

Abstract
We demonstrated that ATP synthase serves the functions of a primary mitochondrial K+ “uniporter,” i.e., the primary way for K+ to enter mitochondria. This K+ entry is proportional to ATP synthesis, regulating matrix volume and energy supply-vs-demand matching. We show that ATP synthase can be upregulated by endogenous survival-related proteins via IF1. We identified a conserved BH3-like domain of IF1 which overlaps its “minimal inhibitory domain” that binds to the β-subunit of F1. Bcl-xL and Mcl-1 possess a BH3-binding-groove that can engage IF1 and exert effects, requiring this interaction, comparable to diazoxide to augment ATP synthase's H+ and K+ flux and ATP synthesis. Bcl-xL and Mcl-1, but not Bcl-2, serve as endogenous regulatory ligands of ATP synthase via interaction with IF1 at this BH3-like domain, to increase its chemo-mechanical efficiency, enabling its function as the recruitable mitochondrial KATP-channel that can limit ischemia-reperfusion injury. Using Bayesian phylogenetic analysis to examine potential bacterial IF1-progenitors, we found that IF1 is likely an ancient (∼2 Gya) Bcl-family member that evolved from primordial bacteria resident in eukaryotes, corresponding to their putative emergence as symbiotic mitochondria, and functioning to prevent their parasitic ATP consumption inside the host cell.
Keywords: ATP synthase regulation, ATPase Inhibitory Factor-1 (IF₁), Bcl-2 family proteins, mitochondrial potassium transport, volume regulation, mitochondrial permeability transition pore
Graphical Abstract
Graphical Abstract.
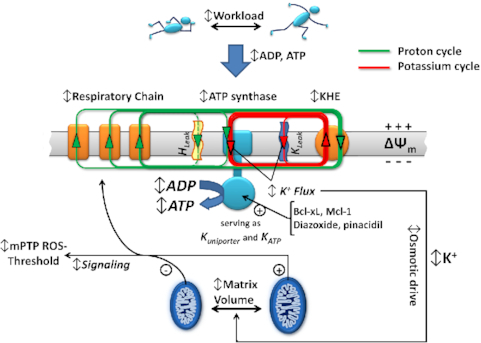
ATP Synthase K+- and H+-fluxes Drive ATP Synthesis and Enable Mitochondrial K+-“Uniporter” Function: II. Ion and ATP Synthase Flux Regulation.
Introduction
Recently, we provided compelling evidence that F1Fo ATP synthase utilizes both H+- and K+-transport to drive ATP synthesis with a high degree of H+: K+ selectivity of ∼106:1.1 Using purified ATP synthase reconstituted in proteoliposomes we assessed ATP synthesis by means of ATP luminescence, determined the contribution of unitary H+ and K+ ion currents from F1Fo by electrophysiological assessment in planar lipid membranes, and measured oxygen consumption flux and the corresponding driving forces in experiments with isolated mitochondria. We concluded that ΔΨm-driven K+ entry through F1Fo is directly proportional to ATP synthesis and regulates matrix volume, and in turn serves the function of directing the matching of cellular energy utilization with its production. Our data revises the central tenet that ATP synthase operates solely on H+ flux to synthesize ATP; however, due to the participation of the mitochondrial K+/H+ exchanger in K+ cycling, our findings remain fully compatible with Mitchell's chemiosmotic mechanism.1
It has been recognized for a long time that a “mitochondrial ATP-dependent K+ channel” (mKATP) activity plays a crucial role in the mitochondrial volume regulation and cellular bioenergetics.2 mKATP is a possible trigger among a multiplicity of distinct upstream pathways that we showed can activate signaling that converges, via inhibition of the master-switch kinase, GSK-3β, on the end effector, the mitochondrial permeability transition pore (mPTP) complex, to increasingly resist pore-opening by ROS (e.g., as occurs during reperfusion after ischemia), and resulting in mitochondrial and cell protection against oxidant stress injury. Opening of mKATP, activated either by repetitive short periods of ischemia (“ischemic preconditioning”) or by K+ channel openers (KCO) such as diazoxide (Dz), serves as a critical link in a cascade of kinases preventing the deleterious effects of mPTP opening, consequently limiting cell damage and death after ischemia/reperfusion injury.3–5
The pharmacological action of selected KCOs (e.g., diazoxide, pinacidil and nicorandil), on the previously empirically/functionally-defined mKATP has been correlated with protection against ischemic/hypoxic injury in intact heart6,7 and brain,8 in isolated cardiomyocytes4,9 and neurons,10 recently reviewed.11 Furthermore, the fact that ischemic preconditioning can be blocked by specific mKATP inhibitors suggests that this entity also occupies a critical locus in the general protection signaling network.4,6,12–16 Unfortunately, as for many of the mitochondrial channels, the difficulties of the molecular identification of the mKATP has largely restricted hypothesis-driven research to the use of pharmacological agents with known properties. Thus, identifying this channel at a molecular level is critical to the development of new treatments limiting the damage of heart attacks and strokes. This endeavor has been a major research priority in top laboratories worldwide but several decades long quest for identifying the mKATP entity had led to multiple largely inconclusive attempts and different candidates.17–20 The results presented in our recent work showed that ATP synthase may function as a recruitable mKATP channel which, when engaged, as we show here, may activate cardioprotective signaling pathways with subsequent increase in mPTP ROS-threshold, a metric of resistance to oxidative stress.1
A significant decrease or loss of mitochondrial membrane potential (ΔΨm) could potentially reverse the ATP synthesis function of F1Fo, resulting instead in ATP hydrolysis. During ischemia, consuming substantial amounts of ATP at a time when its supply is limited would likely be detrimental in energetically-sensitive cells such as cardiomyocytes and neurons. It is known that Inhibitory Factor-1 (IF1), a small ∼12kDa regulatory protein, limits the reversal of F1Fo function, and that during ischemia this helps to prevent excessive (or even futile) ATP consumption by damaged mitochondria to maintain ΔΨm.21 Interestingly, Dz binds to the catalytic domain of βDP in F1, thus inducing the nucleotide stabilization and facilitating IF1 binding in the C-terminal domain of βDP22 and enhances the inhibitory functions of IF1 suggesting a tendency to preserve ATP during ischemia that may lead to enhanced cell survival and resistance to damage.
Certain members of the Bcl-2 family of proteins can protect the heart against ischemia/reperfusion injury,23,24 reviewed in.25 The BH4 domain of Bcl-xL has been shown to be sufficient for protection against mitochondrial dysfunction.3,26 Hearts perfused with a peptide corresponding to residues 4–23 of Bcl-xL conjugated to the protein transduction domain of HIV TAT (TAT-BH4) displayed reduced infarct size after ischemic injury27 and exhibited enhanced mPTP ROS-threshold.3 Importantly, Bcl-xL has been found to be localized not only in the outer mitochondrial membrane but also in the inner membrane28,29 where its interaction with F1Fo regulates metabolic efficiency.30
In this work, building on the findings of our recent manuscript,1 we employed IF1 gene silencing technique in the neonatal cardiac myocytes and used IF1 knockout mouse to investigate the actions of endogenous regulators on the novel functional ability of ATP synthase to harness energy from K+ flux. This property enables K+ uniporter-like function that facilitates energy supply-demand matching, and additionally enables ATP synthase to function as a mKATP. We found that while retaining the high degree of H+-selectivity, the chemo-mechanical efficiency, and the monovalent cation conductance of F1Fo, can be increased by endogenous pro-survival proteins, Bcl-xL and Mcl-1, and certain KCOs. This process requires IF1 and is regulated naturally by the concentration of ATP (i.e., the free energy of ATP provides a natural concentration-dependent inhibitory counter-torque on the synthase-activity of F1Fo). Additionally, we examined the origin of IF1 in relation to the evolution of F1Fo. Interestingly, phylogenetic analysis shows that IF1 is possibly closely related to ancient BH3-containing proteins (e.g., Bad, PUMA, Bcl-xL). This would suggest existence of an evolutionary selection pressure tuned to preventing energy wastage and at the same time to improve the activity and efficiency of the F1Fo machinery.
