

Abstract
Glioblastoma (GBM) is the deadliest primary brain tumor and is generally resistant to immunotherapy because of severe dysfunction of T cells. Novel treatment options are critically needed to overcome the immunotherapy resistance of GBM. Here we demonstrate that Zika virus (ZIKV) treatment improves the efficacy of anti-PD ligand 1 (PD-L1) immunotherapy in GBM. We found that ZIKV induces a strong pro-inflammatory response and increases CD4+ and CD8+ T cell intratumoral infiltration and activation in GBM mouse models. ZIKV treatment of mice bearing GBM tumors inhibits tumor growth and prolongs survival. These therapeutic effects of ZIKV on GBM tumors are negated in mice depleted of T cells. Moreover, ZIKV dramatically promotes activation of the type I interferon signaling pathway in GBM cells. ZIKV treatment potently sensitizes GBM to PD-L1 blockade and provides significant and durable survival benefits. Our findings reveal that ZIKV overcomes the resistance of GBM to immune checkpoint blockade, which may lead to therapeutic applications of ZIKV in individuals with GBM receiving immunotherapy.
Keywords: Zika virus (ZIKV), oncolytic virus, glioblastoma, CD8 T cells, PD-L1 blockade, immunotherapy
Graphical Abstract
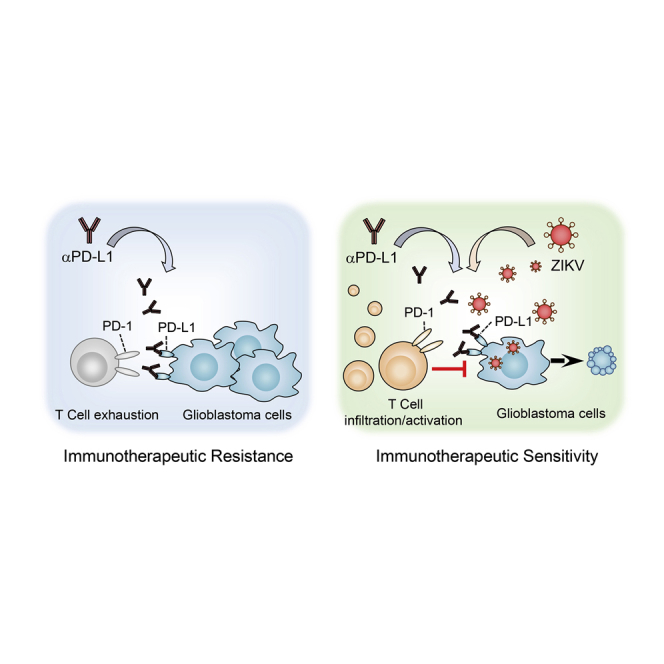
Chen et al. investigate the therapeutic effect of Zika virus in glioblastoma mouse models. They demonstrate that Zika virus treatment promotes CD8+ T cell intratumoral infiltration and activation and sensitizes GBM tumors to PD-L1 blockade. This study suggests that Zika virus may be an adjunctive treatment to improve GBM immunotherapy.
Introduction
Glioblastoma (GBM) is the most common and lethal primary brain tumor, with median survival averaging 14–16 months despite treatment with surgical resection plus chemoradiotherapy.1, 2, 3 Immune checkpoint blockade therapy yields promising tumor regression for various cancers, but its efficacy in GBM is limited. The resistance in GBM likely occurs for multiple reasons, including few mutations that can be targeted immunotherapeutically, severe local and systemic immunosuppression, poor immune infiltration, and low expression of immune checkpoint molecules.4, 5, 6
Tumor-infiltrating lymphocytes (TILs) are central in antitumor immunity, and CD8+ T cells are responsible for recognizing and killing tumor cells. However, profound T cell dysfunction and exhaustion are observed in GBM,5, 6, 7 indicating that immune checkpoint blockade therapy alone, such as anti-programmed death protein (PD-1) or anti-PD ligand 1 (PD-L1) antibodies, is unlikely to work. Thus, strategies to reverse immunosuppression of the tumor microenvironment and induce antitumor T cell infiltration are urgently needed in GBM immunotherapy.
Oncolytic viral anticancer therapy was initially considered a treatment strategy because it can directly kill tumor cells. However, immune responses are observed during oncolytic viral infections, which may improve infiltration of immune cells into tumors and induce an inflamed tumor microenvironment.8, 9, 10 Clinical trials have shown that oncolytic virotherapy with talimogene laherparepvec (T-Vec), a genetically modified herpes simplex virus expressing granulocyte-macrophage colony-stimulating factor (GM-CSF), promotes cytotoxic T cell infiltration and improves the efficacy of anti-PD-1 and anti-CTLA4 therapies in advanced melanoma.11,12 In GBM, an investigation of surgically resected tissue following treatment with an oncolytic adenovirus (tasadenoturev) showed that infiltration of T cells is strongly increased in tumors treated with a virus.13 Additional clinical studies are currently underway. For example, a clinical study suggests that intravenous infection with human orthoreovirus increases intratumoral infiltration of T cells in GBM, although the survival benefit to individuals with GBM will be determined in the ongoing clinical trial.14
Zika virus (ZIKV) is a single-stranded, positive-sense RNA virus of the flavivirus genus.15 A 2015 ZIKV epidemic in Central and South America became a global health emergency because of microcephaly and other congenital anomalies occurring in newborns.15,16 Although it preferentially infects neural progenitor cells, causes cell death, and affects brain development of fetuses, ZIKV shows less severe effects in adult brains.17, 18, 19, 20 Several research groups, including ours, have demonstrated that ZIKV shows promising oncolytic activity in preclinical GBM models by preferentially killing glioma stem cells (GSCs),21,22 which display tumor immune escape and therapeutic resistance in GBM.5,23,24
In this study, we show that ZIKV treatment increases cytotoxic T cell intratumoral infiltration and induces a strong antitumor immune response in the GBM microenvironment. Combination of ZIKV treatment and PD-L1 blockade strongly enhances immunotherapy efficacy and improves survival in immunocompetent mouse GBM models.
Results
ZIKV treatment induces an immune response in the immunocompetent mouse brain
Oncolytic viruses can induce an immune response.8,25 To investigate the effects of ZIKV infection on the immune microenvironment in the brain, we intracranially injected ZIKV-FSS, a historical virus isolated in Cambodia in 2010,26 or ZIKV-GZ01, a contemporary strain isolated in 2016 in Guangzhou, China,27 into immunocompetent mice. qRT-PCR analysis of viral RNA accumulation and immunofluorescence (IF) staining of the ZIKV envelope protein showed that ZIKV was replicated in brains of mice, peaked around day 6 after injection, and then was almost cleaned up after day 12 (Figures S1A and S1B). We only observed apoptosis in a few cells in mouse brains but did not find any weight loss in mice treated with ZIKV (Figures S1C and S1D).
