

Abstract
Thirteen percent of cancers worldwide are associated with viral infections. While many human oncogenic viruses are widely endemic, very few infected individuals develop cancer. This raises the question why oncogenic viruses encode viral oncogenes if they can replicate and spread between human hosts without causing cancer. Interestingly, viral infection triggers innate immune signaling pathways that in turn activate tumor suppressors such as p53, suggesting that tumor suppressors may have evolved not primarily to prevent cancer, but to thwart viral infection. Here, we summarize and compare several major immune evasion strategies used by viral and non-viral cancers, with a focus on oncogenes that play dual roles in promoting tumorigenicity and immune evasion. By highlighting important and illustrative examples of how oncogenic viruses evade the immune system, we aim to shed light on how non-viral cancers avoid immune detection. Further study and understanding of how viral and non-viral oncogenes impact immune function could lead to improved strategies to combine molecular therapies targeting oncoproteins in combination with immunomodulators.
Introduction
When Peyton Rous harvested a tumor from a Long Island hen in 1911 and showed that a cell-free filtrate of the tumor could cause cancer when injected into other chickens, he provided some of the earliest evidence that viruses could cause cancer (1). It was only in 1966 that Rous was awarded the Nobel Prize, but now over a century from his initial findings, seven human cancer-causing viruses have been identified: Epstein-Barr Virus (EBV), Human Papilloma Virus (HPV), Kaposi sarcoma-associated Herpesvirus (KSHV), Hepatitis B Virus (HBV), Hepatitis C Virus (HCV), Human T-lymphotropic Virus-1 (HTLV-1), and Merkel cell polyomavirus (MCPyV) (2). While many of these viruses are widely endemic in humans, relatively few infected individuals go on to develop cancer, indicating that other factors in addition to viral infection are required for cancer induction. Nevertheless, virus-associated cancers account for a significant fraction (13%) of the global cancer burden worldwide, particularly in the developing world (3).
Oncogenic viruses vary widely in structure – from single-stranded RNA viruses to large, double-stranded DNA viruses – as well as in the target cells they infect. All the human oncogenic viruses can cause persistent infections and/or enter a latent phase after initial infection, remaining quiescent in host cells for years (4). None of the human tumor viruses cause cancer acutely, and decades can pass between initial infection and cancer development, which occurs only rarely. Thus, human tumor viruses do not need to induce cancer to replicate, and in patients with virally-associated cancers, infectious virions are not found in cancer cells, but in other non-cancerous cells and tissues (5).
Transformation by oncogenic viruses occurs through either direct or indirect mechanisms. In cancers resulting from direct viral transformation – such as HPV-related malignancies – cancer cells must retain at least one copy of the viral genome to remain malignant (6). In contrast, cancers caused by indirect viral transformation result from prolonged inflammation associated with chronic infection that drives mutagenesis and transformation in infected and non-infected cells (6, 7). Note that while there are non-viral infections associated with cancer induction, e.g. Helicobacter pylori infection, here by non-viral cancers we refer to cancers not associated with infection and presumably driven by inherited and/or stochastic mutations or non-infectious mutagens.
Initially, it was thought that viral oncoproteins served mainly to bypass cell cycle checkpoints such as p53 and pRB, driving host cell proliferation to facilitate viral genome replication and leading to eventual cancer in some cases (8). However, oncogenic viral replication and transmission does not require host cell transformation, evidenced by the fact that most people infected with oncogenic viruses do not develop virus-associated cancers (9). If oncogenic viruses do not need to cause cancer to replicate, the question arises why human tumor viruses encode viral oncogenes? Our growing knowledge of anti-viral immunology suggests an alternative explanation, elegantly put forward by Drs. Patrick Moore and Yuan Chang: viral oncogenes prevent innate immune-induced cell death or cell cycle arrest (6).
The adaptive immune system is critical to control viral infection, including infection by oncogenic viruses, evidenced by the increased incidence of viral-associated cancers in immunocompromised individuals (10). On first infection, T and B cells specific for a particular virus take several days to emerge, expand, eliminate infected cells, and neutralize virus; a subset of these virus-specific T and B cells differentiate into memory cells that can then respond more rapidly and robustly to viral re-challenge. The ability of adaptive cells to form memory has motivated efforts to develop vaccines against oncogenic viruses. Successful vaccines have been developed against HPV and HBV, significantly decreasing the incidence of HPV- and HBV-associated cancers (11).
In addition to the adaptive immune system, the innate immune system plays an important role in anti-viral immunity. In 2003, Taniguchi and colleagues showed that viral infection, through type I interferon signaling, increased TP53 (encoding p53) expression in infected cells, leading to apoptosis (12). Within cells, viruses can trigger innate pathways when cellular pattern recognition receptors (PRR) bind conserved viral structures, such as cytosolic DNA or double-stranded RNA (13). It is becoming increasingly clear that p53 coordinates responses to multiple cellular stresses, including viral infection (14). Thus, viral oncoproteins, by inhibiting critical cell regulators such as p53 and pRB, may serve to prevent virus-trigged innate immune signaling and cell cycle arrest or apoptosis, allowing viral persistence and/or latency. It is long-term persistence that is crucial for viral replication. One hypothesis is that larger DNA viruses, which rely on high-fidelity cellular replication machinery, cannot use antigenic drift to evade immune responses but must rather become latent in hosts, re-activating periodically to allow infection of naïve hosts from generation to generation (9). Given that orthologs of TP53 exist in organisms in which cancers do not occur (15), TP53 likely initially evolved not primarily to prevent cancers, but to respond to cellular stresses such as viral infection (14, 16).
In this review, we discuss several major pathways of viral and non-viral cancer immune evasion with the goal of using selected viral and cellular oncogenes to illustrate specific mechanisms (Table 1). For a comprehensive review of viral oncogenes, we refer the readers to (2), and for cellular oncogenes to (17, 18). Similar to viral oncoproteins, several oncogenes mutated in non-viral cancers not only function to drive proliferation and cell survival but also enhance immune evasion. With the growing clinical use of cancer immunotherapies such as immune checkpoint blockade (ICB), understanding immune evasion strategies in both viral and non-viral cancers will provide important insights into immunotherapy resistance.
Table 1.
Role of cellular oncogenes in immune evasion.
| Oncogene | Role in Immune Evasion | References |
|---|---|---|
| ALK | Inhibits immunogenic cell death | (63, 64) |
| β-catenin | Represses the expression of CCL4, which recruits DCs and T cells | (57) |
| BRAF | Drives internalization and sequestration of MHC-I | (80, 81) |
| EGFR | Contributes to increased PD-L1 expression | (83, 102) |
| EPHA2 | Increases TGFB signaling and COX-2 expression, causing increased proinflammatory PGE2 | (56) |
| FGFR | Drives PD-L1 expression | (104) |
| HER2 | Binds to STING to prevent immune sensing Internalization of MHC-I |
(40, 41, 82) |
| MYC | Induces expression of CCL9 to recruited macrophages Acts with KRAS to upregulate IL-23 to suppress innate immune cells and reduce CTL infiltration Drives PD-L1 expression Inhibits immunogenic cell death |
(49, 58, 59, 103) |
| NOTCH | Suppresses SASP, causing decreased T cell recruitment | (67) |
| RAS | Drives OIS and SASP expression | (51, 58, 59, 105) |
| SMAD4 | Drives TGFB signaling, which inhibits adaptive immune response | (56) |
Countering innate immunity: oncogenes inhibit the cGAS/STING pathway
The innate immune system responds rapidly to infection by recognizing shared features among pathogens, in contrast to the adaptive immune system, which can take hours to days to generate sufficient numbers of activated, antigen-specific lymphocytes. While we typically think of the innate immune system as comprising immune cells such as macrophages and NK cells, all cells can activate cell-intrinsic innate immune pathways when infected. Non-immune and immune cells express PRR, such as Toll-Like Receptors (TLR), which bind to conserved structures on pathogens called pattern-associated molecular patterns (PAMP) or host-derived structures called damage-associated molecular patterns (DAMP). PRR binding to PAMP or DAMP initiates a signaling cascade that ultimately leads to expression of Type I Interferons and other inflammatory cytokines (13, 19). Type I Interferons activate innate immune cells such as macrophages and NK cells (20). The cytokines induced by PAMP and DAMP, particularly Type I Interferons, inhibit cellular proliferation in part through induction of TP53 (12). Evidence suggests that tumor suppressors are also involved in the regulation of innate immune receptors. For example, p53 acts as a transcriptional regulator for expression of TLR family genes (21) while pRB deactivates an inhibitor of TLR3 (22). The lack of innate activation and inflammation causes improper priming of tumor-specific CD8 T cells, and these T cells fail to gain or lose cytolytic effector function and are unable to eliminate cancer cells (23, 24). This leads to chronic stimulation of tumor-specific T cells, leading to a more profound state of dysfunction/exhaustion. One of the hallmarks of T cell dysfunction/exhaustion is expression of multiple inhibitory receptors such as PD-1, and below we discuss oncogene-induced upregulation of inhibitory ligands such as PD-L1 on cancer cells. For a more in-depth review of CD8 T cells differentiation to the dysfunctional/exhausted state in cancer, we refer the reader to (25).
