

Abstract
Most cancer patients receive radiotherapy during the course of their disease. Improvements in the therapeutic index have been based on physical improvements in delivery. Radiosensitizer development targeting tumor cells has not yielded effective agents. Recent investigations have focused on the tumor stroma as a potential target for radiosensitization. We report that depletion of tumor associated macrophages by systemic or local injection of macrophage depleting liposomal clodronate prior to radiotherapy increased the anti-tumor effects of ionizing radiation (IR) as a large single dose (20 Gy) or fractionated dose (2 Gy × 10). Co-implantation of tumor cells with BM-derived macrophages (BMDMφ) increased tumor radioresistance. Studies using mice with germ line deletions of TNF receptors 1 and 2 (TNFR1,2−/−) or TNFα (TNF−/−) and treatment with a soluble TNF receptor fusion protein (EnbrelR) showed that radioresistance mediated by BMDMφ required intact TNFα signaling. Radiation exposure led to the upregulation of VEGF by macrophages, and neutralizing antibodies to VEGF also enhanced the anti-tumor response to IR demonstrating that the radioprotective effect of TNFα was mediated by VEGF production in tumor associated macrophages (TAMφ). These data provide a mechanistic basis for targeting macrophage populations generally, and TNFα induced macrophage VEGF specifically, to improve radiotherapy.
Keywords: BMDMφ, radiation, TNFα signaling, VEGF
INTRODUCTION
Radiation therapy is a common cancer treatment usually delivered in a fractionated schedule (daily 1.8–3Gy) for 6–8 weeks. In some selected lung cancers, or limited forms of metastasis, very large doses (1–5 treatments of 10–20 Gy) may be employed (1). Despite research into the radiobiology and physics of radiotherapy, many patients fail within the irradiated target volume. Currently, the most effective radiosensitizers represent the commonly used chemotherapeutic agents (2). These drugs are not designed as selective radiosensitizers and clinical improvement reflected by increased local control is achieved at the expense of significant normal tissue toxicity. More selective radiosensitizing agents and new treatment strategies are required to improve the therapuetic index in radiotherapy.
Research on the mechanisms of tumor radioresistance and the development of radiosensitizers focuses on tumor cells or exploiting differences in the oxygenation status of tumors and normal tissue (3). Recently the tumor-associated stroma has gained attention as an important component of the response to radiotherapy (4, 5) and investigations have centered on the role of the tumor associated vasculature (6, 7). Several clinical trials are underway to investigate the role of angiogenesis inhibitors in combination with radiotherapy (8–11). The results are preliminary and though promising, show mixed results in terms of improving radiotherapy outcomes and unexpected toxicities have been reported (11–13). Conversely, little is known about other components of the tumor associated stroma and the effect of these cells on the response of tumors to radiotherapy.
Macrophages are multifunctional cells of the myeloid lineage, phenotyped as CD14+. Macrophages engulf microbes and cell debris, are antigen presenting cells and secrete cytokines and chemokines. Macrophage function is described within the context of normal physiological or pathological microenvironments. Macrophages are designated as M1 classically activated macrophages or M2 alternatively activated macrophages. Pro-inflammtory M1 macrophages are activated by LPS and IFNγ, secrete TNFα and IL-12 and support T cell function. M2 macrophages are considered to be anti-inflammatory and immunosuppressive, expressing F4/80, CD11b and CD206, secrete IL-10, IL-4 and TGFβ and down regulate T cell function (14, 15) Tumor associated macrophages (TAMφ) are usually characterized as M2. However, functional overlap exists between characteristics of M1 and M2 macrophages. Additional/expanded definitions of TAMφ populations have been investigated as determinants of tumor progression and metastasis. These populations include CD11b+/Gr1+ myeloid suppressor cells as well as Tie2+ expressing macrophages (15, 16). Emerging data suggest key roles for these various monocyte/macrophage cells in generating pro-angiogenic signals during tumor growth.
Radiation was originally reported to prime the anti-tumor effects of macrophages through production of TNFα and nitric oxide (NO) (17–20). However, Milas, et al., investigated the association between TAMφ and the response of tumors to radiotherapy and noted a trend toward decreasing radiocurability with increasing TAMφ content. These investigators recognized the contradiction between potential beneficial (phagocytic function) and deleterious functions (pro-angiogenic phenotypes) of TAMφ (21–23). Recently, Tsai, et al., reported that irradiated macrophages contributed to tumor growth through increased secretion of COX-2, iNOS and Arg-1 although no direct mechanisms for promotion of tumor growth were described (24). Brown, et al., reported that cells of the myeloid/macrophage lineage contributed to tumor regrowth following radiation by increasing tumor vasculogenesis when tumor angiogenesis was suppressed by prior irradiation of the tumor bed (25). Collectively these studies raise the possibility that macrophage populations contribute to tumor radioprotection. Moreover, radiation of the tumor microenvironment might upregulate macrophage derived cytokines that promote tumor growth, survival, angiogenesis and vasculogenesis.
TNFα, which has anti-tumor effects at high concentrations, also promotes tumor angiogenesis, tumor cell survival and metastases at lower levels (26). A recent report suggested that TNFα mediates the differentiation of monocytes into angiogenic cells that support tumor angiogenesis (27–29). Radiotherapy, while directly inducing tumor cell death, may upregulate pro-angiogenic and pro-survival factors within the tumor microenvironment. We and others have found that radiation upregulates TNFα production by tumor cells and cells of the myeloid lineage (30, 31). Here we report that depletion of macrophages by liposomal chlodronate prior to irradiation increases the anti-tumor effects of IR, whereas co-implantation of tumor cells with BM-derived macrophages (BMDMφ) results in increased tumor radioresistance. The radioprotective effect of BMDMφ requires functional TNFα/TNFR signaling and induction of macrophage secreted VEGF. We report that blockade of macrophage VEGF induction by EnbrelR (soluble TNF receptor dimeric fusion protein) used in several murine studies, increased tumor radiosensitivity (32, 33). We also report that blocking VEGF with neutralizing antibodies improves the anti-tumor effects of IR. These data provide a rationale for targeting macrophage populations generally and TNFα induced VEGF signaling specifically when designing radiotherapeutic strategies.