To determine the biological effect of ZIKV in vivo, we performed an RNA sequencing (RNA-seq) analysis of mouse brains with ZIKV infection. We found that ZIKV treatment resulted in upregulation of numerous genes on days 7 and 14 (Figure S1E). Gene set enrichment analysis (GSEA) coupled with enrichment map visualization28 revealed that the enriched pathways of genes induced by ZIKV were linked to regulation of T cell activation and proliferation, adaptive immune response, and the immune effector process (Figures S1F and S1G), suggesting that ZIKV induces a strong immune response in mouse brains. However, we did not observe any enriched pathways of the downregulated genes in ZIKV treated mouse brains. In addition, expression of genes related to the inflammatory response, T cell activation, and immune effector process were strongly increased on day 7, gradually reduced on day 14, and decreased to basal levels on day 33 after ZIKV infection (Figure S1H), indicating transient induction by ZIKV treatment.
To confirm these results, we performed a flow cytometry analysis of brain-infiltrating immune cells and found that the fraction and total number of CD3+ T cells were increased dramatically in a time-dependent manner in mouse brains treated with ZIKV (Figure S1I). We observed an increase in infiltrating CD8+ and CD4+ T cells in ZIKV-treated mouse brains (Figure S1J), which was confirmed by immunohistochemistry (IHC) staining (Figure S1K). CD8+ T cells induced by ZIKV exhibited an increasing level of interferon-γ (IFN-γ) (Figure S1L), indicative of an activated cytotoxic phenotype. We next examined replication of ZIKV in different organs and body fluids in mice. The results showed viral replication primarily in the mouse brain and spleen after ZIKV treatment for 20 days. On day 30 after infection, ZIKV replication was only found in mouse brains (Figures S1M and S1N). These data suggest that ZIKV infection promotes an immune response in the immunocompetent mouse brain.
ZIKV treatment induces T cell infiltration and activation in GBM models
GBM is a local and systemic immunosuppressive neoplasm with severe T cell dysfunction.4,6 To assess whether ZIKV infection promotes an antitumor immune response in GBM, we established orthotopic GBM models with a mixture of GL261, mouse glioma cells, and different strains of ZIKV in immunocompetent mice. ZIKV was effectively replicated in brain tumor tissue by 5 days after infection (Figure S2A). Administration of ZIKV-GZ01 or ZIKV-FSS significantly suppressed tumor growth and prolonged survival (Figures 1A and S2B). Flow cytometry analysis of tumor single-cell suspensions showed that ZIKV treatment resulted in a significant increase in CD3+ T cells in orthotopic GBM tumors (Figures S2C and S2D). Consistent with ZIKV infection in a normal brain, ZIKV treatment promoted significant intratumoral infiltration of CD4+ and CD8+ T cells in a time-dependent manner (Figure 1B). The infiltrating T cells induced by ZIKV were kept at high levels, compared with control tumors, until day 20 after treatment, although replication of ZIKV was markedly decreased at this time point (Figures 1B and S2A). Infiltration of T cells was validated by IHC and IF staining (Figures 1C and S2E). The percentages of CD8+ and CD4+ T cells expressing IFN-γ were increased significantly in ZIKV-treated tumors relative to control tumors (Figure 1D), suggesting that these infiltrating T cells were functionally improved.
Figure 1.

ZIKV treatment induces T cell infiltration and activation in GBM models
(A–D) C57BL/6N mice were injected intracranially with GL261 cells mixed with ZIKV-FSS or ZIKV-GZ01. Kaplan-Meier survival plots of mice with the indicated treatment are shown (A). The percentages of CD4+ or CD8+ T cells in CD45+ CD3+ T cells in tumors were determined by flow cytometry (B). IHC staining of CD3+ and CD8+ T cells in tumors with the indicated treatment for 15 days are shown (C, left). CD3+ or CD8+ cells were compared with all cells in 5 randomly selected microscope fields from each of 5 tumors, and the percentages of CD3+ and CD8+ T cells are shown (C, right). Scale bars, 100 μm. The percentages of IFN-γ+ in CD4+ or CD8+ T cells in tumors with ZIKV-FSS treatment for 15 days were determined by flow cytometry (D). (E–I) C57BL/6N mice were implanted intracranially with GL261 cells expressing luciferase (E, F, and H) or CT-2A cells (G and I). Mice were grouped randomly and treated with ZIKV-FSS from day 7 after implantation, as shown by the schematic (E, top). The xenografts were tracked by bioluminescence, and representative images are shown (E, bottom). Bioluminescence quantification of tumor growth is shown (E, right). Kaplan-Meier survival plots of mice are shown (F and G; log rank test). 12 days after treatment, the proportions of CD3+ T cells in CD45+ cells, CD4+ or CD8+ T cells in CD45+ CD3+ T cells, and IFN-γ+ or TNF-α+ cells in CD8+or CD4+ T cells (H) in tumors were determined by flow cytometry. The proportions of CD11c+ MHC class II+ cells in CD45+ cells and CD80+ or CD86+ cells in CD45+ CD11c+ MHC class II+ cells in tumors with ZIKV-FSS treatment for 15 days were determined by flow cytometry (I). (J and K) Tumors treated with ZIKV-FSS for 15 days were harvested, and gene expression analysis was performed using RNA-seq. Pathway enrichment analysis using GSEA (K) and visualization using Cytoscape Enrichment Map (J) in tumors with ZIKV infection are shown. Red nodes represent pathways that are upregulated and blue nodes represent pathways that are downregulated in ZIKV-infected tumors compared with control tumors (FDR < 0.05). Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, as assayed by unpaired Student’s t test or Welch’s t test.
We next assessed the effects of ZIKV on already existing tumors because this would more closely recapitulate the clinical situation. We established orthotopic GBM tumors with glioma cells expressing a luciferase reporter. On day 7 after transplantation, we randomly grouped the mice based on tumor size and treated them with ZIKV (Figure 1E, top left). The results showed that ZIKV treatment in two established GBM models significantly inhibited tumor growth (Figure 1E) and prolonged survival (Figures 1F and 1G). In the established tumors, ZIKV treatment also increased T cell intratumoral infiltration and activation (Figures 1H and S2F).
Additionally, we assessed activation of dendritic cells (DCs), which present antigens and contribute to activation of CD8+ T cells. The results showed that ZIKV treatment significantly increased the expression of CD80, CD86, and major histocompatibility complex (MHC) class II in DCs (Figure 1I), suggesting enhanced maturation and an elevated capability for antigen presentation of DCs. The numbers of macrophages, especially the proportion of M1-like macrophages, were increased significantly in tumors with ZIKV treatment (Figure S2G). We also found that intratumoral CD11b+Gr1+ myeloid-derived suppressor cells (MDSCs) and CD4+CD25+FoxP3+ regulatory T (Treg) cells were reduced by ZIKV treatment (Figures S2H and S2I).
RNA-seq analysis of GBM tumors showed that the upregulated genes induced by ZIKV treatment were significantly enriched in positive regulation of T cell activation and adaptive immune response signaling pathways (Figures 1J and 1K). However, the downregulated genes in ZIKV-treated tumors were involved in positive regulation of the cell cycle and chromosome segregation, indicating suppression of tumor growth by ZIKV (Figures 1J and 1K). These results suggest that ZIKV treatment induces an anti-tumoral immune response in the tumor microenvironment.
