

Abstract
Previous studies have shown that different paclitaxel formulations produce distinct anticancer efficacy and safety profiles in animals and humans. This study aimed to investigate the distinct pharmacokinetics and tissue distribution of various nanoformulations of paclitaxel, which may translate into potential differences in safety and efficacy. Four nanoparticle formulations (nab-paclitaxel, mouse albumin nab-paclitaxel [m-nab-paclitaxel], micellar paclitaxel, and polymeric nanoparticle paclitaxel) as well as solvent-based paclitaxel were intravenously administered to mice. Seventeen blood and tissue samples were collected at different time points. The total paclitaxel concentration in each tissue specimen was measured with liquid chromatography–tandem mass spectrometry. Compared with solvent-based paclitaxel, all four nanoformulations demonstrated decreased paclitaxel exposure in plasma. All nanoformulations were associated with paclitaxel blood-cell accumulation in mice; however, m-nab-paclitaxel was associated with the lowest accumulation. Five minutes after dosing, the total paclitaxel in the tissues and blood was approximately 44% to 57% of the administered dose of all paclitaxel formulations. Paclitaxel was primarily distributed to liver, muscle, intestine, kidney, skin, and bone. Compared with solvent-based paclitaxel, the different nanocarriers altered the distribution of paclitaxel in all tissues with distinct paclitaxel concentration–time profiles, nab-paclitaxel was associated with increased delivery efficiency of paclitaxel in the pancreas compared with the other formulations, consistent with the demonstrated efficacy of nab-paclitaxel in pancreatic cancer. All the nanoformulations led to high penetration in the lungs and fat pad, which potentially points to efficacy in lung and breast cancers. Micellar paclitaxel and polymeric nanoparticle paclitaxel were associated with high paclitaxel accumulation in the heart; thus, the risk of cardiovascular toxicity with these formulations may warrant further investigation. The solvent-based formulation was associated with the poorest paclitaxel penetration in all tissues and the lowest tissue-to-plasma ratio. The different nanocarriers of paclitaxel were associated with distinct pharmacokinetics and tissue distribution, which largely align with the observed efficacy and toxicity profiles in clinical trials.
Keywords: paclitaxel, nanoformulation, nanocarriers, pharmacokinetics, tissue penetration and distribution, cancer
Graphical Abstract
1. INTRODUCTION
A Cremophor EL (renamed as Kolliphor EL) solvent–based formulation of paclitaxel (Taxol; pac-T)1,2 is approved for the treatment of ovarian, breast, and non-small cell lung cancers and AIDS-related Kaposi sarcoma.2-5 However, pac-T has not demonstrated efficacy in pancreatic cancer.6,7 The major adverse effects of pac-T are anaphylaxis, severe hypersensitivity, and neutropenia, in addition to other common adverse effects of chemotherapy.8-10 Therefore, novel formulations aimed at improving safety and efficacy have been developed.9,11-15 Thus far, the most successful formulation is nab-paclitaxel (nab-P), an albumin-bound nanoparticle formulation of paclitaxel that has shown improved clinical efficacy compared with pac-T.16-20 nab-P, alone or in combination regimens, has demonstrated clinical efficacy in metastatic breast, non-small cell lung, and pancreatic cancers.21-25 Additionally, a pivotal clinical study in breast cancer showed that nab-P had an improved safety profile compared with pac-T.21
Because of the success of nab-P, other alternative paclitaxel nanoformulations have been developed.9,11,14,15,26-29 Previous studies have shown that different paclitaxel formulations produced distinct efficacy and safety profiles in animals and humans. Polymeric micellar paclitaxel (Genexol-PM and Cynviloq; pac-G) is a monomethoxy poly(ethylene glycol)–block–poly(d,l-lactide) polymer formulation of paclitaxel.30,31 It has been approved for the treatment of metastatic breast cancer and advanced lung cancer in South Korea.32-37 It was found that pac-G had significant antitumor activity and an increased maximum tolerated dosage compared with pac-T.30-33 Another product is a micellar formulation of paclitaxel encapsulated in the proprietary retinoid compound XR-17 (Paclical; pac-P).9,38 It has been approved in Russia for ovarian cancer in combination with carboplatin and obtained orphan drug designation from the U.S. Food and Drug Administration based on its improved toxicity profile compared with pac-T.9,38,39
Although nanoparticle formulations are recognized to have advantages over conventional formulations,40,41 it is still unknown specifically why paclitaxel nanoformulations have different efficacy and toxicity profiles compared with pac-T. Indeed, preclinical studies have demonstrated that nab-P increased tumor accumulation of paclitaxel, which may improve antitumor activity.42,43 A prior study has shown that the rapid decline of circulating paclitaxel below 720 ng/mL associated with nab-P was correlated with a lower incidence of neutropenia.44 Further, Li et al.45 showed that paclitaxel tissue distribution was faster with nab-P vs pac-T. A phase 1 trial showed that unlike pac-T, nab-P displayed linear pharmacokinetics (PK).46 Additionally, paclitaxel clearance and volume of distribution were significantly higher with nab-P than with pac-T.47 Overall, nab-P was associated with distinct paclitaxel tissue distribution and efficacy compared with pac-T, which may be mediated by the drug-carrier complex rather than by free drug alone.45
Much is yet unknown regarding the distribution of different types of nanomaterials.48 Various factors can affect nanoparticle distribution to organs, such as the composition, size, and surface modifications of the particles.49 The preclinical and clinical results on the differences in efficacy and PK of paclitaxel between the nanoformulations and pac-T raise multiple questions about tissue distribution. First, in paclitaxel nanoformulations, do the nanocarrier and paclitaxel travel together, or do they dissociate in plasma? In humans, similar systemic exposure of paclitaxel between nab-P and pac-T has been reported despite a higher initial concentration of paclitaxel with nab-P (as a result of the increased dose and shorter infusion time).42 Thus, the plasma profile alone cannot distinguish between the formulations. Nanoformulations of paclitaxel may have improved efficacy compared with pac-T; for instance, nab-P has demonstrated improved efficacy vs pac-T21-23 in certain types of cancer (eg, nab-P is approved for pancreatic cancer,23,50 in which pac-T has not demonstrated efficacy6,7). Given these different efficacies, are there variations in tissue distribution between the nanoformulations and pac-T? A preliminary study showed that in rodents, nab-P was associated with a much lower concentration of paclitaxel in the blood than pac-T.47 Therefore, are there differences in the plasma profiles between humans and rodents among distinct nanoformulations? While the different nanoformulations have not been directly compared in head-to-head trials, clinical results suggest that the different nanoformulations may have different efficacies;19,21,30,32,33,47 thus, are there differences in tissue distribution among different nanoformulations?
A similar PK profile suggests a similar sum of tissue distribution and elimination but does not indicate a similar tissue distribution or a similar elimination individually. Thus, it is unknown if the nanoformulations have different paclitaxel distribution or elimination profiles, nab-P was associated with different PK profiles in humans and mice,47 which may be due to albumin. One study on the antitumor activity of nab-P in mice used nab-P prepared with mouse albumin instead of human albumin.51 Thus, what is the role of albumin in the PK and tissue distribution of albumin-bound paclitaxel? Drug blood (or plasma) concentrations are thought to be reasonable surrogates of efficacy. However, the PK profiles in plasma produced by pac-T, nab-P, pac-G, and pac-P do not sufficiently explain the different efficacy and toxicity observed in humans.30,46,52 In a study of the drug distribution of three different formulations of doxorubicin (doxorubicin injectable solution,53 unprotected liposomal doxorubicin formulation [Myocet],54 and pegylated liposomal formulation [Doxil]55), each formulation was associated with a distinct tissue distribution. This suggests that the drug-carrier complex may have directly interacted with tissues, affecting drug distribution and associated with efficacy and toxicity.56 Similarly, is the tissue distribution of paclitaxel associated with clinical efficacy and toxicity for different formulations?
