

Abstract
All living organisms synthesize phospholipids as the primary constituent of their cell membranes. Enzymatic synthesis of diacylphospholipids requires preexisting membrane-embedded enzymes. This limitation has led to models of early life in which the first cells used simpler types of membrane building blocks and has hampered integration of phospholipid synthesis into artificial cells. Here we demonstrate an enzyme-free synthesis of natural diacylphospholipids by transacylation in water, which is enabled by a combination of ion pairing and self-assembly between lysophospholipids and acyl donors. A variety of membrane-forming cellular phospholipids have been obtained in high yields. Membrane formation takes place in water from natural alkaline sources such as soda lakes and hydrothermal oceanic vents. When formed vesicles are transferred to more acidic solutions, electrochemical proton gradients are spontaneously established and maintained. This high-yielding non-enzymatic synthesis of natural phospholipids in water opens up new routes for lipid synthesis in artificial cells and sheds light on the origin and evolution of cellular membranes.
Cellular membranes composed of glycerophospholipids are found in all living organisms. Bacterial and eukaryotic membranes consist of diacylphospholipids, in which two ester linkages connect a polar head group to two hydrophobic tails1. Cells synthesize diacylphospholipids through enzymatic acylation of lysophospholipids2. As several enzymes involved in diacylphospholipid biosynthesis must themselves be membrane-bound for enzyme activity, it is unclear whether phospholipid membranes could have formed in the absence of advanced enzymatic machinery3–6. Using wet–dry cycling, an enzyme-free reaction between ether mono-glyceride, a short-chain fatty acid, and phosphate in the presence of dicyanamide can yield glycerophospholipids that possess an ether linkage and a relatively short acyl chain7. Acylation of glycerophosphates can take place in the presence of a large excess of activated acyl imidazole derivatives, but the reactions require organic co-solvent due to the hydrophobicity of the acylating precursors and result in unnatural diacylphospholipids8,9. Here we demonstrate that a combination of ion pairing and self-assembly allows transacylation reactions of lysophospholipids with acyl donors to afford natural diacylphospholipids in water. The high-yielding aqueous synthesis of natural phospholipids provides new possible routes to the origin of cell membranes and suggests that diacylphospholipids may have been incorporated before the evolution of complex biochemical machinery10,11. Furthermore, we believe that the use of ion pairing and self-assembly in water to dramatically accelerate reactions and control reaction selectivity will have applications in green chemistry, for example, in the aqueous synthesis of peptides and glycans.
Results and discussion
In cells, lysophospholipid esterification requires acyl thioesters and the action of an acyltransferase. Inspired by acyltransferase reactions in lipid biosynthesis2, and past hypotheses on the role of thioesters in the origins of life12, we questioned whether synthetic acyl thioesters 2, in lieu of acyl coenzyme A, could acylate 1-acyl-2-hydroxy-sn-glycero-3-phosphocholines 1 in water to give the desired natural phosphatidylcholines (PCs) 3 (Fig. 1a). An ionized polar head group on 2 would facilitate transacylation by increasing the long-chain acyl thioester’s solubility in water and driving the self-assembly of micelles, possibly in combination with the lysophospholipid reactant. We initially investigated the synthesis of a naturally occurring phospholipid, 1,2-dioleoyl-sn-glycero-3-p hosphocholine 3a (DOPC)13, in aqueous solution via acylation of lysophospholipid 1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine 1a (Fig. 1a). A water-soluble anionic thioester, sodium 2-(oleoylthio)ethane-1-sulfonate 2a, was chosen as an oleoyl donor14. Only trace conversion was observed (<1%) and addition of acylation catalysts to accelerate the transacylation reaction did not have a substantial effect (Supplementary Table 1); however, to enable spontaneous membrane formation in water, acylation would have to be high-yielding and occur readily at near stoichiometric ratios of starting materials to prevent amphiphilic precursors from disrupting membrane formation.
Fig. 1 |. Enzyme-free synthesis of natural phospholipids.