To determine which cell types are infected by ZIKV in the tumor microenvironment, we performed coIF staining with ZIKV E protein and CD11b, SOX2 (a stem cell marker), CD3, or Tuj1 (a mature neuron marker) in tumors treated with ZIKV. The results showed that ZIKV E protein was co-localized with CD11b and SOX2 but not with CD3 or Tuj1 (Figure S2J). These results suggest that macrophages and GSCs, but not T cells or differentiated non-stem tumor cells, are infected by ZIKV. Furthermore, the percentages of CD3+, CD4+, or CD8+ T cells in spleens of mice bearing tumors were not changed by ZIKV treatment, but the activities of CD4+ and CD8+ T cells in mice spleens were increased slightly (Figures S2K and S2L).
ZIKV treatment results in immune-mediated tumor control in GBM models
To determine whether tumor-infiltrating T cells are involved in the antitumor activity of ZIKV, we treated the GBM tumor-bearing mice with ZIKV or ZIKV plus depletion antibodies against CD3+, CD4+, or CD8+ T cells (Figure 2A). Cell depletion was confirmed by flow cytometry analysis of peripheral blood mononuclear cells (PBMCs) in mice (Figure S3A). Depletion of CD3+, CD4+, or CD8+ T cells largely abrogated the antitumor activity and survival benefits of ZIKV in mouse GBM models (Figures 2B and 2C). Flow cytometry analysis validated the loss of CD3+, CD4+, or CD8+ T cells in tumors collected from mice treated with ZIKV and immune-cell-depleting antibodies (Figures 2D and S3B). We also confirmed these results in the established GBM tumors (Figures 2E, 2F, and S3C).
Figure 2.

ZIKV infection results in immune-mediated tumor control in GBM models
(A–D) C57BL/6N mice were injected intracranially with GL261 cells plus ZIKV-FSS. Two days before injection, mice were left untreated or treated with depletion Abs against CD3, CD4, or CD8 T cells, as shown by the schematic (A). Representative images of H&E-stained sections of mouse brains collected on day 16 after GL261 cell transplantation are shown (B). Scale bars, 2 mm. A Kaplan-Meier survival plot of mice is shown (C, log rank test). Depletion of T cells in tumors was determined by flow cytometry (D). (E–H) C57BL/6N mice were injected intracranially with CT2A cells. Mice were left untreated or treated with depletion Abs against CD3, CD4, or CD8 T cells on day 5 and then left untreated or treated with ZIKV-FSS on day 7 after implantation, as shown by the schematic (E). A Kaplan-Meier survival plot of mice is shown (F, log rank test). The proportions of CD4+ or CD8+ T cells (G) or activation of CD8+ T cells (H) were determined by flow cytometry. (I) Cell proliferation in tumors from (A) was determined by Ki67 IHC staining. Ki67+ cells were compared with all cells in 4 tumors from each group, and the percentages of Ki67+ cells are shown. Scale bars, 100 μm. Data are presented as means ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001, as assayed by unpaired Student’s t test or Welch’s t test.
Because CD8+ T cells are considered major drivers of antitumor immunity,29 we next investigated the roles of CD4+ T cells in CD8+ T cell activation induced by ZIKV. We observed a compensatory increase in CD4+ T cells in tumors of mice depleted of CD8+ cells and in CD8+ T cells in tumors of mice depleted of CD4+ cells (Figure 2G). However, depletion of CD4+ T cells in tumors significantly decreased activation of CD8+ T cells (Figure 2H), suggesting that CD4+ T cells are crucial for activation of CD8+ T cells in response to ZIKV treatment. Additionally, we observed that expression of PD-1 was increased in CD8+ T cells, but not in CD4+ T cells, in tumors treated with ZIKV for 20 days (Figure S3D). Ki67 staining showed that ZIKV treatment inhibited cell growth in tumors, which was rescued by T cell depletion (Figure 2I), supporting the antitumor T cell immunity induced by ZIKV infection.
To further assess whether the immune response also acted against the virus, we examined replication of ZIKV in tumors with or without depletion of T cells. The results showed that depletion of T cells significantly increased replication of ZIKV in tumor tissues (Figure S3E). To determine whether tumor-bearing mice with T cell depletion died because of the tumor, we depleted T cells in tumor-free mice treated with ZIKV. Compared with all tumor-bearing mice that died within 20 days because of T cell depletion (Figure 2F), T cell depletion only resulted in two of seven deaths until day 40 in ZIKV-treated, tumor-free mice (Figures S3F and S3G). These results suggest that the majority of tumor-bearing mice treated with ZIKV in the T cell depletion groups are dying of tumors.
ZIKV sensitizes GBM to PD-L1 blockade and improves survival
On the basis that intratumoral infiltrating T cells were induced by ZIKV, we tested whether ZIKV treatment sensitizes GBM to immune checkpoint blockade. We first intracranially injected glioma cells mixed with ZIKVs (FSS or GZ01) or without ZIKVs in mice and then treated mice with anti-PD-L1 antibody (once every week, three times in total) (Figure 3A, bottom). We observed that treatment with anti-PD-L1 antibody alone was unable to efficiently control tumor growth, as indicated by bioluminescence monitoring of the tumor response and survival analysis (Figures 3A–3E). ZIKV treatment dramatically sensitized GBM tumors to anti-PD-L1 antibody treatment and conferred the longest survival extension among all experimental groups (Figures 3A–3E). In comparison with control mice, the combined ZIKV-GZ01 and anti-PD-L1 antibody treatment led to an almost 2-fold increase in survival with an added median survival benefit of 18 days (Figure 3D). Mice under combination treatment of ZIKV-FSS and anti-PD-L1 antibody appeared to live significantly longer than mice in other groups because half of the mice in this group survived more than 120 days (Figure 3C). Additionally, the efficacy of combination treatment was validated in two established GBM models (Figures 3F, 3G, and S4A), suggesting that ZIKV treatment could improve the therapeutic benefit of immunotherapy in GBM.
Figure 3.

ZIKV infection sensitizes GBM to immune checkpoint blockade
(A–D) C57BL/6N mice were implanted intracranially with GL261 cells expressing luciferase plus ZIKV-FSS (A–C) or ZIKV-GZ01 (D). Mice were grouped randomly and treated with isotype or anti-PD-L1 Abs from day 7 (once a week, three times in total), as shown by the schematic (A, bottom). The orthotopic xenografts were tracked by bioluminescence, and representative images are shown (A, top). Bioluminescence quantification of tumor growth is shown (B). Data are presented as means ± SEM; unpaired Student’s t test. Kaplan-Meier survival plots of mice are shown (C and D); log rank test. (E) C57BL/6N mice were implanted intracranially with CT-2A cells plus ZIKV-FSS or ZIKV-GZ01. Mice were treated as described in (A). A Kaplan-Meier survival plot of mice is shown; log rank test (F and G) C57BL/6N mice were implanted intracranially with GL261 cells expressing luciferase. Mice were grouped randomly and treated as indicated from day 6 after implantation, as shown by the schematic (F, top). The xenografts were tracked by bioluminescence. Representative images (F, bottom) and bioluminescence quantifications of tumor growth (F, right) are shown. Kaplan-Meier survival plots of mice are shown (G, log rank test). (H) Age-matched C57BL/6N mice and mice surviving in the combination treatment group from (C) were rechallenged with GL261 cells (luciferase). Tumor growth was tracked by bioluminescence. Representative images (left) and bioluminescence quantification of tumor growth (center) are shown. Kaplan-Meier survival plots of mice are shown (right, log rank test). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To test whether combined ZIKV treatment and PD-L1 blockade generates a long-term memory antitumor immune response, we reinjected the same tumor cells into tumor-free mice from the combination treatment group in Figures 3C and 3E. The results showed that all cured mice from the combination therapy completely rejected the re-challenged tumors (Figures 3H and S4B). We examined the effector memory T cells in the surviving mice and found that CD4+CD44+CD62L− T cells and CD8+CD44+CD62L− T cells were increased significantly increased in the brains and spleens of tumor-bearing mice that received the combination treatment (Figure S4C), suggesting that ZIKV combined with PD-L1 blockade induces an adaptive antitumor immune memory response in vivo.
