

Abstract
The iron(II) oxidation kinetic process was studied at 25 stations in coastal seawater of the Macaronesia region (9 around Cape Verde, 11 around the Canary Islands, and 5 around Madeira). In a physicochemical context, experiments were carried out to study the pseudo-first-order oxidation rate constant (k′, min–1) over a range of pH (7.8, 7.9, 8.0, and 8.1) and temperature (10, 15, 20, and 25 °C). Deviations from the calculated kcal′ at the same T, pH, and S were observed for most of the stations. The measured t1/2 (ln 2/k′, min) values at the 25 stations ranged from 1.82 to 3.47 min (mean 1.93 ± 0.76 min) and for all but two stations were lower than the calculated t1/2 of 3.21 ± 0.2 min. In a biogeochemical context, nutrients and variables associated with the organic matter spectral properties (CDOM and FDOM) were analyzed to explain the observed deviations. The application of a multilinear regression model indicated that k′ can be described (R = 0.921 and SEE = 0.064 for pH = 8 and T = 25 °C) from a linear combination of three organic variables, k′OM = kcal −0.11* TDN + 29.9*bDOM + 33.4*C1humic, where TDN is the total dissolved nitrogen, bDOM is the spectral peak obtained from colored dissolved organic matter (DOM) analysis when protein-like or tyrosine-like components are present, and C1humic is the component associated with humic-like compounds obtained from the parallel factor analysis of the fluorescent DOM. Results show that compounds with N in their structures mainly explain the observed k′ increase for most of the samples, although other components could also play a relevant role. Experimentally, k′ provides the net result between the compounds that accelerate the process and those that slow it down.
Keywords: iron(II), oxidation kinetics, Macaronesia, coastal seawater, CDOM, FDOM
Short abstract
Fe(II) persistence in the marine environment is conditioned by the characteristics of the dissolved organic matter.
1. Introduction
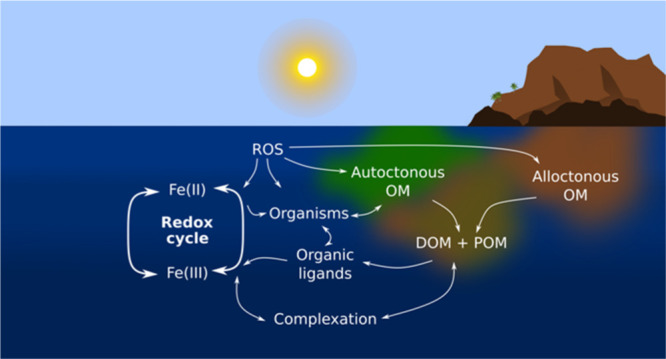
Iron (Fe) is a trace element whose speciation and concentration in the marine environment are affected by multiple abiotic and biotic processes.1 Redox and complexation reactions control its solubility and therefore the fraction of dissolved and bioavailable Fe.2 Processes such as photo-oxidation are a key factor in the control of Fe speciation in surface waters, particularly in areas subject to high irradiation and organic matter content.3 Iron is essential for organisms and plays an important role in the functioning of marine ecosystems.1 Marine organisms have developed different strategies to assimilate Fe, ranging from the reduction of metal on the cell surface to the release of ligands that complex Fe.4 These ligands become part of the organic matter pool. The amount and type of organic matter present in the medium control the speciation of Fe.5 Dissolved organic matter (DOM) affects the Fe organic complexation, with the colloidal and particulate organic matter (COM and POM) acting as net dissolved Fe sinks.6,7
In the ocean, the thermodynamically stable form of Fe is the oxidized ferric iron (Fe(III)), with the soluble fraction in its majority complexed with organic ligands.8 However, ferrous iron (Fe(II)) can also be found in these conditions.9 In surface waters, many authors attribute it to photo-reduction processes in which complexed Fe(III), Fe(III)L, is involved.10 Abiotic redox reactions can also occur in the absence of light, causing the reduction of Fe(III) induced by oxidation of organic matter.11 In anoxic areas and hydrothermal vents, Fe(II) is the predominant form and as soon as these waters are mixed with oxygen-rich waters, Fe(II) tends to oxidize.12,13 The persistence of Fe(II) coming from hydrothermal vents or anoxic sediments is attributed to the complexation with organic ligands and the formation of iron sulfide (FeS).12,14 In the water column, in situ processes such as the remineralization of POM during denitrification are possible sources of Fe(II).15 Particle-bound Fe(III) can be reduced to Fe(II) by reduced sulfur compounds produced within reducing microenvironments of particles.16 In the continental margin, the effect of allochthonous matter is important in the Fe(II) behavior controlling its oxidation.17,18
Knowing how long Fe(II) can remain in solution is one of the challenges in marine chemistry. This knowledge is required to understand the chemical behavior of this trace metal in the marine environment especially when considering that Fe(II) partakes and has played an essential role in the development of marine organisms since primordial times.19 Information on the rate of oxidation of Fe(II) and the half-life time in the ocean is therefore essential for global biogeochemical models.20
This work aimed to study the Fe(II) oxidation kinetic constants in coastal seawater and to investigate the possible factors that could contribute to explain the different persistence of Fe(II) in the marine environment. These studies provide a rate constant (log k′) and a half-life time (t1/2) data set under different pH and temperature conditions, which can be incorporated into ocean biogeochemical Fe models. The k′ and t1/2 values at a fixed pH and T were also determined for every sample. This allows for the identification of non pH and T effects on the oxidation rate constants. Moreover, spectral parameters and ratios obtained from the colored and fluorescent DOM (CDOM and FDOM) were used to provide an equation that accounts for the deviation between measured k′ and calculated kcal′. While the focus of this study is the identification of which variables associated with the organic matter spectral properties (CDOM and FDOM) can explain the deviations of the measured k′ from the calculated kcal at the same T and pH, additional attention should be given in future studies to the effects of other parameters such as Fe(II) and organic ligand concentration ratios and the effect of photogenerated compounds.
2. Materials and Methods
2.1. Sampling Protocol
Samples were collected onboard the RV POSEIDON during the POS533 cruise. The cruise started on the 28th of February 2019 in Mindelo, Sao Vicente (Cape Verde), and ended on the island of Gran Canaria (Canary Islands) on the 22nd of March 2019. The cruise was divided into three sets of stations (Figure 1). The first set was located in the Cape Verde archipelago, the second set was positioned around the Canary archipelago, and the third set encompassed the Madeira area. The number and sampling locations are indicated in Table 1.
Figure 1.
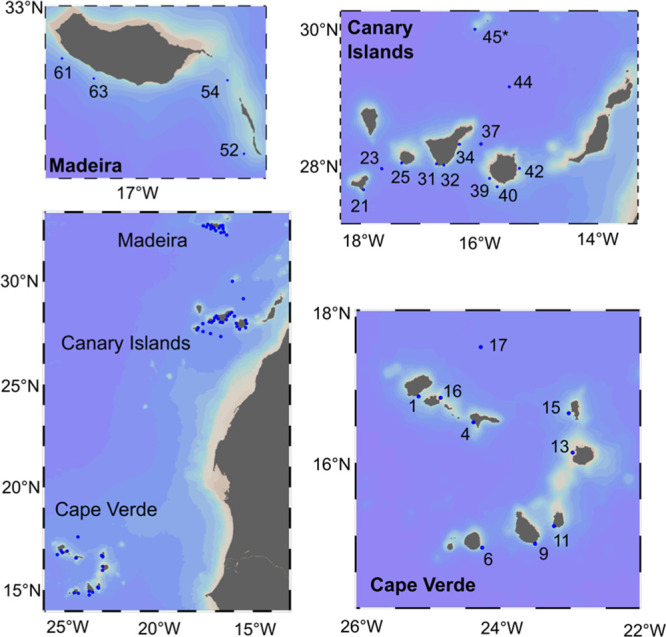
Map of the archipelagos. Cape Verde Island, Canary Islands, and Madeira region. Station 45* (Selvages) belongs to Madeira.
Table 1. Station Number, Name, Location, and Bottom Depth of the sites where Fe(II) oxidation kinetics samples were collected during the POS533 Cruisea.

