

Summary
Asthma is a complex pulmonary disorder with multiple pathological mechanisms. A key pathological feature of chronic asthma is airway remodeling, which is largely attributed to airway smooth muscle (ASM) hyperplasia that contributes to thickening of the airway wall and further drives asthma pathology. The cellular processes that mediate ASM cell proliferation are not completely elucidated. Using multiple approaches, we demonstrate that the adapter protein Abi1 (Abelson interactor 1) is upregulated in ∼50% of ASM cell cultures derived from patients with asthma. Loss-of-function studies demonstrate that Abi1 regulates the activation of Jak2 (Janus kinase 2) and STAT3 (signal transducers and activators of transcription 3) as well as the proliferation of both nonasthmatic and asthmatic human ASM cell cultures. These findings identify Abi1 as a molecular switch that activates Jak2 kinase and STAT3 in ASM cells and demonstrate that a dysfunctional Abi1-associated pathway contributes to the progression of asthma.
Subject areas: Respiratory medicine, Molecular biology, Cell biology
Graphical Abstract
Highlights
-
•
Asthma is a complex pulmonary disorder with multiple pathological mechanisms
-
•
Abi1 expression is upregulated in nearly 50% of human asthmatic ASM cultures
-
•
Abi1 regulates Jak2/STAT3 and proliferation of nonasthmatic and asthmatic ASM cells
Respiratory medicine; Molecular biology; Cell biology
Introduction
Asthma is a complex disease whose pathogenesis likely differs among multiple asthma phenotypes (Borish and Culp, 2008; Carr and Bleecker, 2016; Prakash, 2016; Prakash et al., 2017). A cardinal pathological characteristic of chronic asthma is airway remodeling, a key feature of which is airway smooth muscle (ASM) thickening, which is largely attributed to abnormal ASM cell proliferation (Hershenson et al., 2008; Jiang and Tang, 2015; Penn, 2008; Prakash, 2016; Prakash et al., 2017; Wang et al., 2013a). The increased thickness of the ASM layer within the airway wall exacerbates airway narrowing during asthma attacks. ASM layer thickening also facilitates airway hyperresponsiveness (AHR) to a variety of stimuli (Amrani et al., 2004; Tang, 2015; Wang et al., 2013a). However, the mechanisms that mediate ASM proliferation in asthma are not fully elucidated.
Abi1 (Abelson interactor 1) is an adapter protein that plays a role in actin cytoskeletal remodeling (Innocenti et al., 2005), intercellular adhesion (Ryu et al., 2009), cardiovascular development (Ring et al., 2011), cell migration (Stradal et al., 2001; Wang et al., 2020), and smooth muscle contraction (Wang et al., 2013b). Abi1 has also been implicated in growth signaling and cancer cell growth (Kotula, 2012). The human Abi1 gene is localized in the Chromosome 10p21 region. Genome-wide association studies (GWASs) suggest that Chromosome 10p21 is adjacent to a susceptible locus for asthma and related traits (Akhabir and Sandford, 2011; Myers et al., 2014). In contrast, Abi2, a functional homolog of Abi1, has a negative impact on cancer cell proliferation (Lu et al., 2020), and the human Abi2 gene is localized to Chromosome 2q33.2, which is not proximal to susceptible loci for asthma (Akhabir and Sandford, 2011; Myers et al., 2014). Abi2 has been shown to regulate epithelial-mesenchymal transition of nasopharyngeal carcinoma cells through c-JUN/SLUG signaling (Huang et al., 2020). Moreover, Abi1 affects the activation of Jaks (Janus kinases) and STATs (signal transducers and activators of transcription) in bone marrow (Chorzalska et al., 2018). Nevertheless, the knowledge regarding the role of Abi1 in human asthma and Jaks-associated pathway remains limited.
The Jaks are non-receptor protein tyrosine kinases and consist of Jak1, Jak2, Jak3, and Tyk2 (Ghoreschi et al., 2009; O'Shea et al., 2013). Jaks have been implicated in cytokine-associated signaling in immune cells and cancer cells. Jaks constitutively interact with the cytoplasmic domains of cytokine receptors. Binding of cytokines to corresponding cytokine receptors leads to dimerization of receptors, promoting transphosphorylation and activation of Jaks at tyrosine residues. Activated Jaks mediate phosphorylation at tyrosine residues of cytoplasmic tails of cytokine receptors, which serve as docking sites for STATs. Jaks then phosphorylate STATs, which translocate to the nucleus, by which STATs promote gene expression and cell proliferation (Ghoreschi et al., 2009; Hubbard, 2011; Imada and Leonard, 2000; Shan et al., 2014; Simon et al., 2002).
ASM cells derived from asthmatic patients possess intrinsic biological properties. Cultures of human airway smooth muscle (HASM) cells derived from asthmatic patients proliferate faster than do cultured HASM cells derived from nonasthmatic subjects (Johnson et al., 2001; Liao et al., 2018). This intrinsic property is retained with extensive culture passage. Thus, asthmatic HASM cells serve as an excellent experimental model to investigate whether Abi1 expression or function associates with asthma pathogenesis.
In the current study, we demonstrate that Abi1 is upregulated in ∼50% of cultures of ASM derived from asthmatic patients, while Abi1 is shown to be a critical mediator of HASM cell proliferation, as well as AHR and ASM thickening in an animal model of allergic asthma. Moreover, pro-mitogenic effects of Abi1 are mediated via intracellular activation of Jak2 in cells stimulated with platelet-derived growth factor (PDGF) (a major growth factor that contributes to asthma pathology) (Ammit and Panettieri, 2003; Brzozowska et al., 2015).
Results
Upregulation of Abi1 expression in asthmatic HASM cells and tissues
Because GWASs indicate that the human Abi1 gene is near a susceptible locus for asthma and related traits (Akhabir and Sandford, 2011; Mathias, 2014), we evaluated the expression of Abi1 in HASM cultures derived from nonasthmatic and asthmatic donors. Basic information of these donors was described in Table 1. We found that nonasthmatic HASM cultures exhibited relatively lower and consistent expression of Abi1 mRNA (Figure 1A). In contrast, we found variable Abi1 expression in asthmatic ASM cultures, with 10 of 21 cultures (48%) of asthmatic ASM cells displaying >1.5-fold Abi1 mRNA expression (Figure 1B). The mean of Abi1 mRNA expression was significantly higher in asthmatic versus nonasthmatic ASM cultures (Figure S1A). Abi2 mRNA expression exhibited limited variability and did not differ between nonasthmatic and asthmatic HASM cultures (Figures 1C and 1D; Figure S1B). Abi1 protein levels were similarly upregulated in the same HASM cell cultures exhibiting higher Abi1 mRNA levels (Figures 1E and 1F) (hereafter these cell lines are referred to as Abi1-up cells). Protein expression levels of Abi1 were significantly greater in asthmatic versus nonasthmatic ASM cultures (Figures S1C and S1D). Finally, expression of Abi1 was higher in asthmatic small airway tissues as assessed by immunohistochemistry (Figure S1E). These results demonstrate that Abi1 expression in HASM is upregulated in a large subpopulation of asthmatics, consistent with the notion that asthma is a pulmonary syndrome with distinct pathologic pathways (Borish and Culp, 2008; Carr and Bleecker, 2016).
Table 1.
Donor characteristics
| Control, N = 24 | Asthma, N = 21 | p value | |
|---|---|---|---|
| Age (years) | 37.42 ± 15.35 | 31.90 ± 14.94 | NS |
| Gender, male | 13 (54.16%) | 10 (47.6%) | – |
| Race, White/African American/Hispanic + Asian | 14/7/3 | 14/5/2 |
Figure 1.

Expression of Abi1 is upregulated in asthmatic human airway smooth muscle (HASM) cells
(A and B) Abi1 mRNA in nonasthmatic HASM cultures (n = 24) and asthmatic HASM cultures (n = 21) was evaluated by RT-qPCR. Abi1 mRNA is upregulated in 10 of 21 asthmatic cultures.
(C and D) Abi2 mRNA is similar between nonasthmatic and asthmatic cultures. mRNA normalization is calculated using the following formula: Abi1 RQ (relative quantification) of individual cell culture line divided by the means of Abi1 RQ of all control cell cultures.
(E and F) Abi1 protein in HASM cultures was determined using ELISA. Abi1 protein is enhanced in asthmatic ASM cultures with increased Abi1 mRNA expression.
Regulation of HASM cell proliferation by Abi1
Given that asthmatic HASM cells are known to proliferate faster (Johnson et al., 2001; Liao et al., 2018), and the current finding of Abi1 upregulation in multiple asthmatic ASM cultures, we questioned whether Abi1 plays a role in ASM cell proliferation by determining the effects of Abi1 knockdown (KD) and rescue on mitogen-stimulated HASM cells. Stable Abi1 KD cells were treated with lentivirus encoding Abi1 isoform 8 (NM_001178121.2) to rescue Abi1 expression. Immunoblot analysis verified effective Abi1 KD and rescue in the cells (Figure 2A), consistent with our previous results (Wang et al., 2013b, 2020). Abi1 KD attenuated the PDGF-induced enhancement of HASM cell numbers as well as BrdU incorporation, both of which were restored by Abi1 rescue (Figures 2B and 2C). Moreover, the overexpression of Abi1 sufficiently enhanced cell proliferation (Figures 2D–2F). The results demonstrate that Abi1 positively regulates HASM proliferation and that Abi1 isoform 8 is a major isoform in this cell type.
Figure 2.

Abi1 regulates HASM proliferation
(A) Protein expression of control, stable Abi1 knockdown (KD), and rescue cell cultures from non-asthmatic HASM was evaluated by immunoblotting. Abi1 shRNA inhibits Abi1 protein expression, which is restored by rescue. Data are means ± SEM. n = 5. One-way ANOVA was used for statistical analysis.
