

Abstract
The analysis via density functional theory was employed to understand high photocatalytic activity found on the Au–Ag high-noble alloys catalysts supported on rutile TiO2 during the oxygen evolution of water oxidation reaction (OER). It was indicated that the most thermodynamically stable location of the Au–Ag bimetal-support interface is the bridging row oxygen vacancy site. On the active region of the Au–Ag catalyst, the Au site is the most active for OER catalyzing the reaction with an overpotential of 0.60 V. Whereas the photocatalytic activity of other active sites follows the trend of Au > Ag > Ti. This finding evident from the projected density of states revealed the formation of the trap state that reduces the band gap of the catalyst promoting activity. In addition, the Bader charge analysis revealed the electron relocation from Ag to Au to be the reason behind the activity of the bimetallic that exceeds its monometallic counterparts.
Subject terms: Chemical engineering, Heterogeneous catalysis, Photocatalysis, Density functional theory, Chemical physics, Chemical physics
Introduction
In the current era, solar energy is confined for practical reasons and has increased the attention of both academia and industrial sectors. The essential worldwide goal of solar energy is to create electricity, and it is prospective to generate fuels from the waste of CO2 and H2O by induction of solar photochemistry. In the past 40 years, heterogeneous photocatalysis and photochemistry fields have developed comprehensively in return to demanding energy and environmental issues1–8. One of the most capable solutions for the energy problem is photocatalytic overall water oxidation (POWS), i.e., oxygen and hydrogen evolution9,10. Water electrolysis—one of the most practical routes to produce hydrogen to establish clean and renewable energy cycles, accounted for up to 4% of the global hydrogen production11,12. Water electrolysis entails two half-cell reactions: the cathodic hydrogen evolution (HER) and anodic oxygen evolution (OER) reactions.
The metal/semiconductor photocatalysts have vast POWS potential and have drawn broad attention for a long time13. Since 1972, a typical semiconductor TiO2 exhibited a suitable band structure for POWS as a photocatalyst14. Moreover, the TiO2-supported Pt catalysts are competent of POWS under the range of ultraviolet light15. Evidently, the visible light POWS shows a higher potential for practical applications. However, due to the large bandgap of TiO2, TiO2-based photocatalysts POWS driven by visible light remains a great challenge16–18. Even though the bandgap of TiO2 could be effectively narrowed down by elemental doping to harvest visible light, the energy levels introduced by impurity dopants can act as recombination centers for electrons and holes, resulting in a poor photocatalytic activity19,20.
Moreover, it has been reported that various metals, e.g., Pt, Au, Ag, Pd, Ru, Cu, etc., loaded on semiconductors materials, e.g., TiO2, RuO2, etc. to confine photoexcited electrons through the formed Schottky junction can result in reserved charge recombination for photocatalytic enhancement21–23. When the material is operated in a visible light range, Au, Ag, and Cu can act as metallic plasmons and trigger localized surface plasmon resonance (LSPR)24,25. This energizes the electrons above the Fermi level from the occupied energy levels26,27. These electrons with high energy pass through the Schottky barrier of metal–semiconductor can be located in the semiconductor conduction band (CB), contributing to the competent charge separation and the photocatalytic reduction in the visible light region28,29. In the meantime, the positive charges left behind in the separated energy bands of plasmonic metals can be used to drive various oxidation reactions30.
Therefore, metal-loaded TiO2 (M/TiO2, M = Au, Ag, Cu, etc.) plasmonic photocatalysts can be good candidates for the POWS that work under visible light. The Au, Ag, Cu plasmonic metals act as electron donors and generate electrons into TiO2 under visible light. The differences in electron transfer from TiO2 to the metal or metal to TiO2 could drive different routes of water redox reaction resulting in various products distribution31–33. Silver (Ag) has been regarded as a promising candidate over gold (Au) due to its high catalytic activity. Water oxidation is a sluggish reaction in nature and artificial photosynthetic systems. Nevertheless, the plasmon resonance frequency of Ag is in the near-ultraviolet region, limiting its photocatalysis applications in the visible light region34. Consequently, the application of Ag-based photocatalysts remains challenging35.
A substitute method to stabilize the Ag-based photocatalyst is to alloy the Ag metal with stable metals such as Au. These alloys comprising high content of noble metals are regarded as the high-noble alloys. In dentistry, such materials exhibit increased resistance to the corrosive environment–high stability36. When Ag is alloyed with Au, the stability is still maintained. The alloy could produce optical regulation due to their different plasmon response range and produce a prospect to tune the electronic structure, affecting Schottky barrier height and the plasmon-induced charges potentials37. In heterogeneous catalysis, although bimetallic alloys have been investigated extensively38–40, the reliance of photocatalytic oxygen evolution reactions (OER) on the alloy components for water oxidation reaction is yet to be studied. The DFT calculations can be coupled with the experimental techniques to understand the enhanced activity of heterogeneous catalysts, e.g., Pd–Cu alloy nanoparticles supported on carbon support41 and M4/CeO2 (ZrO2) where M4 is Pt, Pd, and Rh42. In addition, there are other theoretical and experimental works that have been carried out on similar interfaces. For instance, TiO2-nanocluster adsorption on Ag and Au noble metal surfaces compared with that on graphene surfaces was recently investigated using density functional theory calculation43. This demonstrated that the electronic properties of the cluster on Ag and Au are not different, which helps to understand the experimental results44. The bimetallic nanocluster on a finite-size TiO2 nano-wire was designed and presented as electronic properties, which are motivating to explore that in other bimetallic groups45. The importance of TiO2 nanostructure and its energy application in terms of an experimental realization is summarized46.
In this work, using DFT calculations and computational hydrogen electrode (CHE), we have evaluated the catalytic performance of Au–Ag/TiO2 high noble alloys catalysts during water oxidation together with the investigation on roles of Au in such bimetallic cluster via the Au–Ag bimetallic cluster supported on rutile TiO2 (110) models.
Results and discussion
Electronic properties of the catalysts: density of states analyses
We employed the HSE06 hybrid functional in DFT calculations for studying the band structure of the most stable structure, i.e., Au2–Ag2/TiO2 embedded in place of oxygen vacancy. We used the Au2–Ag2 tetrahedral cluster supported on the rutile (110) surface due to its high stability47. The density of states (DOS) profiles for Au2–Ag2/TiO2 are shown in Fig. 1. The valence band comprises O (2p), while the conduction band is majorly contributed by Ti (3d). In the forbidden gap, there are hybrid gap states found composed of Au (5d) and Ag (4d), with a significant contribution from Au (5d). The Ag-5 s orbital is much higher in energy than the Au-6 s orbital energy, so a partial charge transfer from Au to Ag occurs48.
Figure 1.

The density of states (DOS) profiles of (a) Au2–Ag2/TiO2 (four atoms) cluster on bridging row oxygen vacant site and (b) the enlarged view of trap states which is majorly composed of Au(5d) and Ag(4d). (The DOS profile reflects the activity of the catalyst based on the analysis of bonding and antibonding states located at the left side of the EF and right side of the EF regions, respectively, where they are separated by the Fermi level designated at EF = 0 eV.