An important PRR for viral sensing is cyclic GMP-AMP synthase (cGAS). cGAS binds to double stranded DNA in the cytosol and converts ATP and GTP to cyclic GMP-AMP (cGAMP; Fig 1). cGAMP activates Stimulator of Interferon Genes (STING), which acts as a scaffold to bring TANK-binding kinase-1 (TBK1) in proximity to interferon response factor-3 (IRF3) (26). This close association allows TBK1 to phosphorylate IRF3. Phosphorylated IRF3 then enters the nucleus and acts as a transcription factor to induce the expression of Type I Interferons.
Figure 1. Viral and cellular oncoproteins inhibit the cGAS/STING pathway.
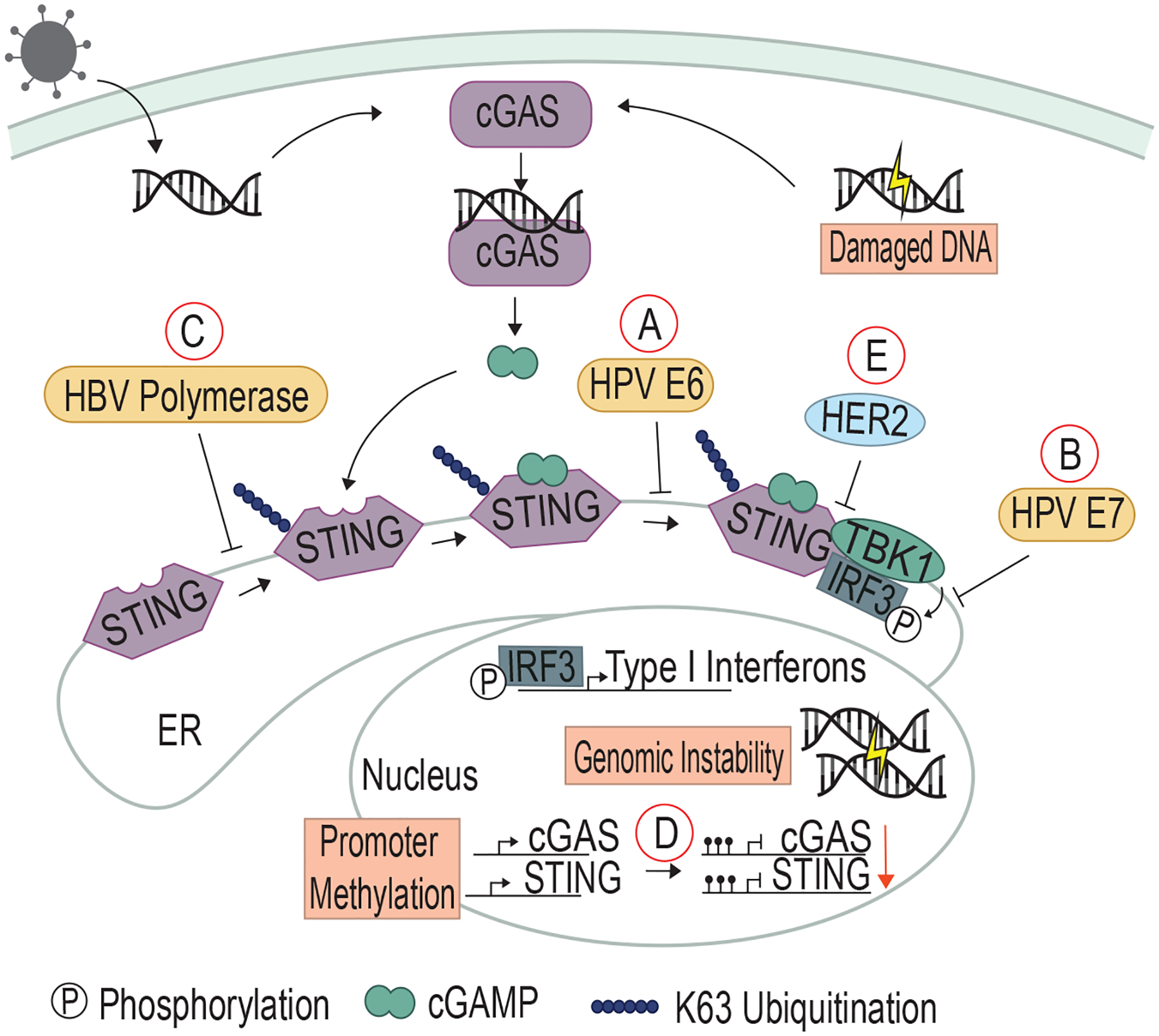
A) HPV E6 binds to STING to inhibit TBK1 and IRF3 binding, and B) HPV E7 binds to IRF3 to prevent phosphorylation. C) HBV polymerase prevents K63 ubiquitination of STING to block STING activation. D) In non-viral cancers such as CRC and melanoma, the promoters of both cGAS and STING are methylated to prevent expression of these proteins. E) HER2 associates with STING and recruits AKT to phosphorylate TBK1, which prevents the kinase from binding to STING and facilitating downstream signaling. Viral proteins are depicted in gold, and cellular proteins in green.
HPV is a small DNA virus that infects epithelial cells through sites of trauma such as cuts and micro-abrasions. HPV is categorized into low-risk and high-risk strains (27), and it is the high-risk strains which have the potential to drive host cell transformation. HPV accounts for nearly all cases of cervical cancer as well as a significant portion of other anogenital cancers and head and neck cancers (27). HPV quickly integrates into the host genome to establish latent infection in host cells (27).
HPV proteins E6 and E7 expressed by high-risk strains bind to and inhibit p53 and pRB, respectively, eventually leading to transformation. Both are also implicated in deactivating the cGAS/STING pathway. High-risk HPV strains encode E6 with a high binding affinity for IRF3. E6 binding to IRF3 prevents TBK1 from phosphorylating IRF3 and thus inhibits IRF3’s ability to function as a transcription factor (28) (Fig 1A). In HPV-transformed cervical cancer cells (HeLa cells), E7 binds STING, preventing STING-mediated TBK1/IRF3 association and leaving IRF3 in an inactivated state. CRISPR/Cas9 knockout of E7 restored Type I Interferon expression (29) (Fig 1B). E7 has been shown to bind to both pRB and STING through the LXCXE motif (29, 30), highlighting the dual role played by E7 in inhibiting cell cycle brakes and innate immune surveillance.
