

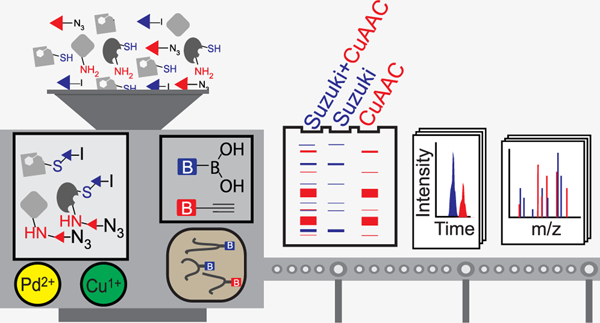
Abstract
Mass-spectrometry-based chemoproteomics has enabled the rapid and proteome-wide discovery of functional and potentially ‘druggable’ hotspots in proteins. While numerous transformations are now available, chemoproteomic studies still rely overwhelmingly on copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) or ‘click’ chemistry. The absence of bio-orthogonal chemistries that are functionally equivalent and complementary to CuAAC for chemoproteomic applications has hindered the development of multiplexed chemoproteomic platforms capable of assaying multiple amino acid side chains in parallel. Here, we identify and optimize Suzuki–Miyaura cross-coupling conditions for activity-based protein profiling and mass-spectrometry-based chemoproteomics, including for target deconvolution and labeling site identification. Uniquely enabled by the observed orthogonality of palladium-catalyzed cross-coupling and CuAAC, we combine both reactions to achieve dual labeling. Multiplexed targeted deconvolution identified the protein targets of bifunctional cysteine- and lysine-reactive probes.
Graphical Abstract
INTRODUCTION
Bio-orthogonal chemistry is a mainstay of chemoproteomic sample preparation workflows. In a typical chemoproteomic experiment, the proteome is decorated with a bifunctional chemical probe that incorporates both a reactive group (e.g., electrophile or photocrosslinker) and a detection handle (e.g., biotin, azide, or alkyne handle). Alkyne and azide moieties are widely used in chemoproteomics, due in large part to their small size, inertness toward biological molecules and aqueous environments, and ease of conjugation to biotin reagents using copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) or ‘click’ chemistry.1–5 After enrichment and proteolytic digestion, the protein targets and sites of probe labeling are identified by tandem mass spectrometry (MS/MS) analysis.
Enabled by innovative new probes, a plethora of protein functions can now be assayed by chemoproteomics, including the reactivity of nearly all nucleophilic amino acid side chains, inclusive of serine,6–8 cysteine,9,10 lysine,11 histidine,12 tyrosine,13 aspartate and glutamate,14–16 and methionine,17 and the interactions between proteins and cofactors,18–20 lipids,21,22 reactive metabolites,23 and druglike molecules.10,24 Recently, bifunctional probes featuring two electrophiles have emerged as a new strategy to induce protein association, functioning as a ‘molecular glue’.25 Assessment of the protein targets of such bifunctional molecules would benefit from the development of new chemoproteomic methods that assay compound labeling at multiple amino acids simultaneously. The identification of additional inert handles compatible with chemoproteomics and complementary to azide and alkyne groups would enable such multiplexed chemoproteomic studies that use two chemical probes simultaneously.
Aryl halides are ideally suited to chemoproteomic applications. Many drugs and probes already incorporate aryl halides,26 and halogenated building blocks are economical and readily available. As the Van der Waals volume27 of the iodo (32.52 Å3/molecule), and bromo (26.52 Å3/molecule) substituents are smaller than those of ethene (39.7 Å3/molecule) and ethyne (38.4 Å3/molecule),28 halogens are suited to applications that cannot tolerate large modifications. Enabled by mild and biocompatible Suzuki–Miyaura cross-coupling, site-specific labeling of halogen-containing proteins and oligonucleotides has been achieved.29–42 However, to our knowledge, the utility of palladium-catalyzed cross-coupling in chemoproteomic applications remains unexplored.
Here, we report multiplexed CuAAC Suzuki–Miyaura chemoproteomics (termed mCSCP) as a novel platform capable of assessing labeling of both cysteines and lysine residues simultaneously. As a key step for mCSCP, we identify SP3 sample cleanup43,44 as an improved method to remove contaminants from chemoproteomic samples. Application of mCSCP to chemoproteomic target deconvolution on a global scale enables identification of residues that can be targeted by bifunctional cysteine- and lysine-reactive molecules.
EXPERIMENTAL SECTION
Gel-Based Activity-Based Protein Profiling (ABPP) with Suzuki–Miyaura Cross-Coupling.
HEK293T proteome (50 μL of 2 mg/mL) was labeled with 1 (1 μL of 10 mM stock solution in DMSO, final concentration = 200 μM) for 1 h at ambient temperature. Suzuki–Miyaura cross-coupling was performed with biotin-boronic acid 2 or 3 (1 μL of 100 mM stock in DMSO, final concentration = 2 mM) and catalyst 4 (1 μL of 50 mM stock in dimethylacetamide (DMA) prepared according to the catalyst preparation procedure described in the Supplementary Methods, final concentration = 1 mM). Samples were allowed to react (3 h at 37 °C or 1 h at 50 °C) at which point the reactions were quenched with 4× Laemmli buffer (20 μL). The samples were then denatured (5 min, 95 °C) and analyzed by SDS-PAGE, using Criterion TGX Stain-Free gels obtained from Bio-Rad. Loading control images were obtained using the stain-free workflow with a Bio-Rad ChemiDoc Imager.45
SP3 Proteomic Sample Preparation Using Suzuki–Miyaura Cross-Coupling.
HEK293T cell lysates were prepared and labeled as described for gel-based ABPP. After Suzuki–Miyaura labeling, each sample was then treated with 0.5 μL of benzonase (Fisher Scientific, 70–664–3) for 30 min at 37 °C. DTT (10 μL of 200 mM stock in water, final concentration = 10 mM) was added to each sample, and the sample was incubated at 65 °C for 15 min. To this, iodoacetamide (10 μL of 400 mM stock in water, final concentration = 20 mM) was added, and the solution was incubated for 30 min at 37 °C with shaking. SP3 sample cleanup was performed as described previously.43,44 See the Supporting Information for a detailed description of sample cleanup.
NeutrAvidin Enrichment of Labeled Peptides.
For each sample, 50 μL of NeutrAvidin Agarose resin slurry (Pierce, 29,200) was washed twice in 10 mL of IAP buffer (50 mM MOPS pH 7.2, 10 mM sodium phosphate, and 50 mM NaCl buffer) and then resuspended in 500 μL of IAP buffer. Peptide solutions eluted from SP3 beads were then transferred to the NeutrAvidin Agarose resin suspension, and the samples were then rotated for 2 h at room temperature (RT). After incubation, the beads were pelleted by centrifugation (21,000 g, 1 min) and washed by centrifugation (6 × 700 μL water). Bound peptides were eluted with 60 μL of 80% acetonitrile in molecular biology grade water containing 0.1% FA (10 min at RT). The samples were then harvested by centrifugation (21,000 g, 1 min), and residual beads were separated from supernatants using Micro Bio-Spin columns (Bio-Rad). The remaining peptides were then eluted from pelleted beads with 60 μL of 80% acetonitrile in water containing 0.1% FA (10 min, 72 °C). The beads were then separated from the eluants using the same Bio-Spin column. The eluants were then collected by centrifugation (21,000 g, 1 min), and the combined eluants were dried (SpeedVac). The samples were then reconstituted in 40 μL of water containing 5% acetonitrile and 1% FA and analyzed by LC–MS/MS.
Data Availability.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE46 partner repository with the dataset identifier PXD022279 and 10.6019/PXD022279.
RESULTS
Bio-orthogonal Suzuki–Miyaura Cross-Coupling Complex Cell Lysates.
