

Summary
The existence of overlapping genes (OLGs) with significant coding overlaps revolutionizes our understanding of genomic complexity. We report two exceptionally long (957 nt and 1536 nt), evolutionarily novel, translated antisense open reading frames (ORFs) embedded within annotated genes in the pathogenic Gram-negative bacterium Pseudomonas aeruginosa. Both OLG pairs show sequence features consistent with being genes and transcriptional signals in RNA sequencing. Translation of both OLGs was confirmed by ribosome profiling and mass spectrometry. Quantitative proteomics of samples taken during different phases of growth revealed regulation of protein abundances, implying biological functionality. Both OLGs are taxonomically restricted, and likely arose by overprinting within the genus. Evidence for purifying selection further supports functionality. The OLGs reported here, designated olg1 and olg2, are the longest yet proposed in prokaryotes and are among the best attested in terms of translation and evolutionary constraint. These results highlight a potentially large unexplored dimension of prokaryotic genomes.
Subject areas: Microbiology, Genomic analysis
Graphical abstract
Highlights
-
•
Two novel, very long, overlapping genes were found in Pseudomonas aeruginosa
-
•
Both overlapping genes, olg1 and olg2, are transcribed, translated, and regulated
-
•
Mass spectrometry verifies translation of the overlapping and their mother genes
-
•
Both overlapping genes are taxonomically restricted, but under purifying selection
Microbiology; Genomic analysis
Introduction
The tri-nucleotide character of the genetic code enables six reading frames in a double-stranded nucleotide sequence. Protein-coding ORFs at the same locus but in different reading frames are referred to as overlapping genes (OLGs). Studies of coding overlaps of more than 90 nucleotides, i.e., nontrivial overlaps, have mainly been restricted to viruses, where the first OLG was found in 1976 (Barrell et al., 1976). An OLG pair with such a nontrivial overlap can be described in terms of an older, typically longer “mother gene” and more recently evolved “daughter” gene by analogy to mother and daughter cells in reproduction.
In prokaryotes, automated genome-annotation algorithms such as Glimmer allow only one open reading frame (ORF) per locus with the exception of only short overlaps (Delcher et al., 2007). This systematically excludes overlapping ORFs from being annotated as genes (Warren et al., 2010). The “inferior” ORFs (e.g., shorter or fewer hits in databases) within overlapping gene pairs have been called “shadow ORFs,” as they are found in the shadow of the annotated coding ORF (Yooseph et al., 2007). Determining which ORF to annotate within such pairs has been described as the most difficult problem in prokaryotic gene annotation (Salzberg et al., 1998). Nevertheless, a few prokaryotic OLGs have been discovered, often serendipitously in the pursuit of other unannotated genes (Baek et al., 2017; Jensen et al., 2006; Weaver et al., 2019). For instance, in some Escherichia coli strains a few nontrivial overlaps have been detected and experimentally analyzed (Behrens et al., 2002; Fellner et al., 2014; Fellner et al., 2015; Hücker et al., 2018a; Hücker et al., 2018b; Vanderhaeghen et al., 2018; Zehentner et al., 2020a; b). Transcriptomic or translatomic evidence for OLGs also exist in genera such as Pseudomonas (Filiatrault et al., 2010) and Mycobacterium (Smith et al., 2019). Recently, evidence for OLGs has also been reported in archaea (Gelsinger et al., 2020) and in mammals (Khan et al., 2020; Loughran et al., 2020). These findings support the hypothesis that OLGs are ubiquitous. Various aspects of OLGs, including potential applications in synthetic biology, have been discussed in a recent review (Wright et al., 2021).
Verifying OLGs using mass spectrometry (MS) is difficult because most OLGs appear to be short and weakly expressed, and proteomics has limited abilities in detecting such proteins (Petruschke et al., 2020). RNA sequencing led to the discovery of many antisense transcripts, but whether many of these are translated is controversial (Ardern et al., 2020). More recently, mRNA protected by ribosomes after enzymatic degradation has been sequenced using “ribosome profiling” (RiboSeq) (Ingolia et al., 2009), showing evidence of translation for antisense transcripts (Ardern et al., 2020; Zehentner et al., 2020a). However, artifacts may occur due to structured RNAs (Fremin and Bhatt, 2020). Moreover, proteins produced from such transcripts have been claimed to be predominantly nonfunctional in Mycobacterium tuberculosis (Smith et al., 2019). Detecting purifying selection would provide evidence for functionality in OLGs; however, this is made difficult by selection pressure on the other reading frame. A number of methods have been developed (Firth, 2014; Nelson et al., 2020b; Sabath et al., 2008; Sealfon et al., 2015; Wei and Zhang, 2015) but so far applied mainly in viruses. Additional reason to expect to find OLGs under selection in bacteria comes from a study showing excess long antisense ORFs in a pathogenic E. coli strain (Mir et al., 2012). Another is the frequent exchange of functional genes between phages and bacteria (Koskella and Brockhurst, 2014).
The evolution and origin of OLGs and their constraints have long been discussed (Miyata and Yasunaga, 1978; Portelli, 1982; Sander and Schulz, 1979; Yockey, 1979) and were briefly mentioned in two early important books (Grassé, 1977; Ohno, 1970). The origin of a new gene within an existing gene is termed “overprinting” (Keese and Gibbs, 1992). Advantageous for same-strand overlaps is that the hydrophobicity profile of a frame-shifted sequence tends to be similar to that of the unshifted sequence (Bartonek et al., 2020), as may have contributed to the origin of an OLG in SARS-CoV-2 (Nelson et al., 2020a). Intriguingly, overlaps of the reference frame “+1” and antisense “−1” tend to have opposite hydrophobicities for their amino acids (Konecny et al., 1993). Further, similar amino acids are conserved in the antisense frame −1 following synonymous mutation in the reference frame (Konecny et al., 1993), facilitating the maintenance of overlapping genes. The developing research area of overlapping gene origins complements recent findings of many taxonomically restricted (“orphan”) genes (Tautz and Domazet-Lošo, 2011) and unannotated short genes in prokaryotes (Storz et al., 2014).
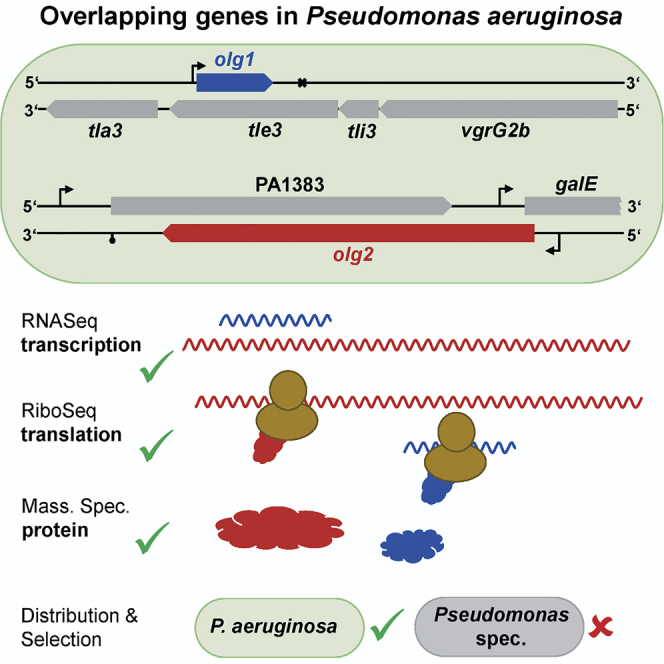
The genus Pseudomonas (Gram-negative, Gammaproteobacteria) is of particular interest regarding long OLGs. Its high GC content of 60%–70% results in longer average lengths for antisense ORFs compared with Escherichia (∼50% GC) (Mir et al., 2012). Previous studies reported OLGs in the well-characterized species Pseudomonas fluorescens and Pseudomonas putida, although based on limited evidence (Silby and Levy, 2004, 2008; Yang et al., 2016). Since then, methods allowing improved discovery and verification of such genes have been developed. Here, we examine Pseudomonas aeruginosa, which is a versatile pathogenic species (Crone et al., 2020). As an opportunistic human pathogen, it predominately causes disease in immunocompromised individuals (Kerr and Snelling, 2009). Hospital-acquired infections with P. aeruginosa are often associated with high morbidity and mortality and may include infections of the respiratory tract, the urinary tract, the bloodstream, the skin, and wounds (Weinstein et al., 2005). The high level of intrinsic and acquired resistance as well as the rapid rise of multidrug-resistant strains limit effective antibiotic treatment options (Bassetti et al., 2018; Livermore, 2009). Thus, it is important to understand the entire genomic complexity of this organism. In P. aeruginosa, we detected two exceptionally long novel ORFs showing large overlaps of 957 nt and 1542 nt in antisense to annotated genes, by employing RiboSeq as well as mass spectrometry. We present evidence that both OLGs indeed encode proteins that evolved only recently by overprinting. Further, we detected purifying selection operating through a depletion of stop codons and nonsynonymous changes.
Results
RNASeq and RiboSeq allow detection of two novel expressed genes
RiboSeq in combination with RNASeq is a powerful technique to analyze transcription and translation for any gene expressed (Ingolia et al., 2009). Here, we studied P. aeruginosa PAO1, which was cultivated under standard conditions (LB, 37°C) in two biological replicates. In the past, we had found overlapping genes in E. coli with a GC content of around 50% (e.g., Zehentner et al., 2020b) and now aimed for the detection of overlapping genes in a bacterium with a high GC genome (Mir et al., 2012). Several signals for novel ORFs were detected in the RNASeq data (not shown), including two relatively long ORFs, later designated olg1 and olg2, which attracted our particular interest. The RPKM values, (i.e., reads per kilobase gene per million sequenced reads) for both novel ORFs in either RNASeq or RiboSeq were comparable to values obtained for annotated genes. Further, these values were also comparably high for the respective annotated genes that overlap olg1 and olg2 (Figures 1A and 1B). Both coverage by RiboSeq reads as well as the ratio of RiboSeq reads to RNAseq reads were also in a similar range to most of the other annotated genes (Figures 1C and 1D). More specifically, RNASeq revealed high RPKMRNASeq values of about 36 for the ORF later named olg1, which is encoded antisense to gene PA0260 (tle3) and had an RPKMRNASeq of about 30 as well. The second ORF, later named olg2, had an RPKMRNASeq of about 22, as compared with approximately 20 for the overlapping (annotated) gene PA1383. These values were comparable to annotated protein-coding genes, which range between 0.4 and 13,563.5 (Figure 1A), indicating substantial transcription for the two novel ORFs and the overlapping annotated genes (Figures 2A and 2B, first track; Table S1). Full-length transcription of both OLGs was confirmed by RT-PCR (Figure S1).
Figure 1.

RNASeq and RiboSeq metrics of the overlapping gene pairs olg1-tle3 and olg2-PA1383 compared with all annotated, protein-coding genes (n = 5,572)
(A–D) Shown are violin plots displaying mean reads per kilobase per million mapped reads (RPKM) values for RNASeq (A) and RiboSeq (B), mean coverage values for RiboSeq (C), and mean ribosome coverage values (D), which are calculated by dividing the RPKM [RiboSeq] by the RPKM [RNASeq] of two biological replicates. Included boxplots indicate 25%, 50%, and 75% quartile values for all annotated, protein-coding genes. Values of the overlapping ORFs are represented by colored symbols and their mother genes by the respective grey-shaded symbol.
Figure 2.

RNASeq, RiboSeq, and mass spectrometry signals of the novel OLGs
(A) Locus olg1-tle3,
(B) locus olg2-PA1383
Shown are the mean normalized rpm values of all transcriptome (first track) and translatome reads (second track) of this study (n = 2) for olg1 (blue), olg2 (red), and their mother genes tle3 and PA1383 (both gray). Transcription start (TSS) and stop sites (termination) as well as the positions of start and stop codons are indicated by arrows. Signals of unknown origin upstream of olg1 are highlighted by a question mark. Track three and four illustrate the position and intensity of all peptides obtained by mass spectrometry. Peptides that were selected for targeted proteomics (PRM) are highlighted with an asterisk. Peptides validated and quantified by PRM are indicated by filled asterisks.
Antisense transcription, reported in diverse bacteria including P. aeruginosa, often plays a role in gene regulation, indicated by a negative correlation between antisense and mRNA levels (Dornenburg et al., 2010; Eckweiler and Häussler, 2018). However, evidence exists that some antisense transcripts are translated (Ardern et al., 2020; Friedman et al., 2017; Stringer et al., 2021; Weaver et al., 2019). Our results (Figures 2A and 2B, second track) strongly support translation in both directions for the loci of PA1383 and tle3. The annotated genes showed RPKMRiboSeq values close to the median annotated gene RPKMRiboSeq of 25.7 (Figure 1B). The expression of olg1 was even higher (RPKMRiboSeq = 40.3), whereas olg2 showed a lower, but still unequivocal signal (RPKMRiboSeq = 14.2). In each case, expression was consistent across replicates, and coverage was excellent across the whole ORF (Figures 1C, 2A and 2B, second track). The sequence features reported earlier overall matched the read distributions. However, reads upstream of the proposed olg1 start codon ATG291556 suggest additional transcription and translation (question mark in Figure 2A), perhaps from an alternative start codon or a different frame ORF upstream (Figure 2A, first and second track).