Hepatitis B Virus (HBV) is another small DNA virus and infects hepatocytes. HBV is most commonly transmitted through vertical transmission from mother to child, which almost always results in chronic infection. In contrast, 90% of individuals infected as adults through exchange of infected bodily fluids are able to clear the HBV infection (31). Those with chronic HBV infection can go on to develop liver cirrhosis and hepatocellular carcinoma secondary to chronic infection/inflammation (31). HBV is unique as it is a non-retrovirus that has retroviral activity. The HBV polymerase can synthesize viral DNA genome from RNA (32) and also associate with STING directly to prevent ubiquitination of STING lysine 63 (K63; Fig 1C). K63 ubiquitination activates STING (33).
cGAS/STING activation also occurs in non-viral cancers. Cells with chromosomal instability or damaged by genotoxic agents such as carcinogens or radiation can leak DNA into the cytosol, triggering cGAS/STING activation (34). Likewise, cancers with DNA mismatch repair defects have increased levels of cytosolic DNA, and these cancers frequently develop alterations to circumvent the cGAS/STING DNA sensing pathway (35). Recent studies have shown that STING activity is often suppressed in colorectal cancer (CRC), particularly the subset of CRC with defects in the DNA mismatch repair mechanisms. The Cancer Genome Atlas (TCGA) analysis of cGAS/STING genes in CRC samples as well as cell lines of colorectal cancer, melanoma, and ovarian cancer showed that cGAS/STING components are frequently downregulated, with evidence of disrupted downstream signaling (36). This was due at least in part to hypermethylation of the promoters of the genes encoding STING and cGAS (37–39), although it is not known what induces this hypermethylation (Fig 1D).
HER2, a commonly altered receptor tyrosine kinase (RTK) in breast, prostate, and ovarian cancers, has recently been shown to be associated with the suppression of the cGAS/STING pathway. HER2 acts as a heterodimer with epidermal growth factor (EGFR) at the plasma membrane, and when altered due to overexpression or mutation, drives cellular proliferation (40). HER2 can additionally act independently of EGFR in the ER and associate with STING. This association recruits AKT, which then phosphorylates TBK1 and prevents it from binding to STING (Fig 1E) (41). Thus, the HER2 oncogene, like the HPV and HBV proteins described above, both promotes cellular proliferation and prevents immune sensing. HER2 is the first known cellular oncoprotein shown to directly inhibit the cGAS/STING pathway, but given that several oncogenic viruses target this pathway and the evidence that cGAS/STING is inhibited in mismatch repair-defective CRC, it is likely that other cellular-derived cancers and oncogenes may inhibit this innate signaling pathway.
The suppression of cGAS and STING expression across several different cancer types has led to development of therapies to re-activate these pathways. The synthetic STING agonist 2’3’-cGAMP has been shown to activate NK cell responses in mice (42). However, many dinucleotide-based cGAS/STING agonists are unstable, limiting effectiveness. Recent studies have identified non-nucleotide agonists that work effectively when delivered orally in subcutaneous mouse models of colon adenocarcinoma and melanoma (43, 44). In current clinical trials, cGAS/STING therapies on their own do not lead to tumor eradication. However, STING agonists have been shown to be an effective adjuvant when given with a tumor-associated antigen (TAA) peptide cancer vaccine in a mouse model of melanoma (45). Therefore, several strategies of combining STING agonists with other immunotherapies are currently being tested in Phases I and II trials (46).
Preventing immune cell recruitment: oncogenes alter cytokine/chemokine production
Upon viral infection, infected cells and their surrounding cells release inflammatory cytokines and chemokines (Fig 2). Chemokines induce immune cell migration to sites of infection, while cytokines regulate immune cell differentiation and activity. Thus, viruses have evolved several mechanisms to alter cytokine/chemokine production to evade both the innate and adaptive immune response (47).
Figure 2. Viral and cellular oncoproteins modulate cytokine production to change the immune landscape.
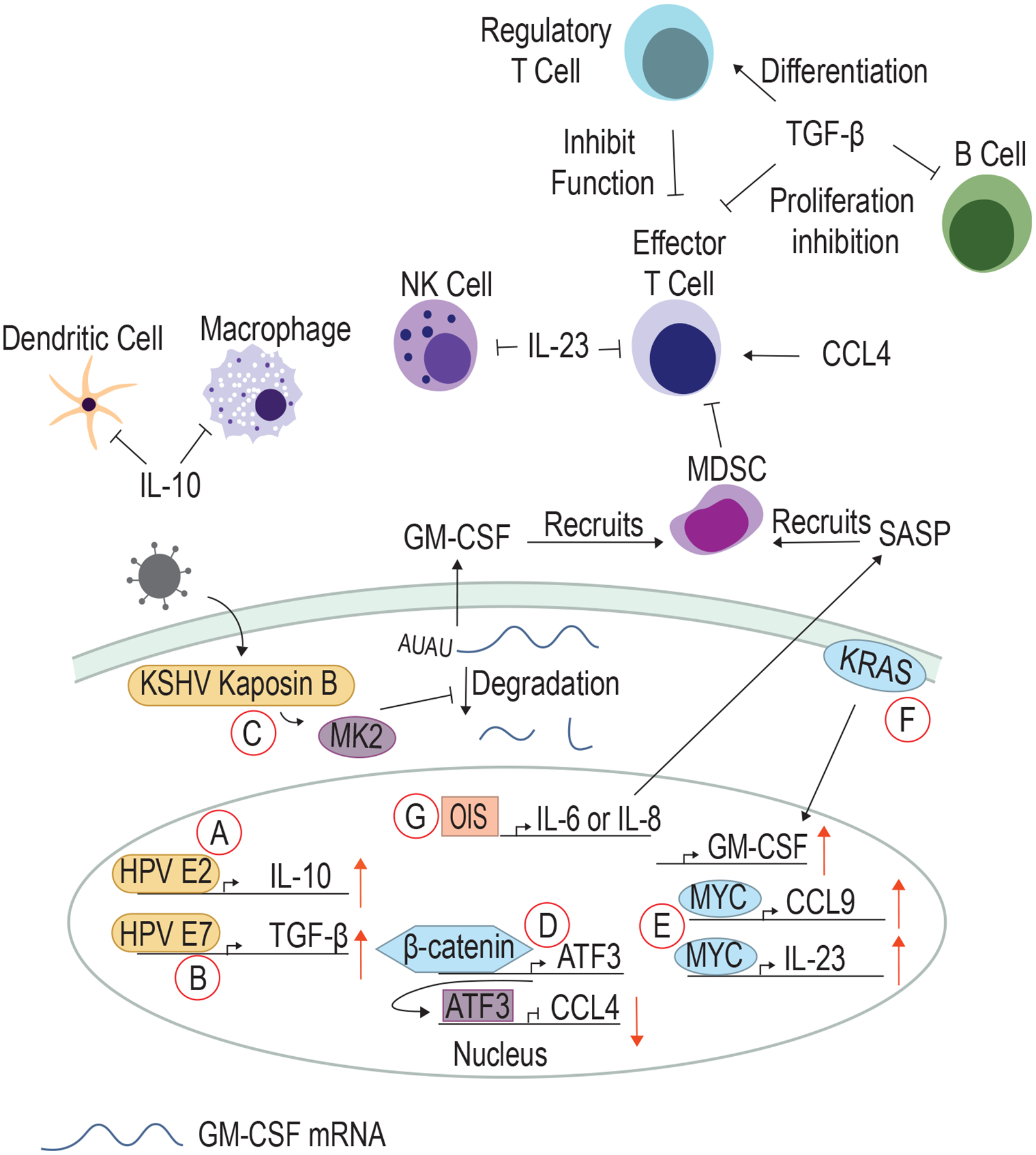
A) HPV E2 upregulates IL-10 to inhibit DCs and macrophages. B) E7 upregulates TGF-β to inhibit B cells and effector T cells and promote regulatory T cell infiltration. C) KSHV Kaposin B activates MK2, which inhibits proteins that promote the degradation of cytokine RNAs such as GM-CSF that recruits MDSC. D) β-catenin upregulates ATF3 that acts as a negative regulator for CCL4. E) MYC upregulates both CCL9 and IL-23, cytokines that inhibit T cells and NK cells. F) KRAS promotes GM-CSF production. G) Oncogene-induced senescence (OIS) induces the upregulation of IL-6 or IL-8, which promotes SASP.