MATERIALS AND METHODS
Mice and Tumor Cell Lines.
C57BL/6 mice WT, C57BL/6–129S-Tnfrsf1atm1ImxTnfrsf1btm1Imx/J (TNFR1,2−/−), B6:−129S-Tnftm1Gkl/J (TNF−/−) and C57BL/6 -Tg (UBC-GFP) 30SchaJ (GFP+/+) breeding pairs were purchased from Jackson Laboratories (Bar Harbor, ME). TNF−/− mice maintained in the 129/SvEv and C57BL/6 backgrounds after 6 generation backcross have been previously used to study the effects of TNFα on tumor promotion and antigen presentation (29, 34). B16F1 melanoma cells were cultured as described (35) B16.SIY melanoma cells expressing model antigen SIY, which can be recognized by CD8+ T cells in the context of Kb were cultured as described. The care and treatment of experimental animals was in accordance with institutional guidelines.
Generation of BM-derived Macrophages (BMDMφ).
Femoral BM cells were obtained from mice and cultured in complete RPMI supplemented 10% FCS and 30% pretested conditioned medium from the L929 cell line as a source of M-CSF for the first 5 days, followed by complete RPMI supplemented with 10% FCS and 10ng/ml recombinant M-CSF (R&D Systems, Minneapolis, MN) for additional 5 days. These BM-derived macrophages were >95% CD14+ and >90% CD11b+F4/80+ analyzed by FACS.
Tumor Induction and Irradiation.
Tumors cells (5×105 B16F1, or B16.SIY) were inoculated subcutaneously (s.c.) into the right hind limb. Tumors were measured with calipers and volume calculated as length × width × depth/2. At day 10–12 local radiotherapy (single dose 20 Gy, or fractional doses 2 Gy × 10) was delivered. In some experiments, B16F1/B16.SIY cells were co-injected with BMDMφ at a 4:1 ratio. Elsewhere macrophages were depleted by i.p. or intratumoral administration of Lip-Clod every 5–7 days (36). Cl2MDP (or clodronate) was a gift of Roche Diagnostics GmbH, Mannheim, GR (37). Other reagents include: phosphatidylcholine (Lipoid GmbH, Ludwigshafen, Germany), and cholesterol (Sigma, St. Louis, MO). Macrophage depletion was confirmed by FACS and immunohistochemistry of spleen and tumor cryosections with greater than 90% reduction compared with the control. Blockade of VEGF using neutralizing IgG against mouse VEGF-164 (R & D Systems) has been previously described (38). Briefly, goat IgG against mouse VEGF-164 was suspended in PBS and administered via i.p. injection (10 μg/mouse, 3 h before IR and 3, 8 days after IR). Control mice received goat IgG (Sigma).
Colony Forming Assay.
200 control or irradiated B16F1 or B16.SIY cells were seeded in 5cm culture dishes in RPMI containing 10% FCS, with or without 30% spent supernatant collected from either WT, TNF−/− or TNFR1,2−/− BMDMφ (5×106 cells). After 7 days, cells were washed and stained with crystal violet. Colonies with more than 50 cells were counted.
Protein Array and Luminex.
Spent culture supernatants were collected and incubated with membranes coated with 62 anti-mouse cytokine antibodies (RayBiotech, Inc., Norcross, GA) according to the manufacturer’s instruction. An antibody labeled with biotin coated on the upper left and lower right corners of the membrane serve as positive control. The film was scanned and spots were quantified by densitometry analysis using UN-SCAN-IT gel automated digitizing system software (Silk Scientific, Orem, UT). 32 cytokines were quantified using Mouse Cytokine/Chemokine Premixed 32 Plex (Milipore, Billerica, MA). Median Fluorescent Intensity (MFI) from each well was acquired and the relative concentration of each cytokine/chemokine was calculated.
FACS and Cell Sorting.
Tumors were excised, sectioned and digested in DMEM supplemented with 2% FCS and 1.5 mg/ml of collagenase D (Sigma) for 30 min in a 37°C shaking incubator to collect single-cell suspension. Cells were stained with anti-CD11b, anti-CD206, anti-F4/80, anti-TNFR1, anti-TNFR2 (Bio Legend, San Diego, CA), washed and analyzed on a LSR flow cytometer. Frequency of CD11b+ and F4/80+ TAMφ were analyzed and sorted on a Mo Flow to collect TAMφ. Purity reached >95% CD11b+ F4/80+ cells.
Western Blot.
Cells were lysed with cell lysis buffer containing 20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin and protease inhibitor mixture (Roche Applied Science, Indianapolis, IN). Protein concentrations were determined using the Bio-Rad protein assay kit and 10 μg from each sample was analyzed on 10% SDS–PAGE gels. Protein bands were transferred onto PVDF membranes, probed with rabbit anti-mouse VEGF (Santa Cruz Biotech, Santa Cruz, CA) for 2 h, then HRP-conujugated anti-rabbit for 1 h. Membranes were washed and immunodetected using ECL Kit (Amersham Biosciences, Pittsburg, PA). Anti-β-actin stained membranes were used as loading control.
Histopathlogy and Immunohistochemistry.
Tissues were fixed in 10% neutral formalin or 4% paraformaldehyde. Sections were blocked with 5% goat serum and incubated with VEGFR2 antibody (Cell Signaling Technology, MA, 1:200 dilution) for 1 h. After washing with TBS, biotinylated anti-rabbit secondary antibody followed by ABC-AP (Vector Labs, Burlingame, CA) was applied for 30 min each. Vector blue substrate was used for color development. For the following NG2 Chondroitin Sulfate Proteoglycan staining, the slides were washed, blocked with Avidin/Biotin blocking kit, then 10% goat serum for 10 min. Slides were incubated in 1:100 dilution of NG2 antibody (Millipore, Billerica, MA) overnight at 4°C. After applying biotinylated secondary antibody and ABC-AP solution, tissues were covered with Vulcan fast red substrate (Biocare Medical, Concorc, CA) for 15 min. Slides were washed and counter stained with methyl green. Sections were dehydrated in 100% ethanol, cleared by Histoclear (Wards Natural Science, Rochester, NY), then mounted with VectaMount (Vector Labs). Images were photographed at 200X magnification using a Zeiss camera operated by Openlab software.