DeepRibo is a software tool combining RiboSeq signals and DNA sequence motifs by using neural networks (Clauwaert et al., 2019). DeepRibo predicts olg1 and olg2 to be translated with the same start codons as we predict from visual assessment in both replicates; it also confirms translation of the mother genes tle3 and PA1383. As a comparison, we analyzed the genes adjacent to tle3—these genes similarly had high coverage and RPKM values (Table S1), implying expression of the operon, but no antisense signals (Figure S2). Finally, ribosome coverage values (RCV) were calculated, i.e. RPKMRiboSeq over RPKMRNASeq (Neuhaus et al., 2016). This value allows a direct estimation of the “translatability” of an ORF. With a value of 1.13, olg1 and PA1383 were within the top 20% of all annotated genes. tle3 and olg2 showed lower RCVs (i.e., 0.79 and 0.64, respectively) but within the range of other annotated genes (Figure 1D).
Mass spectrometry identifies translated peptides
Mass-spectrometry-based proteomics was used to verify the expression of both OLGs as well as the respective mother gene proteins. For that, P. aeruginosa PAO1 was cultivated as described earlier. In total, 4,076 proteins could be detected, including Olg1, Olg2, Tle3, and PA1383 with 12, 5, 10, and 21 peptides, respectively (Table S2). These peptides covered the start, middle, and end regions in each of the four coding sequences (Figures 2A and 2B, last tracks). The measured mass spectrometric intensities of the four target proteins were in a medium to low range compared with all other detected proteins (Figure S3). To exclude false-positive peptide identifications, we validated the fragment ion spectra of all detected peptides from the four target proteins using the artificial intelligence algorithm Prosit (Gessulat et al., 2019). Prosit can predict a peptide’s fragment ion spectrum based on its amino acid sequence. Except for two peptides, correlation scores (dot product) between experimental and predicted spectra were larger than 0.6 (Table S2), which supports correct identification of almost all putative peptides from Olg1, Olg2, Tle3, and PA1383.
For peptide identification with highest confidence, as well as for accurate protein quantification, we next performed targeted proteomic measurements including isotopically labeled reference peptides. Based on our initial mass spectrometric data, we selected four to five peptides per target protein (Figures 2A and 2B, lower tracks, peptides indicated with an asterisk) and purchased those in synthetic and stable isotopically labelled form. Those heavy reference peptides were spiked into P. aeruginosa PAO1 samples taken from various growth time points (Figure 3A) and measured using the targeted proteomic method Parallel Reaction Monitoring (PRM). We successfully validated and quantified four peptides for Olg1, five peptides for PA1383, four peptides for Tle3, and one peptide of Olg2 (Figures 3B and S4A).
Figure 3.

Regulated protein expression of Olg1 and Olg2
(A) Shown are the mean OD600nm values measured for P. aeruginosa PAO1 in three biological replicates. Samples were taken for targeted proteomics and qPCR at 1, 2, 4, 6, 8, and 24 h as well as at OD600nm = 1 (∼160 min) as indicated.
(B) Peptide intensities measured by targeted proteomics (PRM) for the proteins Tle3, Olg1, PA1383, and Olg2 at selected time points.
(C) Mean transcriptome and translatome reads per kilobase per million mapped reads (RPKM) of the datasets “LB” (n = 2; this study) as well as “M9+glycerol” and “M9+alkane” (n = 3 each; published by Grady et al. (2017)) are shown as dots for olg1 (blue) and olg2 (red). Bars indicate minimum and maximum values of the experiments.
Regulation of gene expression suggests functionality
Growth-phase-dependent changes in protein abundances were observed for all four target proteins via PRM (Figures 3B and S4A). High levels of both OLGs were obtained during exponential growth (1 h, 2 h) and at the exponential-stationary transition (OD1). In contrast, protein abundance in late stationary phase (24 h) was significantly lower. Both OLGs exhibited protein kinetics deviating from their respective mother gene proteins Tle3 and PA1383, indicating an independent biological regulation. qPCR analysis confirmed similar kinetics for the mRNAs of olg1 and olg2 (Figure S4C).
Further indication of regulated OLG expression was provided by published RNASeq and RiboSeq data in two P. aeruginosa strains (PAO1 & ATCC33988; SRA accession number PRJNA379630) (Grady et al., 2017). Strains were cultivated in M9 broth with glycerol or n-alkanes. Reanalysis revealed transcriptional and translational signals for both OLGs in strain PAO1 (Figures 3C and S5) and olg1 in strain ATCC33988 (Table S1). Interestingly, RPKMRNASeq values were similar for both OLGs when comparing LB with M9+glycerol but differed in M9+alkane (Figure 3C). This might suggest a carbon-source-dependent regulation. Further, these data indicate regulated translation of olg1 (log_FC = 1.15) and olg2 (log_FC = −0.55) in PAO1 when comparing M9+alkane with M9+glycerol (false discovery rate, FDR ≤0.05). Thus, both OLGs are expressed more weakly in alkane media than LB (Figure 3C).
Temporal control and dependence on growth media for OLG expression strongly implies functionality (Figures 3B and 3C and S4). However, elucidation of the biological role of these overlapping-encoded proteins requires further experiments.
Genomic locations of two long overlapping genes
The novel olg1 of P. aeruginosa PAO1 (NC_002516.2) is located at the coordinates 291,556–292,512 (+) in frame −1 (i.e., directly antisense) with respect to its mother gene (Figure 4A). It has a minimum length of 957 nucleotides (nt), and the most probable start codon is ATG291556 (see Data S1). olg1 completely overlaps with the annotated mother gene tle3 (PA0260), encoding the toxic type VI lipase effector 3 of the vgrG2b-tli3-tle3-tla3 operon. tle3 contains two structural domains, an N-terminal α/β hydrolase fold domain and a C-terminal domain of unknown function (DUF3274) (Berni et al., 2019). olg1 overlaps 39 nt with the α/β fold domain and at least 594 nt with DUF3274.
Figure 4.

Schematic overview of the genomic structure of the tle3-olg1 and PA1383-olg2 locus
(A) olg1 completely overlaps antisense in frame −1 (subpanel a) relative to the annotated gene tle3, which is part of the vgrG2b-tli3-tle3-tla3 operon. Location of the N-terminal α/β hydrolase fold as well as the C-terminal DUF3274 domain of tle3 are displayed in dark gray. olg1 shares structural features of a protein-coding gene including −35 and −10 consensus elements, divided by a 14 bp spacer, of a putative σ70 promoter. A core SD sequence of AGG was identified according to Ma et al. (2002), interacting with the aSD sequence at the 3′ end of the 16S rRNA within the 30S ribosomal subunit. A putative terminator between 219 and 349 nt downstream of the stop codon was identified via RT-PCR using the primer pairs indicated (P4/5 + P6).
(B) olg2 overlaps nontrivially with the hypothetical gene PA1383 and trivially with galE encoding a UDP-glucose 4-epimerase. Structural features of both annotated genes are indicated. The mRNA of olg2 starts probably at a putative σ70 promoter 93 nt upstream of the start codon and terminates at the predicted terminator 218 to 247 nt downstream of the stop codon.
Novel olg2 (Figure 4B) is likewise encoded in frame −1, in the mother gene PA1383 at the coordinates 1501875–1503602 (−). With ATG1503602 as putative start codon, olg2 spans 1728 nt and overlaps with two annotated genes. A region of 1536 nt is shared with PA1383, a hypothetical gene predicted to code for an N-terminal type I signal sequence for cytoplasmic export (Lewenza et al., 2005), which has been shown to be regulated by the transcriptional repressor NrdR (Crespo et al., 2015) and by both the repressors MvaT and MvaU (Lippa et al., 2021). In addition, olg2 overlaps in frame +2 by 34 nt with the galE gene (PA1384), which encodes for a UDP-glucose 4-epimerase.
Both OLGs were discovered by screening RiboSeq data and further characterized using prediction tools, qPCR, transcriptome sequencing, mass spectrometry as well as phylogenetic analyses as described.
Ab-initio prediction indicates gene-like sequence features
Putative σ70 promoter sequences were searched for 300 nt upstream of each OLG's start codon with the tool BPROM (Solovyev and Salamov, 2011). Linear-discriminant function values of 1.94 (olg1) and 1.37 (olg2) clearly exceed 0.2, the threshold distinguishing promoter and nonpromoter sequences. Transcription start sites were localized 37 nt and 94 nt upstream of the start codons, respectively (Figure 4). The observed distances fit the length of 5′UTRs reported for P. aeruginosa PA14 (median: 47 nt) (Wurtzel et al., 2012) and Pseudomonas syringae pv. tomato str. DC3000 (mean: 78 nt) (Filiatrault et al., 2011). To investigate potential ρ-independent terminators, FindTerm (Solovyev and Salamov, 2011) was applied 300 nt downstream of both overlapping ORFs. A terminator was detected for olg2 218 to 247 nt downstream (Figure 4B). For olg1, a ρ-independent terminator was not predicted, but RT-PCR verified termination between 219 and 349 nt downstream of its stop codon (Figure 4A).
In P. aeruginosa, the core anti-Shine-Dalgarno (SD) sequence is CCUCC. It has a mean ΔGSD of −6.5 kcal/mol and an optimal spacing of 7–9 nt to the start codon (Ma et al., 2002). An SD sequence with AGG and a ΔGSD of −3.6 kcal/mol was detected 8 nt upstream of olg1′s proposed start codon (Figure 4A). For the mother gene tle3, an SD sequence (−5.1 kcal/mol) was identified, but neither a σ70 promoter nor a ρ-independent terminator was found. This is consistent with the reported finding that tle3 is part of the vgrG2b-operon (Berni et al., 2019). No SD sequence was detected for olg2. However, SD sequences are similarly absent from 30.8% of the annotated genes in P. aeruginosa (Ma et al., 2002). The upstream region of both PA1383 and galE, the mother genes, harbored a σ70 promoter (transcription start sites 203 nt and 72 nt upstream, respectively) and an SD sequence (−6.1 kcal/mol and −4 kcal/mol, respectively).
Sequence features such as start codon, SD sequences, GC bias, and hexamer coding statistics are used by gene prediction tools, for example Prodigal (Hyatt et al., 2010). However, Prodigal's algorithm prohibits prediction of gene pairs with an overlap larger than 200 nt and annotates only the ORF with the highest score. For P. aeruginosa PAO1, Prodigal predicted 5,681 protein coding genes including the two annotated genes tle3 and PA1383 with total scores of 132.60 and 225.92, respectively (Figure S6). When “hiding” both annotated genes by replacing all start codons by unidentified nucleotides (“N”), Prodigal classified olg1 and olg2 as protein coding genes. Although their total scores of 4.63 and 23.63 were relatively low, some values are comparable to annotated genes, for instance the start-sequence region scores (Figure S6 and Table S3). Furthermore, olg2 showed a confidence score of 99.56, indicating a very high likelihood of being a real protein-coding gene. These overlapping ORFs are not annotated by Prodigal due to their long overlaps but both nonetheless show features associated with protein coding.
Phylogenetic analyses show a relatively recent origin
Almost all detectable homologs of both olg1 and olg2 were found within Pseudomonas spp. according to BLAST searches (Table S4). Homologs outside Pseudomonas clustered within the genus in terms of sequence identity. Thus, we infer that these are best accounted for by relatively recent horizontal gene transfer from the genus Pseudomonas and focus on evolution within this clade.
Homologs of tle3 containing both the α/β hydrolase and DUF3274 domains were found in multiple phyla; however, within the order Pseudomonadales they were found only in the genus Pseudomonas, suggesting an ancient horizontal gene transfer event from another order. The mother gene of olg2, PA1383, in contrast, does not have highly similar homologs outside of Pseudomonas. The one exception was derived from a low-quality genome of Acinetobacter baumanii (noted in RefSeq to be of excess size), which was disregarded. A few distant homologs were found, with the two top hits in E. coli (matching 43% of the PAO1 sequence) and Salmonella enterica (match to 17%). This evolutionary distance again implied a horizontal gene transfer into or out of Pseudomonas with subsequent evolutionary divergence. None of the non-Pseudomonas homologs included the N-terminal signal peptide in PA1383, suggesting functional changes in Pseudomonas compared with other bacteria.
Both olg1 and olg2, in the length present in reference strain PAO1, were highly taxonomically restricted. The taxonomic distribution of both OLGs was limited approximately to the species P. aeruginosa, according to BLASTp hits along with searches of additional metagenome-assembled genomes (MAGs) (Figure 5A). The mother genes tle3 and PA1383 within the order Pseudomonadales resulted in approximately 900 and 300 unique sequences, respectively. For olg1 (in tle3), the intact ORF (i.e., without premature stop codons) was restricted to P. aeruginosa with one exception in Pseudomonas prosekii with a nonstart codon (GTA) at the locus of the start site in PAO1. An intact olg2 was restricted to a few Pseudomonas species, and only P. aeruginosa genomes shared the same stop codon. Pseudomonadales homologs in recent MAG collections (Almeida et al., 2020; Nayfach et al., 2019) supported the inferred taxonomic boundaries. Genomes with intact ORFs of olg1 or olg2 with a stop in the same position were assigned taxonomically to P. aeruginosa in the MAG data (Table S4). Subsequent analyses used the combined GenBank and MAG datasets.
Figure 5.

Phylogenetic distribution of olg1 and olg2 and depletion of stop codons
(A) Homologs of the full olg1 ORF (upper panel), matched to a maximum likelihood tree calculated from the amino acid sequence of the mother gene tle3, down-sampled to 20 genomes. Clade containing genomes with the same start and stop codon as the reference genomes (“OLG genomes”) is highlighted with a blue box. Lower panel: homologs of olg2 overlapping loci, for PA1383. Clade containing genomes with the same stop codon as the reference genomes is highlighted with a red box (start codon is at a nonoverlapping locus outside the sequence shown). The reference genome (NC_002516.2) is underlined in the respective OLG color, and the outgroup used in the evolutionary simulations described below is underlined in gray.