(B and C) Abi1 knockdown (KD) and rescue affect HASM cell numbers (B) and BrdU incorporation (C). Data are normalized to numbers of cells treated with control (Ctrl) shRNA or microplate readings of cells treated with Ctrl shRNA. Data are means ± SEM. n = 5. One-way ANOVA was used for statistical analysis.
(D) Abi1 overexpression in HASM cells. Data are means ± SEM. n = 5. The t test was used for statistical analysis.
(E and F) The overexpression (OE) of Abi1 enhances HASM cell numbers (E) and (F). Data are means ± SEM. n = 5. The t test was used for statistical analysis. ∗p < 0.05; ∗∗p < 0.01. Data normalization is calculated as follows: Data of treated samples/Data of control samples.
Effects of PDGF stimulation on phosphorylation of Jak and STAT isoforms
As described earlier, phosphorylation and activation of Jaks and STATs have been implicated in cytokine-mediated signaling and proliferation. Given the many isoforms of Jaks and STATs (Ghoreschi et al., 2009), we evaluated the effects of PDGF (10 ng/mL, 10 min) on phosphorylation of different Jak and Stat isoforms in ASM. PDGF treatment induced phosphorylation of Jak2 but not that of Jak1, Jak3, and Tyk2 (Figures S2A–S2D). Moreover, PDGF stimulation induced phosphorylation of STAT1 and STAT3 but not that of STAT2 (Figures S2E–S2G). These results demonstrate that PDGF differentially regulates the phosphorylation and activation of Jaks and STATs in this cell type.
Abi1-mediated activation of Jak2 and STAT3 in ASM cells
We next questioned whether Abi1 regulates the phosphorylation of Jak2 and STAT1/3 induced by PDGF. KD of Abi1 attenuated the PDGF-induced phosphorylation of Jak2 at Y1007/Y1008 (indications of Jak2 activation) (Ghoreschi et al., 2009; Imada and Leonard, 2000; Simon et al., 2002) (Figure 3A). In addition, Abi1 KD attenuated the PDGF-induced STAT3 phosphorylation at Y705 (Figure 3B). However, Abi1 KD did not affect STAT1 phosphorylation (Figure S2H). Next, we assessed the effect of Abi1 KD on Jak2 kinase activity. Abi1 KD inhibited the PDGF-induced Jak2 activity in cells (Figure 3C). Using a STAT3 reporter assay (Zhong et al., 1994), we further determined PDGF-induced STAT3 promoter activity to be reduced by Abi1 KD (Figure 3D). Furthermore, overexpression of Abi1 increased the activity of Jak2 and STAT3 (Figures 3E–3F). These results demonstrate that Abi1 positively regulates the activation of Jak2 and STAT3 during stimulation with this growth factor.
Figure 3.

Abi1 modulates phosphorylation and activation of Jak2 and STAT3
(A and B) Abi1 KD attenuates the PDGF-induced Jak2 (A) and STAT3 (B) phosphorylation in HASM cells. Data are means ± SEM. n = 5. Two-way ANOVA was used for statistical analysis.
(C) Abi1 deficiency inhibits the PDGF-induced Jak2 kinase activity. Data are means ± SEM. n = 5. Two-way ANOVA was used for statistical analysis.
(D) The reporter activity of STAT3 is reduced by Abi1 KD. Data are means ± SEM. n = 5. Two-way ANOVA was used for statistical analysis.
(E and F) Overexpression of Abi1 in HASM cells enhances Jak2 kinase activity and STAT3 activity. Data are means ± SEM. n = 5. The t test was used for statistical analysis. ∗p < 0.05; ∗∗p < 0.01. Data normalization is calculated as follows: Data of treated samples/Data of control samples.
Induction of Abi1/Jak2 coupling by PDGF
Because both Abi1 and Jak2 have been implicated in growth signaling (Hubbard, 2017; Kotula, 2012), we questioned whether stimulation with PDGF affects the association of Abi1 with Jak2. First, we found that Jak2 constitutively localized on the membrane identified by the positive staining for PDGF receptor β (Figure 4A). In addition, treatment of cells with PDGF enhanced the interaction of Abi1 with Jak2 as evidenced by coimmunoprecipitation and corroborated by reverse coimmunoprecipitation (Figure 4B). We also generated EGFP-Abi1 (donor) and Jak2-dsRed (acceptor) for use in FRET analysis to assess the protein-protein interaction. PDGF stimulation increased the ratio of acceptor/donor in cells (Figure 4C). In addition, Abi1 translocated to the cell periphery from the cytoplasm and colocalized with Jak2 in response to PDGF stimulation (Figure 5A). However, Abi2 did not redistribute to the cell edge during stimulation with PDGF (Figure 5B). These findings demonstrate that Abi1 associates with Jak2 on the membrane in response to stimulation with this growth factor.
Figure 4.

Stimulation with PDGF promotes the engagement of Abi1 with Jak2
(A) Jak2 constitutively resides on the membrane of HASM cells. PDGF receptor is localized on the membrane.
(B) Coimmunoprecipitation (upper panel) and reverse coimmunoprecipitation (lower panel) demonstrate that treatment with PDGF enhances the engagement of Abi1 with Jak2. Data are means ± SEM. n = 4. The t test was used for statistical analysis.
(C) PDGF stimulation increases FRET signal in cells expressing Abi1-EGFP and Jak2-dsRed. Scale bars, 10 μm ∗∗p < 0.01. The images are representative of four independent experiments. Data normalization is calculated as follows: Data of treated samples/Data of control samples.
Figure 5.

PDGF induces the spatial redistribution of Abi1 in smooth muscle cells
(A) Jak2 is located on the edge of unstimulated HASM cells. PDGF treatment does not affect Jak2 localization. However, Abi1 translocates to the cell periphery upon PDGF stimulation. The arrows indicate a single line scan to analyze the fluorescence intensity for each cell. The inset plots represent fluorescence intensity of the line scan indicated by the arrows. Data are mean ± SEM (n = 32 cells from three independent experiments). The t test was used for statistical analysis.
(B) The cellular localization of Abi2 is not altered in response to PDGF treatment. Data are mean ± SEM (n = 33 cells from three independent experiments). The t test was used for statistical analysis. ∗p < 0.05. NS, not significant
(C) Abi1 KD does not affect the spatial distribution of Jak2 and PDGF receptor β. Data are mean ± SEM (n = 33–34 cells from three independent experiments). The t test was used for statistical analysis. NS, not significant. Protein distribution was analyzed using the methods described in method details. The relative intensity ratios (periphery/interior) are calculated using the following formula: ratio of each cell/average ratios of control cells. Scale bar, 10 μm
Membrane localization of Jak2 not affected by Abi1 KD
Because Abi1 can associate with Jak2, we questioned whether this protein-protein interaction affects Jak2 cellular localization as visualized by confocal immunofluorescence microscopy. Abi1 KD did not influence the membrane localization of Jak2 (Figure 5C). Since the membrane localization of growth factor receptors is essential for growth signaling, we also assessed the impact of Abi1 KD on the spatial distribution of PDGF receptor β (PDGFRβ, a major isoform in smooth muscle [Liao et al., 2015; Walker et al., 1998]) using an antibody against the extracellular domain of PDGFRβ. Abi1 KD did not affect the spatial localization of the receptor (Figure 5C). These results demonstrate that Abi1 regulates Jak2 activation without affecting the spatial localization of Jak2 and PDGFRβ in cells.
Modulation of PDGFRβ autophosphorylation by Abi1 and Jak2
Although growth factor receptors are believed to be autophosphorylated and activated upon ligand binding (Hackel et al., 1999; Seong et al., 2017), it is unknown whether their downstream targets reciprocally affect the activation of growth factor receptors. Thus, we evaluated whether Abi1 or Jak2 KD affects the phosphorylation of PDGFRβ. We unexpectedly found that Abi1 or Jak2 KD reduced the autophosphorylation of PDGFRβ at Y751 (Figure S3), an indication of PDGFRβ activation (Lin et al., 2003; Seong et al., 2017).
STAT3 nuclear translocation mediated by Abi1
Since cytokine activation induces STAT3 nuclear accumulation, which is critical for STAT3-regulated genes in immune cells (O'Shea et al., 2013), we questioned whether PDGF affects STAT3 nuclear import in ASM cells. PDGF treatment caused the time-dependent nuclear translocation of STAT3 in smooth muscle cells (Figures 6A and 6B). Furthermore, we found that the PDGF-induced STAT3 nuclear accumulation (2 h after stimulation) was reduced by Abi1 KD (Figure 6C). Overexpression of Abi1 in HASM cells was able to induce STAT3 nuclear import (Figure 6D). The results indicate that Abi1 regulates the nuclear distribution of STAT3.
Figure 6.

Abi1 regulates STAT3 nuclear import in smooth muscle cells
(A) Cells with perinuclear STAT3 localization are considered “untranslocated cells.” Cells with concentrated STAT3 distribution in the nucleus are considered “translocated cells.” The white arrow points to the nucleus without STAT3 translocation. The green arrows point to the nuclei with STAT3 translocation.
(B) PDGF stimulation induces STAT3 translocation to the nucleus. Data are means ± SEM. n = 5.
(C) Abi1 KD inhibits the PDGF-induced STAT3 nuclear import. Ctrl and Abi1 KD cells were treated with PDGF for 2 h followed by immunostaining. Data are means ± SEM. n = 4. Two-way ANOVA was used for statistical analysis.