Results
Previously, we demonstrated that purified ATP synthase (F1Fo) reconstituted in proteoliposomes can synthesize ATP solely driven by K+ flux using the free energy stored in a K+ gradient, and conducts up to ∼3.5 K+ for every H+ under physiological concentrations of K+ and H+. Purely K+-driven ATP synthesis from single F1Fo molecules reconstituted in a lipid bilayer at the tip of a micropipette was demonstrated by simultaneous luciferase bioluminescence single-photon detection of newly made ATP, and unitary K+ currents by voltage clamp, both blocked by specific inhibitors of ATP synthase. Using a novel technique that we designed for this purpose, this experiment provided unambiguous proof of K+-driven ATP production by single molecules of mammalian ATP synthase under conditions matching the physiological K+ ionic milieu. In the presence of K+, as compared to its absence, intact isolated mitochondria display 3.5-fold higher rates of ATP synthesis at the expense of 2.6-fold higher rates of O2 consumption, and these fluxes are driven by 2.7:1 K+: H+ stoichiometry.1
F1Fo-mediated Mitochondrial K+ Influx Regulates Respiration and Mitochondrial Volume in Cells
As in the presence of KCOs, F1Fo achieves a proportionally greater flux of K+ ions suggesting that it may function as a recruitable mKATP, we decided to investigate whether these findings also apply to living cells where we examined each one of the following manifestations of mKATP activation in cardiomyocytes in response to KCOs, based on previous work by others2, 31, 32 and our group4, 33: (i) flavoprotein (FP) oxidation, (ii) modulation of mitochondrial regulatory swelling (i.e., due to mitochondrial K+ accumulation), (iii) volume activation of respiration (as a consequence of (ii)), (iv) inhibition of GSK-3β activity via ser-9 phosphorylation, and (v) increased mPTP reactive oxygen species (ROS)-threshold. We tested mKATP activation by Dz in myocytes with IF1 knocked down by ∼75% through gene silencing (Figure 1B and C), compared to cells treated with control siRNA (Figure 1A and C). Dz produced an equivalent increase in FP oxidation in control myocytes (Figure 1D; see Figure S1 for the FP signal calibration) as compared to a blunted FP response from IF1 siRNA treated cells (Figure 1E), consistent with F1Fo functioning as a mitoKATP regulated by IF1. KCO-driven activation of mitoKATP causes mitochondrial swelling2 and increases respiration.31, 34 Using a single cardiac myocyte imaging technique,4 we found that KCO Dz, HOE694 (HOE; NHE-1 inhibitor), and the δ-opioid peptide, DADLE, each cause a rapid ∼2.5–4% increase in the average volume of mitochondria throughout the cardiomyocyte and increase in respiration (Figure 1F–K and L). In cardiac myocyte suspension, we found that pharmacologic agents that cause mitochondrial swelling (Dz, HOE, and DADLE) increased oxygen consumption (VO2) over baseline by about 10%, 25–30%, and 35%, respectively, when utilizing glucose, the medium- and long-chain fatty acid octanoate or palmitate, respectively, and that by preventing this volume increase (e.g., using the Cl– channel inhibitor, IAA-94), the accompanying increase in respiration was similarly eliminated.4 Thus, volume activation of respiration is a direct correlate of mitochondrial regulatory volume swelling. Using the same logic as the preceding section, since DADLE causes similar and rapid increases in respiration (as Dz) but is known to not activate the mKATP, it was employed as a negative control in the next series of experiments. We found that Vent completely prevented the Dz-related increase in cardiomyocyte mitochondrial swelling and respiration (Figure 1I, L), while the actions of HOE or DADLE were unaffected (Figure 1J-L). Thus, only the specific effect of Dz acting through the mitoKATP causes mitochondrial swelling leading to an increase in respiration, but not that of DADLE, requires the function of Fo.
Figure 1.

Knockdown of IF1 expression in neonatal cardiomyocytes using RNA interference. (A) IF1 immunocytochemical labeling of control, and (B) IF1 siRNA treated cells. (C) Western blot analysis of control vs siRNA treated samples; positive control corresponds to adult rat heart. (D,E) FP autofluorescence (normalized to dinitrophenol, DNP) as marker of mKATP activity. (D) Dz induced FP oxidation in control, and (E) No effect of Dz was observed in IF1 siRNA-treated cells. (F–K) In situ monitoring of the amplitude and kinetics of regulatory mitochondrial swelling (resulting from increased mitochondrial K+ influx and/or retention) in intact cardiomyocytes, based on Fourier analysis of laser linescan transmittance imaging. (F) KCO, Dz; (G) The NHE-1 inhibitor, HOE, and (H) the δ-opioid peptide, DADLE, induced mitochondrial swelling. (I) The Fo inhibitor, Vent, blocked Dz-induced mitochondrial swelling, while it had no effect on swelling induced by (J) HOE or (K) DADLE. Arrow indicates the time point of drug addition. (L) Mitochondrial respiration (indexed by oxygen consumption, VO2 with respect to Dz and Vent treatment as in (D) in myocytes. (M) mKATP (Dz)-protection signaling via GSK-3β requires IF1 (see text for details). While the KCO, Dz, causes a robust increase in P-GSK-3β in control cells, this was largely prevented in IF1-siRNA treated cells.
Effects on mPTP ROS-threshold
The mPTP is a key end-effector of protection signaling: the threshold for mPTP-induction by ROS being significantly reduced after ischemia-reperfusion injury and contributing to cell death, but beneficially increased by preconditioning, postconditioning and other forms of protection signaling, contributing to cell survival.4, 33 We showed that cell protection involves convergence of a multiplicity of potential and distinct upstream pathways (including opening of mitoKATP), each acting via inhibition of GSK-3β on the end effector, the mPTP complex, to limit its induction (see Figure 1M). We have found that Dz, HOE, Li+ (the direct pharmacologic inhibitor of GSK-3β), and insulin, each cause a significant increase of the ROS-threshold for mPTP induction, tMPT 4 (Figure 2). Since HOE, Li+ and insulin each cause protection via mitoKATP-independent mechanisms they were employed as negative controls in the next series of experiments. The degree of protection (i.e., prolonged tMPT) afforded by HOE, Li+, and insulin was largely unaffected by IF1-knockdown (Figure 2C). In stark contrast, the effect of Dz was decreased in direct relation to the degree of IF1-knockdown, i.e., tMPT-increase was reduced by about half with ∼50% IF1-knockdown, and completely abolished with ∼75% IF1-knockdown (Figure 2B). We conclude that mKATP-related protection signaling to the mPTP requires the functional presence of IF1, thus implicating the role of ATP synthase. Furthermore, similarly to IF1-knockdown, Vent blocked the protection by Dz (Figure 2D). However, while blockage of Fo by Vent prevents mKATP (Dz)-mediated cardioprotection, it does not do so in the case of DADLE or HOE.
Figure 2.
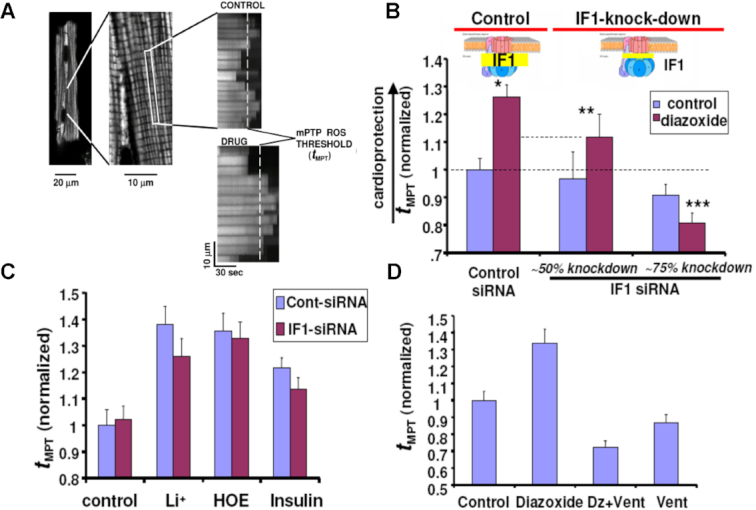
Measurements of the mitochondrial permeability transition ROS threshold (tMPT, the index of cardioprotection) in myocytes. (A) typical positive tMPT effect of a drug is illustrated vs Control. (B) tMPT decreases in proportion to the degree of IF1 knock-down, compared to control cells. (C) GSK-3β-dependent protection signaling which does not require mKATP activated K+ flux (i.e., Li+, HOE, insulin) is unaffected by IF1-knock-down. (D) Block of Fo by Vent prevents mKATP (Dz)-mediated cardioprotection. * P< 0.05 vs paired Control; **, ***P = ns vs paired Control.
