

Summary
The MYC oncogene has been studied for decades, yet there is still intense debate over how this transcription factor controls gene expression. Here, we seek to answer these questions with an in vivo readout of discrete events of gene expression in single cells. We engineered an optogenetic variant of MYC (Pi-MYC) and combined this tool with single-molecule RNA and protein imaging techniques to investigate the role of MYC in modulating transcriptional bursting and transcription factor binding dynamics in human cells. We find that the immediate consequence of MYC overexpression is to increase the duration rather than frequency of bursts, a functional role which is different from the majority of human transcription factors. We further propose that the mechanism by which MYC exerts global effects on the active period of genes is by altering the binding dynamics of transcription factors involved in RNA Polymerase II complex assembly and productive elongation.
Graphical Abstract
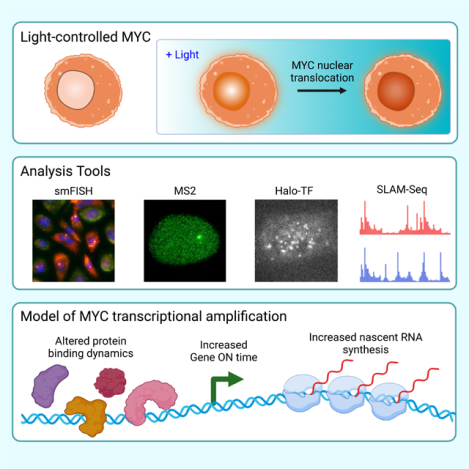
In Brief
Patange et al. engineered a light-inducible form of the MYC oncogene to directly measure how it modulates transcription in living human cells. Their single-molecule studies show direct evidence of MYC functioning as a global amplifier of transcription, across genes and cell types, by increasing the active period of genes.
Introduction
c-MYC (MYC) is an oncogenic transcription factor that is overexpressed in the majority of malignancies (Vita and Henriksson, 2006) and associated with aggressive tumor progression and poor patient survival (Spencer and Groudine, 1991). MYC is widely present in normal proliferating somatic cells and responds to mitogens and growth factors to regulate growth and general cell proliferation. MYC expression is deregulated and elevated in cancer cells and has been shown to be a driver of cancer in mouse models (Adams et al., 1985). The primary mode of MYC deregulation in cancer is overexpression of the normal protein product, and multiple reports indicate that high MYC levels cause a global increase in total RNA levels (Grandori et al., 2005; Hsu et al., 2015; Lin et al., 2012; Nie et al., 2012). As such, MYC has direct interactions with hundreds of proteins involved in a diverse range of functional classes such as RNA processing, ribosome biogenesis, transcription regulation, mitosis, DNA damage and replication, histone modification, and mRNA post-transcriptional modifications (Kalkat et al., 2018).
Given its myriad activities, a unified understanding of MYC function has been elusive. In recent years, several groups have put forth a new hypothesis that describes MYC as a global amplifier of gene expression. Genome-wide ChIP-seq analysis from two groups suggests that MYC is capable of binding all promoters but does so according to their expression output (Lin et al., 2012; Nie et al., 2012). The main findings of the Lin study have since been replicated (Lewis et al., 2018). A further study on MYC occupancy at genes found the effects were limited according to the basal expression level of the gene: promoters already saturated with MYC binding did not increase expression even when MYC expression increases (Lorenzin et al., 2016). A recent study points to the promotion of RNA polymerase II (RNAPII) pause release through direct interactions with SPT5 as the mechanism of MYC amplification (Baluapuri et al., 2019). A contrasting view is that MYC functions as a gene-specific transcription factor, with ubiquitous binding due to indirect effects of MYC binding enhancer regions of non-target genes when it is expressed at high levels (Kress et al., 2015; Sabò et al., 2014). Thus, the scope of the amplifier model remains controversial in part because of direct and indirect consequences on gene expression. Moreover, most studies of MYC consist of cell population measurements obtained at single time-points. A high-temporal, high-spatial precision viewpoint of how MYC acts in single living cells does not exist.
Here, we developed a single-cell assay for precisely controlling MYC and interrogating the effects on transcription in living cells. We engineered an optogenetic variant of MYC that is biologically active, can be visualized under the microscope, and can be controlled with blue light. We used this photo-inducible MYC (Pi-MYC) along with RNA visualization in fixed and living cells to undertake a broad survey of genes that differed with respect to function, DNA features, basal expression level, and cellular context. We found that MYC overexpression consistently increased the duration of active periods of genes population-wide and exerted its greatest effects on genes with low basal expression. These findings provide living, single-cell evidence of MYC as an amplifier of gene expression. Ensemble studies of nascent transcription using SLAM-seq (Herzog et al., 2017) reinforce the pervasiveness of this behavior. Using single-molecule tracking (SMT) experiments to measure the kinetics of MYC, glucocorticoid receptor (GR), TATA-box binding protein (TBP), a subunit of the Mediator complex (MED1), a component of the DSIF complex (SPT5), and the large subunit of RNAPII (POLR2A), we found that MYC globally affects the binding dynamics of core transcription factors involved in RNAPII complex assembly and productive elongation. Our data suggests that MYC increases the period over which productive initiation occurs through multi-faceted interactions with the core transcription machinery. In summary, our single-cell, real-time approach reveals that MYC amplifies gene expression across genes and cell types through direct modulation of the transcriptional active period.
Results
MYC increases the active transcription period of the estrogen-responsive gene TFF1 in breast cancer cells.
Our strategy to dissect the direct effect of MYC on human genes was to use multiple methods for changing MYC levels (transient transfection, induction of stably-integrated genes, siRNA knockdown, and the photo-inducible MYC described later) followed by multiple methods of transcriptional readout (RT-qPCR, nascent RNA counting by smFISH, live-cell-imaging of RNA synthesis by MS2, and nascent RNA sequencing by SLAM-seq). The first candidate gene we chose was TFF1, an estrogen-responsive gene that is upregulated in breast cancer, has one canonical E-box motif in the promoter and another in an enhancer located in TMPRSS3, and is regulated by MYC (Figure 1A) (Al-azzeh et al., 2002). TFF1 RNA is transcribed in bursts, with an average ON time of 16.0 ± 0.5 min and an average OFF time of 66 ± 7 min when fully induced with estradiol (E2) (Rodriguez et al., 2019). The EC50 of TFF1 when induced with E2 is 50pM (May and Westley, 1988), and increasing E2 increases the frequency of TFF1 bursts but has no effect on the duration of bursts.
Figure 1.
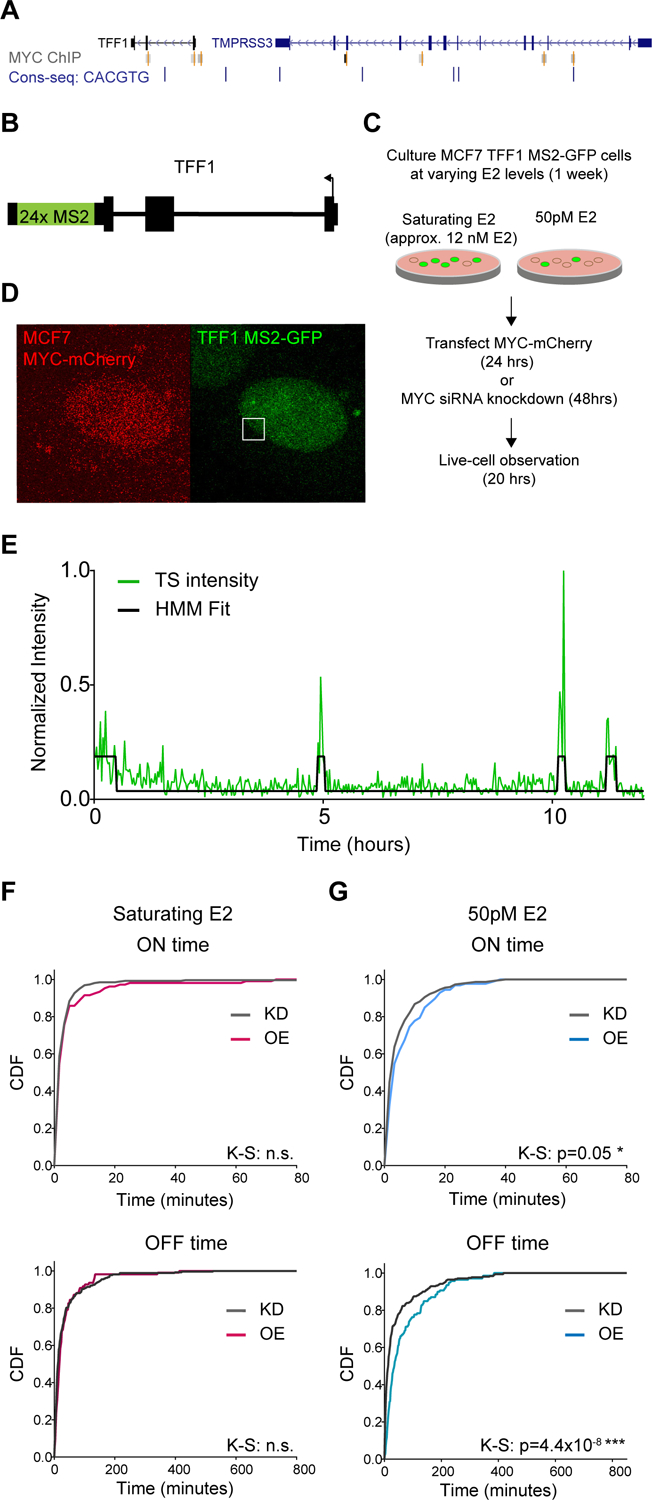
The endogenous gene TFF1 is responsive to MYC perturbations at non-saturating induction levels.
(A) Genome browser screenshot of MYC ChIP-seq peaks (gray) and the E-box consensus motif (blue) at the loci of TFF1 and its enhancer in TMPRSS3.
(B) Schematic of the MS2 insertion in the TFF1 endogenous locus.
(C) Schematic of live-cell experiment with MYC perturbation.
(D) Example frame of a live-cell timeseries of MYC-mCherry overexpression in MCF7 cells. Frames are a maximum projection of a z-stack. TFF1 transcription site indicated by a white square.
(E) Example trace of TFF1 transcription site activity (green line) fit to a 2-state HMM (black line) to categorize gene activity into ‘ON’ and ‘OFF’ periods.
(F) Normalized CDF of TFF1 ON and OFF times with MYC knockdown (KD) or overexpression (OE). Cells were culture at saturating E2 (KD=15 cells, OE=8 cells).
(G) As in (F) for 50pM E2. Cells per condition: KD=22 cells, OE=13 cells.
We evaluated the effect of MYC on TFF1 expression by measuring both total and nascent RNA. First, we overexpressed MYC over a 2-week time course using an inducible MYC-EGFP overexpression system. We observed no detectable changes in TFF1 expression level as measured by RT-qPCR (Figure S1A), agreeing with previous results obtained by a luciferase assay (Al-azzeh et al., 2002). Next, we measured pre-mRNA synthesis of TFF1 at the transcription site (TS) of the gene locus. We used a previously created MCF7 cell line where TFF1 contains 24x repeat of MS2 loops in the 3’UTR, thus enabling live-cell imaging of transcription ((Rodriguez et al., 2019), Figure 1B). MCF7 is polyploid for TFF1, and the MS2 insertion was introduced into 3 of 5 alleles. We subjected MCF7 TFF1-MS2 cells to either 1 week of growth in saturating E2 (~12nM E2) or hormone depletion followed by induction with 50pM E2, followed by MYC plasmid overexpression or MYC siRNA knockdown (Figure 1C, Figure S1B). Cells were imaged for ~14 hours and the TFF1 TS were tracked over that duration (Figure 1D). The TS intensity is a direct readout of the nascent RNA produced at the TFF1 gene locus, and we fit the normalized time traces of TFF1 fluorescence intensity to a two-state Hidden Markov Model to threshold the ON and OFF periods of the gene (Figure 1E). We found no effects on transcription ON and OFF time at saturating E2 conditions (Figure 1F) but did find that MYC affected TFF1 transcription events at 50pM E2, the EC50 for the gene (Figure 1G). We observed a significant increase in the mean ON time from 5.6 ± 0.4 to 7.1 ± 0.7 min, as well as an increase in the OFF time from 44.2 ± 4.3 to 70.2 ± 7.0 min. In summary, despite not seeing changes in total cellular TFF1 mRNA, we observed that MYC increased the duration of TFF1 transcription events, but also decreased the frequency of the events occurring. Yet, this effect was only observed under non-saturating levels of expression.
We also tested our findings for the TFF1 gene transcribing under non-saturating conditions (50pM E2) with an orthogonal method of MYC overexpression based on a MYC-EGFP transgene under a Tet-inducible promoter in MCF7 cells (Porter et al., 2017) (Figure 2A). Induction with doxycycline leads to an increase in both protein and MYC RNA levels over 48 hours. (Figure 2 B–E). Using an smFISH probe set to the intron of TFF1 to identify nascent unspliced pre-mRNA at the TS (Figure 2F,G), we found that TS intensity increased slightly from 0–24 hours of a 48-hour time course (Figure 2H), whereas the fraction of active TS per cell peaked at 6 hours and then decreased below initial levels (Figure 2I). Taken together, two different methods of MYC over-expression and two different methods of transcriptional readout indicate MYC-dependent changes in TFF1 transcription that were not visible at the total mRNA level.
Figure 2.
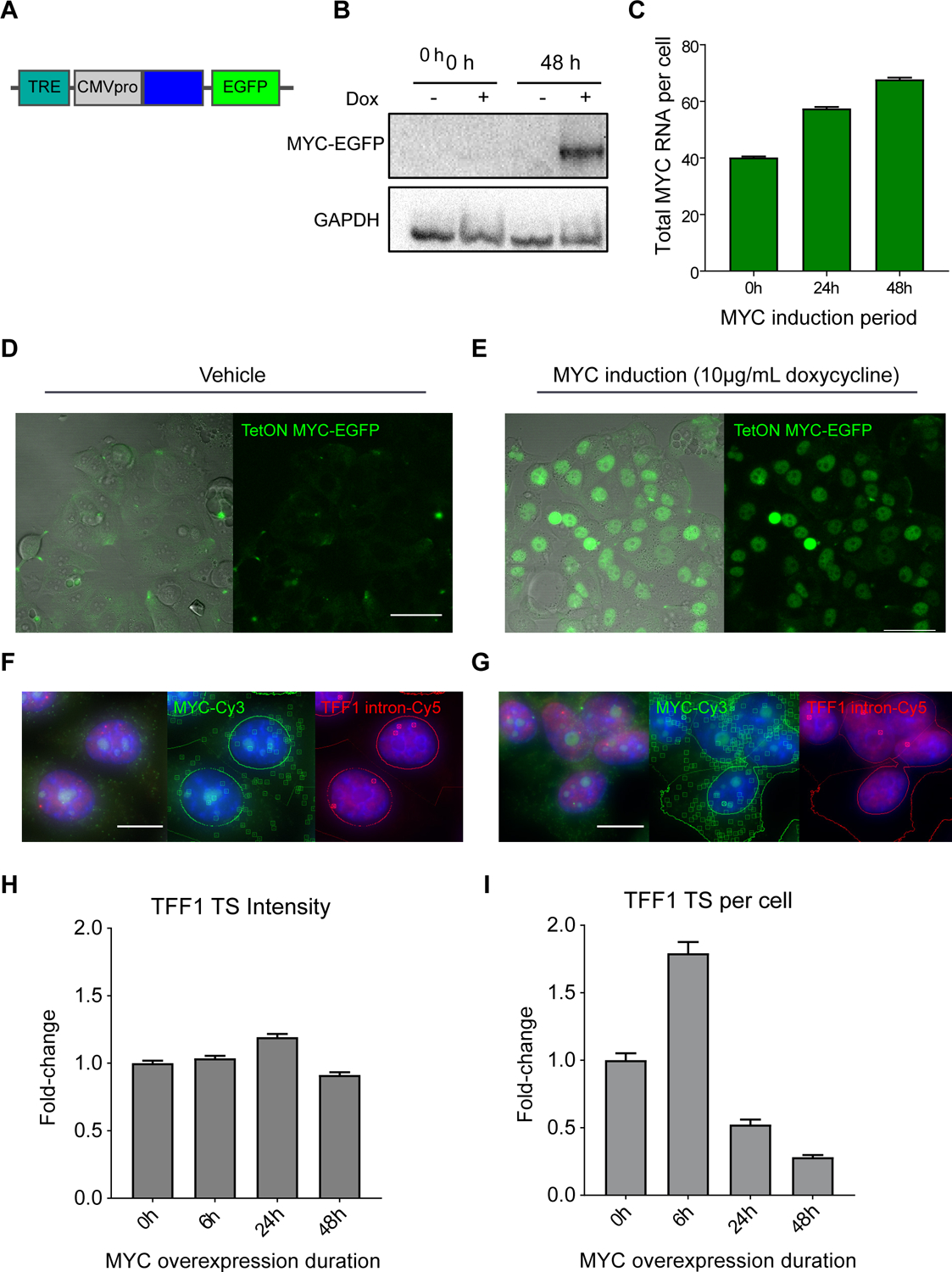
MYC overexpression increases number of nascent RNA at active TFF1 transcription sites.