Consequently, Au atoms tend to be negatively charged, while Ag atoms tend to be positively charged. The partial charge transfer from Au atoms to Ag atoms gives remarkable electrostatic stabilization, making the alloy formation more favorable than pure gold and silver clusters. Therefore, the equivalent mixing between Au and Ag atoms in the alloy formation will likely be more preferential. For Au4/TiO2 and Ag4/TiO2 tetrahedral structures positioned on bridging oxygen rows, vacancy results are shown in S.I (see fig S10 and S11).
It is well known that gold clusters have the lowest spin multiplicity as the ground state49. Previously, the spin effects of Au clusters on TiO2 (110)50 and other metal oxides51 were studied. As a result, spin and electron distributions were assumed to have no effects in the case of noble metal atoms. Figure 1 shows that the spin-up and spin-down states of the DOS profiles of Ag 4d and Au 5d in Ag4Au4/TiO2 are symmetrical, while in the case of Au4/TiO2, it exhibits slightly asymmetrical DOS shown in Figure S10 of the supplementary document.
TiO2 surface can donate electrons and is ascribed to excess electrons from the defect state found inside the bandgap34,52–54. Ag and Au, relatively electronegative atoms, prefer to take the missing oxygen when creating an oxygen vacancy. Pulling an oxygen atom out of the bridging oxygen atom releases electron density, creating a vacancy site. This excess electron is transferred to Ag and Au atoms binding to the vacancy site. This implies that the support plays an active role in catalytic activity because it alters its chemical properties by transferring charge to Ag and Au55. In this work, DFT analyses are employed to understand the water oxidation mechanism on the interface between Au/Ag high noble alloys catalysts supported on rutile TiO2 (110) and such a mechanism on the support itself. Thus, two main issues are focused, (1) determination of active regions on the surface (2) thermodynamically stable size and location of the active site cluster of initial (Au/TiO2, Ag/TiO2, Au–Ag/TiO2) and cluster models (Au4/TiO2, Ag4/TiO2, Au2–Ag2/TiO2).
Catalytic performance of TiO2 support during oxygen evolution reaction (OER)
We first discuss the water oxidation mechanism on clean rutile (110) TiO2 surface. In Fig. 7d, we showed the pure surface of TiO2 rutile (110), which is well investigated by many researchers theoretically and experimentally. The reported overpotential value for the pure surface is 0.80 eV with the rate-determining step OH* as shown with recreation in Fig. 233,56. Furthermore, Malik et al. and Norskov et al. reported that on the clean or pure surface of TiO2 mechanism of water oxidation proceeds through surface-bound peroxo O* species. Thus, Ti was the only metal active site choice for studying water oxidation mechanisms.
Figure 7.

Initial model of one atom (a) Ag on TiO2, (b) Au on TiO2, (c) Au–Ag on TiO2 (d) clean rutile TiO2 (110) surface model.
Figure 2.
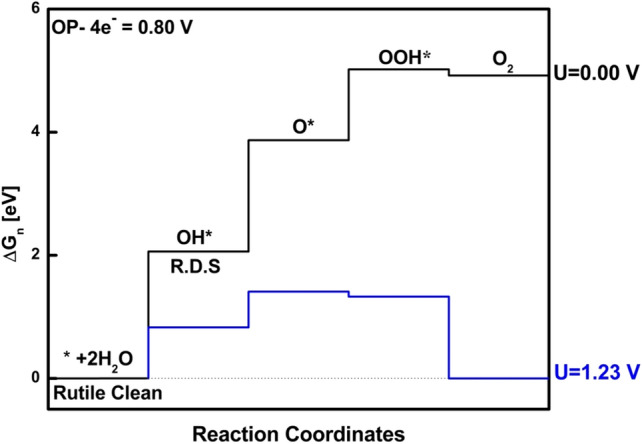
Free energy diagrams for OER through peroxo species on TiO2 rutile (110) under external potential U = 0.00 V. (Black profile = O2 evolution pathways go via the peroxo O* intermediate. The rate-determining steps (R.D.S) are indicated with an arrow. The overpotential is displayed in the upper left corner. (recreated from33).
Determination of active regions on the catalyst
Now we started analyzing with initial models of Au/TiO2, Ag/TiO2, and Au–Ag/TiO2, where the model shown in Fig. 1a comprises one Ag atom replacing the bridging oxygen atom on the TiO2 surface, while in Fig. 7b, one Au atom substituted an oxygen atom on the TiO2 surface. When we studied Au–Ag/TiO2 model (Fig. 7c), we tried different positioning of Au and Ag atoms, such as placing both atoms next to each other in the same bridging row or placing them on the same bridging row with the gap of one bridging oxygen atom in between Au and Ag (In S.I. see figure S1). The most stable position was when Au and Ag atoms were positioned on two bridging oxygen vacant sites in different rows, as shown in Fig. 7c. To study the OER mechanism on the Au–Ag/TiO2 slab structure, the Ti atom was first chosen as an active site, and then the calculations were performed by choosing Au and Ag as an active site. For Au/TiO2 and Ag/TiO2 slab structures, the free energy diagrams are shown in S.I. in Figure S8 (a) and (b). The Ti active site was selected far from noble metal atoms. The OER data of the initial model for Fig. 7c, i.e., Au–Ag/TiO2, is listed in Table 1. The OER data for Fig. 7a,b are shown in S.I. in table S2, and intermediates are shown in Figure S3 and S4.
Table 1.
The relative free energies for each type of active site on the bimetallic Au–Ag/TiO2 system shown in Fig. 7c, starting from the dissociation of a water molecule (H2O + * ↔ H* + HO*) set as the reference point with zero energy.
| Active site | ΔGOH* | ΔGO* | ΔGOOH* | Overpotential (V) | Rate-determining steps |
|---|---|---|---|---|---|
| Au | 1.54 | 3.88* | 4.84 | 1.11 | O* + H2O → HOO* + ½H2 |
| Ag | 0.68 | 3.29* | 4.98 | 1.38 | O* + H2O → HOO* + ½H2 |
| Ti | 0.03 | 0.17 | 3.58* | 2.18 | HOO* → O2 + ½H2 |
The propagating step is denoted as ΔGOH*, ΔGO*, and ΔGOOH* shown together with their overpotentials and rate-determining step. *The rate-determining steps.
From the OER results shown above in Fig. 3, it is indicated that the Au atom is preferably the active site with the lowest overpotential with an initial model. However, since Au and Ag atoms are placed on different bridging rows of oxygen vacancy sites, there is no interaction between noble metal atoms on the TiO2 surface.
Figure 3.
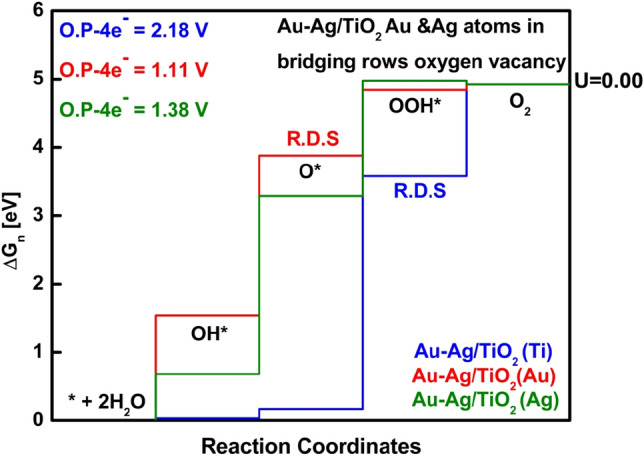
Comparative free energy diagram for Au–Ag/TiO2 (Au and Ag placed in bridging vacant oxygen site) slab structure. Blue, red, and green profiles represent Ti, Au, and Ag as active sites. Overpotential for all energy profiles are shown in the upper left corner. The rate-determining step for Au and Au as active sites is O* while OOH* for Ti atom as an active site. R.D.S is rate-determining step.