To better understand the immune-stimulatory properties of the combination therapy, we examined the transcriptomic effect of ZIKV plus anti-PD-L1 antibody treatment in vivo at 2 and 3 weeks. Interestingly, we did not find any differentially enriched biological signaling pathways for the upregulated genes in the combination treatment groups compared with ZIKV treatment alone at 2 weeks. The combination treatment only resulted in more inhibition of cell cycle and chromosome segregation at this time point (Figure S4D). However, at 3 weeks, compared with tumors with ZIKV monotherapy, upregulated genes in tumors with combination therapy were significantly enriched in regulation of T cell activation, adaptive immune response, cytokine production, and immune effector process signaling pathways (Figure 4A). These analyses suggest that combination treatment with ZIKV and the anti-PD-L1 antibody prolongs the immune response in tumors, which may account for the improved efficacy of immunotherapy by ZIKV.
Figure 4.

Combination ZIKV and anti-PD-L1 treatment improves antitumor T cell immunity
(A–C) C57BL/6N mice were treated as described in Figure 3A. Tumors were harvested, and gene expression analysis was performed using RNA-seq (day 22). Pathway enrichment analysis using GSEA and visualization using Cytoscape Enrichment Map in tumors with combination treatment are shown (A). Red nodes represent pathways that are upregulated in combination treatment tumors compared with ZIKV-FSS treatment tumors (FDR < 0.05). The proportions of CD4+ or CD8+ in CD3+ T cells (B) and IFN-γ+ in CD8+ or in CD4+ T cells (C) in tumors with the indicated treatments for 22 days were determined by flow cytometry. (D) C57BL/6N mice were treated as described in Figure 3F. The proportions of CD3+ T cells in CD45+ cells, CD8+ or CD4+ in CD45+ CD3+ T cells, and IFN-γ+ or TNF-α+ in CD8+ or CD4+ T cells in tumors with the indicated treatments for 15 days were determined by flow cytometry. Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, as assayed by unpaired Student’s t test or Welch’s t test.
To validate these results, we performed flow cytometry analysis with tumors after 3 weeks of therapy. We found that anti-PD-L1 antibody treatment alone did not result in a significant increase in CD3+, CD4+, or CD8+ T cells in tumors (Figures 4B, S4E, and S4F). However, combined ZIKV and anti-PD-L1 antibody treatment potently promoted intratumoral infiltration of CD3+, CD4+, and CD8+ T cells compared with anti-PD-L1 antibody or ZIKV monotherapy groups (Figures 4B, 4C, S4E, and S4F). Importantly, the combination treatment resulted in the highest percentages of CD4+IFN-γ+ and CD8+IFN-γ+ T cells in tumors (Figures 4C, 4D, and S4F). Activation of CD8+ T cells was validated by an increase in CD8+ T cells co-expressing Granzyme B or tumor necrosis factor alpha (TNF-α) in tumors (Figures 4D, S4E, and S4F). Although the combination treatment strongly decreased Treg cells and MDSCs in tumors, there was no significant change in Treg cells or MDSCs between ZIKV monotherapy and combination treatment (Figure S4E).
To evaluate the risk of upregulation of cytokines in the combination therapy, we examined the expression of IFN-regulated genes in brains of mice that received the combination treatment. Expression of the IFN-regulated genes, including IFN-β and IFN-γ, was significantly induced by ZIKV in combination with anti-PD-L1 antibody treatment on day 12, but most relative expression levels returned to the basal levels by day 30 (Figure S5A). However, the combination treatment did not result in significant elevation of immune cytokines in peripheral blood of tumor-bearing mice (Figure S5B). These data suggest that ZIKV treatment overcomes the resistance of GBM to PD-L1 blockade.
ZIKV treatment activates type I IFN signaling in GBM cells
Infection with oncolytic viruses leads to activation of type I IFN signaling pathways, which are crucial in oncolytic virus-mediated antitumor immunity.30 Our results showed that ZIKV treatment significantly promoted IFN-α and IFN-β expression in mouse glioma cells (Figures 5A and S6A). The levels of IFNAR1 and IFNAR2, type I IFN receptors, were unchanged in response to ZIKV (Figure S6B). To extend our finding to human GBM cells, we examined induction of IFN-α and IFN-β by ZIKV using human GSCs (456 GSCs) and fresh isolated primary GBM cells (GBM#27). We observed that ZIKV treatment significantly induced production of IFN-β in human GBM cells (Figures 5B and 5C).
Figure 5.

ZIKV promotes activation of the type I IFN pathway in GBM cells
(A–C) CT-2A (A) or 456 GSCs and GBM#27 cells (B and C) were treated with ZIKV-GZ01 for the indicated time. Levels of IFN-α and IFN-β were assessed by qRT-PCR (A and B). ELISAs of secreted IFN-β in 456 GSCs and GBM#27 tumors are shown (C). (D) Immunoblot (IB) showing phosphorylation of the indicated proteins in 456 GSCs and GBM#27 cells treated with ZIKV-GZ01 for the indicated time. (E) 456 GSCs and GBM#27 cells were treated with ZIKV-GZ01 for 24 and 48 h. The mRNA levels of the indicated genes were analyzed by qRT-PCR. (F and G) 456 GSCs (F) and CT-2A (G) cells were treated as indicated for 48 h. The mRNA levels of the indicated genes were analyzed by qRT-PCR. Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, as assayed by unpaired Student’s t test or Welch’s t test.
The transcription factor IRF3 is a central regulator of type-I IFN signaling.31 In response to virus infection, IRF3 is activated, leading to induction of type I IFNs and upregulation of cytokines and chemokines essential for generating an antiviral immune response.32,33 Our results showed that ZIKV infection led to robust activation of IRF3 and its activator TANK (TRAF family member-associated NFκB activator)-binding kinase 1 (TBK1) in human GBM cells (Figure 5D). Consequently, ZIKV infection resulted in a significant increase in pro-inflammatory chemokines such as CCL5, CXCL9, CXCL10, and CXCL11 in human GBM cells and mouse glioma cells (Figures 5E, S6C, and S6D), which are crucial for regulating T cell recruitment and activity.34 We found that expression of genes such as CXCL10, IFIT1, IFIT2, IFIT3, IRF7, and RSAD2, all belonging to the type I IFN signaling pathway, were increased significantly upon ZIKV treatment in tumor cells (Figures 5F, 5G, and S6E). Blockade of type I IFN signaling rescued induction of these genes by ZIKV (Figures 5F, 5G, and S6E). These data suggest that ZIKV infection triggers a strong pro-inflammatory response in human GBM cells and mouse glioma cells.