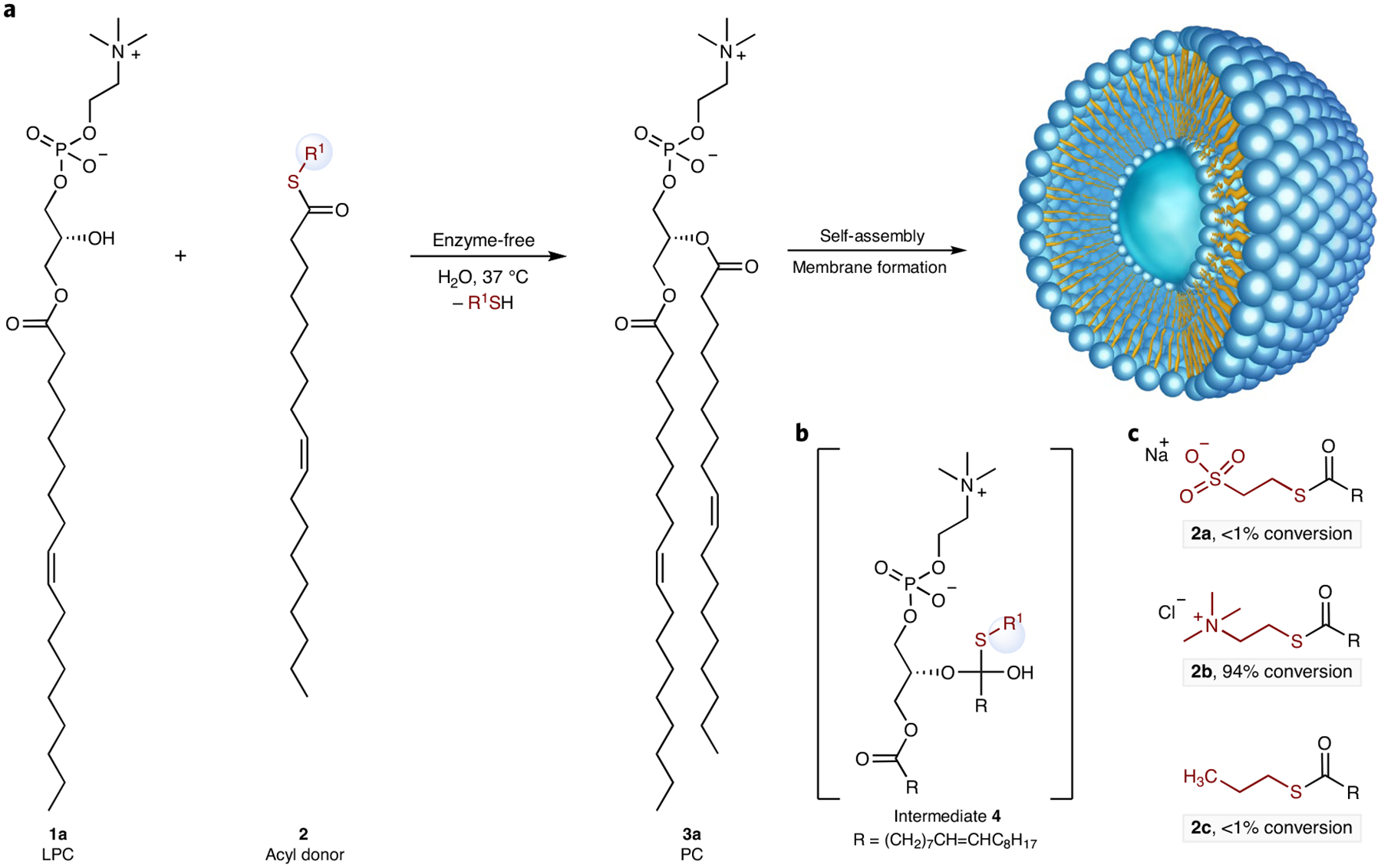
a, De novo synthesis of diacylphospholipids in water leading to an in situ self-assembled membrane. LPC, lysophosphatidylcholine; PC, phosphatidylcholine. The leaving group is shown in red; the structures of r1 are shown in panel c. b, The proposed reaction intermediate. c, reactive fatty acyl derivatives: acyl donors 2 (0.75 mM), with which reactions were carried out with 1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine 1a (0.5 mM) in the presence of Na2CO3/NaHCO3 buffer (pH = 10.6) at 37 °C for 5 h.
To improve the yield of the desired diacylphospholipids, we sought a way to increase the yield of transacylation while minimizing the hydrolysis of the acyl donor. Transacylation reactions between acyl donors and alcohols proceed via a tetrahedral intermediate (Fig. 1b)15,16. We reasoned that the observed poor reactivity in the transacylation reaction is probably due to the inability of acylation reagent 2a to lower ΔG for the tetrahedral intermediate. We therefore sought to synthesize an acyl donor that would stabilize the reaction intermediate. In principle, a positively charged leaving group on the acyl donor 2 would lead to a favourable Coulombic interaction17 with the negatively charged phosphate group of lysophospholipid 1 (Fig. 1b), thus stabilizing intermediate 4 and accelerating the anticipated transacylation reaction through ion pairing. Based on this hypothesis, we prepared 2-(oleoylthio) -N,N,N-trimethylethan-1-aminium chloride 2b, which contains a positively charged quaternary amine head group (Fig. 1c). Precursor 2b is accessible in one synthetic step from oleic acid and thiocholine. Compound 2b could also be obtained using prebiotically relevant condensing agents such as dicyandiamide (Supplementary Fig. 1)18. Lysophospholipids such as lysophosphatidic acid can be obtained by the hydrolysis of cyclophosphate amphiphiles, which have been shown to be prebiotically synthesized from glycerol, fatty acid and diamidophosphate in the presence of imidazole19. Furthermore, modification of phosphatidic acid head group by choline under prebiotically relevant conditions has also been demonstrated20. We evaluated the esterification reaction of lysophospholipid 1a (0.5 mM) with 2b (0.75 mM) in alkaline bicarbonate buffer (pH = 10.6) in the absence of additional catalyst; 94% conversion of lysophospholipid 1a to DOPC 3a was obtained after 5 h at 37 °C (Fig. 1c). By contrast, under the same reaction conditions, we observed only trace acylation of 1a with either anionic 2a or the charge-neutral acylation reagent 2c (<1% conversion).