OER performance of TiO2 supported Au–Ag high-noble alloys catalysts
An initial model is good enough to determine the active site; yet, Au and Ag atoms were placed on different bridging rows, so there was no interaction between Au and Ag atoms. Hence, no charge transfer could take place from Ag to Au. To understand the water oxidation mechanism at the interface of noble metal atoms cluster and TiO2 metal oxide surface, we further extended calculations to use a 3-dimension tetrahedral cluster of (Au and Ag) atoms on the TiO2 surface because of its stability. This structure we refer to as a cluster model. Our previous work34 showed that the Au2–Ag2/TiO2 structure is more suitable when embedded in the location of oxygen vacancy. Nevertheless, we show how the OER activity is affected when the tetrahedral cluster is placed on a stoichiometric surface at the two bridging oxygen rows (a different position), as shown in Fig. 8c. In the supplementary document, we have demonstrated OER intermediates of Au4/TiO2, Ag4/TiO2, and Au2–Ag2/TiO2 when placed on two bridging rows (see figure S5 and S6). The comparative free energy diagram for Au4/TiO2, Ag4/TiO2 on two bridging rows is given in figure S9 (a) and (b) with the OER data (relative free energies) shown in table S3.
Figure 8.

Four atoms cluster on two bridging rows of (a) Ag4 on TiO2, (b) Au4 on TiO2, and (c) Ag2–Au2 on rutile TiO2 (110) surface model.
Figure 4a shows the results for Au2–Ag2/TiO2 on two different bridging rows. The comparative free energy profile shows that the lowest overpotential is when Au was chosen as an active site. However, the OH*, O*, and OOH* steps were strongly bonded for the Ti active site than Au and Ag. Similar findings were observed for the model when Au2–Ag2/TiO2 was placed on a bridging row oxygen vacancy. The O2 evolution was preferred on the Au site, free energy diagram shown in Fig. 4b. From the above-shown results, it has been seen that either Au2–Ag2 cluster adsorb on TiO2 surface by connecting to two bridging oxygen rows or an oxygen vacancy is created, and then the cluster is anchored into the vacant site; the perfect active site is Au. In all cases, Ti is the least preferred due to high overpotential. It was also found that the rate-determining step for OER on Au2–Ag2/TiO2 surface is the O* intermediate, i.e., 2nd step when the active site is noble metal atoms, i.e., Au or Ag. While the rate-determining step for OER with Ti as an active site is OOH* intermediate, i.e., the 3rd step. For Au2–Ag2/TiO2, the perfect active site is Au. When applied bias (U = 0 V), all steps go uphill thermodynamically.
Figure 4.
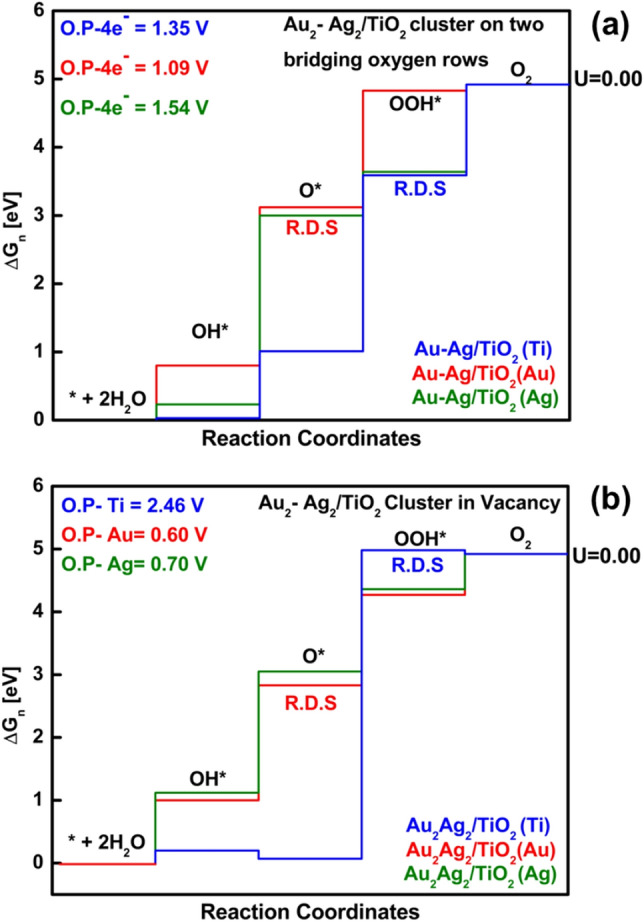
Comparative free energy diagrams for the evolution of O2 on Au2-Ag2 cluster (a) placed on two bridging oxygen atoms, (b) embedded in the place of oxygen vacancy on bridging row34. The blue profile shows the energy for Ti active site, Green and red color shows energy profiles for Ag and Au active sites, respectively. The overpotential is shown in the upper left corner. R.D.S rate-determining step.
When Au2–Ag2/TiO2 cluster is adsorbed on two bridging rows, TiO2 surface is stoichiometric; however, a remarkable change is noticed in overpotential when embedded on a vacant oxygen site. This change is attributed to the presence of oxygen vacancy because Au and Ag atoms are higher in electronegativity and prefer to stay in the place of oxygen vacancy. Therefore, there is excess electron density when the oxygen atom from the bridging row is removed. This "excess electron density" attaches the silver and gold clusters (with the ability of electronegativity) to the vacancy site. Moreover, when gold and silver atoms are bonded together in bimetallic clusters, there is a charge transfer from Ag to Au, which is accountable for making Au negatively charged and expected to be more active.
For the cluster systems, the relative free energies of all the intermediates (OOH*, OH*, and O*) relative to the starting state (* + 2H2O) are shown in Table 2. For all systems, we see that the trend in activity is the same for the active site, i.e., Au > Ag > Ti. For noble metal atoms, the rate-determining step is the O* step, and for transition metal, the rate-determining step is OOH*. Notably, the OOH* step is much less stable when the active site is Ti (blue curves in free energy diagrams) conversely the OH* and O* are more stable and vice versa for the noble metal active sites case (red and green curve for Au and Ag respectively in free energy diagrams).
Table 2.