Statistical Analysis.
A random effects model for longitudinal data was used to obtain an overall estimate of the intercept and slope of linear growth for each group. One-way ANOVA with Dunnett’s post test was performed using GraphPad InStat version 3.05 (p< 0.05).
RESULTS
Depletion of Macrophages Increases the Anti-tumor Effects of Ionizing Radiation.
Macrophages were depleted by i.p. injection of liposomal clodronate (Lip-Clod) (37) one day before injection of 2×106 B16.SIY cells and every 5 days thereafter in WT mice. 90% of the macrophages were specifically depleted in the spleen and tumor microenvironment as analyzed by FACS (36, 39, 40) (Supplemental Figure 1). 20 Gy was administered when tumors reached 150–200 mm3. The combination of Lip-Clod and 20 Gy significantly delayed tumor regrowth compared to 20 Gy alone (335±207mm3 vs 3215±1849mm3, day 22, p=0.002), or Lip-Clod alone (273±198mm3 vs 4987±2556mm3, day 18, p=0.041, Figure 1a). Lip-Clod also enhanced the anti-tumor effects of IR after tumors were established compared to 20 Gy alone (2678±1243mm3 vs 4599±889mm3, day 22, p=0.071, Figure 1b). We also injected Lip-Clod intratumorally and observed that tumor regrowth was significantly delayed compared to IR alone (1669±749mm3 vs 5317±1322mm3, day 26, p=0.037, Figure 1c).
Figure 1. Depletion of macrophages enhances radiation.

(a). Systemic macrophage depletion with Lip-Clod (200μl i.p.) prior to inoculation of B16.SIY cells significantly (p=0.002) enhanced IR (20 Gy) compared with IR alone in WT (n=11–16/group). (b). Systemic depletion of macrophages after tumors were established enhanced IR (p=0.071) compared to IR alone (n=11–16/group). (c). Intratumoral macrophage depletion significantly (p=0.037) delayed tumor regrowth compared to IR alone (n=10–12/group). ♦ Lip-PBS; ◊ 20 Gy + Lip-PBS; ■ Lip-Clod; □ 20 Gy + Lip-Clod
TNFα Signaling in BMDMφ Mediates Tumor Radioresistance.
TNFα is reported to both enhance tumor growth and mediate anti-tumor effects. To study the role of TNFα produced by macrophages, we cultured BMDMφ from WT and TNF−/− mice and co-injected these BMDMφ with 5×105 B16.SIY cells into WT mice. TNF−/− and WT BMDMφ exhibit similar levels of cell surface markers with >95% CD14 and 90% CD11b detected by FACS. They also expressed similar levels of iNOS but TNF−/− BMDMφ expressed significantly lower levels of Arg1 (data not shown). The percentages of F4/80+ TAMφ was similar between the WT and TNF−/− groups, although TNF−/− cells had diminished levels of surface CD206 staining (Supplemental Figure 2). Co-injection of WT BMDMφ with B16.SIY cells significantly accelerated tumor regrowth after 20 Gy compared to 20 Gy alone (1384±553mm3 vs 125±36mm3, day 22, p=0.030, Figure 2a, b). The regrowth of irradiated tumors in which B16.SIY cells were co-injected with WT BMDMφ occurred significantly earlier compared to irradiated tumors in which B16.SIY cells were co-injected with TNF−/− BMDMφ (1384±553mm3 vs 337±261mm3, day 22, P=0.037, Figure 2 b, c). Our data demonstrate that depletion of macrophages enhances the anti-tumor effects of IR and co-implantation of macrophages reverses this effect. These results also implicate macrophage secreted TNFα in the resistance of tumors to IR and suggest that macrophage derived TNFα and/or TNFα signaling in TAMφ contributes in part to B16.SIY tumor radioresistance.
Figure 2. TNFα signaling in BMDMφ mediates tumor radioresistance.

(a). Base line radiation response to 20 Gy of B16.SIY tumors growing in WT (n=12/group). ♦B16; ◊ IR in B16 (b). Co-injection of WT BMDMφ with B16.SIY tumor cells significantly accelerates tumor regrowth following 20 Gy compared with baseline (a), (p=0.03, n=12/group). ♦WT-Mac + B16; ◊ IR in WT-Mac + B16 (c). Co-injection of TNF−/− BMDMφ with B16.SIYcells significantly decreased tumor regrowth following 20 Gy compared to WT (b), (p=0.03, n=12/group). ♦ TNF−/− Mac + B16; ◊ IR in TNF−/− Mac + B16
TNFα Signaling in BMDMφ Promotes Tumor Growth.
To investigate the role of TNFα produced specifically by BMDMφ, we injected B16.SIY cells into TNF−/− mice transplanted with WT BM or TNF−/− BM. FACS analysis demonstrated that the BM was reconstituted with >70% of donor cells (Supplemental Figure 3). Animals underwent macrophage depletion with Lip-Clod and tumor growth was compared to controls. Tumor volume in WT BMT mice was significantly reduced by macrophage depletion (3067±615mm3 vs 825±174mm3, day 60, p=0.017, Supplemental Figure 4a). No tumor growth was observed in TNF−/− BMT mice, demonstrating an essential role for TNFα produced by BMDMφ. We next injected B16.SIY cells into TNFR1,2−/− mice after WT or TNFR1,2−/− BMT, with or without macrophage depletion. WT BMT in TNFR1,2−/− mice promoted tumor growth 30 days earlier than in TNFR1,2−/− BMT mice, which was significantly delayed when macrophages were depleted (867±64mm3 vs 4353±888mm3, day 45, p=0.013, Supplemental Figure 4b). No significant difference was observed in TNFR1,2−/− mice with TNFR1,2−/− BMT when macrophages were depleted (65±15mm3 vs 73±18mm3, day 45, p=0.549). In summary, tumors grew more slowly in TNFR1,2−/− BMT mice while WT BMT permitted tumor growth. However, tumor growth was suppressed when macrophages were depleted. These results suggest that autocrine/paracrine TNFα signaling in BMDMφ is essential in promoting tumor growth.