(D) Overexpression (OE) of Abi1 is sufficient to induce STAT3 nuclear translocation. Cells were infected with lentivirus encoding the constructs for 2 days followed by staining. Data are means ± SEM. n = 4. The t test was used for statistical analysis. ∗∗p < 0.01. The percentage of cells with translocated STAT3 was calculated as follows: numbers of cells with translocated STAT3/numbers of total cells observed × 100.Scale bar, 10 μm
Effects of Abi1 KD on activation of Jak2/STAT3 and proliferation in Abi1-up HASM cells
Because Abi1 is upregulated in approximately half of all asthmatic HASM cultures we screened, we sought to assess whether the activity of Jak2 and STAT3 is also altered in these asthmatic Abi1-up HASM cultures. We found that the basal and the PDGF-induced activities of Jak2 and STAT3 were higher in Abi1-up cultures (Figures 7A and 7B). Abi1 KD inhibited the basal and the PDGF-induced activation of Jak2 and STAT3 in these cells (Figures 7C and 7D). Furthermore, Abi1 KD attenuated the proliferation of asthmatic Abi1-up cells as evidenced by analysis of cell number and BrdU incorporation (Figures 7E and 7F).
Figure 7.

Abi1 knockdown inhibits the activity of Jak2/STAT3 and the proliferation in Abi1-up HASM cells
(A and B) The basal and the PDGF-induced activity of Jak2 (A) and STAT3 (B) is higher in Abi1-up cells. Data are means ± SEM. n = 4 for each group. One-way ANOVA was used for statistical analysis.
(C and D) Abi1 KD inhibits the basal and the PDGF-induced activity of Jak2 (C) and STAT3 (D) in Abi1-up cells. Data are means ± SEM. n = 4 for each group. Two-way ANOVA was used for statistical analysis
(E and F) Abi1 KD reduced the PDGF-induced cell numbers (E) and BrdU incorporation (F). Data are means ± SEM. n = 4 for each group. The t test was used for statistical analysis. ∗p < 0.05. Data normalization is calculated as follows: Data of treated samples/Data of control samples.
Role of Abi1 in allergen-induced ASM thickening and AHR in vivo
Since Abi1 KD reduces proliferation of ASM cells in culture, we questioned whether Abi1 plays a role in ASM thickening in vivo by determining the effects of functional Abi1 knockout (KO) on phenotype in a murine animal model of asthma employing repeated exposure to house dust mite (HDM). HDM is a major perennial allergen source and a significant cause of allergic asthma (Calderon et al., 2015), and mice exposed to HDM develop lung inflammation, AHR, and airway remodeling (Shang et al., 2013; Southam et al., 2007). C57BL/6 mice were exposed to HDM extracts or PBS for 6 weeks using an InExpose system (Figure S4A). The expression of Abi1 in tracheal/bronchial tissues of HDM-treated mice was increased compared with that in PBS-treated mice (Figure S4B). To assess the role of Abi1 in asthma pathogenesis in vivo, we generated smooth muscle-conditional Abi1 KO (Abi1smko) mice by crossing Abi1−lox mice with Myh11-cre,-EGFP mice that express Cre recombinase under control of a smooth muscle-specific myosin heavy chain promoter (Figures S4C–S4E). We treated Abi1−lox and Abi1smko mice with PBS (control) or HDM extracts for 6 weeks and assessed ASM thickening and AHR. Exposure to the allergen increased the thickness of the ASM layer (α-smooth muscle actin positive) in the airways of Abi1−lox mice, which was reduced in Abi1smko mice (Figures 8A and 8B). The fluorescence intensity of α-smooth muscle actin staining was higher in allergen-treated Abi1−lox mice compared with PBS-treated Abi1−lox mice. Conditional KO of Abi1 attenuated the HDM-induced actin expression in the airways (Figure 8A). The fluorescence intensity of proliferating cell nuclear antigen (PCNA) in the airways was also greater in the airways of Abi1−lox mice treated with HDM than in Abi1−lox mice treated with vehicle, which was reduced by conditional Abi1 KO (Figures 8A and 8C). These results demonstrate that Abi1 has a role in allergen-induced ASM proliferation and thickening. In addition, Abi1 KO also attenuated HDM-induced airway inflammation (Figure S5). This is not surprising given ASM cells have been shown to be capable of generating cytokines and functioning in an immunomodulatory capacity (Lazaar and Panettieri, 2006; Sasse et al., 2016).
Figure 8.

Abi1 mediates the allergen-induced airway smooth muscle thickening and airway hyperresponsiveness in vivo
(A) Representative immunofluorescence images of lung sections showing effects of Abi1 conditional knockout (Abi1smko) on increases in airway smooth muscle thickness and proliferation induced by house dust mite (HDM) extracts. Treatment of Abi1−lox mice with HDM extracts increases actin positive area and PCNA staining, which are reduced in Abi1smko. Scale bar, 100 μm
(B and C) Quantification of actin-positive areas and the intensity of PCNA are evaluated by using NIH ImageJ software. Data are means ± SEM (n = 11–12, 6 males and 5–6 females for each group).
(D) Airway resistance (Rrs) was measured for Abi1−lox mice and conditional KO mice treated with PBS or HDM extracts.
(E) The sizes of lungs from Abi1−lox mice and Abi1smko mice are similar. Shown are lung images of two Abi1−lox mice and two Abi1smko mice.
(F) ACh dose-response curves were determined for tracheal rings from Abi1−lox mice and conditional KO mice treated with PBS or HDM extracts (n = 11–12, 6 males and 5–6 females for each group). Two-way ANOVA was used for statistical analysis. Data are means ± SEM. ∗∗p < 0.01; ∗p < 0.05. Data normalization is calculated as follows: Data of treated samples/Data of control samples.
Because ASM thickening affects AHR, we assessed the role of Abi1 in AHR in vivo and smooth muscle hyperreactivity ex vivo. HDM exposure increased airway resistance in Abi1−lox mice, which was attenuated in Abi1smko mice (Figure 8D). Abi1 conditional KO did not affect lung size of the animals (Figure 8E), suggesting that the reduced airway resistance is not due to smaller lung size of the animals. In vivo HDM treatment increased the acetylcholine-stimulated contractile response of tracheal rings isolated from Abi1−lox mice (Figure 8F). However, HDM-sensitized smooth muscle contraction was reduced in Abi1smko mice (Figure 8F). These results suggest that Abi1 contributes to the development of ASM hyperreactivity in allergen-induced asthma. In addition, Abi1 conditional KO did not completely inhibit AHR and contraction. The results indicate that Abi1 partially contributes to AHR and smooth muscle contraction. We also noticed that Abi1 conditional KO reduced airway resistance and tracheal ring contraction in PBS-treated mice (Figures 8D and 8F). This is because Abi depletion inhibits actin cytoskeletal remodeling and smooth muscle contraction (Wang et al., 2013b).
Discussion
Asthma is a complex pulmonary disorder with multiple pathological mechanisms (Borish and Culp, 2008; Carr and Bleecker, 2016). Although airway inflammation plays a role in asthma pathogenesis, there is increasing evidence to suggest that inflammation-independent processes may also contribute to asthma progression. In this report, Abi1 expression is shown to be upregulated in nearly 50% of the HASM cultures derived from asthmatic donor lungs. To the best of our knowledge, this is the first evidence to suggest that an adapter protein is associated with human asthma. Previous studies by our group and others suggest that the dysregulation of cytokines, intracellular Ca2+, enzymes (e.g., protein kinases), extracellular matrix proteins, and other molecules contributes to asthma pathogenesis (Balenga et al., 2015; Cleary et al., 2013; Li et al., 2016; Page et al., 2017; Tang, 2015; Tliba et al., 2003; Wills-Karp, 2004).
In this study, we used cultured primary HASM cells as a highly controllable reductionist model enabling avoidance of complicating factors such as exposure of cells to agents of airway inflammation and interactions with other airway wall cells (e.g., airway epithelium, fibroblasts). This model also helps avoid the acute effects of drugs such as β-agonists and corticosteroids used in the management of patients with asthma. Thus, the increased expression of Abi1 in asthmatic HASM cells in this study may represent an intrinsic property possibly mediated by genetic or epigenetic factors. GWASs demonstrate that the human Abi1 gene is near a susceptible locus for asthma and related traits (Akhabir and Sandford, 2011; Myers et al., 2014). Previous studies have also suggested that epigenetic alterations contribute to the intrinsic property of asthmatic smooth muscle cells (An et al., 2016; Kogan et al., 2018).
Binding of cytokines to their cognate receptors leads to dimerization of receptors, promoting transphosphorylation and activation of Jaks at tyrosine residues. Although there are four members of the Jak tyrosine kinase family, only Jak2 is activated in ASM cells in response to stimulation with the growth factor. Furthermore, our cell-based experiments demonstrate that stimulation with PDGF enhances the association of Abi1 with Jak2 and that Abi1 deficiency has a negative impact on Jak2 activation. Thus, Abi1 may serve as a molecular switch to regulate Jak2 activation in PDGF-mediated signaling.
Although Abi1 is an adapter protein, its presence affects the spatial localization of the actin-regulatory proteins Pfn-1, N-WASP, and c-Abl during cell migration (Wang et al., 2020). Here, Abi1 deficiency affects Jak2 activation without affecting the membrane localization of Jak2. The results suggest that Abi1 influences the functional state of Jak2 rather than its cellular localization. It is not surprising that Abi1 presence does not affect the spatial distribution of PDGFRβ because the receptor is a transmembrane protein.
Jaks constitutively interact with the cytoplasmic domains of cytokine receptors including growth factor receptors (Ghoreschi et al., 2009; Hubbard, 2011; Imada and Leonard, 2000; Shan et al., 2014; Simon et al., 2002). Here, exposure to PDGF increases the engagement of Abi1 with Jak2, which facilitates Jak2 activation. Furthermore, Abi1 and Jak2 conversely affect the autophosphorylation of PDGFRβ. We propose that PDGFRβ, Abi1, and Jak2 form a unique activation loop in response to PDGF activation. The association of Abi1 with Jak2 may stabilize the conformation of PDGFRβ, rendering PDGFRβ in active status during stimulation with this growth factor. This activation loop may assist cells to efficiently utilize their energy upon external activation (Wang et al., 2013b).