To answer the above questions, we investigated the differences in PK and tissue distribution of different formulations of paclitaxel (nab-P, pac-T, pac-P, and pac-G) and discuss the potential effects of these differences in their distinct pharmacology, efficacy, and safety profiles. In addition, m-nab-P was used to assess the role of albumin in tissue distribution of nab-P. The efficiency of paclitaxel delivery by different formulations was assessed by comparing concurrent concentrations in plasma and tissues.
2. EXPERIMENTAL SECTION
2.1. Chemicals and Reagents.
nab-P and m-nab-P were supplied by Celgene Corporation (Summit, NJ, U.S.A.). pac-T was purchased from the University of Michigan Hospital (Ann Arbor, MI, U.S.A.). pac-P was procured from the Russian market courtesy of Celgene Corporation, and pac-G was procured from the Korean market courtesy of Celgene Corporation. Paclitaxel and docetaxel powder were from Thermo Fisher Scientific (Waltham, MA, U.S.A.). Liquid chromatography–mass spectrometry (LC-MS) grade acetonitrile was purchased from Sigma-Aldrich (St Louis, MO, U.S.A.). Formic acid (98%; LC-MS grade) was obtained from Fluka (Morris Plains, NJ, U.S.A.). Ultrapure deionized water was obtained using a Milli-Q water system (Millipore, Bedford, MA, U.S.A.).
2.2. Animal Experiments.
All animal experiments were performed in accordance with University of Michigan guidelines regarding the humane care and use of animals in research. All animal procedures in this study were approved by the University Committee on Use and Care of Animals at the University of Michigan.
Female CD-1 International Genetic Standardization (IGS) mice (strain code 022) that were 6–8 weeks old were purchased from Charles River Laboratories (Wilmington, MA, U.S.A.). The mice were divided into five groups, and each group was intravenously administered pac-T, nab-P, m-nab-P, pac-P, or pac-G at a dose of 10 mg/kg.
At designated time points after drug administration (0.08, 0.17, 0.25, 0.5, 0.75, 1, 2, 4, 7, 16, 24, 48, and 72 h), three mice from each treatment group were euthanized using isoflurane, and blood was immediately collected via cardiac puncture using a 25-G needle and 1 mL syringe (pretreated with sodium heparin). Plasma was collected after the blood was centrifuged at 14 500 rpm for 10 min. Tissues, that is, brain, fat, heart, intestine, kidney, liver, lung, muscle, pancreas, spleen, stomach, bone, fat pad, uterus, and skin, were removed from the mouse and rinsed extensively in phosphate-buffered saline (pH 7.4). The tissues were transferred to a tube from the Precellys CK28 Lysing Kit (Montigny-le-Bretonneux, France) and were stored at −80 °C until further analysis with LC–tandem mass spectrometry (LC-MS/MS).
2.3. Stock Solutions, Working Solutions, and Quality Control Samples.
Paclitaxel and docetaxel (internal standard [IS]) were individually weighed and dissolved in acetonitrile to 9 mg/mL stock solutions and stored at −20 °C. The paclitaxel stock solution was diluted with acetonitrile to a series of working solutions from 2.44 to 5000 ng/mL. Quality control (QC) working solutions were prepared using a separately weighed and prepared stock solution.
2.4. Sample Preparation and Calibration Curve.
Forty microliters of plasma were dispensed into a 96-well plate (Fisher Scientific), followed by 40 μL of ice-cold acetonitrile and 120 μL of IS solution. After vortexing for 10 min, the plate was centrifuged at 3500 rpm for 10 min at 4 °C. The supernatant (5 μL) was analyzed by LC-MS/MS. The blood samples were sonicated before dispensing. Tissue samples were weighed and suspended in 20% acetonitrile at 1:5 mass:volume and then homogenized four times for 20 s each at 6500 rpm in a Precellys Evolution system. Subsequently, paclitaxel in blood and tissue homogenates was extracted in the same manner as plasma for LC-MS/MS analysis. All samples above the upper limit of quantification were diluted with the same matrix before extraction. Calibrator standard samples and QC samples were prepared by mixing 40 μL of blank bio matrix with 40 μL of working solution and 120 μL of IS solution.
2.5. LC-MS/MS Method.
Concentrations of paclitaxel were determined using an AB-5500 Qtrap (Sciex, Concord, ON, Canada) mass spectrometer with electrospray ionization source interfaced with a Shimadzu high-performance liquid chromatography system. Analyst Software (version 1.6) from Applied Biosystems (MDS SCIEX; Carlsbad, CA, U.S.A.) was used. Separation was performed on an Xbridge C18 column (50 × 2.1 mm ID, 3.5 μm; Waters, Milford, MA, U.S.A.) at a flow rate of 0.4 mL/min. The mobile phase consisted of A (100% H2O with 0.1% formic acid) and B (100% acetonitrile with 0.1% formic acid). The gradient started with 25% B for 30 s, linearly increased to 65% B at 2 min, increased to 95% B at 2.5 min, maintained at 95% B for 2 min, decreased to 25% B at 5 min, and maintained at 25% B for 2 min. The multiple reaction monitoring transitions are summarized in Table 1.
Table 1.
Summary of the Multiple Reaction Monitoring Transitions for Paclitaxel and Docetaxela
| Q1 mass (Da) | Q3 mass (Da) | DP (V) | EP (V) | CE (V) | CCEP (V) | |
|---|---|---|---|---|---|---|
| paclitaxel | 854.40 | 286.10 | 190.00 | 14.00 | 21.00 | 13.00 |
| docetaxel (IS) | 808.00 | 226.00 | 173.00 | 12.90 | 18.80 | 13.90 |
CCEP, collision cell exit potential; CE, collision energy; DP, declustering potential; EP, entrance potential; IS, internal standard.
2.6. PK Data Analysis.
The PK parameters of paclitaxel from all formulations were compiled and calculated with Phoenix/WinNonlin (version 6.4; Pharsight, Mountain View, CA, U.S.A.). The plasma/blood and tissue concentration–time data were compiled and plotted using R. Efficiency of paclitaxel delivery by different formulations was evaluated by comparing concentrations of paclitaxel in plasma and tissues at each time point. The amounts of paclitaxel were calculated as the products of the corresponding concentrations and the blood volumes or tissue weights. The relative amount of paclitaxel in each tissue was calculated using the amount of paclitaxel in each tissue per dose. It must be noted that the number of nanoparticles contained in each dose of the distinct nanoformulations may differ, and that it is not possible to know the amount of paclitaxel contained within each individual nanoparticle. To mitigate the impact of these differences in dosing, each tissue concentration was normalized by its plasma concentration.
2.7. Statistical Analysis.
Significant differences among groups were evaluated by one-way ANOVA. Differences between groups were estimated using a Student–Newman–Keuls multiple comparison posthoc test, if needed. A P-value <0.05 was regarded as statistically significant.
3. RESULTS
3.1. Paclitaxel Concentration–Time Profile in Plasma and Blood.
It was previously reported that the plasma exposure of paclitaxel from nab-P and pac-T in humans was similar.45 However, in mice the results were different. Nanoformulations decreased the exposure of paclitaxel in plasma and blood (Figure 1). At 5 min, the plasma concentration of paclitaxel from nab-P, m-nab-P, pac-P, and pac-G decreased to 15.33%, 14.99%, 21.92%, and 26.20% of pac-T, respectively. Subsequently, the estimated AUC of pac-T was 3.93-, 4.43-, 3.26-, and 3.55-fold that of nab-P, m-nab-P, pac-P, and pac-G (P < 0.0001 for all of these comparisons with pac-T; Table 2). The difference in plasma profiles between pac-T and the nanoformulations in mice may suggest that the nanocarriers are directly taken up by tissues before paclitaxel is fully released from the nanocarriers in blood.
Figure 1.

Total paclitaxel concentration–time profiles in (A) blood and (B) plasma. Inset figures show the same data with a logarithmic y-axis scale. pac-T, solvent-based paclitaxel; m-nab-P, nanoparticle mouse albumin-bound paclitaxel; nab-P, nab-paclitaxel; pac-P, micellar paclitaxel; pac-G, polymeric nanoparticle paclitaxel.