Our initial results suggested that ion pairing is involved in the aqueous transacylation of 2-lysophospholipid. Density functional theory calculations were performed to better understand the effect of ion pairing on lysophospholipid acylation21. The n-butyroyl group was employed in place of the oleoyl group as a model system (Fig. 2). Calculations were performed at D3-B3LYP/6–311+ +G(2d,p)//B3LYP/6–31G (d,p) level with the Truhlar–Cramer empirical model (Solvation Model Density, SMD) for water22. Tetrahedral INT1 and INT2 resemble the corresponding 1,2-addition transition state and predict kinetic barriers; ΔG for the formation of the tetrahedral intermediate INT1 from model acylation reagent 2-(butyroylthio) ethane-1-sulfonate was calculated as 24.1 kcal mol−1, whereas for INT2 from 2-(butyroylthio)-N,N,N-trimethylethan-1-aminium it was 16.5 kcal mol−1 (Fig. 2a). These computational results predict a higher reactivity of the quaternary ammonium-containing reagent analogous to 2b in the transacylation reaction, in line with our experimental observations. Further, the optimized structures of the reaction intermediates indicate that intramolecular Coulombic interactions are important for the stability of the tetrahedral intermediate (Fig. 2b). In INT1, the sulfonate side chain is distal to the phosphate group, which results from the electrostatic repulsion of these two negatively charged groups. By contrast, the optimized structure of INT2 shows electrostatic attraction between the negatively charged phosphate group and the two positively charged quaternary ammonium groups. Our experimental and theoretical findings are consistent with the hypothesis that a positively charged head group on 2 stabilizes the reaction intermediate, resulting in efficient transacylation.
Fig. 2 |. Predicted effects of thioester charge on phospholipid synthesis.
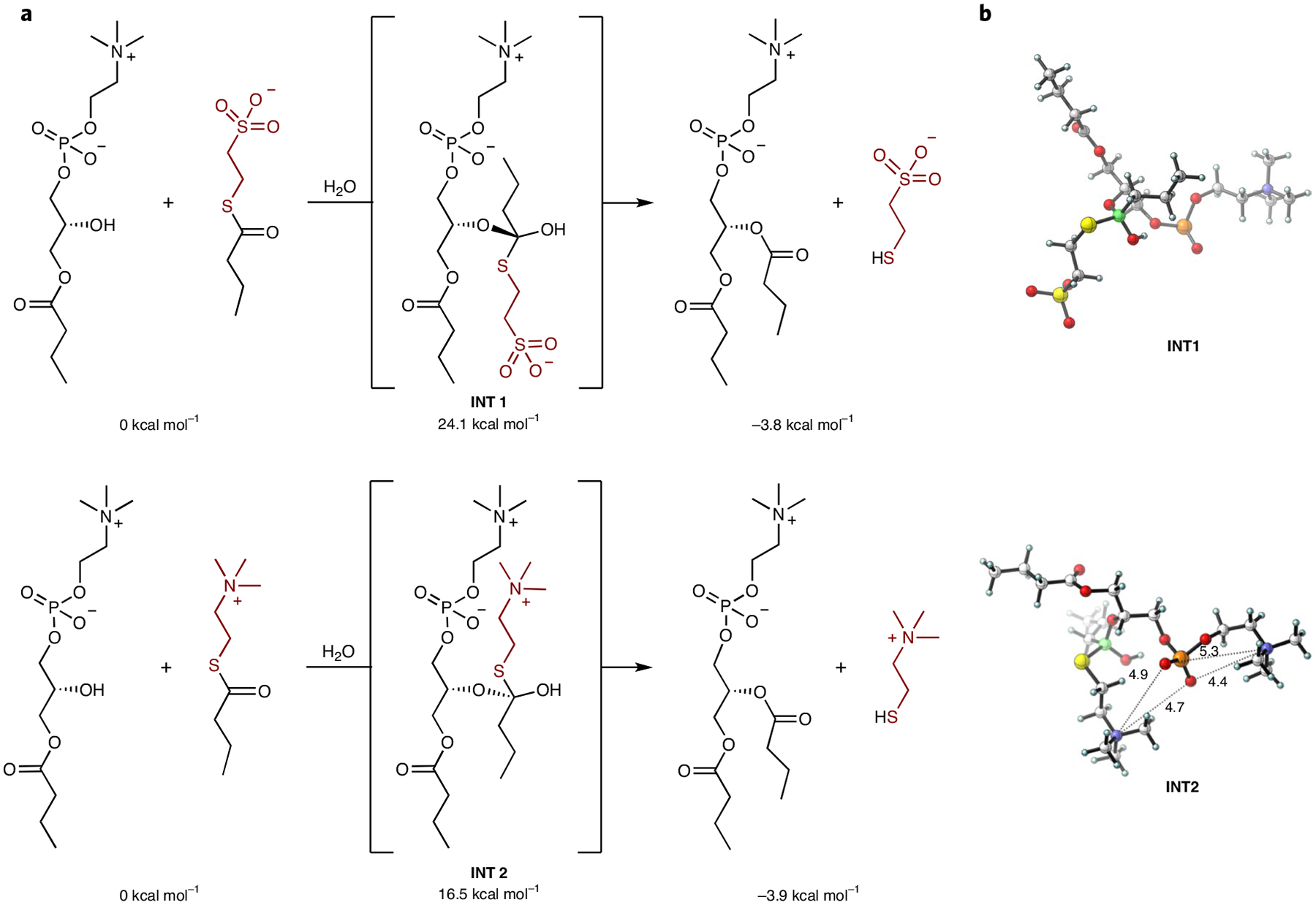
a, Energetics from B3LYP-D3 density functional theory calculations on transacylation reactions. Tetrahedral intermediates iNt1 and iNt2 resemble the transition states of the corresponding addition steps. iNt1 is destabilized by over 7 kcal mol−1 compared with iNt2. b, Optimized structures of reaction intermediates. The negatively charged sulfonate group of iNt1 is distal from the phosphate group to avoid a disfavoured charge repulsion interaction, whereas the positively charged side chain of iNt2 moves into a geometry that will increase the favorable interaction with the phosphate group to stabilize the tetrahedral intermediate. Distances are shown in ångströms.