Our cell-based experiments show that stimulation with the growth factor activates STAT1 and STAT3, but not STAT2, in cells. In addition, Abi1 deficiency blunts the activation of STAT3 but not that of STAT1. Furthermore, Abi1 KD reduces STAT3 nuclear import. STAT3 is known to promote transcription of many genes necessary for cell proliferation (O'Shea et al., 2013). Abi1 KD also inhibits Jak2 activation as well as proliferation. Thus, we propose that activation of the growth factor initiates the coupling of Abi1 with Jak2, and promotes phosphorylation and activation of Jak2 and STAT3, which facilitates STAT3 nuclear transport, and modulates gene expression and cell proliferation. This does not exclude the possibility that Abi1 may regulate other pathways such as phosphoinositide 3-kinases and mitogen-activated protein kinase. In addition, transforming growth factor β (TGF-β) and oxidants have also been implicated in smooth muscle cell proliferation (Chen and Khalil, 2006; Wiegman et al., 2015). We do not rule out the possibility that TGF-β and oxidants may activate the Abi1-dependent pathway. Future studies are required to test the possibility.
The Jak2/STAT3 pathway is constitutively active in cancer cells, which have uncontrolled proliferation (O'Shea et al., 2013). Our findings demonstrate that Abi1 expression and Jak2/STAT3 activation are upregulated in asthmatic HASM cells. Abi1 deficiency inhibits Jak2/STAT3 activation and the proliferation in asthmatic HASM cells. In addition, smooth muscle conditional KO of Abi1 reduces the allergen-induced airway smooth muscle thickening in the animal model of asthma. The results suggest that the expression level of Abi1 in ASM contributes to ASM thickening in vivo. Previous studies have shown that Abi1 participates in the regulation of smooth muscle migration by affecting actin cytoskeletal remodeling (Innocenti et al., 2005; Stradal et al., 2001; Wang et al., 2020). Thus, it is likely that Abi1 contributes to the development of ASM thickening in asthma by controlling both cell proliferation and migration. Moreover, conditional Abi1 KO reduces AHR without affecting lung size. Using lentivirus-mediated shRNA, we previously demonstrated that Abi1 regulates the contraction of ASM ex vivo (Wang et al., 2013b). Airway smooth muscle hyperplasia also facilitates AHR to a variety of stimuli (Amrani et al., 2004; Tang, 2015; Wang et al., 2013a). Therefore, Abi1 mediates AHR likely by affecting both hyperplasia and biochemical regulation of smooth muscle contraction (Amrani et al., 2004; Tang, 2018; Wang et al., 2013b). In this investigation, Abi1 KO also reduces the allergen-induced airway inflammation in vivo. This finding is consistent with the known immunomodulatory capacity of ASM cells through secretion of cytokines/chemokines (Lazaar and Panettieri, 2006; Sasse et al., 2016).
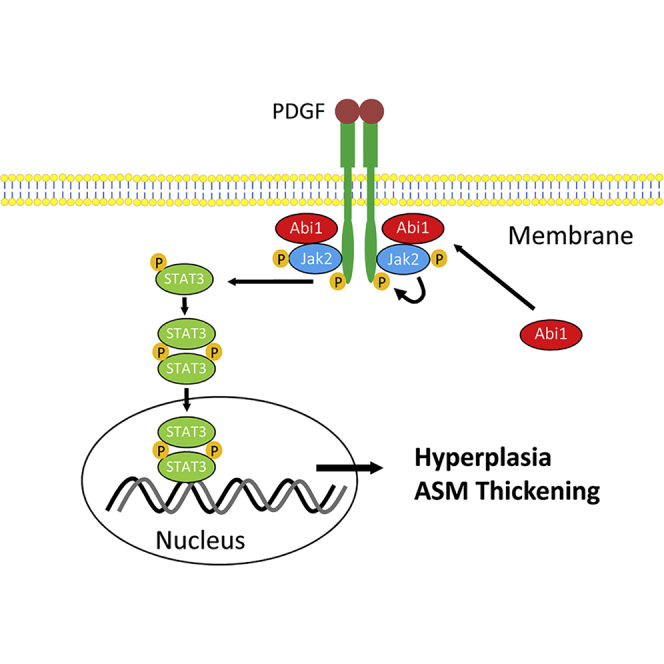
In summary, we demonstrate that Abi1 expression in ASM is increased in a significant subpopulation of asthmatics and regulation of Abi1 expression is shown to regulate ASM/airway remodeling and the asthma phenotype in various reductionist/integrative experimental models. Mechanistically, loss-of-function and/or gain-of-function studies demonstrate that Abi1 mediates ASM cell proliferation by regulating the Jak2-STAT3 pathway. The engagement of Abi1 with Jak2 provides a positive feedback for activation of the growth factor receptor (Figure 9). Furthermore, smooth muscle conditional Abi1 knockout reduces the three cardinal features of the asthma phenotype. Thus, we propose that a dysfunctional Abi1-associated pathway contribute to abnormal ASM proliferation and progression of asthma.
Figure 9.

Proposed mechanism
Stimulation with growth factors such as PDGF induces spatial distribution of Abi1, which facilitates the activation of the Jak2/STAT3 pathway. The association of Abi1 with Jak2 renders the receptor in active state. Abi1 is upregulated in ∼50% of asthmatic airway smooth muscle cultures, promoting the activation of Jak2 and STAT3, hyperplasia, and airway smooth muscle (ASM) thickening.
Limitations of the study
In this study, we focused on the role of Abi1 in the Jak2/STAT3 pathway. We did not evaluate the role of Abi1 in other growth factor-mediated pathways such as phosphoinositide 3-kinases and mitogen-activated protein kinase. Future studies are needed to determine whether Abi1 also affects these pathways.
STAR★Methods
Key resource talbe
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Abi1 | Sigma | #A5106-200UL, L/N 076M4842V:RRID:AB_2220843 |
| GAPDH | Santa Cruz Biotech | #sc-32233, K0315: RRID:AB_627679 |
| JAK2 | Santa Cruz Biotech | #sc-294, G1414:RRID:AB_631854 |
| JAK2 | Thermo | AHO1352, QC216934: RRID:AB_2536334 |
| phospho-Jak2 (Y1007/Y1008) | Cell Signaling | #3771S/10: RRID:AB_33040 |
| phospho-Jak1 (Y1034/Y1035) | Cell Signaling | #74129S/2: RRID:AB_2799851 |
| Jak1 | Cell Signaling | #50996/1: RRID:AB_2716281 |
| phospho-Jak3 (Y980/Y981) | Cell Signaling | #5031S/7: RRID:AB_10612243 |
| Jak3 | Cell Signaling | #5481/1: RRID:AB_10623287 |
| phospho-Tyk2 ((Y1054/Y1055) | Cell Signaling | #68790S/1: RRID:AB_2799752 |
| Tyk2 | Santa Cruz | #SC-5271/C1221: RRID:AB_628419 |
| phospho-STAT1 (Y701) | Cell Signaling | #9167S/25: RRID:AB_561284 |
| STAT1 | Cell Signaling | #9176S/8: RRID:AB_2240087 |
| phospho-STAT2 (Y960) | Cell Signaling | #88410S/4: RRID:AB_2800123 |
| STAT2 | Cell Signaling | #4597S/2: RRID:AB_2198305 |
| phospho-STAT3 (Y705) | Cell Signaling | #9145/1: RRID:AB_2491009 |
| STAT3 | Invitrogen | #MA1-13042/PJ208446: RRID:AB_10985240 |
| phospho-PDGFRβ (Tyr751) | Cell Signaling | #3161/7: RRID:AB_331053 |
| PDGFRβ | Proteintech | #13449-1-AP/00070807: RRID:AB_2162644 |
| Goat anti-Mouse 488 Alexa Fluor 488 | Life Technologies | A11001/1664729: RRID:AB_2534069 |
| Goat anti-Rabbit Alexa Fluor 488 | Life Technologies | A11008/1622775: RRID:AB_143165 |
| Goat anti-rabbit Alexa Fluor 546 | Life Technologies | A11010/1600212: RRID:AB_2534077 |
| Goat anti-Mouse Alexa Fluor 555 | Life Technologies | A21422/1608465: RRID:AB_141822 |
| Bacterial and virus strains | ||
| BL21DE3plysS | Promega | L1195 |
| Dh5α | ThermoFisher | 18258012 |
| Stbl3 | ThermoFisher | C737303 |
| One Shot Top10 | ThermoFisher | C404010 |
| Lentivirus encoding STAT3 luciferase promoter | BPS Bioscience | #79744 |
| Lentivirus encoding Abi1 shRNA | Santa Cruz | sc-40306-V |
| Lentivirus encoding control shRNA | Santa Cruz | sc-108080 |
| Biological samples | ||
| Primary human airway smooth muscle cells | This article | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Ham’s F-12 nutrient mix | GIBCO | 11765-047/2187149 |
| Protein A/G Plus-Agarose | Santa Cruz Biotech. | SC-2003 |
| PDGF-BB | Sigma | # P3201-10UG Lot# SLBW2507 & Lot# SLBZ6214 |
| SDS | Bio-Rad | 161-0302 |
| Human STAT3 | Abcam | Ab268982/GR3352364-3 |
| Trypan blue | Bio-Rad | 145-0013 |
| DAPI | Abnova | U0031/K004-HADBI |
| Triton-X-100 | Fisher | BP151-500 |
| Critical commercial assays | ||
| HiPure Plasmid Maxiprep Kit | ThermoFisher | K210007 |
| BrdU Cell Proliferation Kit | Millipore Sigma | 2750 |
| STAT3 luciferase reporter | BPS Bioscience | #79744 |
| Abi1 ELISA kit | Mybiosource | MBS2018864 |
| Kinase-Glo kinase assay | Promega | V6711 |
| Experimental models: Cell lines | ||
| Primary human airway smooth muscle cells | This article | N/A |
| Experimental models: Organisms/strains | ||
| Abi1smko mouse | This article | N/A |
| Oligonucleotides | ||
| Primer for Abi1 forward 5′-GAGCGCCCTGTAAGGTATATT-3′ |
This article | N/A |
| Primer for Abi1 reverse 5′-GTTCTTGCAGGCTGGTTATTTC-3′ |
This article | N/A |
| Primer for Abi2 forward 5′-CCGATTACTGCGAGAACAACTA-3′ |
This article | N/A |
| Primer for Abi2 reverse 5′-AGATAGGCAACACTTGCTAAGG-3′ |
This article | N/A |
| Primer for B2M forward 5′-TGCTGTCTCCATGTTTGATGTATCT-3′ |
This article | N/A |
| Primer for B2M reverse 5′-TCTCTGCTCCCCACCTCTAAGT-3′ |
This article | N/A |
| Recombinant DNA | ||
| pLenti-puro | Addgene | #39481 |
| pEGFP-Abi1 | Ryu et al. (2009) and this article | N/A |
| pdsRed-Jak2 | This article | N/A |
| pCR2.1 | Invitrogen | 450045/1416345 |
| Software and algorithms | ||
| Leica DMI 6000 software | Leica | http://microscopy.arizona.edu/sites/default/files/sites/default/files/upload/Leica_DMI6000_manual.pdf |
| GE IQTL software | GE | http://www.hhmi.umbc.edu/downloads/Imaging%20support%20GE/IQ%20TL%20collateral/IQTL_UserManual%208.pdf |
| Fuji Multi Gauge Software | Fuji | https://www.ualberta.ca/biological-sciences/media-library/mbsu/fla-5000/mulitgauge20.pdf |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Confocal microscopy LSM 880 with Airyscan | Zeiss | RRID:SCR_020925 |
| Leica TCS SPE | Leica | RRID:SCR_002140 |
| Microscope Leica MDI6000 | Leica | RRID:SCR_008960 |
| GloMax Multi Detection System | Promega | RRID:SCR_015575 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dale D. Tang (tangd@amc.edu).