Discussion
Oncolytic viral therapy in tumors was initially considered as a treatment because of viral replication in and direct killing of tumor cells. Recent studies have shown that oncolytic viral infection promotes an antitumor immune response, suggesting that viral therapies may be an approach to overcome immunosuppression in tumors.8, 9, 10 In this study, we demonstrated that ZIKV induces a strong anti-tumor immune response and improves the therapeutic efficacy of PD-L1 blockade in mouse GBM models.
ZIKV represents the specific tropism for neural progenitor cells and GSCs. We recently showed that ZIKV selectively kills GSCs,21 which has been confirmed by other groups.22,35 A SOX2-Integrin axis was identified as the key signaling pathway in GSCs mediating preferential infection of ZIKV.36,37 Besides direct cell killing, our results here demonstrated that ZIKV infection potently promotes T cell intratumoral infiltration and activation in mouse GBM tumors. We found that ZIKV infection results in significant activation of the type I IFN signaling and increased production, which are crucial in oncolytic virus-mediated T cell recruitment and activation.30,32
The high numbers of T cells induced by ZIKV are potentially directed against the virus because blockade of T cells increases replication of ZIKV. Our findings demonstrate that these T cells also contribute to the anti-tumor immunity of ZIKV because blockade of T cells compromises tumor inhibition by ZIKV. Although the initial T cell response may be virus specific, antigen-presenting cells (APCs) could promote cross-presentation of tumor antigens to T cells.30 We observed enhanced maturation and activation of DCs in response to ZIKV treatment in tumors, indicating that DCs may present tumor antigens and contribute to the anti-tumor immunity of ZIKV. Additionally, CD4+ T cells are also crucial for the antitumor activity of ZIKV because depletion of CD4+ T cells significantly compromises activation of CD8+ T cells and suppression of tumors by ZIKV.
Immunotherapy is a durable clinical response in many types of cancers and largely relies on efficient intratumoral infiltration and activation of T cells.38,39 However, GBM has been studied as a model of resistance to immunotherapy because of extensive immunosuppressive mechanisms, including severe T cell exhaustion.5,40 Our study demonstrated that ZIKV infection strongly elevates production of pro-inflammatory cytokines and induces infiltration of cytotoxic T cells in tumors, which may reverse the severe local immunosuppression in GBM. We demonstrate that ZIKV infection significantly sensitizes GBM tumors to PD-L1 blockade and dramatically improves survival in mouse GBM models. Interestingly, we observed protection when re-challenging cured mice that had undergone combination treatment with the same tumor cells, suggesting that ZIKV plus immune checkpoint blockade induces a long-term antitumor immune memory response.
Although we assessed the risk of using ZIKV as an adjunctive therapy in mouse GBM models and demonstrated that ZIKV treatment does not significantly induce immune cytokines in peripheral blood or result in any weight loss in mice, virus replication remains confined to mouse brains after ZIKV treatment, and we are unable to rule out any potential danger of using ZIKV in individuals with GBM. Determining the safety of using ZIKV in humans remains a paramount concern. To our knowledge, clinically approved vaccines or antiviral antibodies to control or prevent ZIKV infections are currently unavailable.41 Clinical trials testing the safety and efficacy of ZIKV in individuals with GBM need to be conducted. Local delivery of ZIKV into human brain tumors may also be a substantial challenge because of infiltrative disease. A clinical study has investigated the persistence of ZIKV in human body fluids and found that less than 5% of individuals with ZIKV infection had detectable viral RNA in urine for 5 weeks and in serum for 6 weeks after symptom onset.42 This findings, including ours, may have implications for isolation of affected individuals to prevent ZIKV transmission if ZIKV could be used as an adjunctive treatment in human GBM.
Our study demonstrates that ZIKV treatment generates a durable antitumor immune response in the GBM microenvironment and overcomes resistance of GBM to PD-L1 blockade in mouse GBM models. The preclinical evidence in our previous and current studies shows that ZIKV has limited toxicity in adult mice, suggesting that ZIKV may be rapidly translated into clinical use combined with PD-L1 blockade to improve GBM therapy.
Materials and methods
Viruses
ZIKV strain GZ01 (GenBank: KU820898) was originally isolated from a Chinese individual returning from Venezuela in 2016.26,27 ZIKV strain FSS13025 (GenBank: KU955593) was originally isolated from Cambodia in 2016 and recovered from an infectious clone of ZIKV.26 ZIKV was propagated in C6/36 cells after inoculating at an MOI of 0.01 and harvested after 5 days. Virus titers were quantified by standard plaque assay on BHK-21 cells, and virus stocks were separated and stored at −80°C until use. Studies with infectious ZIKV were conducted under biosafety level 2 (BSL-2) conditions at the Beijing Institute of Microbiology and Epidemiology with approval from the Institutional Biosafety Committee.
Cell lines
GL261 (mouse glioma), CT-2A (mouse glioma), and baby hamster kidney (BHK)-21 (ATCC, CCL-10) cells were cultured in DMEM-10% fetal bovine serum (FBS). For human GBM tumor cell isolation, GBM surgical specimens were collected with approval from PLA General Hospital. Informed consent was obtained from all subjects. 456 GSCs and isolated GBM tumor cells were cultured in stem cell medium (Neurobasal-A medium with B27 supplement, 10 ng/mL epidermal growth factor [EGF], 10 ng/mL β fibroblast growth factor [FGF], 1% penicillin/streptomycin, 1 mM sodium pyruvate, and 2 mM L-glutamine). The cells were maintained at 37°C in a humidified incubator with 5% CO2. C6/36 (Aedes albopictus clone, ATCC CRL-1660) cells were cultured in RPMI 1640 medium containing 10% FBS and maintained at 28°C. FBS was purchased from Macgen. EGF and βFGF were purchased from R&D Systems. In bioluminescence studies, GL261-luc cells were obtained by lentivirus transfer of GL261 cells into a luciferase reporter gene and then screened with puromycin (2 μg/mL).
In vivo mouse studies
All animal work was performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the National Center of Biomedical Analysis. Mice used in our studies were 4- to 6-week-old, female C57BL/6N mice purchased from Beijing Vital River Laboratory Animal Technology. Animal care was monitored daily by certified veterinary staffs and laboratory personnel. Every effort was made to minimize discomfort, distress, pain, or injury to the mice. A maximum of 5 mice per cage was allowed.