Autocrine/paracrine TNFα Signaling in BMDMφ Mediates Radioresistance.
We co-injected BMDMφ from WT or TNFR1,2−/− with B16.SIY cells into TNFR1,2−/− mice. Co-injection of WT BMDMφ significantly accelerated tumor regrowth after 20 Gy compared to IR alone (782±179mm3 vs 78±14mm3, day 22, p=0.003, Figure 3a, b). The regrowth of irradiated tumors co-injected with WT BMDMφ was significantly accelerated compared to the response of tumors co-injected with TNFR1,2−/− BMDMφ (782±179mm3 vs 283±157mm3, day 22, p=0.041, Figure 3b, c). These results indicate that intact TNF/TNFR signaling in macrophages is required for accelerated tumor regrowth after IR. We repeated these experiments using fractionated IR and report that the growth of tumors co-injected with WT BMDMφ was significantly increased compared to tumors co-injected with TNFR1,2−/− BMDMφ (384±64mm3 vs 38±7mm3, day 22, p=0.010, Supplemental Figure 5). These data further suggest that TNFα/TNFR signaling in BMDMφ mediates radioresistance.
Figure 3. TNFα signaling in BMDMφ mediates tumor radioresistance through autocrine/paracrine signaling.
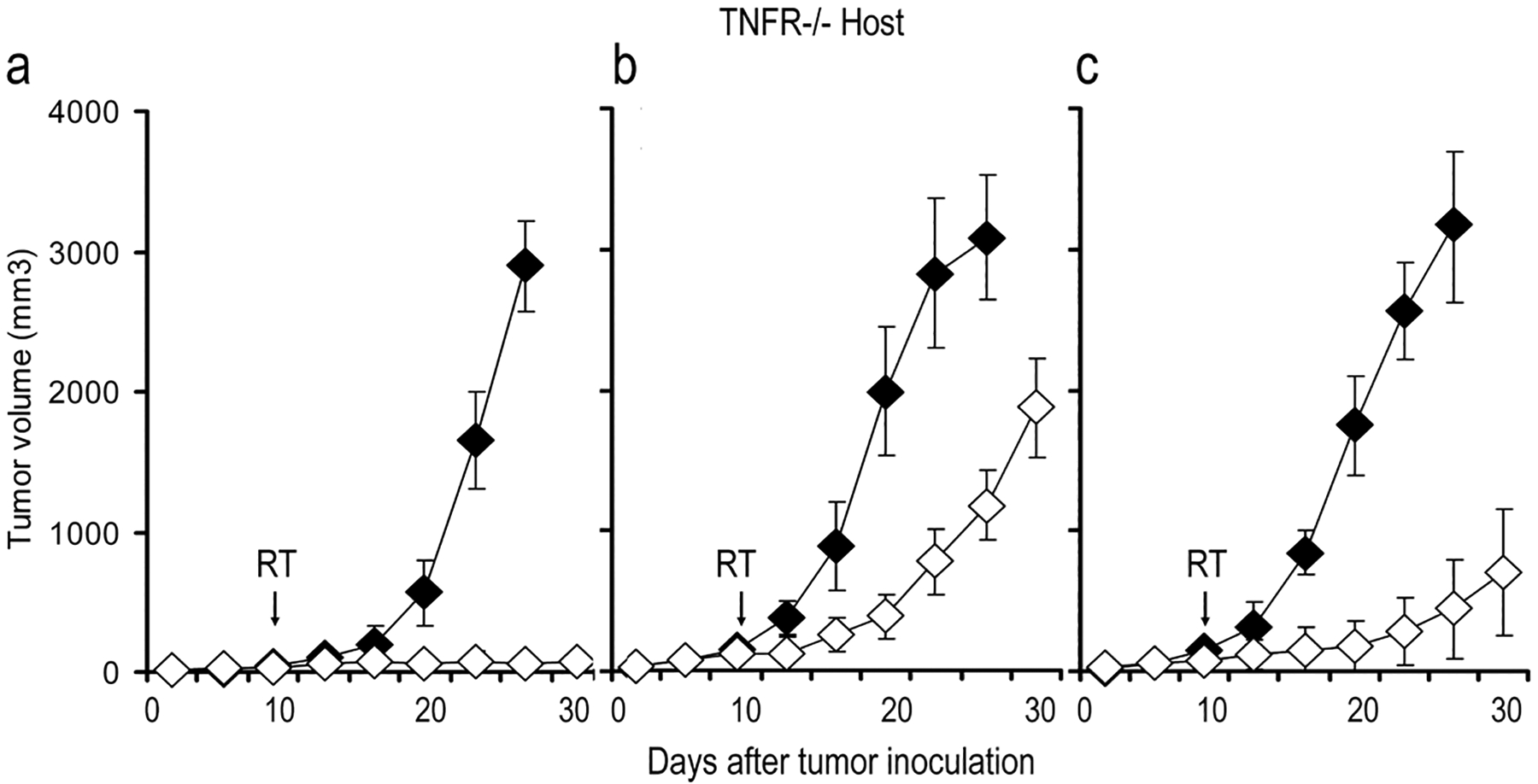
(a). Growth of B16.SIY tumors in TNFR1,2−/− mice (n=15/group). ♦ B16; ◊ IR in B16 (b). Co-injection of WT BMDMφ with B16.SIY cells significantly accelerates tumor regrowth following 20 Gy (p=0.003, n=12/group). ♦ WT-Mac + B16; ◊ IR in WT-Mac + B16 (c). Co-injection of WT BMDMφ with B16.SIY cells significantly accelerates tumor regrowth following 20 Gy compared with co-injection of TNFR1,2−/− BMDMφ and B16.SIY cells (p=0.041). ♦ TNFR−/− Mac + B16; ◊ IR in TNFR−/− Mac + B16
BMDMφ Supernatant Does Not Affect Tumor Cell growth or Radiosensitivity.