(A) Schematic of TetON MYC-EGFP transgene, containing a doxycycline inducible Tet-responsive element and a CMV promoter driving expression of MYC-EGFP.
(B) MYC western blot of MCF7 TetON MYC-EGFP expression at 0 and 48 hours of vehicle or doxycycline induction.
(C) smFISH quantification of MYC RNA/cell with 0hrs (40.2 ± 0.4 RNA/cell, n=3448 cells), 24hrs (n=7890 cells), and 48hrs (67.7 ± 0.7 RNA/cell, n=6599 cells) of TetON MYC-EGFP induction with 10μg/mL doxycycline. Bars represent mean ± SEM.
(D) DIC-merged and fluorescence image of TetON MYC-EGFP expression induced with vehicle. Scale bar = 50μm.
(E) DIC-merged and fluorescence image of TetON MYC-EGFP expression induced with doxycycline. Scale as in (D).
(F) smFISH image of TetON MYC-EGFP expression induced with vehicle (blue=DAPI). RNA transcripts and transcription sites are identified with squares and circles, respectively. Scale bar = 12μm.
(G) smFISH image of TetON MYC-EGFP expression with doxycycline induction. RNA identification and scale as in (F).
(H) smFISH quantification of fold-change in fluorescence intensity (dox/vehicle, normalized to 0h) of TFF1 TS over a 48-hour time course of MYC-EGFP overexpression. Approximately 2000–6000 cells were imaged per condition.
(I) smFISH quantification of fold-change in TFF1 TS per cell (dox/vehicle, normalized to 0h) over a 48-hour time course of MYC-EGFP overexpression.
We verified these findings for another gene in a different transformed cell line. We used a previously developed MS2-PP7 β-globin reporter system expressed in U2OS human osteosarcoma cells (Coulon et al., 2014) (Figure S2). This stable expression system consists of a small tandem array (4 – 7 copies) integrated into chromosome 8. The expression level of the β-globin reporter is tunable by doxycycline and driven by a CMV promoter (Figure S2A). Unlike TFF1, the β-globin reporter is exogenously expressed, present as a multi-copy array, and does not contain any cis-acting elements designed to be MYC-responsive. Interestingly, we saw that MYC overexpression over a 24-hour period (Figure S2B) increased the RNA abundance of the β-globin reporter above the levels achieved by doxycycline (Figure S2C) and increased the mean ON time from 32.1 ± 2.0 to 41.4 ± 5.3 min (Figure S2D). We observed a slight increase in the mean OFF time (70.8 ± 5.5 to 79.3 ± 9.9 min), but this effect was not statistically significant. Thus, even a transgene lacking E-box sequences in the promoter responds to MYC over-expression over long timescales. In summary, single-cell imaging of two genes (TFF1 and a β-globin reporter) in two transformed cell lines (MCF7 and U2OS) shows that MYC increases burst duration.
MYC exerts changes in gene expression in a non-transformed human bronchial epithelial cell line.
We proceeded to expand our single-cell, single-gene survey of MYC transcriptional effects to genes that exhibited a range of functions and expression levels, and also to examine MYC overexpression in a non-cancer context. We studied the role of MYC in a normal human bronchial epithelial cell line (HBEC3-KT, referred to here as ‘HBEC’) (Ramirez et al., 2004). We previously used a ‘gene trap’ to introduce MS2 loops globally into the introns of HBEC genes (Wan et al., 2021). We derived two types of cell lines from this method: An ‘MS2 polyclonal’ cell line, in which the majority of cells contains a single unique gene tagged with MS2; and ‘single-cell clones’ that were derived from the MS2 polyclonal line by single-cell sorting (Figure 3A). We established 10 single-cell clones that each contain an MS2-tagged gene, and from bulk RNA sequencing of the HBEC parental cell line (Palangat et al., 2019) we determined that these genes represent the upper 50% range of expression of the entire transcriptome, varying from ~6 to 230 RPKM (Figure 3B).
Figure 3.
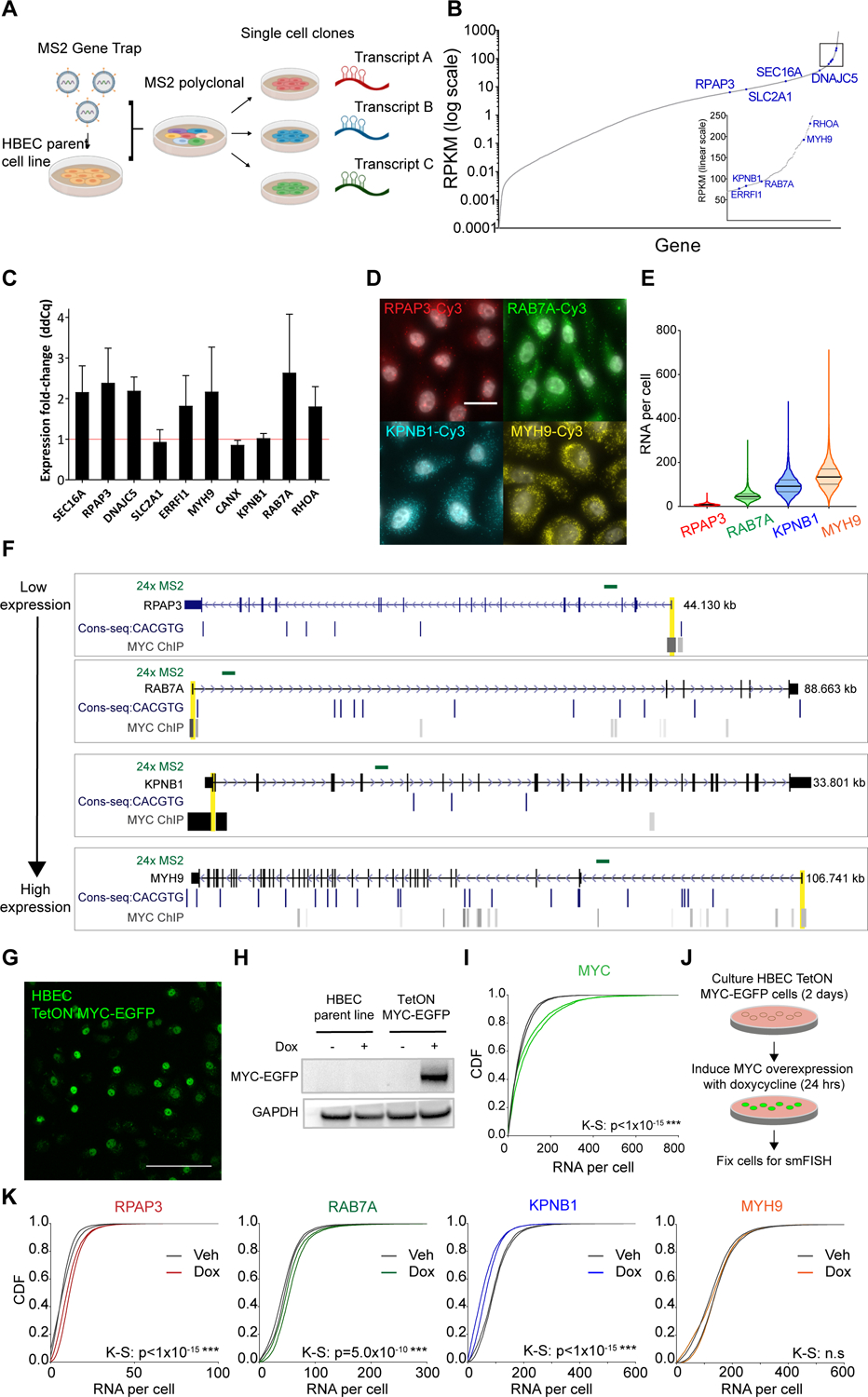
MYC perturbs genes with a range of function and basal expression level in the non-transformed human bronchial epithelial (HBEC) cell line.
(A) Schematic of the gene trap system developed by Wan et al. to insert MS2 stem loops globally into the introns of endogenous genes in the HBEC cell line.
(B) RNA expression profile of the HBEC transcriptome. Indicated in blue are the genes for which MS2 single-cell clones were generated.
(C) qPCR analysis of the effects of 24 hours transient MYC-mCherry overexpression on a panel of genes for which MS2 live cell clones are available. Expression is normalized to a control perturbation of mCherry plasmid.
(D) Representative smFISH images of RPAP3, RAB7A, KPNB1, and MYH9 RNA in HBEC cells in steady state culture (gray = DAPI). Scale bar = 20μm.
(E) Violin plot of distribution of RNA/cell of RPAP3 (n=5955 cells), RAB7A (n=4911 cells), KPNB1 (n=3511 cells), and MYH9 (n=7999 cells) in HBEC steady state culture. Black bar = median; gray bars = 25 and 75 percentiles.
(F) E-box consensus motifs and MYC ChIP-seq binding at four selected genes. Cell lines for which MYC binding at promoters was detected were GM12878, H1-hESC, HeLa, HUVEC, HepG2, K562, MCF7, MCF10A, and NB4. MS2 insertion locations for the single-cell clones are indicated in green. Promoter-proximal regions are indicated in yellow.
(G) Fluorescence image of TetON MYC-EGFP transgene stable expression in HBEC cells induced with 10μg/mL doxycycline for 24 hours. Scale bar = 70μm.
(H) MYC western blot of HBEC parent line and TetON MYC-EGFP stable line with 24 hours vehicle or doxycycline induction.
(I) Normalized CDF of MYC RNA abundance with vehicle or doxycycline induction of TetON MYC-EGFP. Two biological replicates per condition shown. K-S test performed on the replicates combined.
(J) Schematic of MYC overexpression experiment.
(K) Normalized CDF of MYH9, KPNB1, RAB7A, and RPAP3 RNA levels with vehicle and doxycycline induction of TetON MYC-EGFP. Two biological replicates per condition shown. Approximately 6000 cells were imaged on average per replicate. K-S test performed on the replicates combined.
We first assayed whether any of the 10 genes for which we had MS2 live-cell clones responded to MYC perturbations. We performed 24 hours MYC overexpression (MYC-mCherry plasmid) and determined the fold-change in expression compared to an mCherry control with RT-qPCR to total mRNA. We found that increasing MYC levels increased the expression of most genes (Figure 3C). We picked four genes— RPAP3, RAB7A, KPNB1, and MYH9 — for more detailed analysis. We note there is a discrepancy in rank order of expression when comparing RNA-seq and smFISH for RAB7A and KPNB1. All subsequent experiments utilize smFISH, which we find to be a more robust measurement of expression levels.
Our four candidate genes represent a range of features. First, these genes have diverse cellular functions: RPAP3 is an RNAPII associated protein, RAB7A is a GTP-binding protein that is a structural component of lysosomes, KPNB1 is a member of the importin beta family of nuclear chaperones, and MYH9 is a non-muscle myosin. Second, all these genes except KPNB1 showed a response to MYC overexpression detectable by RT-qPCR. Third, these genes represent a range of RNA abundance levels as quantified by smFISH, from RPAP3 with an average of 8.2 ± 0.1 RNA/cell, to MYH9 with 140 ± 0.6 RNA/cell (Figure 3D, E). Fourth, they represent a range of DNA features: all have MYC binding detectable by MYC ChIP-seq in various cell lines (Figure 3F). None of the genes have a canonical E-box sequence in their promoter proximal regions within 200 bp of the transcription start site (Figure 3F, yellow highlight), although there are canonical E-boxes within 1kb of RAB7A and RPAP3.
From the unmodified HBEC parental cell line, we generated a TetON MYC-EGFP stable cell line to look at the effects of MYC overexpression on the four genes. MYC-EGFP overexpression is visible in the nuclei of HBEC cells after a few hours of doxycycline induction (Video S1) Stable expression in the population can be observed by microscopy, Western blot, and smFISH after 24 hours of induction (Figure 3G–I). We conducted smFISH experiments in which we overexpressed MYC for 24 hours, then fixed and labeled the cells with smFISH probes to the 3’UTR of the four genes RPAP3, RAB7A, KPNB1, and MYH9 (Figure 3J, Supplemental Table S1). We found that MYC increased the RNA abundance of the lower expressed genes, RPAP3 and RAB7A (Figure 3K). Curiously, KPNB1 showed a decrease in RNA abundance. The highest expressing gene, MYH9, showed no change. For all four genes, the number of nascent RNA measured by TS intensity did not show significant changes with MYC overexpression: all had changes of less than 1 RNA (Supplemental Table S2). The two genes that showed increased RNA abundance, RPAP3 and RAB7A, also showed a 1.3- and 1.8- fold increase in the number of active TS. Taken together, both population and single-cell methods show that the lower expressed genes increase expression in response to MYC overexpression. The more highly expressed genes responded to MYC knockdown but show discordant results with MYC overexpression between the two assays. However, these assays provide a mixed view as to whether the effects are direct. They are performed at >24 hours of MYC overexpression, potentially resulting in homeostatic compensation which would obscure direct effects on transcription. We thus desired a more acute perturbation of MYC activity.
Optogenetic engineering of Pi-MYC creates a fast, functional form of MYC for initiating acute onset of overexpression in living cells.
To achieve acute overexpression of MYC, we engineered photo-inducible MYC ‘Pi-MYC’ whereby nuclear translocation of MYC is controlled by light (Figure 4A). This capability is achieved using the light oxygen voltage 2 (LOV2) phototrophic domain from the Avena sativa plant (Niopek et al., 2014). The LOV2 domain undergoes a conformational change when exposed to blue-green wavelengths of light (450–500nm). The Pi-MYC transgene consists of MYC-mCherry fused to a nuclear export signal (NES) followed by the LOV2 domain enclosing the c-MYC nuclear localization signal (NLS). MYC-mCherry is constitutively expressed yet retained in the cytoplasm due to the combination of an exposed NES and LOV2-caged NLS. Upon induction with light, the LOV2 domain unhinges and exposes the NLS, allowing MYC to be imported into the nucleus. We found it necessary to inactivate the endogenous MYC NLS located in exon 2 by replacing basic residues with alanine. As a control to make sure any effects we saw in our experiments were specifically due to MYC, we created a ‘Pi-mCherry’ construct which contains all the domains except MYC.
Figure 4.