Table 2.
Calculated AUCs for All Five Formulations of Paclitaxel in All Analyzed Tissuesa
| AUC0-INF mean (SD), h·ng/mL |
|||||
|---|---|---|---|---|---|
| solvent-based paclitaxel (pac-T) |
nab-Paclitaxel (nab-P) | mouse albumin nab-paclitaxel (m-nab-P) |
micellar paclitaxel (pac-P) |
polymeric nanoparticle paclitaxel (pac-G) |
|
| plasma | 10847.53 (232.50) | 2492.37b (276.25) | 2447.38b (144.51) | 3328.42b (276.29) | 3056.56b (105.36) |
| blood | 5566.50 (151.76) | 1904.76b (415.36) | 1326.57b (79.16) | 2325.94b (245.58) | 1983.00b (84.41) |
| liver | 71,131.52 (4952.55) | 56,570.81b (5351.94) | 64,023.61 (1579.83) | 99,394.93b (9391.88) | 75,974.15 (7994.15) |
| stomach | 21,813.99 (972.27) | 17,543.62b (2064.45) | 20,508.37 (1451.79) | 24,321.27 (1705.92) | 19,772.03 (1175.46) |
| muscle | 7122.72 (573.73) | 6165.92 (613.50) | 6282.92 (294.81) | 8279.61 (487.70) | 6931.53 (815.18) |
| skin | 10,150.87 (339.49) | 8505.55 (1271.54) | 7405.45b (579.95) | 11,268.54 (978.75) | 8325.44b (749.37) |
| fat | 3328.60 (422.81) | 2029.48b (49.73) | 3887.34 (266.34) | 5713.56b (93.69) | 4406.78 (689.27) |
| spleen | 15,840.13 (1063.73) | 11,864.65b (2061.66) | 12,850.25b (858.92) | 17,945.36 (1528.89) | 14,126.55 (815.05) |
| lung | 18,851.56 (1492.11) | 12,378.56b (2359.01) | 11,655.31b (513.80) | 16,063.07b (412.86) | 14,147.04b (430.56) |
| brain | 152.84 (25.23) | 91.29 (34.76) | 239.38b (38.57) | 290.32 (122.14) | 195.07 (49.28) |
| uterus | 23,121.74 (5187.14) | 15,543.08 (2837.44) | 13,297.48b (2400.04) | 18,185.78 (5111.12) | 15,638.86 (1821.37) |
| intestine | 34,134.64 (5792.73) | 29,289.10 (2482.30) | 24,402.88b (1360.68) | 39,446.21 (4378.35) | 32,754.36 (969.64) |
| pancreas | 27,975.06 (409.81) | 20,678.54b (1466.13) | 21,803.74b (934.24) | 16,965.35b (576.41) | 16,522.33b (1498.12) |
| kidney | 22,941.88 (1238.78) | 15,602.01b (1425.70) | 17,836.97b (834.99) | 22,770.56 (1664.14) | 20,321.29 (1815.81) |
| fat pad | 5263.13 (97.00) | 4013.45b (286.08) | 4772.09 (554.54) | 6955.02b (392.75) | 5408.70 (787.03) |
| heart | 9028.75 (427.18) | 6286.26b (862.72) | 6149.73b (61.00) | 11,625.11b (959.87) | 8490.81 (362.74) |
| bone | 9147.74 (535.58) | 6240.46b (928.96) | 9803.12 (1286.41) | 10,352.02 (768.61) | 9617.37 (609.13) |
AUC, area under the curve. AUC0-INF, area under the curve extrapolated to infinity.
P < 0.05 for comparison with pac-T.
The nanoformulations demonstrated distinct paclitaxel plasma and blood exposures—paclitaxel exposures of nab-P and m-nab-P were lower than those of pac-P and pac-G (Figure 1). With a similar blood/plasma ratio in concentration and AUC, m-nab-P and pac-T had similar blood-cell accumulation. However, nab-P, pac-P, and pac-G increased the ratio of blood to plasma concentration compared with that of pac-T (Figure 2), indicating that these three formulations enhanced the blood–cell accumulation of paclitaxel. nab-P had the highest blood–cell accumulation in mice, likely because nab-P uses human albumin in the formulation, whereas m-nab-P does not, inferring that in humans nab-P would decrease the accumulation of paclitaxel in blood cells compared with pac-P and pac-G.
Figure 2.

Blood and plasma concentration ratio and area under the curve (AUC) ratio. Ratio (y-axis) of blood concentration scaled to its corresponding plasma concentration at each time point (right) and ratio (y-axis) of accumulated AUC in blood scaled to its corresponding AUC of plasma at each time point (left) for solvent-based paclitaxel (pac-T), nab-paclitaxel (nab-P), mouse albumin nab-paclitaxel (m-nab-P), micellar paclitaxel (pac-P), and polymeric micellar paclitaxel (pac-G) administration.
3.2. Paclitaxel Concentration–Time Profiles in Tissues and Relative Amounts of Paclitaxel from Injected Doses.
Paclitaxel concentration was high in liver, kidney, intestine, heart, spleen, lung, pancreas, and stomach (Figure 3; Supplemental Figure 1). Each nanoformulation resulted in different tissue distributions. Amounts of unchanged paclitaxel in individual tissues were calculated as a product of tissue concentration and tissue weight (Figure 4A). At 5 min after dosing, the total percentage of paclitaxel in the tissues and blood from all formulations was approximately 44% to 57% (Figure 4B); at 1 h, this percentage declined to 20% to 32% of injected dose. This change indicated that nearly half of the injected dose was rapidly eliminated, consistent with the rapid paclitaxel elimination in rodents that was previously reported.47 The distinct formulations had different total amounts of unchanged paclitaxel, which suggests that nanoformulations might be associated with altered elimination of paclitaxel compared with pac-T. pac-P demonstrated the highest total percentage compared with pac-T, and the percentage from pac-G was similar to that from pac-T. nab-P demonstrated a lower percentage of total unchanged paclitaxel, suggesting that nab-P may be associated with increased elimination of paclitaxel compared with pac-T. This finding may explain the decreased exposure of paclitaxel in blood when delivered as nab-P. Paclitaxel was mainly found in the blood, liver, intestine, and kidney. The absolute amounts in blood were lower with nab-P, pac-P, and pac-G than with pac-T at 5 min after dosing, but the amounts in the liver, intestine, and muscle were higher than with pac-T. This difference in tissue distribution may explain the decreased exposure of paclitaxel from nanoformulations compared with pac-T.
Figure 3.

Total paclitaxel concentration–time profile in select tissues (fat pad, heart, lung, pancreas, and stomach). Solvent-based paclitaxel (pac-T), mouse albumin nab-paclitaxel (m-nab-P), nab-paclitaxel (nab-P), micellar paclitaxel (pac-P), and polymeric nanoparticle paclitaxel (pac-G) have distinct tissue distributions.
Figure 4.

Relative amounts of paclitaxel in tissues. Total relative drug amount in distinct tissues (A) and all tissues (B) at different time points from injected doses of solvent-based paclitaxel (pac-T) nab-paclitaxel (nab-P), mouse albumin nab-paclitaxel (m-nab-P), micellar paclitaxel (pac-P), and polymeric nanoparticle paclitaxel (pac-G).
3.2.1. Liver.
In the liver, concentrations of paclitaxel associated with the nanoformulations were higher than that associated with pac-T at early time points. However, after 30 min distinct formulations showed different clearances in liver, nab-P was associated with faster paclitaxel clearance in the liver but pac-P and pac-G were associated with weakened clearance compared with pac-T. These results correlate with the decreased AUC demonstrated by nab-P (P = 0.0258) and increased AUCs demonstrated by pac-P (P = 0.0100) compared with pac-T.