We explored the direct effects of BMDMφ on B16.SIY tumor cell radiosensitivity and/or growth in vitro. Supernatant from WT BMDMφ suppressed B16.SIY colony formation (p=0.015), while supernatant from TNF−/− or TNFR1,2−/− BMDMφ had no effect (p=0.259, p=0.338, Supplemental Figure 6). Unexpectedly, supernatants from TNF−/− and TNFR1,2−/− BMDMφ cultures increased colony formation in irradiated cells (p=0.065, p=0.055). Interestingly, the radioprotective effect of TNF−/− or TNFR1,2−/− supernatants in vitro differs from the in vivo findings with TNF−/−or TNFR1,2−/− macrophages. Supernatant from WT BMDMφ had no effect on irradiated B16.SIY colony formation (p=0.890). Supernatant collected from irradiated WT, TNF−/−or TNFR1,2−/− BMDMφ had no significant effect on either control or irradiated B16.SIY growth. These results suggest that the radioprotective effects of TNFα signaling in BMDMφ are not exerted directly on tumor cells but likely on non-tumor cell constituents of the tumor microenvironment.
Induction of VEGF Through TNFα/TNFR Signaling in TAMφ Mediates Rapid Tumor Regrowth Following Irradiation.
In addition to TAMφ, tumor stroma is also comprised of matrix proteins and various cell types including blood/lymphatic vessels (41). Recent data suggest that TAMφ support tumor growth by contributing to angiogenesis and/or vasculogenesis (41–44) in part mediated by TNFα. We employed a protein array and examined 62 cytokines and chemokines in unirradiated BMDMφ and BMDMφ treated with 5 Gy. Unirradiated WT and TNFR1,2−/− BMDMφ produced similar cytokine/chemokine levels including M-CSF, G-CSF, GM-CSF, CCL2, CCL9, IL-6, CXCL2, IL-10, TNFα, IL-12 and low levels of VEGF. Following 5 Gy there was a significant increase of VEGF in WT BMDMφ but not TNFR1,2−/− BMDMφ, while TNFα was induced in both WT and TNFR1,2−/− BMDMφ (Figure 4a). These results were confirmed by Luminex (Figure 4b) and suggest that the induction of VEGF by IR is dependent on TNFα/TNFR autocrine/paracrine signaling in BMDMφ. Our findings support the hypothesis that VEGF production in TAMφ through TNFα signaling activated by IR might play an important role in tumor vessel repair and tumor regrowth.
Figure 4. Radiation induction of VEGF in BMDMφ.

(a). A significant increase in VEGF was detected in WT BMDMφ compared with TNFR1,2−/− BMDMφ with 5 Gy (b). Luminex assay confirmation of VEGF induction by IR in WT BMDMφ. The mean of triplicates from one representative experiment is shown.
We examined if irradiation leads to TNF/TNFR mediated upregulation of VEGF in tumor macrophages. We injected B16.SIY cells into WT and TNFR1,2−/− mice and 20 Gy was delivered when tumors reached 150–200 mm3. Tumors were excised and digested into single cell suspensions. CD11b+ TAMφ were sorted and VEGF expression was assayed by western blot and Luminex. Significantly higher levels of VEGF were detected in CD11b+ TAMφ isolated from tumors grown in WT mice compared to TNFR1,2−/− mice (Figure 5a, b, p=0.015). Increased TAMφ VEGF from tumors in WT mice was mirrored by increased tumor neovasculature/angiogenesis post IR visualized in H&E and VEGFR2 stained tissue sections (Figure 5c). We quantified functional vascular structures demonstrating intact blood perfusion by the presence of red blood cells in VEFGR2+ vessels. In tumors grown in TNFR1,2−/− mice, there were significantly decreased functional vessels after IR, compared to either untreated control or tumors grown in WT post IR (p<0.0001 and p=0.002, respectively, Figure 5d). Also, more surviving tumor cells exhibiting intact morphology were present in irradiated tumors in WT compared to tumors in TNFR1,2−/− which demonstrated greater radiosensitivity.
Figure 5. Radiation induction of VEGF in TAMφ.

(a). Western blot analysis of VEGF expression of CD11b+F4/80+ TAMφ isolated from tumors grown in WT and TNFR1,2−/−. (b). VEGF levels were significantly elevated in CD11b+F4/80+ TAMφ from WT compared with TNFR1,2−/− (p=0.015, Luminex). (c) Increase neovasculature/angiogenesis in tumors grown in WT compared to TNFR1,2−/− post IR. Arrows indicate functional vessels containing red blood cells. (d). VGFR2+ staining shows a significant decrease in perfused vessels post IR in tumors grown in TNFR1,2−/− compared to WT control (p=0.0001) and WT treated with 20 Gy (p=0.002).
Depleting TNFα with Enbrel® Enhances Tumor Radiosensitivity.
To test this hypothesis that TNFα blockade might be clinically relevant, we injected B16.SIY cells into WT mice and treated them with Enbrel® + 20 Gy (32, 33). Enbrel® treated animals had slightly larger tumors than untreated control animals. Mice treated with Enbrel® + 20 Gy exhibited a reduction in tumor regrowth compared to 20 Gy alone (153±34mm3 vs 440±97mm3, day 28, p=0.022, Figure 6a).
Figure 6. Enbrel® enhances radiosensitivity by blocking the VEGF induction in TAMφ.

(a). Enbrel® + 20 Gy produced a significant decrease in B16.SIY regrowth compared to 20 Gy alone (p=0.022). ♦ PBS; ◊ 20 Gy + PBS; ■ Enbrel; □ 20 Gy + Enbrel (b). Western blot of VEGF expression by CD11b+F4/80+ TAMφ from WT treated with Enbrel® + 20 Gy. (c). Luminex showing inhibition of VEGF induction in CD11b+F4/80+ TAMφ isolated from tumors treated with Enbrel® + 20 Gy in WT. The mean of triplicates from one representative experiment is shown. (d). Representative tumor tissue sections showing a high density of intact neovasculature/angiogenesis in WT mice post IR and was reduced with IR + Enbrel® (upper panel); Blue VEGFR2+ microvessel density and red NG2+ pericyte coverage were compared between groups (lower panel). Enbrel® + 20 Gy inhibited VEGFR2+ microvessel repair/neovasculature with less pericyte coverage noted by NG2+ staining.