Regulation of F1Fo by Bcl-xL and Mcl-1
Suspecting that the effect of Dz and pinacidil via IF1 could be naturally operating under the control of yet-to-be discovered endogenous ligands of IF1 we set out to find them. We examined IF1 for conserved survival protein-related homology domains since IF1 is known to have a “minimal inhibitory domain” sequence of 33 amino acids (AA) that binds to the β-subunit of F1.35 We found that IF1 contains a conserved BH3-like domain (residues 32–46) that significantly overlaps its minimal inhibitory sequence (residues 14–47) (Figure 3A), and that Bcl-xL and Mcl-1, which are each known to have a BH3-binding groove, exert effects comparable to Dz on the H+ and K+ ion currents sustained by ATP synthase (Figures 3B, 4A). Furthermore, the effect of Bcl-xL and remarkably also of Dz, are reversed by a 26 AA peptide consisting of the BH3-domain of Bad (BH3 peptide, known to have nM affinity for Bcl-xL, but 1–2 orders less so for Bcl-236, 37). A single AA substitution in the BH3 peptide (L12A), that reduces the affinity for Bcl-xL by almost 2 orders of magnitude,27 eliminated the inhibitory effects (Figure 4A). Notably, unlike Bcl-xL, Bcl-2 has no effect on F1Fo H+ and K+ ion currents, which agrees with their known affinities for the BH3-domain of Bad, respectively. In binding experiments measuring changes in intrinsic tryptophan fluorescence, we found that Bcl-xL has a high affinity (sub-nM Kd) for the ligand IF1, whereas Bcl-2’s affinity is several orders of magnitude lower (see Supplement, section Protein binding (Kd) measurements). This data suggests that IF1 harbors a functionally-active BH3 domain homologous to that of Bad that overlaps with part of IF1’s inhibitory domain and functions as the area of binding to the β-subunit of F1. Additionally, Bcl-xL and Mcl-1, but not Bcl-2, serve as endogenous regulatory ligands of ATP synthase via interaction with IF1 at the BH3-like domain.
Figure 3.

Regulation of F1Fo current and activity by Dz, Bcl-2 family peptides and proteins mediated by IF1. (A) AA alignment of the 26-residue BAD BH3 peptide and F1Fo inhibitory factor IF1 from mouse, rat, bovine, pig, monkey and human. The consistency of the alignment is indicated in the last row (asterisk shows complete conservation) (see also Figure S4). BH3 peptide L12 aligns with L42 in full length IF1. (B) Effect of Bcl-xL (20 nM) and BH3 peptide (20 nM) on the voltage ramp (from -60 mV to +60 mV) evoked F1Fo currents. Column headers about the red line denote three experimental groups; Bcl-xl and BH3 peptide middle and right columns, respectively; control currents (left column). Bottom traces (left and middle) correspond to 2 mM ATP inhibited F1Fo current.
Figure 4.

Current-time integral of voltage ramp evoked F1Fo currents (A) Augmentation of the current-time integral by Bcl-xL and Dz (30 µM) is reversed by a 26 AA peptide consisting of the BH3-domain of Bad. Control peptide with a single AA substitution L12A has no effect. (B) Reconstitution of F1Fo from IF1–/– mice. Addition of IF1 (100 nM) restores the stimulatory effect of Dz, Bcl-xL and Mcl-1 on the current-time integral of voltage ramp evoked F1Fo currents from IF1–/– mice. The order of addition of IF1 and Dz, Bcl-xL or Mcl-1 is varied among the various groups as indicated. (C) F1Fo activity (ATP synthesis) driven by a H+ or (D) a K+ gradient in PL. (E) ATP production/consumption kinetics (chemiluminescence traces) in a K+ gradient in PL. (F) Dose-response of ATP inhibition of F1Fo (H+) currents and Dz and Bcl-xL activated F1Fo currents: x-axis-(linear) ATP concentration used for inhibition of F1Fo currents, y-axis (log) normalized current-time integral of F1Fo currents. Dz and Bcl-xL produced a parallel shift in the F1Fo activity vs control resulting in the energy of an additional ∼2.8 mM ATP being required to provide sufficient counter-torque to limit F1Fo to the same level of function as under control conditions. The relative change in efficiency was calculated as the ratio of the free energy of ATP hydrolysis during activation by Dz and Bcl-xL over that under basal conditions. * P<0.05, ** P< 0.02.
Regulation of ATP Synthase by Bcl-xL, Mcl-1 and Dz Requires IF1
Thus far, we have discussed the role and function of IF1 in intact cells, organelles and purified single molecules of F1Fo, as well as in IF1-knockdown experiments. Next, we examined the regulation of F1Fo by Bcl-xL, Mcl-1 and Dz in the absence of IF1, and upon reconstitution with IF1. We measured H+ and K+ currents in F1Fo isolated from IF1–/– mice (see Supplement section: Generation of IF1–/– mice, and Figure S2 regarding confirmatory proof of lack of IF1 expression) and the baseline properties of the ionic currents were essentially like that of WT control. Importantly, upon reconstitution with IF1, PH and PK as well as total current reversal potential were found to be unchanged. One notable difference, however, was the ability of graded mM ATP amounts (by the energy transferred via its hydrolysis) to produce sufficient mechanical counter-torque (exerted by F1 on the γ shaft) in excess of the oppositely-directed electrogenic mechanical torque exerted by Fo, causing a net reversal of electrical current in the IF1–/– case (resulting in ATP hydrolysis-generated reverse ion pumping). The latter (i.e., ATP-generated reverse ion pumping) was not observed in parallel experiments with WT and is entirely consistent with the known function of IF1 to limit the waste of futile ATP hydrolysis by impaired mitochondria under circumstances when ΔΨm would drop below levels needed to synthesize ATP.38
Assessing the current-time integral function (CTI, which in the direction of negative current is the direct analog of the amount of ATP synthesized) after reconstitution of F1Fo with IF1 in IF1–/–, we observed a small increase of ∼11–14% in CTI (vs baseline). Thus, it is notable that IF1 does not cause a net inhibitory drag on the energy transfer in F1Fo likely due to frictional losses in the direction of ATP synthesis (see also discussion below regarding BH3 peptide effects). In contrast to WT, neither Dz, Bcl-xL nor Mcl-1 exerted a significant positive augmentation of CTI in the absence of IF1, and subsequent IF1 reconstitution was similarly ineffective (Figure 4B). A likely explanation for the apparent ineffectiveness of IF1 added after Bcl-xL, Mcl-1 or Dz can be given by the possible interference exerted by these molecules on the intrinsic disorder of IF1,39 hindering its interaction at the F1’s binding cleft and γ shaft into a functionally active complex. In one case, the 5-fold excess of IF1 used leaves effectively no free Bcl-xL because of the high affinity of this pair, and presumably only free IF1, rather than bound, can reconstitute into F1Fo. In the case of Dz, this molecule could directly affect the intrinsic disorder of IF1 or the binding cleft preventing effective reconstitution. On the other hand, prior reconstitution of F1Fo with IF1 restores the WT behavior entirely, with Dz, Bcl-xL and Mcl-1 manifesting a robust augmentation of CTI (Figure 4B). Taken together, this data allows us to conclude that IF1 is required for these mediators to augment F1Fo activity (for the same driving force).
As stated earlier, the positive effects of Bcl-xL and Mcl-1 on WT F1Fo currents could be reversed by the BH3 peptide (but not by the L12A variant, null-acting control BH3 peptide; Figure 3). Since this BH3 peptide, as well as IF1, likely binds to the same region of F1-β, but because of its short length is unable to reach to the γ shaft, we examined the functional effects upon binding F1Fo in the absence of IF1. We found that in IF1-deficient F1Fo, the BH3 peptide alone exerts a robust positive effect comparable to that of Dz, Bcl-xL and Mcl-1 (i.e., doubling to tripling the activity; not shown), but the L12A-modified BH3 peptide had no effect, suggesting that IF1 likely produces significant frictional drag via its constitutive contact with the γ shaft that is fully offset by some function-augmenting mechanism achieved by the portion of IF1 bound to F1-β (Figure 4A, B; see below and Discussion).
Regulation of Mechano-Chemical Efficiency of F1Fo
We have shown that F1Fo, conducting univalent cations at a fixed driving energy, Δμ, can be upregulated to increase the total ion flux against a constant load without slip or leak via the IF1-dependent actions of synthetic small molecules such as Dz and pinacidil, and endogenous proteins such as Bcl-xL and Mcl-1. The absence of slip is revealed by complete inhibition of currents at high Δμ by excess ATP. This activity enables ATP synthase to function as a recruitable mKATP, whereby the triggered increase of mitochondrial K+ influx and matrix volume upregulate respiration and produce redox activation of local signaling inhibiting GSK-3β and resulting in desensitization of the mPTP to damaging levels of ROS.4, 40 These data, together with the results showing that Bcl-xL and Dz (and pinacidil and Mcl-1) are each capable of increasing the amount of ATP synthesized by reconstituted F1Fo (WT, IF1-competent) utilizing either K+ or H+ gradients (Figure 4C–E), suggest that these IF1-dependent effectors have increased the mechano-chemical efficiency of the ATP synthase. To investigate this, we examined the titration curve of the CTI (at each of the ion-reversal potentials for H+ and K+, in single ATP synthase molecules) as a function of the counter-torque on the γ shaft applied by F1 resulting from the hydrolysis energy derived from increasing ATP concentrations, in the presence of Dz or Bcl-xL as compared to controls. The data obtained are well described by a log-linear relationship between CTI and [ATP]. Dz and Bcl-xL produced a parallel upward shift of 5.6-fold in the F1Fo activity vs control (Figure 4F) indicating that the hydrolysis energy of an additional ∼2.8 mM ATP is required to provide sufficient counter-torque to constrain the F1Fo to the same level of function as under control conditions. Based on considerations of energy conservation, the additional ATP synthesis might be driven by extra energy that was not lost to viscous drag and intermolecular friction. Together, these results agree with the idea that both Bcl-xL and Dz increase the mechano-chemical efficiency of ATP synthase (e.g., by ∼7% at 1 mM, ∼5% at 2 mM, and ∼3% at 4 mM ambient ATP).