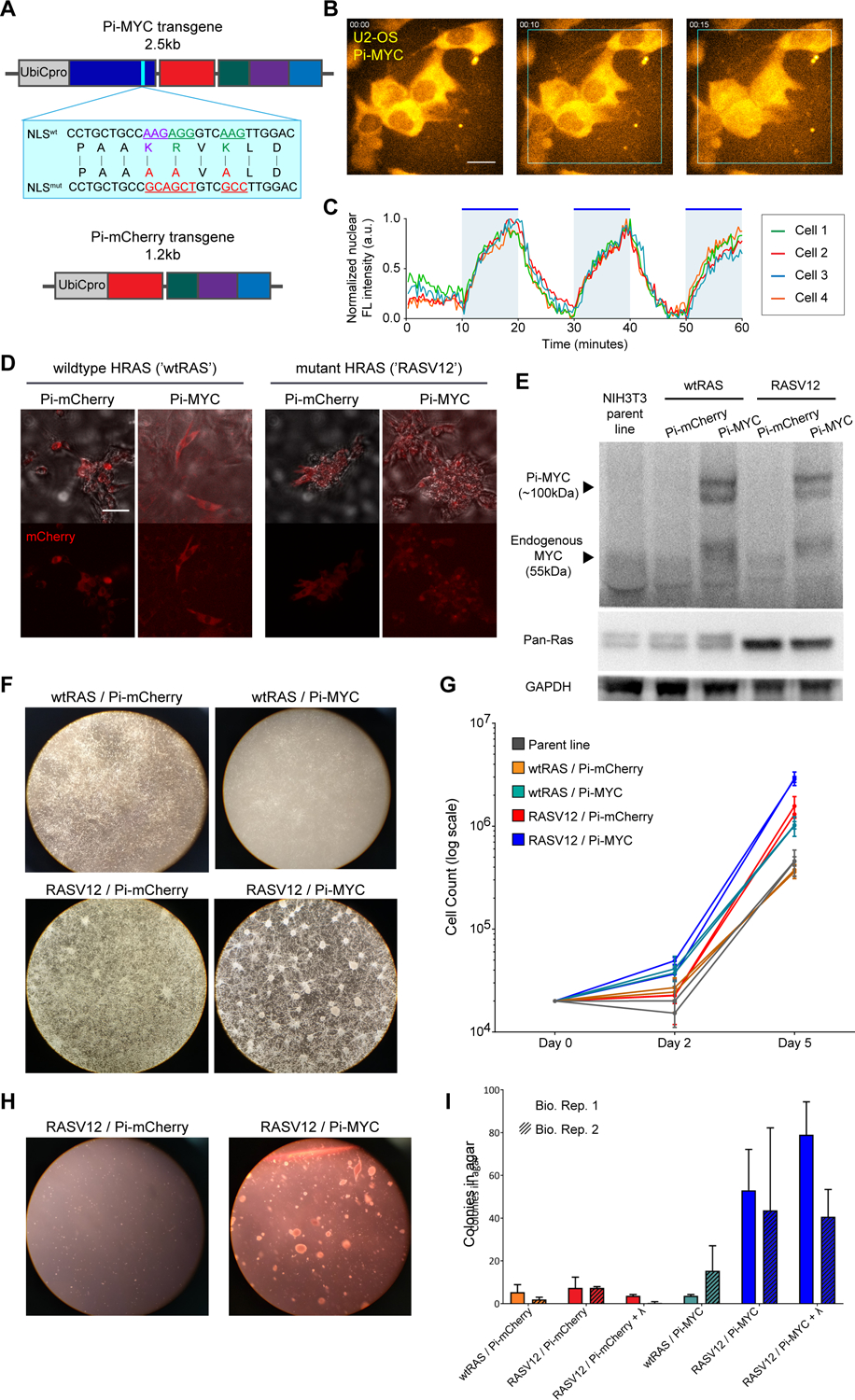
Photo-inducible MYC displays oncogenic capability in NIH3T3 cells.
(A) Schematic of Pi-MYC. The transgene consists of the human Ubiquitin C promoter (UbiCpro), c-myc exons 2 and 3 (MYC), alanine mutations in the native NLS (blue inset shows changed residues), mCherry, a nuclear export signal (NES), and the LOV2 domain followed by the wildtype c-myc NLS sequence. The Pi-mCherry control transgene contains all domains except MYC.
(B) Pi-MYC stable expression in U2OS cells. Scale bar = 15μm. In the absence of irradiating light, the NES allows Pi-MYC retention in the cytoplasm. Upon irradiation with blue-green wavelengths of light (450–500nm), the LOV2 domain exposes the enclosed NLS and allows Pi-MYC to be imported into the nucleus (indicated by white triangles). Pi-mCherry operates via the same mechanism.
(C) Quantification of nuclear fluorescence intensity from four cells numbered in (B) over a 1-hour time series. The field of view was subjected to alternating 10-minute periods of activating light indicated by the blue regions in the plot.
(D) DIC merge and fluorescence images of NIH3T3 fibroblasts stably expressing Pi-mCherry or Pi-MYC (visualized with mCherry) in a background of wildtype HRAS or V12 mutant. Scale bar = 50μm.
(E) Western blot of MYC and RAS expression in the NIH3T3 stable lines.
(F) Growth and focus formation in monolayer culture of four NIH3T3 lines.
(G) Quantification of growth rates, two biological replicates per cell line. Error bars are SD of three technical replicates.
(H) Colony formation in soft agar of Pi-mCherry and Pi-MYC stable lines in the RAS mutant background.
(I) Quantification of colonies visible after 2 weeks growth in soft agar of the four stable NIH3T3 lights, and the RASV12 lines cultured with 24 hours light before embedding in agar. Error bars are SD of two biological replicates.
To test the import and export kinetics, we introduced Pi-MYC via lentiviral integration into U2OS cells. MYC exhibited reversible nuclear translocation within minutes of addition and removal of activating 488nm light (Figure 4B, Video S2). We found the translocation was reversible over repeated induction periods of 1 hour with alternating 10-minute periods of induction light, (Figure 4C) with an entry rate of k1/2 = 0.380 ± 0.018 min−1 and an exit rate of k1/2 = 0.261 ± 0.022 min−1. Analysis of Pi-MYC nuclear fluorescence intensity in cells before and after induction shows a 1.7-fold increase in nuclear MYC levels under these illumination conditions.
We next tested whether Pi-MYC is biologically functional using a classic transformation assay in NIH3T3 cells (Land et al., 1986). Introduction of mutant RASV12 and MYC overexpression transforms murine fibroblasts to yield several hallmark phenotypes of oncogenesis: focus formation, faster growth in culture, and anchorage-independent growth in soft agar. We generated NIH3T3 mouse fibroblast cell lines that expressed either a background of wildtype RAS or mutant RASV12, combined with either Pi-mCherry or Pi-MYC stable expression. Pi-MYC and Pi-mCherry expression and nuclear translocation was visible by fluorescence microscopy (Figure 4D, Video S3), and both MYC and RAS protein levels were detectable by Western blot (Figure 4E). Quantification of the band intensities show that the Pi-MYC and endogenous MYC levels are roughly equivalent, meaning total MYC protein levels were doubled when Pi-MYC was introduced. We found that Pi-MYC co-expressed with mutant RAS transformed NIH3T3 cells, resulting in focus formation (Figure 4F), faster growth in culture (Figure 4G), and anchorage independent growth (Figure 4H, I). We note that the NIH3T3 cells were sensitive enough that the transformation occurred without needing to specifically incubate them with blue light. We reason that this ‘leakiness’ could be due to the LOV2 domain being activated by trace amounts of room light, or some LOV2 domains being improperly folded and not able to completely cage the NLS. Nevertheless, these data indicate that Pi-MYC is biologically functional, allowing us to probe for transcription changes when perturbing cells on short timescales.
Having confirmed that Pi-MYC is both controllable and functional, we introduced Pi-mCherry or Pi-MYC into HBECs. We created five HBEC MS2- labeled cell lines derived from the MS2 polyclonal line and the four single-cell clones RPAP3, RAB7A, KPNB1, MYH9 via lentiviral transduction (Figure 5A). The MS2 polyclonal cell line consists of introns of ~900 unique genes labeled with MS2 (Table S3). Multiple screening steps were employed to isolate single cell clones that were heterozygous for the integration, containing only one MS2 tag and therefore only one visible transcription site per gene (See methods in (Wan et al., 2021). We used a smFISH probe set to MS2 to detect the activity of nascent RNA at transcription sites (Figure 5B,C). We found that even under conditions of no activating light, the Pi-MYC cell line exhibited brighter TS compared to the Pi-mCherry line, indicating the production of more nascent RNA at active TS (Figure 5D). This result may follow from slight leakiness of the LOV2 cage, consistent with what we saw in NIH3T3 cells, even though by fluorescence microscopy MYC appears to be excluded from the nucleus (Figure S3A). Similarly, Pi-MYC induced a growth advantage in HBEC cells over a period of two days in culture (Figure 5E). However, in contrast to NIH3T3 cells, this growth difference was more pronounced when the cell line was cultured under blue light. Interestingly, the Pi-mCherry line also displayed some increased growth when cultured under blue light. It has been previously reported that blue and/or green light may have antioxidant effects (Comorosan et al., 2009), have protective effects on protein subjected to UV irradiation (Espinoza and Mercado-Uribe, 2017), and initiate biophysical processes in cells such as increased wound healing and cell proliferation (Adamskaya et al., 2011; Rohringer et al., 2017).
Figure 5.
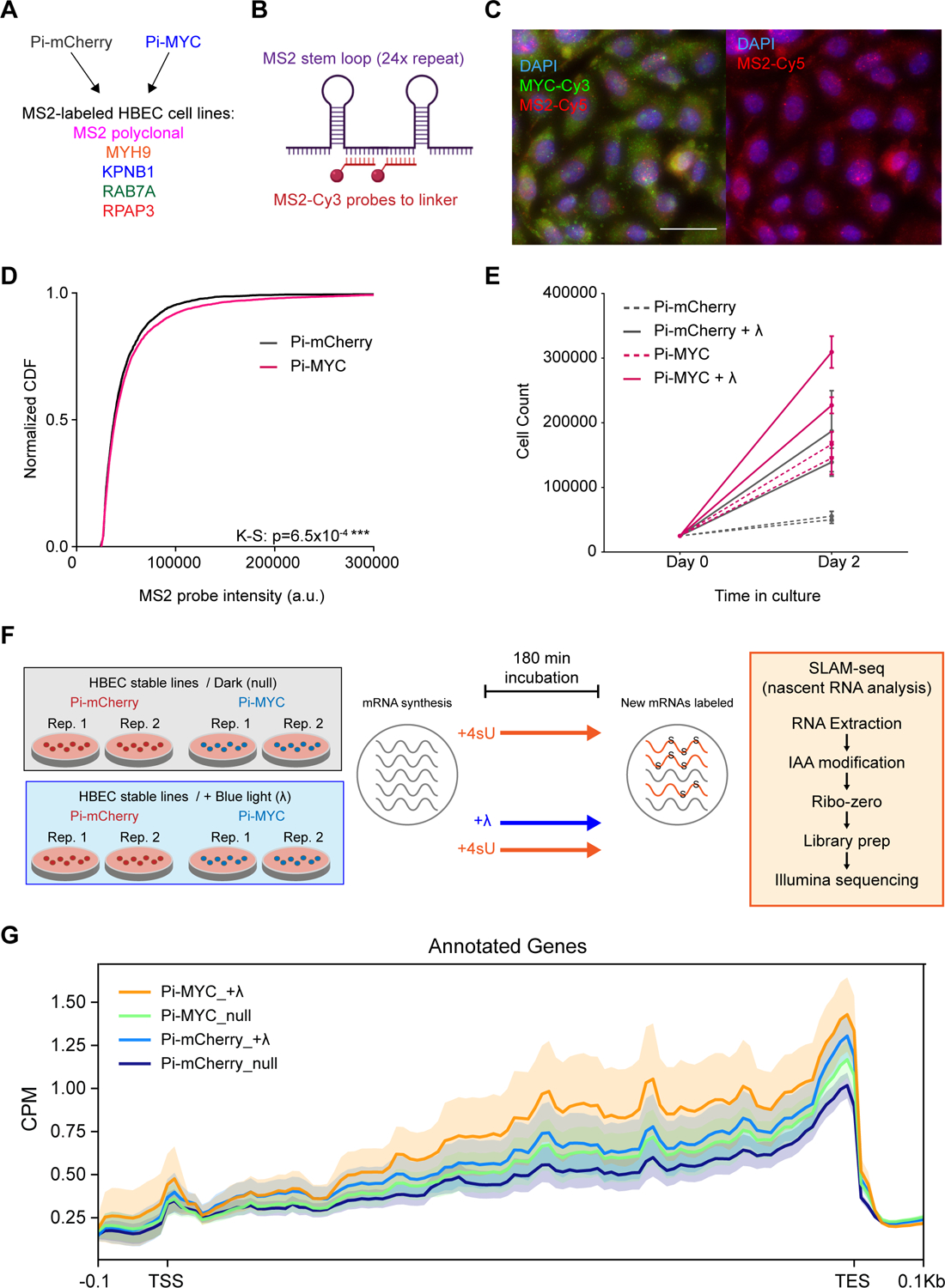
HBEC cells exhibit a growth advantage and more nascent RNA at transcription sites with stable integration of Pi-MYC.
(A) Diagram of HBEC cell lines generated by stably integrating Pi-mCherry or Pi-MYC into the MS2 polyclonal cell line and four single cell clones (10 cell lines total).
(B) MS2-Cy3 smFISH probes are localized to the linker region between a single MS2 stem loop repeat.
(C) smFISH images of the HBEC MS2 polyclonal line with MS2 probes targeting the nascent RNA (introns) of genes. Scale bar = 30μm.
(D) Normalized CDF of MS2 probe intensity in the HBEC polyclonal line, indicating the number of nascent RNA present globally at transcription sites with stable expression of Pi-mCherry (n=3364 cells) or Pi-MYC (n=6670 cells). Shown are two biological replicates combined per condition.
(E) Two-day growth curve of HBEC cells with Pi-mCherry or Pi-MYC stable integration, cultured in the dark or under 455nm light (+λ). Shown are two biological replicates per condition.
(F) Setup of SLAM-seq analysis. Two biological replicate plates of HBEC Pi-mCherry or Pi-MYC cells were incubated with 4sU for 180min in the absence (null) or presence of blue light (+λ). The RNA was extracted and prepared for Illumina sequencing to quantify the abundance of newly synthesized RNAs.
(G) Metagene profile for the abundance of newly synthesized mRNAs (detected in SLAM-seq with T>C conversions). Density diagrams represent the T>C read coverage across genes at transcription start sites (TSS, ±0.1 kb), gene bodies (scaled), and transcription end sites (TES, ±0.1kb). CPM, counts per million mapped reads.
In order to determine whether Pi-MYC had global effects on RNA abundance levels in the cells, we performed SLAM-seq analysis to measure how nascent RNA levels compared between Pi-MYC and the Pi-mCherry control +/− blue light. Cells were incubated with 4sU for 180 min, subjected to incubation in the dark or under blue light (Figure 5F). Pi-MYC with blue light activation showed global increases in nascent RNA synthesis compared to the controls (Figure 5G). Interestingly, we observed differences in the controls similar to what was observed in the growth assay: nascent RNA synthesis increased with blue light alone (translocation of Pi-mCherry), and with Pi-MYC cells incubated in the dark. The latter speaks to possible leakiness of the LOV2 caging mechanism, while the former speaks to how blue light itself may affect proliferation. These data speak to the importance of using the Pi-mCherry control for our experiments where the cell is subjected to all factors involved in the light induction mechanism except translocation of MYC. In total, however, we observe a light- and MYC-dependent increase in nascent transcription using an ensemble measurement across the genome.
Having characterized and biologically validated Pi-MYC, we proceeded to simultaneous MYC perturbation and real-time imaging of RNA synthesis. These experiments were carried out using high-throughput live-cell imaging immediately after Pi-MYC translocation to determine how MYC affected the ON and OFF periods of transcriptional bursts. For a given MS2 cell line, we cultured the Pi-mCherry and Pi-MYC variants in parallel for high-throughput imaging (Figure 6A). We chose an induction/imaging duration of 6 hours to allow us to measure short timescales effects while still capturing the OFF time distribution. By RNA sequencing we found that RPAP3 is expressed 30-fold lower than MYH9 (Figure 3B), so we ultimately extended our perturbation/imaging window to 15 hours in order to more fully capture the OFF time distribution for this gene. We induced Pi-MYC and Pi-mCherry nuclear translocation using the 488nm laser we used to image the MS2-GFP channel; in this way a single wavelength of light could couple the induction/constitutive nuclear expression of the Pi-transgene with imaging of the downstream gene. We normalized the fluorescence intensity traces and fit each dataset to a 2-state Hidden Markov Model (adapted from (Lee, 2009)) threshold the ON and OFF times of the gene (Figure 6B, 6C). The final output of this analysis is a cumulative distribution of ON and OFF times reflecting transcription dynamics immediately (within 100 seconds) after Pi-MYC translocation.