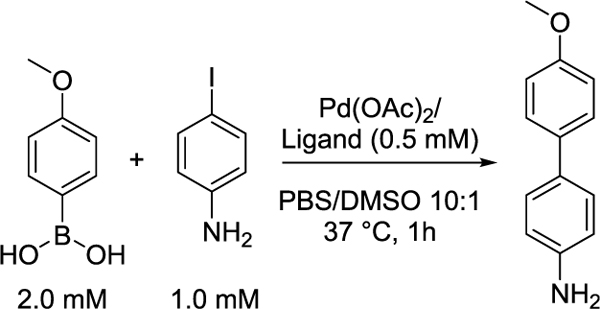
We first evaluated the compatibility of palladium-catalyzed cross-coupling with thiol-rich cell lysates, given the known sensitivity of cross-coupling reactions to free thiols29 and the potential for S-arylation.47 By screening a panel of ligands in the model reaction between 4-iodoaniline and 4-methoxyphenylboronic acid, we determined whether catalysis would be detrimentally impacted by addition of the cellular reductant glutathione (1 mM GSH), the protein bovine serum albumin (1 mg/mL BSA), or the complex milieu of HEK293T cellular lysates (1 mg/mL). In PBS buffer, sSPhos outperformed all other catalysts and afforded high (>80%) conversion (Table 1, Table S1, and Figure S1). Free thiol additives (GSH) dramatically decreased the efficiency of reactions catalyzed by all palladium complexes. In contrast, when the model reaction was catalyzed by palladium complexed with bulky electron-rich phosphine ligands, including sSPhos and P(t-Bu)3, we observed reasonable product formation in the presence of thiol-containing proteins, including the recombinant protein bovine serum albumin (BSA) and bulk cellular lysates.
Table 1.
Sensitivity of Model Suzuki–Miyaura Reaction to Biorelevant Additives
We then tested whether this reactivity would extend to gel-based ABPP (Figure 1A and Figure S2). We obtained a cysteine-reactive iodinated probe, iodophenyl iodoacetamide 1 in 84% yield, and two phenyl boronic acid-substituted biotinylated enrichment handles (biotin-boronic acids 2 and 3) in 84 and 77% yields, respectively (Figure 1B and Scheme S1). We prioritized sSPhos for ABPP studies, as its solubility in aqueous media minimized requirements for cosolvent. Palladium catalyst 4 was generated by stirring one equivalent of palladium acetate (Pd(OAc)2) and one equivalent of sSPhos in degassed DMA at 50 °C for 30 min. Degassed cellular lysates were then labeled with probe 1 (200 μM) for 1 h, subjected to cross-coupling conditions including 2 mM 2 or 3 and 1 mM 4 at 37 °C for 3 h, samples resolved by SDS-PAGE gel, and biotinylation visualized by streptavidin blot. Gratifyingly, labeling was dose-dependent and only observed in the presence of all cross-coupling reagents (Figure 1C, Figure S3, and Figure S4). Comparable labeling intensities were observed when Suzuki–Miyaura cross-coupling was benchmarked against CuAAC, with proteome labeled with the structurally matched cysteine-reactive iodoacetamide alkyne probe 5 (200 μM), which was reacted with biotin-azide 6 (Figure 1B,C).
Figure 1.
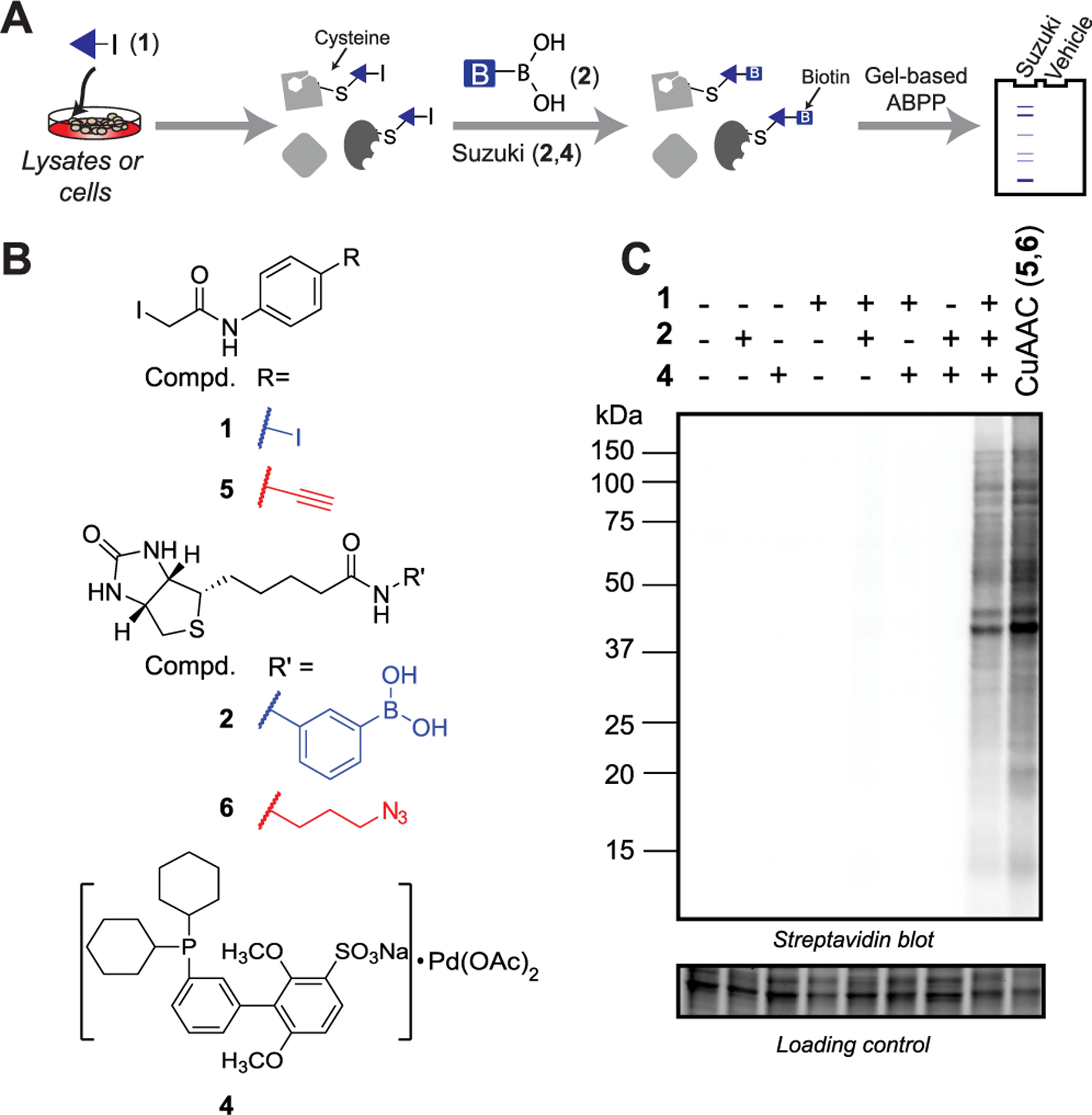
Gel-based ABPP using Suzuki–Miyaura cross-coupling. (A) General workflow. (B) Structures of probes used for Suzuki–Miyaura and CuAAC labeling. (C) Streptavidin detection of Suzuki–Miyaura and CuAAC labeling of degassed HEK293T lysates.
We next determined whether increasing the time, temperature, or concentration of palladium, the ligand, the probe, or biotin-boronic acid would afford increased labeling. While cross-coupling did occur at RT, increasing the temperature to 37 or 50 °C afforded increased labeling intensity (Figure S5). In contrast with increased palladium concentrations, which afforded modestly decreased labeling, we found that increasing the concentration of biotin-boronic acid up to 2 mM significantly enhanced protein labeling (Figure S6). Although cross-coupling reactions are known to be sensitive to oxygen, solution degassing did not significantly alter labeling (Figure S7A,B). Degassing did however completely eliminate low-level background labeling, which was observed in nondegassed lysates subjected to cross-coupling in the absence of probe 1 (Figure S7B) this background labeling can likely be ascribed to the reactivity of boronic acids with peroxo palladium complexes, which are formed in the presence of dioxygen.51 Comparable labeling was observed across catalyst batches, including for the catalyst left on the bench for >2 months (Figure S8A).
As many chemoproteomic experiments incorporate high concentrations of additives, including denaturing reagents, detergents, salts, and other biomolecules, we screened the compatibility of Suzuki–Miyaura labeling against a panel of additives (Figures S9–S11). Cross-coupling was found to be compatible with most common bioadditives, except for high concentrations of reductants, salts, and amines.