Kaposi’s sarcoma-associated herpesvirus (KSHV) is primarily known for causing Kaposi Sarcoma but also causes primary effusion lymphoma and multicentric Castleman’s disease, both B-cell malignancies (48). Transmission occurs primarily through repeated exchange of saliva or sexual contact. KSHV-associated malignancies are most frequently observed in patients infected with human immunodeficiency virus (HIV) and patients on immunosuppression after organ transplant, indicating that KSHV transformation is prevented by the immune system. The KSHV protein Kaposin B promotes pro-tumorigenic angiogenesis by partnering with MYC to inhibit expression of anti-angiogenic miRNAs (49). In addition, Kaposin B activates the kinase MK2, which phosphorylates and deactivates AU-rich-binding proteins that normally degrade cytokine mRNAs such as granulocyte-macrophage colony-stimulating factor (GM-CSF) (50) (Fig 2C). GM-CSF attracts myeloid-derived suppressor cells (MDSC), which in turn induce T cell dysfunction/exhaustion through engagement of inhibitory receptors (described below) and production of inhibitory cytokines such as transforming growth factor-β (TGFβ) (51). TGFβ inhibits proliferation of activated B cells, prevents T cell function through inhibition of both IL-2-induced proliferation and production of cytotoxic molecule perforin, and promotes regulatory T cell differentiation, which further suppress effector T cell function (52).
The HPV E2 protein is expressed early during HPV infection. E2 binds to E2-binding sites to drive transcription of viral genes and as well as several cellular genes. E2 transactivates expression of interleukin-10 (IL-10), an anti-inflammatory cytokine that can suppress the function of both macrophages and dendritic cells (53, 54) (Fig 2A). E6 and E7 have also been shown to upregulate TGFβ expression (55) (Fig 2B).
In non-viral cancers, oncogenes have also been shown to alter cellular cytokine production. EPHA2 works through the SMAD4/TGFβ signaling pathway to exert immune inhibitory effects in the tumor microenvironment. In a model of pancreatic ductal adenocarcinoma, expression of EPHA2 and SMAD4 increases the expression of TGFβ as well as Ptgs2, encoding COX-2, which increases the levels of PGE2, a proinflammatory prostaglandin. TGFβ and Ptgs2 in turn act as positive regulators for EPHA2 and SMAD4 expression, further driving PGE2 expression and inhibiting T cell response in the tumor microenvironment (56).
β-catenin plays a dual role in driving proliferation and immune evasion. Increased β-catenin in human melanomas correlates with a decrease in tumor-infiltrating T cells, and activated β-catenin in a mouse model of melanoma induces expression of the transcriptional repressor ATF3 (57). ATF3 suppresses chemokine (C-C motif) ligand 4 (CCL4) expression, a cytokine that recruits dendritic cells (DC) and is required for T cell infiltration and elimination of melanomas (57) (Fig 2D).
In a KRASG12D-driven model of lung adenocarcinoma, MYC activation drives expression of CCL9, a chemokine that recruits macrophages and plays a role in programmed death ligand-1 (PD-L1) upregulation (58). In the same model, co-expression of KRAS and MYC upregulated interleukin-23 (IL-23), which suppresses innate immune cells such as natural killer (NK) cells and reduces CTL infiltration (58, 59) (Fig 2E). In pancreatic ductal epithelial cells, KRASG12D drives production of GM-CSF (51) (Fig 2F).
A critical determinant of immune cell recruitment to tumors is the manner in which cancer cells die and release antigens. Cells undergoing immunogenic cell death (ICD) release potent immune-stimulatory factors such as DAMPs and antigens, which can robustly activate the adaptive immune response (60). On the other side of the spectrum is tolerogenic cell death, which prevents dying cells from eliciting an unwanted immune response (i.e. autoimmunity or organ-damaging inflammation in an immune-privileged site). Early on, the main recognized forms of cell death were apoptosis, considered a form of tolerogenic cell death, and necrosis, an ICD mechanism, but since then many other cell death pathways have been described (61). For further discussion of immunogenic and tolerogenic cell death, see (62).
Given that ICD is a powerful activator of immune responses, it is perhaps not surprising that oncogenes have been found to inhibit ICD. Anaplastic lymphoma kinase (ALK) is one such oncogene (63), and ALK promotes survival and proliferation of anaplastic large cell lymphoma (ALCL) by signaling through several major downstream pathways, including phosphatidylinositol 3-kinase (PI3K), extracellular signal-regulated kinase (ERK), and signal transducer and activator of transcription (STAT) pathways (64). In a mouse model of ALCL, inhibition of ALK induces ICD, and pharmacologic inhibition of downstream ALK pathways, particularly PI3K, also induces ICD (64).
While ICD is an inflammatory cell death, oncogene-induced senescence (OIS) prevents death of cells harboring oncogenic mutations through cell cycle arrest. OIS can induce IL-6 and IL-8 production, cytokine components of the senescence-associated secretory phenotype (SASP). SASP can be anti-tumorigenic through anti-tumor immune cell recruitment or pro-tumorigenic through promotion of inflammation-driven carcinogenesis (65). In a model of radiation-induced tumorigenesis, IL-6 promoted NKT cell infiltration and inflammation, and IL-6 knockout mice had accelerated development of osteosarcoma (66). In a hepatocellular carcinoma model driven by NRASG12V, NOTCH1 drove malignant hepatocytes to secrete TGFβ, suppressing the SASP response and causing decreased T cell recruitment and proliferation (67). In contrast, mice with Pten−/− prostate cancer exhibited constitutively active JAK2/STAT3 signaling and upregulated SASP, leading to MDSC recruitment and decreased T cell infiltration (68) (Fig 2G). Clearly, the impact of SASP on cancer development is complex and context dependent, and further studies using different in vitro and in vivo models of oncogene activation are needed to dissect the cell-intrinsic and cell-extrinsic impact of SASP on cancer development and immune responses.
Evading adaptive immunity: oncogenes inhibit MHC Class I antigen presentation
While the innate immune system provides the first line of defense for both oncogenic viral infection and cancer induction, adaptive immune cells subsequently mount antigen-specific responses and form long-lasting memory immunity. Thus, both viral and cellular cancers have evolved mechanisms to avoid the activation of the adaptive immune response.
Cytotoxic CD8 T lymphocytes (CTL) recognize and kill virally infected cells through recognition of viral peptides presented on major histocompatibility complex class I (MHC-I), found on all nucleated cells. Thus, oncogenic viruses frequently evade CTL responses through MHC-I downregulation. Viral proteins in the cytosol are processed by proteasomes to generate short peptides that are then transported into the endoplasmic reticulum by the transporter associated with antigen processing (TAP; Fig 3). These viral peptides are loaded onto MHC-I, and the peptide/MHC-I (pMHC-I) complex is transported to the plasma membrane for presentation to T cells. CTL with T cell receptors specific for the viral pMHC-I become activated, proliferate, and directly lyse infected cells and secrete cytotoxic cytokines (69).
Figure 3. Viral and cellular oncoproteins inhibit MHC class I and antigen processing/presentation.