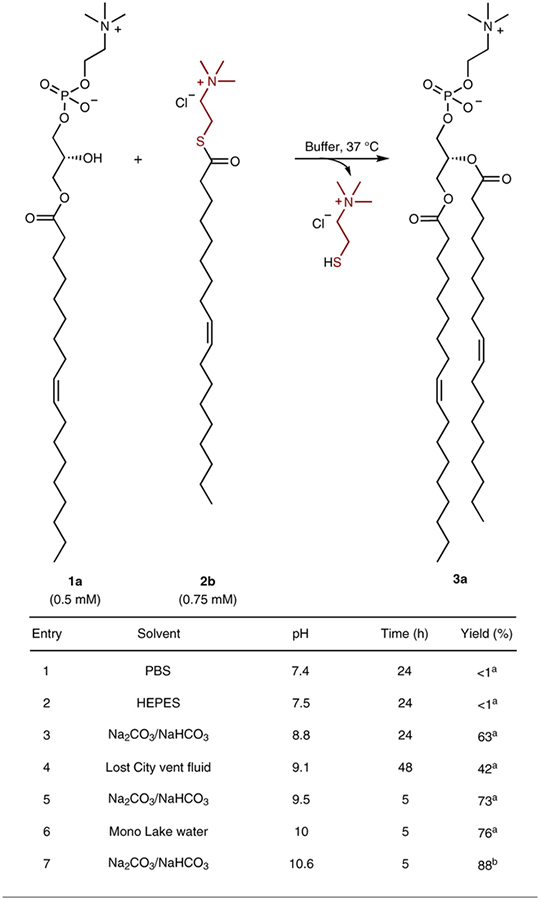
We next explored the range of reaction conditions under which diacylphospholipids could be synthesized. The transacylation reaction between lysophospholipid 1a and acyl donor 2b proceeded rapidly and in high yield in alkaline bicarbonate buffers (pH 9.5–10.6) (Table 1)23. Good reaction yields were also obtained in Na2CO3/NaHCO3 buffer over a longer time period at pH = 8.8 (63% yield, 24 h, entry 3). By contrast, trace product was obtained using phosphate buffered saline at pH = 7.4 (<1% yield, 24 h, entry 1) or HEPES buffer at pH = 7.5 (<1% yield, 24 h, entry 2). Based on these results, we tested if the reaction could proceed in more complex naturally derived alkaline water samples. Numerous models have proposed that life may have started near alkaline hydrothermal vents or soda lakes24,25. We obtained water samples from the Lost City hydrothermal field (LCHF, pH = 9.1) and Mono Lake (pH = 10)—a Californian soda lake—to test whether synthesis of phospholipids could take place in such environments. Acylation of 1a with 2b to form phospholipid 3a took place in water from the LCHF (42% yield, 48 h, entry 4) and Mono Lake water (76% yield, 5 h, entry 6). These results suggest that natural alkaline water sources would have been privileged environments for thioester mediated acylation of lysophospholipids.
Table 1 |.
Aqueous synthesis of phospholipid 3a using different water sources
|
Reactions were carried out with lysophospholipid 1a (0.5 mM) and acylation reagent 2b (0.75 mM) to afford phospholipid 3a in different aqueous sources at 37 °C. PBS, phosphate buffered saline.
HPLC yield.
Isolated yield.