Materials availability
pLenti-puro encoding Abi1, pEGFP-Abi1, dsRed-Jak2, and Abi1smko mice are available to the scientific community from the lead contact upon request.
Experimental model and subject details
Cell culture
Human airway smooth muscle (HASM) cells were prepared from human bronchi and adjacent tracheas obtained from 4 sources-the International Institute for Advanced Medicine, Rutgers University, Thomas Jefferson University, and the University of Chicago as previously described (Long et al., 2019; Wang et al., 2013a, 2013b, 2014a, 2014b, 2015), with studies herein approved by the Albany Medical College Committee on Research Involving Human Subjects. The donor human lungs used to procure tissue and cells were not suitable for transplant, and not identifiable, thus studies were determined to be Not Human Subjects Research. Smooth muscle cells passage 3–10 were used for the studies, as per (Cerutis et al., 1997; Kassel et al., 2008; Liao et al., 2018). The purity of HASM was determined by immunostaining for smooth muscle α-actin. Nearly 100% of these cells expressed α-actin (Liao et al., 2018). Basic characterization of these donors was described in Table 1.
Mice
B6.Cg-Tg (Myh11-cre,-EGFP) 2Mik/J mice were purchased from Jackson Laboratory, genetic background, C57BL/6J). Abi1−lox mice were generated by the Transgenic Mouse and Gene Targeting Core, Emory University School of Medicine; genetic background, C57BL/6). B6.Cg-Tg (Myh11-cre,-EGFP) 2Mik/J mice were crossed with Abi1−lox mice to generate smooth muscle conditional Abi1 knockout mice (Abi1smko mice). The target strategy and genotyping were included in supplemental information. Both male and female mice aging 6–8 weeks were used for the asthma models (See below).
Method details
Immunoblot analysis
Cells were lysed in SDS sample buffer composed of 1.5% dithiothreitol, 2% SDS, 80 mM Tris-HCl (pH 6.8), 10% glycerol and 0.01% bromophenol blue. The lysates were boiled in the buffer for 5 min and separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes. The membranes were blocked with bovine serum albumin or milk for 1 h and probed with use of primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies (Thermo Fisher Scientific). Proteins were visualized by enhanced chemiluminescence (Thermo Fisher Scientific) using the GE Amersham Imager 600 system. The levels of proteins were quantified by scanning densitometry of immunoblots (the Fuji Multi Gauge Software or GE IQTL software). The luminescent signals from all immunoblots were within the linear range (Li et al., 2016; Liao et al., 2018; Rezey et al., 2019; Wang et al., 2015, 2018).
Coimmunoprecipitation analysis
Protein-protein interactions and protein complex formation were evaluated by coimmunoprecipitation analysis as previously described (Li et al., 2016; Liao et al., 2018; Rezey et al., 2019; Wang et al., 2015, 2018) with minor modification. Briefly, cell extracts were incubated overnight with corresponding antibodies and then incubated for 3 h with 20 μL of the Protein A/G Plus-Agarose reagent (Santa Cruz Biotechnology). Immunocomplexes were washed four times in buffer containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl and 0.1% Triton X-100. The immunoprecipitates were separated by SDS-PAGE followed by transfer to nitrocellulose membranes. The membranes of immunoprecipitates were probed with use of corresponding antibodies.
Assessment of cell proliferation
Cells were seeded in 24-well plates with the F12 medium with 10% FBS for at least 18 h. Cells were then serum starved for 24 h. They were subsequently treated with PDGF-BB in the medium containing 0.25% FBS. Numbers of viable cells were counted using the trypan blue exclusion test (Jia et al., 2012). Briefly, cell suspension was incubated with 0.4% trypan blue for 5 min at room temperature. Unstained cells (viable cells) were then counted. The BrdU (5′-bromo-2′-deoxyuridine) cell proliferation assay kit (Millipore) was also used to evaluate DNA synthesis. BrdU is an analog of thymidine, which is able to incorporate into newly-synthesized DNA. Cells in 96-well (1.2 × 104 cells/well) were treated with BrdU for 5 hrs. They were then fixed and reacted with BrdU antibody for 1 h, followed by incubation with secondary antibody conjugated with peroxidase. They were reacted with peroxidase substrates, and the reaction was detected using a Promega GloMax-Multi Microplate reader (Liao et al., 2018; Long et al., 2019).
Antibodies
Antibodies used were anti-Abi1 (1:1000, Sigma #A5106-200UL, L/N 076M4842V), anti-GAPDH (1: 1000, Santa Cruz Biotechnology #sc-32233, K0315), anti-JAK2 (Santa Cruz Biotechnology #sc-294, G1414 and Thermo# AHO1352, QC216934), anti-phospho-Jak2 (Y1007/Y1008) (Cell Signaling, #3771S/10), anti-phospho-Jak1 (Y1034/Y1035) (Cell Signaling, # 74129S/2), anti-Jak1 (Cell Signaling, #50996), anti-phospho-Jak3 (Y980/Y981) (Cell Signaling, # 5031S/7), anti-Jak3 (Cell Signaling, #5481), anti-phospho-Tyk2 ((Y1054/Y1055) (Cell Signaling, # 68790S/1), anti-Tyk2 (SC-5271), anti-phospho-STAT1 (Y701) (Cell Signaling, #9167S/25), anti-STAT1 (Cell Signaling, #9176S), anti-phospho-STAT2 (Y960) (Cell Signaling, # 88410S/4), anti-STAT2 (Cell Signaling, #4597S), anti-phospho-Stat3 (Y705) (Cell Signaling, #9145/1), anti-Stat3 (Invitrogen, #MA1-13042/PJ208446), anti-phospho-PDGFRβ (Tyr751) (Cell Signaling, #3161/7) and anti-PDGFRβ (Proteintech, #13449-1-AP/# 00070807). The antibodies were validated by examining the molecular weight of target proteins. In addition, anti-Abi1 was validated by using Abi1 KD cells. Finally, vendors have provided datasheet to show that antibodies were validated by positive controls. For protein phosphorylation experiments, cells were treated with 10 ng/mL PDGF for 10 min followed by immunoblot analysis (see below).
Lentiviral transduction
Stable KD cells were generated using lentiviruses encoding target shRNA as previously described (Jia et al., 2012; Wang et al., 2013a). Briefly, lentiviruses encoding Abi1 shRNA (sc-40306-V) and control shRNA (sc-108080) were purchased from Santa Cruz Biotechnology. HASM cells were infected with control shRNA lentiviruses or target shRNA lentiviruses for 12 h followed by 3–4 day culture, and selected with puromycin to generate positive clones expressing shRNAs. The expression levels of Abi1 were assessed by immunoblot analysis. Abi1 KD cells and cells expressing control shRNA were stable for at least five passages after initial infection. For the Abi1 rescue experiment, Abi1 KD cells were treated with lentiviruses encoding RNAi-resistant Abi1 construct. Abi1 rescue in the KD cells was verified by immunoblotting.
Overexpression of Abi1
Abi1 cDNA was collected from pCR2.1 T/A cloning plasmid encoding human Abi1 and subcloned to pLenti-puro (Addgene #39481) at EcoRI and XhoI sites. Plasmids (pLenti-puro in BL21DE3plysS, pVSVG in DH5α, and pCMV in Stble3) were purified using the HiPure Plasmid Maxiprep Kit (Invitrogen). To produce viruses, 293FT cells were transfected with pLenti-puro encoding Plk1 plus packaging vector pCMV and envelop vector pVSV-G. Viruses were collected 48 h after transfection. For infection, smooth muscle cells were incubated with viruses 12 h, then cultured in the F12 growth medium for 2 days. Positive clones were selected by puromycin. Protein expression was assessed by immunoblotting.