The relative free energies for each type of active sites on the Au2–Ag2/TiO2 with a cluster on two bridging oxygen rows and bridging row oxygen vacant site shown in Figs. 8c and 9c, respectively, starting from the dissociation of a water molecule (H2O + * ↔ H* + HO*) set as the reference point with zero energy.
| Active site | ΔGOH* | ΔGO* | ΔGOOH* | Overpotential (V) | Rate-determining steps |
|---|---|---|---|---|---|
| System: Au2–Ag2/TiO2 with cluster on two bridging oxygen rows | |||||
| Au | 0.80 | 3.12* | 4.83 | 1.09 | O* + H2O → HOO* + ½H2 |
| Ag | 0.23 | 3.00* | 3.64 | 1.54 | O* + H2O → HOO* + ½H2 |
| Ti | 0.03 | 1.01 | 3.59* | 1.35 | HOO* → O2 + ½H2 |
| System: Au2–Ag2/TiO2 cluster on bridging row oxygen vacant site | |||||
| Au | 1.00 | 2.83* | 4.27 | 0.60 | O* + H2O → HOO* + ½H2 |
| Ag | 1.12 | 3.05* | 4.36 | 0.70 | O* + H2O → HOO* + ½H2 |
| Ti | 0.20 | 0.07 | 3.76* | 2.46 | HOO* → O2 + ½H2 |
The propagating step is denoted as ΔGOH*, ΔGO*, and ΔGOOH* shown together with their overpotentials and rate-determining step. *The rate-determining steps.
OER performance of TiO2 supported Ag and Au catalysts
As explained earlier, adsorption of Ag on TiO2 surface becomes more stable when interacted with Au. Moreover, the photocatalytic property of Ag species is enhanced because Au promotes charge transfer from Ag to Au on the TiO2 surface. To explain this phenomenon in-depth, we aimed to study the effect of only Au on TiO2 and Ag on TiO2 with cluster and initial model. We used the same strategy for choosing the active site and stable structure mentioned earlier for the bimetallic cluster. However, we only discuss the results of Au4/TiO2 and Ag4/TiO2 cluster models shown in Fig. 10a,b, and Table 3, respectively. Free energy diagrams are shown in Fig. 5a,b and for OER intermediates (see S.I. figure S7). For Au4/TiO2 and Ag4/TiO2 clusters placed on two bridging rows, free energy diagrams are given in S.I figure S9.
Figure 10.

OER intermediates on Au2–Ag2/TiO2 semiconductor surface with Ti active site marked blue in (a) OH*-Ti, (b) O*-Ti, (c) OOH*-Ti and with gold as active site shown in (d) OH*-Au, (e) O*-Au, and (f) OOH*-Au.
Table 3.
The relative free energies for each type of active sites on the monometallic Au4/TiO2 and Ag4/TiO2 cluster bridging row oxygen vacant site shown in Fig. 7a,b, starting from the dissociation of a water molecule (H2O + * ↔ H* + HO*) set as the reference point with zero energy.
| Active site | ΔGOH* | ΔGO* | ΔGOOH* | Overpotential (V) | Rate-determining steps |
|---|---|---|---|---|---|
| System: Au4/TiO2, replacing bridging O atoms on the surface with Au4 cluster in vacancy | |||||
| Au | 0.15 | 1.92 | 3.73* | 0.58 V | O* + H2O → HOO* + ½H2 |
| Ti | 0.63 | 2.93* | 4.60 | 1.07 V | HOO* → O2 + ½H2 |
| System: Ag4/TiO2, replacing bridging O atoms on the surface with Ag4 cluster in vacancy | |||||
| Ag | 0.45 | 2.91* | 3.72 | 1.23 V | O* + H2O → HOO* + ½H2 |
| Ti | − 0.13 | 0.67 | 3.55* | 1.65 V | HOO* → O2 + ½H2 |
The propagating step is denoted as ΔGOH*, ΔGO*, and ΔGOOH* shown together with their overpotentials and rate-determining step. *The rate-determining steps.
Figure 5.
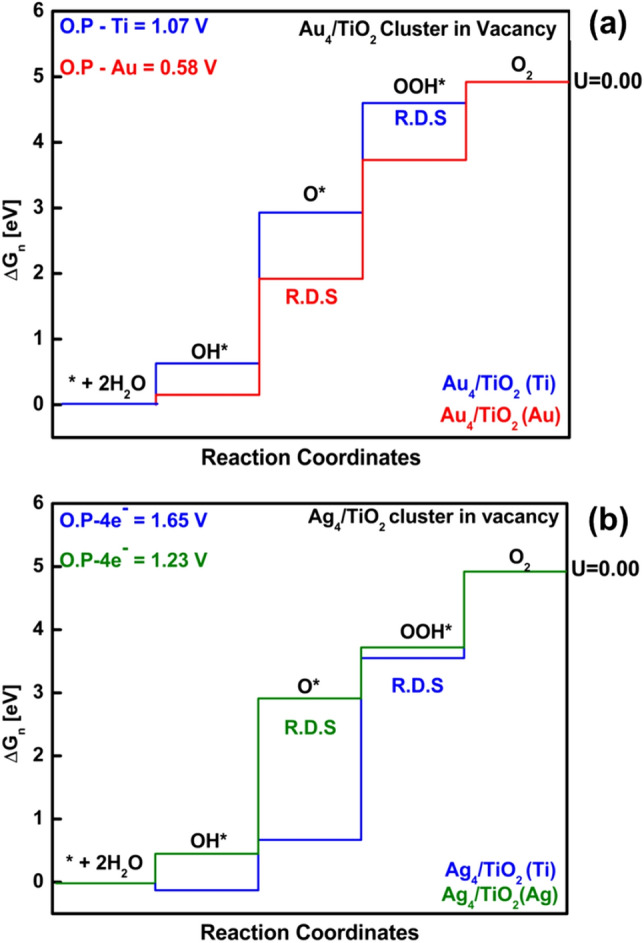
Comparative free energy diagram for (a) Ag4/TiO2 and (b) Au4/TiO2. Overpotential is shown in the upper left corner. The rate-determining step (R.D.S) for Ag and Au as active sites is O* while OOH* for Ti atom as an active site. Thus, the blue profile represents Ti as an active site, the red profile represents Au as an active site, and the green profile represents Ag as an active site.
Studying the OER mechanism on all Au and Ag clusters, i.e., Ag4/TiO2 and (b) Au4/TiO2, placed in bridging row oxygen vacancy, we found that overpotential for Ag active site was 0.53 V higher for Ag4/TiO2 when compared with Ag2–Au2/TiO2, i.e., 0.70 V. However, for the Ti active site, overpotential is remarkably less, and a difference of 0.81 V was observed. If we compare Au4/TiO2 with Ag2–Au2/TiO2 with Au active site, the overpotential remains the same almost. However, there are noticeable changes for the Ti active site, i.e., 1.39 V. So we conclude from our results that, in the case of any model, either initial or cluster, the active site is the preferably Au atom. In terms of overpotential, the trend of photocatalytic activity on all studied models is Au > Ag > Ti.