3.2.2. Stomach and Muscle.
In stomach and muscle, concentrations of paclitaxel associated with pac-P and pac-G were higher than that associated with pac-T at early time points but decreased to levels similar to that of pac-T after 15 min. However, the paclitaxel level associated with nab-P was only slightly higher than that demonstrated by pac-T at 5 and 10 min after dosing and then decreased below that of pac-T. In muscle at 5 min after dosing, the paclitaxel concentrations associated with nab-P, pac-P, and pac-G were higher than that with pac-T; 15 min after dosing, nab-P enhanced the clearance of paclitaxel. The results demonstrated a decreased AUC associated with nab-P compared with pac-T in stomach (P = 0.0316) and a similar AUC associated with pac-G compared with pac-T in both stomach and muscle.
3.2.3. Skin.
In skin, concentrations of paclitaxel as delivered by nab-P, pac-P, and pac-G were higher than that of pac-T at early time points. In addition, the concentrations of paclitaxel produced by nab-P and pac-G increased paclitaxel clearance, resulting in a decreased paclitaxel AUC with the pac-G formulation (P = 0.0184) compared with pac-T.
3.2.4. Fat.
In fat, exposure of paclitaxel associated with pac-P was higher than that associated with pac-T at all time points, accompanied by a 1.71-fold higher estimated AUC (P = 0.0007). However, at all time points except 5 min after dosing, the concentration of paclitaxel delivered as nab-P was lower than that delivered as pac-T, leading to an AUC for nab-P of 60.97% of that for pac-T (P = 0.0061).
3.2.5. Spleen.
In the spleen, the concentrations of paclitaxel produced by nab-P, pac-P, and pac-G were lower than that produced by pac-T. As time passed after dosing, the differences in the concentrations of paclitaxel between the nanoformulations and pac-T narrowed.. As a result, pac-P generated the highest AUC and lowest clearance, whereas nab-P had the lowest estimated AUC and highest clearance.
3.2.6. Brain.
The concentration of paclitaxel delivered by nab-P was lower than that delivered by pac-T at all time points.
3.2.7. Lung.
In the lung, the exposure of paclitaxel associated with all nanoformulations was lower than that of pac-T; the estimated AUCs associated with nab-P, pac-P, and pac-G were 65.66% (P = 0.0159), 85.21% (P = 0.0355), and 75.04% (P = 0.0063), respectively, of the AUC for pac-T.
3.2.8. Uterus.
Estimated AUCs for nab-P, pac-P, and pac-G were 67.22%, 78.65%, and 67.64% of the AUC for pac-T but none of these comparisons were statistically significant.
3.2.9. Intestine.
In the intestine, the most obvious difference in the concentration–time profiles of paclitaxel in all five formulations was the time to maximum concentration (Tmax). Results showed that the pac-T Tmax was 30 min; however, that for nab-P was 5 min and pac-P and pac-G was 1 h. As a result, at 5 min after dosing, the concentration of paclitaxel associated with nab-P was 1.43-, 1.38-, and 1.42-fold higher than those of pac-T, pac-P, and pac-G, respectively. Although the Tmax was different among these formulations, the maximum concentrations (Cmaxs) (Table 3) and estimated AUCs were similar. These results indicate that nab-P was associated with rapid distribution in the intestine.
Table 3.
Calculated Cmaxs for All Five Formulations of Paclitaxel in All Analyzed Tissuesa
|
Cmax, (SD), ng/mL |
|||||
|---|---|---|---|---|---|
| solvent-based paclitaxel (pac-T) |
nab-Paclitaxel (nab-P) | mouse albumin nab-paclitaxel (m-nab-P) |
micellar paclitaxel (pac-P) |
polymeric nanoparticle paclitaxel (pac-G) |
|
| plasma | 17,766.67 (1504.44) | 2876.67b (531.26) | 2753.33b (494.00) | 3895.00b (736.19) | 5073.33b (754.08) |
| blood | 8766.67 (1056.33) | 2093.33b (328.68) | 1390.00b (175.78) | 2485.00b (552.97) | 2653.33b (325.32) |
| liver | 39,083.33 (2961.56) | 48,083.33b (3394.24) | 45,000.00 (6415.61) | 58,000.00b (3905.12) | 67,166.67b (5387.10) |
| stomach | 5250.00 (200.00) | 5445.00 (755.20) | 6016.67b (388.37) | 8283.33b (361.71) | 8350.00b (676.39) |
| muscle | 2163.33 (87.80) | 2246.67 (361.47) | 2230.00 (252.93) | 2741.67b (280.73) | 2493.33 (407.87) |
| skin | 1626.67 (286.37) | 1430.00 (178.96) | 1348.33 (168.55) | 2345.00b (296.35) | 1681.67 (254.08) |
| fat | 1040.00 (383.63) | 745.00 (161.17) | 1260.00 (212.31) | 1961.67b (228.82) | 1446.67 (223.68) |
| spleen | 8400.00 (936.75) | 5383.33b (321.46) | 5466.67b (550.76) | 6050.00b (278.39) | 6466.67b (431.08) |
| lung | 10,700.00 (1300.00) | 6583.33b (378.60) | 6350.00b (132.29) | 11,300.00 (926.01) | 9250.00 (2402.08) |
| brain | 145.83 (19.76) | 56.68b (10.09) | 87.00b (10.64) | 95.83b (14.29) | 94.17b (21.26) |
| uterus | 3358.33 (197.63) | 2915.00b (160.39) | 2261.67b (568.71) | 3020.00 (500.22) | 2271.67b (563.68) |
| intestine | 15,550.00 (1758.55) | 13,533.33 (1382.33) | 8733.33b (652.56) | 13,900.00 (1517.40) | 13,266.67 (2617.41) |
| pancreas | 11,300.00 (1361.07) | 10,116.67 (837.16) | 10,083.33 (301.39) | 8583.33b (956.99) | 10,033.33 (208.17) |
| kidney | 20,166.67 (1286.79) | 14,800.00b (2146.51) | ll,166.67b (682.52) | 20,300.00 (1116.92) | 23,583.33 (5460.16) |
| fat pad | 1595.00 (126.19) | 1543.33 (240.02) | 1825.00 (180.07) | 2128.33b (120.03) | 1826.67 (197.57) |
| heart | 7533.33 (534.63) | 5650.00b (264.58) | 6000.00b (433.01) | 10,700.00b (950.00) | 9150.00 (1650.00) |
| bone | 2163.33 (62.52) | 1940.00 (149.33) | 2310.00 (188.22) | 3166.67b (378.99) | 2871.67b (304.40) |
Cmax, maximum concentration.
P < 0.05 for comparison with pac-T.
3.2.10. Pancreas.
In the pancreas, at 5 min after dosing the concentrations of paclitaxel associated with nab-P and pac-G were both 1.2-fold higher than that produced by pac-T. Subsequently, these concentrations declined below the concentration associated with pac-T after 5 min. The faster decrease associated with pac-G led to a lower AUC compared with that for nab-P. The concentration of paclitaxel as delivered by pac-P was lower than that delivered by pac-T at all time points. The AUC for pac-T was higher than that for nab-P (P = 0.0011), pac-P (P < 0.0001), or pac-G (P = 0.0002). Nanoformulations reduced the Tmax of paclitaxel to 5 min (nab-P and pac-G) or 10 min (pac-P), whereas the Tmax of pac-T was 30 min, indicating that nanoformulations were associated with rapid distribution in the pancreas. Cmax for nab-P and pac-G each was similar to that of pac-T, but the Cmax for pac-P was 78.50% of that for pac-T (P = 0.0474).
3.2.11. Kidney.
In the kidney, the exposure of paclitaxel related to pac-P and pac-G was similar to that of pac-T, whereas the exposure of paclitaxel associated with nab-P was lower than that of pac-T. At 5 min after dosing, the concentration of paclitaxel delivered by nab-P was only 71.40% of that delivered by pac-T; the estimated AUC was 68.00% of that of pac-T (P = 0.0025).