Comparing Pharmacological and Electrophysiological Properties of mKATP Channel and Mitochondrial ATP Synthase
The mitochondrial KATP channel described in the context of myocardial preconditioning and cardioprotection32, 41 displays a typical sensitivity profile to specific activators (e.g., diazoxide and pinacidil) and inhibitors (e.g., glybenclamide, 5-hydroxydecanoate (5 HD), Tertiapin Q [nominally for ROMK/mKATP], and ATP). When the action of those inhibitors and activators was tested on the activity of ATP synthase, based on the prediction that they should exert these same effects as observed for mKATP, all of them display the same pharmacological profile (summarized in Table 1). In the case of the activators their effect on ATP synthase has been described above to require IF1 (see also Figures 1, 2, 4). This IF1-dependent property was demonstrated in single-molecule bioenergetics and reconstituted proteoliposome experiments, and in the protection of cardiomyocytes against oxidative stress-induction of the permeability transition pore (Figure 1). Additionally, the well-characterized sensitivity of ATP synthase to the inhibition by oligomycin and venturicidin A, led to the novel prediction that they should exert similar actions on the mKATP entity. Both inhibitors were shown to inhibit the activity of mito KATP channels in single molecule reconstitution experiments. Remarkably, the permeability for K+ and H+ measured in both the mKATP channel (our analysis1 of electrophysiological data extracted from42–49) and ATP synthase1 display the same values, respectively, for the conduction of each of these cations in both of these molecular entities.
Table 1.
Property and function comparison of ATP synthase vs “mitoKATP”
| Single molecule & reconstituted§ | Cardiomyocytes/Mitochondria | ||||||
|---|---|---|---|---|---|---|---|
| Mito KATP £ | ATP synthase | Cardioprotection | Mitochondria† | ||||
| Control | IF1–/– | Control | IF1 knockdown | Volume | Respiration¶ | ||
| KATPActivators | |||||||
| Diazoxide |
 6,
42,
49,
62,
63
6,
42,
49,
62,
63
|
 ⁑,
1,
64
⁑,
1,
64
|
 ⁑,
1
⁑,
1
|
⁑,
4,
6,
16,
65–69
|
⁑,
70
|
⁑,
4,
71,
72
|
⁑,
4,
9,
16,
32,
71,
73,
74
|
| Pinacidil |
42
|
⁑,
1
|
⁑,
1
|
⁑,
4,
41,
66,
68,
69,
75
|
⁑
|
4,
72
|
32,
72,
73,
76
|
| KATPInhibitors | |||||||
| 5HD§§ |
6,
42,
49,
62
|
⁑,
1
|
⁑,
4,
6,
16,
66–69,
77
|
⁑,
4,
71,
72
|
⁑,
4,
9,
16,
32,
71,
72,
74
|
||
| Glybenclamide§§ |
6,
42,
62,
78
|
1
|
6,
41,
69,
71,
75
|
⁂,
62
|
14,
71
|
||
| Tertiapin Q†† |
17
|
1‡
|
65
|
||||
| ATP |
18,
42,
62
|
⁑,
1
|
71
|
71
|
|||
| ATP synthase Inhibitors | |||||||
| Oligomycin |
(not shown) |
1
|
79
|
1,
80
|
|||
| Venturicidin |
1
|
⁑,
1,
48
|
⁑
|
⁑
|
⁑
|
||
| Mg2+ |
43,
49
|
81,
82
|
|||||
| Permeabilities | |||||||
| PK (m3 s–1) * | 1.9 ± 0.5 × 10–16 1, 42–49 | 1.3 ± 0.3 × 10–16 1 | |||||
| PH (m3 s–1) * | 4.9 ± 1.1 × 10–11 ‡‡1 | 5.2 ± 0.9 × 10–11 1 | |||||
Red symbols and text denote correctly predicted experimental results based on the original proposition that ATP synthase and “mitoKATP” have the same properties and functions.
proteoliposomes containing reconstituted mitoKATP or reconstituted, purified ATP synthase
in situ or isolated
this work
 no effect
no effect
Putative ROMK inhibitor, although Papanicolaou et al. 83 demonstrated that ROMK is likely dispensable to the function of the cardioprotective mitoKATP channel.
at 1 nM (data not shown). Tertiapin Q exhibits nanomolar potency, however, limited specificity, inhibiting multiple Kir channel isoforms (Kir1.1 and Kir3.x) as well as KCa channels 84.
5HD and glybenclamide inhibit activation of mitoKATP by diazoxide or pinacidil
brain compared to heart mitochondria
either respiration as measured directly or inferred from the dynamics of flavoprotein redox state 9,32
P = ns for mitoKATP vs ATP synthase data comparison
Overall, the fact that all the pharmacological and ion-conduction characteristics applied for the characterization of the mKATP channel, are fully recapitulated by the ATP synthase, and vice versa, at the molecular, organelle and cellular levels leads us to establish that the ATP synthase is fully sufficient to serve the principal function(s) of the mKATP channel.
Discussion
Recently, we demonstrated that ATP synthase serves the functions of a primary mitochondrial K+ “uniporter,” i.e., the primary way for K+ to enter mitochondria. This K+ entry is directly proportional to ATP synthesis and regulates matrix volume while driving the matching between energy supply and demand.1 In the present work, we show that the chemo-mechanical efficiency of F1Fo ATP synthase is endogenously upregulated by members of the survival proteins family, Bcl-xL and Mcl-1, but not Bcl-2, acting via IF1, an intrinsic regulatory factor of ATP synthase. This regulation is similar to the pharmacological action by certain K+ channel openers, that acting via IF1, increase the monovalent cation conductance of F1Fo while retaining its high degree H+-selectivity. We show that F1Fo, conducting H+ and K+, can be upregulated (even at the same driving energy, Δμ) to increase the total ion-flux (at constant H+: K+) against a constant load without slip or leak via the IF1-dependent actions of endogenous pro-survival proteins, Bcl-xL and Mcl-1, and of synthetic small molecules, Dz and pinacidil (Figures 3, 4, 5C).
Figure 5.

Scheme of the H+ and K+ transport across the inner mitochondrial membrane. All the energy available for work and to drive ionic movements derives from the original H+ gradient established by proton pumps in the respiratory chain (see also1). A central point is the obligatory preservation of charge and mass balance under the steady state circuits. Panel A, displays the “original view of cation flux cycles” in which the H+ gradient is being harnessed by F1Fo directly to make ATP, whereas a certain amount of K+ enters the matrix through an ordinary K+ channel mechanism (a “mKATP-uniporter” channel), driven by ΔΨ, and extruded via KHE utilizing the energy remaining in the fraction of the H+ gradient not directly harnessed by F1Fo. The equivalent energy of this fraction being used to extrude K+, and a large fraction of that non-ATP-producing energy would essentially be dissipated as heat in the constant cycle of K+ recirculation. Panel B displays the new mechanism in which the same amount of energy available in the original H+ gradient is entirely available to produce ATP, simply by having the mKATP-uniporter mechanism reside inside, and as natural part of, F1Fo with the traffic of H+ or K+ contributing its energy to producing ATP. The remainder of the H+ gradient energy is now utilized to remove all the K+ that entered via F1Fo. However, the gain is that more ATP is produced for the same input energy by not wasting some of that energy on maintaining what was originally thought to be a separate K+ cycle that does not/cannot generate any ATP. This way is a better, tightly coupled system of energy supply-demand matching through the K+ cycle utilizing F1Fo because the matrix influx of K+ is truly directly proportional to ATP synthesis. Any transient increase in F1Fo activity will thus lead to transient K+ accumulation. This will lead to the attraction of a counter-ion and change of the osmotic drive yielding a “volume-activation of respiration” response which previously has been documented in detail.4 The scheme depicted in (C) integrates the implications of modestly enhancing the chemo-mechanical efficiency of F1Fo (by KCO's or Bcl-xL/Mcl-1). For the driving energy of the same H+ gradient the F1Fo flux increases, enabling increased respiration and a directly increased K+ flux cycle (yielding an increased volume signal) and enhanced ATP generation (C) vs the basal conditions (B).