To explore the effects of the ligand and coupling partners on protein labeling, we systematically varied the boronic acid, catalyst, and halogen reaction components. Inspection of the molecular structure of streptavidin52 indicated that the lack of a flexible linker in biotin-boronic acids 2 and 3 might detrimentally affect molecular recognition of the biotin moiety. We therefore synthesized linker biotin-boronic acid 7 in 54% yield. Comparable labeling was observed by streptavidin blot for 2, 3, and 7 biotin boronic acids (Figure S12). Different ratios of sSPhos and Pd(OAc)2 were also examined, revealing that a 1: 1 stoichiometry was optimal (Figure S13). Efficient lysate biotinylation was achieved using a number of commercial ligands complexed with Pd(OAc)2, including XPhos,53 BrettPhos,54 P(t-Bu)3,49 and DavePhos50 (Figure S14). We were also surprised that addition of base (K2CO3), typically obligatory for cross-coupling reactions, did not enhance labeling (Figure S15).
To study whether gel-based ABPP with aryl bromides or chlorides was feasible, we synthesized chloro- and bromo-substituted cysteine-reactive probes 8 and 9 in 82 and 74% yields (Scheme S1). When subjected to sSPhos cross-coupling with biotin-boronic acid 3, neither 8 nor 9 afforded detectable lysate labeling (Figure S16), which we ascribe to the different rates of oxidative addition (I > Br> > Cl)55 and possible halide effects56,57 from the large excess of chloride ions in buffer, which is consistent with our observation that a large excess of sodium chloride (1 M) completely eliminated labeling (Figure S10).
Efficient cross-coupling with aryl bromides and chlorides can be achieved using palladium(0) precatalysts. Therefore, we obtained and screened a set of commercially available precatalysts, including the phosphine-containing sSPhos Pd G2, DavePhos Pd G3, XPhos Pd G3, SPhos Pd G3, XantPhos Pd G3, AdBrettPhos Pd G3, and N-XantPhos G4,58–62 and the N-heterocyclic carbene ligands (NHC)Pd(allyl)Cl63,64 and PEPPSI-IPr.65 Although near quantitative conversion was observed in model cross-coupling reactions between 4-iodoaniline and 4-methoxyphenylboronic acid for all precatalysts in the presence of 2 equivalents of base (K2CO3), 4-bromo- and 4-chloroaniline require an excess of strong base (e.g., 100 equiv of CsOH) to drive the reaction to near completion (Table S2). The general utility of precatalysts was also evaluated for gel-based ABPP with iodo probe 1. Robust protein biotinylation was only observed using sSPhos Pd G2, PEPPSI-IPr, and DavePhos Pd G3 (Figure S17). Use of the sSPhos Pd G2 precatalyst failed to afford significant biotinylation of lysates labeled with bromo- and chloro-substituted probes 8 and 9 (Figure S16 and Table S2).
SP3 sample cleanup enables Suzuki–Miyaura cross-coupling for chemoproteomic applications.
We next determined whether cross-coupling could be extended to chemoproteomic applications. Probe 1-labeled lysates were subjected to cross-coupling with biotin-boronic acid 2, protein-capture on neutravidin resin, on-bead tryptic digest, stage-tip desalting, and tandem MS/MS analysis (see Figure S2 for the workflow). Comparison of the unique proteins and cysteines revealed that the CuAAC substantially outperformed Suzuki–Miyaura cross-coupling (Figure 2, CHCl3/MeOH bars and Table S3). As this finding was inconsistent with our gel-based studies, which indicated similar labeling, we hypothesized that our protein capture may have been confounded by the excess biotin reagents required for Suzuki–Miyaura cross-coupling (vide supra), particularly given that boronic acids are known to interact strongly with the cis 1,2 and 1,3 diols present in glycosylated proteins.66
Figure 2.
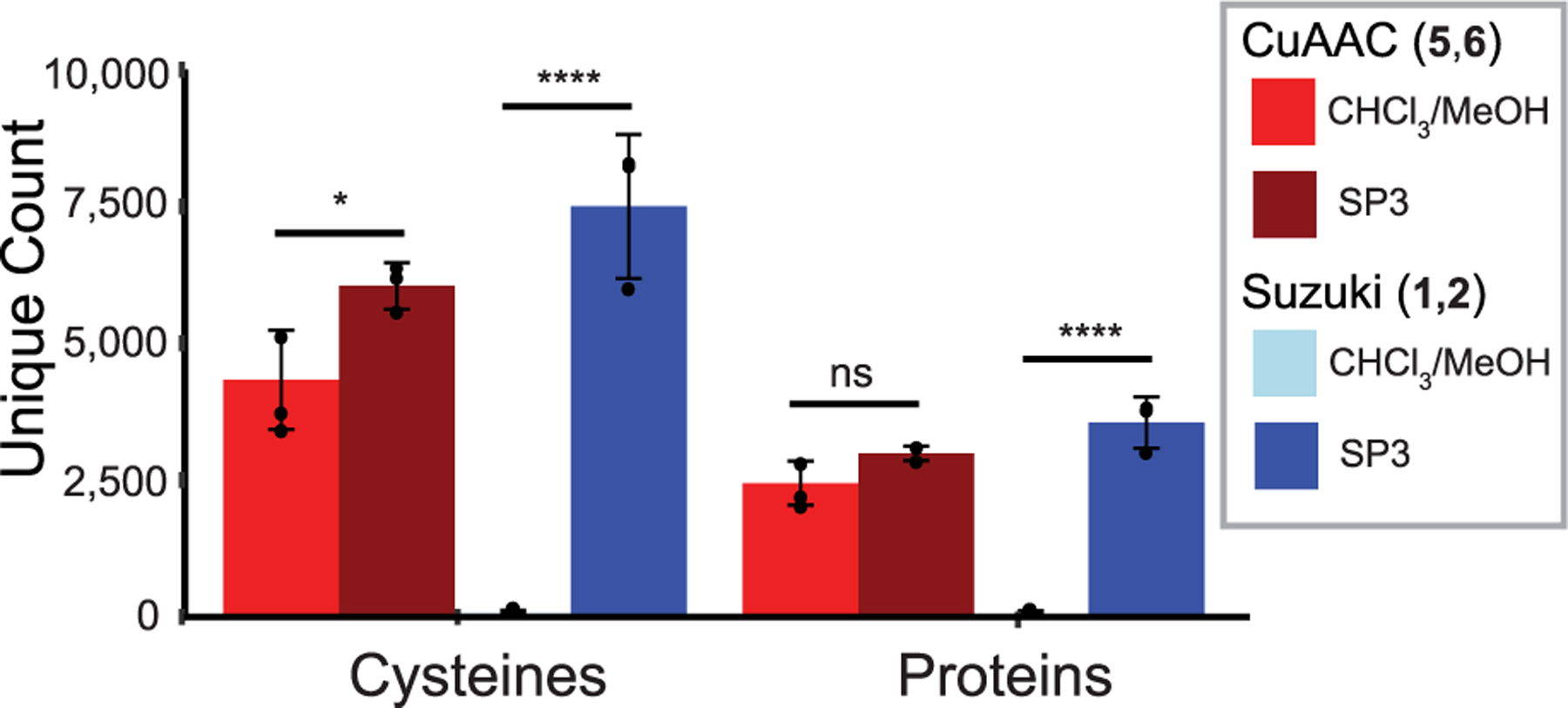
Comparison of CuAAC and Suzuki–Miyaura chemoproteomic experiments shows unique cysteine and protein counts comparing sample cleanup by CHCl3/MeOH precipitation (n = 4 for CuAAC and n = 5 for Suzuki–Miyaura) to solid-phase-enhanced sample-preparation (SP3) using carboxyl magnetic beads43,44 (n = 3 for both). Data represent means ± standard deviation. Statistical significance was calculated with unpaired Student’s t-tests comparing CHCl3/MeOH- to SP3-cleanup, *P < 0.05, ****P < 0.0001.
To test this hypothesis, we first performed labeling studies on BSA. BSA was labeled with the cysteine reactive probes 1 or 5, which were then subject to either Suzuki–Miyaura cross-coupling or CuAAC to biotinylated reagents 2 or 6, respectively. The labeled proteins were then subjected to tryptic digest and LC–MS/MS analysis, which revealed that Suzuki–Miyaura cross-coupling outperformed CuAAC in the biotinylated peptides identified (14 vs 6% of peptides, Figure S18 and Table S3). Incomplete biotinylation was observed for both reactions, and dehalogenation was also observed for 17% of peptides.