3.2.12. Fat pad.
In fat pad tissue, except at 5 min after dosing, the concentration of paclitaxel delivered by nab-P was lower than that associated with pac-T with an AUC of 76.25% of that of pac-T (P = 0.0020). pac-G demonstrated a similar exposure of paclitaxel to pac-T, whereas pac-P demonstrated increased paclitaxel exposure, with a 1.32-fold higher AUC compared with that of pac-T (P = 0.0019).
3.2.13. Heart and Bone.
In heart and bone tissues, pac-P and pac-G demonstrated increased paclitaxel exposure, but nab-P showed decreased paclitaxel exposure compared with pac-T. At 5 min after dosing in the heart, the concentrations of paclitaxel associated with pac-P and pac-G were 1.42- and 1.21-fold, respectively, of that from pac-T, whereas the concentration associated with nab-P was 73.45% of that from pac-T. The estimated AUC in the heart for pac-P was 1.47-fold higher than that for pac-T (P = 0.0128), whereas the AUC for nab-P was 69.63% of that for pac-T (P = 0.0078). At 5 min after dosing, the concentrations of paclitaxel in bone with pac-P and pac-G were 1.64- and 1.43-fold, respectively, of that from pac-T. The estimated AUC for nab-P in bone was 68.22% of that for pac-T (P = 0.0093).
3.2.14. Mouse versus Human Albumin.
m-nab-P demonstrated a trend of altered distribution of paclitaxel compared with nab-P in some tissues. In fat pad, brain, fat, and bone, mouse albumin showed increased paclitaxel exposure compared with human albumin in mice, whereas in kidney, intestine, and uterus mouse albumin was associated with decreased paclitaxel exposure compared with human albumin. In other tissues, these two distinct albumin formulations showed no differences in paclitaxel exposure.
3.3. Efficiency of Paclitaxel Delivery by Different Formulations.
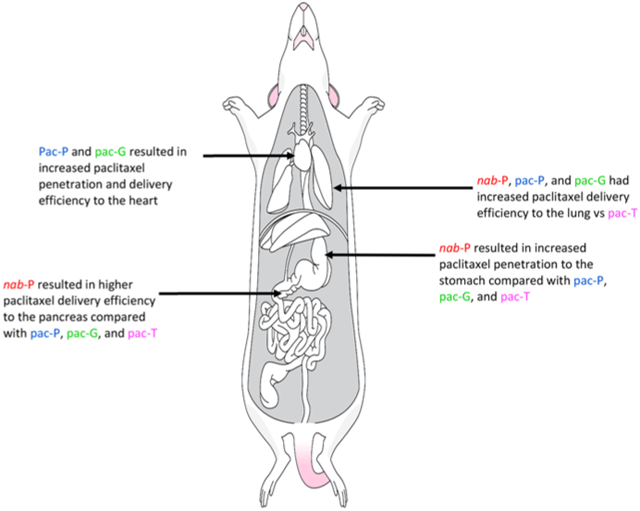
The ratio of AUC0-t between tissue and plasma may be used to indicate delivery efficiency to each tissue (Figure 5; Supplemental Figure 2). Therefore, the delivery efficiency of all formulations in all tissues was compared in this manner. pac-T demonstrated weak delivery efficiency in most tissues with a ratio of <3 after reaching a plateau with the exceptions of intestine (3.15) and fiver (6.59). Each nanoformulations enhanced the delivery efficiency in most tissues with an increased plateau ratio compared with pac-T. Meanwhile, the distinct nanoformulations produced different ratios in all tissues. In the pancreas, the plateau ratios associated with m-nab-P and nab-P were 8.84 and 8.33, respectively, which were higher than those associated with pac-P (4.96), pac-G (5.34), and pac-T (2.58). nab-P was associated with greater delivery efficiency in the pancreas compared with the pac-P and pac-G formulations (P = 0.0248 and P = 0.0331, respectively; Supplemental Table 1), which is in line with the demonstrated efficacy of nab-P in pancreatic cancer.23 In the stomach, kidney, lung, spleen, and bone, paclitaxel delivery efficiency trended higher with m-nab-P than with nab-P. Further, m-nab-P resulted in significantly higher delivery efficiency versus nab-P in bone (P = 0.0147). In the heart, pac-P had improved delivery efficiency versus nab-P (P = 0.0049) as did pac-G (P = 0.0210). Delivery efficiency in the liver trended higher with pac-G vs nab-P and was significantly higher with pac-P versus nab-P (P = 0.0039).
Figure 5.

Ratio of area under the curve (AUC0-t) between select tissues (blood, fat pad, heart, lung, pancreas, and stomach) and plasma. Ratio (y-axis) of accumulated AUC in tissues scaled to its corresponding accumulated AUC of plasma for solvent-based paclitaxel (pac-T), nab-paclitaxel (nab-P), mouse albumin nab-paclitaxel (m-nab-P), micellar paclitaxel (pac-P), and polymeric nanoparticle paclitaxel (pac-G).
The tissue penetration of paclitaxel by different formulations was measured by calculating the ratio of paclitaxel concentrations in tissues versus plasma at each time point (Figure 6). Results showed that the paclitaxel penetration related to pac-T was the poorest in all tissues with the lowest tissue/plasma ratio. All nanoformulations were associated with increased penetration of paclitaxel in all tissues and increased tissue/plasma ratios compared with pac-T. The nanoformulations of paclitaxel resulted in different penetrations in tissues, which may link to distinct anticancer efficacy and safety profiles in animals and humans. Greater penetrations in lung and fat pad associated with all four nanoformulations might be reflective of improved efficacy in lung and breast cancer treatments. pac-P and pac-G had significantly nonselectively higher paclitaxel accumulations in the heart compared with nab-P (P = 0.0085 and P = 0.0146, respectively; Supplemental Table 2), which may point to cardiovascular adverse effects. Paclitaxel tissue penetration in the pancreas trended higher with nab-P than with pac-P or pac-G. In the stomach, nab-P had significantly higher penetration than either pac-P (P = 0.0096) or pac-G (P = 0.0175).
Figure 6.

Ratios of total paclitaxel concentrations in select tissues (blood, fat pad, heart, lung, pancreas, stomach) scaled to corresponding plasma paclitaxel concentration at each time point for solvent-based paclitaxel (pac-T), nab-paclitaxel (nab-P), mouse albumin nab-paclitaxel (m-nab-P), micellar paclitaxel (pac-P), and polymeric nanoparticle paclitaxel (pac-G).
4. DISCUSSION
Actual drug concentrations in tissues are the result of a drug’s absorption, distribution, metabolism, and excretion properties and how the drug is delivered.57 A drug’s therapeutic index, partially reflected by drug exposure in plasma and tissues, is strongly affected by its formulations.58 Drug distribution outside the vasculature is key but difficult to estimate from plasma PK.59 Therefore, an extensive study of the tissue distribution of the distinct formulations, nab-P, pac-G, pac-P, and m-nab-P, was carried out. Tissue distribution predominates over elimination and drives plasma paclitaxel concentration profiles after nab-P administration in humans.44 Because laboratory animals, including mice, usually have a higher rate of drug elimination than humans,60 paclitaxel elimination might predominate over tissue distribution in driving plasma concentration profiles in mice. Similar plasma profiles of paclitaxel in humans could not distinguish between drug and drug carrier delivered together or separately for complex formulations45 but the plasma profile differences in mice between pac-T and the nanoformulations suggests that nanocarriers were directly taken up by tissues before all paclitaxel was fully released in the blood, nab-P, pac-P, and pac-G were associated with increased paclitaxel blood cell accumulation.
Compared with pac-T, the nanoformulations were associated with altered tissue distribution of paclitaxel. Our results indicate that lower concentrations of paclitaxel in blood/plasma after administration of pac-P might primarily be due to the alteration in tissue distribution. The summed relative amount associated with pac-P in all tissues and blood was higher than that with pac-T, inferring that the elimination related to pac-P was lower than that of pac-T. Lower elimination therefore cannot be a reason for the decrease in the plasma concentration.