Stations for OM are the closest stations where organic matter variables were collected.
At each station, trace metal samples were collected at 20 m depth using a Teflon pump (Furon) with a 40 m Teflon tube connected with an AcroPak 1500 capsule w/Supor Memb 0.8/0.2 μm filter. The pump was left for 15 min in flowing seawater to rinse the inner hose, and the 1 L sample was collected in LDPE bottles (Nalgene) and stored at −20 °C until the land-based laboratory analysis. The material was previously cleaned following the trace metal GEOTRACES protocol,21 and the experiments were performed in ISO Class 5 laminar flow hoods within an ISO Class 6 clean lab (QUIMA-IOCAG TM lab).
A lowered SeaBird SBE 9-plus CTD system equipped with two sets of pumped sensors measuring conductivity, temperature, and oxygen at 24 Hz was used. The CTD was mounted on a SeaBird rosette with 12 bottles (10 L) which were used to take discrete water samples for sensor calibration and biogeochemical parameters.22 The biogeochemical parameters sampled were nitrate + nitrite (NO3 + NO2), phosphate (PO4), silicates, dissolved and particulate organic carbon (DOC, POC), total dissolved and particulate nitrogen (TDN, PN), CDOM, and FDOM. The mixed layer depth was deeper than 20 m.22 Information about sampling and analysis is included in the Supporting Information.
2.2. UV Irradiation
An Ace Glass-incorporated ultra-violet photo-oxidation unit which operates at 120 V, 60 Hz was used to UV irradiate the samples. The quartz UV lamp works at 430 mA, 254 nm, and 3.5 W. For the irradiation procedure, each sample was placed in a 100 mL quartz tube and 12 tubes were irradiated at the same time. The samples were irradiated for 4 h. They were then kept in the dark and let to equilibrate with atmospheric air in a laminar flow for more than 72 h before the analysis.
2.3. Oxidation Kinetic Experiments
The oxidation kinetic experiments were carried out in a 60 mL LDPE container within a double-wall glass thermo-regulated cell, connected to a thermostatic bath (Julabo), as described in previous studies.23,24 For each oxidation kinetic study, 25 mL of the seawater sample was used. The initial concentration of added Fe(II) was 0.97 nM. Previous studies have shown that the inorganic Fe(II) oxidation rate constant is independent of the added Fe(II) concentration.25,26 The experiments were carried out at a fixed free-scale pHF = 8 with temperatures of 10, 15, 20, and 25 °C to get the temperature dependence and similarly, at a fixed temperature of 25 °C with pHF of 7.8, 7.9, 8.0, and 8.1 to get the pH dependence. In each oxidation kinetic experiment, the pH was controlled using a Titrino 719 (Metrohm) which automatically added 0.01 M hydrochloric acid (HCl, Panreac Hiperpur-plus). The pH electrodes were calibrated using a Tris buffer solution.27 All the experiments were carried out under O2 saturated conditions to avoid oxygen limitation. Experiments were performed in the dark to avoid the generation of reactive oxygen species (ROS) from photo-oxidation processes and the photo-transformation of Fe.
The concentration of total dissolved Fe(II), TdFe(II), in the samples for the kinetics experiments, was determined using the flow injection analysis by chemiluminescence (FIA-CL) technique with a FeLume system (Waterville Analytical)28 as described in previous studies.12,23 The FIA-CL technique uses luminol as the reagent. 5 L of the luminol reagent was prepared using 2.71 × 10–4 M of 5-amino-2,3-dihydro-1,4-phthalazinedione (Sigma), 4.93 × 10–2 M of Na2CO3 (Sigma-Aldrich), and 0.4 M of previously distilled 25% NH3 (Panreac). The final pH was adjusted to 10.4 by adding 0.06 M Q-HCl. The luminol solution was stored in the dark due to its light sensitivity.
The kinetic studies were carried out in a thermo-regulated cell connected to a thermostatic bath (Julabo) with control to ±0.01 °C. For each study, the seawater sample was acclimated to the desired temperature (in situ and 25 °C). When the temperature was stable, the pHF for the sample was measured.27 For each oxidation kinetic analysis, 50 mL of seawater was used. The seawater was placed in the thermostated cell, and the magnetic stirrer was switched on for 1 h to attain oxygen concentration equilibrium. When the solution stabilized at the desired temperature and pH, the sample line was introduced into the reaction cell. The samples were automatically mixed with the buffer just before being introduced into the detector. The sample plus luminol (1 mL min–1 flux) and the ammonium acetate buffer (0.04 M ammonium acetate adjusted to pH 5.5 with acetic acid at 0.125 mL min–1 flux) were introduced into the detector with a peristaltic pump. This modification provided continuous registration of the measure. This was followed by the Fe(II) addition, and the time was registered.
Iron(II) oxidation kinetic studies in different aqueous media and conditions25,28−37 have shown that the rate of oxidation with O2 can be expressed as an apparent rate constant (kapp), regardless of the mechanism that describes the process (eq 1).
| 1 |
where kapp = k[OH–]2. The brackets indicate the total molar concentration.
The inorganic (i) and organic complexation (L) of Fe influences the kinetics rate constant,29,34 and the apparent rate constant includes the contribution of the inorganic and organic species of Fe(II)
| 2 |
When the reaction is studied in excess O2, the reaction can be considered pseudo-first-order (eq 3).
| 3 |
where k′ = kapp[O2] in s–1 and kapp is expressed in M–1 s–1.
The slope corresponding to the plot of Ln [Fe(II)] versus time determines the k′ (min–1). In the Supporting Information (Figure S1), the lineal relationship is shown for St1.12,23,28
The calculated kcal′ value was obtained from an empirical equation35 for known conditions of temperature, pH, and salinity (eq 4). This equation is valid from 0.526 to 200 nM.35 In this study, the experimental conditions were pH = 8, T = 25 °C, and S = 35.
| 4 |
The extended equation12 calculated for a higher range of low temperatures applicable to deep waters (until 2 °C) can also be used.
2.4. CDOM and FDOM Study
For the CDOM study, the a254 and a325 absorption coefficients at the wavelengths 254 and 325 nm, respectively,38 were selected together with the slope ratio (SR) and the E2/E3 ratios.38,39 The dimensionless SR was calculated from the ratio of the slope of the shorter wavelength region (275–295 nm) to that of the longer wavelength region (350–400 nm). The E2/E3 was calculated as the ratio of absorption at 250 to 365 nm.39−41 From the FDOM study, the fluorescence peaks,42 the humification index (HIX), and the biological index (BIX) were determined43,44 and the parallel factor analysis (PARAFAC) was applied. The variable bDOM was obtained from the fluorescence peak analysis. bDOM is a peak that represents tyrosine-like and protein-like components (with fluorescence Ex/Em = 275/305), and it has been interpreted as a material of autochthonous origin.45
The corrected excitation emission matrices (EEMs), the HIX43 and BIX,44 were used to determine the relative degree of humification and autotrophic productivity of fluorescent CDOM, respectively. HIX is the ratio of the fluorescence over Em 434–480 nm to that over 300–346 nm (at Ex 255 nm). BIX is the ratio of the fluorescence at Em 380 nm to that at 430 nm (at Ex 310 nm). HIX is the estimate of the degree of DOM maturation with an increase in red-shifted emission which presumably arises from increasing conjugation (and possibly aromatization) of DOM.43 BIX is an index for DOM sources. A BIX < 0.7 represents important terrestrial contributions, while a BIX > 1 is characteristic of DOM with a biological/aquatic bacterial origin.44
The PARAFAC analysis for this study contained five components. Components C1humic (Ex/Em: 235(365)/484) and C3humic (Ex/Em: 230/398) had broad excitation and emission spectra, with excitation and emission maxima in the ultraviolet and visible region, respectively. They are traditionally referred to as humic-like components 40. Component C2autoDOM (Ex/Em: 240(300)/342) had a fluorescence signal which is similar to free and protein-bound amino acids and has been ascribed to autochthonous DOM 45. Components C4 (Ex/Em: 230/306) and C5 (Ex/Em: 230(275)/340) were similar to tyrosine-like and tryptophan-like compounds.45 All optical analyses including PARAFAC were conducted using the software R (v4.0.2.) in Rstudio46 with the package staRdom (“spectroscopic analysis of DOM in R”).47 All corrected EEMs of seawater in the POS533 cruise (n = 299) were used for modeling.