These studies complement the main original finding of our recent work1 demonstrating that F1Fo ATP synthase utilizes both H+- and K+-transport (because of > 106-fold K+ excess vs H+) to drive ATP synthesis in spite of a H+: K+ permeability of ∼106:1. Thus, we discovered that ATP synthase also functions as a recruitable mitochondrial ATP-dependent K+ “channel” which serves critical functions in cell protection signaling that can limit the damage of ischemia-reperfusion injury. By harnessing ΔμK, driven essentially by ΔΨm, and continuously converted (restored) from respiratory chain-generated ΔμH through the activity of the KHE (Figure 5), F1Fo generates additional ATP proportional to the amount of energy that would have been dissipated as heat by the same K+ current in passing (in a hypothetical scenario) through a separate entity functioning only as a K+ uniporter. In other words, letting K+ enter via a non-ATP generating process would not be as energetically effective as using the F1Fo as the K+-influx mechanism (Figure 5B, C). Thus, once the K+ is eventually extruded by the KHE using H+ influx, the equivalent energy of that H+ will have been harnessed in form of ATP made by the K+ influx through the F1Fo (Figure 5B).
The K+ uniporter function also enables F1Fo to operate as an on-demand, recruitable mKATP, whereby triggered increases of mitochondrial K+-influx and matrix-volume upregulate the signaling cascade resulting in desensitization of the mPTP, enhancing cell survival.4Table 1 summarizes that all the pharmacological and ion-conduction characteristics applied for the characterization of the mKATP channel and ATP synthase are concordant and fully align in the ability to predict positive (and inhibitory) regulatory changes in cardioprotection, mitochondrial volume and respiration. Not only do the effectors of mKATP exert the same action on ATP synthase activity, but we have shown that the converse is also true: the known functional and pharmacological properties of ATP synthase are also predicted, and without exception, confirmed to be true for the entity conventionally isolated as the mKATP. For example, the well-characterized sensitivity of ATP synthase to the inhibition by oligomycin and venturicidin A, led to the key—but previously unanticipated—novel prediction that they should exert similar actions on the mKATP entity. Both inhibitors were indeed shown to inhibit the activity of mKATP channel. Furthermore, these ATP synthase inhibitors acted in the manner of the classical mKATP inhibitors, by eliminating the ability of K+ channel activators (diazoxide and pinacidil) to exert positive regulatory changes in cardioprotection, mitochondrial volume and respiration.
As reported in our previous publication1 the permeability for K+ and H+ measured in both the mKATP channel and ATP synthase display the same values, respectively, for the conduction of each of these cations in both of these molecular entities. In fact, since ATP synthase is a naturally proton-conducting machine, given the logic described above, we predicted that mKATP must also have similar proton conducting properties. Not only is the existence of the previously unanticipated and undiscovered proton permeability for this entity remarkable, but even more importantly, the magnitude is large and, furthermore, the same as for ATP synthase (Table 1). Overall, considering the weight of solid, prediction-based evidence from the correspondence of all the pharmacological interventions and ion-conduction characteristics applied for the characterization of the mKATP channel, being fully reiterated by the ATP synthase, and vice versa, at the molecular, organelle and cellular levels, leads us to conclude that the ATP synthase is the entity which serves the functions of mKATP. Nevertheless, Nature usually operates important pathways with built-in redundancy so that other mitochondrial K+ channels may contribute to these mechanisms. A recently described protein complex which may also mimic mKATP channel function19 might play a role in this context, but because of the significantly energy-dissipative nature of such ordinary channel function on the inner mitochondrial membrane, these pathways are likely fine-tuning mechanisms. Futhermore, our immunoblotting with ABCB8, CCDC51 (Figure S3) and ROMK1 antibodies ruled out contamination of the isolated F1Fo with these alternative mKATP channel candidates.
Because a transient change in K+ influx would need to be matched by influx (or retention) of a counter-ion (e.g., Cl–) to produce an osmotic imbalance signal, both KHE and the counter-ion transport pathways are also important control steps in matrix volume regulation (Figure 1F-K;4). Dysfunction of mitochondrial KHE activity leads to aberrations in matrix K+ and mitochondrial volume regulation that in turn may affect fission/fusion and mitophagy. Such pathology is evident in the Wolf-Hirschhorn syndrome, a genetic insufficiency of mitochondrial KHE activity ( 1/50000 incidence), characterized by microcephaly, growth retardation, intellectual disability, and epileptic seizures among other severe manifestations.50
Our data also unveil that F1Fo operates at increased efficiency (by up to ∼7% at normal ATP levels) in response to KCOs, Bcl-xL and Mcl-1, yielding both increased ATP output and matrix K+ influx for the same ΔμH (Figure 4; depicted in 5C). Dz and Bcl-xL cause a rightward shift in the ATP-dependence of the CTI (a quantitative index of ATP synthesis), such that the hydrolysis energy of an additional ∼2.8 mM ATP is required to provide enough counter-torque to constrain the F1Fo to the same level of function as in controls. This means that an additional ∼2.8 mM ATP can be produced for the same input energy at normal ambient levels of ATP. This provides quantitative proof that both Bcl-xL and Dz increase the mechano-chemical efficiency of F1Fo (Figure 4F).
Previous work found that Bcl-xL interacts with the F1Fo,30, 51 specifically with the β-subunit of ATP synthase decreasing an ion leak within the F1Fo complex and concluded that this was responsible for increasing net transport of H+ by F1Fo.30 These latter findings and conclusions are non-trivially different from our experiments: (i) we do not observe ion leak (or slip) at all, regulated or otherwise, in F1Fo in the presence or absence of Bcl-xL, i.e., Bcl-xL does not inhibit an ion leak that is not present in ATP synthase, and (ii) the increase in ATP synthetic capacity in response to Bcl-xL is specifically due to an increase in mechano-chemical efficiency of ATP synthase per se, and not by changing an ion leak into useful energy. This evidence leads us to conclude that essential mitochondrial homeostatic and pro-survival mechanisms result from a regulated IF1-mediated increase in chemo-mechanical efficiency of F1Fo conducting both K+ and H+. Our results add a significant dimension to the known, and apparently diverse biological function sets of F1Fo. Additionally, it was proposed that a certain triggered rearrangement of F1Fo dimers is functionally responsible for other major biological functions such as the mitochondrial cristae arrangements52 and possibly the formation of the mPTP.53, 54
Our findings raise the question of how IF1 might control the activity of ATP synthase to engage physiologic/homeostatic and survival-promoting mechanisms. Overall, our data are consistent with a “minimal inhibitory domain” of IF1 (residues 14–47 in bovine IF1 55) binding to the β-subunit of F1 in an “IF1 ligand-binding cleft” (adjacent to the F1 α-subunit interface), forming at its proximal end an α-helix loop that interacts with the F1 γ-rotor shaft which is responsible for limiting ATPase activity. With the evidence of a significant modulatory role by certain Bcl-2 members, we examined this domain for conserved survival protein-related homology domains. Bcl-xL and Mcl-1 are each known to have a BH3-binding groove with high affinity for certain domains of BH3. Together with the result of the high affinity binding of IF1 to Bcl-xL, our data agrees with IF1 harboring a functionally-active BH3-like domain homologous to that of Bad and coincident with IF1’s inhibitory domain that functions as the binding patch to the β-subunit of F1. Binding of Bcl-xL and Mcl-1, but not Bcl-2, via IF1 interaction, endogenously regulate F1Fo activity. This may explain why the effects of Bcl-xL, Mcl-1, and Dz, are reversed by the BH3 peptide, but not by the same peptide with a single AA change (L12A)27, 37 (Figures 3 and 4A). Specifically, the BH3 peptide may compete and displace IF1 from its binding site on F1Fo, as well as interfere with its binding to Bcl-xL or Mcl-1. Moreover, we have shown that, unlike in WT, neither Dz nor Bcl-xL significantly increased CTI in F1Fo from IF1–/–. Alternatively, prior reconstitution of F1Fo in the presence of IF1 entirely rescued the WT behavior, with both Dz and Bcl-xL strongly augmenting the ion currents (Figure 4B). These data allow us to conclude that the higher ATP synthase activity elicited by these effectors (for the same driving force) requires IF1, and that the mere removal from its binding site does not suffice to enhance the enzyme activity. We propose that in the normal basal state IF1 has two mechanical and nearly offsetting effects on the function of ATP synthase operating in the synthesis direction: (i) a net negative, frictional drag-like effect of the IF1 molecule originating at its proximal end where it engages the γ shaft in its natural rotation, and (ii) a net positive effect created somehow by the presence of the long α-helical stretch that engages the IF1 binding cleft on F1-β, the latter effect being mimicked by the BH3-peptide. It has been shown that Bcl-xL can interact forming 3D-domain swapped (3DDS) homodimers56 as well as heterodimers with other survival-regulating proteins. These interactions can significantly affect the residual function of both partners,57 and certain BH3-only proteins can bind to and partially unfold Bcl-xL, changing its interactions with other binding partners and thereby biasing cell survival-signaling.58 Thus, our two-fold proposal implies that (i) Bcl-xL/Mcl-1 (via their intrinsic BH3-binding grooves) tightly bind to IF1 at its minimal inhibitory/BH3-like domain to displace it from its binding cleft at F1-β, and (ii) this interaction triggers a specific unfolding and rearrangement of the Bcl protein's α2 helix, enabling an increase of its potential range-of-motion. This could allow the helix from the Bcl protein to participate in an energetically favorable rearrangement with F1Fo by binding to the empty IF1 binding cleft. We propose a possible model of this interaction (Bcl-protein's α2 helix containing its BH3 domain engaging the IF1 binding cleft on F1-β; Figure 6A-D) that would cause the Bcl-xL/Mcl-1-mediated increase of F1Fo function in the presence of IF1, analogous to that obtained with the BH3 peptide added to the IF1 deficient F1Fo (Figure 4B). The mechanism by which a short IF1/BH3-(like) helical-peptide structure occupying the natural IF1 binding groove can enhance the chemo-mechanical efficiency of F1Fo is of considerable interest, but how it specifically works remains a matter of future study.