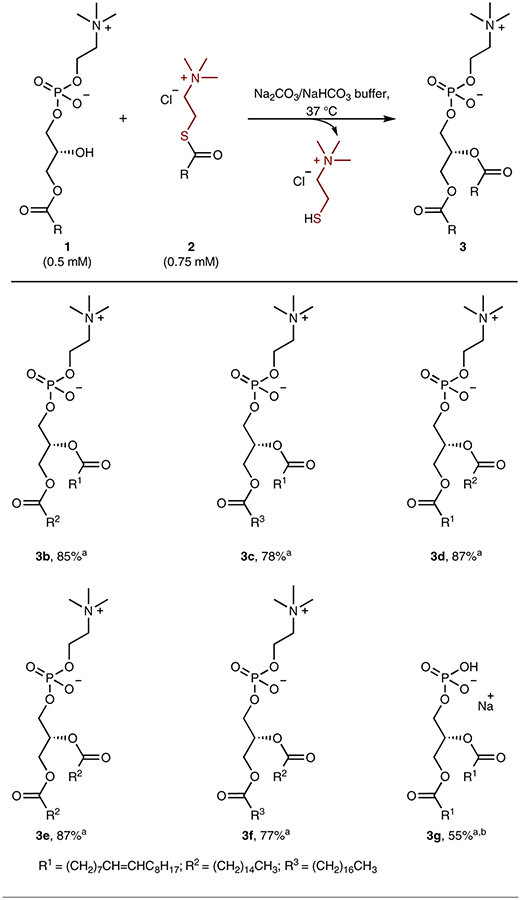
Having optimized reaction conditions (entry 7, Table 1), we investigated the generality of the technique by attempting to synthesize a variety of naturally occurring phospholipids. We explored the acylation of multiple lysophosphatidylcholines with acylation agents of different chain lengths and compositions. Good acylation yields were achieved when 2b was reacted with lysophospholipids that possess either unsaturated (oleoyl) or saturated (palmitoyl and stearoyl) acyl chains (3b and 3c) (Table 2). Changing the acyl donor to a saturated palmitoyl group through the use of 2-(palmitoy lthio)-N,N,N-trimethylethan-1-aminium chloride 2d led to similar reactivities (3d–3f) and enabled the synthesis of dipalmitoylphosphatidylcholine, a key component of pulmonary surfactant26. We also tested whether we could synthesize phosphatidic acids, as they are universal intermediates in glycerophospholipid biosynthesis27. Compared with lysophosphatidylcholine, which has a neutral zwitterionic headgroup, lysophosphatidic acid has a negatively charged phosphate headgroup, and acylation of the sodium salt of 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate with 2b led to a 55% yield of phosphatidic acid 3g. We hypothesized that the lower efficiency might be due to the less facile deprotonation of the 2-hydroxy group of lysophosphatidate, or possibly a higher ΔG for the tetrahedral intermediate. We further attempted to obtain various diacylphospholipids such as phosphatidylethanolamine, phosphatidylserine and phosphatidylglycerol by oleoylating the corresponding lysophospholipids; however, the reactivity of acylation reagent 2b is insufficient for these lysolipids and no considerable yields of the desired diacylphospholipids were achieved (Supplementary Figs. 3–5).
Table 2 |.
Scope of ion pairing-enabled synthesis of natural phospholipids
|
Reactions were carried out with lysophospholipids 1 (0.5 mM) and acylation reagents 2 (0.75 mM) to afford phospholipid 3 in Na2CO3/NaHCO3 buffer (pH = 10.6) at 37 °C for 5 h.
Isolated yield.
1.5 mM of reagent 2b was used.
This transacylation reaction turns out to be highly chemoselective, being unperturbed by the presence of non-amphiphilic biomolecules that possess nucleophilic functional groups. Under the optimized reaction conditions, no oleoylation products were observed when combining reagent 2b with either serine, threonine or glycerophosphocholine as substrates (Supplementary Figs. 6–8), and less than <1% of acetylation product 3o was observed when a non-amphiphilic acyl donor acetylthiocholine chloride was used for the acylation of 1a (Supplementary Fig. 9). Excellent yields of product 3a were obtained in the oleoylation reaction of lysolipid 1a in the presence of serine (88% yield, 5 h, Supplementary Fig. 10) or disodium phosphate (91% yield, 5 h, Supplementary Fig. 11), but less than 10% yield was achieved with an additive sodium dodecyl sulfate, which is a well-known detergent (Supplementary Fig. 12). On the basis of these findings, we believe that the mixed micelles formed by the preorganization of the amphiphilic reagents accelerates the reaction rate and increases the selectivity of lipid synthesis. With this in mind, we next investigated shorter single-chain lipid precursors, which are more prebiotically plausible reactants but require higher concentrations to form micelles than longer chain precursors. When we reacted the ten-carbon chain lysophospholipid 1-decanoyl-2-hydroxy-sn-glycero-3-phosphocholine 1e (0.5 mM) with decanoyl donor 2e 2-(decanoylthio)-N,N, N-trimethylethan-1-aminium chloride (0.75 mM) at 37 °C for 5 h, no 1,2-didecanoyl-sn-glycero-3-phosphocholine was observed (Supplementary Fig. 13); however, when we carried out a reaction between lysophospholipid 1e (5 mM) near its critical micelle concentration (Supplementary Fig. 2C) and decanoyl donor 2e (7.5 mM) above its critical micelle concentration (Supplementary Fig. 2F), 83% yield of 1,2-didecanoyl-sn-glycero-3-phosphocholine was obtained (Supplementary Fig. 13), providing further evidence that mixed micelle formation is necessary for the transacylation reaction to occur.