(B) Distributions of lengths of antisense (−1 frame) ORFs obtained by permutation (green) or synonymous exchanges (orange) of “mother gene” codons, for genes tle3 and PA1383, compared with the lengths of the embedded olg1 (blue, left) and olg2 (red, right). ORF lengths are measured between in-frame stop codons rather than start to stop.
(C) Simulations of evolution of tle3 and PA1383 in the OLG clade rooted on an outgroup with an intact ORF, using an empirical codon model, show that accumulation of stop codons is common; simulated sequences tend to have fewer full-length intact ORFs in the OLG loci and reading frame than real sequences.
Stop codon depletion implies purifying selection
Multiple independent lines of evidence indicated that both OLGs are under purifying (negative) selection, a strong indicator of functionality, particularly when combined with evidence of expression (Cooper and Gardner, 2020). Firstly, the ORFs for olg1 and olg2 were both significantly longer than expected, given the amino acids (AA) in the reference genes tle3 and PA1383, using the synonymous-mutation method from the tool “Frameshift” (Schlub et al., 2018) (Figure 5B). This method substitutes synonymous codons randomly and obtains an empirical cumulative distribution function of the resulting ORF lengths in each alternative reading frame. This resulted in a p-value of <10−10 for both ORFs. olg1 and olg2 were also longer than expected given the overall codon usage (codon-permutation method of “Frameshift”), although not statistically significantly, with p = 0.163 and p = 0.0635, respectively (Table S5). The p-values included a correction for multiple tests, i.e., the number of observed ORFs in this alternate reading frame. The synonymous mutation p-values are still significant after a conservative multiple tests adjustment of multiplying by the total number of genes, arguably appropriate given the OLGs' detection with a genome-wide scan. The nonsignificant results for the codon permutation method are not surprising given that it implicitly depends on stop codons elsewhere in the alternate reading frame, of which there are few due to the length of the OLGs relative to their mother genes. In summary, from these results it can be concluded that the ORF lengths are unexpected, given the overall sequence composition of the mother genes, implying selection for long ORFs via negative selection on stop codons.
Secondly, sequence evolution of each mother gene was modeled without selection for maintaining an overlapping ORF. The presence of stop codons in simulated sequences was then compared with natural sequences. This method was previously used to support an inference to selection on the OLG asp in HIV-1 (Cassan et al., 2016). When evolution was simulated using an empirical codon model in Pyvolve (Spielman and Wilke, 2015) along trees calculated from the mother genes (Figure 5A), stop codons evolved more frequently in the simulated OLG sequences compared with observed natural sequences. As such, fewer simulated than natural sequences had intact full-length ORFs (Figure 5C). Following the originators of the method (Cassan et al., 2016), outgroup sequences without stop codons in the OLG region were chosen to root the tree (Figure S7).
Codon-position-specific constraint supports purifying selection
Synonymous variation in the mother genes tle3 and PA1383 was reduced over a large part of the OLG region (Figure 6A and Table S6), according to results from “FRESCo” (Sealfon et al., 2015). For tle3, a comparison of the rates of synonymous evolution in the OLG-containing genomes versus the rest of the alignment using a paired two-tailed t test in nonoverlapping adjacent windows of 50 codons over the whole OLG sequence showed increased constraint in the OLG region, with p = 0.086. Results for olg2 were similar for the last 350 codons of olg2 (p = 0.03) (Table S6), but there was no synonymous constraint toward the end of the mother gene PA1383 from approximately codon 500 onwards. Because the nonoverlapping start region of olg2 is not well conserved across P. aeruginosa (Table S4), we focused only on the part overlapping PA1383.
Figure 6.

Evidence for evolutionary sequence constraint in olg1 and olg2
(A) Variation in synonymous codons of tle3 (left) and PA1383 (right); sliding windows of 50 codons calculated with FRESCo. Constraint is observed in the OLG regions (blue and red boxes) when compared with the expected rate of 1 (black dotted lines) and that observed in the “non-OLG” genomes, approximately one across the gene (white line). Codon numbers are with respect to an alignment including gaps.
(B) OLGenie’s measure of dN/dS across tle3 (left) and PA1383 (right) in the OLG genomes; sliding windows of 50 codons. A decrease in nonsynonymous changes in the OLG frame is observed in the OLG loci (blue and red boxes) when compared with the expected neutral evolution rate of 1 (black dotted lines) and the non-OLG genomes (white line).
(C) Pairwise comparisons of dNN/dNS (an OLG-appropriate measure of purifying selection calculated with OLGenie). Evidence for purifying selection is found in a wider taxonomic group than the specific ORFs studied here; for olg1, apparent purifying selection is limited to a subclade within Pseudomonas, whereas for olg2 it is found across the genus; for both ORFs however, evidence is strongest in the vicinity of P. aeruginosa. Codon numbers are with respect to an alignment including gaps.
A more precise reading-frame-specific measure of purifying selection against nonsynonymous variants in OLGs is given by the novel tool “OLGenie” (Nelson et al., 2020b). Unlike standard measures of dN/dS, “OLGenie” calculates an OLG-appropriate measure (i.e., dNN/dNS for the OLG) by restricting the analysis to alternative frame sites where variants are nonsynonymous in the reference frame. Within the OLG-containing genome sets for both olg1 and olg2, synonymous variants were favored over variants causing AA changes (Figure 6B), although these tendencies were not statistically significant; for olg1 dNN/dNS = 0.33, p = 0.11 and for olg2, dNN/dNS = 0.52, and p = 0.06. These values contrasted with the genomes without olg1 or olg2 with dNN/dNS = 1.02, p = 0.66 and dNN/dNS = 0.92, p = 0.20 for olg1 and olg2, respectively (Table S7). Both mother genes tle3 and PA1383 were observed to be under significant purifying selection in the sets of Pseudomonas genomes with and without these OLGs. The results from “FRESCo” and “OLGenie” are fully independent, as they depend on synonymous and nonsynonymous sites in the mother genes, respectively. As each measure independently shows a tendency toward constraint, together they provide good evidence of evolutionary constraint in the OLG sequences. Further, some individual pairwise sequence comparisons with “OLGenie” are statistically significant (Figure 6C and Table S7), and these comparisons are informative about the taxonomic extent of functional ORFs. Much of the strongest evidence of purifying selection is taxonomically close to the reference genome PAO1, which supports constraint in these OLGs. This was not guaranteed by the presence of an intact ORF in this clade, as any stop codons are excluded from the “OLGenie” analysis; as such, results are not affected by whether an ORF homologue has premature stops. The pattern of purifying selection on olg1 suggests that a functional ORF may have been found in the common ancestor of P. aeruginosa and Pseudomonas frederiksbergensis; the evidence for positive selection between members of this clade and genomes in the other main Pseudomonas branch also support this hypothesis. In the case of olg2, evidence for purifying selection is taxonomically more widespread across Pseudomonas, fitting the wider distribution of intact olg2 ORFs (some with stop codons downstream of that in the reference strain PAO1).
Discussion
OLGs outside viruses (Chirico et al., 2010; Lebre and Gascuel, 2017) are typically categorically rejected and those already annotated have sometimes been attributed to misannotations (Pallejà et al., 2008). In this study, we describe the detection and characterization of two exceptionally long OLGs, olg1 and olg2, in P. aeruginosa. Although additional signals for further OLGs have been detected in our data (not shown), we concentrated on two strong candidates reported here, namely olg1 and olg2, in order to characterize these in sufficient and convincing depth. The discovery of novel genes, and especially of overlapping genes, is always coupled to the choice of (arbitrary) values that are accepted for verifying a novel gene or protein. These values are discussed for sequencing data, and sometimes unexpected signals are regarded as pervasive or background nonfunctional translation events (e.g., Smith et al., 2019). In mass spectrometry, a single peptide detection event is dismissed as a one-hit-wonder, although methods of cross-validation have been developed to mitigate this problem (Gupta et al., 2008). In any case, we propose that the long overlapping ORFs detected in this study encode functional protein products due to (1) the presence of sequence features necessary for gene expression, (2) successful transcription and translation as indicated by RNASeq and RiboSeq, (3) discovery of several translated peptides via mass spectrometry, (4) validation and confirmation of their regulated expression during growth of P. aeruginosa PAO1 using targeted proteomics and isotopically labeled reference peptides, (5) successful prediction of both ORFs on genomic and translational level by annotation programs, and (6) evidence of purifying selection on both gene candidates from multiple methods. Although these results provide strong evidence for the genuine protein-coding nature and functionality of both ORFs, they can only be designated as OLGs if their respective mother genes (tle3 and PA1383) are correctly annotated and are also genuinely protein coding. The gene tle3 has been confirmed to encode the antibacterial type VI lipase effector 3 (Berni et al., 2019; Russell et al., 2013). PA1383 is annotated as a hypothetical gene, but we show that homologs are widely distributed across bacteria. Further, it contains a signal peptide associated with export, and it is under purifying selection. For both mother genes, we show clear expression in our RNASeq, RiboSeq, and MS experiments. Further, it appears unlikely that the MS-detected peptides represent translation products without function considering the high bioenergetic cost of translation (Lynch and Marinov, 2015). Taken together, it is beyond reasonable doubt that both mother genes encode functional proteins and that the overlapping ORFs presented here are not just annotation errors from artifactual mother genes.
With a minimum length of 957 and 1728 nt, olg1 and olg2 represent the longest known prokaryotic OLGs with extensive experimental evidence. The discovery of such long OLGs is extraordinary considering the short length of most observed OLGs. In E. coli, for instance, several RiboSeq studies (Meydan et al., 2019; Weaver et al., 2019; Zehentner et al., 2020b) revealed translation of antisense OLGs typically encoding for small proteins, rarely exceeding 200 codons. Similarly, Baek et al. (2017) detected 130 unannotated ORFs including overlapping ones with total sizes ranging from 4 to 144 AA in S. enterica Typhimurium. Likewise, Smith et al. (2019) report translation of 274 predominately short OLGs in M. tuberculosis. An equivalent length range was also obtained for antisense OLGs in archaea (Gelsinger et al., 2020). The vast majority of OLGs characterized in detail are less than 200 codons (Behrens et al., 2002; Fellner et al., 2014, 2015; Hücker et al., 2018a, 2018b; Vanderhaeghen et al., 2018). Antisense OLGs encoding for proteins equal or larger than 200 AA have rarely been validated experimentally, e.g., pop in EHEC (200 AA) (Zehentner et al., 2020b), adm in Streptomyces coelicolor (233 AA) (Tunca et al., 2009), or cosA in P. fluorescens Pf0-1 (338 AA) (Silby and Levy, 2008). Even when considering viruses, olg1 and olg2 are of exceptional size and belong to some of the longest yet observed OLGs. In a large-scale study of 5,976 viral genomes (Schlub and Holmes, 2020), the authors noted antisense overlaps ranging from 50 to 2351 nt (median = 212 nt; mean = 244 nt) in all viral groups except + ssRNA viruses. The latter contains an unusual exception constituting an antisense ORF of ∼1000 AA in the family Narnaviridae (DeRisi et al., 2019). This ORF was hypothesized to be protein coding (Dinan et al., 2020), but experimental evidence is lacking.
Almost all proposed antisense OLGs lack a native proof of the encoded protein product, arguably calling their coding potential into question. Proteomic detection of the OLG cosA via MS, for instance, failed, presumably due to its low expression (Kim et al., 2009). In addition to low protein abundance, the generally small size of OLGs also hampers a proteomic proof due to an insufficient amount, or complete absence, of mass spectrometry-detectable peptides (Petruschke et al., 2020). Nevertheless, protein evidence of antisense OLGs was provided in some proteomic studies (Venter et al., 2011) but mainly attributed to a high false-positive rate. Proteomic OLG evidence was found for other bacterial genera, including Helicobacter (Friedman et al., 2017), Salmonella (Willems et al., 2020), or Pseudomonas (Kim et al., 2009; Yang et al., 2016). In P. putida, 44 small antisense-encoded proteins were claimed based on MS data (Yang et al., 2016). For a different species, P. fluorescens, nine protein-coding antisense OLGs were found using MS (Kim et al., 2009). In the latter, eight of nine detected proteins were shorter than 200 AA; but one had a reported length of 530 AA. To our knowledge, the longest antisense OLG with proteomic evidence is a 1644 nt ORF (encoding for 548 AA), located in frame −1 in Deinococcus radiodurans (Willems et al., 2020). However, up until now all prokaryotic OLGs identified via MS have lacked verification. Thus, olg1 and olg2 not only represent antisense OLGs of exceptional sizes across bacteria, archaea, and viruses but constitute the longest known OLGs with reliable proteomic evidence.
Both olg1 and olg2 are phylogenetically young genes under selection. For both olg1 and olg2, the OLG sequence is evolving considerably faster at the AA level than the mother gene protein sequence (approximately 2 and 12 times faster, respectively; Table S8). This appears to have resulted in a long ORF “opening up” in the recent history of Pseudomonas genomes for olg1 and perhaps somewhat earlier for olg2. At some point, they became subject to purifying selection, as shown by depletion of stop codons, nonsynonymous changes, and synonymous variants in the mother gene. The yet unaccounted-for evidence of translation upstream of olg1 shown here raises the possibility of multiple start sites, which have been recently observed for many bacterial proteins (Fijalkowska et al., 2020), including potentially for the OLG pop (Zehentner et al., 2020b). However, in our sliding window analyses of tle3 (Figures 6A and 6B), we found no evidence for selection on upstream sequences.