To study the tissue distribution of ZIKV, sera, major tissues (including brain, liver, spleen, kidney, heart, and lung) and body fluids (including urine, saliva, and feces) of mice injected intracranially with ZIKV-GZ01 (10,000 plaque-forming units [PFUs]/mouse) were harvested on days 3, 6, 9, 12, 20, and 30 after injection (n = 3 per time point), and viral RNA was assessed by qRT-PCR. The primers and probes used in this study have been described previously.20
To verify the effect of the ZIKV on survival and weight of mice with glioma, groups of C57BL/6N mice were inoculated intracranially with 10,000 GL261 cells mixed with 10,000 PFU ZIKV-GZ01. PBS injection was used as a negative control. Mice were weighed and monitored daily to assess weight changes and mortality.
For histological examination, the brains of tumor-bearing mice treated with ZIKV or PBS were harvested at the indicated time after inoculation and then prepared as 6-μm-thick cytosections. The cytosections were stained with hematoxylin and eosin.
For tumor implantation and treatment, a total of 50,000 luciferase-labeled GL261 (or CT-2A) cells were co-implanted with 10,000 PFU of ZIKV intracranially into the right frontal lobe of mice. Bioluminescence imaging was performed on the indicated date using a charge-coupled device camera (Xenogen, Alameda, CA, USA). Briefly, each mouse was injected intraperitoneally (i.p.) with 1.5 mg of the substrate D-luciferin sodium salt (Gold Biotechnology), and images were collected 5 min later for 60 s. To quantify the amount of light emitted from the tumor, regions of interest (ROIs) were defined manually after imaging, and the photon flux was calculated (in photons per second per square centimeter per steradian) using Living Image 3.0 (Caliper Life Sciences, Alameda, CA, USA).
The immune checkpoint inhibitor antibody anti-mPD-L1 (rat clone 10F.9G2, 10 mg/kg) and isotype control antibody rat immunoglobulin G (IgG) 2b (clone LTF-2) were from BioXcell and administered i.p. 3 times. Depletion antibodies (anti-CD3, clone 17A2; anti-CD4, clone YTS 177; anti-CD8, clone 2.43; isotype control antibody rat IgG; 200 μg/mouse, BioXcell) were injected i.p. on the indicated days. On day 22, when mice in the control group showed neurological signs, we randomly selected three mice from each group, collected their brains, and prepared 6-μm-thick cryosections. The cryosections were stained with hematoxylin and eosin and subjected to histological examination.
Flow cytometry
The brains of mice were chopped up and incubated in 6 mL Hank’s balanced salt solution (HBSS) (Thermo Fisher Scientific, 14025092) with 100 U/mL collagenase IV (Thermo Fisher Scientific, 17104019) and 40 U/mL DNase I (Sigma, D5025) for 30 min at 37°C with shaking to form single cells. Immune cells were isolated by 40% and 80% Percoll (GE Healthcare, 17-0891-02) gradient centrifugation.
Single cells from brain or brain tumor tissue were re-suspended in fluorescence-activated cell sorting (FACS) buffer (PBS containing 1% FBS), blocked with anti-CD16/32 antibody (clone 93, BioLegend, 101302) and then stained with the Abs listed below. The Zombie NIR Fixable Viability Kit (BioLegend, 423105) was used to stain dead cells. We followed a “no wash” sequential staining protocol (BioLegend) to stain dead cells and for surface staining. Intracellular FoxP3 staining was performed following the FoxP3 intracellular staining protocol (BioLegend). Intracellular IFN-γ, TNF-α, and Granzyme B staining was performed following the intracellular flow cytometry staining protocol (BioLegend). Fluorescent minus one (FMO) controls were included for each color. All samples were run in a FACSVerse flow cytometer (BD Biosciences). Data were analyzed with FlowJo software.
Abs used for flow cytometry were as follows. CD45 (PerCP-Cy5.5, clone 30-F11, 557235), Gr-1 (PE, P-phycoerythrin, clone RB6-8C5, 553128), and CD62L (APC, clone MEL-14, 553152) were purchased from B&D. CD3 (PE/Cyanine7, clone 145-2C11, 100320/100220), CD4 (fluorescein isothiocyanate [FITC], clone RM4-5, 100510), CD8α (APC, clone 53-6.7, 100712), PD-1 (PE, clone 29F.1A12, 135025), CD11b (FITC, clone M1/70, 101206), CD11c (PE/Cyanine7, clone N418, 117317), CD80 (FITC, clone 16-10A1, 104706), F4/80 (PE, clone BM8, 123110), CD206 (APC, clone C068C2, 141708), CD25 (Alexa Fluor 647, clone PC61, 102019), FoxP3 (PE, clone 150D, 320007), IFN-γ (PE, clone XMG1.2, 505808), TNF-α (PE, clone MP6-XT22, 506305), Granzyme B (PE, clone QA16A02, 372208), and CD44 (PE, clone IM7, 103008) were purchased from BioLegend. CD86 (APC, clone GL1, 17-0862-81) was purchased from Invitrogen.
IF staining
IF staining of tissue sections was performed as described previously.24 The sections were antigen repaired with citrate repair solution, sealed with 1% BSA and 0.3 m glycine in PBST for 1 h, incubated with primary Ab at 4°C overnight, and incubated with the appropriate secondary Ab at room temperature for 1 h. Nuclei were counterstained with Hoechst (Invitrogen). Images were acquired with a Zeiss LSM880 system. The acquisition software was Zen 2.1 SP2. ImageJ was used for image processing after data acquisition. Abs used in IF staining were as follows. CD3 (1:200, 99940S), CD4 (1:400, 25229s), CD8 (1:400, 98941S), cleaved caspase-3 (1:100, 9664S), and CD11c (1:400, 97585S) were purchased from Cell Signaling Technology. F4/80 (1:400, 123122) was purchased from BioLegend. CD11b (1:1,000, ab133357) and Tuj1 (1:1,000, ab18207) were purchased from Abcam. Sox2 (1:100, sc-17320) was purchased from Santa Cruz Biotechnology. E protein of ZIKV (1:400, BF-1176-56) was purchased from Biofront.
IHC
IHC staining of tissue sections was performed as described previously.24 Images were acquired with a Hamamatsu 2.0 HT digital slide scanner. The acquisition software was Nanozoomer 2.0 HT. Abs used for IHC were as follows. CD3 (1:200, 99940S), CD4 (1:200, 25229S), CD8 (1:400, 98941S), and Ki67 (1:100, 9129T) were purchased from Cell Signaling Technology.
qRT-PCR
Cell pellets were collected, and the total RNA was extracted using the RNeasy kit (QIAGEN) and then reverse transcribed to cDNA with PrimeScript RT Master Mix (Takara Bio) according to the manufacturer’s instructions. Real-time PCR was performed with SYBR Green Master Mix (Applied Biosystems) on a cycler (Applied Biosystems). GAPDH or actin was used for normalization. Determination of viral RNA amounts by qRT-PCR was performed using the One Step PrimeScript RT-PCR Kit (Takara, 064A). The primer pairs used to detect the mRNA levels are listed in Table S1.