Figure 6.

Interaction of F1 ATPase with IF1 or a BH3 modeled peptide. (A) Ribbon model of the crystal structure of bovine F1 (PDB ID 4Z1M) emphasizing two of the three β subunits (cyan and magenta) and the γ subunit (blue) in interaction with the long α-helix of the inhibitor protein IF1 (orange). The IF1 domain (residues 18–51 are shown in cyan; residues 23–70 from PDB ID 1GMJ are shown in orange) interacts with the β subunit marked in yellow. (B) Surface representation of subunits β (yellow), γ (blue) and α (magenta) of F1 ATPase interacting with IF1 peptide. (C) As panel (B) with the α2-helix peptide containing BH3 domain from the BAD protein (PDB ID 1G5J, as it binds Bcl-xL) in ribbon representation at the IF1 groove in F1 showing the aliphatic side chain of Leu 42 in IF1 and Leu 12 in BH3-BAD peptide (corresponding to Leu114 in human BAD). (D) Same as (C) at an approximately 90° orientation. (E) Phylogenetic tree of the BH3 extended peptides (35 residues) from Bcl-2 proteins and IF1 across eukaryotes. Sequence alignment was computed with Clustal Omega61 and the tree and divergence times (in Myr) were calculated by MEGA 6.0 (for further details, see Supplemental Information; Figures S4 and S5, and Table S1).
The origin of IF1 in relation to the evolution of F1Fo is also an interesting question. There are conserved “IF1 domains” that can be found embedded in a variety of larger proteins across Archaea, Bacteria, and Eukaryotes,59 suggesting ancient origins for this domain. Although F1Fo exists in all major lifeforms, IF1 as a separate entity is only known to regulate synthase function in Eukaryotes. It is tempting to speculate that when the early bacterium became a mitochondrion as a functional organelle of the eukaryotic cell, some 2 billion years ago, it brought along the genetic information for IF1, which might have evolved to prevent the mitochondrion from wasteful ATP consumption in the host cell. We examined these bacterial IF1-progenitors and they have regions homologous to the BH3-like domains that we found in eukaryotic IF1’s. The Bcl family is also ancient, some 2 billion years extant and resident in eukaryotic lifeforms. Bayesian phylogenetic analysis shows that IF1 is an ancient member of the Bcl family and today may be most closely related to BH3-containing proteins (e.g., Bad, PUMA, Bcl-xL; Figure 6E). This may explain how the Bcl-2 protein family has come to regulate F1Fo function as part of its repertoire of survival-regulating functions.
In conclusion, we demonstrated that mitochondrial ATP synthase utilizes the ion gradient energy not only of H+ but also of K+ to drive ATP synthesis, what is likely to be the primary mechanism by which mitochondrial function matches energy supply with demand for all cells in the body. The essential mitochondrial homeostatic and pro-survival mechanisms discussed here, including F1Fo operation as a primary mitochondrial K+ uniporter to facilitate energy supply-demand matching, and as a recruitable mKATP channel to protect from pathological opening of the mPTP, result from regulated function of ATP synthase conducting both K+ and H+. For the first time, to our knowledge, we have shown that the chemo-mechanical efficiency of ATP synthase can be up-regulated, and that this occurs by certain members of the Bcl-2 family and by certain K+ channel openers acting via an intrinsic regulatory factor of ATP synthase, IF1, which we identified as itself a novel and previously unrecognized member of the Bcl-2 protein family. The specific mechanisms by which KCOs and certain Bcl-2 family proteins engage IF1 to produce an increase in the chemo-mechanical efficiency of ATP synthase will require additional investigation.
Methods
Detailed methods are provided in the the Supplemental Information Section of this manuscript.
Cell Isolation, and Purification, Characterization and Reconstitution of F1Fo
Myocytes were isolated from neonatal and adult rat, and mouse hearts by enzymatic dissociation. Mice carrying an inactivated Atpif1 allele were obtained from the European Mouse Mutant Archive (EMMA), bred to homozygosity and are referred to as IF1–/– throughout the text.
F1Fo was purified according to manufacturer protocol (Mitosciences) and reconstituted into liposomes and planar lipid membranes. Isolated F1Fo was characterized by gel electrophoresis using the Novex Bis-tris gel systems under native and denaturing conditions following the manufacturer protocol (Invitrogen).
ATP Measurements
Bioluminescent assays which employ the luciferin-luciferase ATP-dependent reaction were used to evaluate the ATP production by PL.
Electrophysiological Measurements
The Planar Lipid Bilayer workstations (Warner Instruments) were used to characterize electrophysiological properties of the reconstituted F1Fo.
Confocal Microscopy Experiments
mPTP ROS-threshold induction, mitochondrial volume determination, FP autofluorescence measurements, and immunoflourescence microscopy were performed as described before.4, 60
Statistics
All experiments were performed at least in triplicate, with cell number greater than 12 in each independent experiment unless stated otherwise. All data are mean ± SEM. Comparisons within groups were made by an appropriate one-way ANOVA or Student t test, and P value ˂0.05 was considered as statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank E.G. Lakatta for useful discussions, D. Boyer for animal husbandry, L. Rezanka for mice genotyping and M.J. del Hierro Sanchez for assistance in obtaining the transgenic IF1–/– mice. This work was supported entirely by the Intramural Research Program, National Institute on Aging, NIH.
Notes
Preprint of this paper is available in bioRxiv: Version 2, April 22, 2019, doi: https://doi.org/10.1101/355776
Contributor Information
Magdalena Juhaszova, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Evgeny Kobrinsky, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Dmitry B Zorov, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA; A.N. Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992, Moscow, Russia.
H Bradley Nuss, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Yael Yaniv, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Kenneth W Fishbein, Laboratory of Clinical Investigation, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Rafael de Cabo, Translational Gerontology Branch, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Lluis Montoliu, National Centre for Biotechnology (CNB-CSIC), Biomedical Research Networking Center on Rare Diseases (CIBERER-ISCIII), 28049 Madrid, Spain.
Sandra B Gabelli, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA; Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA; Department of Biophysics and Biophysical Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Miguel A Aon, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Sonia Cortassa, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Steven J Sollott, Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, MD 21224, USA.
Author contributions
Conceptualization, M.J., D.B.Z. and S.J.S.; Methodology, M.J., E.K., H.B.N., K.W.F., L.M., M.A.A., S.C. and S.J.S.; Software, Y.Y., S.B.G. and S.C.; Formal Analysis, Y.Y., S.B.G., S.C. and S.J.S.; Investigation, M.J., E.K., D.B.Z., H.B.N., M.A.A. and S.C.; Resources, R.dC., L.M., S.B.G. and S.J.S.; Writing-Original Draft, S.J.S.; Writing-Review & Editing, M.J., E.K., D.B.Z., K.W.F., R.dC., S.B.G., M.A.A., S.C. and S.J.S.; Visualization, M.J., E.K., Y.Y., S.B.G., M.A.A., S.C. and S.J.S.; Supervision, S.J.S.
Competing Interest Statement
The authors declare that they have no conflict of interest.