Supported by the efficient Suzuki–Miyaura-catalyzed biotinylation of BSA, we sought to identify a suitable method for contaminant removal. We found that single-pot, solid-phase-enhanced sample-preparation (SP3) using carboxyl magnetic beads43,44 far outperformed standard chloroform/ methanol (CHCl3/MeOH) precipitation, affording cross-coupling peptide capture comparable to that obtained using CuAAC (Figure 2, SP3 bars), and the aggregate coverage of CuAAC- and Suzuki-labeled peptides exceeded that reported in previous studies (Figure S19A).9,10 Notably, we found that a modified, steeper chromatography gradient (see the Supporting Information) was required to obtain improved coverage of biotinylated peptides, consistent with the hydrophobic modifications causing increased retention of labeled peptides during reverse-phase chromatography (Figure S20). Surprisingly, SP3 cleanup also increased the coverage of biotinylated peptides in CuAAC-labeled samples. Comparison of Suzuki- and CuAAC-labeled proteins using equimolar concentrations of probes 1 and 5 revealed substantial overlap between the proteins (64%) and peptides (46%) identified by both methods (Figure S19B and Table S3). Open search of the Suzuki-labeled samples using the search algorithm MSFragger67 confirmed the cross-coupling product to be the primary detectable modification. Of note, a handful of peptides (0.2% of all identified peptides) were found to be modified with a mass corresponding to the S-arylation product, indicating that the low-level oxygen-dependent background labeling can likely be ascribed to palladium-catalyzed S-arylation. Showcasing the efficiency of our sample preparation workflow, only a small percentage (17%) of the detected peptides were found to be unlabeled (Figure S21).
We tested whether Suzuki–Miyaura cross-coupling chemistry could robustly identify the protein targets of cysteine-reactive chemical probes. We synthesized and compared iodo-10 and alkyne-11 analogs of a N,N-disubstituted chloroacetamide scaffold KB7 that we previously identified as a modestly potent inhibitor (low micromolar) of procaspase-8 (Figure 3A).10,68 The apparent IC50 of procaspase-8 labeling by 10 was comparable to labeling by unsubstituted probe KB7 (5.4 and 6.9 μM, respectively; Figure 3B and Figure S22). In a competitive gel-based format, we observed that 10 and 11 (each at 10 μM) can both be used to detect blockade of labeling of procaspase-8 by unmodified compound KB7 (50 μM, Figure 3C). Suzuki–Miyaura visualization of procaspase-8 labeled with compound 10 did however result in somewhat lower labeling intensity when compared to CuAAC labeling with compound 11. In contrast, cell lysates treated with both probes showed similar labeling patterns and intensities (Figure 3D), indicating that the reduced labeling was likely restricted to procaspase-8.
Figure 3.
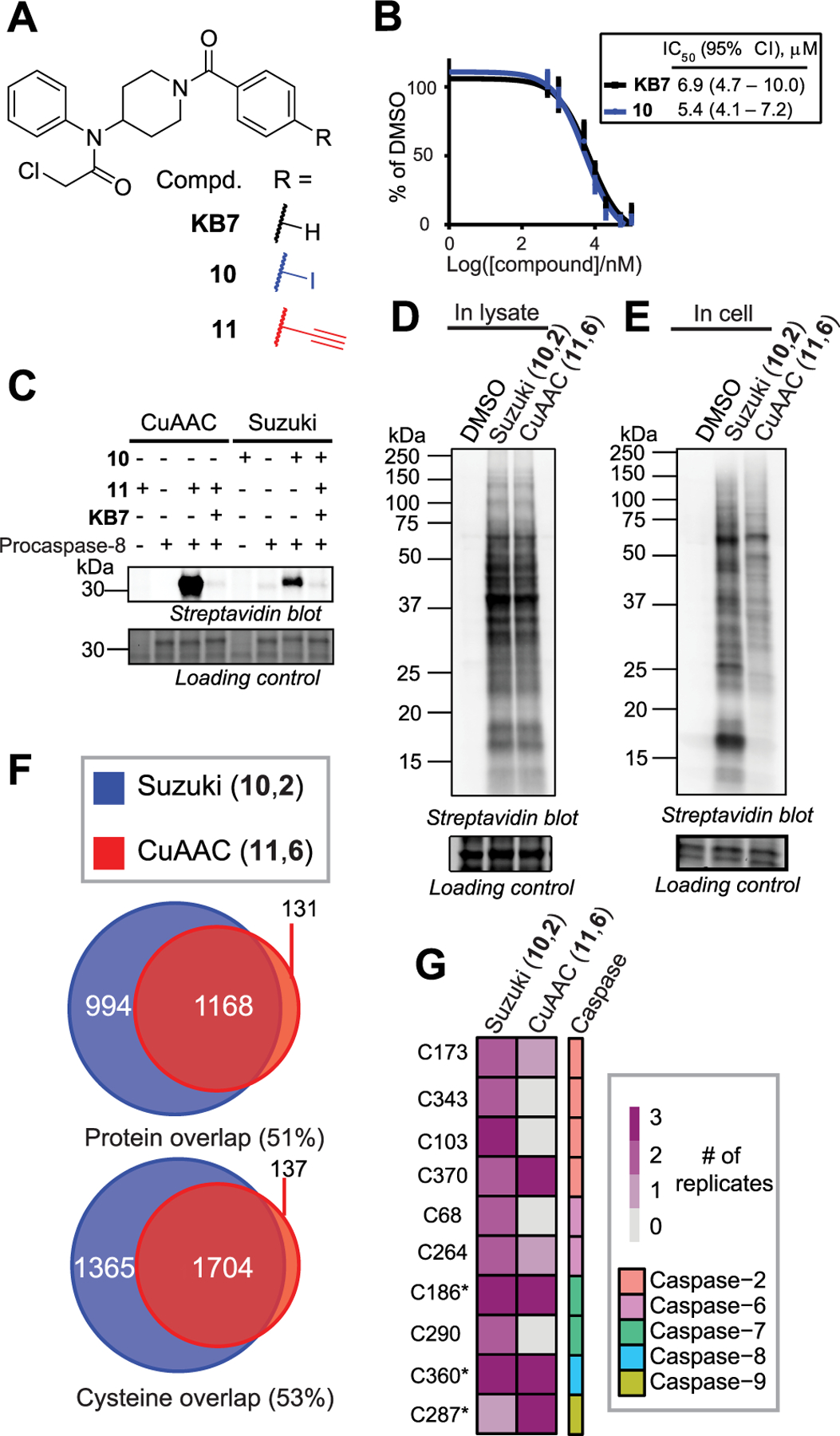
Suzuki–Miyaura cross-coupling for target deconvolution. (A) KB710 and its iodo-10 and alkyne-11 analogs. (B) Apparent IC50s of KB7 and 10 against procaspase-8. (C) Competitive gel-based ABPP profiling of procaspase-8. (D) In lysates and (E) in cells. (F) Chemoproteomic analysis of in-cell labeling of Jurkat cells treated as in 3E (n = 3 for each probe). (G) Cysteines in caspases identified as labeled by chemoproteomics with 10 or 11. * indicates catalytic residues.
We next turned our attention to in-cell labeling. Jurkat T lymphocyte cells were labeled with 10 or 11 (100 μM) followed by lysis, biotinylation, and gel-based analysis. 10 and 11 shared generally similar in-cell labeling profiles, with the exception of an ∼18 kDa band preferentially labeled by 10 (Figure 3E). Proteomics revealed that nearly all peptides labeled by probe 11 were also labeled by 10 (Figure 3F). Gratifyingly, the catalytic cysteine of caspase-8 (Cys360) was identified as labeled by both probes (Figure 3G and Table S3), consistent with our prior studies.10,68 Curiously, we also found that both probes labeled cysteines from additional caspases (Figure 3G), including both catalytic and noncatalytic residues. The relative promiscuity of both compounds can likely be ascribed to the high concentration of probes utilized here (100 μM), which far exceeds the apparent IC50 for caspase-8 labeling, and the fact that direct-labeling studies, such as those described here, do not distinguish between high- and low-affinity labeling events.
Multiplexed CuAAC Suzuki–Miyaura Chemoproteomics (mCSCP).
