

Abstract
Chordomas are rare tumors of notochordal origin, most commonly arising in the sacrum or skull base. Chordomas are considered insensitive to conventional chemotherapy, and their rarity complicates running timely and adequately powered trials to identify effective treatments. Therefore, there is a need for discovery of novel therapeutic approaches. Patient-derived organoids can accelerate drug discovery and development studies and predict patient responses to therapy. In this proof-of-concept study, we successfully established organoids from seven chordoma tumor samples obtained from five patients presenting with tumors in different sites and stages of disease. The organoids recapitulated features of the original parent tumors and inter- as well as intrapatient heterogeneity. High-throughput screenings performed on the organoids highlighted targeted agents such as PI3K/mTOR, EGFR, and JAK2/STAT3 inhibitors among the most effective molecules. Pathway analysis underscored how the NF-κB and IGF-1R pathways are sensitive to perturbations and potential targets to pursue for combination therapy of chordoma.
Chordoma organoid drug screen platform identifies personalized, clinically relevant therapeutic leads.
INTRODUCTION
Chordoma is a rare malignant tumor that arises from the embryonic remnants of the notochord (1). It typically affects older adults (median age, 58.5), is more common in men than in women (5:3), and is diagnosed in about 300 Americans each year, with a median survival of just over 6 years (1). There are three histological subtypes of chordoma: conventional, dedifferentiated, and poorly differentiated (2–5). Conventional chordoma accounts for the vast majority of cases; these are usually indolent, chemoresistant tumors (4, 6, 7). The dedifferentiated subtype is reminiscent of high-grade pleomorphic spindle cell soft tissue sarcomas and typically follows an aggressive course (8). Poorly differentiated chordoma is a rare, aggressive subtype affecting children and young adults and characterized by INI1 (SMARCB1) deletions (1, 2, 4). Unlike conventional chordoma, dedifferentiated and poorly differentiated chordoma patients are typically administered adjuvant chemotherapy (9), with few documented responses (10). Brachyury, a transcription factor that is thought to prevent senescence in the notochord (1, 11), is a useful marker expressed in some conventional and poorly differentiated chordomas (12), but not dedifferentiated chordoma (4).
Treatment for chordoma relies primarily on surgery. Because of the anatomical location, complete resection can be challenging, particularly for clival tumors (7). Even after achieving complete resection, recurrence rates remain high at approximately 40%, often necessitating repeat surgeries (13). If the disease is metastatic or the patient is not a surgical candidate, there are few systemic treatment options available (9, 14). Traditional chemotherapeutic agents have not shown efficacy in this tumor type (6, 7), and there is no preferred regimen for the treatment of either locally recurrent or metastatic chordoma as of March 2021 (9). A small number of targeted agents have shown limited benefits in trials and are National Comprehensive Cancer Network (NCCN) recommended for delaying tumor growth in some patients. These include imatinib with or without cisplatin, sirolimus, dasatinib, sunitinib, erlotinib, sorafenib, and lapatinib for epidermal growth factor receptor (EGFR)–positive chordoma (9). A phase 2 trial of imatinib in 56 patients showed a 70% rate of stable disease at 6 months (14). Sorafenib was associated with a progression-free survival (PFS) of 9 months in 73% of the 27 patients treated in a phase 2 trial (15). The SARC009 study included 32 patients with unresectable chordoma treated with dasatinib and showed a 54% PFS at 6 months (16). However, most of these clinical trials have only extended PFS rather than achieving a partial or complete response (15). Thus, there remains a considerable need to identify efficacious therapies for chordoma (16).
A substantial limitation that continues to hinder the identification of novel therapeutic avenues is the small number of validated chordoma models for preclinical research. Few immortalized chordoma cell lines have been reported (17–19). While helpful, cell lines often fail at recapitulating the heterogeneity of the underlying disease and can deviate substantially from the parental tumor, resulting in changes to drug response (20). As with most slow-growing tumors, the generation of patient-derived xenograft models has lagged for chordoma, with moderate progress in recent years (19, 21–25). An approach to routinely establish chordoma organoids from biopsies or surgical specimen has the potential to power discovery studies to advance our understanding of chordoma and identify new interventions (22, 26).
Patient-derived tumor organoids (PDOs) are ideally suited for modeling rare cancers and investigating their heterogeneity and drug response (26–32). We have developed a high-throughput screening platform to test the response of PDOs to hundreds of therapeutic agents, with results available within a week from surgery (27, 33). Here, we apply our approach and platform to generate and screen chordoma PDOs established from different tumor sites and histologies (27, 33). Determining the clinical efficacy of any new therapeutic approach is challenging in chordoma because of its natural history and slow growth rates. Personalized chordoma models can be rapidly established and effectively screened ex vivo to identify pathways sensitive to perturbations.
RESULTS
Chordoma patient characteristics
In this proof-of-principle study, we aimed to determine whether clinically relevant, viable tumor organoid models can be established routinely from chordoma samples. We obtained n = 7 samples from five patients enrolled in this study (Table 1). Patients were 60:40 male to female with a median age at diagnosis of 61, ranging from 27 to 73 years old. At time of sample procurement, three patients presented with primary conventional chordomas (CHORD001, CHORD004, and CHORD005), one with recurrent chordoma (CHORD003), and one with metastatic chordoma (CHORD002).
Table 1. List of patients and tumor sample characteristics.
Collected attributes for each sample include the patient age at time of diagnosis, sex, tumor location, type of tissue procured, patient diagnosis at time of tissue procurement, pathology report of samples, history of systemic treatment of patient before the procurement of sample, and the current disease status after follow-up.
| Patient ID | Age* | Sex |
Tumor
location |
Tissue
procurement |
Diagnosis† | Pathology |
Systemic
treatment‡ |
Follow-up |
| CHORD001 | 64 | F | T3 vertebrae | Surgical resection of primary tumor |
Primary conventional chordoma |
1.5 cm, differentiated with no necrosis |
No | Disease free at 37 months |
| CHORD002 | 27 | M | Pelvis | Biopsy of pelvis metastasis (a) |
Metastatic conventional chordoma, metastases in bone, liver, and lungs |
N/A | No | Progressive disease |
| L3 vertebrae | Surgical resection of L3 tumor metastasis (b) |
Metastatic conventional chordoma, metastases in bone, liver, and lungs |
1 cm, 15% Ki-67, 15% necrosis |
Yes | Progressive disease |
|||
| Thoracic/ cervical vertebrae |
Resection of thoracical/ cervical spine metastases (c) |
Metastatic conventional chordoma, metastases in bone, liver, and lungs |
35% Ki-67 | Yes | Progressive disease |
|||
| CHORD003 | 61 | F | Sacrum | Surgical resection of recurrence |
Recurrent chordoma, NOS |
3.4 cm, substantial Ki-67 positivity |
No | Residual disease after 4 months |
| CHORD004 | 48 | M | L1-L3 vertebrae |
Surgical resection of primary tumor |
Primary conventional chordoma |
6 cm, 20% necrosis |
No | Progressive disease |
| CHORD005 | 73 | M | Sacrum | Surgical resection of primary tumor |
Primary chordoma, NOS |
11 cm, focal necrosis |
No | Disease free at 21 months |
*Age at time of diagnosis.
†Diagnosis at time of tissue procurement.
‡Before sample procurement.
We obtained a single sample for each patient at the time of surgical resection, with the exception of CHORD002, for which we procured n = 3 samples: one from a biopsy (CHORD002a), and a second (CHORD002b) and third (CHORD002c) from subsequent spinal metastasectomies. Anatomically, of the seven samples we procured, n = 2 originated in the sacrum (CHORD003 and CHORD005), n = 1 in the pelvis (CHORD002a), and n = 4 from the vertebrae: T3 (CHORD001), L3 (CHORD002b), thoracic/cervical vertebrae (CHORD002c), and L1-L3 (CHORD004).