Insights into OER
We mentioned earlier that Ag noble metal could be an excellent approach over Au for plasmonic photocatalytic activity; however, the plasmonic frequency of Ag is near the UV region responsible for limiting the catalytic activity of Ag-based photocatalyst. Therefore, we make a bimetallic alloy of Au and Ag. From our OER calculations, we have concluded that on Ag4/TiO2 over potential is higher for Ag active site, i.e., 1.23 V, for Au4/TiO2 overpotential is lower with Au active site, i.e., 0.58 V. But when Au and Ag are in a bimetallic cluster state, and we perform OER calculations on Au2–Ag2/TiO2, the overpotential with Au and Ag active site is 0.60 V and 0.70 V, respectively. Hence, this proves that the presence of Au noble metal could stabilize the Ag and lead to better photocatalytic activity. Unfortunately, there are fewer reports of OER studies on noble metal-loaded TiO2 structures. Wang et al.57 reported the OER mechanism on Au/TiO2 initial structure. Instead of removing Bridging oxygen, surface oxygen was removed and was further doped with Au atom. They only chose Ti as an active site. The rate-determining step was OOH* with an overpotential of 1.77 V. To provide further insight about noble metal-doped TiO2 surface, the calculations were extended by using a small (four atoms) tetrahedral cluster for determining the OER activities. In the near future, studies with larger bimetallic clusters are needed to check if the trends observed here stay within the range of bimetallic particles.
Bader charge analysis
Oxygen vacancy is one of the vital defects among all the defects known in TiO258. It has been discovered that oxygen vacancies can perform as significant adsorption and active sites for heterogeneous catalysis, which can sturdily sway the reactivity of metal oxides59. Theoretically, oxygen vacancies formation on TiO2 is direct to the creation of unpaired electrons or Ti3+ centers, which possibly will form donor levels in the electronic structure of TiO260. Theoretical and experimental results indicate that the excess electrons are located on the oxygen vacancy states. When noble metal atoms (Au or Ag) are adsorbed on the TiO2 surface, they tend to adsorb on the oxygen vacant site due to the high electronegativity of noble metal atoms.
We performed the Bader charge analysis to investigate the electron transfer between the metal active site (Au, Ag, or Ag–Au) and the catalyst surface for Ag4/TiO2, Au4/TiO2, and Ag2–Au2/TiO2 system, as shown in Fig. 6. The calculated values are given in Table 4. The negative and positive signs refer to electron accumulation and depletion, respectively.
Figure 6.

Configurations of the TiO2-supported monometallic and bimetallic catalysts of (a) Ag4/TiO2, (b) Au4/TiO2, and (c) Au2-Ag2/TiO2. The color code is blue for Ti, red for O, silver for Ag, and gold for Au.
Table 4.
Charge of each atom derived from the Bader charge analysis of Ag4/TiO2, Au4/TiO2 and Ag2–Au2/TiO2 systems.
| System | Cluster | Bader charge (|e|) | |
|---|---|---|---|
| Ag4/TiO2 | Ag4 | Ag4 cluster | + 0.14 |
| Ag1 atom | − 0.23 | ||
| Ag2 atom | + 0.10 | ||
| Ag3 atom | + 0.04 | ||
| Ag4 atom | + 0.23 | ||
| Au4/TiO2 | Au4 | Au4 cluster | − 0.32 |
| Ag1 atom | − 0.41 | ||
| Ag2 atom | + 0.01 | ||
| Ag3 atom | − 0.05 | ||
| Ag4 atom | + 0.13 | ||
| Ag2–Au2/TiO2 | Ag2–Au2 | Ag2–Au2 cluster | − 0.14 |
| Au2 subcluster | − 0.69 | ||
| Ag2 subcluster | + 0.55 | ||
| Au1 atom | − 0.48 | ||
| Au2 atom | − 0.21 | ||
| Ag1 atom | + 0.22 | ||
| Ag2 atom | + 0.33 | ||
For the Ag4/TiO2 structure, the positive value of the Ag4 cluster (+ 0.14 |e|) designates its role as an electron donor during its adsorption on the TiO2 surface with an oxygen vacancy site. Moreover, a negative Bader charge of Ag1 (− 0.23 |e|) together with the positive Bader charge of other surrounding Ag atoms (varies from + 0.04 to + 0.23 |e|) reveal electron transfer from the oxygen vacancy site to other Ag atoms: Ag2, Ag3, and Ag4, to Ag1 atom. Hence, the activity of the Ag1 atom is improved.
For Au4/TiO2 structure, the Au4 cluster plays a role as an electron acceptor, taking electrons from the TiO2 surface. Similar to the Ag4/TiO2 structure, the most negative Bader charge is found on the Au1 site, which specifies that the activity of the Au1 site is enhanced after its adsorption on the TiO2 surface with oxygen vacancy site. In addition, the electron accumulations around the Au1 site is found to be two times higher than that of the Ag1. This suggested a high activity of the Au system.
For the Ag2–Au2/TiO2 system, the whole bimetallic Ag2–Au2 cluster acted as an electron acceptor similar to the Ag cluster in the Ag4/TiO2 system. However, the total Bader charge value of the Ag2–Au2 cluster is less negative (− 0.14 |e|). Considering the two-atom Ag (Ag2) and two-atom Au (Au2) cluster within the Ag2–Au2 cluster, it was found that the Au2 subcluster holds a negative Bader charge of − 0.69|e|, while the positive Bader charge was found in the Ag2 subcluster of + 0.55 |e|. This phenomenon describes the additional electron transfer from the Ag2 to the Au2 subcluster.
In addition, the activity of the Au1 active site in the Ag2–Au2 cluster is promoted via the electron transfer from the Ag site. Moreover, the Au1 site located at the vacancy site of the TiO2 surface still has the most negative Bader charge value of − 0.48 |e|. This Bader charge value of the Au1 atom in the Ag2–Au2 cluster is similar to the Au1 atom in the Au4 cluster. Hence, the presence of Ag species in the Ag2–Au2 bimetallic cluster facilitates electron delocalization around Au1 and Au2, creating another active site—the Au2 site. Therefore, the activity of the bimetallic Ag2–Au2 cluster exceeds the monometallic cluster of Au4 and Ag4 clusters due to the presence of the Ag2 subcluster, which donates additional electrons to the Au2 subcluster.
Conclusion
In summary, we used density functional theory to investigate the role of Au metal in Au–Ag high noble alloys catalysts supported on TiO2 on the performance during the oxygen evolution of water oxidation (OER), in which the catalysts are modeled as Au, Ag, and Au–Ag supported on rutile TiO2 (110). Since doping of noble metal atoms increases the photocatalytic activity of TiO2, the Ag noble metal is shown to have a promising catalytic activity among the groups. However, studying the reaction mechanism on such a surface is essential due to its low stability.
Combining Au (a more stable noble atom) into Ag-based TiO2 forming bimetallic high-noble alloy catalysts can enhance photocatalytic activity. The study investigated the high-noble alloy cluster in terms of the most stable configuration verified by the most stable position of the metal-support interface, stability of the active site, optimal size, and the most active region of the catalyst for the OER during water oxidation. The most stable location of the metal-support interface for the Au–Ag high-noble alloy catalyst modeled by the Au2–Ag2/TiO2 was found at the bridging row oxygen vacant site. On the performance of this high-noble alloy catalyst, the Au atom is always preferred as an active site for OER regardless of the size or position of the cluster. The photocatalytic activity trend indicated by an overpotential and active site preference is Au > Ag > Ti. Hence, in both cases of monometallic (Au and Ag) and bimetallic (Au–Ag), the Ti active site is least preferred.