The observation that IR induced VEGF production by macrophages via TNFα/TNFR signaling pathways led us to examine the impact of pharmacologic TNFα inhibition and radiation on tumor angiogenesis. As with genetic deletion of TNFR1,2, Enbrel® treatment prevented upregulation of VEGF by tumor macrophages in vivo (Figure 6b, c). We next assessed the effect of IR + Enbrel® on neovascularization. Radiation alone led to a significant reduction in microvessel density as measured by VEGFR2+ (PBS 24±1.8 versus PBS + IR 4.1±1.4, p<0.0001). Enbrel® alone led to a modest decrease in microvessel density (PBS 24±1.8 versus Enbrel® 13±1, p<0.01), while Enbrel® + IR significantly inhibited angiogenesis when compared to all groups (2.75±0.35 versus Enbrel® alone 13±1, p<0.0001; versus PBS + IR 4.1±1.4, p<0.01, Figure 6d). NG2+ pericyte coverage was reduced in tumors treated with Enbrel® compared to PBS (98 ±0.2% versus 84±2.3%, p<0.001) and further decreased with Enbrel® + IR (57±22.4%, p=0.03 compared to Enbrel® alone). Vascular size and morphology were also substantially affected by treatment. For example, Enbrel® + IR treated tumors demonstrated narrow lumens suggestive of vascular collapse. Finally, while radiation exposure led to vascular hemorrhage, the addition of Enbrel® intensified hemorrhagic necrosis and regression. These data demonstrate a profound effect of TNFα depletion + IR on angiogenesis and vascular function. Given that irradiated macrophages upregulated VEGF expression, we next hypothesized that blockade of VEGF would likewise enhance radiosensitivity. Treatment of mice with neutralizing antibodies to VEGF led to increased radiosensitivity in B16F1 (806.1±329.1 versus IR alone 2101.8±525.4, p=0.05) and B16.SIY (0.5±0.1 versus IR alone 408.5±65.1, p=0.005) tumors (Supplemental Figure 7) (38).
Discussion
We tested the hypothesis that depletion of TAMφ by Lip-Clod would improve the anti-tumor effects of radiotherapy as determined by a delay of tumor regrowth. The effects of macrophage depletion were most marked when macrophages were depleted before tumors were established. TAMφ have been reported to secrete a variety of cytokines and other proteins that promote tumor growth. Our results demonstrate that the intact TNFα/TNFR signaling in TAMφ is important in the autocrine/paracrine secretion of cytokines by BMDMφ. TNFα is a proinflammatory cytokine which is cytotoxic to some tumor cell lines and tumors in vivo in high concentrations (45, 46). In our study, we were unable to demonstrate that radioresistance was mediated by a direct effect on B16.SIY cells using supernatant from WT BMDMφ. This raised the possibility that TNFα stimulated TAMφ promoted radioresistance by effects on the tumor microenvironment. TNFα at physiological concentrations has recently been implicated in cancer induction and support of tumor angiogenesis, in part, through the attraction of BM derived myeloid precursor cells that contribute to tumor blood vessel formation and stabilization (42, 47). We investigated if TNF/TNFR signaling in macrophages regulates the production of angiogenic cytokines by irradiated macrophages. Our data demonstrate that inhibition of TNFR signaling by both genetic and pharmacologic means prevents increased production of VEGF by irradiated macrophages. Immunohistochemical analyses demonstrate that the combination of TNFα inhibition plus + IR led to both significant decreases in neovascularization as well as vascular function as evidenced by reduced pericyte coverage and increased hemorrhage.
A recent report noted that macrophage secreted VEGF paradoxically slowed tumor growth in part by inducing tumor vessel abnormalities such as tortuosity and leakiness resulting in tumor hypoxia while tumor secreted VEGF “normalized” tumor vasculature (48). Interestingly, when macrophage VEGF was deleted tumors grew more rapidly due to a more “normalized” vasculature but tumors were also more sensitive to cyclophosphamide. Our results are consistent with this report demonstrating the Enbrel® treated tumors were actually more sensitive to IR. In contrast to the report of Stockmann et al (48), TNFα blockade did not increase vessel pericyte coverage, suggesting that TNFR signaling in tumor macrophages likely affects several angiogenic pathways in addition to VEGF. Nonetheless, blockade of VEGF upregulation by inhibition of TNFα signaling represents an alternative clinically relevant method to enhance radiosensitivity via targeting of TAMφ. Paradoxically supernatant from macrophages defective in TNF signaling through germ line deletions in TNF or TNFR1,2 conferred modest radioprotection on B16.SIY cells in vitro through an unknown mechanism(s). Together, these data suggest that macrophage blockade mediates in vivo radiosensitivity predominantly through effects on the microenvironment. Active TNFα signaling in irradiated macrophages appears necessary for upregulation of macrophage derived VEGF resulting in enhanced preservation of the irradiated vasculature.
Tumor angiogenesis is distinguished from post-natal vasculogenesis in that tumor angiogenesis is proposed to occur through endothelial migration and sprouting from preexisting blood vessels, whereas vasculogenesis is proposed to occur by the recruitment of BM cells to the site of tumor angiogenesis or local inflammatory damage. While vasculogenesis was previously described as direct incorporation of progenitor cells into newly emerging vasculature, it also involves the recruitment of bone marrow-derived angiogenic populations that enhance angiogenesis through paracrine mechanisms. The extent to which tumor neovascularization depends on local endothelial cells or infiltrating angiogenic cells is controversial. Recently, tumor regrowth by local myelomonocytic CD11b+ cells were reported to induce tumor vasculogenesis following irradiation through secretion of MMP-9 in a model where tumor angiogenesis was suppressed by prior irradiation of the tumor bed (25). Additionally TAMφ secrete a variety of proangiogenic proteins including IL-8, TNFα and VEGF. Therefore, both angiogenesis and vasculogenesis may be supported by TAMφ derived factors. While our data using EnbrelR suggest that pharmacologic inhibition of macrophage TNFR signaling enhances radiosensitivity, our Lip-Clod data demonstrate that reducing macrophage populations in tumors represents another potential anti-angiogenic and anti-vasculogenic strategy. Systemically delivering liposomal chlodronate to patients may not be feasible. However, CSF-1 receptor kinase inhibitors may block macrophage differentiation and function, providing an alternative biological tool to inhibit pro-inflammatory cytokine production from macrophages (49). The availability of these small molecules and monoclonal antibodies targeting either CSF-1R or CSF-1 to deplete TAMφ number and function may allow us to translate these strategies to promote radiationsensitivity in future studies.
