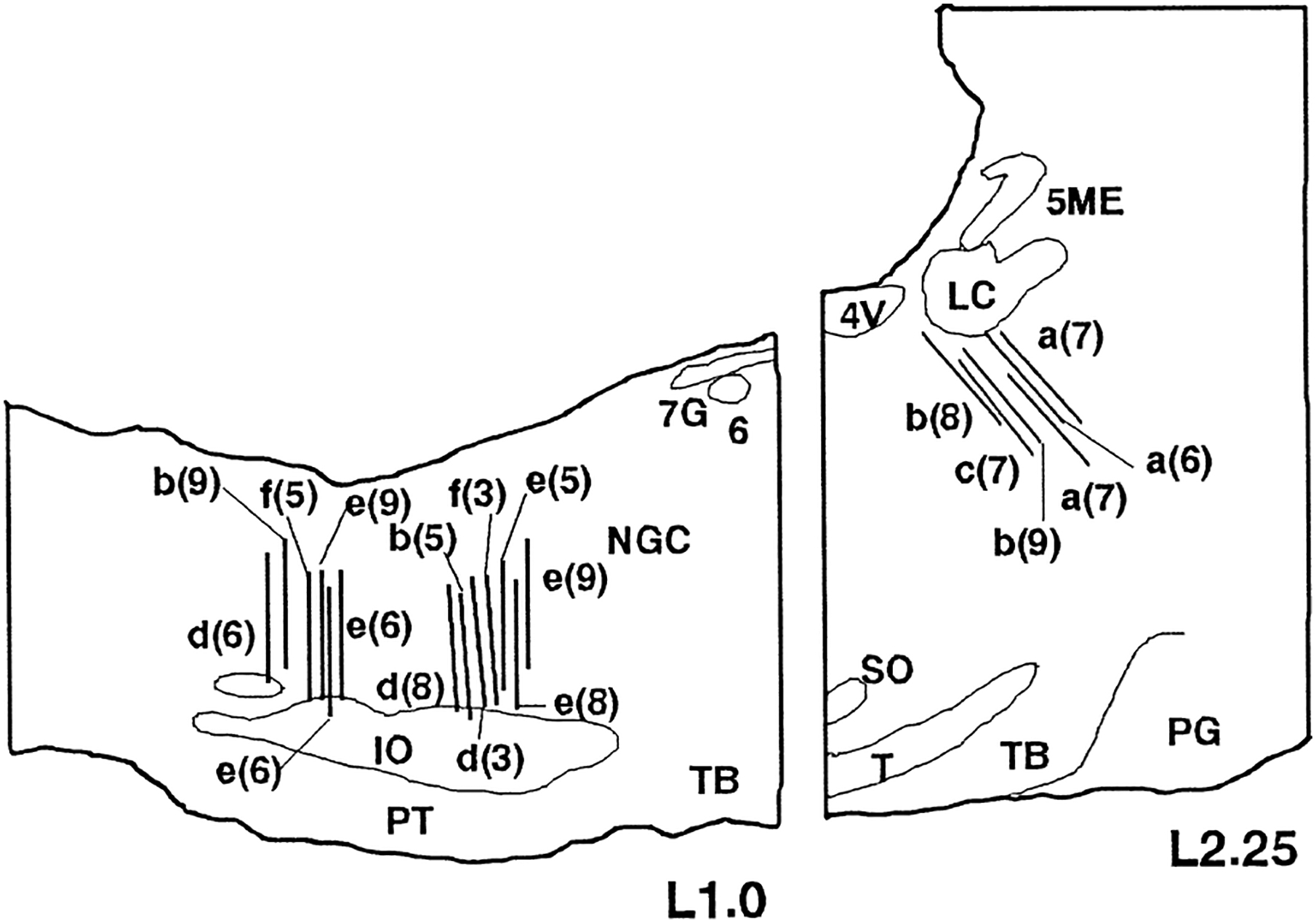
Location of the microdialysis membranes. Each bar indicates the positions of the membrane verified by the histological study. All the positions, which are projected to the L 1.0/L 2.25 sagittal planes, are in the targeted areas (PIA—A: −1.0–−3.0, L: 2.0–2.5, D: −3.5–−5.0; NMC—A: −9.0– −10.5, L: 0.8–1.3, D: −8.5–−11.0; NPM—A: −12.5– −14.0, L: 0.8–1.3, D: −8.5–−11.0.. The letter and number indicate the cat’s identification letter and the number of the dialysate sample sets obtained from the same track, respectively. Abbreviations: 4V, fourth ventricle; 5ME, mesencephalic trigeminal nucleus; 6, abducens nucleus; 7G, genu of the facial nerve; IO, inferior olivary nucleus; LC, locus coerulus nucleus; NGC, nucleus gigantocellularis; PG, pontine gray; PT, pyramidal tract; SO, superior olivary nucleus; T, nucleus of trapezoid body; TB, trapezoid body.