CHORD001 was diagnosed as a conventional chordoma of the thoracic spine in a 64-year-old woman (Table 1). The patient remains disease free at 37 months of follow-up.
CHORD002 is a rare case of aggressive, metastatic conventional type chordoma of the sacrum diagnosed in a 27-year-old male (Table 1; see Supplementary Text for a detailed description). Six months after surgical resection of the primary mass, the patient presented with metastatic recurrence to the lungs, spine, and pubic bone. We procured a first tissue sample from a biopsy of the pubic bone metastasis (CHORD002a), which was confirmed to be conventional chordoma. The patient received several lines of treatment after this biopsy, including nivolumab, a programmed cell death protein 1 (PD-1)–targeting monoclonal antibody (34), in combination with the mammalian target of rapamycin (mTOR) inhibitor nab-rapamycin (34). We then obtained a second sample from the resection of a vertebral metastasis (CHORD002b). After the administration of nivolumab, DeltaRex-G, a retroviral vector encoding a cyclin G1 inhibitor (35), and cetuximab, a monoclonal antibody targeting EGFR in combination with nivolumab (34), we procured a third tissue sample, CHORD002c, from a resection of metastatic lesions in the cervical and thoracic spine. The patient subsequently received cyclophosphamide, the CDK4/5 inhibitor palbociclib, and cisplatin (34).
CHORD003 is a 61-year-old woman diagnosed with a not-otherwise-specified (NOS) chordoma of the sacrum (Table 1). There was a first local recurrence 3 years after diagnosis in the right acetabulum. We obtained a tissue sample from a second local recurrence diagnosed 15 months later (Table 1). Magnetic resonance imaging of the sacrum after 4 months of follow-up showed residual local disease. CHORD004 is a case of conventional chordoma of the lumbar spine in a 48-year-old male. We obtained a specimen from the initial resection, the mass affecting the L1-L3 vertebrae (CHORD004; Table 1). The patient presented with a recurrence of the tumor in the lower thoracic spine and chest metastases after a 19-month follow-up. CHORD005 is a 73-year-old male diagnosed with NOS chordoma of the sacrum (Table 1). The patient has no evidence of recurrence after 21 months of follow-up.
Chordoma organoid establishment and characterization
We set to determine whether viable, tractable chordoma organoid models can be routinely established from tissue obtained from different surgical procedures (primary resection, metastasectomy, and biopsy; Table 1), anatomical sites (cervical, thoracic or lumbar vertebrae, sacrum, and pelvis), and disease characteristics (primary, recurrent, or metastatic chordoma). Tumor tissue was fast-tracked to the laboratory for dissociation into a cell suspension containing single cells and small cell clusters (Fig. 1). We obtained sufficient cells in all cases, including a biopsy (CHORD002a), and established viable organoids for characterization and screening for all n = 7 samples (Figs. 1 and 2 and figs. S1 to S3). After seeding cells in a ring format according to our published protocols (27, 33) (see also Materials and Methods), we incubated them in serum-free medium for a total of 5 days and imaged them daily (Fig. 1) (27, 33).
Fig. 1. Morphology of the PDOs established as visualized by brightfield imaging of maxi-rings in 24-well plates.

CHORD002 to CHORD005 were imaged daily over a 5-day incubation period, while CHORD001 was imaged for 4 days. The organoids displayed morphological features consistent with chordoma such as vacuolated cells arranged in clusters or nests. The sample shown for patient CHORD002 is CHORD002a. Scale bar, 50 μm.
Fig. 2. Histology and immunohistopathology characterization of chordoma samples and derived organoids.

Formalin-fixed paraffin-embedded sections from both the parent tumor and organoid were stained with H&E, Ki-67, and brachyury. All organoids recapitulated features of the parent tumor. The sample shown for patient CHORD002 is CHORD002a. Scale bar, 20 μm.
Organoids cultured in both maxi- and mini-ring formats (27, 33) showed similar morphologies consistent with chordoma (fig. S1A). Individual PDOs showed a variety of behaviors in culture. We quantified the growth patterns in the brightfield images (Fig. 1) using a convolutional neural network (36) to calculate the number of organoids, total area, and average organoid area (fig. S1B; see also Materials and Methods). The normalized average areas show that CHORD003 organoids exhibited the most robust growth pattern, increasing on average by 50% in 4 days (fig. S1B). While CHORD005 showed a small increase in average organoid area after 5 days, samples from patient CHORD002 and CHORD004 were more indolent and mostly rearranged without sustained increase in area (fig. S1B and movie S1). Despite the limited growth, CHORD002a had several vacuolated chordoma cells that assumed a spindle-like morphology and migrated in culture (Fig. 1). To note, CHORD003 was obtained from a clinically rapidly growing recurrence, while CHORD002 was established from a diffusely metastatic chordoma, features that are reminiscent of the observed growth patterns.
To better investigate features of the chordoma PDOs, we fixed and embedded cells for downstream analysis (27, 33). When compared to the histopathology and immunohistochemical staining patterns of the parental tumors, PDOs retained all major features (Fig. 2 and figs. S2 and S3). For instance, both patient tumors and derived organoid cells show similar arrangements with cells in clusters and abundant eosinophilic vacuolated cytoplasm, as well as round nuclei containing small single or multiple nucleoli (Fig. 2 and fig. S2).
Matched chordoma parental tumors and organoids exhibited similar immunohistochemical staining patterns (Fig. 2 and fig. S3). We determined the expression of Ki-67, a marker of cell proliferation (37). We observed positive Ki-67 nuclear staining in both tumor and organoid samples in all cases, except for CHORD001. CHORD001 did not express Ki-67 in regions of the tumor and PDOs (Fig. 2). Of note, CHORD003 had an overall higher percentage of Ki-67–positive cells in the tumor of origin, in line with the clinical characteristics of this tumor (Table 1). This feature is conserved in CHORD003 PDOs (Fig. 2). A correlation between high Ki-67 positivity and aggressive behavior has been reported in chordoma (37).
Next, we investigated expression of brachyury, a protein involved in notochordal development and a well-established chordoma marker (1). CHORD001 showed variable expression for brachyury in the parent tissue with positive, negative, and mixed areas. Organoids from CHORD001 were largely negative for brachyury, likely due to the area that was sampled for organoid establishment (Fig. 2). CHORD002a had positive and negative cells in the parental tissue and was weakly positive in the PDOs (Fig. 2). CHORD003, CHORD004, and CHORD005 organoids and their parent tumor were positive for brachyury staining (Fig. 2). We then performed an expanded immunohistochemistry panel for pan-cytokeratin, epithelial membrane antigen (EMA), and S100 (fig. S3) (38). As noted above, heterogeneity in expression was observed in the parent tissue from CHORD001, similar to what we observed for brachyury (fig. S3), with both the parent tumor and organoids showing positive and negative cells for pan-cytokeratin, EMA, and S100 (fig. S3). Samples and PDOs from CHORD002 were positive for all markers (fig. S3). For all other samples, parent tissue stained positively for pan-cytokeratin and EMA, with organoids accurately mirroring the staining profile (fig. S3). S100 had sporadic positivity in CHORD003 and CHORD005 and was negative in CHORD004, in both the tumor tissue and derived PDOs (fig. S3). The expression of pan-cytokeratin, EMA, and S100 in addition to brachyury provides further evidence of how chordoma organoids recapitulate the features of the tumors they are derived from (1, 12). Overall, our tissue processing protocol and culturing conditions yielded viable chordoma organoids with a 100% success rate (7 of 7) and immunohistopathology characteristics of the parental tumor.