Enabling the synthesis of diacylphospholipids through esterification reactions in water should result in the spontaneous de novo formation of cell-like lipid membranes. This is due to the expected micelle to lamellar transition as single-chain lysolipids are converted to diacylphospholipids. We used time-lapse fluorescence microscopy to observe the de novo formation of vesicles during the optimized reaction (Fig. 3a). Neither lysophospholipid 1a nor the oleoyl thioester 2b formed vesicles when incubated alone in Na2CO3/NaHCO3 buffer of pH = 8.8 at 37 °C (Supplementary Fig. 14). No visible structures were observed after initial mixing of 1a (0.5 mM) and 2b (0.75 mM) in Na2CO3/NaHCO3 buffer of pH = 8.8 at 37 °C (Fig. 3a and Supplementary Fig. 15); however, after 10 min tubular structures appeared, and after 30 min spherical vesicles were observed (Supplementary Figs. 15 and 16). Likewise, large vesicles were formed when 1a (0.5 mM) and 2b (0.75 mM) were mixed in water collected from the LCHF (Fig. 3b and Supplementary Fig. 17A). Membrane-bound vesicles were also detected on mixing the reactants in Mono Lake water, and transmission electron microscopy was able to confirm the generation of vesicles (Fig. 3c and Supplementary Fig. 18C). We reasoned that both fusion of submicron-scale vesicles and growth of small vesicles through addition of nearby newly formed phospholipid led to the observed cell-sized vesicles (Supplementary Fig. 19).
Fig. 3 |. Enzyme-free formation of phospholipid membranes.
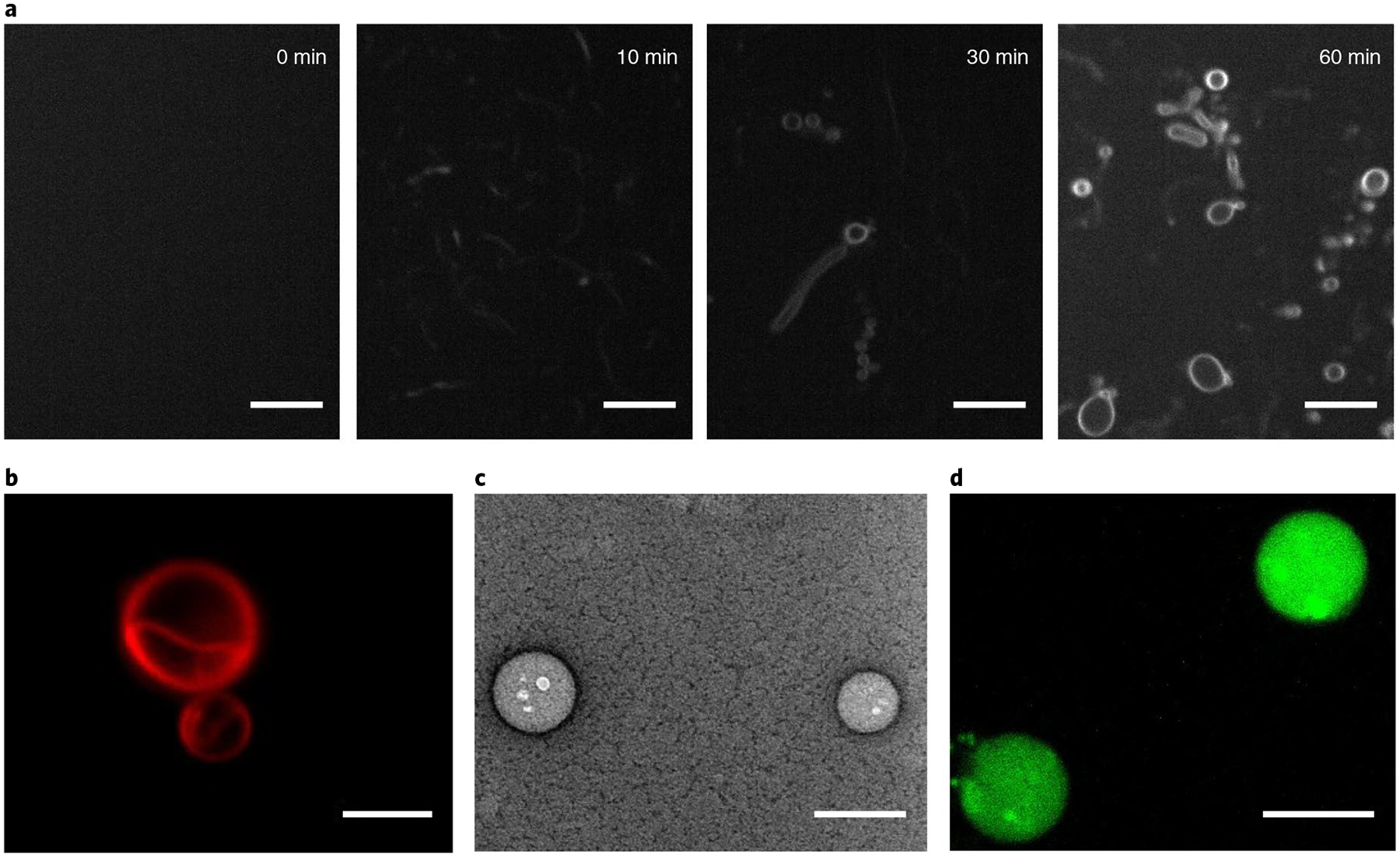
The reaction was carried out by mixing 1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine 1a (0.5 mM) and oleoylation reagent 2b (0.75 mM) in different solvents at 37 °C. a, Fluorescence micrographs are shown: Na2CO3/NaHCO3 buffer (pH = 8.8) was used as a solvent; samples were taken from the reaction mixture at different time points and were stained by using 0.1 mol% Nile red dye. Scale bars, 10 μm. b, A fluorescence micrograph is shown: Lost City vent fluid was used as a solvent; the sample was taken from the reaction mixture after 48 h and was stained by using 0.1 mol% Nile red dye. Scale bar, 5 μm. c, A negative staining transmission electron micrograph is shown: mono Lake water was used as a solvent; the sample was taken from the reaction mixture after 5 h. Scale bar, 200 nm. d, A fluorescence micrograph of vesicles formed in mono Lake water (pH = 10) containing the pH indicator dye HPTS, two hours after the external media was exchanged to citrate buffer (pH = 4.6). Scale bar, 10 μm.