Data Availability
All study data are included in this article and/or in its Supplemental Information. Any other data request will be shared by the corresponding author, Dr. Sollott SJ, upon reasonable request
References
- 1.Juhaszova M, Kobrinsky E, Zorov DBet al. ATP synthase K+- and H+-flux drive ATP synthesis and enable mitochondrial K+-uniporter function: I. Characterization of ion fluxes. Function. 2021. 10.1093/function/zqab065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garlid KD, Dos Santos P, Xie ZJ, Costa AD, Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K(+) channel in cardiac function and cardioprotection. Biochim Biophys Acta. 2003;1606(1-3):1–21. [DOI] [PubMed] [Google Scholar]
- 3.Juhaszova M, Wang S, Zorov DBet al. The identity and regulation of the mitochondrial permeability transition pore: where the known meets the unknown. Ann NY Acad Sci. 2008;1123(1):197–212.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18375592. [DOI] [PubMed] [Google Scholar]
- 4.Juhaszova M, Zorov DB, Kim SHet al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113(11):1535–1549.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15173880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ.. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83(2):213–225.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19447775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garlid KD, Paucek P, Yarov-Yarovoy Vet al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81(6):1072–1082.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9400389 [DOI] [PubMed] [Google Scholar]
- 7.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM.. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning?. Cardiovasc Res. 2002;55(3):534–543.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12160950 [DOI] [PubMed] [Google Scholar]
- 8.Caparrelli DJ, Cattaneo SM 2nd, Bethea BTet al. Pharmacological preconditioning ameliorates neurological injury in a model of spinal cord ischemia. Ann Thorac Surg. 2002;74(3):838–844.; discussion 844-835. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12238848 [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Sato T, O'Rourke B, Marban E.. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection?. Circulation. 1998;97(24):2463–2469.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9641699 [DOI] [PubMed] [Google Scholar]
- 10.Kis B, Rajapakse NC, Snipes JA, Nagy K, Horiguchi T, Busija DW.. Diazoxide induces delayed pre-conditioning in cultured rat cortical neurons. J Neurochem. 2003;87(4):969–980.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14622127 [DOI] [PubMed] [Google Scholar]
- 11.Lotz C, Herrmann J, Notz Q, Meybohm P, Kehl F.. Mitochondria and Pharmacologic Cardiac Conditioning-At the Heart of Ischemic Injury. Int J Mol Sci. 2021;22(6):3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armstrong SC, Kao R, Gao Wet al. Comparison of in vitro preconditioning responses of isolated pig and rabbit cardiomyocytes: effects of a protein phosphatase inhibitor, fostriecin. J Mol Cell Cardiol. 1997;29(11):3009–3024.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9405176 [DOI] [PubMed] [Google Scholar]
- 13.Ganote CE, Armstrong SC.. Effects of CCCP-induced mitochondrial uncoupling and cyclosporin A on cell volume, cell injury and preconditioning protection of isolated rabbit cardiomyocytes. J Mol Cell Cardiol. 2003;35(7):749–759.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12818565 [DOI] [PubMed] [Google Scholar]
- 14.Jaburek M, Yarov-Yarovoy V, Paucek P, Garlid KD.. State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J Biol Chem. 1998;273(22):13578–13582.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9593694 [PubMed] [Google Scholar]
- 15.Lim KH, Javadov SA, Das M, Clarke SJ, Suleiman MS, Halestrap AP.. The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J Physiol. 2002;545(Pt 3):961–974.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12482899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato T, Sasaki N, O'Rourke B, Marban E.. Adenosine primes the opening of mitochondrial ATP-sensitive potassium channels: a key step in ischemic preconditioning?. Circulation. 2000;102(7):800–805.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10942750 [DOI] [PubMed] [Google Scholar]
- 17.Foster DB, Ho AS, Rucker Jet al. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ Res. 2012;111(4):446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mironova GD, Negoda AE, Marinov BSet al. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). J Biol Chem. 2004;279(31):32562–32568.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15138282 [DOI] [PubMed] [Google Scholar]
- 19.Paggio A, Checchetto V, Campo Aet al. Identification of an ATP-sensitive potassium channel in mitochondria. Nature. 2019;572(7771):609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu W, Liu Y, Wang Set al. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science. 2002;298(5595):1029–1033.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12411707 [DOI] [PubMed] [Google Scholar]
- 21.Cabezon E, Runswick MJ, Leslie AG, Walker JE.. The structure of bovine IF(1), the regulatory subunit of mitochondrial F-ATPase. EMBO J. 2001;20(24):6990–6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Contessi S, Haraux F, Mavelli I, Lippe G.. Identification of a conserved calmodulin-binding motif in the sequence of F0F1 ATPsynthase inhibitor protein. J Bioenerg Biomembr. 2005;37(5):317–326.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16341776 [DOI] [PubMed] [Google Scholar]
- 23.Brocheriou V, Hagege AA, Oubenaissa Aet al. Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J Gene MedJ Gene Med. 2000;2(5):326–333. [DOI] [PubMed] [Google Scholar]
- 24.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH.. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280(5):H2313–2320. [DOI] [PubMed] [Google Scholar]
- 25.Gustafsson AB, Gottlieb RA.. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2007;292(1):C45–51. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu S, Konishi A, Kodama T, Tsujimoto Y.. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A. 2000;97(7):3100–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang JL, Zhang ZJ, Choksi Set al. Cell permeable Bcl-2 binding peptides: a chemical approach to apoptosis induction in tumor cells. Cancer Res. 2000;60(6):1498–1502.. http://www.ncbi.nlm.nih.gov/pubmed/10749111. [PubMed] [Google Scholar]
- 28.Gotow T, Shibata M, Kanamori Set al. Selective localization of Bcl-2 to the inner mitochondrial and smooth endoplasmic reticulum membranes in mammalian cells. Cell Death Differ. 2000;7(7):666–674. [DOI] [PubMed] [Google Scholar]
- 29.Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ.. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348(6299):334–336. [DOI] [PubMed] [Google Scholar]
- 30.Alavian KN, Li H, Collis Let al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13(10):1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halestrap AP. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim Biophys Acta. 1989;973(3):355–382.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2647140 [DOI] [PubMed] [Google Scholar]
- 32.Sato T, O'Rourke B, Marban E.. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase C. Circ Res. 1998;83(1):110–114.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9670924 [DOI] [PubMed] [Google Scholar]
- 33.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ.. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104(11):1240–1252.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19498210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korge P, Honda HM, Weiss JN.. K+-dependent regulation of matrix volume improves mitochondrial function under conditions mimicking ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2005;289(1):H66–H77.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15764674 [DOI] [PubMed] [Google Scholar]
- 35.Gledhill JR, Montgomery MG, Leslie AG, Walker JE.. How the regulatory protein, IF(1), inhibits F(1)-ATPase from bovine mitochondria. Proc Natl Acad Sci USA. 2007;104(40):15671–15676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petros AM, Medek A, Nettesheim DGet al. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci USA. 2001;98(6):3012–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelekar A, Chang BS, Harlan JE, Fesik SW, Thompson CB.. Bad is a BH3 domain-containing protein that forms an inactivating dimer with Bcl-XL. Mol Cell Biol. 1997;17(12):7040–7046.. http://www.ncbi.nlm.nih.gov/pubmed/9372935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker JE. The regulation of catalysis in ATP synthase. Curr Opin Struct Biol. 1994;4(6):912–918.. http://nihlibrarysfx.nih.gov:9003/sfx_local?sid=Entrez%3APubMed&id=pmid%3A7712295. [DOI] [PubMed] [Google Scholar]
- 39.Bason JV, Montgomery MG, Leslie AG, Walker JE.. Pathway of binding of the intrinsically disordered mitochondrial inhibitor protein to F1-ATPase. Proc Natl Acad Sci USA. 2014;111(31):11305–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zorov DB, Juhaszova M, Sollott SJ.. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armstrong SC, Liu GS, Downey JM, Ganote CE.. Potassium channels and preconditioning of isolated rabbit cardiomyocytes: effects of glyburide and pinacidil. J Mol Cell Cardiol. 1995;27(8):1765–1774. [DOI] [PubMed] [Google Scholar]