High-throughput drug screening of chordoma PDOs
Large-scale screenings of chordomas have been few and limited to immortalized cell lines so far (39–41). To validate that our PDO mini-ring platform (27, 33) is suitable for high-throughput screening of chordomas, we performed proof-of-principle single-concentration drug discovery screenings of up to 230 compounds on organoids established from CHORD001 to CHORD005. Cells seeded in mini-ring format in 96-well plates (27, 33) were incubated for 3 days, followed by drug treatments every 24 hours over two consecutive days. Viability was measured 24 hours after the last treatment by adenosine triphosphate (ATP) release assay following our established protocols (27, 33).
We compared normalized viability and Z score results for the subset of overlapping drugs (n = 124) tested on all seven samples (Fig. 3). We used a distance matrix to perform hierarchical clustering of the tested drug based on target similarity, as annotated in the PubChem database (42). Clinical samples were clustered by Euclidean distance of their drug response profiles (Fig. 3). Dot maps showing additional molecules tested are visible in Fig. 4 and figs. S5 and S6.
Fig. 3. Dot map of high-throughput screening results for 124 overlapping small molecules tested on all PDOs.
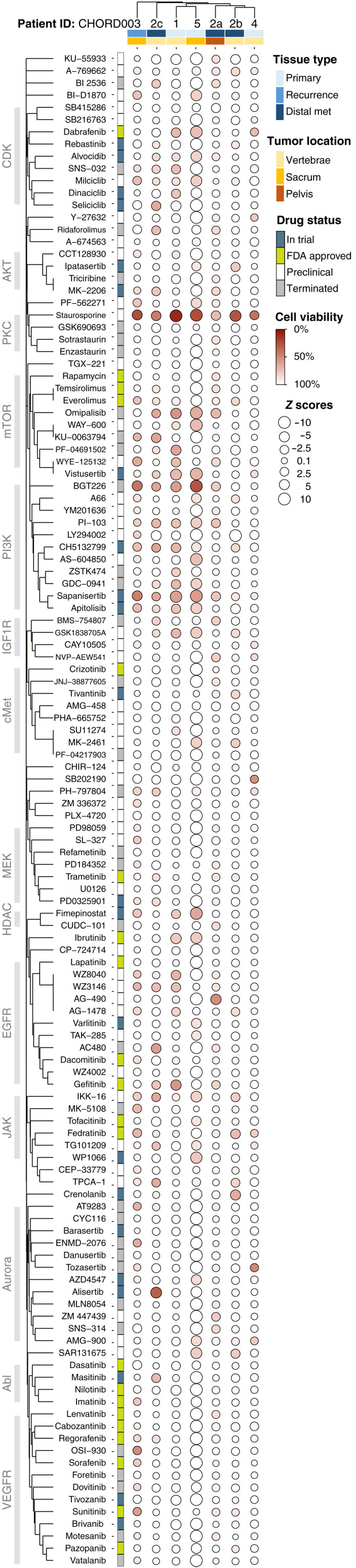
The size of each circle represents the Z score, with larger ones indicating a higher Z score value. The color of each point represents the normalized cell viability %. The drugs are clustered using the Jaccard distance based on common protein targets, and cases are clustered on the basis of their similarities in drug responses. Covariates included represent the type of tissue and location of each sample as well as FDA status for the various drugs.
Fig. 4. Dot map of drug screening on all three samples procured from patient CHORD002.

(A and B) Drugs that induce 15% cell death in at least two of the samples are visualized (51 total). The size of each circle represents the Z score, with larger ones indicating a higher Z score value. The color of each point represents the normalized cell viability %. The drugs are clustered using the Jaccard distance based on common protein targets. Samples are ordered by date of acquisition. Covariates included represent the type of tissue, anatomic location of each sample, and the number of lines of treatment the patient was exposed to before procurement of sample as well as FDA status at the time of publication.
Overall, the chordomas we tested showed marginal level of responses, with only the positive control staurosporine, a potent multikinase inhibitor, showing consistent albeit moderate efficacy across all samples (9 to 71% residual cell viability; table S2). For 5 of n = 7 samples tested, our positive control (staurosporine) was the only drug with a measured residual cell viability of ≤25%. Response rates defined as residual cell viability ≤25% varied between 0.4 and 3% (median, 0.65%) including staurosporine and 0 and 3% excluding the control. This is markedly lower than the responses we observed for ovarian cancer PDOs we tested following the same protocol (0.8 to 6%; median, 1.9%; n = 4; excludes staurosporine) (27), and in line with clinical findings of high therapeutic resistance of chordomas (43).
Given the generally low response rates, we investigated drugs causing residual cell viability of ≤50 and ≤75% (table S1). Response rates were 0 to 6% for the first group of drugs inducing at least 50% cell death (median, 1%) and 2 to 25% for the second, causing at least 25% cell death (median, 8%; table S1). We observed sensitivity toward inhibitors targeting the phosphatidylinositol 3-kinase (PI3K) and mTOR pathways, specifically for samples CHORD001, CHORD005, CHORD003, and CHORD002c, with average residual cell viability between 60% (CHORD001) and 71% (CHORD002c). Molecules such as BGT226, sapanisertib, omipalisib, and vistusertib were effective in inducing ≥25% cell death on most samples (Fig. 3 and table S2). Validation screenings for sapanisertib and several other molecules tested at concentrations between 0 to 10 μM largely confirmed the results of our discovery screenings (fig. S4, A and B).
Besides mTOR/PI3K inhibitors, the Janus kinase 2 (JAK2)–targeting molecule fedratinib also showed partial efficacy, inducing ≥20% cell death in three of seven samples (CHORD002b, CHORD003, and CHORD004; table S2). Inhibition of the JAK2/signal transducer and activator of transcription 3 (STAT3) pathway in vitro has been shown to reduce viability of chordoma cell lines (44, 45). Gefitinib, a U.S. Food and Drug Administration (FDA)–approved EGFR inhibitor, showed responses in three of seven samples (CHORD002a, CHORD002c, and CHORD005; table S2). Gefitinib has been identified as a potentially effective agent against chordoma on large-scale screenings of chordoma cell lines (40). The Abl-targeting drug imatinib and multi-tyrosine kinase inhibitor sunitinib showed efficacy in a single sample, CHORD003, with 27% cell death, Z score −1.6 and 44% cell death, Z score −2.7, respectively (table S2). Both molecules are NCCN recommended as systemic therapy for some chordomas (46).
Drug response evolution of CHORD002
Metachronous procurement of tumors from one patient with metastatic chordoma, CHORD002, allowed us to investigate the evolution of drug responses over time and therapy pressure (Fig. 4 and fig. S5). All organoid samples had similar histology (Fig. 4A). CHORD002a organoids were established from a biopsy of a pelvic metastatic lesion obtained prior to the initiation of any systemic therapy. This sample displays moderate sensitivity to drugs targeting the mTOR and EGFR pathways (Fig. 4).
CHORD002b organoids were generated from tissue obtained at the time of surgical resection of a lumbar spine metastasis. This specimen was obtained after three lines of systemic treatment, which included immune checkpoint blockade and the mTOR inhibitor rapamycin, as well as immune checkpoint blockade in combination with trabectedin (34) and palliative radiotherapy to the lumbar spine. Organoids established from this sample showed diminished sensitivity to the mTOR family of drugs (Fig. 4).
Last, we generated CHORD002c organoids from a surgical resection of cervical/thoracic spine metastasis, obtained after an additional three lines of treatment with the EGFR-targeting monoclonal antibody cetuximab in combination with immune checkpoint blockade, and a trial of the retroviral vector DeltaRex-G. The organoids established from this third sample were responsive to AC480, gefitinib, and WZ3146 treatment, but not AG-490. In addition, sample CHORD002c showed restored sensitivity to the mTOR-targeting drugs (Fig. 4).
Biological pathways associated with metabolism, bone signaling, and inflammation are sensitive to perturbation in chordoma PDOs
The data collected over a large chemical space allowed us to evaluate which pathways were most affected in our screenings. To accomplish this, we compiled a comprehensive list of known targets for each drug we screened according to the PubChem database (42) and overlaid these with well-characterized pathways described by WikiPathways (47).