With this positioning, the overpotential is much lower for the Au and Ag active site, i.e., 0.60 V and 0.70 V, indicating that the Au atom stabilizes Ag. Therefore, utilizing the high-noble alloy catalyst of Au–Ag can improve the oxygen evolution activity on the rutile TiO2 (110) surface during the water oxidation reaction, promoting efficient hydrogen production and supporting the hydrogen economy.
In addition, the future design of high photocatalytic performance catalyst based on this study must consider the profiles from the Bader charge analysis, which suggested that the presence of the Ag atom can stabilize the Au atom via the electron transfer to the Au, where this generated the trap state between the valence band maximum (VBM) and the conduction band minimum (CBM) reducing the band gap of the catalyst promoting activity as supported by the projected density of states (DOS) profile of this Au–Ag/TiO2 catalyst.
Methods
Computational details
Density functional theory-based calculations
We employed the first-principles spin-polarized density functional theory (DFT) calculations using the projector augmented wave (PAW) method applied in the Vienna Ab initio simulation package (VASP)61,62. For the exchange–correlation functional, the generalized gradient approximation (GGA)63 of Perdew − Burke − Ernzerhof (PBE)64 was used with the plane-wave cutoff energy of 400 eV. Ultra-soft pseudo-potential, the interaction between valence electrons and the ionic core65, is used with 2s2 2p4, 3d2 4s2,4d105s1, 5d106s1 as the valence electrons configuration for the O, Ti, Ag, and Au atoms, respectively. The Monkhorst and Pack scheme of k-point sampling was employed to integrate the first Brillouin zone66, and a 3 × 2 × 1 grid was used to obtain the geometry optimization and total energies for the rutile TiO2 (110) surface. The applied residual forces for geometry optimization and convergence on the atoms were 0.01 eV/Å, and the self-consistent iteration awaits the tolerance for total energy to get to 10–4 eV. The optimized lattice parameters a = 11.83 Å, b = 12.99 Å, c = 30.08 Å, and α = β = γ = 90.00° for rutile (110). The calculations were done on a four-layered rutile TiO2 (110) surface slab with a super-cell of 3 × 2 × 1 where the bottom layer was fixed at bulk-truncated positions and the others were fully relaxed. Dipole correction was applied to all of the calculations. We calculated the energy position, valence band maximum (VBM), conduction band minimum (CBM), and trap states level for band structure calculations. For these properties, we carried out single-point energy calculations with the HSE06 functional67 because they are known to provide a better description of the valence band (VB) and conduction band (CB), band edges, and band gaps (that are underestimated with PBE).
Catalyst model constructions
The OER mechanism was studied on the Au/TiO2, Ag/TiO2, and Au–Ag/TiO2. For the initial model: We created an oxygen vacancy in the bridging row on the surface which was filled by gold atom in Au/TiO2 model (Fig. 7a) and similarly Ag atom for Ag/TiO2 (Fig. 7b); however, in the case of the Au–Ag/TiO2 model, we replaced two bridging oxygen atoms from different bridging rows on the surface to replace Au and Ag atoms shown in Fig. 7c. Figure 7d shows the clean rutile (110) surface. We have demonstrated the bridging oxygen, which was pulled out to create oxygen vacancies. These configurations are shown to be at the most stable metal-support interface location. For the cluster model: We placed the tetrahedral cluster of 4 atoms Au4, Ag4, and Au2–Ag2 with different orientations on the surface, as shown in Figs. 8 and 9. We chose the cluster as a model because of its stability68. The result exhibited that the Au2–Ag2 alloy anchored on bridging oxygen on top of the rutile TiO2 (110) surface is the most stable, wherein Fig. 7d, a clean surface, is shown with the chosen oxygen vacancies marked as Ovac.
Figure 9.

Four atoms cluster on bridging row oxygen vacant site of (a) Ag4 on TiO2, (b) Au4 on TiO2, and (c) Ag2–Au2 on rutile TiO2 (110) surface model.
Evaluation of water oxidation performance
The reaction mechanism of water oxidation can occur with four proton-coupled electron transfer steps, where the initial step is the water adsorption process. Our calculations showed that water molecules tend to adsorb on the TiO2 surface. We used the mechanism proposed by Rossmeisl et al.69. The OER mechanism is progressed in the following four steps as shown in equations 1–4, where ½ H2 ↔ H+ + e− is the half electrode reaction.
| 1 |
| 2 |
| 3 |
| 4 |
The OER intermediates are shown in Fig. 10a–f on Ti and Au as an active site, for Ag active site (see figure S2) in S.I. To evaluate the steps as mentioned above, it is expedient to characterize the free energies ΔGOH*, ΔGO* and ΔGOOH* of the OH*, O*, OOH* intermediate states, and ΔGO2 for the O2 final stage, all determined relative to the resting state (* + 2H2O). In Sect. 1 of supplementary information (S.I), we have details of the determination of the free energies (including vibrational and entropy corrections). We can extract the reaction step-free energies from the relative free energies; ΔGn (n = I, II, III, and IV) are written as equations 5–6.
| 5 |
| 6 |
| 7 |
| 8 |
The overpotential of reactions could be associated reliably to the proton and electron transfer to adsorbed species strongly bonded to the surface at the electrode potential. This reaction series starts with (* + 2H2O) on a metal active site leading to OH*, shown in steps I–IV. The diffusion of species and other surface reactions most likely depends weakly on the potential.
The theoretical overpotential is defined as:
| 9 |
Supplementary Information
Acknowledgements
This research is supported by The Second Century Fund (C2F), Chulalongkorn University, Thailand. We would also like to acknowledge the 503 Can Li group, Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences (CAS), China, and the CECC-HCU server from the High-Performance Computing Unit (CECC-HCU), Center of Excellence on Catalysis and Catalytic Reaction Engineering (CECC), Chulalongkorn University for the computing resource.
Author contributions
S.P., A.M., M.R., and T.S. conceived the computational simulations. A.M. performed computational simulations. All authors performed data analyses, wrote, and reviewed the manuscript.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Taifeng Liu, Email: tfliu@vip.henu.edu.cn.
Supareak Praserthdam, Email: supareak.p@chula.ac.th.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-06608-7.