- 42.Ardehali H, Chen Z, Ko Y, Mejia-Alvarez R, Marban E.. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc Natl Acad Sci USA. 2004;101(32):11880–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bednarczyk P, Dolowy K, Szewczyk A.. Matrix Mg2+ regulates mitochondrial ATP-dependent potassium channel from heart. FEBS Lett. 2005;579(7):1625–1632. [DOI] [PubMed] [Google Scholar]
- 44.Bednarczyk P, Kicinska A, Kominkova V, Ondrias K, Dolowy K, Szewczyk A.. Quinine inhibits mitochondrial ATP-regulated potassium channel from bovine heart. J Membr Biol. 2004;199(2):63–72. [DOI] [PubMed] [Google Scholar]
- 45.Inoue I, Nagase H, Kishi K, Higuti T.. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352(6332):244–247. [DOI] [PubMed] [Google Scholar]
- 46.Kicinska A, Swida A, Bednarczyk Pet al. ATP-sensitive potassium channel in mitochondria of the eukaryotic microorganism Acanthamoeba castellanii. J Biol Chem. 2007;282(24):17433–17441. [DOI] [PubMed] [Google Scholar]
- 47.Kulawiak B, Bednarczyk P.. Reconstitution of brain mitochondria inner membrane into planar lipid bilayer. Acta Neurobiol Exp (Wars). 2005;65(3):271–276.. http://www.ncbi.nlm.nih.gov/pubmed/16130801. [DOI] [PubMed] [Google Scholar]
- 48.Miedema H, van Walraven HS, de Boer AH.. Potassium selective and venturicidin sensitive conductances of Fo purified from bovine heart mitochondria, reconstituted in planar lipid bilayers. Biochem Biophys Res Commun. 1994;203(2):1005–1012.. http://www.ncbi.nlm.nih.gov/pubmed/8093018. [DOI] [PubMed] [Google Scholar]
- 49.Zhang DX, Chen YF, Campbell WB, Zou AP, Gross GJ, Li PL.. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ Res. 2001;89(12):1177–1183.. http://www.ncbi.nlm.nih.gov/pubmed/11739283. [DOI] [PubMed] [Google Scholar]
- 50.Zotova L, Aleschko M, Sponder Get al. Novel components of an active mitochondrial K(+)/H(+) exchange. J Biol Chem. 2010;285(19):14399–14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Formentini L, Pereira MP, Sanchez-Cenizo Let al. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J. 2014;33(7):762–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strauss M, Hofhaus G, Schroder RR, Kuhlbrandt W.. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27(7):1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giorgio V, Stockum S, Antoniel Met al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110(15):5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carraro M, Carrer A, Urbani A, Bernardi P.. Molecular nature and regulation of the mitochondrial permeability transition pore(s), drug target(s) in cardioprotection. J Mol Cell Cardiol. 2020;144:76–86. [DOI] [PubMed] [Google Scholar]
- 55.van Raaij MJ, Orriss GL, Montgomery MGet al. The ATPase inhibitor protein from bovine heart mitochondria: the minimal inhibitory sequence. Biochemistry. 1996;35(49):15618–15625. [DOI] [PubMed] [Google Scholar]
- 56.O'Neill JW, Manion MK, Maguire B, Hockenbery DM.. BCL-XL dimerization by three-dimensional domain swapping. J Mol Biol. 2006;356(2):367–381. [DOI] [PubMed] [Google Scholar]
- 57.Rajan S, Choi M, Nguyen QTet al. Structural transition in Bcl-xL and its potential association with mitochondrial calcium ion transport. Sci Rep. 2015;5(1):10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Follis AV, Chipuk JE, Fisher JCet al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat Chem Biol. 2013;9(3):163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geer LY, Domrachev M, Lipman DJ, Bryant SH.. CDART: protein homology by domain architecture. Genome Res. 2002;12(10):1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ.. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–1014.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11015441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sievers F, Wilm A, Dineen Det al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7(1):539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bajgar R, Seetharaman S, Kowaltowski AJ, Garlid KD, Paucek P.. Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J Biol Chem. 2001;276(36):33369–33374. [DOI] [PubMed] [Google Scholar]
- 63.Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA.. The mitochondrial KATP channel as a receptor for potassium channel openers. J Biol Chem. 1996;271(15):8796–8799. [DOI] [PubMed] [Google Scholar]
- 64.Contessi S, Metelli G, Mavelli I, Lippe G.. Diazoxide affects the IF1 inhibitor protein binding to F1 sector of beef heart F0F1ATPsynthase. Biochem Pharmacol. 2004;67(10):1843–1851.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15130761 [DOI] [PubMed] [Google Scholar]
- 65.Henn MC, Janjua MB, Kanter EMet al. Adenosine Triphosphate-Sensitive Potassium Channel Kir Subunits Implicated in Cardioprotection by Diazoxide. J Am Heart Assoc. 2015;4(8):e002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holmuhamedov EL, Wang L, Terzic A.. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J PhysiolJ Physiol. 1999;519 Pt 2:347–360.. ://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10457054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Korge P, Honda HM, Weiss JN.. Protection of cardiac mitochondria by diazoxide and protein kinase C: implications for ischemic preconditioning. Proc Natl Acad Sci USA. 2002;99(5):3312–3317.. ://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11867760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murata M, Akao M, O'Rourke B, Marban E.. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89(10):891–898. [DOI] [PubMed] [Google Scholar]
- 69.Pain T, Yang XM, Critz SDet al. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ ResCirc Res. 2000;87(6):460–466.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10988237 [DOI] [PubMed] [Google Scholar]
- 70.Comelli M, Metelli G, Mavelli I.. Downmodulation of mitochondrial F0F1 ATP synthase by diazoxide in cardiac myoblasts: a dual effect of the drug. Am J Physiol Heart Circ Physiol. 2007;292(2):H820–829. [DOI] [PubMed] [Google Scholar]
- 71.Costa AD, Garlid KD, West ICet al. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res. 2005;97(4):329–336. [DOI] [PubMed] [Google Scholar]
- 72.Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD.. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am J Physiol Heart Circ Physiol. 2001;280(2):H649–H657. [DOI] [PubMed] [Google Scholar]
- 73.Kopustinskiene DM, Liobikas J, Skemiene K, Malinauskas F, Toleikis A.. Direct effects of K(ATP) channel openers pinacidil and diazoxide on oxidative phosphorylation of mitochondria in situ. Cell Physiol Biochem. 2010;25(2-3):181–186. [DOI] [PubMed] [Google Scholar]
- 74.Ozcan C, Bienengraeber M, Dzeja PP, Terzic A.. Potassium channel openers protect cardiac mitochondria by attenuating oxidant stress at reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282(2):H531–H539. [DOI] [PubMed] [Google Scholar]
- 75.Grover GJ, McCullough JR, Henry DE, Conder ML, Sleph PG.. Anti-ischemic effects of the potassium channel activators pinacidil and cromakalim and the reversal of these effects with the potassium channel blocker glyburide. J Pharmacol Exp Ther. 1989;251(1):98–104.. https://www.ncbi.nlm.nih.gov/pubmed/2507775. Published 1989/10/01. [PubMed] [Google Scholar]
- 76.Crestanello JA, Doliba NM, Babsky AMet al. Opening of potassium channels protects mitochondrial function from calcium overload. J Surg Res. 2000;94(2):116–123. [DOI] [PubMed] [Google Scholar]
- 77.An J, Camara AK, Rhodes SS, Riess ML, Stowe DF.. Warm ischemic preconditioning improves mitochondrial redox balance during and after mild hypothermic ischemia in guinea pig isolated hearts. Am J Physiol Heart Circ Physiol. 2005;288(6):H2620–H2627. [DOI] [PubMed] [Google Scholar]
- 78.Paucek P, Mironova G, Mahdi F, Beavis AD, Woldegiorgis G, Garlid KD.. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J Biol Chem. 1992;267(36):26062–26069.. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1464617 [PubMed] [Google Scholar]
- 79.Grover GJ, Atwal KS, Sleph PGet al. Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. Am J Physiol Heart Circ Physiol. 2004;287(4):H1747–H1755. [DOI] [PubMed] [Google Scholar]
- 80.Tzagoloff A, Byington KH, MacLennan DH.. Studies on the mitochondrial adenosine triphosphatase system. II. The isolation and characterization of an oligomycin-sensitive adenosine triphosphatase from bovine heart mitochondria. J Biol Chem. 1968;243(9):2405–2412.. https://www.ncbi.nlm.nih.gov/pubmed/4231099. Published 1968/05/10. [PubMed] [Google Scholar]
- 81.Kanazawa H, Horiuchi Y, Takagi M, Ishino Y, Futai M.. Coupling factor F1 ATPase with defective beta subunit from a mutant of Escherichia coli. J Biochem. 1980;88(3):695–703. [DOI] [PubMed] [Google Scholar]
- 82.Sekiya M, Hosokawa H, Nakanishi-Matsui M, Al-Shawi MK, Nakamoto RK, Futai M.. Single molecule behavior of inhibited and active states of Escherichia coli ATP synthase F1 rotation. J Biol Chem. 2010;285(53):42058–42067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Papanicolaou KN, Ashok D, Liu Tet al. Global knockout of ROMK potassium channel worsens cardiac ischemia-reperfusion injury but cardiomyocyte-specific knockout does not: implications for the identity of mitoKATP. J Mol Cell Cardiol. 2020;139:176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patel D, Kuyucak S, Doupnik CA.. Structural Determinants Mediating Tertiapin Block of Neuronal Kir3.2 Channels. Biochemistry. 2020;59(7):836–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in this article and/or in its Supplemental Information. Any other data request will be shared by the corresponding author, Dr. Sollott SJ, upon reasonable request