For each sample, we first generated a matrix of drugs and corresponding protein targets weighted by the measured cell viability (see Materials and Methods). We then compiled a pathway-protein matrix, which was overlaid with the drug-target interaction matrix to investigate the effect of the tested drugs on these pathways and generate a pathway score. We finally selected the top 30 scored pathways for each sample and visualized those shared between a minimum of two chordomas in Fig. 5, with the size of each circle representing the pathway score, and the color representing the proportion of the genes in a pathway that are targeted by the tested drugs. The pathways are clustered on the basis of protein overlap (Fig. 5).
Fig. 5. Pathway analysis for all chordoma PDOs.

The top 30 ranked pathways that are affected in at least two samples are visualized (53). For each dot, the amount of surface filled in (in black) corresponds to the percentage of proteins within a pathway that were targeted, while the size corresponds to the pathway score. The pathways are clustered using the distance based on their shared proteins. The samples are clustered by the Euclidian distance of their pathway scores. Covariates included represent the type of tissue and site of each sample.
Our analysis highlighted how metabolism-related pathways were mostly sensitive to perturbation, with drugs targeting these pathways reducing chordoma PDO viability in most samples. Affected pathways include insulin signaling (five of seven samples), tricarboxylic acid cycle (five of seven samples), and estrogen signaling pathways (six of seven samples) (Fig. 5). Bone signaling pathways such as the RANK/RANKL and osteopontin signaling pathways were also affected in three of seven samples (Fig. 5).
In addition, pathways related to inflammation, and involving nuclear factor κB (NF-κB), MUC1, HMGB1, and resistin, were affected in most of our samples (>5 of 7; Fig. 5). Targeting the proteasome pathway had also a profound effect in three of seven samples; this is likely due to the large effect of the drugs bortezomib and carfilzomib that were screened on samples CHORD002b, CHORD002c, and CHORD005, and tend to elicit large in vitro responses (Fig. 5).
DISCUSSION
Chordoma remains an understudied rare cancer with limited therapeutic options and few patient-specific models (48). We propose an approach to develop personalized chordoma organoid models for biological characterization and high-throughput drug screenings from tissue obtained through a variety of surgical procedures and tumor sites. The chordoma organoids we have grown recapitulate features of the original tissue, such as histopathology, positivity for brachyury, S100, EMA, and pan-cytokeratin, as well as rate of Ki-67 staining (Fig. 2 and figs. S2 and S3). PDOs demonstrate growth patterns that are representative of specific clinical features (Fig. 1). While the PDO models shared the immunohistopathological and growth profile of their parent tumor sample, they maintained individual differences between patients. For instance, CHORD002 is a very aggressive metastatic chordoma with widely disseminated metastases and cells had unique migration patterns in culture (Fig. 1 and movie S1). Furthermore, patient CHORD003 had a highly proliferative tumor with substantial Ki-67 positivity (Table 1) and developed multiple local recurrences. The organoids established from CHORD003 showed robust proliferation by growth pattern (Fig. 1 and fig. S1B) and Ki-67 staining (Fig. 2). We observed striking heterogeneity in the staining patterns of patient CHORD001, with differences not only when examining different sections but also occasionally within different areas of the same section, as in case of brachyury and S100. Given the observed expression patterns (Fig. 2 and fig. S3), CHORD001 organoids were established from a largely brachyury- and EMA-negative portion of the tumor. As with all approaches that generate models from surgical samples, PDOs are highly dependent on sampling, which can underestimate tumor heterogeneity and lead to bias (49). Studies that synchronously sample multiple areas of the same tumor will help to fully elucidate the extent of chordoma spatial heterogeneity.
The PDOs we developed through our established platform (27, 33) can be effectively screened to identify promising drugs and sensitive pathways (Figs. 3 to 5). Drugs targeting some of the pathways we identified have been evaluated in preclinical models involving cell lines, zebrafish, xenografts (21–25, 50, 51), and clinical trials (15, 16, 52, 53).
For instance, PI3K/mTOR inhibitors have been evaluated in chordoma both as monotherapy and in combination with imatinib (53). The PI3K/mTOR-targeting drugs sapanisertib and vistusertib showed efficacy in our organoid screenings (Fig. 3). Both mTOR- and PI3K-targeting drugs were effective in chordoma cell lines (22, 54), and mTOR inhibitors alone showed partial responses in zebrafish (51). Further evidence supporting the targeting of this pathway includes the finding that mutations and alterations are found in PI3K signaling genes in a subset of chordoma cases and considered as driver events (11). Our screening identified other PI3K/mTOR-targeting drugs such as apitolisib and omipalisib as possible leads (Fig. 3). The clinical efficacy of these drugs in chordoma remains to be determined in trials.
EGFR activation, measured by phosphorylation, is frequently seen in chordomas (55). EGFR-targeting drugs such as lapatinib have shown some efficacy in controlling disease and are approved for use in EGFR-positive chordomas (9, 56), although xenograft models of chordoma have shown negative results (56). Additional EGFR-targeting drugs that have been tested in preclinical models and clinical trials for chordoma include erlotinib (50), linsitinib, afatinib, and the monoclonal antibody cetuximab (53, 54). We observed moderate efficacy of the EGFR-targeting drug gefitinib in three of seven samples. While gefitinib has not been tested in clinical trials as monotherapy in chordoma, it has been tested in combination therapy with cetuximab. In two patients, the combination of cetuximab and gefitinib showed a partial response with a duration of 9 months for a single patient (57) and a reduction of tumor bulk by 44% in another (58). In vitro screenings on chordoma cell lines have also identified gefitinib as an effective EGFR-targeting drug (40).
The STAT3 transcriptional pathway has been found to be implicated in the development of chordomas (44, 59). The efficacy of JAK2/STAT3-targeting drugs on chordoma has only been evaluated in in vitro studies that showed efficacy on chordoma cell lines (45, 60). We found the JAK2-targeting drug fedratinib to be effective on a subset of our samples (three of seven; Fig. 3). Fedratinib has not been tested clinically in chordoma yet.
Our analysis identified pathways that are likely to be dysregulated in chordoma, such as the NF-κB pathway. For instance, IKK-16 (an inhibitor of NF-κB) was active in most of our samples (Figs. 3 and 5). Other drugs that target NF-κB, such as IMD-0354, have also shown efficacy in chordoma xenografts (23). We identified other pathways likely to be dysregulated and targetable in chordoma, such as the insulin signaling pathway (Fig. 5). Previous studies have found increased expression of insulin-like growth factor 1 receptor (IGF-1R) in chordomas (40). Clinical evidence that this pathway can be targeted in chordoma includes a case report of a chordoma patient treated with the combination of the IGF-1R–inhibiting drug linsitinib and the EGFR inhibitor erlotinib for 18 months, who achieved a partial response (61). Further clinical evidence was seen in a phase 1 clinical trial of the same combinatorial regimen, with one chordoma patient experiencing sustained partial response (62). Our pathway analysis also highlighted the MUC1 pathway, which promotes therapy resistance (Fig. 5) (63, 64). Expression of MUC1 plays a protective role in tumors against immune attacks, and inhibition of MUC1 can overcome the resistance to immune processes (64). The association between MUC1 and chordoma is believed to be mediated by brachyury, as MUC1 was found to be up-regulated in tumors that highly express brachyury (64). Overall, the identification of specific pathways highlights avenues for future combinatorial studies.
Last, our observations from PDOs collectively confirm previous findings in chordoma patients and in vitro models. Yet, PDOs were highly patient specific, with characteristics differing not just between patients but also between samples obtained from the same patient at different time points and sites (Figs. 1 to 5). Furthermore, none of the therapies identified worked on each and every one of the PDO models tested (Fig. 3). This suggests that a personalized approach to the treatment of chordoma would allow for better stratification of patients to efficiently target pathways that are susceptible to therapy in each case. In conclusion, our organoid-based functional sensitivity profile may be used to tailor therapy to each individual, both improving the efficacy of treatment and sparing ineffective therapies and their associated toxicities.