The concentration–time profile results showed that pac-G was associated with increased paclitaxel concentration in many tissues. In addition, the estimated AUC for paclitaxel delivered by pac-G trended larger than that delivered by pac-T in the liver, fat, and brain and similar in the stomach, muscle, spleen, fat pad, intestine, and bone. These results indicate that lower concentrations of paclitaxel in blood/plasma after administration of pac-G might be due to the alteration in tissue distribution. The summed relative amount of paclitaxel associated with pac-G in all tissues and blood was similar to that with pac-T, suggesting that elimination could also be excluded as a reason for the decrease in the blood/plasma concentration.
The estimated AUC for nab-P was less than that for pac-T in all tissues with significant differences observed in many tissues. The percentage of the total relative amount of paclitaxel delivered by nab-P was similar to that of pac-T at early time points but later decreased. These results indicate that the decrease in blood/plasma paclitaxel concentration associated with nab-P was the result of altered tissue distribution and increased elimination compared with pac-T.
In humans, reported differences in the plasma concentration of paclitaxel related to nab-P and pac-T have been modest.45 Clinical trials have shown that pac-G and pac-P have similar paclitaxel PK profiles to that of nab-P.30,61 However, in mice all four different nanoformulations, especially nab-P, were associated with decreased paclitaxel exposure in plasma and blood compared with pac-T. The paclitaxel plasma exposures of pac-G and pac-P were higher than that of nab-P. Thus, large species differences exist between humans and mice in paclitaxel PK of different nanoformulations.
Increasing tissue to plasma paclitaxel concentration ratios over time by all paclitaxel formulations demonstrated that paclitaxel PK and tissue distribution were far from equilibrium. Plasma drug concentration is not a good surrogate of tissue concentration under nonequilibrium distribution,45 which accentuated the disconnection between plasma and tissue drug levels. This could explain the different tissue distribution profiles with similar plasma profiles in humans. The delivery efficiency of different formulations into tissues was reflected by comparing the AUC between each tissue and plasma, and the tissue penetration associated with different formulations was determined by comparing the concurrent concentrations in tissues versus plasma at each time points. These two methods showed consistent results. Paclitaxel by itself did not penetrate tissue membranes effectively. Nanoformulations demonstrated increased delivery efficiency of paclitaxel in all tissues, and different nanoformulations resulted in distinct paclitaxel penetration, which may result in distinct efficacy and safety profiles.
Although the nanoformulations have not all been compared with pac-T in head-to-head clinical trials, nab-P, pac-G, and pac-P have shown improved efficacy and safety profiles compared with pac-T.19,21,30,32,33,46 However, in humans42 the plasma profiles did not correlate with the clinical outcome. In humans, one model showed that although the paclitaxel plasma profiles between nab-P and pac-T were similar nab-P displayed rapid paclitaxel tissue distribution, suggesting that the pharmacology, efficacy, and safety profiles between nab-P and pac-T differed because of different nanoformulations.62
Greater penetration in the lung and fat pad of all 4 nanoformulations might point to improved efficacy in lung cancer treatment. nab-P and m-nab-P were associated with high paclitaxel accumulations in the pancreas compared with other formulations, consistent with the efficacy profile of nab-P in pancreatic cancer. Furthermore, there was high paclitaxel penetration when delivered by pac-P and pac-G in the heart compared with delivery by nab-P, which might be associated with adverse effects in heart.
It is important to acknowledge that this experiment was not executed in a cancer animal model. However, the accumulation of anticancer nanoformulations as demonstrated in xenograft tumor models is not necessarily consistent with what has been observed in humans.63-67 Indeed, the distribution data in this noncancer model are stih useful because these data show quantitative distribution differences in organs among the paclitaxel formulations which may reflect the potential efficacies a nd toxicities of the formulations in the particular organs. Therefore, it is reasonable to examine the efficacy/safety of nab-P in gastric, colon, and bladder cancers in addition to pancreatic, lung, and breast cancers. All of these results suggest that the pharmacology of paclitaxel delivery systems is predominately determined by the nanoformulations. Notably, the trend in differences between m-nab-P and nab-P suggest that albumin plays an important role in the PK and tissue distribution of albumin-bound paclitaxel. Finally, the results of this preclinical experiment can guide the design of clinical studies investigating the activity of paclitaxel nanoformulations.
5. CONCLUSION
The results of this study indicate that different nanoformulations have distinct paclitaxel PK profiles and tissue distributions, which may be associated with distinct efficacy and toxicity profiles. Each of the nanoformulations has its own tissue distribution and pharmacology that are distinct from those of the dissolved drug and may be explored further to optimize risk-benefit profiles of existing anticancer agents.
Supplementary Material
ACKNOWLEDGMENTS
Editorial assistance was provided by MediTech Media, Ltd, through funding by Celgene Corporation. The authors are fully responsible for all content and editorial decisions for this manuscript. The authors acknowledge the financial support for this study from Celgene Corporation.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b00527.
Supplemental Figures 1-3; Supplemental Tables 1 and 2 (PDF)
REFERENCES
- (1).Adams JD; Flora KP; Goldspiel BR; Wilson JW; Arbuck SG; Finley R Taxol: A History of Pharmaceutical Development and Current Pharmaceutical Concerns. J. Natl. Cancer Inst Monogr 1993, 15, 141–147. [PubMed] [Google Scholar]
- (2).Taxol (paclitaxel) injection [package insert]; Bristol-Myers Squibb Co.: Princeton, NJ, 2011. [Google Scholar]
- (3).Crown J; O’Leary M The Taxanes: An Update. Lancet 2000, 355, 1176–1178. [DOI] [PubMed] [Google Scholar]
- (4).Marupudi NI; Han JE; Li KW; Renard VM; Tyler BM; Brem H Paclitaxel: A Review of Adverse Toxicities and Novel Delivery Strategies. Expert Opin. Drug Saf 2007, 6, 609–621. [DOI] [PubMed] [Google Scholar]
- (5).Paclitaxel [package insert]; Hospira, Inc.: Lake Forest, IL, 2018. [Google Scholar]
- (6).Whitehead RP; Jacobson J; Brown TD; Taylor SA; Weiss GR; Macdonald JS Phase II Trial of Paclitaxel and Granulocyte Colony-Stimulating Factor in Patients With Pancreatic Carcinoma: A Southwest Oncology Group Study. J. Clin. Oncol 1997, 15, 2414–2419. [DOI] [PubMed] [Google Scholar]
- (7).Rajeshkumar NV; Yabuuchi S; Pai SG; Tong Z; Hou S; Bateman S; Pierce DW; Heise C; Von Hoff DD; Maitra A; et al. Superior Therapeutic Efficacy of nab-Paclitaxel over Cremophor-Based Paclitaxel in Locally Advanced and Metastatic Models of Human Pancreatic Cancer. Br. J. Cancer 2016, 115, 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rowinsky EK; Eisenhauer EA; Chaudhry V; Arbuck SG; Donehower RC Clinical Toxicities Encountered With Paclitaxel (Taxol). Semin Oncol 1993, 20, 1–15. [PubMed] [Google Scholar]