Immunoblot
Immunoblotting was performed following standard methods as described previously.24 Briefly, cells were lysed in lysis buffer (20 mM Tris-HCl [pH 7.4], 0.5% NP-40, 250 mM NaCl, 3 mM EGTA, 3 mM EDTA) supplemented with protease inhibitors (Roche) and incubated on ice for 30 min. A Bradford assay (Bio-Rad) was utilized for determination of protein concentration. Equal amounts of protein were mixed with reducing Laemmli loading buffer, boiled 10 min, resolved by SDS-PAGE, and then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA). Blots were incubated with primary Abs overnight at 4°C, followed by horseradish peroxidase (HRP)-conjugated species-specific Abs (Jackson ImmunoResearch, 1:5,000) at room temperature for 1 h. Abs used in the immunoblot were as follows. p-TBK1 (5483S) and TBK1 (3504S) were purchased from Cell Signaling Technology. p-IRF3 (ab76493) and IRF3 (ab68481) were purchased from Abcam. Tubulin (T5148) was purchased from Sigma.
Cytokine analysis
Mouse serum was collected on the appointed day and inactivated at 56°C. The samples were stored frozen until analysis for cytokine production. A total of 25 μL serum from each mouse was adopt for cytokine analysis with the Bio-Plex Pro Mouse Cytokine Grp I Panel 23-Plex (Bio-Rad, M60009RDPD) or ProcartaPlex Mo Cytokine/Chemokine Panel 1A 36-Plex (Invitrogen, EPX360-26092-901) according to the manufacturer’s instructions. The data were collected on a Luminex 200 and analyzed by Luminex PONENT (Thermo Fisher Scientific).
RNA-seq
Total RNA was isolated from cells using Trizol (Sigma-Aldrich). We fed 1 μg RNA into the NEBNext PolyA mRNA Magnetic Isolation Kit (New England Biolabs, catalog number E7490L) and then constructed the specific chain RNA library using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, catalog number E7420L). We performed library construction according to the vendor’s instructions, starting with the chapter “Protocol for use with NEBNext Poly (A) mRNA Magnetic Isolation Module.” Library quality was evaluated on an Agilent 2100 bioanalyzer and quantified by qPCR using the VAHTS Library Quantification Kit for Illumina (Low ROX Premixed) (Vazyme, catalog number NQ103). Libraries were sequenced on the HiSeq ×10 using the paired-end 2 × 150 bp, single-index format. The RNA-seq data have been deposited in the National Center for Biotechnology Information (NCBI) under accession code PRJNA759363 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA759363/).
Bioinformatics statistics
FastQC software was used to verify the quality of Fastq data. Connectors, primers, and low-quality reads were removed by Trimmomatic. Cleanreads were compared with the mus_musculus G38.p5 genome using STAR software. Finally, the expression of each gene was quantified by RSEM (RNA-Sequence by Expectation-Maximization). DESeq2 software was used for data standardization and differential gene analysis. The screening criteria of differential gene were adjusted to p < 0.05 and fold change greater than 2. GSEA was used to examine differentially expressed genes (DEGs). The output of the GSEA is a normalized enrichment score (NES), which accounts for the size of the gene set being tested, a p value, and an estimated false discovery rate (FDR). The Cytoscape plugin EnrichmentMap was used to generate gene set enrichment bubble plots with an FDR q value threshold of 0.05 as a default.
Quantification and statistical analysis data
All grouped data are presented as mean ± SD or SEM from studies performed at least in triplicate unless otherwise specified. A probability value of less than 0.05 was considered significant. For bar graphs, unpaired Student’s t test (two-tailed) or Welch’s t test was used for comparison of two unpaired two groups. For the survival analysis, Kaplan-Meier survival curves were analyzed by using log rank statistics comparing the different individual or mouse groups. GraphPad Prism Software was used for all statistical analyses.
Acknowledgments
We thank Dr. Jeremy Rich (University of California, San Diego, San Diego, USA) for providing 456 GSCs. We thank Dr. Yu Shi (Army Medical University, Chongqing, China) for GL261 cells and Dr. Qi Xie (Westlake University, Hangzhou, China) for CT-2A cells. This research was supported by grants from the National Science and Technology Major Project of China (2018ZX09711003), the National Key Research and Development Program of China (2017YFA0505602), the Research Fund from the Chinese Academy of Medical Sciences (2016-I2M-1-005), and the National Natural Science Foundation of China (3200129, 31770190, U1702282, 81872408, and 31900472). C.Q. was supported by the National Science Fund for Distinguished Young Scholars (81925025), the Innovative Research Group (81621005) from the NSFC, and the Innovation Fund for Medical Sciences (2019-I2M-5-049) from the Chinese Academy of Medical Sciences.
Author contributions
J.M. and C.Q. co-supervised the project and acquired funding for the study. L.C., C.Z., and Q.C. designed and performed the majority of the experiments. J.S. performed the bioinformatics analysis. Z.L., Y.G., C.L., H.J.W., S.Z., Q.Y. and X.L. contributed to specific experiments. F.L. isolated GBM tumor cells. C.W., Q.X., T.Z., A.L., A.W., and Y.C. contributed to specific experiments and data analyses. J.M., L.C., C.Z., and C.Q wrote the manuscript with contributions from all authors.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.01.011.
Contributor Information
Yun Chen, Email: chenyun@njmu.edu.cn.
Aiping Wu, Email: wap@ism.cams.cn.
Chengfeng Qin, Email: qincf@bmi.ac.cn.
Jianghong Man, Email: jhman@ncba.ac.cn.