The ratios of glutamate content in dialysates during REM sleep and wakefulness to the values during SWS were calculated and submitted to further analysis. Planned comparisons for each area using the paired t-test indicated that: In NMC (n=41), the ratio REM/SWS (1.114±0.056) was significantly higher (two-tailed t-test, p=0.0252) than that of W/SWS (0.978±0.040), while there was no significant difference between the ratio of REM/SWS (1.034±0.043) and W/SWS (1.003±0.046) in NPM (n=41) (Table 1). Contrary to our expectations, the ratio of REM/SWS (1.026±0.036) in PIA (n=44) was almost the same as that of W/SWS (1.029±0.044) (Table 1). As a preliminary experiment, glutamate content was measured by a 1-mm long microdialysis probe in the oral part of pontine reticular formation, but we failed to find any point of a REM specific glutamate increase. In the dorsocaudal part of PIA, glutamate release tended to increase during wakefulness (W/SWS ratio, mean ± S.E.M.=1.170±0.071, n=22) compared to that during REM sleep (REM/SWS ratio=0.994±0.052, n=22). In the ventral part of PIA, glutamate release increased both during REM (REM/SWS=1.188±0.095, n=21) and wakefulness (W/SWS=1.197±0.078, n=21). In the center of PIA, there was little change of glutamate content across sleep cycle (REM/SWS=1.038±0.075; W/SWS=1.075±0.076, n=13).
Table 1.
Mean and standard error of glutamate change compared to SWS level in PIA, NMC and NPM
| REM/SWS | W/SWS | t-test | |
|---|---|---|---|
| PIA (n=44) | 1.026±0.036 | 1.029±0.044 | n.s. |
| NMC (n=41) | 1.114±0.056 | 0.978±0.040 | p=0.0252 |
| NPM (n=41) | 1.034±+0.043 | 1.003±+0.046 | n.s. |
n.s.=Not significant.
Five sets of samples were obtained in the microinjection experiment. Each set has one 5-min pre-injection control, followed by one 5-min sample during stimulation and six 5-min periods following the ACh microinjection in PIA. The glutamate level was enhanced for 10 min after the ACh microinjection period. The glutamate content during the first (554.48±178.01 fmol/min sample; mean±S.E.M.) and second (404.82±80.19) 5 min was significantly higher (two-tailed t-test, p=0.0372) than the glutamate level in the pre-stimulation period (262.59±27.55). (Fig. 2) The ACh microinjection produced muscle atonia lasting a mean of 11.0±4.25 min.
Fig. 2.