Multiplexed ABPP assays using fluorophores of different wavelengths can improve the throughput and coverage of gel-based inhibitor screens.69 However, such multiplexing has not been extended to MS-based proteomics due to the absence of orthogonal labeling and capture methods. To determine whether multiplexed CuAAC Suzuki–Miyaura labeling of the same sample was feasible, we sequentially labeled lysates with cysteine- and lysine-reactive probes, containing iodo and azido moieties, 1 (200 μM) and 12 (50 μM), respectively. Gratifyingly, we observed concurrent labeling by both probes with minimal cross-talk between the reactions (Figure 4A,B and Figure S22). Order of reactions did affect labeling, with CuAAC followed by Suzuki–Miyaura cross-coupling as the preferred reaction sequence. In a parallel set of chemoproteomic experiments, lysates labeled with probes 1 and 12 were labeled with biotinylated capture reagents, 2 and 13, respectively, and the labeled lysates were subjected to SP3-chemoproteomics analysis, as described above. A total of 3930 cysteine-labeled peptides and 7704 lysine-labeled peptides were detected across three replicate experiments (Figure 4A,C and Table S3). Overlap between the proteins identified by probes 1 and 12 was modest (Figure 4D). Surprisingly, only a handful of peptides (62 in total) were found to be labeled by both probes in the same sequence (Table S3). These dual labeling sites may point to privileged binding sites. For example, in the protein elongation factor 1-alpha 1 (EEF1A1), both Cys234 and Lys219 were labeled in the same peptide sequence and were also proximally located in the X-ray crystal structure (Figure 4E).
Figure 4.
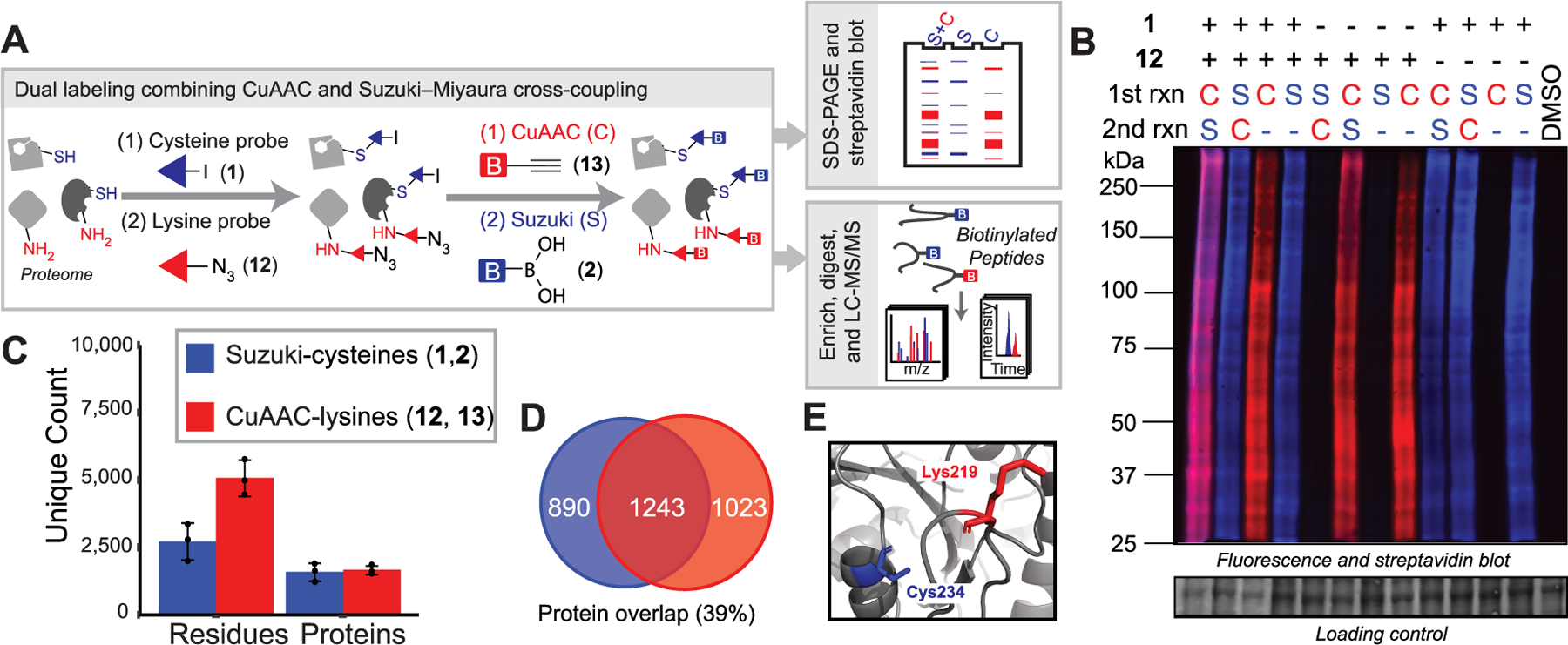
Dual labeling of cysteines and lysines by combining Suzuki–Miyaura cross-coupling and CuAAC. (A) General workflow. (B) Gel-based visualization of tandem labeling by 1 and 12. C-CuAAC with AF594 alkyne and S-Suzuki with 2 and 4. (C) Unique proteins, peptides, and residues identified by tandem chemoproteomics. (D) Overlap of proteins labeled by 1 (blue) and 12 (red) (n = 3). (E) Molecular structure of EEF1A1 (PDB ID 5LZS).
Next, we sought to determine whether our mCSCP platform could be applied to assess target engagement for bifunctional chemical crosslinkers. We synthesized a small library of compounds functionalized with both cysteine-reactive (acryl-amide- or chloroacetamide-) lysine-reactive (pentafluorophenyl ester) and dual lysine- and tyrosine-reactive electrophiles (sulfonyl fluoride). We also obtained compounds with only single electrophiles for comparison (Figure 5A and Scheme S2).
Figure 5.
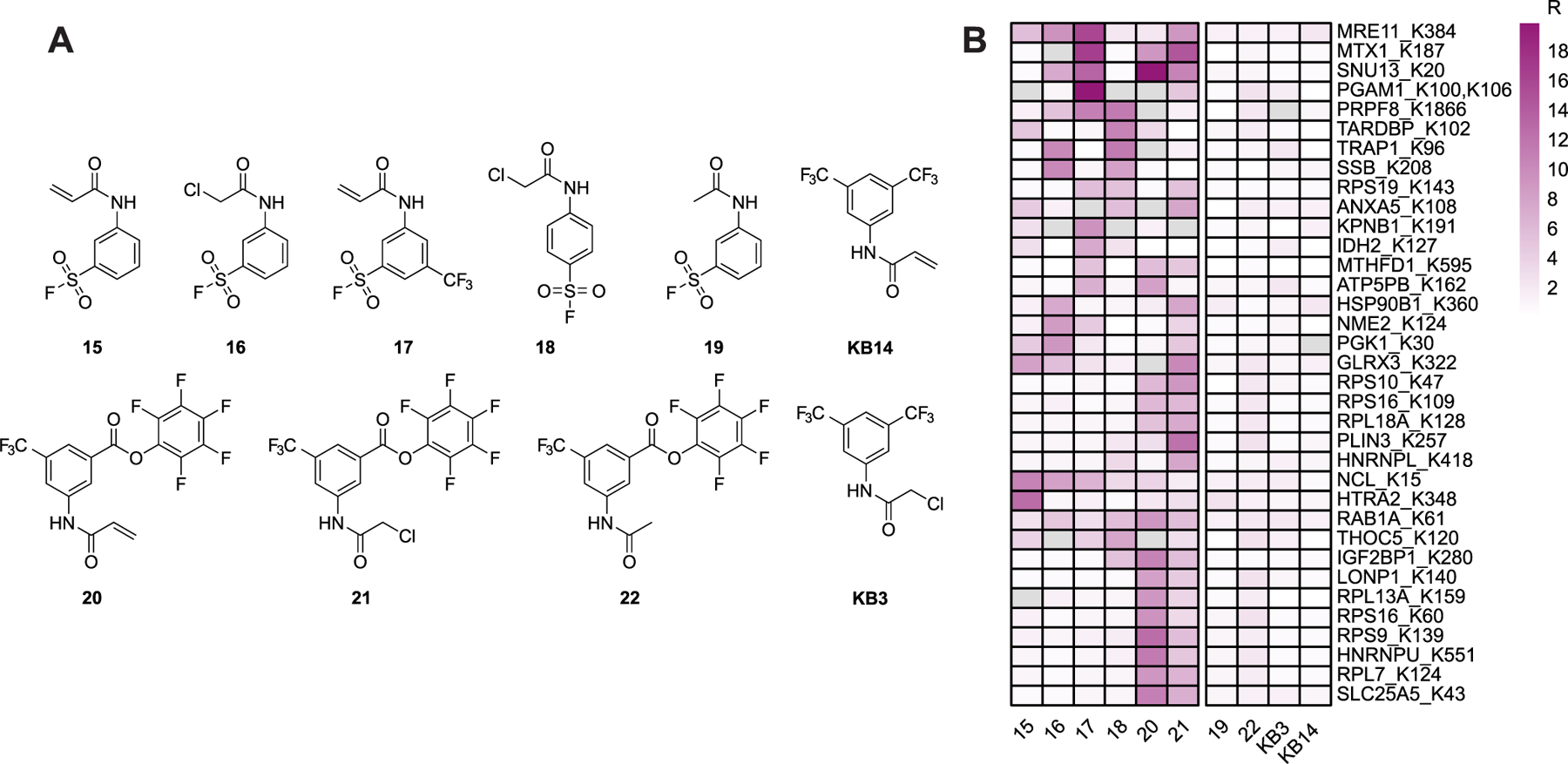
(A) Chemical structures of bifunctional cysteine- and lysine-reactive probes profiled by multiplexed CuAAC Suzuki–Miyaura chemoproteomics (mCSCP). (B) Heatmap shows representative lysines labeled preferentially by bifunctional cysteine- and lysine-reactive compounds 15–18, 20, and 21 and not by single-reactive compounds 19, 22, KB3, and KB14.