Figure 6.
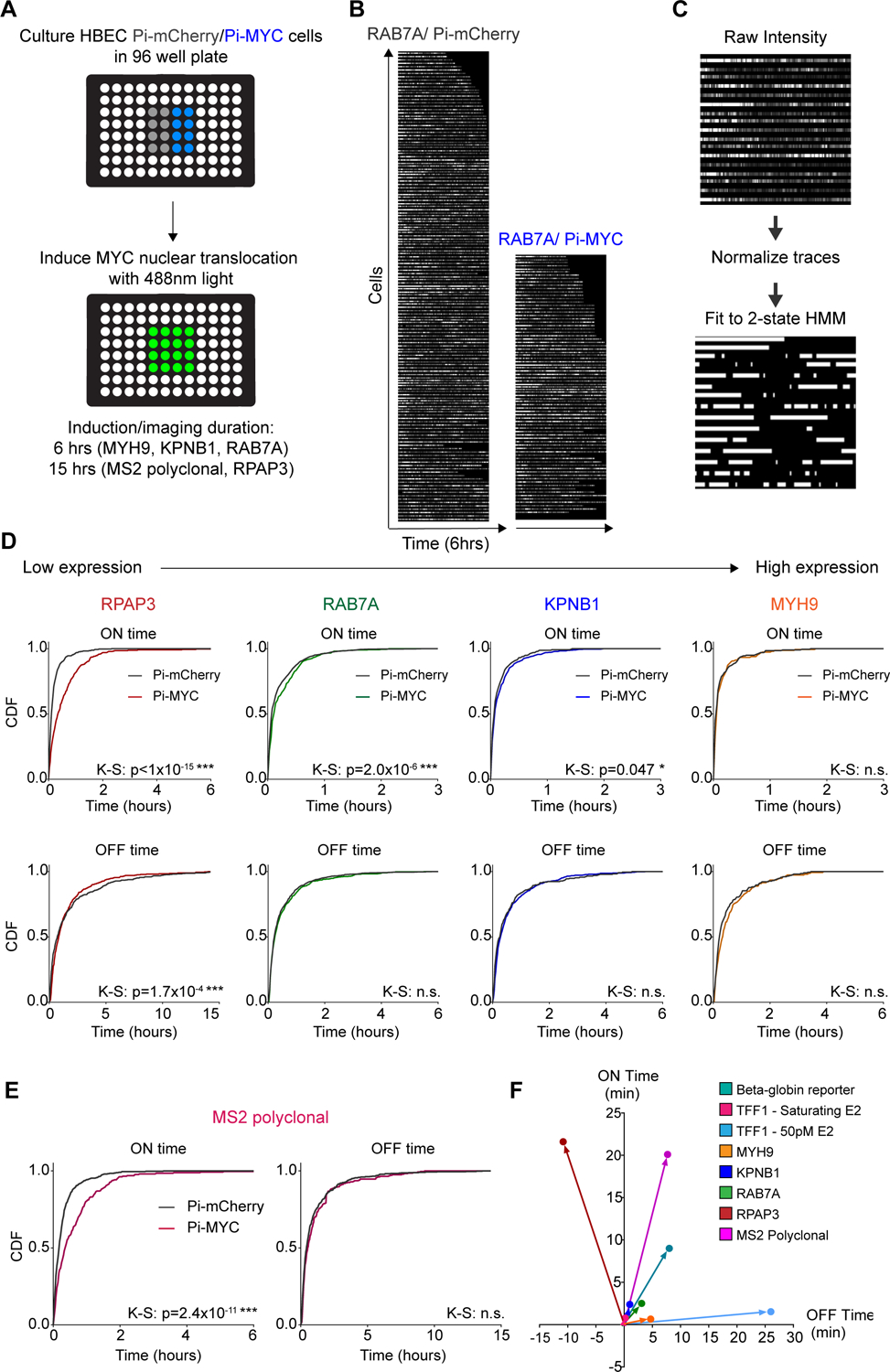
Pi-MYC overexpression on short timescales reveals genome-wide increases in transcription ON time.
(A) Schematic of HBEC cells grown in 96-well plates for high-throughput live cell imaging of transcription site activity.
(B) Example raw data of RAB7A transcription site activity with 6 hours of Pi-mCherry or Pi-MYC overexpression.
(C) Raw fluorescence intensity traces are normalized and fit to a 2-state HMM to yield ‘ON’ and ‘OFF’ periods of each gene.
(D) Normalized CDF of transcription ON and OFF times for the RPAP3, RAB7A, KPNB1, and MYH9 single cell clones with Pi-mCherry or Pi-MYC overexpression.
(E) Normalized CDF of transcription ON and OFF times in the MS2 polyclonal cell line Pi-mCherry n=83 cells, Pi-MYC n=52 cells).
(F) Summary delta plot showing the change in ON and OFF times for all genes studied.
We found that three of four genes showed an increase in burst duration ranging from 2.4 to 21.6 minutes. For the single-cell clones, the extent of the ON time increase was related to the expression level (Figure 6D). The burst duration of the lowest expressed gene RPAP3 increased from 11.5 ± 0.8 to 33.1 ± 2.3 min upon Pi-MYC translocation. The highest expressed gene, MYH9, showed no detectable change in burst duration (8.9 ± 1.1 min with Pi-mCherry and 9.5 ± 1.3 min with Pi-MYC). These results are in agreement with the fixed-cell data measured earlier, in that the genes showing the greatest increases in expression by RT-qPCR and smFISH at 24 hours were RPAP3 and RAB7A. Interestingly, although KPNB1 expression showed no change by RT-qPCR and a decrease in expression by smFISH, we saw a statistically significant increase in KPNB1 burst duration by the live-cell assay. With the exception of RPAP3, we observed no change in the OFF periods (i.e. burst frequency) under Pi-MYC translocation conditions. Thus, live-cell imaging with Pi-MYC shows that the duration of transcription active periods is longer immediately after translocation of Pi-MYC, but that this effect can be masked by downstream effects on the overall abundance of the cellular mRNA.
Our studies on single-cell clones demonstrated the feasibility of an optogenetic real-time.approach to dissecting MYC function. We next extended these experiments to the MS2 polyclonal cell line. The polyclonal population allows us to obtain a quasi-global measurement of gene expression, although we cannot ascertain which particular gene is labeled in each cell. When averaged together, these data represent a unique live-cell ‘metagene’ analysis. We used a 15-hour experimental window since this cell line represents a distribution of transcription dynamics that also includes infrequently transcribed genes. Since the median burst interval for human genes is ~ 1 hour (Wan et al., 2021), we collect data over a sufficiently long window to accurately measure the time between bursts. Under these conditions, we observed a substantial global increase in ON time of 20 min, but the OFF time was unchanged (Figure 6E). Thus, the average response to acute Pi-MYC translocation is an increase in burst duration. Finally, we summarize the live-cell imaging data of all our genes across cell type and measurement approach as a ‘delta plot’ that indicates the change MYC overexpression had on the average ON and OFF time of a gene compared to the control (Figure 6F). Regardless of the cell type, method of overexpression, or the gene studied, we see that at no point did MYC ever reduce the ON time of a gene. Intriguingly, MYC also appears to generally increase the time between bursts (reduced burst frequency), but this change was not statistically significant under all experimental conditions. The relative balance of these countervailing effects – increased burst duration and reduced burst frequency –determines the net RNA synthesis rate.
MYC globally affects residence times of transcription factors involved in pre-initiation complex formation and RNAPII pause release
During our experiments, we observed that Pi-MYC in HBECs exhibited a variable nuclear entry/exit profile compared to previous conditions (i.e. Figure 4C). We found that HBEC appeared to display nuclear retention of Pi-MYC even when the induction light was removed (Figure 7A, Video S4), unlike the U2OS Pi-MYC cells that displayed more obvious reversible translocation (Figure 4B, Video S2). We replicated the induction experiment of Figure 4C with the HBEC stable lines and found that Pi-MYC displayed slower exit kinetics compared to the Pi-mCherry control (Figure 7B). Notably, there was a decreased export rate of Pi-MYC (0.257 ± 0.037 min−1) compared to Pi-mCherry (0.347 ± 0.015 min−1).
Figure 7.
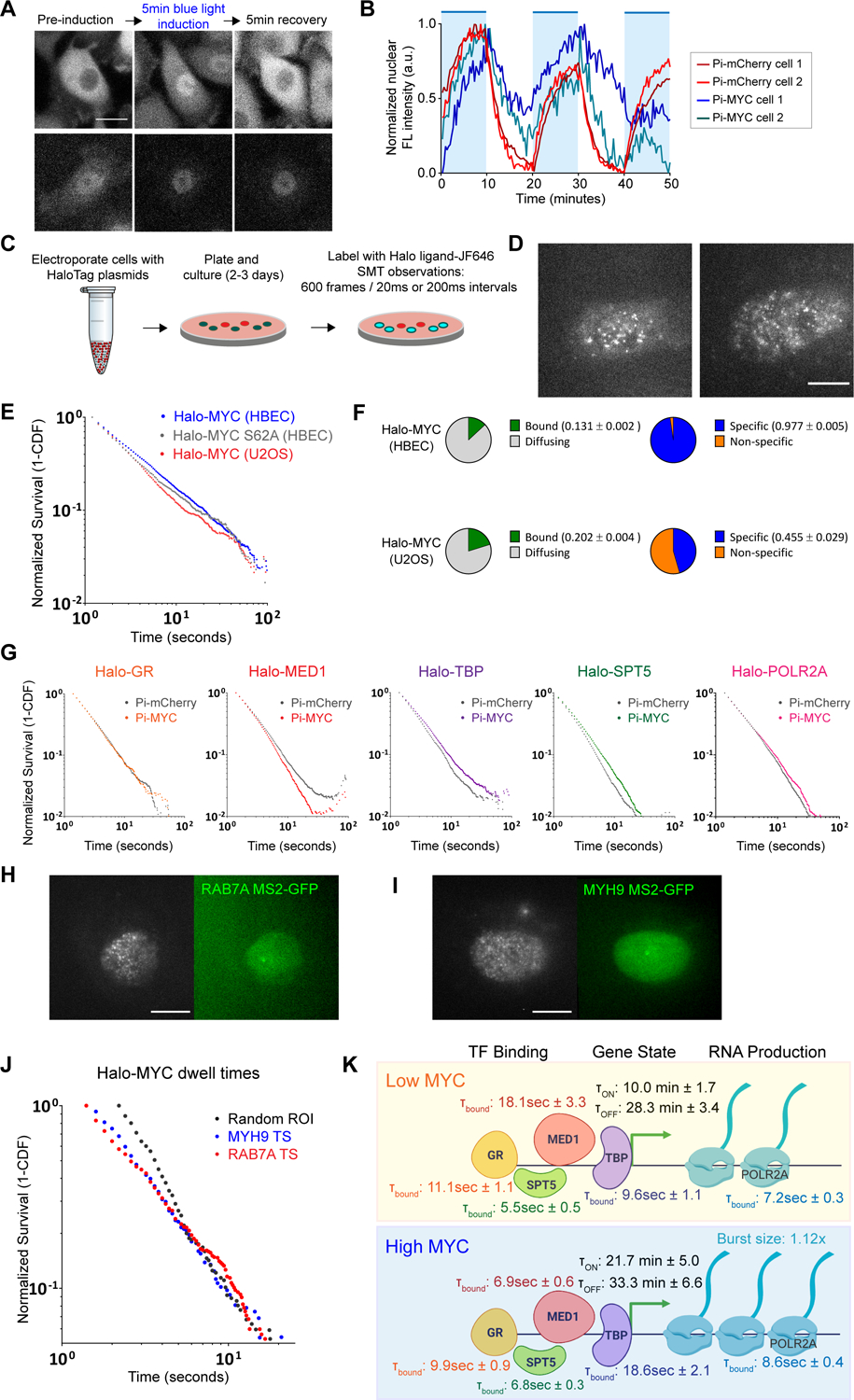
MYC effects on residence time of basal transcription factors.
(A) Example fluorescence images comparing nuclear entry and exit between Pi-mCherry and Pi-MYC stably expressed in HBEC cells. Scale bar = 15μm.
(B) Quantification of nuclear fluorescence intensity over a one-hour time course of HBEC Pi-mCherry or Pi-MYC cells with repeated 10-minute periods of activating light (indicated by blue regions).
(C) Schematic representation of SMT experiments.
(D) Representative SMT images of Halo-MYC expressed in HBEC and U2OS. Scale bar = 10μm.
(E) Survival plot of Halo-MYC dwell times in HBEC (blue, n=33224 tracks) and U2OS (red, n=14845 tracks), and Halo-MYCS62A dwell times in HBEC (gray, n=18522 tracks).
(F) Fraction of Halo-MYC bound vs. diffusing population (green/gray) and specific vs. non-specific bound populations (blue/orange) in HBEC and U2OS.
(G) Survival plots of transcription factor dwell times with nuclear translocation of Pi-mCherry (GR=24662 tracks, MED1=66960 tracks, TBP=34205 tracks, SPT5=41957 tracks, POLR2A=50112 tracks) or Pi-MYC (GR=14666 tracks, MED1=30811 tracks, TBP=33008, SPT5=39618 tracks, POLR2A=49528 tracks).
(H) Representative images of dual-color SMT experiments tracking Halo-MYC in a cell actively transcribing RAB7A. Active TS indicated by white arrow. Scale bar = 10μm.
(I) As in (H), Halo-MYC tracked in a cell actively transcribing MYH9. Scale bar = 10μm.
(J) Survival plot comparing the dwell time distribution of Halo-MYC in HBEC nuclei at a random ROI (gray, n= 3361 tracks), MYH9 active TS (blue, n=1303 tracks), and RAB7A active TS (red, n=2304 tracks).
(K) Model of MYC mechanism of action. Rates and characteristic dwell times of transcription factor binding, gene ON/OFF states, and RNA production are from measurements taken in the HBEC MS2 polyclonal cell line.
We hypothesized that the slower export kinetics of Pi-MYC compared to the Pi-mCherry control could be due to MYC actively binding available sites on the genome and modulating transcription activity across the nucleus. For example, lower endogenous MYC levels in HBEC compared to U2OS might create a physically different environment in which there is less competition for exogenous MYC to bind and exert effects on transcription activity. We explored this hypothesis by engineering a new MYC overexpression system, ‘Halo-MYC’, to enable single-molecule tracking (SMT) measurements of MYC dwell times in the nucleus, an approach which has been previously used to detect protein residence times on DNA (Cho et al., 2016; Garcia et al., 2021a; Hansen et al., 2017; Khan et al., 2019; Paakinaho et al., 2017; Swinstead et al., 2016). Halo-MYC was engineered by fusing the Halo-Tag (Los et al., 2008) to the C-terminus of the MYC transgene sequence and transiently expressing the plasmid in wildtype HBEC and U2OS (Figure 7C). Halo-MYC expressing cells were subsequently labeled with Halo ligand conjugated to a JaneliaFluor dye (JF646) which allowed single MYC proteins to be tracked in live cells with HILO microscopy (Figure 7D). In addition to engineering Halo-MYC which contains the wildtype MYC coding sequence, we created a Halo-MYCS62A mutant. The S62A mutation has been previously been shown to reduce MYC stability and promote degradation of the protein (Sears et al., 2000; Wang et al., 2011).