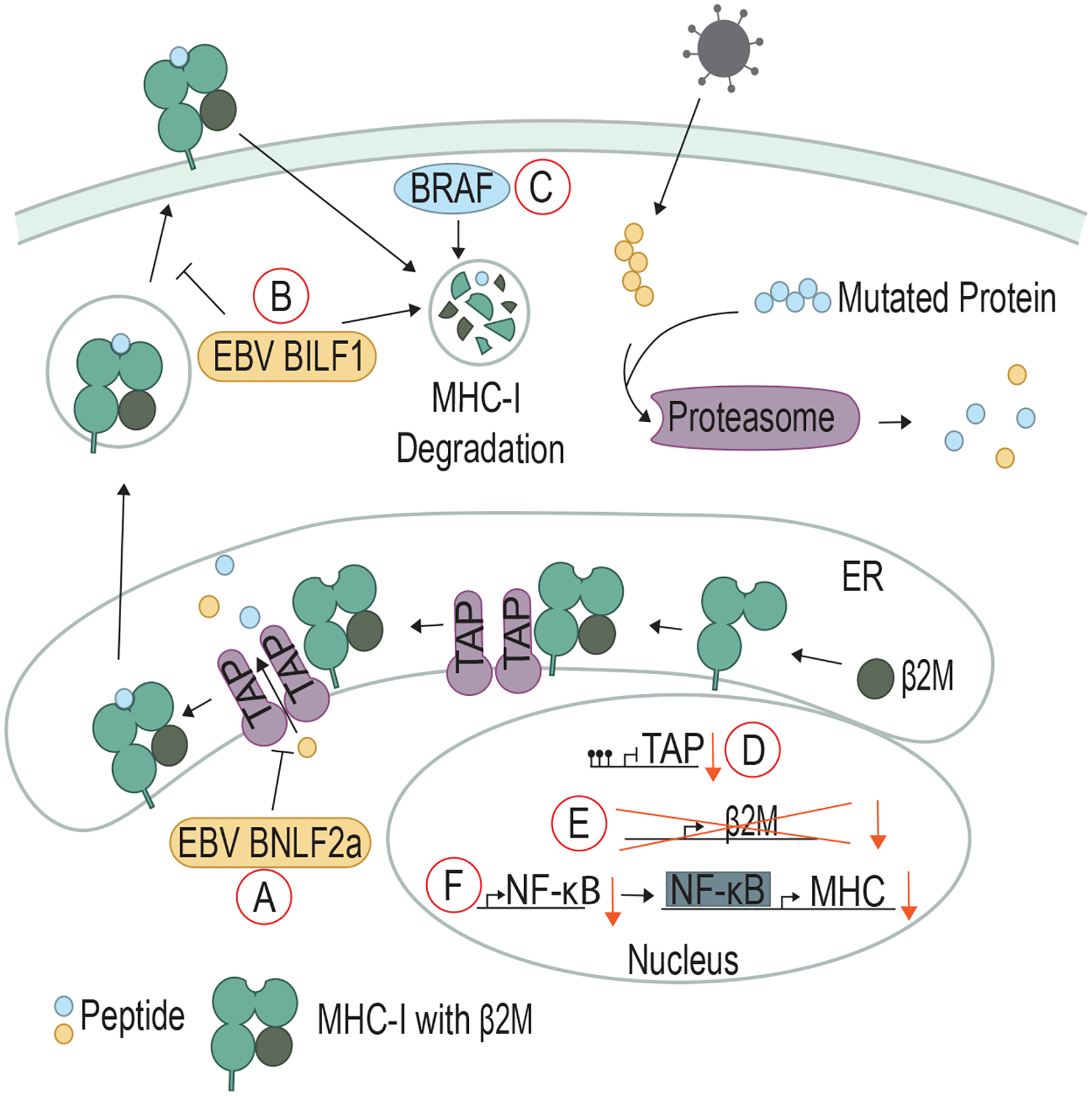
A) EBV BNFL2a binds to TAP and inhibits the transport of peptides into the ER. B) EBV BILF1 promotes the degradation of surface MHC-I and can also prevent MHC-I from reaching the cell surface. C) Mutated BRAF can promote degradation of surface MHC-I. Non-viral cancers D) exhibit methylation of the promoter of TAP to prevent expression, E) LOH of β2-microglobulin (β2M), and F) downregulate NF-kB to lower the expression of MHC-I.
Epstein-Barr Virus (EBV), the first human oncogenic virus discovered in 1964, is a large DNA herpesvirus that is nearly ubiquitous in humans (70). EBV is transmitted through contact with bodily fluids and infects both epithelial and lymphoid cells, establishing latency most commonly in B cells. While EBV infection is generally asymptomatic, it has been linked to epithelial and lymphoid cancers, including gastric cancer, nasopharyngeal cancer, Burkitt Lymphoma, and Hodgkin lymphoma, with these 4 cancer types responsible for about 17% of global cancer deaths each year (71). EBV undergoes several different latent stages as well as a lytic phase in which it actively replicates and packages its genome into virions to infect other cells (72). Because actively replicating EBV results in host cell lysis, EBV-driven malignancies mainly occur in latently-infected cells (73).
The presentation of viral peptides on MHC-I is a critical signal to the adaptive immune system as to whether a cell is healthy or has been virally infected. Melanoma cells were transduced to express the EBV protein BamHI-N leftward frame 2a (BNLF2a) (74), resulting in T cells failing to recognize and lyse BNLF2a-expressing cells (75). BNLF2a’s cytosolic domain associates with TAP to inhibit ATP and peptide binding and prevent viral peptide transport into the ER (74, 75) (Fig 3A).
Even during EBV infection stages when BNLF2a is not expressed, T cell recognition of EBV-infected cells is poor, indicating that other proteins may be involved in immune evasion during EBV infection (76). BILF1, an EBV lytic phase protein, functions as a constitutively-active G-protein coupled receptor (GPCR) and oncogene (73). Independent of its GPCR activity, BILF1 associates with MHC-I molecules at the cell surface and rapidly enhances MHC-I internalization and degradation (77). In addition, BILF1 can divert MHC-I molecules from being transported to the plasma membrane, further inhibiting viral peptide presentation on MHC-I to T cells (78) (Fig 3B). Thus, the lack of viral peptides on MHC-I prevents CD8 T cells from recognizing the EBV-infected cell.
Many non-viral cancers also employ the strategy of downregulating MHC-I presentation of neoepitopes generated from mutated proteins. An estimated 40–90% of human tumors present with MHC-I downregulation (79). BRAF, a serine/threonine protein kinase, is mutated in 50% of melanoma patients and induces downstream mitogen-activated protein kinase (MAPK) signaling to activate the cell cycle. More recent studies have shown that BRAFV600E has a secondary function: driving internalization and sequestration of MHC-I into endocytic compartments (80) (Fig 3C). Treatment with the BRAF inhibitor vemurafenib increased MHC-I expression on the cell surface and led to increased T cell recognition (80, 81). Expression of the breast cancer oncoprotein HER2, an upstream receptor in the MAPK pathway, is inversely correlated with MHC-I expression (82). While the mechanism of HER2 and BRAFV600E internalization of MHC-I is not yet known, a screening study showed that other components of the MAPK pathway, namely MAP kinase/ERK kinase 1 (MEK1) and epithelial growth factor receptor (EGFR), negatively regulate MHC-I expression in a mesothelioma cell line (83).
Non-viral cancer cells can also block T cell pMHC-I recognition by interfering with peptide processing and presentation. A subset of primary triple-negative breast cancer cells exhibits TAP downregulation along with MHC-I downregulation, and this phenotype is strongly associated with poor clinical outcome (84). In addition, some cervical cancers exhibit promoter methylation and downregulated expression of multiple genes encoding antigen presentation proteins, including TAP (85) (Fig 3D). The inflammation-induced transcription factor NF-κB directly binds to the HLA gene promoters (encoding MHC-I in humans) to induce MHC-I expression (86). In neuroblastoma tumors, downregulation of the NF-κB subunit p65 occurs frequently, reducing MHC-I expression (86) (Fig 3E). Patients with metastatic melanoma often undergo loss of heterozygosity (LOH) in the locus encoding β2-microglobulin (B2M). β2-microglobulin is an essential subunit of MHC-I molecule; therefore, loss of β2-microglobulin prevents pMHC-I complex formation (87) (Fig 3F).
Studies have also uncovered MHC-I downregulation as a common response in ICB resistance. Metastatic melanoma patients with B2M LOH had reduced response to ICB and worse overall survival (88). Additionally, B2M LOH was found in lung tumor samples from patients that had acquired resistance to ICB (89). Thus, more research into the restoration of MHC-I on cancer cells is needed to boost primary immune responses and effective ICB treatment.
Dampening T cell responses: oncogenes upregulate checkpoint molecules
Immune checkpoint molecules, such as programmed death-1 (PD-1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4), are expressed on T lymphocytes and other immune cells and negatively regulate TCR and immune receptor-driven signaling. As mentioned above, inhibitory receptors are a hallmark of T cell dysfunction/exhaustion. Inhibitory ligands such as PD-L1 are expressed on immunosuppressive cells, including regulatory T cells as well as on non-immune tissues in response to inflammatory cytokines such as interferon gamma (IFNγ) (90). Immune checkpoints are an essential dampening mechanism that prevent autoimmune disease or excessive immunopathology during chronic viral inflammation (91). However, these inhibitory mechanisms can prevent effective anti-cancer immune responses, prompting intense interest in immune checkpoint blockade (ICB) therapy (reviewed in (91)).