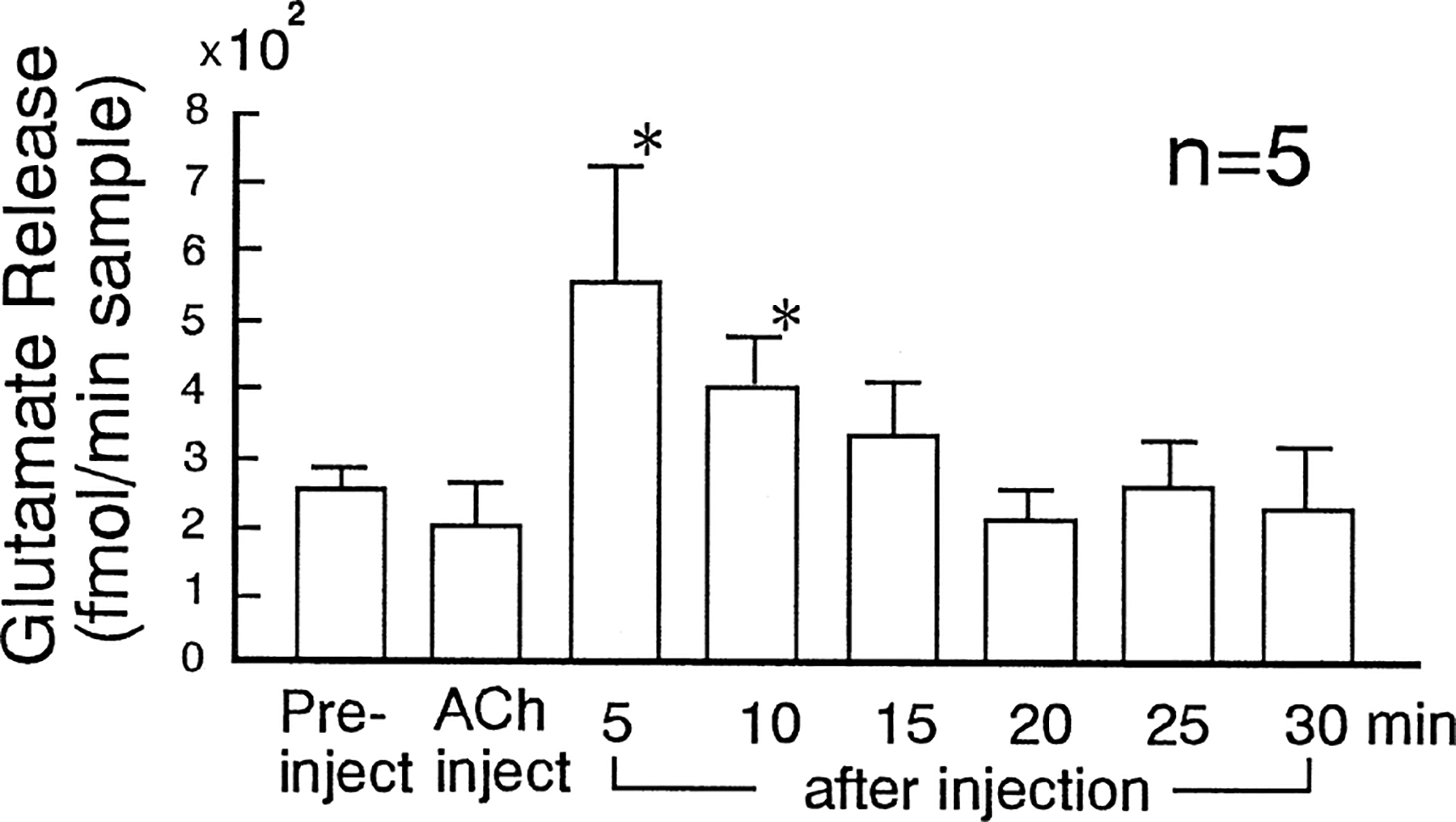
Glutamate release in 5-min period of pre-, during and after ACh (100 μg/0.5 μl) microinjection into the PIA. Glutamate release was enhanced for 10 min after ACh microinjection. Asterisk indicates a significant change (p<0.05).
Acetylcholine and glutamate are two major excitatory inputs to the brainstem neurons and may work synergistically. Lai and Siegel [19] first reported that activation of non-NMDA glutamate receptors in PIA triggered atonia. Elazar et al. [4] found that injection of glutamate antagonists in this region disrupted REM sleep. Onoe and Sakai [24] showed that application of kinate through a microdialysis probe in PIA (peri-locus coeruleus α) produced increased REM sleep. On the other hand, it is well known that cholinergic system in PIA is important for REM related mechanisms [2,5,9,12,23,26]. The REM sleep-generating region is responsive to both ACh [5,26] and glutamate [21,24]. Furthermore, some cholinergic neurons in the laterodorsal tegmental (LDT) and pedunculopontine (PPT) nuclei [14], which project to PIA, have both ACh and glutamate as neurotransmitters. Therefore, we expected to observe the sleep specific increase of glutamate in PIA such as was observed in ACh [12]. However, the changes in levels of glutamate in PIA detected by dialysis were smaller than those of ACh and showed no significance across sleep stages. The most likely explanation for this is that synaptically released glutamate in this area is being obscured by large glial release. The use of smaller probes to better localize the collection site or larger sample sizes might allow one to detect changes in glutamate release in PIA during REM sleep.