Confocal immunofluorescence microscopy
Cells were plated in dishes containing coverslips followed by fixation and permeabilization (Jia et al., 2012; Li et al., 2006; Tang et al., 2005; Wang et al., 2013a). These cells were blocked with 2% BSA in PBS buffer for 30 min at room temperature. These cells were then reacted with primary antibody (1:20 dilution for anti-Jak2, anti-PDGFRβ, anti-Abi2 and anti-STAT3; 1:50 dilution for Anti-Abi1) 1 hour followed by PBS buffer wash three times. They were incubated with appropriate secondary antibody conjugated to Alexa 488 or Alexa 555 (Invitrogen, ThermoFisher, 1:200 dilution) for 1 hour. If double staining was needed. These cells were reacted with second primary antibody followed by second secondary antibody. To visualize nuclei, cells were incubated with 200 ng/mL DAPI (4′,6-diamidino-2-phenylindole) for 45 min. The cellular localization of fluorescently labeled proteins was viewed by using a Leica TCS SPE confocal microscope (Buffalo Grove, IL, United States) or a Zeiss LSM 880 NLO confocal microscope with Fast Airyscan module (Carl Zeiss Microscopy Jena, Germany).
For assessment of spatial distribution of proteins, cells were treated with 10 ng/mL PDGF for 10 min followed by immune fluorescence microscopy. Image analysis for protein localization was performed using the previously-described method with minor modification (Wang et al., 2007, 2021). By using Leica DMI 6000 software, the pixel intensity was quantified for minimal five line scans across the periphery of cells (nuclei excluded). Ratios of pixel intensity at the cell edge to pixel intensity at the cell interior were determined for each line scan as follows: ratios of the average maximal pixel intensity at the cell periphery to minimal pixel intensity in the cell interior. The ratios of pixel intensity at the cell border to that in the cell interior for all the line scans performed on a given cell were averaged to obtain a single value for the ratio of each cell. For data analysis of STAT3 nuclear import, the percentage of cells with translocated STAT3 was calculated as follows: numbers of cells with translocated STAT3/numbers of total cells observed × 100.
For live-cell experiments, cells were transfected with pEGFP encoding Abi1 (D, donor) and plasmids for dsRed-Jak2 (A, acceptor). After treatment with PDGF, the cells were excited at 488 nm and emission of dsRed (578–650 nm) (A) and EGFP (500–550 nm) (D) was collected using the Zeiss LSM 880 NLO confocal microscope. The Fluorescence Resonance Energy Transfer (FRET) signal was calculated using the formula: Erel = IA/(ID + IA), where IA and ID are the total A and D fluorescence intensities, respectively, both following D excitation. The FRET signal after PDGF stimulation was normalized to unstimulated cells.
Determination of STAT3 reporter activity
Cells were treated with lentivirus encoding STAT3 luciferase reporter (BPS Bioscience, #79744) for 12 h followed by 3-4 culture. Positive cells were then selected by using puromycin. To assess the STAT3 reporter activity, cells were seeded in 96-well plates followed by serum free treatment. The cells were then treated with or without 10 ng/mL PDGF for 18 h. The luciferase activity was evaluated using the One-step Luciferase Assay System kit (BPS Bioscience) according to the manual of the manufacture. Cells without the lentivirus infection were used as a control.
Assessment of Jak2 kinase activity
Cells were treated with 10 ng/mL PDGF for 10 min Jak2 immunoprecipitates were added to kinase buffer containing 50 mM Tris-Cl (pH7.5), 10 mM MgCl2, 1 mM DTT, 0.1 μg STAT3 and 5 μM ATP at 37°C for 30 min. The reaction solution was mixed with the Kinase-Glo reagent (Promega). The reaction was measured using a Glomax Multi-detection system (Promega).
Assessment of mRNA
Total RNA was extracted using the GeneJet RNA Purification kit (Thermo Scientific). The levels of mRNA were determined by reverse transcription quantitative real-time PCR (RT-qPCR). For the detection of human Abi1 mRNA, the 5′-primer sequence was 5′-GAGCGCCCTGTAAGGTATATT-3′; the 3′-primer sequence was 5′-GTTCTTGCAGGCTGGTTATTTC-3′. The 5′-primer sequence of human Abi2 was 5′-CCGATTACTGCGAGAACAACTA-3′; the 3′-primer sequence of human Abi2 was 5′-AGATAGGCAACACTTGCTAAGG-3′. Human β2-microglobulin (B2M) mRNA was used as a reference gene. The 5′-primer sequence of B2M was 5′-TGCTGTCTCCATGTTTGATGTATCT-3′; the 3′-primer sequence of B2M was 5′-TCTCTGCTCCCCACCTCTAAGT-3′. Briefly, total RNA and primers were mixed with the iTaq Universal SYBR Green One-Step Kit (Bio-Rad) and the mRNA levels were detected using a real-time PCR detection system (Bio-Rad). The expression level of target mRNA was calculated using the following formulas (Schmittgen et al., 2000):
-
a.
ΔCt (test samples) = Ct (target in test) - Ct (reference gene in test)
-
b.
ΔCt (calibrator samples) = Ct (target in calibrator) – Ct (reference gene in calibrator)
-
c.
ΔΔCt = ΔCt (test samples) - ΔCt (calibrator samples)
-
d.
Relative quantification (RQ) = 2-ΔΔCt
Assessment of Abi1 protein by ELISA
Proteins in cells were extracted using the buffer containing 2% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH7.6) and 0.2% SDS. Abi1 protein in extracts was determined using the ELISA kit (Mybiosource.com) according to the manual of the manufacture. Protein concentration was evaluated by the BCA assay. Readings of ELISA and BCA was collected using a Glomax Multi-detection system (Promega). Triplicates of each sample were used for data analysis.
Assessment of airway hyperresponsiveness
All animal protocols were reviewed and approved by the Institutional Animal Care and Usage Committee (IACUC) of Albany Medical College. All experiments were strictly performed in accordance with approved protocols and regulations of IACUC. Animals were bred in the specific pathogen free housing of the Animal Research Facility, Albany Medical College. Age- and sex-matched Abi1−lox and Abi1smko mice (6–8 weeks old) were exposed to HDM extract (50 μg/100 μL, d. pteronyssinus, Greer) or PBS (control) by using the InExpose system (SCIREQ, Montreal, Canada) for 5 days followed by every other day exposures weekly for 5 weeks. On Day 43, mice were anesthetized with intraperitoneal injection of ketamine/xylazine cocktail, tracheotomized, and connected to the FlexiVent system (SCIREQ, Montreal, Canada). Mice were mechanically ventilated at 150 breaths/minute with a tidal volume of 10 mL/kg and a positive end-expiratory pressure (PEEP) of 3.35 cm H2O. Following baseline measurements, mice were challenged with methacholine (MCh) aerosol for 10 seconds at different doses. Airway resistance was measured for each mouse after inhalation of the aerosol.
Assessment of tracheal ring contraction
Mice were euthanized by intraperitoneal injection of euthanasia solution (VEDCO, 0.1 mL/25 g). All experimental protocols were approved by the Institutional Animal Care and Usage Committee. A segment of tracheas (4–5 mm in length) was immediately removed and placed in physiological saline solution (PSS) containing 110 mM NaCl, 3.4 mM KCl, 2.4 mM CaCl2, 0.8 mM MgSO4, 25.8 mM NaHCO3, 1.2 mM KH2PO4, and 5.6 mM glucose. The solution was aerated with 95%O2-5%CO2 to maintain a pH of 7.4. Two stainless steel wires were passed through the lumen of tracheal rings. One of the wires was connected to the bottom of organ baths and the other was attached to a Grass force transducer that had been connected to a computer with A/D converter (Grass). Tracheal segments were then placed in PSS at 37°C. At the beginning of each experiment, 0.5 g passive tension was applied to tracheal rings. After 60 min equilibrium they were stimulated with 80 mM KCl repeatedly until contractile responses and passive tension reached a steady state. Contractile force in response to acetylcholine was then measured.
Immunohistochemistry
Mouse lungs were placed in frozen tissue-embedding medium (Neg 52, Richard-Allen Scientific) and cryosectioned using Cryostats (Richard-Allen Scientific). Tissue sections were fixed for 15 min in 4% paraformaldehyde, and were then washed three times in PBS buffer followed by permeabilization with 0.2% Triton X-100 dissolved in PBS for 5 min. These tissues were incubated with α-smooth muscle actin antibody (Sigma) or proliferating cell nuclear antigen (PCNA) antibody (Thermo Scientific) followed by appropriate secondary antibody conjugated to Alexa 488 or Alex-543 (Molecular Probes/Life Technologies). The sections were also counterstained with 4′,6-diamidino-2-phenylindole to visualize the nucleus. The samples were viewed and digitally captured using a Leica microscope system (MDI 6000) (Cleary et al., 2013; Li et al., 2016). All immunohistochemical measurements were performed by using the NIH ImageJ software.
Analysis of airway inflammation
Lungs from sacrificed mice were lavaged three times with 1 mL sterile Hanks balanced salt solution (HBSS) containing 3 mM EDTA. Bronchoalveolar lavage fluid (BALF) was collected after centrifugation and, the supernatant was removed and frozen at −80 °C for cytokine/chemokine measurements (See below). The cell pellet was resuspended in HBSS, and total number of inflammatory cells in the BALF was counted by using a hemocytometer. Differential cell counts (macrophages, neutrophils, lymphocytes, and eosinophils) were performed by counting 100 cells from cytospin preparations stained with DiffQuick stain. The levels of IL-13 in the BALF were determined using the ELISA kits (R&D systems) according to the manufacturer’s instructions.