References
- 1.Chen X, Shen S, Guo L, Mao SS. Semiconductor-based photocatalytic hydrogen generation. Chem. Rev. 2010;110:6503–6570. doi: 10.1021/cr1001645. [DOI] [PubMed] [Google Scholar]
- 2.Fujishima A, Rao TN, Tryk DA. Titanium dioxide photocatalysis. J. Photochem. Photobiol., C. 2000;1:1–21. [Google Scholar]
- 3.Henderson MA. A surface science perspective on TiO2 photocatalysis. Surf Sci Rep. 2011;66:185–297. [Google Scholar]
- 4.Hoffmann MR, Martin ST, Choi W, Bahnemann DW. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995;95:69–96. [Google Scholar]
- 5.Kamat PV. Meeting the clean energy demand: Nanostructure architectures for solar energy conversion. J. Phys. Chem. C. 2007;111:2834–2860. [Google Scholar]
- 6.Linsebigler AL, Lu G, Yates JT., Jr Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995;95:735–758. [Google Scholar]
- 7.Matsuoka M, et al. Photocatalysis for new energy production: Recent advances in photocatalytic water splitting reactions for hydrogen production. Catal Today. 2007;122:51–61. [Google Scholar]
- 8.Mills A, Davies RH, Worsley D. Water purification by semiconductor photocatalysis. Chem. Soc. Rev. 1993;22:417–425. [Google Scholar]
- 9.Ida S, et al. A cocatalyst that stabilizes a hydride intermediate during photocatalytic hydrogen evolution over a rhodium-doped TiO2 nanosheet. Angew. Chem. 2018;130:9211–9215. doi: 10.1002/anie.201803214. [DOI] [PubMed] [Google Scholar]
- 10.Fu Y, et al. Phase-modulated band alignment in CdS nanorod/SnSx nanosheet hierarchical heterojunctions toward efficient water splitting. Adv. Funct. Mater. 2018;28:1706785. [Google Scholar]
- 11.Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science355 (2017). [DOI] [PubMed]
- 12.Schalenbach M, Zeradjanin AR, Kasian O, Cherevko S, Mayrhofer KJ. A perspective on low-temperature water electrolysis–challenges in alkaline and acidic technology. Int. J. Electrochem. Sci. 2018;13:1173–1226. [Google Scholar]
- 13.Lee B-H, et al. Reversible and cooperative photoactivation of single-atom Cu/TiO 2 photocatalysts. Nat. Mater. 2019;18:620–626. doi: 10.1038/s41563-019-0344-1. [DOI] [PubMed] [Google Scholar]
- 14.Fujishima A, Honda K. Electrochemical photolysis of water at a semiconductor electrode. Nature. 1972;238:37–38. doi: 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- 15.Cao S, et al. Photocatalytic pure water splitting with high efficiency and value by Pt/porous brookite TiO2 nanoflutes. Nano Energy. 2020;67:104. [Google Scholar]
- 16.Wu T, et al. Chlorine capped SnO2 quantum-dots modified TiO2 electron selective layer to enhance the performance of planar perovskite solar cells. Sci. Bull. 2019;64:547–552. doi: 10.1016/j.scib.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Fang S, Hu YH. Recent progress in photocatalysts for overall water splitting. Int. J. Energy Res. 2019;43:1082–1098. [Google Scholar]
- 18.Mao L, Huang Y-C, Fu Y, Dong C-L, Shen S. Surface sulfurization activating hematite nanorods for efficient photoelectrochemical water splitting. Sci. Bull. 2019;64:1262–1271. doi: 10.1016/j.scib.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Zhao D, et al. Synergy of dopants and defects in graphitic carbon nitride with exceptionally modulated band structures for efficient photocatalytic oxygen evolution. Adv. Mater. 2019;31:1903545. doi: 10.1002/adma.201903545. [DOI] [PubMed] [Google Scholar]
- 20.Dozzi MV, Selli E. Doping TiO2 with p-block elements: Effects on photocatalytic activity. J. Photochem. Photobiol. C. 2013;14:13–28. [Google Scholar]
- 21.Li J, et al. Solar hydrogen generation by a CdS-Au-TiO2 sandwich nanorod array enhanced with Au nanoparticle as electron relay and plasmonic photosensitizer. J. Am. Chem. Soc. 2014;136:8438–8449. doi: 10.1021/ja503508g. [DOI] [PubMed] [Google Scholar]
- 22.Xiao JD, Han L, Luo J, Yu SH, Jiang HL. Integration of plasmonic effects and schottky junctions into metal-organic framework composites: Steering charge flow for enhanced visible-light photocatalysis. Angew. Chem. Int. Ed. 2018;57:1103–1107. doi: 10.1002/anie.201711725. [DOI] [PubMed] [Google Scholar]
- 23.Xue F, et al. Interfacial and dimensional effects of Pd co-catalyst for efficient photocatalytic hydrogen generation. J. Phys. Chem. C. 2018;122:25165–25173. [Google Scholar]
- 24.Zhang, P., Wang, T. & Gong, J. Solar Water Splitting: Mechanistic Understanding of the Plasmonic Enhancement for Solar Water Splitting (Adv. Mater. 36/2015). Adv. Mater.27, 5444–5444 (2015). [DOI] [PubMed]
- 25.Lou Z, et al. Plasmonic heterostructure TiO2-MCs/WO3− x-NWs with continuous photoelectron injection boosting hot electron for methane generation. Adv. Funct. Mater. 2019;29:1808696. [Google Scholar]
- 26.Clavero C. Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices. Nat. Photon. 2014;8:95–103. [Google Scholar]
- 27.Brongersma ML, Halas NJ, Nordlander P. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 2015;10:25–34. doi: 10.1038/nnano.2014.311. [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Dong CL, Du Y, Zhao D, Shen S. Nanogap engineered plasmon-enhancement in photocatalytic solar hydrogen conversion. Adv. Mater. Interfaces. 2015;2:1500280. [Google Scholar]
- 29.Linic S, Christopher P, Ingram DB. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011;10:911–921. doi: 10.1038/nmat3151. [DOI] [PubMed] [Google Scholar]
- 30.Feng B, et al. Achieving infrared detection by all-Si plasmonic hot-electron detectors with high detectivity. ACS Nano. 2019;13:8433–8441. doi: 10.1021/acsnano.9b04236. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Hu H, Huang X, Bi Y. Photo-controlled bond changes on Pt/TiO 2 for promoting overall water splitting and restraining hydrogen–oxygen recombination. J. Mater. Chem. A. 2019;7:5938–5942. [Google Scholar]
- 32.Xue F, Si Y, Wang M, Liu M, Guo L. Toward efficient photocatalytic pure water splitting for simultaneous H2 and H2O2 production. Nano Energy. 2019;62:823–831. [Google Scholar]
- 33.Malik AS, Liu T, Dupuis M, Li R, Li C. Water oxidation on TiO2: A comparative DFT study of 1e–, 2e–, and 4e—processes on rutile, anatase, and brookite. J. Phys. Chem. C. 2020;124:8094–8100. doi: 10.1021/acs.jpcc.9b11450. [DOI] [Google Scholar]
- 34.Haider, R. S. et al. Boosting photocatalytic water oxidation by surface plasmon resonance of AgxAu1-x alloy nanoparticles. Nano Energy, 106189 (2021).