We also report that blocking VEGF enhances the effect of IR (38, 50). Though anti-VEGF therapy could block VEGF derived from either vasculogenic myeloid populations or tumor cells, our results confirm that direct VEGF blockade is another therapeutic strategy to increase radiosensitivity. Despite the clear correlation between tumor vascularization and VEGF expression, TNF can also modulate the expression of other antiangiogenic factors, including thrombospondin and angiostatic chemokines like CXCL10, CXCL9. Therefore, targeting the TNFα/TNFR signaling in TAMφ may enhance radiosensitivity through additional pathways beyond VEGF signaling.
In summary, we utilized both genetic and pharmacological inhibition of TNFα signaling to study the role of tumor macrophages in promoting tumor radioresistance. Rather than directly affecting tumor cells, active TNFα signaling in irradiated macrophages results in the production of angiogenic cytokines such as VEGF. We have used co-implantation models to demonstrate that macrophages are sufficient for this effect, and Lip-Clod to demonstrate that macrophages are required. While Lip-Clod might have off-target cellular affects, previous studies suggest that Lip-Clod selectively depletes macrophages as opposed to other hematopoietic cells. Since the effects of Lip-Clod on other stromal components remains unclear, we also utilized bone marrow transplantation studies together with Lip-Clod to study the effects of depletion of bone marrow-derived cells. It nonetheless remains possible that active TNFα signaling in cellular components of the microenvironment might mediate radioresistance as well. Other limitations of our study include 1) the lack of a tissue specific promoter such as LysM to delete TNFα specifically in myeloid cells, 2) identification of which BM derived macrophage populations (M1 versus M2) mediate tumor vascular formation/stabilization following IR, and 3) whether VEGF secreted by macrophages attracts additional local angiogenic cells which contribute to tumor vascularization and regrowth following radiotherapy. In spite of these caveats, our results suggest that blockade of TNF/TNFR signaling in TAMφ is an attractive target to improve the efficacy of radiotherapy.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded in part by grant number CA111423 from the NCI (RRW) and grant K08HL071938 from the NHLBI (KSC).
REFERENCES
- 1.Salama JK CS, Mehta N, Yenice KM, Stadler WM, Vokes EE, Haraf DJ, Hellman S, and Weichselbaum RR. An initial report of a radiation dose escalation trial in patients with one to five sites of metastatic disease. Clinical Cancer Research. 2008. [DOI] [PubMed] [Google Scholar]
- 2.Vokes EE, Panje WR, Mick R, Kozloff MF, Moran WJ, Sutton HG, Goldman MD, Tybor AG, Weichselbaum RR. A randomized study comparing two regimens of neoadjuvant and adjuvant chemotherapy in multimodal therapy for locally advanced head and neck cancer. Cancer. 1990;66:206–13. [DOI] [PubMed] [Google Scholar]
- 3.Masunaga S, Hirayama R, Uzawa A, Kashino G, Suzuki M, Kinashi Y, Liu Y, Koike S, Ando K, Ono K. The effect of post-irradiation tumor oxygenation status on recovery from radiation-induced damage in vivo: with reference to that in quiescent cell populations. J Cancer Res Clin Oncol. 2009;135:1109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roses RE, Xu M, Koski GK, Czerniecki BJ. Radiation therapy and Toll-like receptor signaling: implications for the treatment of cancer. Oncogene. 2008;27:200–7. [DOI] [PubMed] [Google Scholar]
- 6.Jain RK. Lessons from multidisciplinary translational trials on anti-angiogenic therapy of cancer. Nat Rev Cancer. 2008;8:309–16. [DOI] [PubMed] [Google Scholar]
- 7.Senan S, Smit EF. Design of clinical trials of radiation combined with antiangiogenic therapy. Oncologist. 2007;12:465–77. [DOI] [PubMed] [Google Scholar]
- 8.Crane CH, Ellis LM, Abbruzzese JL, Amos C, Xiong HQ, Ho L, Evans DB, Tamm EP, Ng C, Pisters PW, Charnsangavej C, Delclos ME, O’Reilly M, Lee JE, Wolff RA. Phase I trial evaluating the safety of bevacizumab with concurrent radiotherapy and capecitabine in locally advanced pancreatic cancer. J Clin Oncol. 2006;24:1145–51. [DOI] [PubMed] [Google Scholar]
- 9.Duda DG, Jain RK, Willett CG. Antiangiogenics: the potential role of integrating this novel treatment modality with chemoradiation for solid cancers. J Clin Oncol. 2007;25:4033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gutin PH, Iwamoto FM, Beal K, Mohile NA, Karimi S, Hou BL, Lymberis S, Yamada Y, Chang J, Abrey LE. Safety and Efficacy of Bevacizumab with Hypofractionated Stereotactic Irradiation for Recurrent Malignant Gliomas. Int J Radiat Oncol Biol Phys. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seiwert TY, Haraf DJ, Cohen EE, Stenson K, Witt ME, Dekker A, Kocherginsky M, Weichselbaum RR, Chen HX, Vokes EE. Phase I study of bevacizumab added to fluorouracil- and hydroxyurea-based concomitant chemoradiotherapy for poor-prognosis head and neck cancer. J Clin Oncol. 2008;26:1732–41. [DOI] [PubMed] [Google Scholar]
- 12.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9. [DOI] [PubMed] [Google Scholar]
- 15.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31. [DOI] [PubMed] [Google Scholar]
- 16.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. [DOI] [PubMed] [Google Scholar]
- 17.Lambert LE, Paulnock DM. Modulation of macrophage function by gamma-irradiation. Acquisition of the primed cell intermediate stage of the macrophage tumoricidal activation pathway. J Immunol. 1987;139:2834–41. [PubMed] [Google Scholar]