Quantification and statistical analysis
All statistical analysis was performed using Prism software (GraphPad Software, San Diego, CA). Differences between pairs of groups were analyzed by a 2-tailed Student’|'s t-test for 2-group comparisons for normally distributed continuous data. A comparison among multiple groups was performed by one-way or two-way ANOVA followed by a post hoc test (Tukey’s multiple comparisons) for normally distributed continuous data. Values of n refer to the number of experiments used to obtain each value. p < 0.05 was considered to be significant.
Acknowledgments
The authors thank Alyssa C. Rezey, Brennan Gerlach, Olivia Gannon, Eylon Arbel, Saiyang Hu, and Thomas Brown of Albany Medical College for technical assistance.
This work was supported by NHLBI Grants HL-110951 (to D.D.T.), HL-130304 (to D.D.T.), HL-145392 (to R.B.P. and D.D.T.), and PO1-HL114471 (to R.A.P. and R.B.P.).
Author contributions
R.W., Y.W., G.L., and B.C. performed experiments. R.A.P. and R.B.P. provided research materials and analyzed and interpreted the data. D.D.T. conceived the research direction and coordinated the study. R.W., D.D.T., and R.B.P. wrote the manuscript. All authors approved the final version of the manuscript.
Declaration of interests
The authors have declared that no conflict of interest exists.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103833.
Supplemental information
Data and code availability
-
•
All data are included in the published paper and supplemental information files or available from the lead contact up request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Akhabir L., Sandford A.J. Genome-wide association studies for discovery of genes involved in asthma. Respirology. 2011;16:396–406. doi: 10.1111/j.1440-1843.2011.01939.x. [DOI] [PubMed] [Google Scholar]
- Ammit A.J., Panettieri R.A., Jr. Airway smooth muscle cell hyperplasia: a therapeutic target in airway remodeling in asthma? Prog. Cell Cycle Res. 2003;5:49–57. [PubMed] [Google Scholar]
- Amrani Y., Tliba O., Deshpande D.A., Walseth T.F., Kannan M.S., Panettieri R.A., Jr. Bronchial hyperresponsiveness: insights into new signaling molecules. Curr. Opin. Pharmacol. 2004;4:230–234. doi: 10.1016/j.coph.2004.02.004. [DOI] [PubMed] [Google Scholar]
- An S.S., Mitzner W., Tang W.Y., Ahn K., Yoon A.R., Huang J., Kilic O., Yong H.M., Fahey J.W., Kumar S., et al. An inflammation-independent contraction mechanophenotype of airway smooth muscle in asthma. J. Allergy Clin. Immunol. 2016;138:294–297.e4. doi: 10.1016/j.jaci.2015.12.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balenga N.A., Klichinsky M., Xie Z., Chan E.C., Zhao M., Jude J., Laviolette M., Panettieri R.A., Jr., Druey K.M. A fungal protease allergen provokes airway hyper-responsiveness in asthma. Nat. Commun. 2015;6:6763. doi: 10.1038/ncomms7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borish L., Culp J.A. Asthma: a syndrome composed of heterogeneous diseases. Ann. Allergy Asthma Immunol. 2008;101:1–8. doi: 10.1016/S1081-1206(10)60826-5. [quiz 8–11, 50] [DOI] [PubMed] [Google Scholar]
- Brzozowska A., Majak P., Jerzynska J., Smejda K., Bobrowska-Korzeniowska M., Stelmach W., Koczkowska M., Stelmach I. Exhaled nitric oxide correlates with IL-2, MCP-1, PDGF-BB and TIMP-2 in exhaled breath condensate of children with refractory asthma. Postepy. Dermatol. Alergol. 2015;32:107–113. doi: 10.5114/pdia.2014.40953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon M.A., Linneberg A., Kleine-Tebbe J., De Blay F., Hernandez Fernandez de Rojas D., Virchow J.C., Demoly P. Respiratory allergy caused by house dust mites: what do we really know? J. Allergy Clin. Immunol. 2015;136:38–48. doi: 10.1016/j.jaci.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Carr T.F., Bleecker E. Asthma heterogeneity and severity. World Allergy Organ. J. 2016;9:41. doi: 10.1186/s40413-016-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutis D.R., Nogami M., Anderson J.L., Churchill J.D., Romberger D.J., Rennard S.I., Toews M.L. Lysophosphatidic acid and EGF stimulate mitogenesis in human airway smooth muscle cells. Am. J. Physiol. 1997;273:L10–L15. doi: 10.1152/ajplung.1997.273.1.L10. [DOI] [PubMed] [Google Scholar]
- Chen G., Khalil N. TGF-beta1 increases proliferation of airway smooth muscle cells by phosphorylation of map kinases. Respir. Res. 2006;7:2. doi: 10.1186/1465-9921-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorzalska A., Morgan J., Ahsan N., Treaba D.O., Olszewski A.J., Petersen M., Kingston N., Cheng Y., Lombardo K., Schorl C., et al. Bone marrow-specific loss of ABI1 induces myeloproliferative neoplasm with features resembling human myelofibrosis. Blood. 2018;132:2053–2066. doi: 10.1182/blood-2018-05-848408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary R.A., Wang R., Wang T., Tang D.D. Role of Abl in airway hyperresponsiveness and airway remodeling. Respir. Res. 2013;14:105. doi: 10.1186/1465-9921-14-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K., Laurence A., O'Shea J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel P.O., Zwick E., Prenzel N., Ullrich A. Epidermal growth factor receptors: critical mediators of multiple receptor pathways. Curr. Opin. Cell Biol. 1999;11:184–189. doi: 10.1016/s0955-0674(99)80024-6. [DOI] [PubMed] [Google Scholar]
- Hershenson M.B., Brown M., Camoretti-Mercado B., Solway J. Airway smooth muscle in asthma. Annu. Rev. Pathol. 2008;3:523–555. doi: 10.1146/annurev.pathmechdis.1.110304.100213. [DOI] [PubMed] [Google Scholar]
- Huang J., Qin Y., Yang C., Wan C., Dai X., Sun Y., Meng J., Lu Y., Li Y., Zhang Z., et al. Downregulation of ABI2 expression by EBV-miR-BART13-3p induces epithelial-mesenchymal transition of nasopharyngeal carcinoma cells through upregulation of c-JUN/SLUG signaling. Aging (Albany NY) 2020;12:340–358. doi: 10.18632/aging.102618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S.R. Cytokine signaling exposed. Structure. 2011;19:1–2. doi: 10.1016/j.str.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Hubbard S.R. Mechanistic insights into regulation of JAK2 tyrosine kinase. Front. Endocrinol. (Lausanne) 2017;8:361. doi: 10.3389/fendo.2017.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imada K., Leonard W.J. The Jak-STAT pathway. Mol. Immunol. 2000;37:1–11. doi: 10.1016/s0161-5890(00)00018-3. [DOI] [PubMed] [Google Scholar]
- Innocenti M., Gerboth S., Rottner K., Lai F.P., Hertzog M., Stradal T.E., Frittoli E., Didry D., Polo S., Disanza A., et al. Abi1 regulates the activity of N-WASP and WAVE in distinct actin-based processes. Nat. Cell Biol. 2005;7:969–976. doi: 10.1038/ncb1304. [DOI] [PubMed] [Google Scholar]
- Jia L., Wang R., Tang D.D. Abl regulates smooth muscle cell proliferation by modulating actin dynamics and ERK1/2 activation. Am. J. Physiol. Cell Physiol. 2012;302:C1026–C1034. doi: 10.1152/ajpcell.00373.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S., Tang D.D. Plk1 regulates MEK1/2 and proliferation in airway smooth muscle cells. Respir. Res. 2015;16:93. doi: 10.1186/s12931-015-0257-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P.R., Roth M., Tamm M., Hughes M., Ge Q., King G., Burgess J.K., Black J.L. Airway smooth muscle cell proliferation is increased in asthma. Am. J. Respir. Crit. Care Med. 2001;164:474–477. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- Kassel K.M., Wyatt T.A., Panettieri R.A., Jr., Toews M.L. Inhibition of human airway smooth muscle cell proliferation by beta 2-adrenergic receptors and cAMP is PKA independent: evidence for EPAC involvement. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;294:L131–L138. doi: 10.1152/ajplung.00381.2007. [DOI] [PubMed] [Google Scholar]
- Kogan V., Millstein J., London S.J., Ober C., White S.R., Naureckas E.T., Gauderman W.J., Jackson D.J., Barraza-Villarreal A., Romieu I., et al. Genetic-epigenetic interactions in asthma revealed by a genome-wide gene-centric search. Hum. Hered. 2018;83:130–152. doi: 10.1159/000489765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotula L. Abi1, a critical molecule coordinating actin cytoskeleton reorganization with PI-3 kinase and growth signaling. FEBS Lett. 2012;586:2790–2794. doi: 10.1016/j.febslet.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaar A.L., Panettieri R.A., Jr. Airway smooth muscle as a regulator of immune responses and bronchomotor tone. Clin. Chest Med. 2006;27:53–69. doi: 10.1016/j.ccm.2005.10.003. vi. [DOI] [PubMed] [Google Scholar]
- Li J., Wang R., Gannon O.J., Rezey A.C., Jiang S., Gerlach B.D., Liao G., Tang D.D. Polo-like kinase 1 regulates vimentin phosphorylation at Ser-56 and contraction in smooth muscle. J. Biol. Chem. 2016;291:23693–23703. doi: 10.1074/jbc.M116.749341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.F., Spinelli A.M., Wang R., Anfinogenova Y., Singer H.A., Tang D.D. Critical role of vimentin phosphorylation at Ser-56 by p21-activated kinase in vimentin cytoskeleton signaling. J. Biol. Chem. 2006;281:34716–34724. doi: 10.1074/jbc.M607715200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G., Panettieri R.A., Tang D.D. MicroRNA-203 negatively regulates c-Abl, ERK1/2 phosphorylation, and proliferation in smooth muscle cells. Physiol. Rep. 2015;3:e12541. doi: 10.14814/phy2.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G., Wang R., Rezey A.C., Gerlach B.D., Tang D.D. MicroRNA miR-509 regulates ERK1/2, the vimentin network, and focal adhesions by targeting Plk1. Sci. Rep. 2018;8:12635. doi: 10.1038/s41598-018-30895-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.Y., Ballou L.M., Lin R.Z. Stimulation of the alpha1A adrenergic receptor inhibits PDGF-induced PDGF beta receptor Tyr751 phosphorylation and PI 3-kinase activation. FEBS Lett. 2003;540:106–110. doi: 10.1016/s0014-5793(03)00233-3. [DOI] [PubMed] [Google Scholar]