- 35.Plodinec M, et al. Black TiO2 nanotube arrays decorated with Ag nanoparticles for enhanced visible-light photocatalytic oxidation of salicylic acid. J Alloy Compd. 2019;776:883–896. [Google Scholar]
- 36.Wataha JC. Biocompatibility of dental casting alloys: A review. J. Prosthet. Dent. 2000;83:223–234. doi: 10.1016/S0022-3913(00)80016-5. [DOI] [PubMed] [Google Scholar]
- 37.Strauss I, et al. Characterization of multi-channel intraneural stimulation in transradial amputees. Sci Rep UK. 2019;9:1–11. doi: 10.1038/s41598-019-55591-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Link S, Wang ZL, El-Sayed M. Alloy formation of gold−silver nanoparticles and the dependence of the plasmon absorption on their composition. J. Phys. Chem. B. 1999;103:3529–3533. [Google Scholar]
- 39.Papavassiliou GC. Surface plasmons in small Au-Ag alloy particles. J. Phys. F Met. Phys. 1976;6:L103. [Google Scholar]
- 40.Liu J-H, Wang A-Q, Chi Y-S, Lin H-P, Mou C-Y. Synergistic effect in an Au− Ag alloy nanocatalyst: CO oxidation. J. Phys. Chem. B. 2005;109:40–43. doi: 10.1021/jp044938g. [DOI] [PubMed] [Google Scholar]
- 41.Goswami C, et al. Boosting the electrocatalytic activity of Pd/C by Cu alloying: Insight on Pd/Cu composition and reaction pathway. J. Colloid Interface Sci. 2021;587:446–456. doi: 10.1016/j.jcis.2020.11.104. [DOI] [PubMed] [Google Scholar]
- 42.Koga H, et al. Effect of ceria and zirconia supports on NO reduction over platinum-group metal catalysts: A DFT study with comparative experiments. Catal. Today. 2019;332:236–244. doi: 10.1016/j.cattod.2018.07.023. [DOI] [Google Scholar]
- 43.Aguilera-Granja, F., Aguilera–del–Toro, R. H., Vogel, E. E. & Cisternas, E. TiO2 nano-clusters adsorbed on surfaces: A density-functional-theoretic study. J. Phys. Chem. Solids150, 109716. 10.1016/j.jpcs.2020.109716 (2021).
- 44.Biener J, Farfan-Arribas E, Biener M, Friend CM, Madix RJ. Synthesis of TiO2 nanoparticles on the Au(111) surface. J. Chem. Phys. 2005;123:094705. doi: 10.1063/1.1999607. [DOI] [PubMed] [Google Scholar]
- 45.Aguilera-Granja, F., Aguilera–del–Toro, R. H., Vogel, E. E. & Cisternas, E. Bimetalic (AuPt)4 nano-clusters adsorbed on TiO2 nano-wires: A density-functional-theoretic study. J. Phys. Chem. Solids159, 110275, 10.1016/j.jpcs.2021.110275 (2021).
- 46.Zhang X-F, et al. TiO2 nanorods loaded with AuPt alloy nanoparticles for the photocatalytic oxidation of benzyl alcohol. J. Phys. Chem. Solids. 2019;126:27–32. doi: 10.1016/j.jpcs.2018.10.026. [DOI] [Google Scholar]
- 47.Vijay A, Mills G, Metiu H. Adsorption of gold on stoichiometric and reduced rutile TiO2 (110) surfaces. J. Chem. Phys. 2003;118:6536–6551. doi: 10.1063/1.1557919. [DOI] [Google Scholar]
- 48.Lee HM, Ge M, Sahu BR, Tarakeshwar P, Kim KS. Geometrical and electronic structures of gold, silver, and gold−silver binary clusters: Origins of ductility of gold and gold−silver alloy formation. J. Phys. Chem. B. 2003;107:9994–10005. doi: 10.1021/jp034826+. [DOI] [Google Scholar]
- 49.Assadollahzadeh B, Schwerdtfeger P. A systematic search for minimum structures of small gold clusters Aun (n=2–20) and their electronic properties. J. Chem. Phys. 2009;131:064306. doi: 10.1063/1.3204488. [DOI] [PubMed] [Google Scholar]
- 50.Tada K, et al. Effects of halogens on interactions between a reduced TiO2 (110) surface and noble metal atoms: A DFT study. Appl. Surf. Sci. 2017;411:149–162. doi: 10.1016/j.apsusc.2017.03.113. [DOI] [Google Scholar]
- 51.Tada K, et al. Effect of surface interactions on spin contamination errors of homogeneous spin dimers, chains, and films: model calculations of Au/MgO and Au/BaO systems. Mol. Phys. 2021;119:e1791989. doi: 10.1080/00268976.2020.1791989. [DOI] [Google Scholar]
- 52.Diebold U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003;48:53–229. doi: 10.1016/S0167-5729(02)00100-0. [DOI] [Google Scholar]
- 53.Chrétien, S. & Metiu, H. Density functional study of the interaction between small Au clusters, Aun (n=1–7) and the rutile TiO2 surface. I. Adsorption on the stoichiometric surface. J. Chem. Phys.127, 084704. 10.1063/1.2770462 (2007). [DOI] [PubMed]
- 54.Madsen GKH, Hammer B. Effect of subsurface Ti-interstitials on the bonding of small gold clusters on rutile TiO2(110) J. Chem. Phys. 2009;130:044704. doi: 10.1063/1.3055419. [DOI] [PubMed] [Google Scholar]
- 55.Vijay, A., Mills, G. & Metiu, H. Adsorption of gold on stoichiometric and reduced rutile TiO2 (110) surfaces. Vol. 118 (2003).
- 56.Valdés Á, Qu ZW, Kroes GJ, Rossmeisl J, Nørskov JK. Oxidation and photo-oxidation of water on TiO2 surface. J. Phys. Chem. C. 2008;112:9872–9879. doi: 10.1021/jp711929d. [DOI] [Google Scholar]
- 57.Wang S, et al. Positioning the water oxidation reaction sites in plasmonic photocatalysts. J. Am. Chem. Soc. 2017;139:11771–11778. doi: 10.1021/jacs.7b04470. [DOI] [PubMed] [Google Scholar]
- 58.Pacchioni G. Oxygen vacancy: the invisible agent on oxide surfaces. ChemPhysChem. 2003;4:1041–1047. doi: 10.1002/cphc.200300835. [DOI] [PubMed] [Google Scholar]
- 59.Polarz S, et al. On the role of oxygen defects in the catalytic performance of zinc oxide. Angew. Chem. Int. Ed. 2006;45:2965–2969. doi: 10.1002/anie.200503068. [DOI] [PubMed] [Google Scholar]
- 60.Nowotny, M. K., Sheppard, L. R., Bak, T. & Nowotny, J. Defect chemistry of titanium dioxide. Application of defect engineering in processing of TiO2-based photocatalysts. J. Phys. Chem. C112, 5275–5300 (2008).
- 61.Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54:11169. doi: 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- 62.Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999;59:1758. [Google Scholar]
- 63.Perdew JP, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B. 1992;45:13244. doi: 10.1103/physrevb.45.13244. [DOI] [PubMed] [Google Scholar]
- 64.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 65.Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B. 1990;41:7892. doi: 10.1103/physrevb.41.7892. [DOI] [PubMed] [Google Scholar]
- 66.Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys. Rev. B. 1976;13:5188. [Google Scholar]
- 67.Heyd J, Scuseria GE, Ernzerhof M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003;118:8207–8215. doi: 10.1063/1.1564060. [DOI] [Google Scholar]
- 68.Heiles S, Logsdail AJ, Schäfer R, Johnston RL. Dopant-induced 2D–3D transition in small Au-containing clusters: DFT-global optimisation of 8-atom Au–Ag nanoalloys. Nanoscale. 2012;4:1109–1115. doi: 10.1039/c1nr11053e. [DOI] [PubMed] [Google Scholar]
- 69.Rossmeisl J, Qu ZW, Zhu H, Kroes GJ, Nørskov JK. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007;607:83–89. doi: 10.1016/j.jelechem.2006.11.008. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).