The identified fluorescent components were compared with the published data on an open-access spectral database48 (OpenFluor, https://openfluor.lablicate.com). The database compares fluorescence data sets and determines Tucker congruence (set at 95%) as the similarity criteria between pairs of excitation and emission spectra. It provides a rapid way to test new PARAFAC models against those in the literature.
2.5. Statistical Analysis
The Pearson product-moment correlation and Spearman rank-order correlation statistical analysis tests between the oxidation rate constant (k′ at pH = 8 and T = 25 °C) and the measured biochemical variables were performed. A multiple linear regression (MLR) model was also applied to predict if the variability observed in the oxidation rate constants at a fixed temperature, pH, and oxygen saturated condition could be described by two or more organic spectral properties.
3. Results and Discussion
The Fe(II) oxidation kinetics process was studied in coastal seawater considering both a physicochemical and a biogeochemical framework. In the physicochemical context, experiments were carried out to study k′ over a range of pH and T. The goal was to verify if there were differences in the calculated kcal′ value from eq 4.12,35 In the biogeochemical context, variables associated with the presence of OM were analyzed. The goal was to explain the deviations observed for k′ in the physicochemical context. These deviations are a consequence of the interaction between Fe and OM, expressed as ligands, L
| 5 |
| 6 |
| 7 |
| 8 |
3.1. pH Effect
The dependence of log k′ on the pH obtained for each station was plotted (Supporting Information Figure S2) together with the expected calculated log kcal′ under different pH conditions. In all the experiments, linear dependences of log k′ with pH were observed. Deviations from log kcal as a function of pH were observed for some stations.
The slope of the dependence of log k′ with the pH is shown in Figure 2. In Cape Verde, only St4 (Sao Nicolau, 1.68 ± 0.03) presented a value similar to the theoretical one (1.78 ± 0.03). The other stations presented lower slopes ranging between 0.60 ± 0.03 (St1, Santo Antao) and 1.50 ± 0.05 (St6, CVAO).
Figure 2.

Representation of the slope of log k′–pH for the stations in Cape Verde, the Canary Islands, and the Madeira region. The slope was obtained from the plot of log k′ (min–1) vs pH (7.8, 7.9, 8, and 8.1) (Supporting Information Figure S2). The slope for kcal′ is included as St70.
In the Canary Islands, St25 (Gomera), St32 (Tenerife-S), St39 (Gran Canaria-W), and St40 (Gran Canaria-S) presented a log k′–pH slope value close to the theoretical value. The other stations presented lower slopes ranging between 0.54 ± 0.03 (St37, between Tenerife and Gran Canaria) and 1.39 ± 0.08 (St34, Tenerife-E). In the north of the Canary Islands, the ESTOC site presented a slope of 0.97 ± 0.06.
Five stations were studied in the Madeira region. Station 45 (Selvagens) presented the lowest log k′–pH slope of 0.53 ± 0.03. At St54 (Ridge), the value was 1.56 ± 0.09, and St63 presented a slope (1.85 ± 0.03) similar to the theoretical one, and St61 presented the highest slope of the cruise (2.39 ± 0.06).
Two factors must be considered when interpreting the results. First, pH can change the speciation of both Fe(II) and organic ligands. Second, pH can affect the Fe(II)L complexation. The oxidation of Fe(II) in the absence of ligands is affected by pH as a result of speciation changes as demonstrated in previous research.34 In seawater, degraded and recently produced organic compounds are present and can also interact with Fe(II). The log k′–pH slope shows the net effect of organic ligands on k′. If there is an interaction between the ligands and Fe(II), forming complexes with different strengths, there will be a displacement of the log k′–pH slope with respect to the calculated value. The slope will shift up or down if the net result produces an acceleration or a slow-down of the oxidation kinetics rate. Moreover, if the pH changes the speciation of these complexes, a change in slope will occur. This change is due to the net effect of the organic matter matrix. However, the scientific community is currently not able to define the specific functional groups involved. In this sense, previous studies18,49 found differences between the effects of allochthonous and autochthonous DOM on the Fe(II) oxidation rate constant.
3.2. Temperature Effect
The k′ dependence with temperature was studied, and the activation energy (Ea) was calculated using the Arrhenius equation (eq 9).
| 9 |
The ln k′ versus 1/T for ESTOC, CVOO, and Selvagens is shown in the Supporting Information (Figure S3). The Ea for each studied station was plotted in Figure 3 and compared to the calculated Ea,cal (103 ± 3 kJ mol–1) obtained from eq 4. For Cape Verde, the Ea ranged from 20.7 kJ mol–1 in St1 (Santo Antao) to 106.4 kJ mol–1 in St13 (Boa Vista W), the latter having an Ea within the theoretically calculated Ea. In the Canary Islands, St21 (El Hierro-SE) and St44 (ESTOC) presented an Ea (124.7 and 124.0 kJ mol–1, respectively) higher than the theoretical Ea. The other stations presented low Ea values ranging from 20.8 kJ mol–1 in St31 (Tenerife-W) to 97.5 kJ mol–1 in St34 (Tenerife-E). In the Madeira area, St54 (Ridge) presented a high Ea value (117.7 kJ mol–1), with coastal stations presenting 79.8 and 96.2 kJ mol–1 (St63 and St61, respectively).
Figure 3.

Activation energy for each station. The calculated value is also included as St70.
If the rate-controlling process is always the same, then the Ea obtained from the Arrhenius equation should not change for the same experimental conditions. Different Ea may involve different mechanisms or at least, different Fe(II)-organic matter species involved in the oxidation process.
For artificial seawater, without organic ligands, or seawater in which k′ is not affected by the organic matter present,35 the average slope is −5362 ± 162 and the activation energy is 103 ± 3 kJ mol–1. The same results (−5434 ± 183) 104 ± 3 kJ were obtained in non-hydrothermally affected stations within the Mid-North Atlantic Ridge.12 According to these studies, changes in the Ea are probably caused by the interaction of organic compounds with the Fe(II) species which affected the limiting Fe(II) oxidation step. Therefore, it may have a different oxidation reaction mechanism.
3.3. The t1/2 at a Fixed pH and Temperature
Previous studies showed log k′ differences between stations when experiments were carried out under the same pH and T conditions. From the pseudo-first-order rate constant (k′), the t1/2 was calculated as t1/2 = ln 2/k′. This variable represents the persistence time of Fe(II) in each station under the studied conditions. The t1/2 was calculated for pH = 8 and T = 25 °C conditions (Figure 4). The mean t1/2 for the three archipelagos was 1.93 ± 0.76 min for Cape Verde, 1.82 ± 0.45 min for the Canary Islands, and 2.86 ± 0.66 min for Madeira, and the theoretical t1/2 was 3.21 ± 0.2 min. Overall, all stations presented values below the theoretical t1/2 except for St11 (3.09 min) in Cape Verde and St61 (3.10 min) in Madeira which presented values similar to the theoretical t1/2. Only two stations presented t1/2 values higher than the theoretical: St46 (3.47 min) and St52 (3.41 min).
Figure 4.

The t1/2 (min) for the stations in Cape Verde, the Canary Islands, and the Madeira region. The t1/2 theoretical value is included as St70.
Although factors such as O2, pH, and temperature are the main variables that control the oxidation kinetics of Fe(II), photo-generated compounds (organic radicals and ROS)50,51 and other biochemical variables can play a relevant role. When these other variables affect the oxidation process, either by accelerating or delaying it, the value obtained for k′ is different from the calculated kcal′. Photochemical processes should be important in samples from the top few centimeters of the seawater column, while thermal processes control Fe transformation in deeper waters.50
Variations in nutrient or DOC concentrations, the nature of the OM, and colloids present in the solution can lead to changes in the Fe(II) oxidation rate.12,23,52,53 These factors were analyzed for their relationship with the observed changes in t1/2 in different stations at fixed pH and T.