We then treated isotopically differentiated (stable isotope labeling by amino acids in cell culture or SILAC70) HEK293T cells with each compound (light cells) or DMSO vehicle (heavy cells) and subjected the labeled lysates to our mCSCP platform (see Figure S24 for the workflow). In aggregates, we detected 2632 cysteines on 1482 proteins and 9452 lysines on 2248 proteins across all compound-treatment experiments with 890 cysteines on 651 proteins and 607 lysines on 479 proteins showing MS1 peak area ratios >3, indicative of irreversible compound labeling (Table S4). Activated ester containing compounds 20–22 competed STP probe labeling for 479 aggregate lysines. Sulfonyl fluoride containing compounds 15–20 competed STP probe labeling for 396 aggregate lysines. A total of 128 lysines showed elevated MS1 area ratios (R > 3) only upon sulfonyl fluoride compound treatment and 211 lysines only upon activated ester compound treatment, indicating preferential labeling by a subset of lysine-reactive electrophiles. Surprisingly, mCSCP analysis with activated ester 22, which lacks a cysteine-reactive warhead, identified 324 cysteines with MS1 peak areas >3. These data point to widespread cysteine-reactivity of activated esters as a potential liability for the design of lysine-specific probes using activated esters and related warheads. In contrast, sulfonyl fluoride 19, which also lacks a cysteine-reactive electrophile, only showed elevated MS1 area ratios for 70 cysteines. It remains to be seen whether this small number of putative cysteines labeled by sulfonyl fluorides represent bona fide cysteine labeling or ratio changes that stem from labeling alternative proximal residues (e.g., lysines or tyrosines). Gratifyingly, we also detected 140 lysine residues on 116 proteins that are labeled by bifunctional compounds 15–18, 20, and 21 and not by parent cysteine- or lysine-reactive compounds KB3, KB14, 19, and 22 (Figure 5B), indicating that for these residues, cysteine labeling is likely a requisite for lysine labeling. For 53 of these proteins, we also identified at least one ligandable cysteine residue.
We next sought to confirm that the dual proteomic data could be faithfully recapitulated by competitive gel-based ABPP analysis. We selected the protein IMPDH2 for these studies, as our dataset indicated labeling at Cys140 by all dual electrophile library members as well as cysteine-reactive probes KB3 and KB14. We recombinantly expressed IMPDH2 in E coli and subjected the protein to gel-based ABPP using the previously identified cysteine-selective probe KB1810 (Scheme S2). Consistent with our dual chemoproteomic studies, all cysteine-reactive compounds showed substantial competition of KB18 labeling (Figure S25). Using IMPDH2 as a model protein, we also assessed the number of peptide crosslinks generated by treatment with sulfonyl fluorides 15–19. Compound-labeled protein was subjected to tryptic digest and MS/MS analysis. Search with the algorithm SIM-XL71 revealed a number of crosslinks for each compound, including both shared and unique crosslinks between cysteine residues and both lysine and tyrosine residues (Tables S5 and S6). Surprisingly, our competitive mCSCP dataset showed modest (Ratio ≈ 1.9) competition of STP-probe labeling at only one of these residues (K134), which indicates generally modest to low efficiency of labeling lysine residues by bifunctional sulfonyl-fluoride containing bifunctional compounds.
DISCUSSION
Taken together, our study expands the toolbox of reactions compatible with gel-based ABPP and chemoproteomics. We find that Suzuki–Miyaura cross-coupling is functionally equivalent and complementary to CuAAC. Building upon prior studies that demonstrated the utility of cross-coupling chemistry for labeling proteins, oligonucleotides, and cell surfaces, we show here that Suzuki–Miyaura cross-coupling will also proceed efficiently in cell lysates under mild conditions that afford excellent bio-orthogonality. We observed only trace probe-independent background labeling, which likely stems from palladium-catalyzed cysteine S-arylation. While solution degassing further reduces this background labeling, we acknowledge that a requirement for degassing may limit the broad applicability of our method. We anticipate that additional optimization of the palladium catalyst and reaction conditions will likely further reduce the observed low-level background reactivity.
We then applied Suzuki–Miyaura cross-coupling to activity-based chemoproteomics. Using a pan-cysteine-reactive iodophenyl probe 1, we showed that the single iodo atom could function as a bio-orthogonal enrichment handle for chemoproteomics, even outperforming structurally matched alkyne-containing probes for numbers of proteins and peptides identified. Key to these findings was our discovery that SP3 sample cleanup can be applied to chemoproteomic sample preparation workflows, which enabled enrichment of biotinylated peptides from samples labeled with excess of the biotin-boronic acid reagent. SP3 sample preparation offers the advantages of reduced sample size, near quantitative sample recovery, and streamlined sample preparation workflows. We expect that, based on our findings, SP3 cleanup will likely become an integral part of future chemoproteomic sample preparation workflows.
Our target deconvolution studies comparing structurally matched iodo and alkyne probes revealed that the protein targets and sites of labeling of electrophilic probes are readily identified by our Suzuki–Miyaura platform. Strikingly, nearly all proteins and cysteines identified by the alkyne-probe 11 were also identified by the iodo probe 10. Interestingly, for in-cell labeling studies, we found that iodo probe 10 also labeled a significant number of proteins and peptides not captured by alkyne probe 11. We ascribe this increased labeling to the greater lipophilicity of the iodo probe, which likely leads to increased in-cell probe accumulation and possibly altered subcellular localization. Of note, our in vitro gel-based ABPP studies showed comparable labeling profiles and near-identical banding patterns, indicating that the increased labeling observed for the iodo probes is likely limited to cell-based studies.
Uniquely enabled by the observed orthogonality of Suzuki–Miyaura cross-coupling and CuAAC, we then applied our findings to develop an innovative multiplexed chemoproteomic platform (mCSCP) capable of assaying the ligandability of both cysteine and lysine residues in single experiments. We first showed that these reactions can function orthogonally using gel-based ABPP and then extended our findings to mass spectrometry-based chemoproteomics. Application of our multiplexed profiling method to the analysis of bifunctional chemical crosslinkers revealed that the ligandability of both lysine and cysteine residues can be assayed in a single experiment, including for dual amino acid-reactive probes, enabling identification of lysines preferentially labeled by compounds that also feature a cysteine-reactive warhead. Decreased coverage of labeled peptides due to the increased sample complexity caused by multiplexing is one potential limitation of our approach that could, in part, be addressed with isobaric labeling reagents.