Bioinformatic analysis of OLGs is still in its infancy. For instance, for evolutionary simulation, it would be ideal to start with the actual ancestral sequence, but accurate ancestral-sequence reconstruction for OLGs is yet unsolved. Thus, for the simulation method, rather than introducing new biases with imperfect reconstruction, we instead followed the approach of Cassan et al. (2016) of using a known leaf sequence in place of the root sequence. Further, choosing an outgroup with intact ORF to root the tree implicitly assumes that the ancestor of the outgroup and OLG clade contained an intact OLG, and the results are sensitive to the choice of sequence on which the tree is rooted (Figure S7B). Here also, ancestral-sequence construction would assist with realistic simulations. Further, another limitation with all existing methods is that they all use only a subset of the sequence information, e.g., “Frameshift” only considers stop codons in one genome, “FRESCo” only considers synonymous sites in the mother gene, and “OLGenie” is restricted to the nonsynonymous mother gene sites. Future developments combining features should increase accuracy. Additional considerations such as masking out RNA secondary structures, using machine-learning methods to find subtle signatures of selection, or including sequences from metagenomes studies of different niches should improve our understanding of the evolution of OLGs and other taxonomically restricted genes. Until recently it was thought that almost all modern genes arose through duplication and divergence from ancient genes (Ohno, 1970). Many taxonomically restricted genes, found only in one strain or relatively few closely related genomes, have recently been discovered. The origin of few of these “orphan” genes, however, has been explicated in molecular detail. Young OLGs have some important advantages in the study of gene evolution. In particular, the genetic context is fixed due to the presence of the mother gene. This dramatically reduces the major problems associated with false homologs and failure to detect true homologs (Vakirlis et al., 2020; Weisman et al., 2020). The evolutionary processes involved in the initial expression and neo-functionalization of these ORFs deserve further attention. For instance, a shift in function in PA1383 appears to have involved substantial sequence change, including gain of a signal peptide. We hypothesize that during this process of positive selection on the mother gene many possible sequences were explored in the antisense −1 frame, facilitating the origin of the ORF encoding olg2.
Our results demonstrate that bacterial genomics after decades of advance still has additional fundamental secrets to reveal (Grainger, 2016; Kirchberger et al., 2020), potentially including many more long OLGs, which were until now hiding in the shadows of known, annotated genes. These elements have not been rigorously searched for before at a whole genome level, as appropriate detection methods are still in development, and if found they are often disregarded. In this discovery of long OLGs, new research opportunities are opened for genomics, proteomics, and translatomics, as well as in the study of evolutionary novelty and bacterial gene function. These findings together shine a spotlight on the remarkable multi-layer coding potential enabled by the redundancy in the standard genetic code.
Limitations of the study
Although we report two novel overlapping genes from P. aeruginosa, we omitted many other putative overlapping genes observed in our data. Mainly, our limited resources did not allow detailing more overlapping genes. For instance, so-called “one-hit-wonders,” i.e., proteins only found represented by a single peptide, are widely discounted and so were also not examined. Furthermore, we do not have data on biological function of the two genes. Here, one would need, e.g., strand-specific knockouts, overexpression phenotypes, or many other experiments classically used to elucidate protein function. Regarding their evolution, we currently do not understand well how such genes originate “de novo” through overprinting.
Data availability
Scripts for evolutionary and taxonomic analyses are available in the GitHub repository (https://github.com/ZacharyArdern/Pseudomonas_long_OLGs). Sequencing data have been deposited in Sequence Read Archive (NCBI) under the BioProject accession number PRJNA716268 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA716268). The proteomics raw data, MaxQuant search results, and used protein sequence databases have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019) and can be accessed using the dataset identifier PXD023992 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD023992). All targeted proteomic raw data and Skyline analysis files have been deposited using the identifier OLG_PSE to Panorama Public (https://panoramaweb.org/OLG_PSE.url) (Sharma et al., 2018).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Pseudomonas aeruginosa PAO1 | DSMZ - Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany | DSM 19880 |
| Biological samples | ||
| Ribosome profiling of P. aeruginoasa PAO1, sample 1 | This paper | SRA: SRR14029804 |
| RNA sequencing of P. aeruginoasa PAO1, sample 1 | This paper | SRA: SRR14029803 |
| Ribosome profiling of P. aeruginoasa PAO1, sample 2 | This paper | SRA: SRR14029802 |
| RNA sequencing of P. aeruginoasa PAO1, sample 2 | This paper | SRA: SRR14029801 |
| Chemicals, peptides, and recombinant proteins | ||
| Trizol | Thermo Fisher Scientific | Cat# 15596026 |
| Zirconia beads | Carl Roth GmbH, Karlsruhe, Germany | N033.1 |
| TURBO DNase | Thermo Fisher | Cat# AM2238 |
| SUPERase·In RNase Inhibitor | Thermo Fisher | Cat# AM2696 |
| Taq polymerase | NEB | Cat# M0273 |
| Q5 High-Fidelity DNA Polymerase | NEB | Cat# M0491 |
| Antarctic phosphatase | NEB | Cat# M0289 |
| T4 Polynucleotide Kinase | NEB | Cat# M0201 |
| Synthetic peptides (see Table S9) | JPT Peptide Technologies | N/A |
| SuperScript III Reverse Transcriptase | Thermo Fisher | Cat# 18080093 |
| MNase | Thermo Fisher | Cat# EN0181 |
| RNase R | Lucigen | Cat# RNR07250 |
| RNase T (Exonuclease T) | NEB | Cat# M0265 |
| XRN-1 | NEB | Cat# M0338 |
| Buffer 4 | NEB | Cat# B7004 |
| SYBR Gold | Invitrogen | Cat# S11494 |
| Trifluor acetic acid (TFA) absolute | Sigma-Aldrich | Cat# 91707 |
| Bradford reagent | Sigma-Aldrich | Cat# B6916 |
| Tris(2-carboxyethyl)phosphine (TCEP) | Sigma-Aldrich | Cat# 68957 |
| 2-Chloracetamid (CAA) | Sigma-Aldrich | Cat# 22790 |
| Empore C18 disks | 3M | Cat# 2215 |
| Procal peptides | JPT | N/A |
| Critical commercial assays | ||
| Agilent RNA 6000 Nano Kit | Agilent | Cat# 5067-1511 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad Laboratories | Cat# 1725270 |
| riboPOOL kit (version v1-5) | siTOOLs Biotech | Cat# Pseudomonas aeruginosa 18 |
| miRNeasy Mini Kit | Qiagen | Cat# 217084 |
| TruSeq Small RNA Library Prep Kit | Illumina | Cat# RS-200-0012 |
| HiSeq Rapid SBS Kit v2 (50 cycles) | Illumina | Cat# FC-402-4022 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Cat# Q32851 |
| Deposited data | ||
| RefSeq file for P. aeruginosa PAO1 GCF_000006765.1_ASM676v1_protein.faa | PathoGenesis Corporation | https://www.ncbi.nlm.nih.gov/assembly/GCF_000006765.1/ |
| Sequencing data | This paper | SRA: PRJNA716268 |
| Proteomics raw data, MaxQuant search results and used protein sequence databases | ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019) | http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD023992 |
| Targeted proteomic raw data and Skyline analysis files | Panorama Public (Sharma et al., 2018) | https://panoramaweb.org/OLG_PSE.url |
| Oligonucleotides | ||
| 1: PA_16S_F: GATGTTGGGTTAAGTCCCGT | Biomers | N/A |
| 2: PA_16S_R: CCCCTACGGCTACCTTGTTA | Biomers | N/A |
| 3: olg1+730R_RT: GCTGCCAGACGACCATCGAC | Biomers | N/A |
| 4: PA0260+575F : GCACCATGACCCATCCGCTGT | Biomers | N/A |
| 5: PA0260+423F : CGAATGGCTGGACCGCAACG | Biomers | N/A |
| 6: olg1+524F: TCGCCATTGCGCTTGCGTAC | Biomers | N/A |
| 7: PA1383+172F : GTGGAAAATGGTGCCAACCT | Biomers | N/A |
| 8: PA0260+800F : ACGGCCTGTTCGAACCCCTC | Biomers | N/A |
| 9: olg1+34F: CTCGTAGGGAGTTTCCGCGCG | Biomers | N/A |
| 10: PA1383+278F: CCTACACCATCGATCCAGTG | Biomers | N/A |
| 11: olg2+20F: TGACCAATACGCGCATCTCG | Biomers | N/A |
| 12: PA_gyrA+346F: AACGCCG CAGCCATGCGATA |
Biomers | N/A |
| 13: PA_gyrA+458R: CATGACCG CCGGGATCTGCT |
Biomers | N/A |
| 14: olg1+640R: AGACGGTGGGACTTGCCAAC | Biomers | N/A |
| 15: PA0260+1773R: CTCCGG GTTGTTGGGTATGGCC |
Biomers | N/A |
| 16: PA0260+1838R: ACCACA TCATCCACGCTGTCCC |
Biomers | N/A |
| 17: PA0260+1890R: GCCTTCC AATCCAACCGCCAA |
Biomers | N/A |
| 18: PA0260+1969R: CGCACGG TACAAAGCACGCAC |
Biomers | N/A |
| Random nonamer | Sigma Aldrich | Cat# R7647-100UL |
| Software and algorithms | ||
| MaxQuant v1.6.3.4 (includes Andromeda) | Tyanova et al. (2016) and Cox et al. (2011) | https://www.maxquant.org/ |
| Skyline daily (64-bit, v20.1.9.234) | MacLean et al. (2010) | https://skyline.ms/project/home/software/Skyline/begin.view |
| Prosit | Gessulat et al. (2019) | https://www.proteomicsdb.org/prosit/ |
| iBAQ | Schwanhäusser et al. (2011) | N/A |
| BPROM | Solovyev and Salamov (2011) | http://www.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb |
| Shine-Dalgarno sequence identification | Ma et al. (2002) | N/A |
| FindTerm | Solovyev and Salamov (2011) | http://www.softberry.com/berry.phtml?topic=findterm&group=help&subgroup=gfindb |
| Mfold | Zuker (2003) | http://www.unafold.org/mfold/applications/dna-folding-form.php |
| FastQC | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| fastp | Chen et al. (2018) | https://github.com/OpenGene/fastp |
| Bowtie2 | Langmead and Salzberg (2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al. (2009) | http://samtools.sourceforge.net/ |
| BEDTools | Quinlan and Hall (2010) | https://github.com/arq5x/bedtools2 |
| pyGenomeTracks | Ramírez et al. (2018) | https://github.com/deeptools/pyGenomeTracks |
| Ribosome coverage value | Neuhaus et al. (2017) | N/A |
| RiboSeq datasets “M9+n-alkane” and “M9+glycerol” | Grady et al. (2017) | SRA: PRJNA379630 |
| edgeR | Robinson et al. (2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Prodigal | Hyatt et al. (2010) | https://github.com/hyattpd/Prodigal |
| DeepRibo | Clauwaert et al. (2019) | https://github.com/Biobix/DeepRibo |
| blast(n/p) | Altschul et al. (1990) | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Scripts for evolutionary and taxonomic analyses | This paper | https://github.com/ZacharyArdern/Pseudomonas_long_OLGs |
| Entrez Programming Utilities | Kans, 2021 | https://www.ncbi.nlm.nih.gov/books/NBK25501/ |
| Diamond | Buchfink et al. (2014) | https://github.com/bbuchfink/diamond |
| QuickProbs 2 | Gudyś and Deorowicz (2017) | https://github.com/refresh-bio/QuickProbs |
| Pal2Nal | Suyama et al. (2006) | http://www.bork.embl.de/pal2nal/ |
| IQ-TREE | Minh et al. (2020) and Nguyen et al. (2015) | http://www.iqtree.org/ |
| Treemmer | Menardo et al. (2018) | https://github.com/fmenardo/Treemmer |
| Newick Utilities | Junier and Zdobnov (2010) | https://bio.tools/newick_utilities |
| ETE3 packages | Huerta-Cepas et al. (2016) | http://etetoolkit.org/ |
| OLGenie | Nelson et al. (2020b) | https://github.com/chasewnelson/OLGenie |
| Frameshift | Schlub et al. (2018) | https://github.com/TimSchlub/Frameshift |
| Evolutionary simulation method | Cassan et al. (2016) | https://figshare.com/s/9668ef62e84488d4787a |
| Pyvolve | Spielman and Wilke (2015) | https://github.com/sjspielman/pyvolve |
| FRESCo | Sealfon et al. (2015) | https://www.broadinstitute.org/fresco/running-fresco |
| FastTree 2 | Price et al. (2010) | http://www.microbesonline.org/fasttree/ |
| custom Bash scripts | This paper | https://github.com/ZacharyArdern/Pseudomonas_long_OLGs |
| Other | ||
| Polysome-lysis buffer | Woolstenhulme et al. (2015) | N/A |
| Cell crusher | Cellcrusher Ltd, Schull, Ireland | Cellcrusher Kit + Drill-bit accessory |
| FastPrep | MP Biomedicals™ | FastPrep®-24 |
| Bioanalyzer | Agilent | Cat# G2939BA |
| Ultrasonicator system S220 | Covaris | Cat# 500217 |
| Speedvac concentrator | Eppendorf | Cat# EP5305000100 |
| Qbit T Fluorometer | Thermo Fisher | Cat# Q33238 |
| HiSeq1500 | Illumina | N/A |
| Gel breaker tube | IST Engineering | Cat# 3388-100 |
| Agilent 1100 HPLC | Agilent | Cat# G1380-90000 |
| UltiMate™ 3000 RSLCnano System | Thermo Fisher | Cat# ULTIM3000RSLCNANO |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | Thermo Fisher | Cat# IQLAAEGAAPFADBMBHQ |
| XBridge Peptide BEH C18 Column, 130Å, 3.5 μm, 2.1 mm X 250 mm, 1K - 15K | Waters | Cat# 186003566 |
| ReproSil-pur C18-AQ, 5 μm, 20 mm × 75 μm | Dr. Maisch | Cat# r15.aq |
| ReproSil Gold C18-AQ, 3 μm, 450 mm × 75 μm | Dr. Maisch | Cat# r13.b9 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Klaus Neuhaus (neuhaus@tum.de).