3.4. Nutrients
The stations in the Cape Verde archipelago presented the highest and most variable nutrient concentrations, ranging from 0.03 to 1.16 μM for total inorganic nitrogen (nitrate and nitrite), 0.07 to 0.18 μM for phosphates, and 0.04 to 0.63 μM for silicates. The Canary Islands presented the lowest total inorganic nitrogen (0.08 ± 0.11 μM) while Madeira was characterized by the lowest phosphate (0.04 ± 0.01 μM) and silicate (0.03 ± 0.04 μM) concentrations. Concentrations for each station are shown in Supporting Information (Figure S4). The effects of nutrient concentrations, in particular N and Si, in the t1/2 are generally observed when nutrient concentrations are high, for example, in phytoplankton growing media or eutrophic environments.52,53 The concentrations found in these areas were low and did not exert an appreciable effect on k′.
3.5. Total Dissolved Organic C and N
The DOC and TDN concentrations varied by archipelago and oceanic stations (Supporting Information Figure S5). The mean DOC and TDN concentrations were, respectively, 91.3 ± 3.5 and 6.98 ± 0.7 μM in Cape Verde, 78.2 ± 0.4 and 5.2 ± 0.1 μM in the Canary Islands, and 73.8 ± 0.2 and 5.1 ± 0.1 μM in Madeira. A slight gradient was observed which increased from south to north between the archipelagos.
3.6. Particulate Organic C and N
Unlike DOC and TDN, the particulate organic C (POC) and N (PN) showed significant variations between stations of the same region (Supporting Information Figure S5). The south-to-north gradient was also observed for POC and PN with the lowest concentrations measured in Madeira, followed by the Canary Islands. The highest POC and PN concentrations were measured in Cape Verde. The differences observed between archipelagos in the particulate material suggested that variations in the content and distribution of the colloidal material could also occur.54 However, the samples for Fe(II) were filtered through a 0.2 μm pore size filter, where the particulate material should not affect the obtained t1/2.12 The colloidal material which has been demonstrated to influence the oxidation process12 was not analyzed due to time constraints but should be taken into account in future work.
3.7. Bulk of DOC and the UV-Irradiation Effect
A 20 m sea-surface water sample of ESTOC was used to carry out studies with a known initial DOC content of 78 μM. The sample was divided into two sub-samples: one was irradiated, while the other was kept in the dark (non-irradiated). The evolution of k′ over time was studied for 100 days at a fixed pH = 8 and T = 20 °C (Figure 5). For the non-irradiated seawater, k′ was similar during all the experiments with a value of 0.092 ± 0.003 min–1 and the t1/2(non-IR) = 7.5 ± 0.2 min. For the irradiated samples, high values were obtained the first few days after the irradiation, presenting an exponential decay described by eq 10. k′ changed from 3.59 ± 0.10 min–1 on day 0 to 0.92 ± 0.02 min–1 on day 30 at a rate of 0.088 ± 0.01 min–1, after which it reached a plateau. The reduction of the initial k′ by 50% was reached on day 7.
| 10 |
std dev = ±0.14 min–1.
Figure 5.

Time evolution of k′ for non-UV-irradiated seawater and UV-irradiated seawater for an ESTOC sample with 78 μM of DOC [(Fe(II))0 = 0.97 nM, pH = 8, T = 20 °C, S = 36]. The continuous line defines the behavior for a sample that contains a DOC of 78 μM as in the ESTOC. The dashed red lines represent the calculated variation that would be obtained if the DOC content changes. The longest dashed line in green corresponds to a sample that has 91 μM DOC, as in the samples from Cape Verde. The dashed line in red corresponds to a sample that has 73 μM DOC, as in the samples from Madeira.
The study carried out with a UV-irradiated and a non-irradiated sample presented differences between the two conditions. When the organic matter undergoes photo-oxidation, ROS intermediates, ROS (H2O2, O2•–, and OH•) are generated and these affect the Fe(II) oxidation kinetics rate constant.55,56 Organic radicals such as semiquinone radicals and/or peroxyl radicals can also be generated, becoming even more important than ROS under certain conditions.50 Consequently, UV-irradiated samples should be left in the dark for at least 30 days to reach the plateau and measure a consistent k′.
The difference of k(day 30)′ – k(day 0) = 2.674 min–1 is a measure of the photo-generated compound effect due to the irradiation process. The effect of OM present in the sample can be calculated from the difference between the non-irradiated and irradiated k′. The k′ value changed by 0.82 min–1, which accounted for an increase in the t1/2 of 6.78 min at pH = 8 and T = 20 °C conditions. The effect of three DOC concentrations (73, 78, and 91 μM for Madeira, ESTOC, and Cape Verde, respectively) is represented in Figure 5 assuming a change only in the amount but not in general composition.
The competitive effect between O2 and H2O2 was studied in previous work,36,57 concluding that the oxidation of Fe(II) with H2O2 plays a relatively minor role in most natural waters. At the pH of seawater, O2 is the most important oxidant when [H2O2] is below 200 nM and [Fe(II)] is at nanomolar concentrations. Previous studies in the area showed that H2O2 concentrations are below 100 nM.58
The nature of OM should also be considered. Culture studies carried out in laboratories show a high dependence of k′ on the content of DOC.59,60 However, the same does not occur with oceanic samples12,23 presumably because the dilution factor in the ocean may be important.
The properties of DOM are diverse and depend on its source (terrestrial or aquatic) and diagenetic state. The colorimetric properties of DOM give information about its origin and diagenetic status.38,44 The analysis carried out considered the fraction of OM that absorbs ultraviolet and visible light, through the CDOM, and considered the fluorescent properties of the FDOM. The CDOM and FDOM sampling did not always coincide with the kinetic study sampling. As a result, the analysis only compares common stations or geographically close stations (Table 1).
3.8. CDOM and FDOM
Using spectral parameters and ratios obtained from CDOM and FDOM, different information about the characteristics of DOM was obtained (Supporting Information Figures S6–S11). The absorbances a254 and a325 are respectively proportional to the abundance of conjugated carbon double bonds61 and aromatic substances.38,40 The comparison of a254 and a325 measured for different samples did not show significant variations (Supporting Information Figure S6). For the entire study region, the a254 average value was 1.07 ± 0.23 m–1, with minimum and maximum values of 0.62–1.25 m–1. The a254 absorbances were in agreement with those obtained for temperate Atlantic waters61 where the higher a254 absorbances were found in surface waters (1.43 ± 0.18 m–1) decreasing to 0.87 ± 0.78 m–1 in deep waters. In this study, the a325 average was 0.11 ± 0.02 m–1 and ranged between 0.06 and 0.15 m–1. The determined coastal zone absorbances were slightly higher than those observed in oceanic waters of the North Atlantic subtropical gyre.38 This may be due to the increased production in coastal waters.
The SR and E2/E3 ratios are independent of the CDOM concentration and provide information on the average characteristics (chemistry, source, and diagenesis) of CDOM.39 The average value for SR (Supporting Information Figure S7) was 2.21 ± 1.29, ranging from 1.18 to 6.61, and was within the marine origin DOM SR range. In open ocean waters, SR varies from 1.5 to 4, while it reaches values lower than one when it has a terrestrial origin DOM.39,62
The E2/E3 ratio is used to track changes in the relative size of DOM molecules (Supporting Information Figure S8). As the molecular size increases, E2/E3 decreases due to stronger light absorption by high-molecular-weight CDOM at longer wavelengths.41 The E2/E3 ratio changed from 16 to 40 with an average of 24.39 ± 6.73. The highest ratios were obtained at St41, located close to St42 (Gran Canaria-E), while the lowest ratio was calculated at the oceanic St18.