- 18.Fulton AM, Chong YC. The role of macrophage-derived TNFa in the induction of sublethal tumor cell DNA damage. Carcinogenesis. 1992;13:77–81. [DOI] [PubMed] [Google Scholar]
- 19.Ibuki Y, Goto R. Augmentation of NO production and cytolytic activity of M phi obtained from mice irradiated with a low dose of gamma-rays. J Radiat Res (Tokyo). 1995;36:209–20. [DOI] [PubMed] [Google Scholar]
- 20.Weichselbaum RR, Kufe DW, Hellman S, Rasmussen HS, King CR, Fischer PH, Mauceri HJ. Radiation-induced tumour necrosis factor-alpha expression: clinical application of transcriptional and physical targeting of gene therapy. Lancet Oncol. 2002;3:665–71. [DOI] [PubMed] [Google Scholar]
- 21.Milas L, Wike J, Hunter N, Volpe J, Basic I. Macrophage content of murine sarcomas and carcinomas: associations with tumor growth parameters and tumor radiocurability. Cancer Res. 1987;47:1069–75. [PubMed] [Google Scholar]
- 22.Milas L Tumor bed effect in murine tumors: relationship to tumor take and tumor macrophage content. Radiat Res. 1990;123:232–6. [PubMed] [Google Scholar]
- 23.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ, Lee CC, McBride WH, Chiang CS, Hong JH. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys. 2007;68:499–507. [DOI] [PubMed] [Google Scholar]
- 25.Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Vincent A, Cates J, Brantley-Sieders DM, Polk DB, Young PP. Low levels of tumor necrosis factor alpha increase tumor growth by inducing an endothelial phenotype of monocytes recruited to the tumor site. Cancer Res. 2009;69:338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. [DOI] [PubMed] [Google Scholar]
- 29.Balkwill F Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–71. [DOI] [PubMed] [Google Scholar]
- 30.Sherman ML, Datta R, Hallahan DE, Weichselbaum RR, Kufe DW. Regulation of tumor necrosis factor gene expression by ionizing radiation in human myeloid leukemia cells and peripheral blood monocytes. J Clin Invest. 1991;87:1794–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weichselbaum RR, Hallahan DE, Sukhatme VP, Kufe DW. Gene therapy targeted by ionizing radiation. Int J Radiat Oncol Biol Phys. 1992;24:565–7. [DOI] [PubMed] [Google Scholar]
- 32.Grounds MD, Davies M, Torrisi J, Shavlakadze T, White J, Hodgetts S. Silencing TNFalpha activity by using Remicade or Enbrel blocks inflammation in whole muscle grafts: an in vivo bioassay to assess the efficacy of anti-cytokine drugs in mice. Cell Tissue Res. 2005;320:509–15. [DOI] [PubMed] [Google Scholar]
- 33.Wolthuis EK, Vlaar AP, Choi G, Roelofs JJ, Haitsma JJ, van der Poll T, Juffermans NP, Zweers MM, Schultz MJ. Recombinant human soluble tumor necrosis factor-alpha receptor fusion protein partly attenuates ventilator-induced lung injury. Shock. 2009;31:262–6. [DOI] [PubMed] [Google Scholar]
- 34.Scott KA, Moore RJ, Arnott CH, East N, Thompson RG, Scallon BJ, Shealy DJ, Balkwill FR. An anti-tumor necrosis factor-alpha antibody inhibits the development of experimental skin tumors. Mol Cancer Ther. 2003;2:445–51. [PubMed] [Google Scholar]
- 35.Mauceri HJ, Beckett MA, Liang H, Sutton HG, Pitroda S, Galka E, Efimova E, Darga T, Khodarev NN, King CR, Posner MC, Hellman S, Kufe DW, Weichselbaum RR. Translational strategies exploiting TNF-alpha that sensitize tumors to radiation therapy. Cancer Gene Ther. 2009;16:373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J, Wainstok R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol. 2007;127:2031–41. [DOI] [PubMed] [Google Scholar]
- 37.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. [DOI] [PubMed] [Google Scholar]
- 38.Gorski DH, Beckett MA, Jaskowiak NT, Calvin DP, Mauceri HJ, Salloum RM, Seetharam S, Koons A, Hari DM, Kufe DW, Weichselbaum RR. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999;59:3374–8. [PubMed] [Google Scholar]
- 39.van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods. 1996;193:93–9. [DOI] [PubMed] [Google Scholar]
- 40.Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther. 2008;7:788–99. [DOI] [PubMed] [Google Scholar]
- 41.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. [DOI] [PubMed] [Google Scholar]
- 43.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. [DOI] [PubMed] [Google Scholar]
- 44.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. [DOI] [PubMed] [Google Scholar]
- 45.Menon C, Bauer TW, Kelley ST, Raz DJ, Bleier JI, Patel K, Steele K, Prabakaran I, Shifrin A, Buerk DG, Sehgal CM, Fraker DL. Tumoricidal activity of high-dose tumor necrosis factor-alpha is mediated by macrophage-derived nitric oxide burst and permanent blood flow shutdown. Int J Cancer. 2008;123:464–75. [DOI] [PubMed] [Google Scholar]
- 46.Havell EA, Fiers W, North RJ. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J Exp Med. 1988;167:1067–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. 1999;5:828–31. [DOI] [PubMed] [Google Scholar]
- 48.Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irvine KM, Burns CJ, Wilks AF, Su S, Hume DA, Sweet MJ. A CSF-1 receptor kinase inhibitor targets effector functions and inhibits pro-inflammatory cytokine production from murine macrophage populations. Faseb J. 2006;20:1921–3. [DOI] [PubMed] [Google Scholar]
- 50.Zwolak P, Dudek AZ, Bodempudi VD, Nguyen J, Hebbel RP, Gallus NJ, Ericson ME, Goblirsch MJ, Clohisy DR. Local irradiation in combination with bevacizumab enhances radiation control of bone destruction and cancer-induced pain in a model of bone metastases. Int J Cancer. 2008;122:681–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.