Patients with chronic HBV infection have been shown to have higher percentages of PD-1+ T cells in peripheral blood, and infected cells have lower levels of CD274 (encoding PD-L1) methylation (92). There is evidence that a higher EBV load correlates with an increase in PD-L1 expression in gastric carcinomas and non-small cell lung cancer (93, 94). One of the first proteins expressed in EBV infection is Epstein-Barr virus nuclear antigen 2 (EBNA2), which is capable of immortalizing B cells (95). In Burkitt lymphoma cells, EBNA2 forms a complex with Early B-cell Factor 1 (EBF1), a transcription factor important in B cell signal transduction and differentiation (96). The EBNA2-EBF1 complex binds to the microRNA mir-34a promoter and downregulates its expression. miR-34a binds to the 3’UTR of the CD274 mRNA, preventing PD-L1 translation (97). Thus, EBNA2 inhibition of miR-34a leads to high PD-L1 expression and T cell inhibition (96) (Fig 4A).
Figure 4. Viral and cellular oncoproteins upregulate checkpoint molecules such as PD-L1.
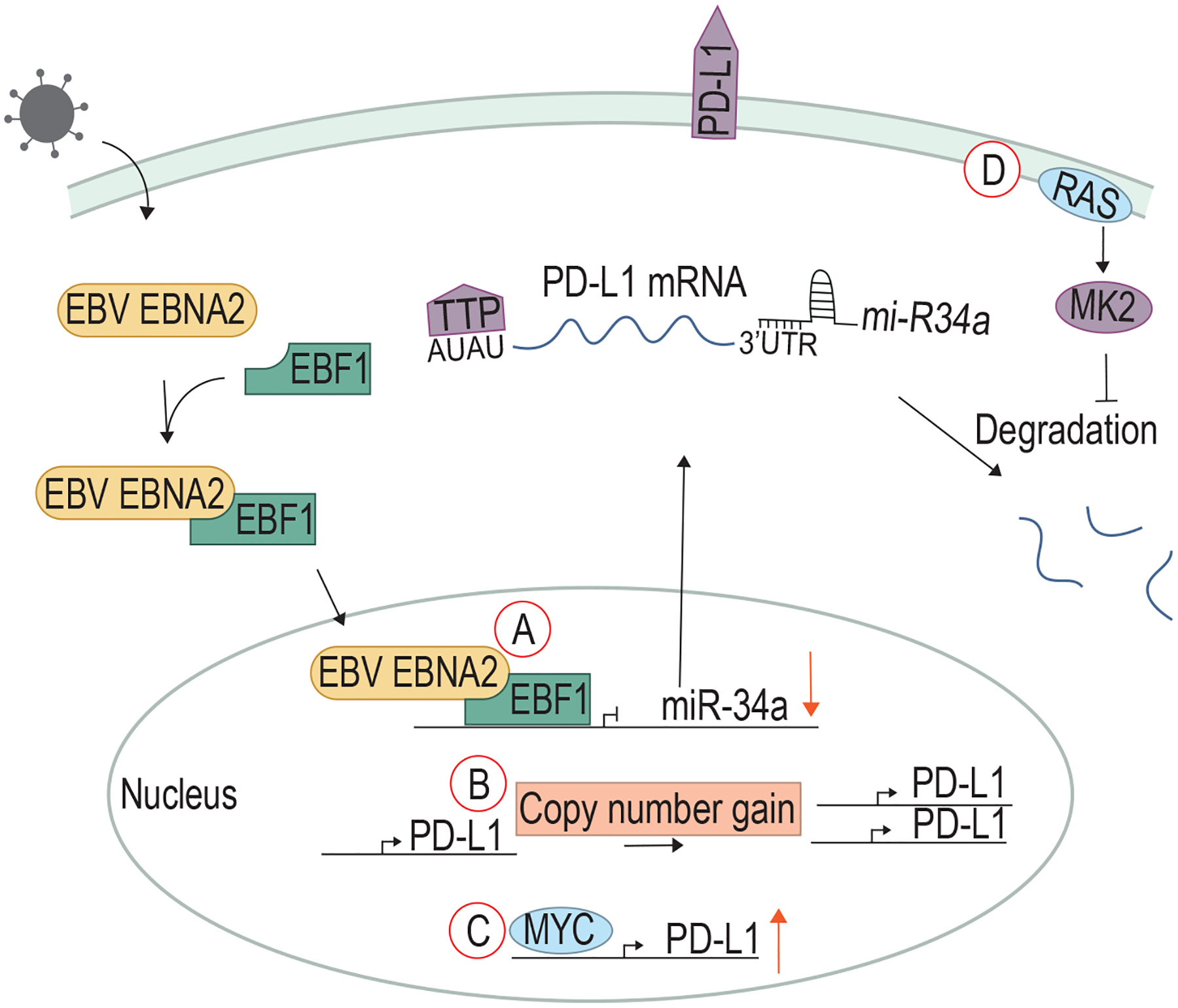
A) EBV EBNA2 stabilizes PD-L1 mRNA by forming a complex with EBF1 to downregulation miR-34a, a microRNA that promotes the degradation of PD-L1 mRNA. Some non-viral cancers have B) copy number gains in the PD-L1 gene. C) Oncogenic MYC drives upregulation of PD-L1. D) Mutated RAS activates MK2, which promotes the phosphorylation of TTP to inhibit the degradation of PD-L1 mRNA.
Non-viral cancers make use of several mechanisms to upregulate PD-L1. An inverse correlation between miR-34a and PD-L1 expression has been found in Acute Myeloid Leukemia (AML), and downregulation of miR-34a is associated with poor clinical outcomes (97). The highest response rate to PD-1/PD-L1 ICB occurs in patients with Hodgkin Lymphoma (HL) (98). HL cases often have a copy number gain of chromosome 9p, containing CD274 (99, 100), which leads to increased PD-L1 expression (Fig 4B). The gene encoding Janus kinase 2 (JAK2) is also located on 9p, and increased JAK2 signaling was shown to drive further upregulation of PD-L1 (100). Additionally, dysregulation of other pathways such as NF-κB, JAK/STAT, and PI3K may also contribute to the upregulation of inhibitory molecules (101) within malignant cells.
Several oncogenes have been shown to regulate PD-L1 expression. Lung cancers with mutated EGFR exhibit increased PD-L1 as compared to EGFR wild-type lung cancers. EGFR inhibition abrogated the increased PD-L1, indicating that mutant EGFR signals through a yet-unknown mechanism to contribute to the immunosuppressive environment of lung cancer, in addition to its role in driving growth and proliferation (102). Oncogenic MYC can bind directly to the CD274 promoter to drive PD-L1 expression in T cell acute lymphoblastic leukemia (T-ALL; Fig 4C). MYC inhibition in T-ALL cells lowered PD-L1 expression and improved T cell responses against these cancers (103). FGFR amplification or mutation in colorectal cancer causes downstream proliferation and transformation through the activation of the MAPK and PI3K pathways. In addition, FGFR signals through the JAK/STAT pathway, which causes upregulation of PD-L1 expression (104).
Analyses of lung and colon adenocarcinomas in TCGA suggest an association between RAS activation and PD-L1 upregulation (105). Studies in a human epithelial cell model with inducible RASG12V expression showed that RAS regulates PD-L1 expression through tristetraprolin (TTP), an RNA-binding protein that binds to AU-rich elements. PD-L1 mRNA contains AU-rich regions, and TTP binding causes degradation of PD-L1 mRNA. However, aberrant RAS signaling activates the kinase MAPK-activated protein kinase 2 (MK2), which directly phosphorylates and inhibits TTP, stabilizing PD-L1 mRNA, increasing PD-L1 expression, and inhibiting T cell responses (105) (Fig 3D).
Clinically, ICB has been a substantial advancement in cancer treatment. However, primary non-responsiveness to ICB therapies as well as secondary resistance (reviewed in (106)) in patients who initially respond remains a significant clinical obstacle. We are just beginning to understand the mechanisms for ICB treatment failures, paving a clear direction for future research. By studying mechanisms of resistance in both viral and non-viral cancers, we can develop strategies to make ICB therapy effective in more patients.