The diagenetic state of DOM can be deduced from the BIX and HIX indexes.44 The BIX index varied between 0.8 and 8.1 with an average of 1.80 ± 1.81 for the studied regions. In the Canary Islands, St31 (Tenerife-W) had the highest BIX index (8.1 ± 0.75). Stations 9 (Santiago) in Cape Verde and 21 (El Hierro-SE) in the Canary Islands presented values of 0.85 ± 0.39 and 0.96 ± 0.1, respectively (Supporting Information Figure S9). However, most of the stations measured presented BIX indices greater than 1. Consequently, DOM predominantly had an autochthonous origin. Increases in the BIX index indicated a recently reworked one by bacteria DOM.44 The HIX index ranged between 0.09 and 0.62 with an average of 0.48 ± 0.12. In this study, HIX indexes were always below 4, indicating autochthonous DOM from a biological origin.44
The PARAFAC analysis characterized five components (Supporting Information Figures S10 and S11).63 C1 varied between 0.008 and 0.015 RU. C1 was significantly higher in the Cape Verde region (ANOVA, Tukey, p < 0.05). C3 ranged from 0.004 to 0.025 RU. The Cape Verde region was significantly higher than the Canary Islands.45,64 C2autoDOM ranged from 0.004 to 0.018 RU. Two exceptions were found in the Canary Island region, at St31 and St33 (Tenerife W and E), where concentrations reached 0.54 and 0.06 RU, respectively. C4 was not significantly different between the regions (ANOVA p < 0.05) with an average of 0.018 ± 0.005 RU. C5 fluctuated from 0.012 to 0.12 RU with higher values at St31 (Tenerife-W) and St63 (Madeira-W) of 0.12 and 0.10 RU, respectively.
3.9. Statistical Analysis
The measured biochemical variables (nutrients, DOC, TDN, CDOM-a254, a325, E2/E3, SR, b–t–a–m–c peaks, FDOM-BIX, HIX, and C1–C5 from PARAFAC) at 13 stations were tested to identify any correlation with the oxidation rate constant k′ at pH = 8 and T = 25 °C. In the previous work,12,35 the main controlling factors in the determination of kcal′ were pH, temperature, salinity, and oxygen. The statistic results indicated a lack of significant correlations (p < 0.05) between k′ and the biogeochemical variables. From the MLR model, the organic variable TDN and the spectral organic variables bDOM and C1humic were able to predict k′ (eq 11) with an R-value of 0.921 and a standard error of estimate for k′ of 0.064 min–1.
| 11 |
where TDN is the total dissolved nitrogen (μM) and bDOM is the absorbance peak that appears when protein-like or tyrosine-like components are present,42 and C1humic is associated with humic-like components63 in RU (Raman units). Equation 11 was able to explain 84 ± 10% of k′ for the Macaronesia region (data available in Supporting Information Table S1). At fixed pH = 8, T = 25 °C, and k′ = 0.218 min–1, the statistical p-values were p < 0.001 for TDN, p = 0.015 for bDOM, and p = 0.009 for C1humic.
The equation indicated that within the highly variable DOM, compounds containing nitrogen in their structure or nitrogen functional groups could exert an important effect on k′. The overall effect was that increasing concentrations of tyrosine-, protein- or humic-like compounds resulted in a higher observed k′ than the kcal′. As a result, Fe(II) would have a lower t1/2 which would affect the permanence of Fe(II) in the ocean. This does not mean that only nitrogen-containing compounds have an impact on the Fe(II) oxidation process. Other functional groups have also been described in the literature.34 It may be that in the study area, they are the most active.
In the presence of organic ligands L with N in the structure, eq 11 can be considered and explained by eqs 5 and 6
When the Fe(III)L (ferric chelate) stability constant is higher than that of Fe(II)L (ferrous chelate), oxidation of Fe(II)L to Fe(III)L by O2 is a highly favored reaction.65,66 Moreover, for ligands that have a weaker nitrogen donor, the stability of Fe(II)L increases with basicity, but not as much as that of Fe(III)L.65 Iron(II) adsorbed onto mineral surfaces and soluble Fe(II) chelates are important natural reductants. Studies with both Fe-goethite and Fe(II)-tiron as models of Fe(II) adsorbed and soluble Fe(II) chelates with different N–O containing compounds indicated that both the amino-functional groups and the pyridine ring are involved in complexation. Ring-N is more strongly involved than ring-O.66
Specific components of the DON pool in the ocean include urea, dissolved combined and free amino acids, proteins, nucleic acids, amino sugars, and humic substances.67 The chemical identity of most of these compounds and the mechanisms by which they are cycled are unknown.68 Furthermore, DON originates from both allochthonous and autochthonous sources.67 In this study, DON had a higher autochthonous origin (deduced from SR). Previous studies indicated that a substantial fraction of DON in the ocean has bacterial origin such as glutamine and glutamate, with the largest proportion released by planktonic marine cyanobacteria.69,70
Other compounds may also interact with Fe, such as bacterial siderophores and planktonic exudates such as polysaccharides and transparent exopolymers.5 In the same way that microorganisms follow a seasonal cycle in surface waters, the composition and concentration of autochthonous ligands will be conditioned by seasonal variability and the characteristic organism of each marine environment.
4. Environmental Implications
The sources and molecular identities of DOM in the ocean are not yet fully understood, and the parameterizations of organic ligands in ocean biogeochemistry models still have significant uncertainties.7 It is known that the presence of DOM in seawater can affect the Fe(II) oxidation rate constant.17,34,37 Previous laboratory studies35,71 show that organic compounds can accelerate, reduce, or have no effect on Fe(II) oxidation kinetics. The k′ variability in the presence of organic ligands suggests that the effect of DOM on Fe(II) oxidation is dependent on the molecules and their properties. This implies that specific ligands or groups of organic compounds have to be known to understand the effect that those ligands produce on Fe(II). Studies using environmental samples show that DOM from different sources presents varying effects on Fe(II) oxidation kinetics.17,18,37 Oxidation rates for freshwater (e.g., river water and wastewater effluent) are generally higher than those for coastal waters.18 The humic-type DOM (allochthonous origin) was defined as the key factor that accelerates the Fe(II) oxidation in freshwater samples. The lower oxidation rates of coastal seawater compared with those of freshwater and organic ligand-free seawater were thought to be associated with microbially derived autochthonous DOM. Few studies have considered the redox activity of compounds that makes up part of the DOM.72,73 DOM is capable of acting as both an electron donor and an acceptor, keeping Fe in a redox cycle.2 In any case, the results show the net effect of the different organic compounds that may be present in seawater.
This study remarks the important role of DOM in the Fe(II) oxidation kinetic process and the consequences of Fe(II) persistence in the marine environment. Although the physicochemical variables pH and temperature control the Fe(II) oxidation rate in a non-oxygen limited medium, the biogeochemical context is important. The k′ deviation from kcal′ was explained through the spectral characterization of the organic matter. The observed variability of k′ was correlated with the TDN and two spectral variables, bDOM and C1humic. However, it is necessary to indicate that the ratio of the organic ligand and Fe(II) will influence the fraction of Fe(II) complexed by organics and thereby the Fe(II) oxidation kinetics. Although we have obtained a correlation and it is an important advancement, studies related to the concentration ratio between Fe (II) and organics are necessary to the extent of the validity of the empirical equation to a range of Fe(II) concentration.
The nature of DOM in the medium may control the redox cycle of Fe. In the continental margin, Fe(II) is influenced by allochthonous contributions (i.e., rivers, marshes, and estuaries). However, around the volcanic islands, DOM has an autochthonous origin. The studied archipelagos presented common characteristics: they have a volcanic origin and are generally arid. There are no fluvial contributions in these islands, but sporadic contributions through the ravines. Rainfall is quite scarce in Cape Verde and the Canary Islands. This produces a predominantly autochthonous DOM. Little is known about the compounds or structures that make up autochthonous CDOM in the ocean. This study demonstrated that a higher degree of specificity in the OM characterization is required if we want to determine the role that organic compounds play in the persistence of Fe(II) in seawater and the biogeochemical cycle of Fe..
Acknowledgments
We want to express our gratitude to B.Q. from GEOMAR for inviting us to participate in the AIMAC project. Thanks to the master and crew of the R/V Poseidon for the support during the cruise. Special thanks go to Rui Caldeira, Cátia Azevedo, Claudio Cardoso, Ricardo Faria, and Jesus Reis from the Oceanic Observatory of Madeira for the CTD deployments and data. Dr. Gerd Krahmann (GEOMAR) for the evaluation of CTD-data. Kastriot Qelaj (GEOMAR) for analyzing the nutrient samples and Jon Roa, Tania Klüver, and Vivien Floren (all GEOMAR) for sample analysis of DOM and POM components.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.1c04512.