In NMC, we detected a sleep-related change (11%) of glutamate release. We reported the sleep stage related increases of glutamate in LDT and basal forebrain [1,10], but they were also as small as in NMC. Tetrodotoxin (TTX) studies have shown that the neuronal release of glutamate amounts to only 10% (our observation) or less [28] of the ambient level as measured by HPLC. The major source of basal glutamate content is presumably glia cells surrounding neurons [3] and metabolic sources, rather than synaptic releases, which makes it difficult to determine the significant glutamate changes. Under the condition of TTX perfusion, we observed little glutamate changes across sleep stages, which suggested most of the changes of glutamate content came from neuronal release. From these aspects, 11% increase observed in NMC is equal to almost twice the basal neuronal release, which could be interpreted as sufficiently large change.
In the present study, a REM sleep-related increase of glutamate release was observed in NMC. Anatomical studies indicated the existence of glutamate projections from PIA to NMC and cholinergic projections from PIA to NPM [14,15,27]. Electrical stimulation in the PIA induces muscle atonia and activates medullary reticulospinal units activities [13]. In the present study, an increase of glutamate release in NMC was elicited by ACh microinjection into PIA. In our prior study, we found a REM sleep-related increase in ACh release in NPM but not in NMC [11]. These observations support our hypothesis that muscle atonia is mediated by two distinct pathways. One pathway is via cholinoceptive glutamatergic neurons in the pons projecting to glutamate sensitive non-cholinergic neurons in NMC [16]. These NMC neurons then project to the spinal cord to either activate interneurons responsible for muscle atonia, or to directly hyperpolarize motoneurons. The other pathway is via cholinergic neurons in the pons, such as the LDT and PPT, projecting to cholinoceptive neurons in NPM [16,25] which in turn project to the spinal cord producing motoneuron hyperpolarization directly or through interneurons.
Acknowledgements
This study was supported by the Medical Research Service of the Department of Veterans Affairs, USPHS HL41370 and NS14610.
References
- [1].Azuma S, Kodama T, Honda K, Inoué S, State-dependent changes of extracellular glutamate in the medial pre-optic area in freely behaving rats, Neurosci. Lett 214 (1996) 179–182. [DOI] [PubMed] [Google Scholar]
- [2].Baghdoyan HA, Rodrigo-Angula ML, McCarley RW, Hobson JA, Site-specific enhancement and suppression of desynchronized sleep signs following cholinergic stimulation of three brainstem regions, Brain Res. 306 (1984) 39–52. [DOI] [PubMed] [Google Scholar]
- [3].Bernath S, Calcium-independent release of amino acid neurotransmitters: fact or artifact?, Prog. Neurobiol 38 (1992) 57–91. [DOI] [PubMed] [Google Scholar]
- [4].Elazar Z, Fahringer HM, Baldwin C, Camelleri C, Siegel JM, Effect of pontine injection of glutamate antagonists on REM sleep, Sleep Res. 19 (1990) 12. [Google Scholar]
- [5].George R, Haslett WL, Jenden DJ, A cholinergic mechanism in the brainstem reticular formation: induction of paradoxical sleep, Int. J. Neuropharmacol 3 (1964) 541–552. [DOI] [PubMed] [Google Scholar]
- [6].Henley K, Morrison AR, A reevaluation of the effects of lesions of the pontine tegmentum and locus coeruleus on phenomena of paradoxical sleep in the cat, Acta Neurobiol. Exp 34 (1974) 215–232. [PubMed] [Google Scholar]
- [7].Holmes CJ, Jones BE, Importance of cholinergic, GABAergic, serotonergic and other neurons in the medial medullary reticular formation for sleep–wake states studied by cytotoxic lesions in the cat, Neuroscience 62 (1994) 1179–1200. [DOI] [PubMed] [Google Scholar]
- [8].Jouvet M, Delorme F, Locus coeruleus et sommeil paradoxal, C. R. Soc. Biol 159 (1965) 895–899. [Google Scholar]