- Long J., Liao G., Wang Y., Tang D.D. Specific protein 1, c-Abl and ERK1/2 form a regulatory loop. J. Cell Sci. 2019;132:jcs222380. doi: 10.1242/jcs.222380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Zhang L., Lu S., Yang D., Ye J., Li M., Hu W. miR-25 expression is upregulated in pancreatic ductal adenocarcinoma and promotes cell proliferation by targeting ABI2. Exp. Ther. Med. 2020;19:3384–3390. doi: 10.3892/etm.2020.8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias R.A. Introduction to genetics and genomics in asthma: genetics of asthma. Adv. Exp. Med. Biol. 2014;795:125–155. doi: 10.1007/978-1-4614-8603-9_9. [DOI] [PubMed] [Google Scholar]
- Myers R.A., Scott N.M., Gauderman W.J., Qiu W., Mathias R.A., Romieu I., Levin A.M., Pino-Yanes M., Graves P.E., Villarreal A.B., et al. Genome-wide interaction studies reveal sex-specific asthma risk alleles. Hum. Mol. Genet. 2014;23:5251–5259. doi: 10.1093/hmg/ddu222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea J.J., Holland S.M., Staudt L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013;368:161–170. doi: 10.1056/NEJMra1202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page C., O'Shaughnessy B., Barnes P. Pathogenesis of COPD and asthma. Handb. Exp. Pharmacol. 2017;237:1–21. doi: 10.1007/164_2016_61. [DOI] [PubMed] [Google Scholar]
- Penn R.B. Embracing emerging paradigms of G protein-coupled receptor agonism and signaling to address airway smooth muscle pathobiology in asthma. Naunyn Schmiedebergs Arch. Pharmacol. 2008;378:149–169. doi: 10.1007/s00210-008-0263-1. [DOI] [PubMed] [Google Scholar]
- Prakash Y.S. Emerging concepts in smooth muscle contributions to airway structure and function: implications for health and disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016;311:L1113–L1140. doi: 10.1152/ajplung.00370.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash Y.S., Halayko A.J., Gosens R., Panettieri R.A., Jr., Camoretti-Mercado B., Penn R.B., Structure, A.T.S.A.o.R., and Function An official American thoracic society research statement: current challenges facing research and therapeutic advances in airway remodeling. Am. J. Respir. Crit. Care Med. 2017;195:e4–e19. doi: 10.1164/rccm.201611-2248ST. [DOI] [PubMed] [Google Scholar]
- Rezey A.C., Gerlach B.D., Wang R., Liao G., Tang D.D. Plk1 mediates paxillin phosphorylation (Ser-272), centrosome maturation, and airway smooth muscle layer thickening in allergic asthma. Sci. Rep. 2019;9:7555. doi: 10.1038/s41598-019-43927-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring C., Ginsberg M.H., Haling J., Pendergast A.M. Abl-interactor-1 (Abi1) has a role in cardiovascular and placental development and is a binding partner of the alpha4 integrin. Proc. Natl. Acad. Sci. U S A. 2011;108:149–154. doi: 10.1073/pnas.1012316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J.R., Echarri A., Li R., Pendergast A.M. Regulation of cell-cell adhesion by Abi/Diaphanous complexes. Mol. Cell Biol. 2009;29:1735–1748. doi: 10.1128/MCB.01483-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse S.K., Altonsy M.O., Kadiyala V., Cao G., Panettieri R.A., Jr., Gerber A.N. Glucocorticoid and TNF signaling converge at A20 (TNFAIP3) to repress airway smooth muscle cytokine expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016;311:L421–L432. doi: 10.1152/ajplung.00179.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen T.D., Zakrajsek B.A., Mills A.G., Gorn V., Singer M.J., Reed M.W. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal. Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- Seong J., Huang M., Sim K.M., Kim H., Wang Y. FRET-based visualization of PDGF receptor activation at membrane microdomains. Sci. Rep. 2017;7:1593. doi: 10.1038/s41598-017-01789-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y., Gnanasambandan K., Ungureanu D., Kim E.T., Hammaren H., Yamashita K., Silvennoinen O., Shaw D.E., Hubbard S.R. Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat. Struct. Mol. Biol. 2014;21:579–584. doi: 10.1038/nsmb.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y., Das S., Rabold R., Sham J.S., Mitzner W., Tang W.Y. Epigenetic alterations by DNA methylation in house dust mite-induced airway hyperresponsiveness. Am. J. Respir. Cell Mol. Biol. 2013;49:279–287. doi: 10.1165/rcmb.2012-0403OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A.R., Takahashi S., Severgnini M., Fanburg B.L., Cochran B.H. Role of the JAK-STAT pathway in PDGF-stimulated proliferation of human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282:L1296–L1304. doi: 10.1152/ajplung.00315.2001. [DOI] [PubMed] [Google Scholar]
- Southam D.S., Ellis R., Wattie J., Inman M.D. Components of airway hyperresponsiveness and their associations with inflammation and remodeling in mice. J. Allergy Clin. Immunol. 2007;119:848–854. doi: 10.1016/j.jaci.2006.12.623. [DOI] [PubMed] [Google Scholar]
- Stradal T., Courtney K.D., Rottner K., Hahne P., Small J.V., Pendergast A.M. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr. Biol. 2001;11:891–895. doi: 10.1016/s0960-9822(01)00239-1. [DOI] [PubMed] [Google Scholar]
- Tang D.D. Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir. Res. 2015;16:134. doi: 10.1186/s12931-015-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D.D. The dynamic actin cytoskeleton in smooth muscle. Adv. Pharmacol. 2018;81:1–38. doi: 10.1016/bs.apha.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Tang D.D., Bai Y., Gunst S.J. Silencing of p21-activated kinase attenuates vimentin phosphorylation on Ser-56 and reorientation of the vimentin network during stimulation of smooth muscle cells by 5-hydroxytryptamine. Biochem. J. 2005;388:773–783. doi: 10.1042/BJ20050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tliba O., Deshpande D., Chen H., Van B.C., Kannan M., Panettieri R.A., Jr., Amrani Y. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br. J Pharmacol. 2003;140:1159–1162. doi: 10.1038/sj.bjp.0705558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker T.R., Moore S.M., Lawson M.F., Panettieri R.A., Jr., Chilvers E.R. Platelet-derived growth factor-BB and thrombin activate phosphoinositide 3-kinase and protein kinase B: role in mediating airway smooth muscle proliferation. Mol. Pharmacol. 1998;54:1007–1015. doi: 10.1124/mol.54.6.1007. [DOI] [PubMed] [Google Scholar]
- Wang R., Cleary R.A., Wang T., Li J., Tang D.D. The association of cortactin with profilin-1 is critical for smooth muscle contraction. J. Biol. Chem. 2014;289:14157–14169. doi: 10.1074/jbc.M114.548099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Li Q.F., Anfinogenova Y., Tang D.D. Dissociation of Crk-associated substrate from the vimentin network is regulated by p21-activated kinase on ACh activation of airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L240–L248. doi: 10.1152/ajplung.00199.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Liao G., Wang Y., Tang D.D. Distinctive roles of Abi1 in regulating actin-associated proteins during human smooth muscle cell migration. Sci. Rep. 2020;10:10667. doi: 10.1038/s41598-020-67781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Mercaitis O.P., Jia L., Panettieri R.A., Tang D.D. Raf-1, actin dynamics and Abl in human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2013;48:172–178. doi: 10.1165/rcmb.2012-0315OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Cleary R.A., Wang R., Tang D.D. Role of the adapter protein Abi1 in actin-associated signaling and smooth muscle contraction. J. Biol. Chem. 2013;288:20713–20722. doi: 10.1074/jbc.M112.439877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Cleary R.A., Wang R., Tang D.D. Glia maturation factor-gamma phosphorylation at Tyr-104 regulates actin dynamics and contraction in human airway smooth muscle. Am. J. Respir. Cell Mol. Biol. 2014;51:652–659. doi: 10.1165/rcmb.2014-0125OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wang R., Cleary R.A., Gannon O.J., Tang D.D. Recruitment of beta-catenin to N-cadherin is necessary for smooth muscle contraction. J. Biol. Chem. 2015;290:8913–8924. doi: 10.1074/jbc.M114.621003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Liao G., Wang R., Tang D.D. Acetylation of Abelson interactor 1 at K416 regulates actin cytoskeleton and smooth muscle contraction. FASEB J. 2021;35:e21811. doi: 10.1096/fj.202100415R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Rezey A.C., Wang R., Tang D.D. Role and regulation of Abelson tyrosine kinase in Crk-associated substrate/profilin-1 interaction and airway smooth muscle contraction. Respir. Res. 2018;19:4. doi: 10.1186/s12931-017-0709-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegman C.H., Michaeloudes C., Haji G., Narang P., Clarke C.J., Russell K.E., Bao W., Pavlidis S., Barnes P.J., Kanerva J., et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015;136:769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol. Rev. 2004;202:175–190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- Zhong Z., Wen Z., Darnell J.E., Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data are included in the published paper and supplemental information files or available from the lead contact up request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.