MATERIALS AND METHODS
Tissue procurement and processing
Patients were consented on the University of California Los Angeles Institutional Review Board–approved protocol 18-000980. Samples obtained in the operating room were stored in RPMI 1640 and immediately transferred to the laboratory. Tumors were cut into small, 1- to 3-mm3 fragments and dissociated to single or small cell clusters by adding collagenase IV (200 U/ml) and incubating at 37°C with 5% CO2. Samples were vortexed every 15 min, and cells were collected every 2 hours. After red blood cell lysis, tumor cells were filtered through a 70-μm cell strainer and then counted (33).
High-throughput drug screening: Mini-ring establishment
We followed our established methods to generate organoids amenable to high-throughput drug screening (27, 33). Briefly, a suspension of 5000 cells per well (single cells or small clusters) was plated at the rim of white 96-well white plates (Corning, 3610) in a 3:4 mixture of medium (STEMCELL Technologies, 05620) and Matrigel (BD Biosciences, CB-40324). Plates were incubated at 37°C with 5% CO2 for 30 min to solidify the gel before adding 100 μl of prewarmed MammoCult medium to each well using a liquid handler. Three days after seeding mini-rings, cells were treated by replacing the medium with fresh MammoCult containing the indicated drugs. The same procedure is repeated after 24 hours.
High-throughput drug screening: ATP assay
Twenty-four hours after the last treatment, the medium was removed and wells were washed with 100 μl of prewarmed phosphate-buffered saline (PBS). Organoids were released from Matrigel by incubating at 37°C for 25 min in 50 μl of dispase (5 mg/ml; Life Technologies, 17105-041). Plates were vigorously shaken at 80 rpm for 5 min before adding 75 μl of CellTiter-Glo 3D Reagent (Promega, G968B) to each well. Luminescence was measured after a total 30-min incubation at room temperature (RT) using SpectraMax iD3 (Molecular Devices) over 500 ms of integration time. For each 96-well plate, eight wells are dedicated to staurosporine-positive controls and eight wells for dimethyl sulfoxide (DMSO)–negative controls. The rest of the wells were used to plate the samples with varying drugs. The drugs are tested at single concentration (1 μM) for discovery screening as established by Phan et al. (27) or at 0.1, 1, and 10 μM in duplicates for validation screenings.
Maxi-ring preparation
Suspensions of single cells and small clusters were plated at the rims of 24-well plates (Corning, 3527) in the same 3:4 mixture of medium (STEMCELL Technologies, 05620) and Matrigel (BD Biosciences, CB-40324). For this setup, we plated 100,000 cells per well in a 70-μl mixture per ring as previously reported (27, 33, 65). Plates were incubated at 37°C with 5% CO2 for 30 min to solidify the gel before adding 1 ml of prewarmed MammoCult medium to each well. Medium was fully removed and replaced with fresh prewarmed one after an initial 3-day incubation period. After 5 days of growing, maxi-rings were washed with 1 ml of prewarmed PBS and fixed in 500 μl of 10% buffered formalin (VWR, 89370-094). Organoids were then transferred to a 15-ml Falcon tube the following day, washed twice in PBS followed by the addition of 5 μl of Histogel (Thermo Fisher Scientific, HG-40000-012), and transferred to a cassette. We then proceed with standard embedding, sectioning, and hematoxylin and eosin (H&E) staining.
Immunohistochemistry
For Ki-67 staining, slides were baked at 45°C for 20 min and deparaffinized in xylene followed by washes in ethanol and deionized water. Endogenous peroxidases were blocked with Peroxidazed 1 (Biocare Medical, PX968M) at RT for 5 min. Antigen retrieval was performed in the NxGEN Decloaking Chamber (Biocare Medical) using Diva Decloacker (Biocare Medical, DV2004LX) at 110°C for 15 min for Ki-67/caspase-3 staining. The combo Ki-67/caspase-3 (Biocare Medical, PPM240DSAA) solution is prediluted and was added to the sample for 60 min at RT. Secondary antibody staining was performed with MACH 2 Double Stain 2 (Biocare Medical, MRCT525G). All secondary antibodies were incubated at RT for 30 min. Chromogen development was performed with a Betazoid DAB kit (Biocare Medical, BDB2004). The reaction was quenched by dipping the slides in deionized water. Hematoxylin 1 (Thermo Fisher Scientific, 7221) was used for counterstaining. The slides were mounted with Permount (Thermo Fisher Scientific, SP15-100). For brachyury staining, an anti-brachyury antibody (Abcam, catalog number ab209665) was used at a 1:6000 dilution and incubated for 50 min. The BOND-III fully automated immunohistochemical staining was detected using the BOND Polymer Refine Detection (Leica, DS9800). Images were acquired with the Revolve Upright and Inverted Microscope System (Echo Laboratories).
For pan-cytokeratin, EMA, and S100 staining, primary antibody incubation was performed for 60 min with the following antibodies: S100 (Cell Marque, catalog number 330M 1-400), cytokeratin (Agilent, M3515 1-200), and EMA (Agilent, M0613, 1-2000). Automated detection was performed using the Leica Bond RX processor with steps based on Protocol F using the Bond Polymer Refine Detection Kit (Leica Biosystems, catalog number DS9800). For the S100 antibody, the heat-induced antigen retrieval step was performed at 100°C for 20 min using the ER2 buffer (BOND Epitope Retrieval Solution 2, Leica Biosystems, catalog number AR9640). For the pan-cytokeratin and EMA antibodies, the same step was performed at 100°C for 20 min using the ER1 buffer instead (BOND Epitope Retrieval Solution 1, Leica Biosystems, catalog number AR9961).
Image segmentation
We formulated the image segmentation as a binary, pixel-level classification problem, where each pixel in the image belongs to one of two classes: cell or no cell. For our predictor, we used a U-Net architecture (36), a type of fully convolutional neural network, with a ResNet-34 model as its encoder (66). We initialized the architecture with weights that were pretrained on the ImageNet dataset (67), and fine-tuned their values using a cross-entropy loss function over 80 epochs, on a training dataset composed of seventy-one 512 by 512 pixel patches created from brightfield images of our maxi-rings in 24-well plates (that have a total resolution of 16,896 by 16,896 pixels; 1 pixel = 1 μm). For each sample, we produced four representative whole-well images by examining two maxi-rings in two distinct focal planes. For each image, we used the trained network to infer the organoid segmentation. Our algorithm detected a total number of organoids within a range of 1576 to 18,186 per image, with a median of 5383.
Organoid area calculation
We used the OpenCV’s Python binding (68) to calculate the centroids and the areas of the organoid masks that were detected by U-Net inference. We then summed the areas of all the inferred regions to calculate a total area and tracked this by day for each of the patient samples. Last, we divided the daily area measurements by the number of organoids and normalized to the day 1 measurement to quantify growth/time. We plotted the results using GraphPad Prism.
Cell viability calculation
The luminescence values are normalized to the control DMSO vehicle wells, and Z scores are calculated as (viability of drug X – viability of vehicle)/SD of vehicle. For the dot map visualization and pathway analysis, we used drug viability values at 1 μM. PubChem and the FDA orange book databases were used to determine the FDA approval status of the screened drugs, and results were categorized as either preclinical, in trial, FDA-approved, or terminated in Figs. 3 and 4 and figs. S5 and S6.
Target and pathway analysis
The BioAssay results from the PubChem database for each of the screened drugs were obtained using the PubChem API. To create a list of the most active targets for each drug, we selected only protein targets that are within 10-fold of the second-lowest reported value for Kd (dissociation constant) or IC50 (median inhibitory concentration) in the PubChem database. For drugs that did not have any BioAssay data in PubChem, protein targets were manually curated from the literature.