- [9].Katayama Y, DeWitt DS, Becker DP, Hayes RL, Behavioral evidence for cholinoceptive pontine inhibitory area: descending control of spinal motor output and sensory input, Brain Res. 296 (1984) 241–262. [DOI] [PubMed] [Google Scholar]
- [10].Kadama T, Honda Y, Koyama Y, Glutamate–noradrenergic interaction in the laterodorsal tegmentum neurons mediated by nitric oxide, Psychiatry Clin. Neurosci 51 (1997) S45. [Google Scholar]
- [11].Kodama T, Lai YY, Siegel JM, Enhancement of acetylcholine release during REM sleep in the caudomedical medulla as measured by in vivo microdialysis, Brain Res. 580 (1992) 348–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kodama T, Takahashi Y, Honda Y, Enhancement of acetylcholine release during paradoxical sleep in the dorsal tegmental field of the cat brain stem, Neurosci. Lett 114 (1990) 277–282. [DOI] [PubMed] [Google Scholar]
- [13].Kohyama J, Lai YY, Siegel JM, Medullary reticular unit activity during muscle atonia induced by rostral pontine electrical stimulation in decerebrate cats, Sleep Res. 25 (1996) 13. [Google Scholar]
- [14].Lai YY, Clements JR, Siegel JM, Glutamatergic and cholinergic projections to the pontine inhibitory area identified with horseradish peroxidase retrograde transport and immunohistochemistry, J. Comp. Neurol 336 (1993) 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lai YY, Clements JR, Siegel JM, Afferents to the nucleus magnocellularis of the medulla, Soc. Neurosci. Abstr 19 (1993) 1437. [Google Scholar]
- [16].Lai YY, Siegel JM, Medullary region mediating atonia, J. Neurosci 8 (1988) 4790–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lai YY, Siegel JM, Cardiovascular and muscle tone changes produced by microinjection of cholinergic and glutamatergic agonists in dorsolateral pons and medial medulla, Brain Res 514 (1990) 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lai YY, Siegel JM, Muscle tone suppression and stepping produced by stimulation of midbrain and rostral pontine reticular formation, J. Neurosci 10 (1990) 2727–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lai YY, Siegel JM, Ponto–medullary glutamate receptors mediating locomotion and muscle tone suppression, J. Neurosci 11 (1991) 2931–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lai YY, Siegel JM, Corticotropin-releasing factor-mediated muscle atonia in pons and medulla, Brain Res. 575 (1992) 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lai YY, Siegel JM, Wilson WJ, Effect of blood pressure on changes in muscle tone produced by stimulation of the medial medulla, Am. J. Physiol 252 (1987) H1249–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Magoun HW, Bulbar inhibition and facilitation of motor activity, Science 100 (1944) 549–550. [DOI] [PubMed] [Google Scholar]
- [23].Mitler MM, Dement WC, Cataplectic-like behavior in cats after micro-injection of carbachol in the pontine reticular formation, Brain Res. 68 (1974. 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Onoe H, Sakai K, Kinate receptors: a novel mechanism in paradoxical (REM) sleep generation, Neuroreport 6 (1995) 353–356. [PubMed] [Google Scholar]
- [25].Schenkel E, Siegel JM, REM sleep without atonia after lesions of the medial medulla, Neurosci. Lett 98 (1989) 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shiromani PJ, Siegel JM, Tomaszewski KS, McGinty DJ, Alternations in blood pressure and REM sleep after pontine carbachol microinjection, Exp. Neurol 91 (1986) 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shiromani PJ, Lai Y-Y, Siegel JM, Descending projections from the dorsolateral pontine tegmentum to the paramedian reticular nucleus of the caudal medulla in the cat, Brain Res. 517 (1990) 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Werterink BHC, Damsma G, Rollema H, DeVries JB, Horn AS, Scope and limitation of in vitro brain dialysis: a comparison of its application to various neurotransmitter systems, Life Sci. 41 (1987) 1763–1776. [DOI] [PubMed] [Google Scholar]