We measured the chromatin dwell times of Halo-MYC in HBEC and U2OS, and Halo-MYCS62A in HBEC (Figure 7E). We show this data as a survival probability, which is computed from the cumulative distribution function and reflects the frequency of dwell time events in the time-series analysis. In general, we find that Halo-MYC in HBEC shows the longest dwell times on chromatin, with Halo-MYCS62A in HBEC, and Halo-MYC in U2OS showing progressively faster turnover (shorter dwell times). By fitting the raw dwell time data to a bi-exponential distribution, we can distinguish the fraction of total bound molecules from the diffusing population. Of the total bound molecules, the bi-exponential fit enables us to parse out the populations that exhibit ‘long’ versus ‘short’ dwell times, conventionally interpreted to be the ‘specific’ and ‘non-specific’ bound fraction on chromatin (Patange et al., 2021). We observed a slightly greater fraction of total bound Halo-MYC in U2OS compared to HBEC (0.202 ± 0.004 vs. 0.131 ± 0.002). More significantly, we observed that a greater fraction of Halo-MYC bound molecules exhibited longer dwell times in HBEC compared to U2OS, interpreted to be a greater specific bound fraction in HBEC (0.977 ± 0.005 vs. 0.455 ± 0.029) (Figure 7F). Compared to wildtype HaloMYC, the Halo-MYCS62A mutant exhibited both a modest increase in the total bound fraction (0.144 ± 0.003) and decrease in the specific bound fraction (0.941 ± 0.005) (Supplementary Figure S5C). We observed similar residence times for the two populations of bound molecules in both HBEC and U2OS (Supplementary Figure S5E). The larger specific bound fraction in HBEC means the average time that a Halo-MYC molecule remains bound is approximately twice as long than in U2OS (26.3 ± 1.4 s vs. 15.2 ± 1.7 s). Taken together, the nuclear translocation kinetics of Pi-MYC and the dwell time of Halo-MYC indicate the dynamic range of MYC binding in a transformed and non-transformed cell line. Further, the S62A mutation appears to slightly de-stabilize binding to chromatin.
Having observed both the global effects of Pi-MYC on nascent transcription and the changes in MYC dynamics in different cell lines, we next asked whether the effects of MYC overexpression might alter global residence times of core transcription factors on DNA. We tested this hypothesis using SMT measurements on a panel of five HaloTag-labeled transcription factors: Glucocorticoid receptor (GR), a gene-specific transcription factor; TATA binding protein (TBP), a core component of the pre-initiation complex (PIC) shown to have direct interactions with MYC (Wei et al., 2019); MED1, a subunit of the mediator complex; SPT5, a subunit of the elongation factor DSIF that is recruited by MYC to initiate RNAPII pause release and productive elongation (Baluapuri et al., 2019); and POLR2A (Rpb1), the largest subunit of RNAPII.
We initiated fast SMT measurements consisting of a 6-min induction period of Pi-mCherry or Pi-MYC nuclear translocation with 488nm light, and a Halo-SMT measurement in the last 2 min of the induction after translocation had visibly occurred (Video S4). In this manner, single TFs could be observed diffusing or exhibiting various states of immobility (Video S5), and the molecules were tracked to quantify the dwell time distribution (Figure 7G). We measured increased residence times of TBP, SPT5, and POLR2A (indicated by right-ward shifts of the survival probabilities) and decreased residence time of MED1 (left-ward shift) with Pi-MYC overexpression. The residence time of the sequence-specific factor GR was not affected. Thus, MYC overexpression changes the residence times of transcription factors related to PIC assembly and pause release throughout the nucleus.
There are multiple approaches for fitting residence time data, but we relied on two models which have been widely used. The first is a power law model (Garcia et al., 2021b) which assumes a continuous distribution of residence times, where the slope of the line in log-log space α indicates differences in the distributions. The second is a bi-exponential model (Ball et al., 2016; Mazza et al., 2012; Mehta et al., 2018; Presman et al., 2017), which assumes two populations that are conventionally interpreted as a ‘nonspecifically bound’ population with a faster off rate k1, and a ‘specifically-bound’ fraction with a slower off rate k2. We found that the fitting parameters reflect the statistical significance of the differences in the raw data (Figure S4). Strikingly, TBP specific residence time nearly doubles after Pi-MYC translocation, increasing from 9.6s ± 1.1 to 18.6 ± 2.1s with only a small change in the non-specific binding. POLR2A also exhibited a statistically significant increase in mean dwell time of the specific bound fraction (7.2 ± 0.3s to 8.6 ± 0.4s). In contrast, MED1 residence time decreases from 18.1 ± 3.3s to 6.9 ± 0.6s. SPT5 appears to show comparable increases in the residence times of both the specific and non-specific bound fraction after Pi-MYC translocation. GR, which is involved in the early steps of transcription initiation for its target genes, was not affected by Pi-MYC overexpression. Taken together, these data suggest that MYC primarily serves to affect the mobility of transcription factors involved later in the transcription cycle, and that global changes in the dynamics of the core transcription machinery are observable seconds after Pi-MYC translocation.
Finally, we attempted to measure MYC dynamics at specific gene loci. We evaluated two genes from our earlier studies that showed different transcriptional responses to MYC overexpression: RAB7A, a gene that showed increased RNA abundance and an increase in transcription burst duration, and MYH9, a high-expressing gene that did not change expression. We initiated dual-color SMT experiments in which we introduced Halo-MYC JF646 to the HBEC single cell clones RAB7A MS2-GFP (Figure 7H) and MYH9 MS2-GFP (Figure 7I) and tracked MYC molecules inside a 10 pixel radius of either an active transcription site or a random ROI in another region of the same nucleus (Figure 7J). The most pronounced difference we observed was a higher bound fraction of Halo-MYC at the RAB7A active TS than at the MYH9 active TS or a random ROI. This change is seen in the raw data primarily as a shoulder in the longer dwell times of RAB7A but also by a corresponding change at fast time-scales. We could further determine the specific bound fraction of Halo-MYC at RAB7A TS (0.961 ± 0.039), MYH9 TS (0.378 ± 0.113) and at random ROI (0.218 ± 0.091) (Figure S5A). These results provide single-molecule evidence that the effects on transcription burst duration caused by MYC overexpression (as seen by our Pi-MYC studies) can be attributed to physical interactions of MYC at active transcription sites that are not already saturating in expression, thereby facilitating changes in the dwell times of RNAPII and other core factors involved in PIC assembly and pause release.
Discussion
Single-cell imaging provides a window into the molecular mechanisms of gene expression. Although the MYC oncogene has been studied for many decades, an outstanding question remains as to how MYC acts directly on the discrete events of gene expression — transcriptional bursts — in single living cells. To answer this question, we undertook a systematic survey of MYC using single-cell imaging approaches to quantify RNA production, gene active and inactive periods, and transcription factor dwell times. This approach allowed us to build an empirical model that relates the effects of MYC nuclear localization to discrete quantities and rates of transcription factor binding, gene state, and RNA production (Figure 7K).
We find that MYC increases the active period of transcription and exerts global changes in the dynamics of the basal transcription machinery. In breast carcinoma cells, MYC extended the transcriptional bursts of the estrogen responsive gene TFF1, but only when estradiol levels were non-saturating. In immortalized lung epithelial cells, we again observed that MYC increased gene active periods for a panel of genes, and that the significance of the increase corresponded to the basal expression level of the gene: the higher the gene was expressed, the less the active period changed. Finally, by using a photo-inducible MYC (Pi-MYC) in an MS2 polyclonal cell line created with a novel gene trap, we observed that the increase in gene active period was a global effect. This result was further supported by nascent RNA-sequencing. The global increase in the transcription burst duration was accompanied by an increase in the dwell time for TBP, SPT5, and POLR2A, but a decrease in dwell time for MED1. We thus achieved the first real-time readout of how MYC affects the discrete events of gene expression in living human cells.
Our findings from live-cell imaging across genes and cell lines show that MYC increases the active period of a gene, which is unusual among transcription factors and provides clues to the mechanism of activity. The Notch transcription factor has been recently shown to change burst duration in Drosophila (Falo-Sanjuan et al., 2019), however the majority of experimental and computational studies show that gene regulation occurs predominantly through burst frequency modulation (Bartman et al., 2016; Cai et al., 2008; Fritzsch et al., 2018; Fukaya et al., 2016; Hansen and O’Shea, 2015; Rodriguez et al., 2019; Wan et al., 2021). Thus, MYC plays an orthogonal role to most gene regulatory factors, modulating burst duration rather than burst frequency. The robust increase in the duration of transcriptional bursts suggested to us that MYC is acting on transcription throughout initiation and early elongation and may be exerting effects through multiple interactions with core transcription machinery. Our survey of the effects of acute MYC translocation on transcription factor residence times with SMT supports this hypothesis, as we saw changes in the basal machinery such as TBP, MED1, SPT5, and POLR2A, but not the sequence-specific activator GR. MYC overexpression caused increased residence times for TBP and POLR2A and decreased residence times for MED1, suggesting stabilization of the PIC and recruitment of RNAPII while facilitating dissociation of the Mediator complex to allow for promoter clearance. Increased residence time of SPT5 may coincide with a greater proportion of productive elongation events. In terms of order of recruitment, there appears to be a trend where the early steps of activation (binding of a sequence-specific activator and recruitment of Mediator) are either unaffected or destabilized but later steps (recruitment of TBP, RNAPII, and SPT5) are stabilized. There is evidence for physical interaction between MYC and each of these factors (Baluapuri et al., 2019; Liu et al., 2008; McEwan et al., 1996), but our assay reports on functional outcomes and does not distinguish between direct and indirect mechanisms. Based on this evidence, we predict that MYC increases gene expression through factors that affect the ON time (i.e. core transcription machinery) rather than factors that affect the OFF time (i.e. pioneer transcription factors, chromatin remodelers).
Surprisingly, single-molecule tracking of Halo-MYC revealed slower turnover of MYC on chromatin in HBEC cells compared to U2OS cells. This observation is reminiscent of a previous in vivo single-molecule tracking study in which MYC exhibited non-compact ‘global’ exploration of nuclear space (Izeddin et al., 2014). However, that study was based on over-expression of MYC in U2OS cells. In our case, we find the bound fraction of exogenous MYC is substantial– greater than 97% -- suggesting to us that MYC may be limiting in non-transformed cells and perhaps not capable of global exploration to the same extent.
An outstanding question to emerge from these studies is: How do changes in dwell time on the order of seconds lead to changes in burst duration on the order of minutes? One possibility which is hard to exclude is that SMT misses a population of extremely long-lived events. Our SMT acquisition parameters introduce a bias in sampling the full distribution of residence times; our short measurement window of two minutes causes an underestimation of the dwell times of the highly stable population of transcription factors. A second technical limitation is that the position of the stem loops in the target gene can determine the resolution of bursting analysis (Rodriguez and Larson, 2020). One biological possibility is that cooperative effects on many transcription factors leads to assembly of a more stable permissive environment for sustained transcription. In some cases, such as the GAL locus in budding yeast, there is a clear relationship between activator dwell time and burst duration (Donovan et al., 2019). However, a general model in metazoans has not emerged.
Limitations of this study
We note several caveats when working with a phototrophic domain. First, it can be sensitive to trace amounts of light found in a normal laboratory setting. Room light itself may cause the LOV2 domain to react and uncage the NLS, and cause trace amounts of nuclear translocation during passaging, culture, and transport of the cell lines. We made every effort to reduce leaky activation by covering cell culture plates, working with the cell lines in minimal light, and keeping the samples in the dark for 1 hour prior to beginning an imaging acquisition. Second, under certain conditions that we document here, blue light can change growth properties in tissue culture. Although we were able to control for these effects in our biological studies, future technical efforts will be directed toward developing tighter cages, possibly through simultaneous caging of the NLS and re-directing to other cellular compartments as demonstrated previously (Wang et al., 2016).
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel R. Larson (dan.larson@nih.gov)
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The SLAM-Seq dataset generated in this paper has been deposited at GEO and is publicly available as of the date of publication. The accession number is listed in the key resources table.
No new custom code was generated for this paper.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Myc Y69 | Abcam | Cat# ab32072 |
| Mouse monoclonal Pan-Ras | Sigma-Aldrich | Cat# OP40 |
| Mouse monoclonal anti-GAPDH | Ambion | Cat# AM4300 |
| Anti-rabbit HRP | GE Healthcare Life Sciences | Cat# NA934 |
| Anti-mouse HRP | GE Healthcare Life Sciences | Cat# NA931V |
| Chemicals, peptides, and recombinant proteins | ||
| β-estradiol (E2) | Sigma-Aldrich | Cat# E8875 |
| Doxycycline | Sigma-Aldrich | Cat# D3447 |
| Zeocin | InvivoGen | Cat# ant-ZN |
| Dexamethasone | Sigma-Aldrich | Cat# D1756 |
| Agar (for colony formation assay) | Sigma-Aldrich | Cat# A1296 |
| JF646 HaloTag ligand | Grimm et al., 2017 | N/A |
| 4-thiouridine (4sU) | Sigma-Aldrich | Cat# T4509 |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat# I1149 |
| TRIzol Reagent | Invitrogen | Cat# 15596018 |
| Critical commercial assays | ||
| Virabind Lentivirus Purification Kit | CellBioLabs | Cat# VPK-104 |
| iQ SYBR Green Supermix | Bio-Rad | Cat# 170880 |
| Deposited data | ||
| SLAM-Seq | This manuscript | GEO: GSE189943 |
| Experimental models: Cell lines | ||
| U2OS MS2-PP7 Beta-globin reporter | Coulon et al., 2014 | N/A |
| MCF7-TFF1 MS2 | Rodriguez et al., 2019 | N/A |
| MCF7 TetON MYC-EGFP | Eric Batchelor Lab | N/A |
| NIH3T3 | ATCC | Cat# CRL-1658 |
| NIH3T3 HRAS V12/Pi-MYC | This manuscript | N/A |
| NIH3T3 HRAS V12/Pi-mCherry | This manuscript | N/A |
| HBEC3-KT | ATCC | Cat# CRL-4051 |
| HBEC3-KT with intronic MS2 stem loops (polyclonal population) | Wan et al., 2021 | N/A |
| HBEC3-KT with intronic MS2 stem loops (single cell clones) | Wan et al., 2021 | N/A |
| HBEC3-KT TetON MYC-EGFP | This manuscript | N/A |
| HBEC3-KT MS2 polyclonal / Pi-MYC | This manuscript | N/A |
| HBEC3-KT MS2 polyclonal / Pi-mCherry | This manuscript | N/A |
| HBEC3-KT MS2 single cell clones / Pi-MYC | This manuscript | N/A |
| HBEC3-KT MS2 single cell clones / Pi-mCherry | This manuscript | N/A |
| Oligonucleotides | ||
| qPCR primers | IDT | SeeTable S1 |
| Single-molecule RNA FISH probes | LGC Biosearch Technologies | SeeTable S1 |
| Recombinant DNA | ||
| pTRIEX-mCherry-LOV2 | Addgene | Cat# 81041 |
| pd4-MYC-EGFP | Nie et al., 2012 | N/A |
| pBABE-neo-HRAS-V12 | Scaffidi and Misteli, 2011 | N/A |
| pTRIPZ TetON MYC-EGFP | David Levens Lab | N/A |
| pLenti-Pi-Myc | This manuscript | N/A |
| pLenti-Pi-mCherry | This manuscript | N/A |
| pHaloTag-GR | Morisaki et al., 2014 | N/A |
| pHaloTag-H2B | Mazza et al., 2012 | N/A |
| pFC15A-HaloTag-MED1 | Gordon Hager Lab | N/A |
| pFN21A-HaloTag-TBP | Promega | Cat# FHC11099 |
| pFN21A-HaloTag-SUPT5H (SPT5) | Promega | Cat# FHC02125 |
| pFN21A-HaloTag-POLR2A | Promega | Cat# FHC28428 |
| pFC15A-HaloTag-MYC | This manuscript | N/A |
| pFC15A-HaloTag-MYC-S62A | This manuscript | N/A |
| Software and algorithms | ||
| CellProfiler | Carpenter et al., 2006 | https://cellprofiler.org |
| Localize | Larson et al., 2013 | https://github.com/CBIIT/Larson-Lab-CCR-NCI/tree/master |
| FISHAuxiliary | Larson et al., 2013 | https://github.com/CBIIT/Larson-Lab-CCR-NCI/tree/master |
| KNIME-based image analysis pipeline | Wan et al., 2021 | https://github.com/CBIIT/Larson-Lab-CCR-NCI/tree/master |
| MATLAB | The MathWorks, Inc. | https://www.mathworks.com |
| MatlabTrack_v6 | Ball et al., 2016 | https://sourceforge.net/projects/single-molecule-tracking |
Experimental model and subject details
All cell lines described were cultured under temperature-controlled conditions at 37C and 5% CO with humidity. The cell lines have not been authenticated.