All living organisms harness proton gradients across phospholipid membranes to generate energy. Vesicles formed from simpler amphiphiles such as fatty acids are unable to maintain proton gradients, suggesting that diacylphospholipid membranes appeared early in the origin of life28. De novo phospholipid vesicle formation in alkaline solution results in an alkaline interior of formed vesicles. If the exterior solution is then exchanged for a more acidic media, a proton gradient might spontaneously form. To test whether this was possible, a water-soluble pH indicator dye, 8-hydroxypyrene-1, 3,6-trisulfonic acid (HPTS), was added before the reaction of 1a with 2b in the alkaline Mono Lake water (pH = 10); HPTS is highly fluorescent at alkaline pH, but >99% fluorescence is quenched at pH = 4.6 (Supplementary Fig. 20). After 5 h of reaction, fluorescence microscopy indicated that HPTS was successfully encapsulated in the in situ formed membrane vesicles (Supplementary Fig. 21A). To test whether a proton gradient could be maintained, the vesicle media was exchanged to citrate buffer (pH = 4.6) using spin-filtration. Fluorescence intensity was initially maintained, indicating the spontaneous formation of a proton gradient. Fluorescence intensity slowly diminished by 80% over 2 h (Fig. 3d). This finding demonstrates that diacylphospholipid membranes generated in alkaline water sources are capable of forming and maintaining a proton gradient over hours. It is tempting to speculate that similar phenomena may have occurred in the early origin of membranes, perhaps as alkaline hydrothermal vent water was diluted in the more acidic water of the Hadean ocean29,30. Such a scenario may have driven selection pressures for developing primitive catalysts for maintaining such proton gradients, which could have eventually been utilized to form chemical energy.
Methods
Synthesis of acyl thioesters.
4-Dimethylaminopyridine (15 mg, 0.12 mmol) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC•HCl) (248 mg, 1.3 mmol) were added to a solution of fatty acid (1.3 mmol) in CH2Cl2 (10 ml) at 0 °C under argon; the reaction mixture was stirred at 0 °C for 30 min. The resulting solution was added dropwise to thiocholine chloride (188 mg, 1.2 mmol) at −78 °C under argon and the reaction was warmed to room temperature and stirred for 4 h. The reaction solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel. Full experimental details and characterization of new compounds can be found in the Supplementary Information.
Enzyme-free transacylation reactions between lysophospholipids and acyl donors to afford diacylphospholipids in water.
A freshly prepared Na2CO3/NaHCO3 buffer of pH = 10.6 (8 ml) was added to a mixture of lysophospholipid (4 μmol) and acylation reagent (6 μmol) at 37 °C unless specified otherwise. The reaction mixture was tumbled at 37 °C for 5 h. The resulting mixture was adjusted to pH 7.0 and concentrated in vacuo. Purification was performed by column chromatography or preparative thin layer chromatography on silica gel. Full experimental details and characterization of new compounds can be found in the Supplementary Information.
pH gradient decay in the de novo formed vesicles.
The reaction was carried out by adding HPTS (0.25 mM) before the reaction of lysophospholipid 1a (0.5 mM) with 2-(oleoylthio)-N,N,N-trimethylethan-1-aminium chloride 2b (0.75 mM) in the presence of Mono Lake water (pH = 10). The reaction mixture was tumbled at 37 °C for 5 h. The media of vesicles was then exchanged with citrate buffer (pH = 4.6, in the same osmolarity as Mono Lake water) using spin-filtration, spontaneously generating a transmembrane pH gradient. Samples of the reaction mixture were taken at different time points, and fluorescence micrographs of vesicles were collected to monitor the maintenance of the pH gradient in the de novo-formed vesicles. Full experimental details and characterization of vesicles can be found in the Supplementary Information.