[Fe(II)] and ln [Fe(II)] versus time under different pH and T conditions; dependence of log k′ with the pH; ln k′ with 1/T; detailed sampling protocol analysis; figures for nutrients, DOC, POC, TDN, PON, CDOM, and FDOM; and TDN (μM), b (RU), and C1 (RU) data (PDF)
This work was financially supported by the ATOPFe project (CTM2017-83476-P) from the Ministerio de Ciencia e Innovación (Spain). This study also received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 820989 (project COMFORT, Our common future ocean in the Earth system-quantifying coupled cycles of carbon, oxygen, and nutrients for determining and achieving safe operating spaces with respect to tipping points). The work reflects only the authors’ view; the European Commission and their executive agency are not responsible for any use that may be made of the information the work contains.
The authors declare no competing financial interest.
Notes
The Fe(II) database is available in the PANGAEA database as J.M.S.-C; B.Q. (2021): Fe(II) kinetic data from water samples during POSEIDON cruise POS533 (AIMAC). PANGAEA, https://doi.pangaea.de/10.1594/PANGAEA.934051. Online database OpenFluor is available at https://openfluor.lablicate.com. The PARAFAC model for this study is named “Macaronesia_POS533”.
Supplementary Material
References
- Boyd P. W.; Ellwood M. J.; Tagliabue A.; Twining B. S. Biotic and Abiotic Retention, Recycling and Remineralization of Metals in the Ocean. Nat. Geosci. 2017, 10, 167–173. 10.1038/ngeo2876. [DOI] [Google Scholar]
- Daugherty E. E.; Gilbert B.; Nico P. S.; Borch T. Complexation and Redox Buffering of Iron(II) by Dissolved Organic Matter. Environ. Sci. Technol. 2017, 51, 11096–11104. 10.1021/acs.est.7b03152. [DOI] [PubMed] [Google Scholar]
- Voelker B. M.; Morel F. M. M.; Sulzberger B. Iron Redox Cycling in Surface Waters: Effects of Humic Substances and Light. Environ. Sci. Technol. 1997, 31, 1004–1011. 10.1021/es9604018. [DOI] [Google Scholar]
- Shaked Y.; Kustka A. B.; Morel F. M. M. A General Kinetic Model for Iron Acquisition by Eukaryotic Phytoplankton. Limnol. Oceanogr. 2005, 50, 872–882. 10.4319/lo.2005.50.3.0872. [DOI] [Google Scholar]
- Hassler C. S.; van den Berg C. M. G.; Boyd P. W. Toward a Regional Classification to Provide a More Inclusive Examination of the Ocean Biogeochemistry of Iron-Binding Ligands. Front. Mar. Sci. 2017, 4, 19. 10.3389/fmars.2017.00019. [DOI] [Google Scholar]
- Lønborg C.; Carreira C.; Jickells T.; Álvarez-Salgado X. A. Impacts of Global Change on Ocean Dissolved Organic Carbon (DOC) Cycling. Front. Mar. Sci. 2020, 7, 466. 10.3389/fmars.2020.00466. [DOI] [Google Scholar]
- Pham A. L. D.; Ito T. Formation and Maintenance of the GEOTRACES Subsurface-Dissolved Iron Maxima in an Ocean Biogeochemistry Model. Global Biogeochem. Cycles 2018, 32, 932–953. 10.1029/2017GB005852. [DOI] [Google Scholar]
- Gledhill M. The Organic Complexation of Iron in the Marine Environment: A Review. Front. Microbiol. 2012, 3, 69. 10.3389/fmicb.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye M.; Aldrich A.; van den Berg C. M. G.; de Jong J. T. M.; Nirmaier H.; Veldhuis M.; Timmermans K. R.; de Baar H. J. W. The Chemical Speciation of Iron in the North-East Atlantic Ocean. Deep Sea Res., Part I 2006, 53, 667–683. 10.1016/j.dsr.2005.12.015. [DOI] [Google Scholar]
- Fan S.-M. Photochemical and Biochemical Controls on Reactive Oxygen and Iron Speciation in the Pelagic Surface Ocean. Mar. Chem. 2008, 109, 152–164. 10.1016/j.marchem.2008.01.005. [DOI] [Google Scholar]
- Santana-Casiano J. M.; González-Dávila M.; González A. G.; Millero F. J. Fe(III) Reduction in the Presence of Catechol in Seawater. Aquat. Geochem. 2010, 16, 467–482. 10.1007/s10498-009-9088-x. [DOI] [Google Scholar]
- González-Santana D.; González-Dávila M.; Lohan M. C.; Artigue L.; Planquette H.; Sarthou G.; Tagliabue A.; Santana-Casiano J. M. Variability in Iron (II) Oxidation Kinetics across Diverse Hydrothermal Sites on the Northern Mid Atlantic Ridge. Geochim. Cosmochim. Acta 2021, 297, 143–157. 10.1016/j.gca.2021.01.013. [DOI] [Google Scholar]
- González-Santana D.; Planquette H.; Cheize M.; Whitby H.; Gourain A.; Holmes T.; Guyader V.; Cathalot C.; Pelleter E.; Fouquet Y.; Sarthou G. Processes Driving Iron and Manganese Dispersal From the TAG Hydrothermal Plume (Mid-Atlantic Ridge): Results From a GEOTRACES Process Study. Front. Mar. Sci. 2020, 7, 568. 10.3389/fmars.2020.00568. [DOI] [Google Scholar]
- Yücel M.; Gartman A.; Chan C. S.; Luther G. W. Hydrothermal Vents as a Kinetically Stable Source of Iron-Sulphide-Bearing Nanoparticles to the Ocean. Nat. Geosci. 2011, 4, 367–371. 10.1038/ngeo1148. [DOI] [Google Scholar]
- Moffett J. W. Iron(II) in the world’s oxygen deficient zones. Chem. Geol. 2021, 580, 120314. 10.1016/j.chemgeo.2021.120314. [DOI] [Google Scholar]
- Canfield D. E.; Stewart F. J.; Thamdrup B.; De Brabandere L.; Dalsgaard T.; Delong E. F.; Revsbech N. P.; Ulloa O. A Cryptic Sulfur Cycle in Oxygen-Minimum-Zone Waters off the Chilean Coast. Science 2010, 330, 1375–1378. 10.1126/science.1196889. [DOI] [PubMed] [Google Scholar]
- Rose A. L.; Waite T. D. Kinetics of Iron Complexation by Dissolved Natural Organic Matter in Coastal Waters. Mar. Chem. 2003, 84, 85–103. 10.1016/S0304-4203(03)00113-0. [DOI] [Google Scholar]
- Lee Y. P.; Fujii M.; Kikuchi T.; Terao K.; Yoshimura C. Variation of Iron Redox Kinetics and Its Relation with Molecular Composition of Standard Humic Substances at Circumneutral pH. PLoS One 2017, 12, e0176484 10.1371/journal.pone.0176484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilbert M.; Bonnefoy V. Insight into the Evolution of the Iron Oxidation Pathways. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 161–175. 10.1016/j.bbabio.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Tagliabue A.; Völker C. Towards Accounting for Dissolved Iron Speciation in Global Ocean Models. Biogeosciences 2011, 8, 3025–3039. 10.5194/bg-8-3025-2011. [DOI] [Google Scholar]
- Cutter G. A.; Andersson P.; Codispoti L.; Croot P. L.; Place P.; Hoe T.; Kingdom U.; Francois R.; Sciences O.; Lohan M. C.; Circus D.; Obata H.. Sampling and Sample-Handling Protocols for GEOTRACES Cruises, 2010.