Clinical therapeutics: oncoprotein inhibitors enhance the immune response
While the effects of oncoproteins on the immune system described above prevent efficient activation of the immune system, therapeutic oncoprotein inhibition can not only lead to slowed cancer cell growth and death but can boost immune responses by bypassing or counteracting oncogene-induced immune evasion strategies.
Recently, several existing chemotherapies have been shown to activate the cGAS/STING pathway and thus promote the infiltration and activation of innate immune cells. For example, drugs that target topoisomerase and DNA damage repair proteins (e.g. ATM) cause enough DNA damage to activate cGAS/STING signaling (107). These therapeutics induced increased immune cell infiltration, including of NK and T cells, which was reduced in STING-deficient mice. For a more extensive review of chemotherapy-induced cGAS/STING activation, see (107).
Inhibitors of cell surface growth factor receptors have been part of cancer therapy for well over a decade. For example, lapatinib inhibits both EGFR and HER2 intracellular tyrosine kinase domains. This inhibition prevents downstream signaling while also preventing the ubiquitination and downregulation of HER2. The accumulation of HER2 promotes antibody-dependent cellular cytotoxicity, which increases cancer cell lysis, typically by NK cells. Lapatinib also promotes infiltration of T cells and their subsequent production of IFNγ through STAT1-dependent expression of T-cell chemotactic cytokines, including CXCL9, CXCL10, and CXCL11 (108, 109).
CDK4/6 inhibitors used to treat breast cancers have been shown in both mice and humans to recruit T cells to otherwise immunologically cold tumors. Inhibitors palbociclib, ribociclib, and abemaciclib all enhance the production of CCL5, CXCL9, and CXCL10, cytokines that attract T cells (110). When treated with only CDK4 or CDK6 inhibitors, this effect was abrogated, suggesting that both cyclins need to be inhibited to produce these cytokines. Patients with these cytokines present have also shown better prognoses than those without (110).
Further improving our understanding of how oncogenes alter immune responses will allow clinicians and researchers to design synergistic combinations of oncogene-targeted therapies and immunomodulatory agents.
Concluding Remarks
The study of oncogenic viruses historically provided many valuable insights and paved the way for our understanding of cellular oncogenes and tumor suppressor genes. In 2000, Hanahan and Weinberg described six hallmark pathways altered in cells that lead to carcinogenesis, including many pathways first identified through the study of viral oncogenes such as evading apoptosis and growth factor-independent proliferation. In the years since then, the critical role of the immune system in carcinogenesis has become clear, and in 2011, Hanahan and Weinberg added a new hallmark pathway: evasion of the immune system (111). As we have learned more about how oncogenes alter key hallmark pathways in cells to cause cancer, it has become clear that oncogenes can impact several pathways at once. Here we have illustrated how viral and cellular oncogenes not only bypass cellular proliferation and apoptotic checkpoints, but also play an important role in viral immune evasion. Through continued study of how viral and cellular oncogenes promote immune evasion and carcinogenesis, we may identify novel opportunities to activate both innate signaling and adaptive immune responses to improve cancer therapy (112).
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Rous P A SARCOMA OF THE FOWL TRANSMISSIBLE BY AN AGENT SEPARABLE FROM THE TUMOR CELLS. J Exp Med. 1911;13(4):397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krump NA, You J. Molecular mechanisms of viral oncogenesis in humans. Nat Rev Microbiol. 2018;16(11):684–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. 2020;8(2):e180–e90. [DOI] [PubMed] [Google Scholar]
- 4.Chang Y, Moore PS, Weiss RA. Human oncogenic viruses: nature and discovery. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zur Hausen H The search for infectious causes of human cancers: where and why. Virology. 2009;392(1):1–10. [DOI] [PubMed] [Google Scholar]
- 6.Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10(12):878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14(6):433–9. [DOI] [PubMed] [Google Scholar]
- 8.Gaglia MM, Munger K. More than just oncogenes: mechanisms of tumorigenesis by human viruses. Curr Opin Virol. 2018;32:48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandey NV. DNA viruses and cancer: insights from evolutionary biology. Virusdisease. 2020;31(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arroyo Muhr LS, Bzhalava Z, Hortlund M, Lagheden C, Nordqvist Kleppe S, Bzhalava D, et al. Viruses in cancers among the immunosuppressed. Int J Cancer. 2017;141(12):2498–504. [DOI] [PubMed] [Google Scholar]
- 11.Stanley M Tumour virus vaccines: hepatitis B virus and human papillomavirus. Philos Trans R Soc Lond B Biol Sci. 372 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516–23. [DOI] [PubMed] [Google Scholar]
- 13.Ma Z, Damania B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe. 2016;19(2):150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rivas C, Aaronson SA, Munoz-Fontela C. Dual Role of p53 in Innate Antiviral Immunity. Viruses. 2010;2(1):298–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu WJ, Abrams JM. Lessons from p53 in non-mammalian models. Cell death and differentiation. 2006;13(6):909–12. [DOI] [PubMed] [Google Scholar]
- 16.Munoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol. 2016;16(12):741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Croce CM. Oncogenes and cancer. N Engl J Med. 2008;358(5):502–11. [DOI] [PubMed] [Google Scholar]
- 18.Kontomanolis EN, Koutras A, Syllaios A, Schizas D, Mastoraki A, Garmpis N, et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020;40(11):6009–15. [DOI] [PubMed] [Google Scholar]
- 19.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Q, Lavorgna A, Bowman M, Hiscott J, Harhaj EW. Aryl Hydrocarbon Receptor Interacting Protein Targets IRF7 to Suppress Antiviral Signaling and the Induction of Type I Interferon*. J Biol Chem. 290 2015. p. 14729–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menendez D, Shatz M, Resnick MA. Interactions between the tumor suppressor p53 and immune responses. Curr Opin Oncol. 2013;25(1):85–92. [DOI] [PubMed] [Google Scholar]
- 22.Taura M, Suico MA, Koyama K, Komatsu K, Miyakita R, Matsumoto C, et al. Rb/E2F1 regulates the innate immune receptor Toll-like receptor 3 in epithelial cells. Mol Cell Biol. 2012;32(8):1581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437(7055):141–6. [DOI] [PubMed] [Google Scholar]
- 24.Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity. 2016;45(2):389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philip M, Schietinger A. CD8(+) T cell differentiation and dysfunction in cancer. Nature reviews Immunology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5(214):ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–60. [DOI] [PubMed] [Google Scholar]
- 28.Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12(13):2061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science. 2015;350(6260):568–71. [DOI] [PubMed] [Google Scholar]
- 30.Shaikh MH, Bortnik V, McMillan NA, Idris A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb Pathog. 2019;132:162–5. [DOI] [PubMed] [Google Scholar]
- 31.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nature reviews Immunology. 2005;5(3):215–29. [DOI] [PubMed] [Google Scholar]
- 32.Clark DN, Hu J. Unveiling the roles of HBV polymerase for new antiviral strategies. Future Virol. 2015;10(3):283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Li J, Chen J, Li Y, Wang W, Du X, et al. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J Virol. 2015;89(4):2287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng KW, Marshall EA, Bell JC, Lam WL. cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends in immunology. 2018;39(1):44–54. [DOI] [PubMed] [Google Scholar]
- 36.Yang CA, Huang HY, Chang YS, Lin CL, Lai IL, Chang JG. DNA-Sensing and Nuclease Gene Expressions as Markers for Colorectal Cancer Progression. Oncology. 2017;92(2):115–24. [DOI] [PubMed] [Google Scholar]
- 37.Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016;14(2):282–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer research. 2016;76(22):6747–59. [DOI] [PubMed] [Google Scholar]
- 39.de Queiroz N, Xia T, Konno H, Barber GN. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol Cancer Res. 2019;17(4):974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26(45):6469–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu S, Zhang Q, Zhang F, Meng F, Liu S, Zhou R, et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat Cell Biol. 2019;21(8):1027–40. [DOI] [PubMed] [Google Scholar]
- 42.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity. 2018;49(4):754–63.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science. 2020;369(6506):993–9. [DOI] [PubMed] [Google Scholar]