Our sulfonyl fluoride-substituted compounds represent a useful advance for the field of crosslinking mass spectrometry. While sulfonyl fluorides have been used in crosslinking mass spectrometry experiments previously,72 to our knowledge, this chemotype has not yet been incorporated into cysteine-reactive crosslinkers. As sulfonyl fluorides are known to also react with tyrosine residues, future studies should integrate tyrosine-reactive probes13 into such multiplexed chemoproteomic workflows. While competive chemoproteomic studies are well-suited to identify high-affinity crosslinking sites, our findings indicate that such studies may miss lower efficient crosslinking events, as shown by our crosslinking MS data using the protein IMPDH2. Given the widespread interest in chemical probes that function as covalent molecular glues, our data point to possible challenges associated with generating compounds that react intermolecularly with high efficiency simultaneously at two amino acids.
Looking to the future, we can envision a wide range of additional applications for this chemistry. Multiplexed gel-based and chemoproteomic studies, such as those described here, should prove useful for cases where samples are limited, such as patient-derived samples or rare cell types. Suzuki–Miyaura cross-coupling chemistry should also extend to the labeling and enrichment of additional classes of biomolecules, such as RNA, that are known to be degraded by the copper employed in click chemistry.73,74 As halogens are ubiquitous in bioactive molecules, clinical candidates, and drugs, we foresee the widespread utility of Suzuki–Miyaura cross-coupling chemistry in target deconvolution studies, particularly when biological activity is precluded by bulkier substitutions. The development of more specialized ligands that reduce requirements for excess palladium and boronic acid reagents and that are compatible with Suzuki–Miyaura cross-coupling of aryl bromide and chloride probes in cell lysates will further increase the general utility of this chemistry. CuAAC is the gold standard bio-orthogonal reaction, which has been widely adopted by diverse communities and applications. The fact that Suzuki–Miyaura cross-coupling performs comparably to CuAAC is a testament to the robustness of the reaction. We do not anticipate that cross-coupling will supplant CuAAC and instead expect, as shown here, that bio-orthogonal cross-coupling reactions, including Suzuki–Miyaura, as well as Sonagashira,75 oxidative Heck,76 and likely additional reactions, can complement CuAAC labeling and add to the bio-orthogonal toolbox of reactions for ABPP and chemoproteomics.
Supplementary Material
ACKNOWLEDGMENTS
K.M.B. was supported by the DOD-Advanced Research Projects Agency (DARPA) D19AP00041, L.M.B by University of California Los Angeles System and Integrative Biology Training Program (NIH T32 5T32GM008185–33), N.R.B. by University of California Los Angeles Biotechnology Training in Biomedical Sciences and Engineering (NIH T32 GM067555–11), and E.A. by University of California Los Angeles Cellular and Molecular Biology Training Program (NIH T32 GM007185–42). We gratefully acknowledge James Wohlschlegel, Weixian Deng, Jihui Sha, Xiaorui Fan, Yasaman Jami-Alahmadi, and all members of the Backus lab for helpful suggestions and the UCLA Proteome Research Center for assistance with mass-spectrometry-based proteomic data collection.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c04726
The authors declare no competing financial interest.
Contributor Information
Jian Cao, Department of Biological Chemistry, David Geffen School of Medicine, UCLA, Los Angeles, California 90095, United States.
Lisa M. Boatner, Department of Biological Chemistry, David Geffen School of Medicine and Department of Chemistry and Biochemistry, UCLA, Los Angeles, California 90095, United States.
Heta S. Desai, Department of Biological Chemistry, David Geffen School of Medicine, UCLA, Los Angeles, California 90095, United States; Molecular Biology Institute, UCLA, Los Angeles, California 90095, United States
Nikolas R. Burton, Department of Biological Chemistry, David Geffen School of Medicine and Department of Chemistry and Biochemistry, UCLA, Los Angeles, California 90095, United States
Ernest Armenta, Department of Biological Chemistry, David Geffen School of Medicine and Department of Chemistry and Biochemistry, UCLA, Los Angeles, California 90095, United States.
Neil J. Chan, Department of Biological Chemistry, David Geffen School of Medicine and Department of Chemistry and Biochemistry, UCLA, Los Angeles, California 90095, United States
José O. Castellón, Department of Biological Chemistry, David Geffen School of Medicine, UCLA, Los Angeles, California 90095, United States; Molecular Biology Institute, UCLA, Los Angeles, California 90095, United States
Keriann M. Backus, Department of Biological Chemistry, David Geffen School of Medicine and Department of Chemistry and Biochemistry, UCLA, Los Angeles, California 90095, United States; Molecular Biology Institute, DOE Institute for Genomics and Proteomics, and Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, UCLA, Los Angeles, California 90095, United States; Jonsson Comprehensive Cancer Center, UCLA, Los Angeles, California 90095, United States.
REFERENCES
- (1).Speers AE; Adam GC; Cravatt BF J. Am. Chem. Soc 2003, 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- (2).Speers AE; Cravatt BF Curr. Protoc. Chem. Biol 2009, 1, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Willems LI; van der Linden WA; Li N; Li KY; Liu N; Hoogendoorn S; van der Marel GA; Florea BI; Overkleeft HS Acc. Chem. Res 2011, 44, 718–729. [DOI] [PubMed] [Google Scholar]
- (4).Martell J; Weerapana E Molecules 2014, 19, 1378–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Parker CG; Pratt MR Cell 2020, 180, 605–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li W; Blankman JL; Cravatt BF J. Am. Chem. Soc 2007, 129, 9594–9595. [DOI] [PubMed] [Google Scholar]
- (7).Simon GM; Cravatt BF J. Biol. Chem 2010, 285, 11051–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Adibekian A; Martin BR; Wang C; Hsu K-L; Bachovchin DA; Niessen S; Hoover H; Cravatt BF Nat. Chem. Biol 2011, 7, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MB; Bachovchin DA; Mowen K; Baker D; Cravatt BF Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; Gonzalez-Paez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Nature 2016, 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF Nat. Chem 2017, 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jia S; He D; Chang CJ J. Am. Chem. Soc 2019, 141, 7294–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hahm HS; Toroitich EK; Borne AL; Brulet JW; Libby AH; Yuan K; Ware TB; McCloud RL; Ciancone AM; Hsu KL Nat. Chem. Biol 2020, 16, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cheng K; Lee J-S; Hao P; Yao SQ; Ding K; Li Z Angew. Chem., Int. Ed 2017, 56, 15044–15048. [DOI] [PubMed] [Google Scholar]
- (15).Ma N; Hu J; Zhang Z-M; Liu W; Huang M; Fan Y; Yin X; Wang J; Ding K; Ye W; Li Z J. Am. Chem. Soc 2020, 142, 6051–6059. [DOI] [PubMed] [Google Scholar]
- (16).Bach K; Beerkens BLH; Zanon PRA; Hacker SM ACS Cent. Sci 2020, 6, 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Science 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Montgomery DC; Sorum AW; Meier JL J. Am. Chem. Soc 2014, 136, 8669–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Horning BD; Suciu RM; Ghadiri DA; Ulanovskaya OA; Matthews ML; Lum KM; Backus KM; Brown SJ; Rosen H; Cravatt BF J. Am. Chem. Soc 2016, 138, 13335–13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).George Cisar EA; Nguyen N; Rosen HJ Am. Chem. Soc 2013, 135, 4676–4679. [DOI] [PubMed] [Google Scholar]