- Mehlmann M.; Quack B.; Atlas E.; Hepach H.; Tegtmeier S. Natural and Anthropogenic Sources of Bromoform and Dibromomethane in the Oceanographic and Biogeochemical Regime of the Subtropical North East Atlantic. Environ. Sci.: Processes Impacts 2020, 22, 679–707. 10.1039/c9em00599d. [DOI] [PubMed] [Google Scholar]
- Santana-González C.; González-Dávila M.; Santana-Casiano J. M.; Gladyshev S.; Sokov A. Organic Matter Effect on Fe(II) Oxidation Kinetics in the Labrador Sea. Chem. Geol. 2019, 511, 238–255. 10.1016/j.chemgeo.2018.12.019. [DOI] [Google Scholar]
- Santana-González C.; Santana-Casiano J. M.; González-Dávila M.; Santana-del Pino A.; Gladyshev S.; Sokov A. Fe(II) oxidation kinetics in the North Atlantic along the 59.5° N during 2016. Mar. Chem. 2018, 203, 64–77. 10.1016/j.marchem.2018.05.002. [DOI] [Google Scholar]
- Millero F. J.; Sotolongo S.; Izaguirre M. The Oxidation Kinetics of Fe(II) in Seawater. Geochim. Cosmochim. Acta 1987, 51, 793–801. 10.1016/0016-7037(87)90093-7. [DOI] [Google Scholar]
- Santana-González C.; Santana-Casiano J. M.; González-Dávila M.; Fraile-Nuez E. Emissions of Fe(II) and Its Kinetic of Oxidation at Tagoro Submarine Volcano, El Hierro. Mar. Chem. 2017, 195, 129–137. 10.1016/j.marchem.2017.02.001. [DOI] [Google Scholar]
- Millero F. J. The pH of Estuarine Waters. Limnol. Oceanogr. 1986, 31, 839–847. 10.4319/LO.1986.31.4.0839. [DOI] [Google Scholar]
- King D. W.; Lounsbury H. A.; Millero F. J. Rates and Mechanism of Fe(II) Oxidation at Nanomolar Total Iron Concentrations. Environ. Sci. Technol. 1995, 29, 818–824. 10.1021/es00003a033. [DOI] [PubMed] [Google Scholar]
- Santana-Casiano J. M.; González-Dávila M.; Millero F. J. The Oxidation of Fe (II) in NaCl–HCO3– and Seawater Solutions in the Presence of Phthalate and Salicylate Ions: A Kinetic Model. Mar. Chem. 2004, 85, 27–40. 10.1016/j.marchem.2003.09.001. [DOI] [Google Scholar]
- Stumm W.; Lee G. F. Oxygenation of Ferrous Iron. Ind. Eng. Chem. 1961, 53, 143–146. 10.1021/ie50614a030. [DOI] [Google Scholar]
- Kester D. R.; Byrne R. H. Jr.; Liang Y.-J.. Redox Reactions and Solution Complexes of Iron in Marine Systems; ACS Publications, 1975. [Google Scholar]
- Tamura H.; Goto K.; Yotsuyanagi T.; Nagayama M. Spectrophotometric determination of iron(II) with 1,10-phenanthroline in the presence of large amounts of iron(III). Talanta 1974, 21, 314–318. 10.1016/0039-9140(74)80012-3. [DOI] [PubMed] [Google Scholar]
- Emmenegger L.; King D. W.; Sigg L.; Sulzberger B. Oxidation Kinetics of Fe(II) in a Eutrophic Swiss Lake. Environ. Sci. Technol. 1998, 32, 2990–2996. 10.1021/es980207g. [DOI] [Google Scholar]
- Santana-Casiano J. M.; González-Dávila M.; Rodríguez M. J.; Millero F. J. The Effect of Organic Compounds in the Oxidation Kinetics of Fe(II). Mar. Chem. 2000, 70, 211–222. 10.1016/S0304-4203(00)00027-X. [DOI] [Google Scholar]
- Santana-Casiano J. M.; González-Dávila M.; Millero F. J. Oxidation of Nanomolar Levels of Fe(II) with Oxygen in Natural Waters. Environ. Sci. Technol. 2005, 39, 2073–2079. 10.1021/es049748y. [DOI] [PubMed] [Google Scholar]
- González-Dávila M.; Santana-Casiano J. M.; Millero F. J. Competition between O2 and H2O2 in the Oxidation of Fe (II) in Natural Waters. J. Solution Chem. 2006, 35, 95–111. 10.1007/s10953-006-8942-3. [DOI] [Google Scholar]
- Rose A. L.; Waite T. D. Kinetic Model for Fe(II) Oxidation in Seawater in the Absence and Presence of Natural Organic Matter. Environ. Sci. Technol. 2002, 36, 433–444. 10.1021/es0109242. [DOI] [PubMed] [Google Scholar]
- Nelson N. B.; Siegel D. A. The Global Distribution and Dynamics of Chromophoric Dissolved Organic Matter. Ann. Rev. Mar. Sci 2013, 5, 447–476. 10.1146/annurev-marine-120710-100751. [DOI] [PubMed] [Google Scholar]
- Helms J. R.; Stubbins A.; Ritchie J. D.; Minor E. C.; Kieber D. J.; Mopper K. Absorption Spectral Slopes and Slope Ratios as Indicators of Molecular Weight, Source, and Photobleaching of Chromophoric Dissolved Organic Matter. Limnol. Oceanogr. 2008, 53, 955–969. 10.4319/lo.2008.53.3.0955. [DOI] [Google Scholar]
- Catalá T. S.; Martínez-Pérez A. M.; Nieto-Cid M.; Álvarez M.; Otero J.; Emelianov M.; Reche I.; Arístegui J.; Álvarez-Salgado X. A. Dissolved Organic Matter (DOM) in the open Mediterranean Sea. I. Basin-wide distribution and drivers of chromophoric DOM. Prog. Oceanogr. 2018, 165, 35–51. 10.1016/j.pocean.2018.05.002. [DOI] [Google Scholar]
- De Haan H.; De Boer T. Applicability of Light Absorbance and Fluorescence as Measures of Concentration and Molecular Size of Dissolved Organic Carbon in Humic Lake Tjeukemeer. Water Res. 1987, 21, 731–734. 10.1016/0043-1354(87)90086-8. [DOI] [Google Scholar]
- Coble P. G. Marine Optical Biogeochemistry: The Chemistry of Ocean Color. Chem. Rev. 2007, 107, 402–418. 10.1021/cr050350+. [DOI] [PubMed] [Google Scholar]
- Zsolnay A.; Baigar E.; Jimenez M.; Steinweg B.; Saccomandi F. Differentiating with Fluorescence Spectroscopy the Sources of Dissolved Organic Matter in Soils Subjected to Drying. Chemosphere 1999, 38, 45–50. 10.1016/S0045-6535(98)00166-0. [DOI] [PubMed] [Google Scholar]
- Huguet A.; Vacher L.; Relexans S.; Saubusse S.; Froidefond J. M.; Parlanti E. Properties of Fluorescent Dissolved Organic Matter in the Gironde Estuary. Org. Geochem. 2009, 40, 706–719. 10.1016/j.orggeochem.2009.03.002. [DOI] [Google Scholar]
- Coble P. G. Characterization of Marine and Terrestrial DOM in Seawater Using Excitation-Emission Matrix Spectroscopy. Mar. Chem. 1996, 51, 325–346. 10.1016/0304-4203(95)00062-3. [DOI] [Google Scholar]
- Ihaka R.; Gentleman R. R: A Language for Data Analysis and Graphics. J. Comput. Graph Stat. 1996, 5, 299–314. 10.1080/10618600.1996.10474713. [DOI] [Google Scholar]
- Pucher M.; Wünsch U.; Weigelhofer G.; Murphy K.; Hein T.; Graeber D. StaRdom: Versatile Software for Analyzing Spectroscopic Data of Dissolved Organic Matter in R. Water 2019, 11, 2366. 10.3390/w11112366. [DOI] [Google Scholar]
- Murphy K. R.; Stedmon C. A.; Wenig P.; Bro R. OpenFluor- An Online Spectral Library of Auto-Fluorescence by Organic Compounds in the Environment. Anal. Methods 2014, 6, 658–661. 10.1039/c3ay41935e. [DOI] [Google Scholar]