Supplementary Material
Acknowledgements
N.K.D. acknowledges financial support for this work provided by the National Science Foundation (grant no. CHE-1254611) and the National Institutes of Health (grant no. DP2DK111801). K.N.H. acknowledges National Science Foundation (grant no. CHE-1764328), the National Institutes of Health, National Institute of General Medical Sciences (grant no. R01 GM109078), for financial support of this research. S.Q.L. acknowledges financial support for this work provided by National Science Foundation (grant no. OCE-1536702). Computation time was provided by the UCLA Institute for Digital Research and Education (IDRE).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-020-00559-0.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41557-020-00559-0.
Data availability
The data that support the findings of this study are available within the paper and its Supplementary Information.
References
- 1.Jackowski S, Cronan JE Jr & Rock CO Biochemistry of Lipids, Lipoproteins and Membranes Vol. 20, 80–81 (Elsevier, 1991). [Google Scholar]
- 2.Lands WEM Metabolism of glycerolipides: a comparison of lecithin and triglyceride synthesis. J. Biol. Chem 231, 883–888 (1958). [PubMed] [Google Scholar]
- 3.Schmidli PK, Schurtenberger P & Luisi PL Liposome-mediated enzymatic synthesis of phosphatidylcholine as an approach to self-replicating liposomes. J. Am. Chem. Soc 113, 8127–8130 (1991). [Google Scholar]
- 4.Deamer DW & Boatman DE An enzymatically driven membrane reconstitution from solubilized components. J. Cell Biol 84, 461–467 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morris-Natschke SL et al. Synthesis of phosphocholine and quaternary amine ether lipids and evaluation of in vitro antineoplastic activity. J. Med. Chem 36, 2018–2025 (1993). [DOI] [PubMed] [Google Scholar]
- 6.Harayama T et al. Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell Metabol. 20, 295–305 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Hargreaves WR, Mulvihill SJ & Deamer DW Synthesis of phospholipids and membranes in prebiotic conditions. Nature 266, 78–80 (1977). [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Garcia C & Powner MW Selective acylation of nucleosides, nucleotides, and glycerol-3-phosphocholine in water. Synlett 28, 78–83 (2017). [Google Scholar]
- 9.Bonfio C et al. Length-selective synthesis of acylglycerol-phosphates through energy-dissipative cycling. J. Am. Chem. Soc 141, 3934–3939 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szostak JW, Bartel DP & Luisi PL Synthesizing life. Nature 409, 387–390 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Budin I & Szostak JW Physical effects underlying the transition from primitive to modern cell membranes. Proc. Natl Acad. Sci. USA 108, 5249–5254 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Ouve C The beginnings of life on earth. American Scientist 83, 428–437 (1995). [Google Scholar]
- 13.Ejsinga CS et al. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl Acad. Sci. USA 106, 2136–2141 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brea RJ, Cole CM & Devaraj NK In situ vesicle formation by native chemical ligation. Angew. Chem. Int. Ed 53, 14102–14105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bender ML Mechanisms of catalysis of nucleophilic reactions of carboxylic acid derivatives. Chem. Rev 60, 53–113 (1960). [Google Scholar]
- 16.McClelland RA & Santry LJ Reactivity of tetrahedral intermediates. Acc. Chem. Res 16, 394–399 (1983). [Google Scholar]
- 17.Raynal M, Ballester P, Vidal-Ferran A & van Leeuwen PWNM Supramolecular catalysis. Part 1: non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev 43, 1660–1733 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Steinman G & Cole MN Synthesis of biologically pertinent peptides under possible primordial conditions. Proc. Natl Acad. Sci. USA 58, 735–742 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toparlak D, Karki M, Egas Ortuno V, Krishnamurthy R & Mansy SS Cyclophospholipids increase protocellular stability to metal ions. Small 16, 1903381 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Rao M, Eichberg J & Oró J Synthesis of phosphatidylcholine under possible primitive Earth conditions. J. Mol. Evol 18, 196–202 (1982). [DOI] [PubMed] [Google Scholar]
- 21.Frisch MJ et al. Gaussian 09, Revision A.02 (Gaussian Inc, 2016). [Google Scholar]
- 22.Marenich AV, Cramer CJ & Truhlar DG Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Delory DE & King EJ A sodium carbonate-bicarbonate buffer for alkaline phosphatases. Biochem. J 39, 245 (1945). [PubMed] [Google Scholar]
- 24.Kempe S & Degens ET An early soda ocean? Chem. Geol 53, 95–108 (1985). [Google Scholar]
- 25.Martin W & Russell MJ On the origin of biochemistry at an alkaline hydrothermal vent. Phil. Trans. R. Soc. B 362, 1887–1925 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkins WR et al. Role of lipid polymorphism in pulmonary surfactant. Science 273, 330–332 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Athenstaedt K & Daum G Phosphatidic acid, a key intermediate in lipid metabolism. Eur. J. Biochem 266, 1–16 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Chen IA & Szostak JW Membrane growth can generate a transmembrane pH gradient in fatty acid vesicles. Proc. Natl Acad. Sci. USA 101, 7965–7970 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morese JW & Mackenzie FT Hadean ocean carbonate geochemistry. Aquat. Geochem 4, 301–319 (1998). [Google Scholar]
- 30.Macleod G, McKeown C, Hall AJ & Russell MJ Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Orig. Life Evol. Biospheres 24, 19–41 (1994). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within the paper and its Supplementary Information.