U2OS human osteosarcoma cells
U2OS cells were cultured in DMEM with low glucose, GlutaMAX, and pyruvate (Gibco 10567) supplemented with 1% Penicillin-Streptomycin (ThermoFisher) and 10% FBS (Sigma).
MCF7 breast carcinoma cells
MCF7 TFF1-MS2 GFP cells were cultured in MEM (Corning 15–010) supplemented with 1% Pen-Strep, 10% FBS, and 2mM L-glutamine (HyClone SH30034). Pi-MYC and Pi-mCherry stable lines were cultured the same media.
MCF7 TetON MYC-EGFP cells were cultured in MEM supplemented with 1% Pen-Strep, 2mM L-glutamine, 10% Tetracycline-free FBS (Clontech) and 0.5 micrograms/mL Puromycin to maintain selection.
Saturating E2 (~12nM E2) imaging media is phenol-free MEM (Corning 17–305) supplemented with 1x Pen-Strep, 2mM L-Glutamine, and 10% FBS. Hormone depletion media is phenol-free MEM supplemented with 1% Pen-Strep, 2mM L-Glutamine, and charcoal/dextran treated FBS (HyClone). E2 (β-estradiol, Sigma) is added to this media to achieve the desired concentration.
HBEC3-KT normal human bronchial epithelial cells
HBEC3-KT were cultured and imaged in Keratinocyte Serum-Free Media supplemented with bovine pituitary extract and EGF (Gibco 17005) and 1% Pen-Strep.
NIH3T3 Mouse Fibroblasts
NIH3T3 fibroblasts were cultured in DMEM (Gibco 10564) supplemented with 1% pen-strep and 10% FBS.
Harvest Cells for Virus generation
HEK293 and Phoenix-AMPHO cells were cultured in DMEM with high glucose, GlutaMAX and pyruvate (Gibco 10569), supplemented with 1% pen-strep and 10% FBS.
Method Details
Treatment vehicles
β-estradiol (E2) was dissolved in 100% ethanol at a stock concentration of 1mM and stored in aliquots at −20C. Doxycycline was dissolved in water at a stock concentration of 9μg/mL and stored in aliquots at −20C. HBEC and MCF7 TetON MYC-EGFP stable lines were induced with 1μg/mL doxycycline to induce MYC overexpression.
Transient MYC overexpression and knockdown in MCF7
MCF7 TFF1-MS2 cells were plated in a 2-well glass bottom chamber (Nunc Lab-Tek 155379) in culture media and allowed to recover. The saturating E2 condition had a media change on Day 2 and changed into Saturating E2 imaging media along transfections of 1.5μg MYC-mCherry plasmid per well or 40nM MYC siRNA (ThermoFisher) on Day 5. Cells were imaged at Day 6 (24 hours MYC overexpression) and Day 7 (48 hours MYC knockdown). For the 50pM E2 condition, cells were hormone depleted with 2 washes and a replacement into hormone depletion media on Day 2. On Day 5, cells were changed into hormone depletion media containing 50pM E2. On Day 7 the 50pM E2/hormone depletion media was replaced and cells were transfected with MYC-mCherry plasmid or MYC siRNA and imaged after 24 hours (Day 8) or 48 hours (Day 9), respectively.
TetON MYC-EGFP stable expression in HBEC
Frozen stock of purified pTRIPZ TetON MYC-EGFP virus was obtained from the Levens lab and transduced with the Lenti-X system (Takara Bio USA, Inc.). HBEC cells were plated in T-75 flasks in antibiotic free media and grown to 80% confluency. A viral transduction mixture was prepared with 100μL virus, 40μL Lenti-X beads, and 260μL 1x PBS. The viral mixture was incubated for 30min at RT, tapping briefly every 5min to mix. The mixture was then added dropwise to the flask of cells and placed on a Lenti-X magnetic plate for 5 min and a media exchange afterwards. The flask was then incubated on the magnetic plate overnight at 37C. After 1 week, positive cells were enriched by induction 5μg/mL doxycycline for 24 hours to stimulate MYC-EGFP expression, and then sorted by FACS for GFP fluorescence. Two rounds of induction and sorting were required to fully enrich for the transduced cells.
qRT-PCR
qRT-PCR was performed in HBEC cells to measure effects of MYC overexpression and knockdown for 11 genes identified in (Wan et al., 2021). RNA was purified from cell pellets using a Quick-RNA Microprep kit (Zymo). cDNA was generated using Protoscript II (NEB) with Random Hexamers (IDT). qPCR was performed with iQ SYBR Green Supermix (BioRad) on 10 genes (CANX, DNAJC5, ERRFI1, KPNB1, MYH9, PFN1, RAB7A, RHOA, RPAP3, SEC16A, SLC2A1), using beta-actin (ACTB) as a control (primer sequences in Table S1).
Western blotting
Cells were resuspended in 20uL RIPA buffer (50mM TrisHCl pH 7.5, 150mM NaCl, 0.05% Tween-20, 1% sodium deoxycholate, 1 tablet protease inhibitor cocktail in H20) per 350,000 cells. Lysates were incubated in RIPA buffer on ice for 30min, with a brief vortex every 10 min. Lysates were spun at 14,000rpm at 4C for 10 min and supernatant was collected into new tubes. 4x LDS Sample Buffer (Invitrogen) was added to lysates and boiled for 10 min at 70C. Samples were loaded onto 10-, 12-, or 15-lane 1.0mm NuPAGE 4–12% Bis-Tris gel (Invitrogen) and run with Spectra BR protein ladder (ThermoFisher Scientific). Sample was transferred to a PDVF membrane with a Trans-Blot Turbo Transfer System (BioRad). Transfer of protein bands were verified with Ponceau stain, and membrane was then blocked in 5% milk /TBST (19.98mM Tris, 136mM NaCl, pH 7.4) for 1 hour at RT. Membrane was incubated with primary antibodies at 4C overnight at 1:1000 in milk block. Primary antibodies used were Rabbit monoclonal anti-Myc Y69 (Abcam), Mouse monoclonal Pan-Ras (Sigma-Aldrich), and Mouse monoclonal anti-GAPDH (Ambion). The next day, membranes were washed 3x in TBST and incubated with secondary antibodies 1:10,000 in milk block using anti-rabbit HRP (GE Healthcare Life Sciences) or anti-mouse HRP (GE Healthcare Life Sciences) for 1 hour at RT. The membranes were then washed 3x with TBST and developed with SuperSignal ECL reagent (ThermoFisher Scientific).
Pi-MYC design and cloning
The mCherry-NES-LOV2-NLS sequence from pTRIEX-mCherry-LOV2 (Addgene plasmid 81041) was PCR amplified with NotI-5’ and AgeI-3’ ends. The fragment was cloned into a pd4-MYC-EGFP plasmid where the GFP had been removed via restriction digest with AgeI-NotI. The resulting plasmid (pd4-MYC-LOV2) was digested with SacII-AgeI to remove the c-myc wildtype NLS and was replaced with a GeneBlock (IDT) in which the lysine and arginine residues of the c-MYC NLS sequence were replaced with alanine. The entire sequence was then PCR amplified with NotI-5’ and NdeI-3’ ends, with the 5’ primer starting at either MYC exon 2 (for Pi-MYC) or mCherry (for Pi-mCherry), and the 3’ primer at the end of the NLS which followed the LOV2 sequence. The PCR fragments were ligated into lentiviral vectors containing ampicillin and bleomycin resistance cassettes, resulting in the pLenti-Pi-MYC and pLenti-Pi-mCherry plasmids. All plasmids were purified with Qiagen maxiprep kit and verified with Sanger sequencing.
Additional control plasmids were made without photo-inducible capability. pLenti-wtMYC-mCherry (with the wildtype NLS sequence in c-myc exon 3) was cloned by PCR amplifying and inserting the MYC-mCherry sequence from pd4-MYC-LOV2 into a lentiviral vector. The resulting plasmid was digested to remove the MYC sequence and re-ligated with a short linker to create the pLenti-mCherry plasmid.
Pi-MYC and Pi-mCherry lentivirus generation
HEK293 cells were plated at 162cm flasks and grown to 80% confluency. The cells were exchanged into media with no antibiotics for 4.5 hours prior to transfection. Cells were transfected with Fugene HD (Promega) containing a mixture of 1.5μg each of Tat, Rev, Gag/Pol, and VsvG plasmids, and 30μg of the pLenti-Pi-Myc or pLenti-Pi-mCherry plasmid. Cells were incubated overnight at 37C and moved to a 32C incubator the next day to facilitate more efficient virus production. 25mL supernatant was harvested every 24 hours for two days and purified with the Virabind Lentivirus Purification Kit (Cell BioLabs) in 100μL aliquots of glycerol and stored at −80C.
NIH3T3 Pi-MYC and Pi-mCherry stable lines
NIH3T3 cells were transduced with pBABE-HRAS-V12 retrovirus (Scaffidi and Misteli, 2011) and Pi-mCherry or Pi-MYC lentivirus to create the following cell lines for stable expression: 1) wtRAS/Pi-mCherry, 2) wtRAS/Pi-MYC, 3) RASV12/Pi-mCherry, 4) RASV12/Pi-MYC. We generated the RASV12 lines first; briefly, Phoenix-AMPHO harvest cells were plated at a density of 5 million cells in 10cm plates. Cells were exchanged into media without serum or antibiotics and transfected with 10μg DNA per plate with JetPrime (VWR). Plates were moved to a 32C incubator to improve virus stability. The next day, the target cells (NIH3T3) were plated at 350,000 cells/well in a 6-well plate to prepare for transduction. Phoenix-AMPHO cell media containing viral supernatant was collected at 48 hours post transfection and applied directly to target cells at a ratio of 1:1 with NIH3T3 media, and polybrene at a concentration of 5.4μg/mL. The target cell plate was spun at 2900rpm for 2 hours at RT to facilitate efficient viral transduction and incubated at 32C overnight. The next day, cells were expanded into a T-75 flasks and returned to incubation at 37C. The following day, G418 antibiotic selection was applied for 7 days. After NIH3T3 wtRAS / RASV12 backgrounds were established, the cells were plated in 6-well plates at 200,000 cells per well in 2mL media. The next day, cells were transduced with virus solution containing 100μL glycerol aliquot of Pi-Myc or Pi-mCherry virus and 10μg polybrene in 2mL media per well. The plates were spun at 2900rpm at 32C for 2 hours to facilitate efficient transduction, and then placed into a 32C incubator overnight. Media was replaced the next day and the plates were moved to a 37C incubator. After reaching confluency, cells were transferred to T-75 flasks and subjected to antibiotic selection for 7 days with zeocin (InvivoGen). The same lentivirus transduction method was used for generating stable expression of Pi-mCherry and Pi-MYC in HBEC cells.
NIH3T3 MYC/RAS growth assay
NIH3T3 stable lines and the parental line were plated at 20,000 cells per well for two timepoints with biological replicates for. Cells were trypsinized and resuspended in < 2mL media, and the total count per well was calculated with an automated cell counter at Days 2 and 5.
Colony formation in soft agar
NIH3T3 cells were seeded in agar (Sigma) at a concentration of 40,000 cells/well in 6-well plates, with three biological replicates per condition, and NIH3T3 culture media containing 20% FBS. Each well consisted of a bottom layer of 1.5mL 1% agar/media, a middle layer containing cells suspended in 1mL 0.35% agar/media, and a top feeder layer of 1mL media. Media was added to the feeder layer twice a week for two weeks. After two weeks, cells were stained with crystal violet and imaged with Epson scanner (RGB, 1200dpi) and scored for colonies. For the light induction condition, cells were incubated in pulses of blue light (10 sec on/10 sec off) with a 455nm collimated LED (Thorlabs M455L4-C1) for 24 hours prior seeding in agar.
Photo-induction conditions
Nuclear translocation of Pi-mCherry and Pi-MYC was achieved with either a 488nm laser to illuminate a single field of view in a microscope, or a 455nm collimated blue LED to illuminate a 35mm area of a culture dish. Translocation kinetics of Pi-mCherry and Pi-MYC in U2OS cells were measured by inducing translocation with a 488nm laser at 10% power with 150ms pulses at 20 sec intervals. The nuclear intensity values for each cell were baseline corrected and then normalized. The nuclear intensity values for each cell were baseline corrected by quantifying the mean intensity of an area in the field of view that did not contain cells and subtracting the value from the nuclear fluorescence intensity values of the cells, for every timepoint in the movie. The nuclear fluorescence intensity of each cell was then normalized to its highest value over the 60-minute time series. This was to account for photo-bleaching and avoid biases in different total Pi-MYC expression levels across cells. For the HBEC growth assay, cells were seeded at 20,000 cells per well in two 12-well plates separated by a barrier in a 37C, 5% CO2 incubator. One plate was cultured under constant 455nm light at 50mW for two days (+λ condition). For the HBEC live-cell imaging experiments, cells were induced with a 488nm laser at 30% power for approximately 15 seconds at 100 second intervals.
smFISH probe design
Stellaris probe sets were ordered from LGC Biosearch Technologies and use Quasar570 and Quasar670 dyes (see Table S1 for full sequences). Probe sets labeling the 3’UTR for RAB7A, KPNB1, MYH9 and RPAP3 were designed using Stellaris Probe Designer (biosearchtech.com) with the following parameters: Masking level = 5, Max number of probes = 48, Oligo length = 20, and Minimum spacing = 2. In order to minimize off-target labeling, probe sequences were screened by BLAT to confirm they were unique to the target gene. TFF1 exon and intron probes (Rodriguez et al., 2019) were designed similarly. MS2 probes were designed to target the linker region between stem loops. Probes are reconstituted in TE buffer to a stock concentration of 25μM and stored at −20C.
smFISH labeling protocol
Cells were grown on glass coverslips (18mm #1.5 thickness, Electron Microscopy Sciences 72222-01) in 12-well plates for the experiments described in this paper. Briefly, samples were washed 3 times with Hank’s Balanced Salt Solution (HBSS) and then fixed for 10min in 4% PFA/PBS. After fixation, samples were washed 2× 10 min in PBS and permeabilized in 70% ethanol at 4C for two days.
We performed smFISH according to the Stellaris RNA FISH protocol for adherent cells, using custom made wash and hybridization buffers. Hybridization buffer is made with 10% dextran sulfate/10% formamide/2x Saline Sodium Citrate (SSC)/H2O and stored in 1mL aliquots at −20C. Wash buffer is made with 2x SSC/10% formamide/H20 and prepared fresh. On the day of labeling, cells are removed from permeabilization buffer and incubated in wash buffer for 5min at RT. Probe stocks are diluted to 100nM in hybridization buffer to a volume of 55μL per coverslip. Coverslips are placed face-down on 50μL drops of probe in a humidified container and incubated in the dark for 4 hours at 37C. Probe incubation can go overnight to increase labeling efficiency. After incubation, coverslips are placed back in a 12-well plate and washed 2× 30 min at 37C with wash buffer. Samples are then moved to RT and washed quickly with 2xSSC/H20 and then with PBS for 5min. Samples are then mounted face down on glass slides with Prolong Gold Antifade Mountant with DAPI (Invitrogen).
HaloTag labeling protocol
HBEC cells (MS2-polyclonal background) expressing Pi-mCherry or Pi-MYC were electroporated with HaloTag plasmids at a ratio of 50,000 cells : 500ng DNA : 10μL total volume with a Neon Transfection System (Invitrogen). The electroporation pulse settings were: Voltage = 1400V, Width = 20ms, Pulses = 2. After electroporation, cells were plated into an 8-well glass bottom chamber (Cellvis C8-1.5H-N) at a density of 50,000 cells/well in HBEC media and were cultured for 2–3 days to allow for recovery and expression of HaloTag plasmid.