To perform the pathway analysis, we first generated a matrix of drug and protein target interactions ndrugs × mprotein with values of 0 or 1, with 1 indicating the presence of an interaction between a drug and a certain protein target, and 0 indicating its absence. We multiplied each row by a weight proportional to the mean viability of the organoids treated by each drug [1 − (mean viability/100)]. We then multiplied this matrix by a vector of 1s to obtain a row-wise summation of the protein target viability values. Subsequently, we normalized the row vector by dividing each element by the sum of non-zero column entries in the ndrugs × mprotein matrix. For example, if the total number of drugs that targets a given protein is five, we divide the elements of the matrix corresponding to that protein by five. This analysis produced a row-wise vector of weighed targets causing organoid viability changes.
We then mapped this list of proteins to the canonical pathways defined by the WikiPathways Database [version 20210210 (47)]. We used a subset of pathways that excluded those not biologically pertinent to cancer, such pathways related to microorganisms and pathogens, as well as the newly added coronavirus disease (COVID)–related pathways. To create this map, we populated a new npathway × mprotein matrix, with 1s indicating the presence of a protein in a particular pathway or 0s indicating its absence. We then normalized the rows in the npathway × mprotein matrix to account for the differences in the number of proteins included in each pathway. To obtain the relative effect that targeting a specific pathway has on the viability of each chordoma organoid case, we multiplied the npathway × mprotein mapping matrix by the normalized ndrugs × mprotein vector. The resulting vector represents the relative impact that targeting a given pathway has on the viability of the organoids.
Dot map generation
We used R and the tidyverse package to plot the viability data or pathway data for each case and the arrays of drugs tested. We clustered the drugs (Figs. 3 and 4 and fig. S5 and S6) and pathways (Fig. 5) by the number of overlapping elements. This was done using the Jaccard distance function in R. Cases that had similar responses to a given drug or similar affected pathways were clustered together using the distance function, as parameterized by a continuous Euclidean distance. The visualizations for all dot maps were produced using the ggplot2 package and finalized with Adobe Illustrator.
Acknowledgments
We thank P. Krawczuk for guidance in selecting the appropriate neural network for the image segmentation of the organoids, the Translational Pathology Core Laboratory at UCLA for assistance in performing some of the immunohistochemistry studies, and R. Damoiseaux and the UCLA MSSR Core for providing part of the drug libraries included in this study. We also thank the Neuropathology Laboratory at UCLA and J. Yi for technical assistance with the brachyury staining.
Funding: This project was supported by the NIH (R01CA244729 to A.S.) and a seed grant from UCLA DGSOM (to A.S., N.F., and J.Y.).
Author contributions: A.S. designed and supervised the project; F.H., N.F., N.B., and J.Y. consented the patients and obtained the surgical specimens. S.D.N. performed all pathology assessments; H.T.L.N., N.T., and A.S. performed organoid establishment, characterization, and screening experiments; A.A.S., H.T.L.N., and N.T. performed immunohistochemistry experiments; A.D. and A.A.S. created the database of samples and drugs, performed the clustering analysis and pathway analysis, and generated visualizations; A.D. trained the neural net and performed the image segmentation experiment; and A.A.S., A.D., and A.S. wrote the paper with contributions from all authors.
Competing interests: A.S., J.Y., and N.F. are founders and owners of Icona BioDx. The authors declare no other competing interests.
Data availability and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Supplementary Materials
This PDF file includes:
Supplementary Text
Figs. S1 to S6
Tables S1 and S2
Other Supplementary Material for this manuscript includes the following:
Movie S1
REFERENCES AND NOTES
- 1.Karpathiou G., Dumollard J. M., Dridi M., Dal Col P., Barral F.-G., Boutonnat J., Peoc’h M., Chordomas: A review with emphasis on their pathophysiology, pathology, molecular biology, and genetics. Pathol. Res. Pract. 216, 153089 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Shih A. R., Cote G. M., Chebib I., Choy E., DeLaney T., Deshpande V., Hornicek F. J., Miao R., Schwab J. H., Nielsen G. P., Chen Y.-L., Clinicopathologic characteristics of poorly differentiated chordoma. Mod. Pathol. 31, 1237–1245 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Hasselblatt M., Thomas C., Hovestadt V., Schrimpf D., Johann P., Bens S., Oyen F., Peetz-Dienhart S., Crede Y., Wefers A., Vogel H., Riemenschneider M. J., Antonelli M., Giangaspero F., Bernardo M. C., Giannini C., Din N. U., Perry A., Keyvani K., van Landeghem F., Sumerauer D., Hauser P., Capper D., Korshunov A., Jones D. T. W., Pfister S. M., Schneppenheim R., Siebert R., Frühwald M. C., Kool M., Poorly differentiated chordoma with SMARCB1/INI1 loss: A distinct molecular entity with dismal prognosis. Acta Neuropathol. 132, 149–151 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Choi J. H., Ro J. Y., The 2020 WHO classification of tumors of bone: An updated review. Adv. Anat. Pathol. 28, 119–138 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Hung Y. P., Diaz-Perez J. A., Cote G. M., Wejde J., Schwab J. H., Nardi V., Chebib I. A., Deshpande V., Selig M. K., Bredella M. A., Rosenberg A. E., Nielsen G. P., Dedifferentiated chordoma: Clinicopathologic and molecular characteristics with integrative analysis. Am. J. Surg. Pathol. 44, 1213–1223 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Stacchiotti S., Sommer J.; Chordoma Global Consensus Group , Building a global consensus approach to chordoma: A position paper from the medical and patient community. Lancet Oncol. 16, e71–e83 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Walcott B. P., Nahed B. V., Mohyeldin A., Coumans J.-V., Kahle K. T., Ferreira M. J., Chordoma: Current concepts, management, and future directions. Lancet Oncol. 13, e69–e76 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Hanna S. A., Tirabosco R., Amin A., Pollock R. C., Skinner J. A., Cannon S. R., Saifuddin A., Briggs T. W. R., Dedifferentiated chordoma. J. Bone Joint Surg. 90-B, 652–656 (2008). [DOI] [PubMed] [Google Scholar]
- 9.NCCN Clinical Practice Guidelines in Oncology (2021); https://nccn.org/professionals/physician_gls/default.aspx.