Materials availability
Reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
P. aeruginosa PAO1 (DSM 19880) has been used in this work.
Method details
Cultivation of bacteria
Lysogeny broth (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) was inoculated 1:100 using an overnight culture of P. aeruginosa PAO1 (DSM 19880) and aerobically incubated (37°C, 150 rpm). After 1 h, 2 h, 4 h, 6 h, 8 h, and 24 h and at OD600nm = 1, samples were taken by centrifugation (10 min, 12,000×g, 4°C). For transcriptomes and translatomes, cellular processes were stalled at OD600nm = 1 by adding dry ice reaching 4°C. Next, cells were centrifuged (8,000×g, 4°C, 5 min) and resuspended in polysome-lysis-buffer (Woolstenhulme et al., 2015) (325 μL per 100 mL initial culture). Cells were lysed in a cell crusher with liquid nitrogen. After centrifugation as before, the supernatant was used for transcriptomes and translatomes. Thus, the supernatant was split and the two portions were used to isolate total RNA and ribosomes for RIBOseq, respectively.
RNA isolation
Total RNA was extracted from cells (qPCR) or cell lysate (transcriptomes and translatomes) using Trizol (Thermo Fisher Scientific). For qPCR, cell pellets were resuspended in 1 mL cooled Trizol and subjected to bead beating (0.1 mm zirconia beads) using a FastPrep (3 cycles, 6.5 ms-1, 45 s; with 5 min incubation on ice after each cycle). Cell lysates (see above) were incubated each 5 min with first cooled Trizol and next 200 μL chloroform. After centrifugation (15 min, 12,000×g, 4°C), RNA was precipitated (500 μL 2-propanol, 1 μL glycogen, 30 min). RNA was pelleted (10 min, 12,000×g, 4 °C) and washed twice with cold 70% ethanol. Air-dried RNA was dissolved in RNase-free water. Integrity was verified by agarose gel electrophoresis (1.5%, 100 V, 45 min; Carl Roth) and Bioanalyzer measurements (RNA 6000 Nano Assay, Agilent Technologies).
RNA samples were incubated with TURBO DNase (Thermo Fisher Scientific; 1 h, 60°C) and 25 U SUPERase·In RNase Inhibitor (Thermo) for removing residual DNA. After inactivation (15 mM EDTA, 10 min, 65°C), the RNA was precipitated overnight (-20°C) using ethanol, 3 M sodium acetate, and glycogen (690, 27.6, and 1 μL, respectively). Precipitated RNA was pelleted, washed, dried and dissolved as before. DNA absence was confirmed with PCR using Taq polymerase (NEB) with primer 1 & 2.
Oligonucleotides and peptides
Oligonucleotides and synthetic peptides are listed in Table S9. For Olg1, Olg2, Tle3, and PA1383 in total eighteen optimal peptides were selected for isotopically-labeled reference peptides (SpikeTidesL) purchased from JPT Peptide Technologies. Either the C-terminal lysine (Lys8) or arginine residue (Arg10) were 13C- and 15N-labeled. Isotope-labelled peptides were not purified and, thus, concentrations represent only estimates.
cDNA synthesis for PCR
RNA (500 μg) was reverse-transcribed using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). Random nonamer (50 pmol; Sigma Aldrich) or 10 pmol primer 3 were used for reverse transcription of gyrA (reference gene) or olg1, respectively, in the presence of 20 U SUPERase·In RNase Inhibitor. Samples without reverse transcriptase served as negative controls. For transcriptional termination sites of olg1, reverse transcription was performed with primer 4 or 5. Of the latter, 1 μL cDNA was used in a 30-cycle PCR using Taq DNA Polymerase (NEB) with primer 4 & 6 or 5 & 6. For olg2, RNA was reversed transcribed using primer 7. Subsequently, 1 μL cDNA was used in a 30-cycle PCR using Q5 DNA Polymerase (NEB) with primer 8 & 9 or 10 & 11. cDNA of olg1 was additionally used testing for alternative start sites by PCR using the primer 8 & 15, 8 & 16, 8 & 17, and 8 &18. Primer functionalities were verified with genomic DNA before (not shown).
Quantitative PCR
Expression levels of olg1, olg2 and gyrA were quantified by qPCR. Each 20-μL reaction contained 10 μL SsoAdvanced Universal SYBR Green Supermix (Bio-Rad Laboratories), 500 nM forward and reverse primer, and 1 μL cDNA (or water for “No Template Control”). For gyrA and olg1, primer 12 & 13 and 6 & 14 were used, respectively. Cycling was as follows: 95°C for 30 s; 40 cycles of 95°C for 15 s and 60°C for 30 s. A melt curve analysis (65 to 95°C with 0.5°C increments) confirmed the correct product. Each reaction was conducted in three biological and technical replicates. Data were analysed using the ΔΔCt method (Livak and Schmittgen, 2001). Significance was evaluated with a two-tailed Welch two-sample t-test (p value ≤0.05).
Transcriptome sequencing
rRNA was depleted from total RNA (of 200 μL cell-extract, DNase treated, as above) using the P. aeruginosa-specific riboPOOL kit (version v1-5, siTOOLs Biotech) followed by RNA precipitation and DNase digestion of the probes. One μg depleted RNA was fragmented (Ultrasonicator system S220, Covaris; 175 W, 10% duty cycle, 200 cycles for 180 s), dephosphorylated (Antarctic phosphatase, NEB), and phosphorylated (T4 Polynucleotide Kinase, NEB). Fragments were purified after each step using the miRNeasy Mini Kit (Qiagen). Finally, the volume was reduced to 5 μL in a Speedvac concentrator (Eppendorf) and sequencing libraries were prepared using the TruSeq Small RNA Library Prep Kit (Illumina). cDNA concentration and length were measured using a Qubit (dsDNA HS Assay Kit, Thermo Fisher) and Bioanalyzer (High Sensitivity DNA Kit, Agilent). Libraries were diluted to 2 nM in 10 μL 10 mM Tris-HCl (pH 8.5) and sequenced on a HiSeq1500 (Illumina) using a v2 Rapid SR50 cartridge (Illumina) for two biological replicates.
Ribosome profiling
Translatome sequencing was conducted as described (Hücker et al., 2017) with a few modifications. Briefly, 25 absorption units of the cell lysate were incubated (1 h, 25 °C, shaking at 850 rpm) with 62.5 U MNase (Thermo Fisher), 18.75 U RNase R (Lucigen), 4.375 U RNase T (NEB), 1.875 U XRN-1 (NEB), 1 mM CaCl2, and 1×Buffer 4 (NEB). The reaction was stopped (6 mM EGTA, 50 U SUPERase·In, 10 min). Monosomes were isolated by sucrose density gradient centrifugation (104,000×g, 4 °C, 3 h) followed by RNA isolation and DNase treatment as described. Ribosomal footprints were size selected using a 16% denaturing urea polyacrylamide gel (200 V, 1.5 h). After staining (SYBR Gold, Invitrogen), ribosomal footprints (19 – 27 nt) were excised. Gel pieces were crushed in gel breaker tubes (15,700 ×g, 2 min). Gel debris was incubated overnight in extraction buffer (300 mM NaOAc pH 5.5, 1 mM EDTA, 0.1 U/μL SUPERase·In). After centrifugation (2 min, 9,300×g, RT) in 0.22-μm pore cellulose-acetate filter tubes, footprints were precipitated with ethanol and transformed in a sequencing library as above for two biological replicates.
Cell lysis and protein digest for mass spectrometry
Cells were lysed in 100 μL absolute TFA (Sigma-Aldrich; 5 min, 55 °C, shaking at 1,000 rpm) and neutralized with 900 μL 2 M Tris (Doellinger et al., 2020). Protein concentration was determined using Bradford reagent (B6916, Sigma-Aldrich). For offline high-pH reversed-phase (hpH RP) fractionation and for targeted proteomics, 75 μg and 20 μg of total protein amount were reduced and alkylated (10 mM TCEP, 55 mM CAA; 5 min, 95°C), respectively. Water-diluted samples (1:1) were subjected to proteolysis with trypsin (enzyme to protein ratio 1:50, 30°C, overnight, shaking at 400 rpm) and then stopped (3% formic acid, FA).
Offline high pH reversed-phase fractionation for full proteome analysis
Three discs of Empore C18 (3M) material were packed in 200-μL pipette tips. The resulting desalting columns were conditioned (100% acetonitrile, ACN) and equilibrated (40% ACN/0.1% FA) followed by 2% ACN/0.1% FA. Peptides of the 75-μg protein digest were loaded, washed (2% ACN/0.1% FA) and eluted (40% ACN/0.1% FA). Next, peptides were fractionated using an Agilent 1100 series HPLC system operating a XBridge BEH130 C18 3.5 μm 2.1 × 250 mm column (Waters) at a flow rate of 200 μL/min. Buffer A was 25 mM ammonium bicarbonate (pH 8.0), buffer B was 80% ACN. Fractions were collected every minute into a 96 well plate. Peptides were separated by a linear gradient from 4% to 32% buffer B over 45 min, followed by a gradient from 32% to 85% buffer B over 6 min. Samples were collected in 30 s steps between minute 3 and 51. The solvent was evaporated and samples were redissolved in 2% ACN/0.1% FA. To increase sensitivity, all samples were fractionated into 4 fractions and all samples were measured using the targeted proteomic method Parallel Reaction Monitoring (PRM).
High pH reversed-phase fractionation for targeted proteomics
C18-packed 200-μl tips (see above) were loaded with peptides from the 20 μg digest. A pH switch was performed using 25 mM ammonium formate (pH 10) and varying ACN concentrations for each of six fractions. ACN was added at concentrations of 0, 5, 10, 15, 25, and 50%, respectively. Fraction 1 and 5 and fraction 2 and 6 were combined. The solvent was each evaporated (1+5, 2+6, 3, and 4), and samples were dissolved in 2% ACN/0.1% FA.
LC-MS/MS measurements - full proteomes
Peptides were analysed on a Dionex Ultimate 3000 RSLCnano system coupled to a Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific). For full proteome analyses, 0.5 μg of peptides were delivered to a trap column (ReproSil-pur C18-AQ, 5 μm, 20 mm × 75 μm, Dr. Maisch, self-packed) at a flow rate of 5 μL/min of solvent A (HPLC-grade water with 0.1% FA). After loading (10 min), peptides were transferred to an analytical column (ReproSil Gold C18-AQ, 3 μm, 450 mm × 75 μm, Dr. Maisch, self-packed) and separated using a 50-min linear gradient from 4% to 32% of solvent B (ACN/0.1% FA/5% dimethyl sulfoxide, DMSO) in solvent A (HPLC-grade water with 0.1% FA/5% DMSO) at 300 nL/min flow rate. Both solvents contain DMSO boosting MS intensity. The Fusion Lumos Tribrid mass spectrometer was operated in data-dependent acquisition (DDA) and positive ionization mode. MS1 spectra (360–1300 m/z) were recorded at a resolution of 60,000 using an automatic gain control (AGC) target value of 4×105 and maximum injection time (MaxIT) of 50 ms. Up to 20 peptide precursors were selected for fragmentation in case of the full proteome analyses. Only precursors with charge state 2 to 6 were selected and dynamic exclusion of 20 s was enabled. Peptide fragmentation was performed using higher energy collision induced dissociation (HCD) and a normalized collision energy (NCE) of 30%. The precursor isolation window width was set to 1.3 m/z. MS2 spectra were acquired in the orbitrap with a resolution of 15.000 and an AGC target value of 5x104. MaxIT was set to 22 ms.
Mass spectrometric data analysis - full proteomes
Peptide identification and quantification was performed using MaxQuant (Tyanova et al., 2016) (v1.6.3.4) with Andromeda (Cox et al., 2011). MS2 spectra were searched against the RefSeq (O'Leary et al., 2016) file downloaded for P. aeruginosa PAO1 (GCF_000006765.1_ASM676v1_protein.faa, 5,572 reviewed entries, 7 Feburary 2020), supplemented with common contaminants (by MaxQuant) and Olg1 and Olg2 AA sequences. Trypsin/P was specified as proteolytic enzyme. Precursor tolerance was set to 4.5 ppm and fragment ion tolerance to 20 ppm. Results were adjusted to 1% FDR on peptide spectrum match level and protein level employing a target-decoy approach using reversed protein sequences. Minimal peptide length was defined as 7 AA; the “match-between-run” function disabled. For full proteome analyses, carbamidomethylated cysteine was set as fixed and oxidation of methionine and N-terminal protein acetylation as variable modifications. Correlation scores (dot product) between experimental and predicted spectra were calculated via Skyline daily (64-bit, v20.1.9.234) (MacLean et al., 2010) that supports Prosit (Gessulat et al., 2019) spectra predictions. For data analysis, protein intensities and iBAQ (Schwanhäusser et al., 2011) values were calculated.