On the day of imaging, cells were labeled with Halo ligand conjugated to JaneliaFluor-646 (JF646) dye (Grimm et al., 2017). Cells were labeled and incubated for 15min then rinsed with HBEC media with a 3x wash, 10-minute incubation, and a final 3x wash. For the Halo-GR condition, cells were induced with 100nm dexamethasone to stimulate GR nuclear translocation immediately prior to imaging.
smFISH microscope setup
Samples were imaged on a custom ‘RAMM’ Rapid Automated Modular Microscope (ASI Imaging, OR, USA). The microscope consists of the RAMM chassis with a quad bandpass filter for imaging DAPI, GFP, Cy3, and Cy5 (VCGR-SPX-P01-PC, Chroma Technology Corp., VT, USA). The RAMM is coupled to the following hardware: MS-2000 motorized XY stage (ASI Imaging), ORCA-Flash4 V2 CMOS camera (Hamamatsu Photonics K.K., Japan), 40x/1.4 N.A. oil immersion objective (Zeiss, Germany), SpectraX Light Engine (Lumencor, OR, USA), FW-1000 High Speed Emission Filter Wheel (ASI Imaging), and TG-1000 Tiger Controller (ASI Imaging). The filter wheel contains additional emission filters for Cy3 and Cy5 (605/70, 700/75, Semrock, Inc., NY, USA). Automated control of the microscope is programmed through a MicroManager computer interface (Edelstein et al., 2010). The following excitation wavelengths/camera exposures were used for each channel: 395nm/25ms (DAPI), 550nm/500ms (Cy3), and 640nm/500ms (Cy5). Z-stacks with a 0.5μm step size were used to image the whole volume of the cell (U2OS and HBEC = ~11 slices, MCF7= ~14 slices). The stack is then converted into a maximum intensity projection to produce a single image containing all the volume information for a given field of view.
Live-cell microscope setup
MCF7 cells were imaged on a Zeiss LSM780 confocal microscope with a 37C, 5% CO2 environment. Imaging was performed with 488nm and 594nm excitation at 2% power, 63× 1.4 N.A. oil immersion objective, 2.5μm pinhole size, 1.5x zoom, and 1024×1024 frame size acquired at 16-bit depth. Z-stacks were acquired at 14 slices/0.5μm step size. The imaging duration was 512 frames / 100 sec intervals (~14.2 hours), allowing for 1 well with 4/FOV per well to be imaged per acquisition.
For high-throughput imaging, HBEC cells were cultured in 96-well plates (Brooks Life Science Systems MGB096-1-2-LG-L) and microscopy was performed on an automated Yokogawa Cell Voyager 7000S dual spinning disk microscope with a 37C, 5% CO2 and 80% humidity environment. The microscope was equipped with a quad-bandpass filter (405/488/561/604 nm) and an additional GFP 525/50 emission filter. Imaging was performed with 488nm excitation at 30% power and a 60x/1.2 N.A. water immersion objective. Fluorescence was detected with an Andor Neo 5.5 sCMOS camera with 250ms exposure time, 1278×1078 frame size, and 2×2 binning acquired at 16-bit depth. Z-stacks were acquired with 14 slices/0.5μm step size. The imaging duration was 6 or 15 hours with 100 second frame intervals, allowing for 8 wells with 5 FOV per well to be imaged in a single time course. Flat-field correction and maximum intensity projections were processed on-the-fly by Yokogawa acquisition software.
HILO microscope setup
For SMT, we used a custom wide-field microscope capable of simultaneous imaging in three channels that is based on previously described designs (English and Singer, 2015; Morisaki et al., 2016). Briefly, three solid-state excitation lasers at wavelengths 488, 561, and 647 nm (OBIS, Coherent Inc., CA, USA) are combined, expanded to provide more even illumination at the sample, and focused at the back focal plane of a 100x, 1.49 N.A., oil immersion objective (Olympus Scientific Solutions, MA, USA). To reduce background, HILO illumination (Tokunaga et al., 2008) is achieved by moving the radial position of the beam in the objective back aperture with a movable mirror, and the thickness of the excitation is adjusted by use of a manual diaphragm. Fluorescence emission is separated from scattered laser light by use of a quad-band dichroic (ZT405/488/561/647, Chroma Technology Corp., VT, USA), and the emission bands are separated by two long-pass filters (T588lpxr, T660lpxr), and emission filters (525/50, 609/58 and 736/128, Semrock, Inc., NY, USA) before being focused on separate EMCCD cameras (Evolve 512 Delta, Teledyne Photometrics, Tucson, AZ), with 200 mm tube lenses. The combination of objective lens and tube lens results in a total magnification of 111x, corresponding to a XY pixel size of 144 nm. The sample is held on a motorized XY translation stage with piezo Z (PZ-2000 XYZ, Applied Scientific Instrumentation, OR, USA), which is mounted on a Rapid Automated Modular Microscope System (RAMM, Applied Scientific Instrumentation). All lasers and cameras are synchronized using a microcontroller board (Arduino UNO), and images are collected using the open source microscope control software, Micro-Manager (Edelstein et al., 2014). Time-lapse images were collected at intervals of 200ms for 2 min (600 frames total). Prior to imaging, the field-of-view was exposed to 100μW of 488 nm laser light for 4 min to induce translocation of either Pi-mCherry or Pi-Myc into the nucleus. The 488 nm laser was left on during imaging of the JF646 to retain Pi-mCherry or Pi-Myc in the nucleus (Video S5). The JF646 dye attached to the protein of interest was excited with 647 nm with a laser power of 1 mW measured at the back aperture of the objective.
SLAM-Seq sample preparation
RNA isolation
Note: DTT is added during RNA isolation to keep the sample under reducing conditions.
Thaw lysate and incubate 5 min at room temperature.
Add 200μl chloroform per 1 ml of TRIzol®.
Shake tube vigorously for 15 sec.
Incubate at room temperature for 2–3 min.
Spin down at 16,000 × g for 15 min at 4C.
Transfer aqueous phase to new tube.
Add 1/100 volume of 10 mM DTT (0.1 mM final concentration), 1 volume 2-propanol and optionally 1 μl glycogen (20 mg/ml). Vortex well.
Incubate 10 min at room temperature.
Spin down at 16,000 × g for 20 min at 4C.
Discard supernatant.
Add 500 μl 75% EtOH and 5 μl of 10 mM DTT, vortex well.
Spin down at 7,500 × g for 5 min at room temperature.
Remove supernatant, let the pellet dry for 5–10 min and resuspend in appropriate amount of water supplemented with 1 mM DTT (final concentration).
Incubate for 10 min at 5C.
Measure concentration by Nanodrop and freeze RNA at −80C or proceed to the next step.
Thiol modification (Iodoacetamide treatment)
Prepare the reaction mix for each Iodoacetamide (IAA) treatment reaction: 15μL RNA, 5μL IAA (100mM freshly prepared), 5μL NaPO4 (500 mM, pH8), 25μL DMSO.
Incubate reaction at 50°C for 15 min.
Stop reaction by quenching the reaction with 1μl 1M DTT.
Add 1μl glycogen (20 mg/ml), 5 μl NaOAc (3M, pH 5.2), 125 μl EtOH 100%, vortex and precipitate for 30 min at −80°C.
Spin down at 16,000 × g for 30 min.
Wash with 1 ml 75% EtOH, vortex.
Spin down at 16,000 × g for 10 min.
Remove supernatant and let the pellet dry for 5–10 min.
Resuspend in 10μl H2O.
Proceed with RNA quality control and library preparation.
Quantification and Statistical Analysis
The statistical details for the data described in this paper, including the value and definition of n and dispersion and precision measures, can be found in the figure legends.
smFISH image analysis
Cell and nuclear masks were generated for each image using the open source software CellProfiler (Carpenter et al., 2006) with Otsu’s method for thresholding. Using custom software Localize and FishAuxiliary (LarsonLab GitHub), RNA spots were identified and merged with the cell and nuclear masks to define single mRNA and TS within each cell. The basic forms of output data were: 1) Number of cellular and nuclear RNA/cell. 2) Number of TS per cell, and 3) Integrated fluorescence intensity of each identified spot. Intron probes identify TS and unspliced RNA, whereas 3’UTR/exon probes identify TS, unspliced RNA, and mRNA. To yield the discrete number of nascent RNA at a TS, we use the 3’UTR/exon probe set for a given gene and divide TS intensity values by the median intensity of RNA in the dataset (the majority of values coming from single mRNA in the cytosol).
Live-cell image analysis
Automated image analysis of live-cell time series data was carried out in the KNIME open-source workflow environment (64-bit Version 3.5.1) (Berthold et al., 2008) containing the following image processing and scripting nodes: KNIP, Version 1.5.3.201611190650 (Dietz and Berthold, 2016); R, 64-bit Version 3.3.1; and Python, 64-bit Version 2.7.12. Our analysis modules consisted of five steps: 1) Nucleus segmentation and tracking, 2) Registration of tracked nuclei, 3) TS tracking within registered nuclei, 4) Filtering correct TS tracks, and 5) Normalizing tracks and fitting to a 2-state HMM to classify ON and OFF periods.
Nucleus segmentation was performed on the MS2-GFP channel using a Random Forest Classifier. A training set with supervised learning of a given cell type (MCF7 or HBEC) was conducted in which correctly and incorrectly segmented nuclei were manually classified in single frames of a representative time series movie. The RF model was tested on unseen data and then applied across all acquisitions of the given cell type to segment the nuclei. Registration of tracked nuclei in X-Y was done after segmentation. The reason we limited the movement of the nuclei by registration is that cell movement and long transcription OFF periods that can occur over a 6–15 hour range. This makes it difficult to set an adequate search radius that would allow us to successfully link bursts of the same allele into the same trace. TS tracking was performed automatedly using previously developed deep learning methods (Gudla et al., 2017). All TS tracks were then manually inspected and filtered for correct tracking before analysis. All tracks of a given acquisition were batch analyzed in KNIME. The pixel intensity values of filtered traces were normalized to the highest and lowest value on a per track basis, and then fit to a 2-state HMM based on custom IDL software previously described (Coulon et al., 2014). The outputs of the analysis were the raw and normalized intensity values of each track, the HMM fit, and a distribution of the ON and OFF periods of each track.
SMT image analysis
Tracking was performed automatically with the MATLAB-based (The Mathworks, Natick, MA). Track Record software (freely available at https://sourceforge.net/projects/single-molecule-tracking). Track Record links molecule positions in two consecutive planes using a nearest-neighbor approach (Mazza et al., 2012). Molecules are considered bound if they move less than rmax = 530 nm from one frame to the next (Ball et al., 2016) for at least Nmin = 8 frames. The value for rmax is the distance that 99% of tagged histone H2 molecules moved in the same imaging conditions. Assuming a moderate diffusion coefficient, Df, of 0.5 μm2/s, with our frame interval, Δt, of 200 ms, The minimum number of frames, Nmin, is calculated such that the probability, P, that a freely diffusing molecule will be classified as bound is below 1% (Mazza et al., 2012):
We used two different methods to analyze the residence times of the various target proteins, both of which attempt to correct for photobleaching. The first method relies on correcting for photobleaching separately in each experiment by fitting the number of particles over time to a bi-exponential. The survival curve is then divided by this photobleaching bi-exponential before being fit to both mono- and bi-exponentials (Mazza et al., 2012). An F-test is performed to determine if the added parameters in the bi-exponential provide a statistically significant improvement in the fit.
The second method requires a separate dataset where a very stable molecule, such as the histone H2B, is measured (Garcia et al., 2021b). For our studies, we used Halo-H2B expressed in the HBEC Pi-mCherry background as our control for the photobleaching analysis. We induced Pi-mCherry translocation followed by an SMT measurement as described earlier, and fit the resulting survival curve to a triexponential model,
where the slowest decay rate k3 is assumed to be loss of fluorescence due to photobleaching, and is used as the correction to the survival curves of the samples of interest. This latter method fits the data to alternate models to the mono- and bi-exponential including a power law, and a combination of a power law and an exponential. The Bayesian Information Criterion (BIC) is calculated for each of the four fits. The two models with the lowest BIC that are described in this paper are the bi-exponential model:
And the power law with exponential:
SLAM-Seq analysis
The sample preparation protocol for SLAM-Seq is described in Supplemental Experimental Procedures. SLAM-seq analysis was done according to a previous publication (Herzog et al., 2017) with minor changes. Briefly, we performed whole transcriptome sequencing instead of 3’ end sequencing. Reads are pre-processed with fastp (https://github.com/OpenGene/fastp) and then mapped by SlamDunk map, filter and snp (http://t-neumann.github.io/slamdunk/). T>C converted reads were separated by Alleyoop. The reads with T>C conversion were normalized by total mapped reads in each group. Due to the multimapper reconciliation strategy used in SlamDunk (multimappers are assigned towards 3’ end), there is a distort of the signal to the 3’ end of the gene. Metagene coverage matrix and profile were generated through deepTools (https://deeptools.readthedocs.io/en/develop/).
Supplementary Material
Table S1. Related to Figure 3. Oligo sequences for qRT-PCR experiments and smFISH probe sets.
Table S3. Related to Figure 3. MS2-labeled genes identified HBEC MS2-GFP polyclonal population.
Video S1. Related to Figure 2. 48-hour time course of TetON MYC-EGFP expression in HBEC cells with induction at T=0. 6 fields of view are shown.
Video S2. Related to Figure 4. 1-hour time course of Pi-MYC nuclear translocation in U2OS cells with alternating 10-minute periods of light induction (indicated by blue square). Time stamp is hh:mm.
Video S3. Related to Figure 4. Representative movie of blue light-induced nuclear translocation in NIH3T3. 10-minute time course of NIH3T3 RASV12-Pi-mCherry stable cell line; 5 min activation followed by 5 min recovery.
Video S4. Related to Figure 7. 30-minute time course of Pi-MYC translocation in HBEC cells with 15 min of light induction (indicated by blue square). 2 fields of view are shown.
Video S5. Related to Figure 7. Example of induction/imaging time course used for SMT experiments. HBEC cells are induced with light for 6 min to translocate Pi-mCherry or Pi-MYC, with an SMT measurement taking place in the last 2 min.
Video S6. Related to Figure 7. Example 2-minute time courses of HBEC cells that stably express Pi-mCherry or Pi-MYC (channel not shown) and transiently express Halo-tag transcription factors measured by SMT.
Highlights.