Supplemental information
References
- 1.Gilbert M.R., Dignam J.J., Armstrong T.S., Wefel J.S., Blumenthal D.T., Vogelbaum M.A., Colman H., Chakravarti A., Pugh S., Won M., et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R., Taillibert S., Kanner A., Read W., Steinberg D., Lhermitte B., Toms S., Idbaih A., Ahluwalia M.S., Fink K., et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318:2306–2316. doi: 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldape K., Brindle K.M., Chesler L., Chopra R., Gajjar A., Gilbert M.R., Gottardo N., Gutmann D.H., Hargrave D., Holland E.C., et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019;16:509–520. doi: 10.1038/s41571-019-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim M., Xia Y., Bettegowda C., Weller M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018;15:422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 5.Sampson J.H., Gunn M.D., Fecci P.E., Ashley D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer. 2020;20:12–25. doi: 10.1038/s41568-019-0224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson C.M., Choi J., Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat. Immunol. 2019;20:1100–1109. doi: 10.1038/s41590-019-0433-y. [DOI] [PubMed] [Google Scholar]
- 7.Woroniecka K., Chongsathidkiet P., Rhodin K., Kemeny H., Dechant C., Farber S.H., Elsamadicy A.A., Cui X., Koyama S., Jackson C., et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 2018;24:4175–4186. doi: 10.1158/1078-0432.CCR-17-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lichty B.D., Breitbach C.J., Stojdl D.F., Bell J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 10.Prestwich R.J., Harrington K.J., Pandha H.S., Vile R.G., Melcher A.A., Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev. Anticancer Ther. 2008;8:1581–1588. doi: 10.1586/14737140.8.10.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ribas A., Dummer R., Puzanov I., VanderWalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E., et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chesney J., Puzanov I., Collichio F., Singh P., Milhem M.M., Glaspy J., Hamid O., Ross M., Friedlander P., Garbe C., et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J. Clin. Oncol. 2018;36:1658–1667. doi: 10.1200/JCO.2017.73.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lang F.F., Conrad C., Gomez-Manzano C., Yung W.K.A., Sawaya R., Weinberg J.S., Prabhu S.S., Rao G., Fuller G.N., Aldape K.D., et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018;36:1419–1427. doi: 10.1200/JCO.2017.75.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samson A., Scott K.J., Taggart D., West E.J., Wilson E., Nuovo G.J., Thomson S., Corns R., Mathew R.K., Fuller M.J., et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018;10:eaam7577. doi: 10.1126/scitranslmed.aam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heymann D.L., Hodgson A., Sall A.A., Freedman D.O., Staples J.E., Althabe F., Baruah K., Mahmud G., Kandun N., Vasconcelos P.F., et al. Zika virus and microcephaly: why is this situation a PHEIC? Lancet. 2016;387:719–721. doi: 10.1016/S0140-6736(16)00320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mlakar J., Korva M., Tul N., Popovic M., Poljsak-Prijatelj M., Mraz J., Kolenc M., Resman Rus K., Vesnaver Vipotnik T., Fabjan Vodusek V., et al. Zika virus associated with microcephaly. N. Engl. J. Med. 2016;374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 17.Ming G.L., Tang H., Song H. Advances in Zika virus research: stem cell models, challenges, and opportunities. Cell Stem Cell. 2016;19:690–702. doi: 10.1016/j.stem.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parra B., Lizarazo J., Jimenez-Arango J.A., Zea-Vera A.F., Gonzalez-Manrique G., Vargas J., Angarita J.A., Zuniga G., Lopez-Gonzalez R., Beltran C.L., et al. Guillain-barre syndrome associated with Zika virus infection in Colombia. N. Engl. J. Med. 2016;375:1513–1523. doi: 10.1056/NEJMoa1605564. [DOI] [PubMed] [Google Scholar]
- 19.Petersen E., Wilson M.E., Touch S., McCloskey B., Mwaba P., Bates M., Dar O., Mattes F., Kidd M., Ippolito G., et al. Rapid spread of Zika virus in the Americas--implications for public health preparedness for mass gatherings at the 2016 Brazil olympic games. Int. J. Infect. Dis. 2016;44:11–15. doi: 10.1016/j.ijid.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Li C., Xu D., Ye Q., Hong S., Jiang Y., Liu X., Zhang N., Shi L., Qin C.F., Xu Z. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell. 2016;19:120–126. doi: 10.1016/j.stem.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Chen Q., Wu J., Ye Q., Ma F., Zhu Q., Wu Y., Shan C., Xie X., Li D., Zhan X., et al. Treatment of human glioblastoma with a live attenuated Zika virus vaccine candidate. MBio. 2018;9:e01683. doi: 10.1128/mBio.01683-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Z., Gorman M.J., McKenzie L.D., Chai J.N., Hubert C.G., Prager B.C., Fernandez E., Richner J.M., Zhang R., Shan C., et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017;214:2843–2857. doi: 10.1084/jem.20171093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., Dewhirst M.W., Bigner D.D., Rich J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 24.Zhan X., Guo S., Li Y., Ran H., Huang H., Mi L., Wu J., Wang X., Xiao D., Chen L., et al. Glioma stem-like cells evade interferon suppression through MBD3/NuRD complex-mediated STAT1 downregulation. J. Exp. Med. 2020;217:e20191340. doi: 10.1084/jem.20191340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanerva A., Nokisalmi P., Diaconu I., Koski A., Cerullo V., Liikanen I., Tahtinen S., Oksanen M., Heiskanen R., Pesonen S., et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin. Cancer Res. 2013;19:2734–2744. doi: 10.1158/1078-0432.CCR-12-2546. [DOI] [PubMed] [Google Scholar]
- 26.Shan C., Xie X., Muruato A.E., Rossi S.L., Roundy C.M., Azar S.R., Yang Y., Tesh R.B., Bourne N., Barrett A.D., et al. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe. 2016;19:891–900. doi: 10.1016/j.chom.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang F.C., Li X.F., Deng Y.Q., Tong Y.G., Qin C.F. Excretion of infectious Zika virus in urine. Lancet Infect. Dis. 2016;16:641–642. doi: 10.1016/S1473-3099(16)30070-6. [DOI] [PubMed] [Google Scholar]
- 28.Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Leun A.M., Thommen D.S., Schumacher T.N. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer. 2020;20:218–232. doi: 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bommareddy P.K., Shettigar M., Kaufman H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018;18:498–513. doi: 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 31.Long L., Deng Y., Yao F., Guan D., Feng Y., Jiang H., Li X., Hu P., Lu X., Wang H., et al. Recruitment of phosphatase PP2A by RACK1 adaptor protein deactivates transcription factor IRF3 and limits type I interferon signaling. Immunity. 2014;40:515–529. doi: 10.1016/j.immuni.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Zitvogel L., Galluzzi L., Kepp O., Smyth M.J., Kroemer G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 33.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spranger S., Dai D., Horton B., Gajewski T.F. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31:711–723.e4. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaid C., Goulart E., Caires-Junior L.C., Araujo B.H.S., Soares-Schanoski A., Bueno H.M.S., Telles-Silva K.A., Astray R.M., Assoni A.F., Junior A.F.R., et al. Zika virus selectively kills aggressive human embryonal CNS tumor cells in vitro and in vivo. Cancer Res. 2018;78:3363–3374. doi: 10.1158/0008-5472.CAN-17-3201. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Z., Mesci P., Bernatchez J.A., Gimple R.C., Wang X., Schafer S.T., Wettersten H.I., Beck S., Clark A.E., Wu Q., et al. Zika virus targets glioblastoma stem cells through a SOX2-integrin alphavbeta5 Axis. Cell Stem Cell. 2020;26:187–204.e10. doi: 10.1016/j.stem.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S., Zhang Q., Tiwari S.K., Lichinchi G., Yau E.H., Hui H., Li W., Furnari F., Rana T.M. Integrin alphavbeta5 internalizes Zika virus during neural stem cells infection and provides a promising target for antiviral therapy. Cell Rep. 2020;30:969–983.e4. doi: 10.1016/j.celrep.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoenfeld A.J., Hellmann M.D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443–455. doi: 10.1016/j.ccell.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8:328rv324. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nduom E.K., Wei J., Yaghi N.K., Huang N., Kong L.Y., Gabrusiewicz K., Ling X., Zhou S., Ivan C., Chen J.Q., et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18:195–205. doi: 10.1093/neuonc/nov172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou K., Li C., Shi W., Hu X., Nandakumar K.S., Jiang S., Zhang N. Current progress in the development of Zika virus vaccines. Vaccines (Basel) 2021;9:1004. doi: 10.3390/vaccines9091004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paz-Bailey G., Rosenberg E.S., Sharp T.M. Persistence of Zika virus in body fluids - final report. N. Engl. J. Med. 2019;380:198–199. doi: 10.1056/NEJMc1814416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.