Targeted LC-MS/MS measurements
Targeted measurements using Parallel Reaction Monitoring (PRM) were performed with a 50-min linear gradient on a Dionex Ultimate 3000 RSLCnano system coupled to a Q-Exactive HF-X mass spectrometer (Thermo Fisher Scientific). The spectrometer was operated in PRM and positive ionization mode. MS1 spectra (360–1300 m/z) were recorded at a resolution of 60,000 using an AGC target value of 3×106 and a MaxIT of 100 ms. Targeted MS2 spectra were acquired at 60,000 resolution with a fixed first mass of 100 m/z, after HCD with 26% NCE, and using an AGC target value of 1×106, a MaxIT of 118 ms and an isolation window of 0.7 m/z. For the PRM analysis of the growth phase samples, 18 OLG and mother gene peptides plus 12 retention time reference peptides (subset of Procal peptides synthesized by JPT (Zolg et al., 2017) were targeted within a single PRM run and with a 5 min scheduled retention time window. The cycle time was ∼2.1 s, which leads to ∼10 data points per chromatographic peak.
Selection and validation of target peptides
Isotope-labelled internal reference peptides were used for confident identification and quantification. Peptide selections were based on results of DDA measurements of the deep proteome at OD600nm = 1. Peptides were selected based on intensity, location within the protein, Andromeda score, excluding modification, and charge state. All isotopically-labeled synthetic peptides were pooled and targeted proteomic measurements (PRM) showed confident detection of all 18 peptides (MaxQuant score >90, Table S9). Skyline-daily (MacLean et al., 2010) was used to build an experimental spectral library from the generated PRM data.
Targeted mass spectrometric data analysis
PRM data was analysed using Skyline-daily (MacLean et al., 2010). Peak integration, transition interferences and integration boundaries were reviewed manually, considering four to six transitions per peptide. To discriminate between true or false peptide detection, filtering according to correlation of fragment ion intensities between endogenous (light) and spike-in (heavy) peptides was applied (“Library Dot Product” ≥0.8). Additionally, a good correlation of fragment ion intensities between light and heavy peptide (“DotProductLightToHeavy” of >0.9) and a mass accuracy of below ±20 ppm (“Average Mass Error PPM”) was required. Total protein intensity was computed by summing up all light peptide intensities in each sample. Uniqueness of peptides was assessed against the RefSeq database for P. aeruginosa PAO1.
Bioinformatic analyses
Putative σ70 promoters within a 300-nt region upstream of the start codon were predicted by BPROM (Solovyev and Salamov, 2011) with minimum LDF scores of 0.2.
Shine-Dalgarno sequence identification was performed as described (Ma et al., 2002) within a region of 30 nt upstream of the start codon and a minimum free energy (ΔGSD) threshold of −2.9 kcal/mol.
To predict ρ-independent terminators, a 300-nt region downstream of the respective stop codon was analysed using FindTerm (Solovyev and Salamov, 2011) with an threshold of −3. Predicted terminator regions were read in non-overlapping sliding windows of 30 nt and folded with Mfold (Zuker, 2003), identifying stem loops.
FASTQ files were processed using a custom perl script. In short, FastQC (available online at http://www.bioinformatics.babraham.ac.uk/projects/fastqc) was used to assess raw read quality and adapter sequences were trimmed with fastp (Chen et al., 2018). Trimmed reads were aligned to the reference (GCF_000006765.1_ASM676v1) with Bowtie2 v2.2.6 (Langmead and Salzberg, 2012) using “--very-sensitive end-to-end” with a seed length of 17 nt. Reads mapping to rRNAs and tRNAs were filtered with SAMTools (Li et al., 2009) and BEDTools (Quinlan and Hall, 2010). Remaining reads were normalized to gene length and sequencing depth (RPKM: “reads per kilobase per million mapped reads”). For each genomic nucleotide position, reads per million mapped reads (RPM) were calculated, averaged over biological replicates and visualized with pyGenomeTracks (Ramírez et al., 2018). Ribosome coverage values (RCV) (Neuhaus et al., 2017) were calculated by dividing the RPKM of the translatome by the RPKM of the transcriptome for evaluating ‘translatability’.
Significant changes in translation were determined between the published RiboSeq datasets “M9+n-alkane” and “M9+glycerol” (Grady et al., 2017; SRA accession number PRJNA379630). Read counts were scaled to the smallest library size and differential expression analysis was performed using an exact test implemented in edgeR (Robinson et al., 2010).
Gene prediction was performed with Prodigal (Hyatt et al., 2010) using default parameters. In order to detect overlapping ORFs, all possible start codons within and in the upstream-vicinity of the coding regions for tle3 and PA1383 were masked by N (any nucleotide). Further, protein-coding ORFs were predicted based on RiboSeq data using DeepRibo (Clauwaert et al., 2019) with default settings.
BLAST searches (blast) (Altschul et al., 1990) against NCBI databases were used to find homologous nucleotide (blastn) and protein sequences (blastp).
Evolutionary and taxonomic analyses
Scripts for evolutionary and taxonomic analyses are available in the GitHub repository https://github.com/ZacharyArdern/Pseudomonas_long_OLGs.
Phylostratigraphy – taxonomic distribution
Homologs of tle3 and PA1383 (NC_002516.2) were detected using BLASTp in annotated proteins from genomes in Pseudomonadales, and from the Identical Protein Groups database using the Entrez Programming Utilities (Kans, 2021). Sequences from MAG collections were added using Diamond blastp (Buchfink et al., 2014) finding homologs within genomes annotated as being within Pseudomonadales. The combined collection was downsampled to unique nucleotide sequences with a length of ≥50% compared to tle3 or PA1383 in the reference strain PAO1.
Protein sequences were aligned using QuickProbs2 (Gudyś and Deorowicz, 2017), and nucleotide sequences subsequently converted to codon alignments using Pal2Nal (Suyama et al., 2006). Maximum likelihood trees of tle3 and PA1383 alignments were calculated using IQ-TREE (Minh et al., 2020; Nguyen et al., 2015) with default settings and 1000 bootstrap iterations. Trees were downsampled to 20 genomes using Treemmer (Menardo et al., 2018), and edited using Newick Utilities (Junier and Zdobnov, 2010) for OLG-region visualisation using a Python script written with ETE3 packages (Huerta-Cepas et al., 2016), and subsequently also used for pairwise ‘OLGenie’ comparisons (Nelson et al., 2020b).
ORF length and stop codon analyses
Whether overlapping ORF lengths exceeded background was tested using ‘Frameshift’ (Schlub et al., 2018), modified to run from the Unix command line, as well as to print the scores obtained for each method (codon permutation and synonymous codon mutation), and to print the simulated ORF lengths.
An evolutionary simulation method (Cassan et al., 2016) was re-implemented using IQ-TREE and Pyvolve (Spielman and Wilke, 2015), with sequence evolution simulated along trees calculated from the OLG-containing genomes and a selected outgroup sequence without stop codons in the OLG loci. In the original study, an outgroup with an intact ORF was used for rooting the tree (Cassan et al., 2016). A sequence from P. prosekii, the only intact homolog outside the OLG clade of P. aeruginosa, was chosen as outgroup for olg1. For olg2, multiple non-P. aeruginosa intact ORFs were available. A more distant outgroup was chosen as tests of purifying selection described below suggest more taxonomically widespread functionality. Omega values (approximately equivalent to dN/dS) of 0.5 for both genes were chosen based on alignments of the two mother genes, using results from ‘OLGenie’. The empirical codon model (ECM) was used with default parameters except for the omega values specified.
Codon-position constraint analyses
Constraints in synonymous sites of tle3 and PA1383 were assessed using ‘FRESCo’ (Sealfon et al., 2015). Approximate maximum-likelihood nucleotide trees were calculated using FastTree 2 (Price et al., 2010) for the full sets of “OLG” and “non-OLG” genomes, and ‘FRESCo’ was run on codon alignments (described above) with a sliding window size (50 codons).
Constraint on non-synonymous codon changes in the OLGs was assessed using ‘OLGenie’ (Nelson et al., 2020b). Analysis for each mother-gene codon alignment (created using PAL2NAL, described above) of OLG and non-OLG genomes was conducted with standard settings. Sliding window analyses of 50 codons were conducted using a minimum number of defined codons of 2. Pairwise whole-gene comparisons of olg1 and olg2 were conducted using standard settings, and a custom Bash script available at the Github repository cited in the key resources table, producing a pairwise matrix.
Quantification and statistical analyses
Concerning sequencing data, absolute RPKM values were determined, i.e., calculating the number of reads per kilobase gene per million reads sequenced. Ribosome coverage values (RCV) were calculated by dividing the RPKM of the translatome by the RPKM of the transcriptome for evaluating ‘translatability’. For published data sets used, read counts were scaled to the smallest library size and differential expression analysis was performed using an exact test implemented in edgeR. In qPCR, data were analysed using the ΔΔCt method. Significance was evaluated with a two-tailed Welch two-sample t-test (p value ≤0.05). For mass spectrometry, peptide identification and quantification was performed using MaxQuant (v1.6.3.4) with Andromeda. Precursor tolerance was set to 4.5 ppm and fragment ion tolerance to 20 ppm. Results were adjusted to 1% FDR on peptide spectrum match level and protein level employing a target-decoy approach using reversed protein sequences. Correlation scores (dot product) between experimental and predicted spectra were calculated via Skyline daily (64-bit, v20.1.9.234) that supports Prosit spectra predictions. For data analysis, protein intensities and iBAQ values were calculated. Peptides for validation were selected based on intensity, location within the protein, Andromeda score, excluding modification, and charge state. PRM data was analysed using Skyline-daily (MacLean et al., 2010). Peak integration, transition interferences and integration boundaries were reviewed manually, considering four to six transitions per peptide. To discriminate between true or false peptide detection, filtering according to correlation of fragment ion intensities between endogenous (light) and spike-in (heavy) peptides was applied (“Library Dot Product” ≥0.8). Additionally, a good correlation of fragment ion intensities between light and heavy peptide (“DotProductLightToHeavy” of >0.9) and a mass accuracy of below ±20 ppm (“Average Mass Error PPM”) was required. Total protein intensity was computed by summing up all light peptide intensities in each sample. Concerning selection tests, omega values (approximately equivalent to dN/dS) of 0.5 were chosen based on alignments of the mother genes, using results from ‘OLGenie’. The empirical codon model (ECM) was used with default parameters except for the omega values specified.
Acknowledgments
We thank Romy Wecko, Verena Breitner, Lara Wanner, Hermine Kienberger, and Franziska Hackbarth for technical assistance and Christopher Huptas for bioinformatic support. We also thank Siddhanth Rao for assistance with scripts for the use of “FRESCo” and Chase Nelson and April Wei for helpful comments on the manuscript. TUM University Library Publishing Fund helped covering publishing costs, given to K.N.
Author contributions
M.K. performed the sequencing experiments, analyses thereof, and wrote the first draft of the manuscript. Z.A. conducted the evolutionary analyses and critically revised the manuscript. M.A. performed the mass spectrometry experiments and analyses thereof under supervision of C.L. The study was conceived, designed, and coordinated by S.S. and K.N. All authors helped with writing and editing.
Declaration of interests
The authors declare no competing interests.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103844.
Contributor Information
Zachary Ardern, Email: zachary.ardern@sanger.ac.uk.
Klaus Neuhaus, Email: neuhaus@tum.de.
Supporting citations
The following reference appears in the Supplemental Information: Bachvarov et al., 2008, Bagag et al., 2013, Baldwin, 2004, , Landry et al., 2015, Nakahigashi et al., 2016, Potvin et al., 2008,Slavoff et al., 2013, Smollett et al., 2009, West and Iglewski, 1988
Supplemental information
Data and code availability
-
•
This paper analyses data produced for this publication. These accession numbers for the datasets are listed in the key resources table.