- (9).Bernabeu E; Cagel M; Lagomarsino E; Moretton M; Chiappetta DA Paclitaxel: What Has Been Done and the Challenges Remain Ahead. Int. J. Pharm 2017, 526, 474–495. [DOI] [PubMed] [Google Scholar]
- (10).Rowinsky EK; Donehower RC. Paclitaxel (Taxol). N. Engl. J. Med 1995, 332, 1004–1014. [DOI] [PubMed] [Google Scholar]
- (11).Sofias AM; Dunne M; Storm G; Allen C The Battle of “Nano” Paclitaxel. Adv. Drug Delivery Rev 2017, 122, 20–30. [DOI] [PubMed] [Google Scholar]
- (12).Caster JM; Patel AN; Zhang T; Wang A Investigational Nanomedicines in 2016: A Review of Nanotherapeutics Currently Undergoing Clinical Trials. Wiley Interdiscip Rev. Nanomed Nanobiotechnol 2017, 9, 1416. [DOI] [PubMed] [Google Scholar]
- (13).Egusquiaguirre SP; Igartua M; Hernandez RM; Pedraz JL Nanoparticle Delivery Systems for Cancer Therapy: Advances in Clinical and Preclinical Research. Clin. Transl. Oncol 2012, 14, 83–93. [DOI] [PubMed] [Google Scholar]
- (14).Luo C; Wang Y; Chen Q; Han X; Liu X; Sun J; He Z Advances of Paclitaxel Formulations Based on Nanosystem Delivery Technology. Mini-Rev. Med. Chem 2012, 12, 434–444. [DOI] [PubMed] [Google Scholar]
- (15).Perez EA Novel Enhanced Delivery Taxanes: An Update. Semin Oncol 2007, 34, 163. [DOI] [PubMed] [Google Scholar]
- (16).Furlanetto J; Jackisch C; Untch M; Schneeweiss A; Schmatloch S; Aktas B; Denkert C; Wiebringhaus H; Kummel S; Warm M; et al. Efficacy and Safety of nab-Paclitaxel 125 mg/m2 and nab-Paclitaxel 150 mg/m2 Compared to Paclitaxel in Early High-Risk Breast Cancer. Results From the Neoadjuvant Randomized GeparSepto Study (GBG 69). Breast Cancer Res. Treat 2017, 163, 495–506. [DOI] [PubMed] [Google Scholar]
- (17).Henderson IC; Bhatia V nab-Paclitaxel for Breast Cancer: A New Formulation With an Improved Safety Profile and Greater Efficacy. Expert Rev. Anticancer Ther 2007, 7, 919–943. [DOI] [PubMed] [Google Scholar]
- (18).Montero AJ Adams B; Diaz-Montero CM; Gluck S nab-Paclitaxel in the Treatment of Metastatic Breast Cancer: A Comprehensive Review. Expert Rev. Clin. Pharmacol 2011, 4, 329–334. [DOI] [PubMed] [Google Scholar]
- (19).Foote M Using Nanotechnology to Improve the Characteristics of Antineoplastic Drugs: Improved Characteristics of nab-Paclitaxel Compared With Solvent-Based Paclitaxel. Biotechnol. Annu. Rev 2007, 13, 345–357. [DOI] [PubMed] [Google Scholar]
- (20).Miele E; Spinelli GP; Miele E; Tomao F; Tomao S Albumin-Bound Formulation of Paclitaxel (Abraxane ABI-007) in the Treatment of Breast Cancer. Int. J. Nanomed 2009, 4, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gradishar WJ; Tjulandin S; Davidson N; Shaw H; Desai N; Bhar P; Hawkins M; O’Shaughnessy J Phase III Trial of Nanoparticle Albumin-Bound Paclitaxel Compared With Polyethylated Castor Oil-Based Paclitaxel in Women With Breast Cancer. J. Clin. Oncol 2005, 23, 7794–7803. [DOI] [PubMed] [Google Scholar]
- (22).Socinski MA; Bondarenko I; Karaseva NA; Makhson AM; Vynnychenko I; Okamoto I; Hon JK; Hirsh V; Bhar P; Zhang H; et al. Weekly nab-Paclitaxel in Combination With Carboplatin Versus Solvent-Based Paclitaxel Plus Carboplatin as First-line Therapy in Patients With Advanced Non-Small-Cell Lung Cancer: Final Results of a Phase III Trial. J. Clin. Oncol 2012, 30, 2055–2062. [DOI] [PubMed] [Google Scholar]
- (23).Von Hoff DD; Ervin T; Arena FP; Chiorean EG; Infante J; Moore M; Seay T; Tjulandin SA; Ma WW; Saleh MN; et al. Increased Survival in Pancreatic Cancer With nab-Paclitaxel Plus Gemcitabine. N. Engl. J. Med 2013, 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Matsui A; Tatibana A; Suzuki N; Hirata M; Oishi Y; Hamaguchi Y; Murata Y; Nagayama A; Iwata Y; Okamoto Y Evaluation of Efficacy and Safety of Upfront Weekly Nanoparticle Albumin-Bound Paclitaxel for HER2-negative Breast Cancer. Anticancer Res 2016, 27, 6481–6488. [DOI] [PubMed] [Google Scholar]
- (25).Zong Y; Wu J; Shen K Nanoparticle Albumin-Bound Paclitaxel as Neoadjuvant Chemotherapy of Breast Cancer: A Systematic Review and Meta-Analysis. Oncotarget. 2017, 8, 17360–17372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khanna C; Rosenberg M; Vail DM A Review of Paclitaxel and Novel Formulations Including Those Suitable for Use in Dogs. J. Vet. Intern. Med 2015, 29, 1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nehate C; Jain S; Saneja A; Khare V; Alam N; Dubey RD; Gupta PN Paclitaxel Formulations: Challenges and Novel Delivery Options. Curr. Drug Delivery 2014, 11, 666–686. [DOI] [PubMed] [Google Scholar]
- (28).Koudelka S; Turanek J Liposomal Paclitaxel Formulations. J. Controlled Release 2012, 163, 322–334. [DOI] [PubMed] [Google Scholar]
- (29).Singla AK; Garg A; Aggarwal D Paclitaxel and Its Formulations. Int. J. Pharm 2002, 235, 179–192. [DOI] [PubMed] [Google Scholar]
- (30).Kim TY; Kim DW; Chung JY; Shin SG; Kim SC; Heo DS; Kim NK; Bang YJ Phase I and Pharmacokinetic Study of Genexol-PM, A Cremophor-Free, Polymeric Micelle-Formulated Paclitaxel, in Patients With Advanced Malignancies. Clin. Cancer Res 2004, 10, 3708–3716. [DOI] [PubMed] [Google Scholar]
- (31).Kim SC; Kim DW; Shim YH; Bang JS; Oh HS; Wan Kim S; Seo MH In Vivo Evaluation of Polymeric Micellar Paclitaxel Formulation: Toxicity and Efficacy. J. Controlled Release 2001, 72, 191–202. [DOI] [PubMed] [Google Scholar]
- (32).Lee KS; Chung HC; Im SA; Park YH; Kim CS; Kim SB; Rha SY; Lee MY; Ro J Multicenter Phase II Trial of Genexol-PM, A Cremophor-Free, Polymeric Micelle Formulation of Paclitaxel, in Patients With Metastatic Breast Cancer. Breast Cancer Res. Treat 2008, 108, 241–250. [DOI] [PubMed] [Google Scholar]
- (33).Kim DW; Kim SY; Kim HK; Kim SW; Shin SW; Kim JS; Park K; Lee MY; Heo DS Multicenter Phase II Trial of Genexol-PM, A Novel Cremophor-Free, Polymeric Micelle Formulation of Paclitaxel, With Cisplatin in Patients With Advanced Non-Small-Cell Lung Cancer. Ann. Oncol 2007, 18, 2009–2014. [DOI] [PubMed] [Google Scholar]
- (34).van der Meel R; Lammers T; Hennink WE Cancer Nanomedicines: Oversold or Underappreciated? Expert Opin. Drug Delivery 2017, 14, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Samyang Biopharm. History. https://www.samyangbiopharm.com/eng/Aboutus/history (accessed April 27, 2018).
- (36).Samyang Biopharm. Genexol-PM Was Launched in the Korean Market for Breast Cancer and Non-Small Cell Lung Cancer. https://www.samyangbiopharm.com/eng/BP04/Details/199 (accessed April 27, 2018).
- (37).Park IH; Sohn JH; Kim SB; Lee KS; Chung JS; Lee SH; Kim TY; Jung KH; Cho EK; Kim YS; et al. An Open-Label, Randomized, Parallel, Phase III Trial Evaluating the Efficacy and Safety of Polymeric Micelle-Formulated Paclitaxel Compared to Conventional Cremophor EL-Based Paclitaxel for Recurrent or Metastatic HER2-Negative Breast Cancer. Cancer Res. Treat 2017, 49, 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Oasmia. Oasmia’s Lead Human Oncology Product Paclical Shows A Positive Risk/Benefit Profile Versus Standard Treatment in Pivotal Phase III Clinical Study. https://oasmia.com/en/press-release/oasmias-lead-human-oncology-product-paclical-shows-positive-riskbenefit-profile-versus-standard-treatment-pivotal-phase-iii-clinical-study/ (accessed April 27, 2018).