MYC overexpression leads to genome-wide increases in transcription burst duration
MYC globally alters dwell times of general transcription factors
Coupled photoactivation and RNA imaging enables precise measurements of gene regulation
Acknowledgements
We thank Zuqin Nie and Ying Zheng for providing protocols and reagents for MYC biochemical characterization. The pTRIEX-mCherry-LOV2 plasmid was a gift from Klaus Hahn (Addgene #81041). The HBEC3-KT cell line was a gift from John Minna (ATCC #CRL-4051). We thank the NCI HiTIF Core for use of the Yokogawa CV7000 microscope and assistance with the KNIME analysis pipelines. David Garcia and Arpita Upadhyaya provided guidance and critical feedback on the SMT experiments. We thank R. Louis Schiltz from the Gordon Hager lab (NCI) for providing additional reagents used in this paper. This work utilized computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). This work was supported by the NIH Intramural Research Program, the NCI-UMD Graduate Partnership Program, and NSF Award DGE-1632976.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, and Brinster RL (1985). The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318, 533–538. [DOI] [PubMed] [Google Scholar]
- Adamskaya N, Dungel P, Mittermayr R, Hartinger J, Feichtinger G, Wassermann K, Redl H, and Van Griensven M (2011). Light therapy by blue LED improves wound healing in an excision model in rats. Injury 42, 917–921. [DOI] [PubMed] [Google Scholar]
- Al-azzeh E, Dittrich O, Vervoorts J, Blin N, Gött P, and Lüscher B (2002). Gastroprotective peptide trefoil factor family 2 gene is activated by upstream stimulating factor but not by c-Myc in gastrointestinal cancer cells. Gut 51, 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball DA, Mehta GD, Salomon-Kent R, Mazza D, Morisaki T, Mueller F, Mcnally JG, and Karpova TS (2016). Single molecule tracking of Ace1p in Saccharomyces cerevisiae defines a characteristic residence time for non-specific interactions of transcription factors with chromatin. Nucleic Acids Res. 44, e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluapuri A, Hofstetter J, Dudvarski Stankovic N, Endres T, Bhandare P, Vos SM, Adhikari B, Schwarz JD, Narain A, Vogt M, et al. (2019). MYC Recruits SPT5 to RNA Polymerase II to Promote Processive Transcription Elongation. Mol. Cell 74, 674–687.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman CR, Hsu SC, Hsiung CC-S, Raj A, and Blobel GA (2016). Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol. Cell 62, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, and Wiswedel B (2008). KNIME: The Konstanz Information Miner. (Springer, Berlin, Heidelberg: ), pp. 319–326. [Google Scholar]
- Cai L, Dalal CK, and Elowitz MB (2008). Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature 455, 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WK, Jayanth N, Mullen S, Tan TH, Jung YJ, and Cissé II (2016). Super-resolution imaging of fluorescently labeled, endogenous RNA Polymerase II in living cells with CRISPR/Cas9mediated gene editing. Sci. Rep 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comorosan S, Kappel W, Constantinescu I, Gheorghe M, Ionescu E, Pîrvu C, Cinca S, and Cristache L (2009). Green light effects on biological systems: a new biophysical phenomenon. J. Biol. Phys 35, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon A, Ferguson ML, de Turris V, Palangat M, Chow CC, and Larson DR (2014). Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife 3, e03939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz C, and Berthold MR (2016). KNIME for open-source bioimage analysis: A tutorial. Adv. Anat. Embryol. Cell Biol 219, 179–197. [DOI] [PubMed] [Google Scholar]
- Donovan BT, Huynh A, Ball DA, Patel HP, Poirier MG, Larson DR, Ferguson ML, and Lenstra TL (2019). Live cell imaging reveals the interplay between transcription factors, nucleosomes, and bursting. EMBO J. 38, e100809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English BP, and Singer RH (2015). A three-camera imaging microscope for high-speed single-molecule tracking and super-resolution imaging in living cells. In Biosensing and Nanomedicine VIII, (SPIE; ), p. 955008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza JH, and Mercado-Uribe H (2017). Visible light neutralizes the effect produced by ultraviolet radiation in proteins. J. Photochem. Photobiol. B Biol 167, 15–19. [DOI] [PubMed] [Google Scholar]
- Falo-Sanjuan J, Lammers NC, Garcia HG, and Bray SJ (2019). Enhancer Priming Enables Fast and Sustained Transcriptional Responses to Notch Signaling. Dev. Cell 50, 411–425.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch C, Baumgärtner S, Kuban M, Steinshorn D, Reid G, and Legewie S (2018). Estrogen dependent control and cell to cell variability of transcriptional bursting. Mol. Syst. Biol 14, e7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya T, Lim B, and Levine M (2016). Enhancer Control of Transcriptional Bursting. Cell 166, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DA, Johnson TA, Presman DM, Fettweis G, Wagh K, Rinaldi L, Stavreva DA, Paakinaho V, Jensen RAM, Mandrup S, et al. (2021a). An intrinsically disordered region-mediated confinement state contributes to the dynamics and function of transcription factors. Mol. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DA, Fettweis G, Presman DM, Paakinaho V, Jarzynski C, Upadhyaya A, and Hager GL (2021b). Power-law behavior of transcription factor dynamics at the single-molecule level implies a continuum affinity model. Nucleic Acids Res. 49, 6605–6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, and White RJ (2005). c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol 7, 311–318. [DOI] [PubMed] [Google Scholar]
- Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, et al. (2017). A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudla PR, Nakayama K, Pegoraro G, and Misteli T (2017). SpotLearn: Convolutional Neural Network for Detection of Fluorescence In Situ Hybridization (FISH) Signals in High-Throughput Imaging Approaches. Cold Spring Harb. Symp. Quant. Biol 82, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, and O’Shea EK (2015). Limits on information transduction through amplitude and frequency regulation of transcription factor activity. Elife 4, e06559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, and Darzacq X (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6, e25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, Von Haeseler A, Zuber J, and Ameres SL (2017). Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TY-T, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. (2015). The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeddin I, Récamier V, Bosanac L, Cissé II, Boudarene L, Dugast-Darzacq C, Proux F, Bénichou O, Voituriez R, Bensaude O, et al. (2014). Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife 3, e02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkat M, Resetca D, Lourenco C, Chan PK, Wei Y, Shiah YJ, Vitkin N, Tong Y, Sunnerhagen M, Done SJ, et al. (2018). MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 72, 836–848.e7. [DOI] [PubMed] [Google Scholar]
- Khan AO, White CW, Pike JA, Yule J, Slater A, Hill SJ, Poulter NS, Thomas SG, and Morgan NV (2019). Optimised insert design for improved single-molecule imaging and quantification through CRISPR-Cas9 mediated knock-in. Sci. Rep 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Sabò A, and Amati B (2015). MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607. [DOI] [PubMed] [Google Scholar]
- Land H, Chen AC, Morgenstern JP, Parada LF, and Weinberg RA (1986). Behavior of myc and ras oncogenes in transformation of rat embryo fibroblasts. Mol. Cell. Biol 6, 1917–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH (2009). Extracting kinetics information from single-molecule fluorescence resonance energy transfer data using hidden markov models. J. Phys. Chem. B 113, 11535–11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LM, Edwards MC, Meyers ZR, Talbot CC, Hao H, Blum D, Iorns E, Tsui R, Denis A, Perfito N, et al. (2018). Replication study: Transcriptional amplification in tumor cells with elevated c-Myc. Elife 7, e30274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Vorontchikhina M, Wang Y-L, Faiola F, and Martinez E (2008). STAGA Recruits Mediator to the MYC Oncoprotein To Stimulate Transcription and Cell Proliferation. Mol. Cell. Biol 28, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, Kisker C, Wolf J, Eilers M, and Wolf E (2016). Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 5, e15161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- May FE, and Westley BR (1988). Identification and characterization of estrogen-regulated RNAs in human breast cancer cells. J. Biol. Chem 263, 12901–12908. [PubMed] [Google Scholar]
- Mazza D, Abernathy A, Golob N, Morisaki T, and McNally JG (2012). A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 40, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan IJ, Dahlman-Wright K, Ford J, and Wright APH (1996). Functional interaction of the cMyc transactivation domain with the TATA binding protein: Evidence for an induced fit model of transactivation domain folding. Biochemistry 35, 9584–9593. [DOI] [PubMed] [Google Scholar]
- Mehta GD, Ball DA, Eriksson PR, Chereji RV, Clark DJ, McNally JG, and Karpova TS (2018). Single-Molecule Analysis Reveals Linked Cycles of RSC Chromatin Remodeling and Ace1p Transcription Factor Binding in Yeast. Mol. Cell 72, 875–887.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T, Lyon K, DeLuca KF, DeLuca JG, English BP, Zhang Z, Lavis LD, Grimm JB, Viswanathan S, Looger LL, et al. (2016). Real-time quantification of single RNA translation dynamics in living cells. Science 352, 1425–1429. [DOI] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niopek D, Benzinger D, Roensch J, Draebing T, Wehler P, Eils R, and Di Ventura B (2014). Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat. Commun 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paakinaho V, Presman DM, Ball DA, Johnson TA, Schiltz RL, Levitt P, Mazza D, Morisaki T, Karpova TS, and Hager GL (2017). Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat. Commun 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palangat M, Anastasakis DG, Fei DL, Lindblad KE, Bradley R, Hourigan CS, Hafner M, and Larson DR (2019). The splicing factor U2AF1 contributes to cancer progression through a noncanonical role in translation regulation. Genes Dev. 33, 482–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patange S, Ball DA, Karpova TS, and Larson DR (2021). Towards a ‘Spot On’ Understanding of Transcription in the Nucleus. J. Mol. Biol 433, 167016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JR, Fisher BE, Baranello L, Liu JC, Kambach DM, Nie Z, Koh WS, Luo J, Stommel JM, Levens D, et al. (2017). Global Inhibition with Specific Activation: How p53 and MYC Redistribute the Transcriptome in the DNA Double-Strand Break Response. Mol. Cell 67, 1013–1025.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presman DM, Ball DA, Paakinaho V, Grimm JB, Lavis LD, Karpova TS, and Hager GL (2017). Quantifying transcription factor binding dynamics at the single-molecule level in live cells. Methods 123, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, DiMaio JM, et al. (2004). Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 64, 9027–9034. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, and Larson DR (2020). Transcription in Living Cells: Molecular Mechanisms of Bursting. Annu. Rev. Biochem 89, 189–212. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, Ren G, Day CR, Zhao K, Chow CC, and Larson DR (2019). Intrinsic Dynamics of a Human Gene Reveal the Basis of Expression Heterogeneity. Cell 176, 213–226.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohringer S, Holnthoner W, Chaudary S, Slezak P, Priglinger E, Strassl M, Pill K, Mühleder S, Redl H, and Dungel P (2017). The impact of wavelengths of LED light-therapy on endothelial cells. Sci. Rep 7, 10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabò A, Kress TR, Pelizzola M, De Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al. (2014). Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, and Misteli T (2011). In vitro generation of human cells with cancer stem cell properties. Nat. Cell Biol 13, 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, and Nevins JR (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14, 2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CA, and Groudine M (1991). Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res 56, 1–48. [DOI] [PubMed] [Google Scholar]
- Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD, et al. (2016). Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell 165, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, and Sakata-Sogawa K (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 159–161. [DOI] [PubMed] [Google Scholar]
- Vita M, and Henriksson M (2006). The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol 16, 318–330. [DOI] [PubMed] [Google Scholar]
- Wan Y, Anastasakis DG, Rodriguez J, Palangat M, Gudla P, Zaki G, Tandon M, Pegoraro G, Chow CC, Hafner M, et al. (2021). Dynamic imaging of nascent RNA reveals general principles of transcription dynamics and stochastic splice site selection. Cell 184, 2878–2895.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Vilela M, Winkler A, Tarnawski M, Schlichting I, Yumerefendi H, Kuhlman B, Liu R, Danuser G, and Hahn KM (2016). LOVTRAP: An optogenetic system for photoinduced protein dissociation. Nat. Methods 13, 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, and Sears RC (2011). Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 71, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Resetca D, Li Z, Johansson-Åkhe I, Ahlner A, Helander S, Wallenhammar A, Morad V, Raught B, Wallner B, et al. (2019). Multiple direct interactions of TBP with the MYC oncoprotein. Nat. Struct. Mol. Biol 26, 1035–1043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 3. Oligo sequences for qRT-PCR experiments and smFISH probe sets.
Table S3. Related to Figure 3. MS2-labeled genes identified HBEC MS2-GFP polyclonal population.
Video S1. Related to Figure 2. 48-hour time course of TetON MYC-EGFP expression in HBEC cells with induction at T=0. 6 fields of view are shown.
Video S2. Related to Figure 4. 1-hour time course of Pi-MYC nuclear translocation in U2OS cells with alternating 10-minute periods of light induction (indicated by blue square). Time stamp is hh:mm.
Video S3. Related to Figure 4. Representative movie of blue light-induced nuclear translocation in NIH3T3. 10-minute time course of NIH3T3 RASV12-Pi-mCherry stable cell line; 5 min activation followed by 5 min recovery.
Video S4. Related to Figure 7. 30-minute time course of Pi-MYC translocation in HBEC cells with 15 min of light induction (indicated by blue square). 2 fields of view are shown.
Video S5. Related to Figure 7. Example of induction/imaging time course used for SMT experiments. HBEC cells are induced with light for 6 min to translocate Pi-mCherry or Pi-MYC, with an SMT measurement taking place in the last 2 min.
Video S6. Related to Figure 7. Example 2-minute time courses of HBEC cells that stably express Pi-mCherry or Pi-MYC (channel not shown) and transiently express Halo-tag transcription factors measured by SMT.
Data Availability Statement
The SLAM-Seq dataset generated in this paper has been deposited at GEO and is publicly available as of the date of publication. The accession number is listed in the key resources table.
No new custom code was generated for this paper.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Myc Y69 | Abcam | Cat# ab32072 |
| Mouse monoclonal Pan-Ras | Sigma-Aldrich | Cat# OP40 |
| Mouse monoclonal anti-GAPDH | Ambion | Cat# AM4300 |
| Anti-rabbit HRP | GE Healthcare Life Sciences | Cat# NA934 |
| Anti-mouse HRP | GE Healthcare Life Sciences | Cat# NA931V |
| Chemicals, peptides, and recombinant proteins | ||
| β-estradiol (E2) | Sigma-Aldrich | Cat# E8875 |
| Doxycycline | Sigma-Aldrich | Cat# D3447 |
| Zeocin | InvivoGen | Cat# ant-ZN |
| Dexamethasone | Sigma-Aldrich | Cat# D1756 |
| Agar (for colony formation assay) | Sigma-Aldrich | Cat# A1296 |
| JF646 HaloTag ligand | Grimm et al., 2017 | N/A |
| 4-thiouridine (4sU) | Sigma-Aldrich | Cat# T4509 |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat# I1149 |
| TRIzol Reagent | Invitrogen | Cat# 15596018 |
| Critical commercial assays | ||
| Virabind Lentivirus Purification Kit | CellBioLabs | Cat# VPK-104 |
| iQ SYBR Green Supermix | Bio-Rad | Cat# 170880 |
| Deposited data | ||
| SLAM-Seq | This manuscript | GEO: GSE189943 |
| Experimental models: Cell lines | ||
| U2OS MS2-PP7 Beta-globin reporter | Coulon et al., 2014 | N/A |
| MCF7-TFF1 MS2 | Rodriguez et al., 2019 | N/A |
| MCF7 TetON MYC-EGFP | Eric Batchelor Lab | N/A |
| NIH3T3 | ATCC | Cat# CRL-1658 |
| NIH3T3 HRAS V12/Pi-MYC | This manuscript | N/A |
| NIH3T3 HRAS V12/Pi-mCherry | This manuscript | N/A |
| HBEC3-KT | ATCC | Cat# CRL-4051 |
| HBEC3-KT with intronic MS2 stem loops (polyclonal population) | Wan et al., 2021 | N/A |
| HBEC3-KT with intronic MS2 stem loops (single cell clones) | Wan et al., 2021 | N/A |
| HBEC3-KT TetON MYC-EGFP | This manuscript | N/A |
| HBEC3-KT MS2 polyclonal / Pi-MYC | This manuscript | N/A |
| HBEC3-KT MS2 polyclonal / Pi-mCherry | This manuscript | N/A |
| HBEC3-KT MS2 single cell clones / Pi-MYC | This manuscript | N/A |
| HBEC3-KT MS2 single cell clones / Pi-mCherry | This manuscript | N/A |
| Oligonucleotides | ||
| qPCR primers | IDT | SeeTable S1 |
| Single-molecule RNA FISH probes | LGC Biosearch Technologies | SeeTable S1 |
| Recombinant DNA | ||
| pTRIEX-mCherry-LOV2 | Addgene | Cat# 81041 |
| pd4-MYC-EGFP | Nie et al., 2012 | N/A |
| pBABE-neo-HRAS-V12 | Scaffidi and Misteli, 2011 | N/A |
| pTRIPZ TetON MYC-EGFP | David Levens Lab | N/A |
| pLenti-Pi-Myc | This manuscript | N/A |
| pLenti-Pi-mCherry | This manuscript | N/A |
| pHaloTag-GR | Morisaki et al., 2014 | N/A |
| pHaloTag-H2B | Mazza et al., 2012 | N/A |
| pFC15A-HaloTag-MED1 | Gordon Hager Lab | N/A |
| pFN21A-HaloTag-TBP | Promega | Cat# FHC11099 |
| pFN21A-HaloTag-SUPT5H (SPT5) | Promega | Cat# FHC02125 |
| pFN21A-HaloTag-POLR2A | Promega | Cat# FHC28428 |
| pFC15A-HaloTag-MYC | This manuscript | N/A |
| pFC15A-HaloTag-MYC-S62A | This manuscript | N/A |
| Software and algorithms | ||
| CellProfiler | Carpenter et al., 2006 | https://cellprofiler.org |
| Localize | Larson et al., 2013 | https://github.com/CBIIT/Larson-Lab-CCR-NCI/tree/master |
| FISHAuxiliary | Larson et al., 2013 | https://github.com/CBIIT/Larson-Lab-CCR-NCI/tree/master |
| KNIME-based image analysis pipeline | Wan et al., 2021 | https://github.com/CBIIT/Larson-Lab-CCR-NCI/tree/master |
| MATLAB | The MathWorks, Inc. | https://www.mathworks.com |
| MatlabTrack_v6 | Ball et al., 2016 | https://sourceforge.net/projects/single-molecule-tracking |