-
•
Any original code reported is available via github, as listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Almeida A., Nayfach S., Boland M., Strozzi F., Beracochea M., Shi Z.J., Pollard K.S., Sakharova E., Parks D.H., Hugenholtz P., et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2020;39:105–114. doi: 10.1038/s41587-020-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/s0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ardern Z., Neuhaus K., Scherer S. Are antisense proteins in prokaryotes functional? Front. Mol. Biosci. 2020;7:187. doi: 10.3389/fmolb.2020.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachvarov B., Kirilov K., Ivanov I. Codon usage in prokaryotes. Biotechnol. Biotechnological Equipment. 2008;22:669–682. doi: 10.1080/13102818.2008.10817533. [DOI] [Google Scholar]
- Baek J., Lee J., Yoon K., Lee H. Identification of unannotated small genes in Salmonella. G3. 2017;7:983–989. doi: 10.1534/g3.116.036939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagag A., Jault J.-M., Sidahmed-Adrar N., Réfrégiers M., Giuliani A., Le Naour F. Characterization of hydrophobic peptides in the presence of detergent by photoionization mass spectrometry. PLoS ONE. 2013;8:e79033. doi: 10.1371/journal.pone.0079033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin M.A. Protein identification by mass spectrometry: issues to be considered. Mol. Cell Proteomics. 2004;3:1–9. doi: 10.1074/mcp.R300012-MCP200. [DOI] [PubMed] [Google Scholar]
- Barrell B.G., Air G.M., Hutchison C.A., 3rd Overlapping genes in bacteriophage phiX174. Nature. 1976;264:34–41. doi: 10.1038/264034a0. [DOI] [PubMed] [Google Scholar]
- Bartonek L., Braun D., Zagrovic B. Frameshifting preserves key physicochemical properties of proteins. Proc. Natl. Acad. Sci. U S A. 2020;117:5907. doi: 10.1073/pnas.1911203117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetti M., Vena A., Croxatto A., Righi E., Guery B. How to manage Pseudomonas aeruginosa infections. Drugs Context. 2018;7:212527. doi: 10.7573/dic.212527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens M., Sheikh J., Nataro J.P. Regulation of the overlapping pic/set locus in Shigella flexneri and enteroaggregative Escherichia coli. Infect Immun. 2002;70:2915–2925. doi: 10.1128/iai.70.6.2915-2925.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berni B., Soscia C., Djermoun S., Ize B., Bleves S. A type VI secretion system trans-kingdom effector is required for the delivery of a novel antibacterial toxin in Pseudomonas aeruginosa. Front. Microbiol. 2019;10:1218. doi: 10.3389/fmicb.2019.01218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2014;12:59. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Cassan E., Arigon-Chifolleau A.M., Mesnard J.M., Gross A., Gascuel O. Concomitant emergence of the antisense protein gene of HIV-1 and of the pandemic. Proc. Natl. Acad. Sci. U S A. 2016;113:11537–11542. doi: 10.1073/pnas.1605739113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirico N., Vianelli A., Belshaw R. Why genes overlap in viruses. Proc. Biol. Sci. 2010;277:3809–3817. doi: 10.1098/rspb.2010.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauwaert J., Menschaert G., Waegeman W. DeepRibo: a neural network for precise gene annotation of prokaryotes by combining ribosome profiling signal and binding site patterns. Nucleic Acids Res. 2019;47:e36. doi: 10.1093/nar/gkz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper H.B., Gardner P.P. Features of functional human genes. bioRxiv. 2020 doi: 10.1101/2020.10.10.334193. [DOI] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Crespo A., Pedraz L., Torrents E. Function of the Pseudomonas aeruginosa NrdR transcription factor: global transcriptomic analysis and its role on ribonucleotide reductase gene expression. PLoS ONE. 2015;10:e0123571. doi: 10.1371/journal.pone.0123571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone S., Vives-Flórez M., Kvich L., Saunders A.M., Malone M., Nicolaisen M.H., Martínez-García E., Rojas-Acosta C., Catalina Gomez-Puerto M., Calum H., et al. The environmental occurrence of Pseudomonas aeruginosa. APMIS. 2020;128:220–231. doi: 10.1111/apm.13010. [DOI] [PubMed] [Google Scholar]
- Delcher A.L., Bratke K.A., Powers E.C., Salzberg S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi J.L., Huber G., Kistler A., Retallack H., Wilkinson M., Yllanes D. An exploration of ambigrammatic sequences in narnaviruses. Sci. Rep. 2019;9:17982. doi: 10.1038/s41598-019-54181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan A.M., Lukhovitskaya N.I., Olendraite I., Firth A.E. A case for a negative-strand coding sequence in a group of positive-sense RNA viruses. Virus Evol. 2020;6:veaa007. doi: 10.1093/ve/veaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doellinger J., Schneider A., Hoeller M., Lasch P. Sample preparation by easy extraction and digestion (SPEED) - a universal, rapid, and detergent-free protocol for proteomics based on acid extraction. Mol. Cell Proteomics. 2020;19:209. doi: 10.1074/mcp.TIR119.001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornenburg J.E., Devita A.M., Palumbo M.J., Wade J.T. Widespread antisense transcription in Escherichia coli. mBio. 2010;1 doi: 10.1128/mBio.00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckweiler D., Häussler S. Antisense transcription in Pseudomonas aeruginosa. Microbiology. 2018;164:889–895. doi: 10.1099/mic.0.000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner L., Bechtel N., Witting M.A., Simon S., Schmitt-Kopplin P., Keim D., Scherer S., Neuhaus K. Phenotype of htgA (mbiA), a recently evolved orphan gene of Escherichia coli and Shigella, completely overlapping in antisense to yaaW. FEMS Microbiol. Lett. 2014;350:57–64. doi: 10.1111/1574-6968.12288. [DOI] [PubMed] [Google Scholar]
- Fellner L., Simon S., Scherling C., Witting M., Schober S., Polte C., Schmitt-Kopplin P., Keim D.A., Scherer S., Neuhaus K. Evidence for the recent origin of a bacterial protein-coding, overlapping orphan gene by evolutionary overprinting. BMC Evol. Biol. 2015;15:283. doi: 10.1186/s12862-015-0558-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska D., Fijalkowski I., Willems P., Van Damme P. Bacterial riboproteogenomics: the era of N-terminal proteoform existence revealed. FEMS Microbiol. Rev. 2020;44:418–431. doi: 10.1093/femsre/fuaa013. [DOI] [PubMed] [Google Scholar]
- Filiatrault M.J., Stodghill P.V., Bronstein P.A., Moll S., Lindeberg M., Grills G., Schweitzer P., Wang W., Schroth G.P., Luo S., et al. Transcriptome analysis of Pseudomonas syringae Identifies new genes, noncoding RNAs, and antisense activity. J. Bacteriol. 2010;192:2359. doi: 10.1128/JB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiatrault M.J., Stodghill P.V., Myers C.R., Bronstein P.A., Butcher B.G., Lam H., Grills G., Schweitzer P., Wang W., Schneider D.J., Cartinhour S.W. Genome-wide identification of transcriptional start sites in the plant pathogen Pseudomonas syringae pv. tomato str. DC3000. PLoS ONE. 2011;6:e29335. doi: 10.1371/journal.pone.0029335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth A.E. Mapping overlapping functional elements embedded within the protein-coding regions of RNA viruses. Nucleic Acids Res. 2014;42:12425–12439. doi: 10.1093/nar/gku981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremin B.J., Bhatt A.S. Structured RNA contaminants in bacterial Ribo-Seq. Msphere. 2020;5 doi: 10.1128/mSphere.00855-20. e00855–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R.C., Kalkhof S., Doppelt-Azeroual O., Mueller S.A., Chovancová M., von Bergen M., Schwikowski B. Common and phylogenetically widespread coding for peptides by bacterial small RNAs. BMC Genomics. 2017;18:553. doi: 10.1186/s12864-017-3932-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsinger D.R., Dallon E., Reddy R., Mohammad F., Buskirk A.R., DiRuggiero J. Ribosome profiling in archaea reveals leaderless translation, novel translational initiation sites, and ribosome pausing at single codon resolution. Nucleic Acids Res. 2020;48:5201–5216. doi: 10.1093/nar/gkaa304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessulat S., Schmidt T., Zolg D.P., Samaras P., Schnatbaum K., Zerweck J., Knaute T., Rechenberger J., Delanghe B., Huhmer A., et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods. 2019;16:509–518. doi: 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- Grady S.L., Malfatti S.A., Gunasekera T.S., Dalley B.K., Lyman M.G., Striebich R.C., Mayhew M.B., Zhou C.L., Ruiz O.N., Dugan L.C. A comprehensive multi-omics approach uncovers adaptations for growth and survival of Pseudomonas aeruginosa on n-alkanes. BMC Genomics. 2017;18:334. doi: 10.1186/s12864-017-3708-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger D.C. The unexpected complexity of bacterial genomes. Microbiology. 2016;162:1167–1172. doi: 10.1099/mic.0.000309. [DOI] [PubMed] [Google Scholar]
- Grassé P.P. Evolution of Living Organisms: Evidence for a New Theory of Transformation. Academic Press; 1977. A new concept of the gene and gene overprinting; pp. 231–237. [Google Scholar]
- Gudyś A., Deorowicz S. QuickProbs 2: towards rapid construction of high-quality alignments of large protein families. Sci. Rep. 2017;7:1–12. doi: 10.1038/srep41553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N., Benhamida J., Bhargava V., Goodman D., Kain E., Kerman I., Nguyen N., Ollikainen N., Rodriguez J., Wang J. Comparative proteogenomics: combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res. 2008;18:1133–1142. doi: 10.1101/gr.074344.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hücker S.M., Ardern Z., Goldberg T., Schafferhans A., Bernhofer M., Vestergaard G., Nelson C.W., Schloter M., Rost B., Scherer S., Neuhaus K. Discovery of numerous novel small genes in the intergenic regions of the Escherichia coli O157:H7 Sakai genome. PLoS ONE. 2017;12:e0184119. doi: 10.1371/journal.pone.0184119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hücker S.M., Vanderhaeghen S., Abellan-Schneyder I., Scherer S., Neuhaus K. The novel anaerobiosis-responsive overlapping gene ano is overlapping antisense to the annotated gene ECs2385 of Escherichia coli O157:H7 Sakai. Front. Microbiol. 2018;9:931. doi: 10.3389/fmicb.2018.00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hücker S.M., Vanderhaeghen S., Abellan-Schneyder I., Wecko R., Simon S., Scherer S., Neuhaus K. A novel short L-arginine responsive protein-coding gene (laoB) antiparallel overlapping to a CadC-like transcriptional regulator in Escherichia coli O157:H7 Sakai originated by overprinting. BMC Evol. Biol. 2018;18:21. doi: 10.1186/s12862-018-1134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J., Serra F., Bork P. Ete 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016;33:1635–1638. doi: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D., Chen G.-L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.T., Petersen L., Falk S., Iversen P., Andersen P., Theisen M., Krogh A. Novel overlapping coding sequences in Chlamydia trachomatis. FEMS Microbiol. Lett. 2006;265:106–117. doi: 10.1111/j.1574-6968.2006.00480.x. [DOI] [PubMed] [Google Scholar]
- Junier T., Zdobnov E.M. The Newick utilities: high-throughput phylogenetic tree processing in the UNIX shell. Bioinformatics. 2010;26:1669–1670. doi: 10.1093/bioinformatics/btq243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kans J. Entrez Programming Utilities Help. National Center for Biotechnology Information (US); 2021. Entrez direct: E-utilities on the UNIX command line. [Google Scholar]
- Keese P.K., Gibbs A. Origins of genes: “big bang” or continuous creation? Proc. Natl. Acad. Sci. U S A. 1992;89:9489–9493. doi: 10.1073/pnas.89.20.9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr K.G., Snelling A.M. Pseudomonas aeruginosa: a formidable and ever-present adversary. J. Hosp. Infect. 2009;73:338–344. doi: 10.1016/j.jhin.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Khan Y.A., Jungreis I., Wright J.C., Mudge J.M., Choudhary J.S., Firth A.E., Kellis M. Evidence for a novel overlapping coding sequence in POLG initiated at a CUG start codon. BMC Genet. 2020;21:25. doi: 10.1186/s12863-020-0828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W., Silby M.W., Purvine S.O., Nicoll J.S., Hixson K.K., Monroe M., Nicora C.D., Lipton M.S., Levy S.B. Proteomic detection of non-annotated protein-coding genes in Pseudomonas fluorescens Pf0-1. PLoS ONE. 2009;4:e8455. doi: 10.1371/journal.pone.0008455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchberger P.C., Schmidt M.L., Ochman H. The ingenuity of bacterial genomes. Annu. Rev. Microbiol. 2020;74:815–834. doi: 10.1146/annurev-micro-020518-115822. [DOI] [PubMed] [Google Scholar]
- Konecny J., Eckert M., Schöniger M., Hofacker G.L. Neutral adaptation of the genetic code to double-strand coding. J. Mol. Evol. 1993;36:407. doi: 10.1007/BF02406718. [DOI] [PubMed] [Google Scholar]
- Koskella B., Brockhurst M.A. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014;38:916–931. doi: 10.1111/1574-6976.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry C.R., Zhong X., Nielly-Thibault L., Roucou X. Found in translation: functions and evolution of a recently discovered alternative proteome. Curr. Opin. Struct. Biol. 2015;32:74–80. doi: 10.1016/j.sbi.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebre S., Gascuel O. The combinatorics of overlapping genes. J. Theor. Biol. 2017;415:90–101. doi: 10.1016/j.jtbi.2016.09.018. [DOI] [PubMed] [Google Scholar]