- Lee Y. P.; Fujii M.; Kikuchi T.; Natsuike M.; Ito H.; Watanabe T.; Yoshimura C. Importance of Allochthonous and Autochthonous Dissolved Organic Matter in Fe(II) Oxidation: A Case Study in Shizugawa Bay Watershed, Japan. Chemosphere 2017, 180, 221–228. 10.1016/j.chemosphere.2017.04.008. [DOI] [PubMed] [Google Scholar]
- Garg S.; Jiang C.; Waite T. D. Impact of pH on Iron Redox Transformations in Simulated Freshwaters Containing Natural Organic Matter. Environ. Sci. Technol. 2018, 52, 13184–13194. 10.1021/acs.est.8b03855. [DOI] [PubMed] [Google Scholar]
- Garg S.; Rose A. L.; Waite T. D. Photochemical Production of Superoxide and Hydrogen Peroxide from Natural Organic Matter. Geochim. Cosmochim. Acta 2011, 75, 4310–4320. 10.1016/j.gca.2011.05.014. [DOI] [Google Scholar]
- Samperio-Ramos G.; Santana-Casiano J. M.; González-Dávila M. Effect of Ocean Warming and Acidification on the Fe(II) Oxidation Rate in Oligotrophic and Eutrophic Natural Waters. Biogeochemistry 2016, 128, 19–34. 10.1007/s10533-016-0192-x. [DOI] [Google Scholar]
- González A. G.; Santana-Casiano J. M.; Pérez N.; González-Dávila M. Oxidation of Fe(II) in Natural Waters at High Nutrient Concentrations. Environ. Sci. Technol. 2010, 44, 8095–8101. 10.1021/es1009218. [DOI] [PubMed] [Google Scholar]
- Boye M.; Nishioka J.; Croot P.; Laan P.; Timmermans K. R.; Strass V. H.; Takeda S.; de Baar H. J. W. Significant Portion of Dissolved Organic Fe Complexes in Fact Is Fe Colloids. Mar. Chem. 2010, 122, 20–27. 10.1016/j.marchem.2010.09.001. [DOI] [Google Scholar]
- González-Davila M.; Santana-Casiano J. M.; Millero F. J. Oxidation of Iron (II) Nanomolar with H2O2 in Seawater. Geochim. Cosmochim. Acta 2005, 69, 83–93. 10.1016/j.gca.2004.05.043. [DOI] [Google Scholar]
- Millero F. J.; Sotolongo S. The Oxidation of Fe(II) with H2O2 in Seawater. Geochim. Cosmochim. Acta 1989, 53, 1867–1873. 10.1016/0016-7037(89)90307-4. [DOI] [Google Scholar]
- Santana-Casiano J. M.; González-Dávila M.; Millero F. J. The Role of Fe(II) Species on the Oxidation of Fe(II) in Natural Waters in the Presence of O2 and H2O2. Mar. Chem. 2006, 99, 70–82. 10.1016/j.marchem.2005.03.010. [DOI] [Google Scholar]
- Heller M. I.; Gaiero D. M.; Croot P. L. Basin Scale Survey of Marine Humic Fluorescence in the Atlantic: Relationship to Iron Solubility and H2O2. Global Biogeochem. Cycles 2013, 27, 88–100. 10.1029/2012GB004427. [DOI] [Google Scholar]
- González A. G.; Santana-Casiano J. M.; González-Dávila M.; Pérez-Almeida N.; Suárez de Tangil M. Effect of Dunaliella Tertiolecta Organic Exudates on the Fe(II) Oxidation Kinetics in Seawater. Environ. Sci. Technol. 2014, 48, 7933–7941. 10.1021/es5013092. [DOI] [PubMed] [Google Scholar]
- Samperio-Ramos G.; Santana-Casiano J. M.; González-Dávila M. Variability in the Production of Organic Ligands, by Synechococcus PCC 7002, under Different Iron Scenarios. J. Oceanogr. 2017, 74, 277–286. 10.1007/s10872-017-0457-6. [DOI] [Google Scholar]
- Lønborg C.; Álvarez-Salgado X. A. Tracing Dissolved Organic Matter Cycling in the Eastern Boundary of the Temperate North Atlantic Using Absorption and Fluorescence Spectroscopy. Deep Sea Res., Part I 2014, 85, 35–46. 10.1016/j.dsr.2013.11.002. [DOI] [Google Scholar]
- Loginova A. N.; Thomsen S.; Engel A. Chromophoric and Fluorescent Dissolved Organic Matter in and above the Oxygen Minimum Zone off Peru. J. Geophys. Res.: Oceans 2016, 121, 7973–7990. 10.1002/2016JC011906. [DOI] [Google Scholar]
- Catalá T. S.; Reche I.; Fuentes-Lema A.; Romera-Castillo C.; Nieto-Cid M.; Ortega-Retuerta E.; Calvo E.; Álvarez M.; Marrasé C.; Stedmon C. A.; Álvarez-Salgado X. A. Turnover Time of Fluorescent Dissolved Organic Matter in the Dark Global Ocean. Nat. Commun. 2015, 6, 5986. 10.1038/ncomms6986. [DOI] [PubMed] [Google Scholar]
- Stedmon C. A.; Thomas D. N.; Granskog M.; Kaartokallio H.; Papadimitriou S.; Kuosa H. Characteristics of Dissolved Organic Matter in Baltic Coastal Sea Ice: Allochthonous or Autochthonous Origins?. Environ. Sci. Technol. 2007, 41, 7273–7279. 10.1021/es071210f. [DOI] [PubMed] [Google Scholar]
- Martell A. E.; Motekaitis R. J.; Chen D.; Hancock R. D.; McManus D. Selection of New Fe(III)/Fe(II) Chelating Agents as Catalysts for the Oxidation of Hydrogen Sulfide to Sulfur by Air. Can. J. Chem. 1996, 74, 1872–1879. 10.1139/v96-210. [DOI] [Google Scholar]
- Li X.; Chen Y.; Zhang H. Reduction of Nitrogen-Oxygen Containing Compounds (NOCs) by Surface-Associated Fe(II) and Comparison with Soluble Fe(II) Complexes. Chem. Eng. J. 2019, 370, 782–791. 10.1016/j.cej.2019.03.203. [DOI] [Google Scholar]
- Berman T.; Bronk D. Dissolved Organic Nitrogen: A Dynamic Participant in Aquatic Ecosystems. Aquat. Microb. Ecol. 2003, 31, 279–305. 10.3354/ame031279. [DOI] [Google Scholar]
- McCarthy M. D.; Hedges J. I.; Benner R. Major Bacterial Contribution to Marine Dissolved Organic Nitrogen. Science 1998, 281, 231–234. 10.1126/science.281.5374.231. [DOI] [PubMed] [Google Scholar]
- Capone D. G.; Ferrier M. D.; Carpenter E. J. Amino Acid Cycling in Colonies of the Planktonic Marine Cyanobacterium Trichodesmium Thiebautii. Appl. Environ. Microbiol. 1994, 60, 3989–3995. 10.1128/aem.60.11.3989-3995.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glibert P. M.; Bronk D. A. Release of Dissolved Organic Nitrogen by Marine Diazotrophic Cyanobacteria, Trichodesmium Spp. Appl. Environ. Microbiol. 1994, 60, 3996–4000. 10.1128/aem.60.11.3996-4000.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis T. L.; Singer P. C. Complexation of Iron(II) by Organic Matter and Its Effect on Iron(II) Oxygenation. Environ. Sci. Technol. 1974, 8, 569–573. 10.1021/es60091a008. [DOI] [Google Scholar]
- Garg S.; Jiang C.; David Waite T. Mechanistic Insights into Iron Redox Transformations in the Presence of Natural Organic Matter: Impact of PH and Light. Geochim. Cosmochim. Acta 2015, 165, 14–34. 10.1016/j.gca.2015.05.010. [DOI] [Google Scholar]
- Jiang C.; Garg S.; Waite T. D. Hydroquinone-Mediated Redox Cycling of Iron and Concomitant Oxidation of Hydroquinone in Oxic Waters under Acidic Conditions: Comparison with Iron-Natural Organic Matter Interactions. Environ. Sci. Technol. 2015, 49, 14076–14084. 10.1021/acs.est.5b03189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.