- 10.Dhall G., Traverso M., Finlay J. L., Shane L., Gonzalez-Gomez I., Jubran R., The role of chemotherapy in pediatric clival chordomas. J. Neuro-Oncol. 103, 657–662 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Tarpey P. S., Behjati S., Young M. D., Martincorena I., Alexandrov L. B., Farndon S. J., Guzzo C., Hardy C., Latimer C., Butler A. P., Teague J. W., Shlien A., Futreal P. A., Shah S., Bashashati A., Jamshidi F., Nielsen T. O., Huntsman D., Baumhoer D., Brandner S., Wunder J., Dickson B., Cogswell P., Sommer J., Phillips J. J., Amary M. F., Tirabosco R., Pillay N., Yip S., Stratton M. R., Flanagan A. M., Campbell P. J., The driver landscape of sporadic chordoma. Nat. Commun. 8, 890 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miettinen M., Wang Z., Lasota J., Heery C., Schlom J., Palena C., Nuclear brachyury expression is consistent in chordoma, common in germ cell tumors and small cell carcinomas, and rare in other carcinomas and sarcomas: An immunohistochemical study of 5229 cases. Am. J. Surg. Pathol. 39, 1305–1312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchs B., Dickey I. D., Yaszemski M. J., Inwards C. Y., Sim F. H., Operative management of sacral chordoma. J. Bone Joint Surg. Am. 87, 2211–2216 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Stacchiotti S., Longhi A., Ferraresi V., Grignani G., Comandone A., Stupp R., Bertuzzi A., Tamborini E., Pilotti S., Messina A., Spreafico C., Gronchi A., Amore P., Vinaccia V., Casali P. G., Phase II study of imatinib in advanced chordoma. J. Clin. Oncol. 30, 914–920 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Bompas E., Le Cesne A., Tresch-Bruneel E., Lebellec L., Laurence V., Collard O., Saada-Bouzid E., Isambert N., Blay J. Y., Amela E. Y., Salas S., Chevreau C., Bertucci F., Italiano A., Clisant S., Penel N., Sorafenib in patients with locally advanced and metastatic chordomas: A phase II trial of the French Sarcoma Group (GSF/GETO). Ann. Oncol. 26, 2168–2173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuetze S. M., Bolejack V., Choy E., Ganjoo K. N., Staddon A. P., Chow W. A., Tawbi H. A., Samuels B. L., Patel S. R., von Mehren M., D’Amato G., Leu K. M., Loeb D. M., Forscher C. A., Milhem M. M., Rushing D. A., Lucas D. R., Chugh R., Reinke D. K., Baker L. H., Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer 123, 90–97 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Yang C., Hornicek F. J., Wood K. B., Schwab J. H., Choy E., Iafrate J., Rosenberg A., Nielsen G. P., Xavier R. J., Mankin H., Duan Z., Characterization and analysis of human chordoma cell lines. Spine 35, 1257–1264 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosotti R., Magnaghi P., Di Bella S., Cozzi L., Cusi C., Bozzi F., Beltrami N., Carapezza G., Ballinari D., Amboldi N., Lupi R., Somaschini A., Raddrizzani L., Salom B., Galvani A., Stacchiotti S., Tamborini E., Isacchi A., Establishment and genomic characterization of the new chordoma cell line Chor-IN-1. Sci. Rep. 7, 9226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ricci-Vitiani L., Runci D., D’Alessandris Q. G., Cenci T., Martini M., Bianchi F., Maira G., Stancato L., De Maria R., Larocca L. M., Pallini R., Chemotherapy of skull base chordoma tailored on responsiveness of patient-derived tumor cells to rapamycin. Neoplasia 15, 773–782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ben-David U., Siranosian B., Ha G., Tang H., Oren Y., Hinohara K., Strathdee C. A., Dempster J., Lyons N. J., Burns R., Nag A., Kugener G., Cimini B., Tsvetkov P., Maruvka Y. E., O’Rourke R., Garrity A., Tubelli A. A., Bandopadhayay P., Tsherniak A., Vazquez F., Wong B., Birger C., Ghandi M., Thorner A. R., Bittker J. A., Meyerson M., Getz G., Beroukhim R., Golub T. R., Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz R. J., Luck A., Bondoc A., Golbourn B., Picard D., Remke M., Loukides J., Sabha N., Smith C., Cusimano M. D., Rutka J. T., Characterization of a clival chordoma xenograft model reveals tumor genomic instability. Am. J. Pathol. 188, 2902–2911 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Davies J. M., Robinson A. E., Cowdrey C., Mummaneni P. V., Ducker G. S., Shokat K. M., Bollen A., Hann B., Phillips J. J., Generation of a patient-derived chordoma xenograft and characterization of the phosphoproteome in a recurrent chordoma. J. Neurosurg. 120, 331–336 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trucco M. M., Awad O., Wilky B. A., Goldstein S. D., Huang R., Walker R. L., Shah P., Katuri V., Gul N., Zhu Y. J., McCarthy E. F., Paz-Priel I., Meltzer P. S., Austin C. P., Xia M., Loeb D. M., A novel chordoma xenograft allows in vivo drug testing and reveals the importance of NF-κB signaling in chordoma biology. PLOS ONE 8, e79950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bozzi F., Manenti G., Conca E., Stacchiotti S., Messina A., Dagrada G., Gronchi A., Panizza P., Pierotti M. A., Tamborini E., Pilotti S., Development of transplantable human chordoma xenograft for preclinical assessment of novel therapeutic strategies. Neuro Oncol. 16, 72–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siu I.-M., Salmasi V., Orr B. A., Zhao Q., Binder Z. A., Tran C., Ishii M., Riggins G. J., Hann C. L., Gallia G. L., Establishment and characterization of a primary human chordoma xenograft model. J. Neurosurg. 116, 801–809 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barker H. E., Scott C. L., Preclinical rare cancer research to inform clinical trial design. Nat. Rev. Cancer 19, 481–482 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Phan N., Hong J. J., Tofig B., Mapua M., Elashoff D., Moatamed N. A., Huang J., Memarzadeh S., Damoiseaux R., Soragni A., A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun. Biol. 2, 78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Witte C. J., Valle-Inclan J. E., Hami N., Lõhmussaar K., Kopper O., Vreuls C. P. H., Jonges G. N., van Diest P., Nguyen L., Clevers H., Kloosterman W. P., Cuppen E., Snippert H. J. G., Zweemer R. P., Witteveen P. O., Stelloo E., Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter- and intrapatient drug responses. Cell Rep. 31, 107762 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Letai A., Functional precision cancer medicine-moving beyond pure genomics. Nat. Med. 23, 1028–1035 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernández-Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R., Rata M., Koh D.-M., Tunariu N., Collins D., Hulkki-Wilson S., Ragulan C., Spiteri I., Moorcraft S. Y., Chau I., Rao S., Watkins D., Fotiadis N., Bali M., Darvish-Damavandi M., Lote H., Eltahir Z., Smyth E. C., Begum R., Clarke P. A., Hahne J. C., Dowsett M., de Bono J., Workman P., Sadanandam A., Fassan M., Sansom O. J., Eccles S., Starling N., Braconi C., Sottoriva A., Robinson S. P., Cunningham D., Valeri N., Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saltsman J. A., Hammond W. J., Narayan N. J. C., Requena D., Gehart H., Lalazar G., LaQuaglia M. P., Clevers H., Simon S., A human organoid model of aggressive hepatoblastoma for disease modeling and drug testing. Cancer 12, 2668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Driehuis E., van Hoeck A., Moore K., Kolders S., Francies H. E., Gulersonmez M. C., Stigter E. C. A., Burgering B., Geurts V., Gracanin A., Bounova G., Morsink F. H., Vries R., Boj S., van Es J., Offerhaus G. J. A., Kranenburg O., Garnett M. J., Wessels L., Cuppen E., Brosens L. A. A., Clevers H., Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. U.S.A. 116, 26580–26590 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen H. T. L., Soragni A., Patient-derived tumor organoid rings for histologic characterization and high-throughput screening. STAR Protoc. 1, 100056 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.A. Wellstein, G. Giaccone, M. B. Atkins, E. A. Sausville, in Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13e (McGraw-Hill Education, 2017). [Google Scholar]
- 35.E. M. Gordon, “BLESSED: Expanded access for DeltaRex-G for advanced pancreatic cancer and sarcoma” (clinical trial registration NCT04091295, clinicaltrials.gov, 2020). [DOI] [PMC free article] [PubMed]
- 36.O. Ronneberger, P. Fischer, T. Brox, U-Net: Convolutional networks for biomedical image segmentation. arXiv:1505.04597 [cs.CV] (18 May 2015).