- Lewenza S., Gardy J.L., Brinkman F.S.L., Hancock R.E.W. Genome-wide identification of Pseudomonas aeruginosa exported proteins using a consensus computational strategy combined with a laboratory-based PhoA fusion screen. Genome Res. 2005;15:321–329. doi: 10.1101/gr.3257305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa A.M., Gebhardt M.J., Dove S.L. H-NS-like proteins in Pseudomonas aeruginosa coordinately silence intragenic transcription. Mol. Microbiol. 2021;115:1138–1151. doi: 10.1111/mmi.14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Livermore D.M. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 2009;64:i29–i36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- Loughran G., Zhdanov A.V., Mikhaylova M.S., Rozov F.N., Datskevich P.N., Kovalchuk S.I., Serebryakova M.V., Kiniry S.J., Michel A.M., O’Connor P.B.F., et al. Unusually efficient CUG initiation of an overlapping reading frame in POLG mRNA yields novel protein POLGARF. Proc. Natl. Acad. Sci. U S A. 2020;117:24936. doi: 10.1073/pnas.2001433117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M., Marinov G.K. The bioenergetic costs of a gene. Proc. Natl. Acad. Sci. U S A. 2015;112:15690. doi: 10.1073/pnas.1514974112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Campbell A., Karlin S. Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J. Bacteriol. 2002;184:5733–5745. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menardo F., Loiseau C., Brites D., Coscolla M., Gygli S.M., Rutaihwa L.K., Trauner A., Beisel C., Borrell S., Gagneux S. Treemmer: a tool to reduce large phylogenetic datasets with minimal loss of diversity. BMC Bioinformatics. 2018;19:1–8. doi: 10.1186/s12859-018-2164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meydan S., Marks J., Klepacki D., Sharma V., Baranov P.V., Firth A.E., Margus T., Kefi A., Vazquez-Laslop N., Mankin A.S. Retapamulin-assisted ribosome profiling reveals the alternative bacterial proteome. Mol. Cell. 2019;74:481–493.e6. doi: 10.1016/j.molcel.2019.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., Von Haeseler A., Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir K., Neuhaus K., Scherer S., Bossert M., Schober S. Predicting statistical properties of open reading frames in bacterial genomes. PLoS ONE. 2012;7:e45103. doi: 10.1371/journal.pone.0045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T., Yasunaga T. Evolution of overlapping genes. Nature. 1978;272:532. doi: 10.1038/272532a0. [DOI] [PubMed] [Google Scholar]
- Nakahigashi K., Takai Y., Kimura M., Abe N., Nakayashiki T., Shiwa Y., Yoshikawa H., Wanner B.L., Ishihama Y., Mori H. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res. 2016;23:193–201. doi: 10.1093/dnares/dsw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S., Shi Z.J., Seshadri R., Pollard K.S., Kyrpides N.C. New insights from uncultivated genomes of the global human gut microbiome. Nature. 2019;568:505–510. doi: 10.1038/s41586-019-1058-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C.W., Ardern Z., Goldberg T.L., Meng C., Kuo C.-H., Ludwig C., Kolokotronis S.-O., Wei X. Dynamically evolving novel overlapping gene as a factor in the SARS-CoV-2 pandemic. eLife. 2020;9:e59633. doi: 10.7554/eLife.59633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C.W., Ardern Z., Wei X. OLGenie: estimating natural selection to predict functional overlapping genes. Mol. Biol. Evol. 2020;37:2440–2449. doi: 10.1093/molbev/msaa087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus K., Landstorfer R., Fellner L., Simon S., Schafferhans A., Goldberg T., Marx H., Ozoline O.N., Rost B., Kuster B. Translatomics combined with transcriptomics and proteomics reveals novel functional, recently evolved orphan genes in Escherichia coli O157: H7 (EHEC) BMC Genomics. 2016;17:133. doi: 10.1186/s12864-016-2456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus K., Landstorfer R., Simon S., Schober S., Wright P.R., Smith C., Backofen R., Wecko R., Keim D.A., Scherer S. Differentiation of ncRNAs from small mRNAs in Escherichia coli O157:H7 EDL933 (EHEC) by combined RNAseq and RIBOseq - ryhB encodes the regulatory RNA RyhB and a peptide, RyhP. BMC Genomics. 2017;18:216. doi: 10.1186/s12864-017-3586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T., Schmidt H.A., Von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Allen & Unwin; Springer-Verlag; 1970. Evolution by Gene Duplication. [Google Scholar]
- Pallejà A., Harrington E.D., Bork P. Large gene overlaps in prokaryotic genomes: result of functional constraints or mispredictions? BMC Genomics. 2008;9:335. doi: 10.1186/1471-2164-9-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:442–450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruschke H., Anders J., Stadler P.F., Jehmlich N., von Bergen M. Enrichment and identification of small proteins in a simplified human gut microbiome. J. Proteomics. 2020;213:103604. doi: 10.1016/j.jprot.2019.103604. [DOI] [PubMed] [Google Scholar]
- Portelli G. The relations between the precodons of overlapping genes. J. Theor. Biol. 1982;95:345–350. doi: 10.1016/0022-5193(82)90249-1. [DOI] [PubMed] [Google Scholar]
- Potvin E., Sanschagrin F., Levesque R.C. Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 2008;32:38–55. doi: 10.1111/j.1574-6976.2007.00092.x. [DOI] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Bhardwaj V., Arrigoni L., Lam K.C., Grüning B.A., Villaveces J., Habermann B., Akhtar A., Manke T. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 2018;9:189. doi: 10.1038/s41467-017-02525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell A.B., LeRoux M., Hathazi K., Agnello D.M., Ishikawa T., Wiggins P.A., Wai S.N., Mougous J.D. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature. 2013;496:508–512. doi: 10.1038/nature12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath N., Landan G., Graur D. A method for the simultaneous estimation of selection intensities in overlapping genes. PLoS ONE. 2008;3:e3996. doi: 10.1371/journal.pone.0003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg S.L., Delcher A.L., Kasif S., White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander C., Schulz G.E. Degeneracy of the information contained in amino acid sequences: evidence from overlaid genes. J. Mol. Evol. 1979;13:245–252. doi: 10.1007/BF01739483. [DOI] [PubMed] [Google Scholar]
- Schlub T.E., Buchmann J.P., Holmes E.C. A simple method to detect candidate overlapping genes in viruses using single genome sequences. Mol. Biol. Evol. 2018;35:2572–2581. doi: 10.1093/molbev/msy155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlub T.E., Holmes E.C. Properties and abundance of overlapping genes in viruses. Virus Evol. 2020;6:veaa009. doi: 10.1093/ve/veaa009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Sealfon R.S., Lin M.F., Jungreis I., Wolf M.Y., Kellis M., Sabeti P.C. FRESCo: finding regions of excess synonymous constraint in diverse viruses. Genome Biol. 2015;16:38. doi: 10.1186/s13059-015-0603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V., Eckels J., Schilling B., Ludwig C., Jaffe J.D., MacCoss M.J., MacLean B. Panorama public: a public repository for quantitative data sets processed in skyline. Mol. Cell Proteomics. 2018;17:1239–1244. doi: 10.1074/mcp.RA117.000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silby M.W., Levy S.B. Use of in vivo expression technology to identify genes important in growth and survival of Pseudomonas fluorescens Pf0-1 in soil: discovery of expressed sequences with novel genetic organization. J. Bacteriol. 2004;186:7411–7419. doi: 10.1128/JB.186.21.7411-7419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silby M.W., Levy S.B. Overlapping protein-encoding genes in Pseudomonas fluorescens Pf0-1. PLoS Genet. 2008;4:e1000094. doi: 10.1371/journal.pgen.1000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff S.A., Mitchell A.J., Schwaid A.G., Cabili M.N., Ma J., Levin J.Z., Karger A.D., Budnik B.A., Rinn J.L., Saghatelian A. Peptidomic discovery of short open reading frame–encoded peptides in human cells. Nat. Chem. Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C., Canestrari J.G., Wang J., Derbyshire K.M., Gray T.A., Wade J.T. Pervasive translation in Mycobacterium tuberculosis. bioRxiv. 2019 doi: 10.1101/665208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smollett K.L., Fivian-Hughes A.S., Smith J.E., Chang A., Rao T., Davis E.O. Experimental determination of translational start sites resolves uncertainties in genomic open reading frame predictions–application to Mycobacterium tuberculosis. Microbiology. 2009;155:186. doi: 10.1099/mic.0.022889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V., Salamov A. In: Metagenomics and Its Applications in Agriculture, Biomedicine and Environmental Studies. Li R.W., editor. Nova Science Publishers, Inc.; 2011. Automatic annotation of microbial genomes and metagenomic sequences; pp. 61–78. [Google Scholar]
- Spielman S.J., Wilke C.O. Pyvolve: a flexible Python module for simulating sequences along phylogenies. PLoS ONE. 2015;10:e0139047. doi: 10.1371/journal.pone.0139047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G., Wolf Y.I., Ramamurthi K.S. Small proteins can no longer be ignored. Annu. Rev. Biochem. 2014;83:753–777. doi: 10.1146/annurev-biochem-070611-102400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer A., Smith C., Mangano K., Wade J.T. Identification of novel translated small ORFs in Escherichia coli using complementary ribosome profiling approaches. bioRxiv. 2021 doi: 10.1101/2021.07.02.450978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M., Torrents D., Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D., Domazet-Lošo T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011;12:692–702. doi: 10.1038/nrg3053. [DOI] [PubMed] [Google Scholar]
- Tunca S., Barreiro C., Coque J.J., Martin J.F. Two overlapping antiparallel genes encoding the iron regulator DmdR1 and the Adm proteins control siderophore and antibiotic biosynthesis in Streptomyces coelicolor A3(2) FEBS J. 2009;276:4814–4827. doi: 10.1111/j.1742-4658.2009.07182.x. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Vakirlis N., Carvunis A.-R., McLysaght A. Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes. Elife. 2020;9:e53500. doi: 10.7554/eLife.53500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderhaeghen S., Zehentner B., Scherer S., Neuhaus K., Ardern Z. The novel EHEC gene asa overlaps the TEGT transporter gene in antisense and is regulated by NaCl and growth phase. Sci. Rep. 2018;8:17875. doi: 10.1038/s41598-018-35756-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter E., Smith R.D., Payne S.H. Proteogenomic analysis of bacteria and archaea: a 46 organism case study. PLoS ONE. 2011;6:e27587. doi: 10.1371/journal.pone.0027587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren A.S., Archuleta J., Feng W.C., Setubal J.C. Missing genes in the annotation of prokaryotic genomes. BMC Bioinformatics. 2010;11:131. doi: 10.1186/1471-2105-11-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver J., Mohammad F., Buskirk A.R., Storz G. Identifying small proteins by ribosome profiling with stalled initiation complexes. mBio. 2019;10 doi: 10.1128/mBio.02819-18. e02819–02818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X., Zhang J. A simple method for estimating the strength of natural selection on overlapping genes. Genome Biol. Evol. 2015;7:381–390. doi: 10.1093/gbe/evu294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein R.A., Gaynes R., Edwards J.R., National Nosocomial Infections Surveillance, System Overview of nosocomial infections caused by gram-negative bacilli. Clin. Infect. Dis. 2005;41:848–854. doi: 10.1086/432803. [DOI] [PubMed] [Google Scholar]
- Weisman C.M., Murray A.W., Eddy S.R. Many, but not all, lineage-specific genes can be explained by homology detection failure. PLoS Biol. 2020;18:e3000862. doi: 10.1371/journal.pbio.3000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S.E., Iglewski B.H. Codon usage in Pseudomonas aeruginosa. Nucleic Acids Res. 1988;16:9323–9335. doi: 10.1093/nar/16.19.9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems P., Fijalkowski I., Van Damme P. Lost and found: Re-searching and Re-scoring proteomics data aids genome annotation and improves proteome coverage. mSystems. 2020;5:e00833-20. doi: 10.1128/mSystems.00833-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolstenhulme C.J., Guydosh N.R., Green R., Buskirk A.R. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015;11:13–21. doi: 10.1016/j.celrep.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright B.W., Molloy M.P., Jaschke P.R. Overlapping genes in natural and engineered genomes. Nat. Rev. Genet. 2021;5:1–15. doi: 10.1038/s41576-021-00417-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel O., Yoder-Himes D.R., Han K., Dandekar A.A., Edelheit S., Greenberg E.P., Sorek R., Lory S. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog. 2012;8:e1002945. doi: 10.1371/journal.ppat.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Jensen S.I., Wulff T., Harrison S.J., Long K.S. Identification and validation of novel small proteins in Pseudomonas putida. Environ. Microbiol. Rep. 2016;8:966–974. doi: 10.1111/1758-2229.12473. [DOI] [PubMed] [Google Scholar]
- Yockey H.P. Do overlapping genes violate molecular biology and the theory of evolution? J. Theor. Biol. 1979;80:21–26. doi: 10.1016/0022-5193(79)90176-0. [DOI] [PubMed] [Google Scholar]
- Yooseph S., Sutton G., Rusch D.B., Halpern A.L., Williamson S.J., Remington K., Eisen J.A., Heidelberg K.B., Manning G., Li W., et al. The Sorcerer II global ocean sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5:e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehentner B., Ardern Z., Kreitmeier M., Scherer S., Neuhaus K. Evidence for numerous embedded antisense overlapping genes in diverse E. coli strains. bioRxiv. 2020 doi: 10.1101/2020.11.18.388249. [DOI] [Google Scholar]
- Zehentner B., Ardern Z., Kreitmeier M., Scherer S., Neuhaus K. A novel pH-regulated, unusual 603 bp overlapping protein coding gene pop is encoded antisense to ompA in Escherichia coli O157: H7 (EHEC) Front. Microbiol. 2020;11:377. doi: 10.3389/fmicb.2020.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolg D.P., Wilhelm M., Yu P., Knaute T., Zerweck J., Wenschuh H., Reimer U., Schnatbaum K., Kuster B. PROCAL: a set of 40 peptide standards for retention time indexing, column performance monitoring, and collision energy calibration. Proteomics. 2017;17:1700263. doi: 10.1002/pmic.201700263. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts for evolutionary and taxonomic analyses are available in the GitHub repository (https://github.com/ZacharyArdern/Pseudomonas_long_OLGs). Sequencing data have been deposited in Sequence Read Archive (NCBI) under the BioProject accession number PRJNA716268 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA716268). The proteomics raw data, MaxQuant search results, and used protein sequence databases have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019) and can be accessed using the dataset identifier PXD023992 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD023992). All targeted proteomic raw data and Skyline analysis files have been deposited using the identifier OLG_PSE to Panorama Public (https://panoramaweb.org/OLG_PSE.url) (Sharma et al., 2018).
-
•
This paper analyses data produced for this publication. These accession numbers for the datasets are listed in the key resources table.
-
•
Any original code reported is available via github, as listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.