- (21).Hulce JJ; Cognetta AB; Niphakis MJ; Tully SE; Cravatt BF Nat. Methods 2013, 10, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Niphakis MJ; Lum KM; Cognetta AB 3rd; Correia BE; Ichu T-A; Olucha J; Brown SJ; Kundu S; Piscitelli F; Rosen H; Cravatt BF Cell 2015, 161, 1668–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kulkarni RA; Bak DW; Wei D; Bergholtz SE; Briney CA; Shrimp JH; Alpsoy A; Thorpe AL; Bavari AE; Crooks DR; Levy M; Florens L; Washburn MP; Frizzell N; Dykhuizen EC; Weerapana E; Linehan WM; Meier JL Nat. Chem. Biol 2019, 15, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF Cell 2017, 168, 527–541.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Isobe Y; Okumura M; McGregor LM; Brittain SM; Jones MD; Liang X; White R; Forrester W; McKenna JM; Tallarico JA; Schirle M; Maimone TJ; Nomura DK Nat. Chem. Biol 2020, 16, 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).McGrath NA; Brichacek M; Njardarson JT J. Chem. Educ 2010, 87, 1348–1349. [Google Scholar]
- (27).Bondi A J. Phys. Chem 1964, 68, 441–451. [Google Scholar]
- (28).Zhao YH; Abraham MH; Zissimos AM J. Org. Chem 2003, 68, 7368–7373. [DOI] [PubMed] [Google Scholar]
- (29).Chalker JM; Wood CSC; Davis BG J. Am. Chem. Soc 2009, 131, 16346–16347. [DOI] [PubMed] [Google Scholar]
- (30).Omumi A; Beach DG; Baker M; Gabryelski W; Manderville RA J. Am. Chem. Soc 2011, 133, 42–50. [DOI] [PubMed] [Google Scholar]
- (31).Spicer CD; Davis BG Chem. Commun 2011, 47, 1698–1700. [DOI] [PubMed] [Google Scholar]
- (32).Lercher L; McGouran JF; Kessler BM; Schofield CJ; Davis BG Angew. Chem., Int. Ed 2013, 52, 10553–10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dumas A; Spicer CD; Gao Z; Takehana T; Lin YA; Yasukohchi T; Davis BG Angew. Chem., Int. Ed. Engl 2013, 52, 3916–3921. [DOI] [PubMed] [Google Scholar]
- (34).Ding Y; Clark MA ACS Comb. Sci 2014, 17, 1–4. [DOI] [PubMed] [Google Scholar]
- (35).Walunj MB; Tanpure AA; Srivatsan SG Nucleic Acids Res 2018, 46, No. e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li JY; Huang H Bioconjugate Chem 2018, 29, 3841–3846. [DOI] [PubMed] [Google Scholar]
- (37).Kugele A; Braun TS; Widder P; Williams L; Schmidt MJ; Summerer D; Drescher M Chem. Commun 2019, 55, 1923–1926. [DOI] [PubMed] [Google Scholar]
- (38).Chen Y-C; Faver JC; Ku AF; Miklossy G; Riehle K; Bohren KM; Ucisik MN; Matzuk MM; Yu Z; Simmons N Bioconjugate Chem 2020, 31, 770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Spicer CD; Triemer T; Davis BG J. Am. Chem. Soc 2011, 134, 800–803. [DOI] [PubMed] [Google Scholar]
- (40).Spicer CD; Davis BG Chem. Commun 2013, 49, 2747–2749. [DOI] [PubMed] [Google Scholar]
- (41).Bilyard MK; Bailey HJ; Raich L; Gafitescu MA; Machida T; Igleśias-Fernańdez J; Lee SS; Spicer CD; Rovira C; Yue WW; Davis BG Nature 2018, 563, 235–240. [DOI] [PubMed] [Google Scholar]
- (42).Gao Z; Gouverneur V; Davis BG J. Am. Chem. Soc 2013, 135, 13612–13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Mol. Syst. Biol 2014, 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hughes CS; Moggridge S; Müller T; Sorensen PH; Morin GB; Krijgsveld J Nat. Protoc 2019, 14, 68–85. [DOI] [PubMed] [Google Scholar]
- (45).Posch A; Kohn J; Oh K; Hammond M; Liu NJ Visualized Exp 2013, 82, No. 50948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Peŕez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaíno JA Nucleic Acids Res 2019, 47, D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vinogradova EV; Zhang C; Spokoyny AM; Pentelute BL; Buchwald SL Nature 2015, 526, 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Anderson KW; Buchwald SL Angew. Chem., Int. Ed. Engl 2005, 44, 6173–6177. [DOI] [PubMed] [Google Scholar]
- (49).Littke AF; Fu GC Angew. Chem., Int. Ed. Engl 1998, 37, 3387–3388. [DOI] [PubMed] [Google Scholar]
- (50).Old DW; Wolfe JP; Buchwald SL J. Am. Chem. Soc 1998, 120, 9722–9723. [Google Scholar]
- (51).Adamo C; Amatore C; Ciofini I; Jutand A; Lakmini H J. Am. Chem. Soc 2006, 128, 6829–6836. [DOI] [PubMed] [Google Scholar]
- (52).Weber PC; Ohlendorf DH; Wendoloski JJ; Salemme FR Science 1989, 243, 85–88. [DOI] [PubMed] [Google Scholar]
- (53).Huang X; Anderson KW; Zim D; Jiang L; Klapars A; Buchwald SL J. Am. Chem. Soc 2003, 125, 6653–6655. [DOI] [PubMed] [Google Scholar]
- (54).Fors BP; Watson DA; Biscoe MR; Buchwald SL J. Am. Chem. Soc 2008, 130, 13552–13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Amatore C; Azzabi M; Jutand A J. Am. Chem. Soc 1991, 113, 8375–8384. [Google Scholar]
- (56).Fagnou K; Lautens M Angew. Chem., Int. Ed. Engl 2002, 41, 26–47. [DOI] [PubMed] [Google Scholar]
- (57).Fairlamb IJS; Taylor RJK; Serrano JL; Sanchez G New J. Chem 2006, 30, 1695–1704. [Google Scholar]
- (58).Kinzel T; Zhang Y; Buchwald SL J. Am. Chem. Soc 2010, 132, 14073–14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Biscoe MR; Fors BP; Buchwald SL J. Am. Chem. Soc 2008, 130, 6686–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Bruno NC; Tudge MT; Buchwald SL Chem. Sci 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Bruno NC; Niljianskul N; Buchwald SL J. Org. Chem 2014, 79, 4161–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Bruneau A; Roche M; Alami M; Messaoudi S ACS Catal 2015, 5, 1386–1396. [Google Scholar]
- (63).Viciu MS; Germaneau RF; Navarro-Fernandez O; Stevens ED; Nolan SP Organometallics 2002, 21, 5470–5472. [Google Scholar]
- (64).Viciu MS; Kissling RM; Stevens ED; Nolan SP Org. Lett 2002, 4, 2229–2231. [DOI] [PubMed] [Google Scholar]
- (65).O’Brien CJ; Kantchev EA; Valente C; Hadei N; Chass GA; Lough A; Hopkinson AC; Organ MG Chem. Eur. J 2006, 12, 4743–4748. [DOI] [PubMed] [Google Scholar]
- (66).Kuivila HG; Keough AH; Soboczenski EJ J. Org. Chem 1954, 19, 780–783. [Google Scholar]
- (67).Yu F; Teo GC; Kong AT; Haynes SE; Avtonomov DM; Geiszler DJ; Nesvizhskii AI Nat. Commun 2020, 11, 4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Xu JH; Eberhardt J; Hill-Payne B; Gonzaĺez-Paéz GE; Castelloń JO; Cravatt BF; Forli S; Wolan DW; Backus KM ACS Chem. Biol 2020, 15, 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Janssen APA; van der Vliet D; Bakker AT; Jiang M; Grimm SH; Campiani G; Butini S; van der Stelt M ACS Chem. Biol 2018, 13, 2406–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ong SE; Mann M Methods Mol. Biol 2007, 359, 37–52. [DOI] [PubMed] [Google Scholar]
- (71).Lima DB; de Lima TB; Balbuena TS; Neves-Ferreira AGC; Barbosa VC; Gozzo FC; Carvalho PC J. Proteomics 2015, 129, 51–55. [DOI] [PubMed] [Google Scholar]
- (72).Yang B; Wu H; Schnier PD; Liu Y; Liu J; Wang N; DeGrado WF; Wang L Proximity-enhanced SuFEx chemical cross-linker for specific and multitargeting cross-linking mass spectrometry 2018, 115 (44), 11162–11167, DOI: 10.1073/pnas.1813574115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Hermann T; Heumann H Methods Enzymol 2000, 318, 33–43. [DOI] [PubMed] [Google Scholar]
- (74).Paredes E; Das SR ChemBioChem 2011, 12, 125–131. [DOI] [PubMed] [Google Scholar]
- (75).Li N; Lim RKV; Edwardraja S; Lin QJ Am. Chem. Soc 2011, 133, 15316–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Ourailidou ME; van der Meer JY; Baas BJ; Jeronimus-Stratingh M; Gottumukkala AL; Poelarends GJ; Minnaard AJ; Dekker FJ ChemBioChem 2014, 15, 209–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE46 partner repository with the dataset identifier PXD022279 and 10.6019/PXD022279.