- (39).Oasmia. Oasmia’s Lead Cancer Product Paclical Receives Market Approval in the Russian Federation. https://oasmia.com/en/press-release/oasmias-lead-cancer-product-paclical-receives-market-approval-russian-federation/ (accessed April 27, 2018).
- (40).Kamaly N; Xiao Z; Valencia PM; Radovic-Moreno AF; Farokhzad OC Targeted Polymeric Therapeutic Nanoparticles: Design, Development and Clinical Translation. Chem. Soc. Rev 2012, 41, 2971–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Blanco E; Shen H; Ferrari M Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol 2015, 33, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gardner ER; Dahut WL; Scripture CD; Jones J; Aragon-Ching JB; Desai N; Hawkins MJ; Sparreboom A; Figg WD Randomized Crossover Pharmacokinetic Study of Solvent-Based Paclitaxel and nab-Paclitaxel. Clin. Cancer Res 2008, 14, 4200–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Desai N; Trieu V; Yao Z; Louie L; Ci S; Yang A; Tao C; De T; Beals B; Dykes D; et al. Increased Antitumor Activity, Intratumor Paclitaxel Concentrations, and Endothelial Cell Transport of Cremophor-Free, Albumin-Bound Paclitaxel, ABI-007, Compared With Cremophor–Based Paclitaxel. Clin. Cancer Res 2006, 12, 1317–1324. [DOI] [PubMed] [Google Scholar]
- (44).Chen N; Li Y; Ye Y; Palmisano M; Chopra R; Zhou S Pharmacokinetics and Pharmacodynamics of nab-Paclitaxel in Patients With Solid Tumors: Disposition Kinetics and Pharmacology Distinct from Solvent-Based Paclitaxel. J. Clin. Pharmacol 2014, 54, 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Li Y; Chen N; Palmisano M; Zhou S Pharmacologic Sensitivity of Paclitaxel to Its Delivery Vehicles Drives Distinct Clinical Outcomes of Paclitaxel Formulations. Mol. Pharmaceutics 2015, 12, 1308–1317. [DOI] [PubMed] [Google Scholar]
- (46).Nyman DW; Campbell KJ Hersh E; Long K; Richardson K; Trieu V; Desai N; Hawkins MJ; Von Hoff DD Phase I and Pharmacokinetics Trial of ABI-007, A Novel Nanoparticle Formulation of Paclitaxel in Patients With Advanced Nonhematologic Malignancies. J. Clin. Oncol 2005, 23, 7785–7793. [DOI] [PubMed] [Google Scholar]
- (47).Sparreboom A; Scripture CD; Trieu V; Williams PJ; De T; Yang A; Beals B; Figg WD; Hawkins M; Desai N Comparative Preclinical and Clinical Pharmacokinetics of A Cremophor-Free, Nanoparticle Albumin-Bound Paclitaxel (ABI-007) and Paclitaxel Formulated in Cremophor (Taxol). Clin. Cancer Res 2005, 11, 4136–4143. [DOI] [PubMed] [Google Scholar]
- (48).Bourquin J; Milosevic A; Hauser D; Lehner K; Blank F; Petri-Fink A; Rothen-Rutishauser B Biodistribution, Clearance, and Long-Term Fate of Clinically Relevant Nanomaterials. Adv. Mater 2018, 30, 1704307. [DOI] [PubMed] [Google Scholar]
- (49).Alexis F; Pridgen E; Molnar LK; Farokhzad OC Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharmaceutics 2008, 5, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Abraxane [package insert]; Celgene Corporation: Summit, NJ, 2018. [Google Scholar]
- (51).Neesse A; Frese KK; Chan DS; Bapiro TE; Howat WJ; Richards FM; Ellenrieder V; Jodrell DI; Tuveson DA SPARC Independent Drug Delivery and Antitumour Effects of nab-Paclitaxel in Genetically Engineered Mice. Gut 2014, 63, 974–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Trieu V; Hwang L; Motamed K; Hsiao C IG-001 - Evaluation As Next Generation Nanoparticle Paclitaxel Against Poorly Perfused Tumors. Cancer Res 2013, 73, 4526. [Google Scholar]
- (53).Minotti G; Menna P; Salvatorelli E; Cairo G; Gianni L Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol Rev 2004, 56 (2), 185–229. [DOI] [PubMed] [Google Scholar]
- (54).Batist G; Barton J; Chaikin P; Swenson C; Welles L Myocet (Liposome-Encapsulated Doxorubicin Citrate): A New Approach in Breast Cancer Therapy. Expert Opin. Pharmacother 2002, 3, 1739–1751. [DOI] [PubMed] [Google Scholar]
- (55).Barenholz Y Doxil–the First FDA-Approved Nano-Drug: Lessons Learned. J. Controlled Release 2012, 160, 117–134. [DOI] [PubMed] [Google Scholar]
- (56).Luo R; Li Y; He M; Zhang H; Yuan H; Johnson M; Palmisano M; Zhou S; Sun D Distinct Biodistribution of Doxorubicin and the Altered Dispositions Mediated by Different Liposomal Formulations. Int. J. Pharm 2017, 519, 1–10. [DOI] [PubMed] [Google Scholar]
- (57).Doogue MP; Polasek TM The ABCD of Clinical Pharmacokinetics. Ther. Adv. Drug Saf 2013, 4, 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wen H; Jung H; Li X Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges. AAPS J 2015, 17, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Fish MB; Thompson AJ; Fromen CA; Eniola-Adefeso O Emergence and Utility of Nonspherical Particles in Biomedicine. Ind. Eng. Chem. Res 2015, 54, 4043–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Nau H Species Differences in Pharmacokinetics and Drug Teratogenesis. Environ. Health Perspect 1986, 70, 113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Oasmia. Oasmia Pharmaceutical Announces Positive Top-line Results for Paclical From Head-to-Head Comparison Study with Abraxane. https://oasmia.com/en/press-release/oasmia-pharmaceutical-announces-positive-top-line-results-paclical-head-head-comparison-study-abraxane/ (accessed April 27, 2018).
- (62).Chen N; Brachmann C; Liu X; Pierce DW; Dey J; Kerwin WS; Li Y; Zhou S; Hou S; Carleton M; et al. Albumin-Bound Nanoparticle (nab) Paclitaxel Exhibits Enhanced Paclitaxel Tissue Distribution and Tumor Penetration. Cancer Chemother. Pharmacol 2015, 76, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Taurin S; Nehoff H; Greish K Anticancer Nanomedicine and Tumor Vascular Permeability; Where is the Missing Link? J. Controlled Release 2012, 164, 265–275. [DOI] [PubMed] [Google Scholar]
- (64).Stylianopoulos T; Jain RK Design Considerations for Nanotherapeutics in Oncology. Nanomedicine 2015, 11, 1893–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nichols JW; Bae YH EPR: Evidence and Fallacy. J. Controlled Release 2014, 190, 451–464. [DOI] [PubMed] [Google Scholar]
- (66).Maeda H; Tsukigawa K; Fang J A Retrospective 30 Years After Discovery of the Enhanced Permeability and Retention Effect of Solid Tumors: Next-Generation Chemotherapeutics and Photodynamic Therapy–Problems, Solutions, and Prospects. Microcirculation 2016, 23, 173–182. [DOI] [PubMed] [Google Scholar]
- (67).Hare JI; Lammers T; Ashford MB; Puri S; Storm G; Barry ST Challenges and Strategies in Anti-Cancer Nanomedicine Development: An Industry Perspective. Adv. Drug Delivery Rev 2017, 108, 25–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.