- 37.Horbinski C., Oakley G. J., Cieply K., Mantha G. S., Nikiforova M. N., Dacic S., Seethala R. R., The prognostic value of Ki-67, p53, epidermal growth factor receptor, 1p36, 9p21, 10q23, and 17p13 in skull base chordomas. Arch. Pathol. Lab. Med. 134, 1170–1176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Hara B. J., Paetau A., Miettinen M., Keratin subsets and monoclonal antibody HBME-1 in chordoma: Immunohistochemical differential diagnosis between tumors simulating chordoma. Hum. Pathol. 29, 119–126 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Xia M., Huang R., Sakamuru S., Alcorta D., Cho M.-H., Lee D.-H., Park D. M., Kelley M. J., Sommer J., Austin C. P., Identification of repurposed small molecule drugs for chordoma therapy. Cancer Biol. Ther. 14, 638–647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheipl S., Barnard M., Cottone L., Jorgensen M., Drewry D. H., Zuercher W. J., Turlais F., Ye H., Leite A. P., Smith J. A., Leithner A., Möller P., Brüderlein S., Guppy N., Amary F., Tirabosco R., Strauss S. J., Pillay N., Flanagan A. M., EGFR inhibitors identified as a potential treatment for chordoma in a focused compound screen. J. Pathol. 239, 320–334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neal M. E. H., Michmerhuizen N. L., Kovatch K. J., Owen J. H. J., Zhai J., Jiang H., McKean E. L., Prince M. E. P., Brenner J. C., Advancement of PI3 kinase inhibitor combination therapies for PI3K-aberrant chordoma. J. Neurol. Surg. Part B Skull Base , (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B. A., Thiessen P. A., Yu B., Zaslavsky L., Zhang J., Bolton E. E., PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 49, D1388–D1395 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colia V., Stacchiotti S., Medical treatment of advanced chordomas. Eur. J. Cancer 83, 220–228 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Yang C., Schwab J. H., Schoenfeld A. J., Hornicek F. J., Wood K. B., Nielsen G. P., Choy E., Mankin H., Duan Z., A novel target for treatment of chordoma: Signal transducers and activators of transcription 3. Mol. Cancer Ther. 8, 2597–2605 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Wang A. C., Owen J. H., Abuzeid W. M., Hervey-Jumper S. L., He X., Gurrea M., Lin M., Altshuler D. B., Keep R. F., Prince M. E., Carey T. E., Fan X., McKean E. L., Sullivan S. E., STAT3 inhibition as a therapeutic strategy for chordoma. J. Neurol. Surg. Part B Skull Base. 77, 510–520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biermann J. S., Chow W., Reed D. R., Lucas D., Adkins D. R., Agulnik M., Benjamin R. S., Brigman B., Budd G. T., Curry W. T., Didwania A., Fabbri N., Hornicek F. J., Kuechle J. B., Lindskog D., Mayerson J., McGarry S. V., Million L., Morris C. D., Movva S., O’Donnell R. J., Randall R. L., Rose P., Santana V. M., Satcher R. L., Schwartz H., Siegel H. J., Thornton K., Villalobos V., Bergman M. A., Scavone J. L., NCCN Guidelines Insights: Bone Cancer, Version 2.2017. J. Natl. Compr. Canc. Netw. 15, 155–167 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Martens M., Ammar A., Riutta A., Waagmeester A., Slenter D. N., Hanspers K., Miller R. A., Digles D., Lopes E. N., Ehrhart F., Dupuis L. J., Winckers L. A., Coort S. L., Willighagen E. L., Evelo C. T., Pico A. R., Kutmon M., WikiPathways: Connecting communities. Nucleic Acids Res. 49, D613–D621 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heery C. R., Chordoma: The quest for better treatment options. Oncol. Ther. 4, 35–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honkala A., Malhotra S. V., Kummar S., Junttila M. R., Harnessing the predictive power of preclinical models for oncology drug development. Nat. Rev. Drug Discov. 1–16 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Siu I.-M., Ruzevick J., Zhao Q., Connis N., Jiao Y., Bettegowda C., Xia X., Burger P. C., Hann C. L., Gallia G. L., Erlotinib inhibits growth of a patient-derived chordoma xenograft. PLOS ONE 8, e78895 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burger A., Vasilyev A., Tomar R., Selig M. K., Nielsen G. P., Peterson R. T., Drummond I. A., Haber D. A., A zebrafish model of chordoma initiated by notochord-driven expression of HRASV12. Dis. Model. Mech. 7, 907–913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dolly S. O., Wagner A. J., Bendell J. C., Kindler H. L., Krug L. M., Seiwert T. Y., Zauderer M. G., Lolkema M. P., Apt D., Yeh R.-F., Fredrickson J. O., Spoerke J. M., Koeppen H., Ware J. A., Lauchle J. O., Burris H. A., de Bono J. S., Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 22, 2874–2884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng T., Jin J., Jiang C., Huang R., Yin H., Song D., Cheng L., Molecular targeted therapy in the treatment of chordoma: A systematic review. Front. Oncol. 9, 30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderson E., Havener T. M., Zorn K. M., Foil D. H., Lane T. R., Capuzzi S. J., Morris D., Hickey A. J., Drewry D. H., Ekins S., Synergistic drug combinations and machine learning for drug repurposing in chordoma. Sci. Rep. 10, 12982 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dewaele B., Maggiani F., Floris G., Ampe M., Vanspauwen V., Wozniak A., Debiec-Rychter M., Sciot R., Frequent activation of EGFR in advanced chordomas. Clin. Sarcoma Res. 1, 4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stacchiotti S., Tamborini E., Vullo S. L., Bozzi F., Messina A., Morosi C., Casale A., Crippa F., Conca E., Negri T., Palassini E., Marrari A., Palmerini E., Mariani L., Gronchi A., Pilotti S., Casali P. G., Phase II study on lapatinib in advanced EGFR-positive chordoma†. Ann. Oncol. 24, 1931–1936 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Hof H., Welzel T., Debus J., Effectiveness of cetuximab/gefitinib in the therapy of a sacral chordoma. Oncol. Res. Treat. 29, 572–574 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Lindèn O., Stenberg L., Kjellén E., Regression of cervical spinal cord compression in a patient with chordoma following treatment with cetuximab and gefitinib. Acta Oncol. 48, 158–159 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Diaz R. J., Cusimano M. D., The biological basis for modern treatment of chordoma. J. Neuro-Oncol. 104, 411–422 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Yang C., Hornicek F. J., Wood K. B., Schwab J. H., Choy E., Mankin H., Duan Z., Blockage of Stat3 With CDDO-me inhibits tumor cell growth in chordoma. Spine 35, 1668–1675 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Aleksic T., Browning L., Woodward M., Phillips R., Page S., Henderson S., Athanasou N., Ansorge O., Whitwell D., Pratap S., Hassan A. B., Middleton M. R., Macaulay V. M., Durable response of spinal chordoma to combined inhibition of IGF-1R and EGFR. Front. Oncol. 6, 98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Macaulay V. M., Middleton M. R., Eckhardt S. G., Rudin C. M., Juergens R. A., Gedrich R., Gogov S., McCarthy S., Poondru S., Stephens A. W., Gadgeel S. M., Phase I dose-escalation study of linsitinib (OSI-906) and erlotinib in patients with advanced solid tumors. Clin. Cancer Res. 22, 2897–2907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nibu Y., José-Edwards D. S., Di Gregorio A., From notochord formation to hereditary chordoma: The many roles of brachyury. Biomed. Res. Int. 2013, e826435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.David J. M., Hamilton D. H., Palena C., MUC1 upregulation promotes immune resistance in tumor cells undergoing brachyury-mediated epithelial-mesenchymal transition. OncoImmunology 5, e1117738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soragni A., Janzen D. M., Johnson L. M., Lindgren A. G., Thai-Quynh Nguyen A., Tiourin E., Soriaga A. B., Lu J., Jiang L., Faull K. F., Pellegrini M., Memarzadeh S., Eisenberg D. S., A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 29, 90–103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.K. He, X. Zhang, S. Ren, J. Sun, in 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) (IEEE, 2016), pp. 770–778. [Google Scholar]
- 67.J. Deng, W. Dong, R. Socher, L.-J. Li, Kai Li, Li Fei-Fei, in 2009 IEEE Conference on Computer Vision and Pattern Recognition (IEEE, 2009), pp. 248–255. [Google Scholar]
- 68.Bradski G., The OpenCV Library. J-DDJ 25, 122–125 (2000). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text
Figs. S1 to S6
Tables S1 and S2
Movie S1