- 44.Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An orally available non-nucleotide STING agonist with antitumor activity. Science. 2020;369(6506). [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Celis E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol Immunother. 2015;64(8):1057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial watch: STING agonists in cancer therapy. Oncoimmunology. 2020;9(1):1777624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mogensen TH, Paludan SR. Molecular Pathways in Virus-Induced Cytokine Production. Microbiol Mol Biol Rev. 65 2001. p. 131–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider JW, Dittmer DP. Diagnosis and Treatment of Kaposi Sarcoma. Am J Clin Dermatol. 2017;18(4):529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang HC, Hsieh TH, Lee YW, Tsai CF, Tsai YN, Cheng CC, et al. c-Myc and viral cofactor Kaposin B co-operate to elicit angiogenesis through modulating miRNome traits of endothelial cells. BMC Syst Biol. 2016;10 Suppl 1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307(5710):739–41. [DOI] [PubMed] [Google Scholar]
- 51.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. [DOI] [PubMed] [Google Scholar]
- 53.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180(9):5771–7. [DOI] [PubMed] [Google Scholar]
- 54.Bermudez-Morales VH, Peralta-Zaragoza O, Alcocer-Gonzalez JM, Moreno J, Madrid-Marina V. IL-10 expression is regulated by HPV E2 protein in cervical cancer cells. Mol Med Rep. 2011;4(2):369–75. [DOI] [PubMed] [Google Scholar]
- 55.Alcocer-Gonzalez JM, Berumen J, Tamez-Guerra R, Bermudez-Morales V, Peralta-Zaragoza O, Hernandez-Pando R, et al. In vivo expression of immunosuppressive cytokines in human papillomavirus-transformed cervical cancer cells. Viral Immunol. 2006;19(3):481–91. [DOI] [PubMed] [Google Scholar]
- 56.Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J Clin Invest. 2019;129(9):3594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. [DOI] [PubMed] [Google Scholar]
- 58.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell. 2017;171(6):1301–15.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461–5. [DOI] [PubMed] [Google Scholar]
- 60.Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nature reviews Immunology. 2009;9(5):353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang YJ, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018;5(3):194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petrazzuolo A, Perez-Lanzon M, Martins I, Liu P, Kepp O, Minard-Colin V, et al. Pharmacological inhibitors of anaplastic lymphoma kinase (ALK) induce immunogenic cell death through on-target effects. Cell Death Dis. 2021;12(8):713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016;30:533–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kansara M, Leong HS, Lin DM, Popkiss S, Pang P, Garsed DW, et al. Immune response to RB1-regulated senescence limits radiation-induced osteosarcoma formation. J Clin Invest. 2013;123(12):5351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18(9):979–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toso A, Revandkar A, DiMitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Reports. 2014. [DOI] [PubMed] [Google Scholar]
- 69.Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nature reviews Immunology. 2009;9(7):503–13. [DOI] [PubMed] [Google Scholar]
- 70.Young LS, Yap LF, Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer. 2016;16(12):789–802. [DOI] [PubMed] [Google Scholar]
- 71.Khan G, Fitzmaurice C, Naghavi M, Ahmed LA. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-attributable malignancies, 1990–2017. BMJ Open. 2020;10(8):e037505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsurumi T, Fujita M, Kudoh A. Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol. 2005;15(1):3–15. [DOI] [PubMed] [Google Scholar]
- 73.Paulsen SJ, Rosenkilde MM, Eugen-Olsen J, Kledal TN. Epstein-Barr virus-encoded BILF1 is a constitutively active G protein-coupled receptor. J Virol. 2005;79(1):536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strong MJ, Laskow T, Nakhoul H, Blanchard E, Liu Y, Wang X, et al. Latent Expression of the Epstein-Barr Virus (EBV)-Encoded Major Histocompatibility Complex Class I TAP Inhibitor, BNLF2a, in EBV-Positive Gastric Carcinomas. J Virol. 2015;89(19):10110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hislop AD, Ressing ME, van Leeuwen D, Pudney VA, Horst D, Koppers-Lalic D, et al. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J Exp Med. 2007;204(8):1863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Croft NP, Shannon-Lowe C, Bell AI, Horst D, Kremmer E, Ressing ME, et al. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. 2009;5(6):e1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME, et al. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009;5(1):e1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuo J, Quinn LL, Tamblyn J, Thomas WA, Feederle R, Delecluse HJ, et al. The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J Virol. 2011;85(4):1604–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cornel AM, Mimpen IL, Nierkens S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers (Basel). 2020;12(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol Res. 2015;3(6):602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF(V600E) homozygous melanoma cells. Oncoimmunology. 2013;2(1):e22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Inoue M, Mimura K, Izawa S, Shiraishi K, Inoue A, Shiba S, et al. Expression of MHC Class I on breast cancer cells correlates inversely with HER2 expression. Oncoimmunology. 2012;1(7):1104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brea EJ, Oh CY, Manchado E, Budhu S, R SG, Mo G, et al. Kinase regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol Res. 2016;4(11):936–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pedersen MH, Hood BL, Beck HC, Conrads TP, Ditzel HJ, Leth-Larsen R. Downregulation of antigen presentation-associated pathway proteins is linked to poor outcome in triple-negative breast cancer patient tumors. Oncoimmunology. 2017;6(5):e1305531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hasim A, Abudula M, Aimiduo R, Ma JQ, Jiao Z, Akula G, et al. Post-transcriptional and epigenetic regulation of antigen processing machinery (APM) components and HLA-I in cervical cancers from Uighur women. PloS one. 2012;7(9):e44952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Forloni M, Albini S, Limongi MZ, Cifaldi L, Boldrini R, Nicotra MR, et al. NF-kappaB, and not MYCN, regulates MHC class I and endoplasmic reticulum aminopeptidases in human neuroblastoma cells. Cancer research. 2010;70(3):916–24. [DOI] [PubMed] [Google Scholar]
- 87.del Campo AB, Kyte JA, Carretero J, Zinchencko S, Mendez R, Gonzalez-Aseguinolaza G, et al. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int J Cancer. 2014;134(1):102–13. [DOI] [PubMed] [Google Scholar]
- 88.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017;7(12):1420–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual Faces of IFNgamma in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin Cancer Res. 2016;22(10):2329–34. [DOI] [PubMed] [Google Scholar]
- 91.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiao L, Chen J, Wu X, Cai B, Su Z, Wang L. Correlation of CpG methylation of the Pdcd1 gene with PD-1 expression on CD8(+) T cells and medical laboratory indicators in chronic hepatitis B infection. J Gene Med. 2020;22(2):e3148. [DOI] [PubMed] [Google Scholar]
- 93.Nakayama A, Abe H, Kunita A, Saito R, Kanda T, Yamashita H, et al. Viral loads correlate with upregulation of PD-L1 and worse patient prognosis in Epstein-Barr Virus-associated gastric carcinoma. PloS one. 2019;14(1):e0211358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sugiyama E, Togashi Y, Takeuchi Y, Shinya S, Tada Y, Kataoka K, et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci Immunol. 2020;5(43). [DOI] [PubMed] [Google Scholar]
- 95.Zimber-Strobl U, Strobl L, Hofelmayr H, Kempkes B, Staege MS, Laux G, et al. EBNA2 and c-myc in B cell immortalization by Epstein-Barr virus and in the pathogenesis of Burkitt’s lymphoma. Curr Top Microbiol Immunol. 1999;246:315–20; discussion 21. [DOI] [PubMed] [Google Scholar]
- 96.Anastasiadou E, Stroopinsky D, Alimperti S, Jiao AL, Pyzer AR, Cippitelli C, et al. Epstein−Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia. 33 2019. p. 132–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015;27(3):443–52. [DOI] [PubMed] [Google Scholar]
- 98.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol. 2016;34(23):2690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wienand K, Chapuy B, Stewart C, Dunford AJ, Wu D, Kim J, et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019;3(23):4065–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352(6282):227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li P, Huang T, Zou Q, Liu D, Wang Y, Tan X, et al. FGFR2 Promotes Expression of PD-L1 in Colorectal Cancer via the JAK/STAT3 Signaling Pathway. J Immunol. 2019;202(10):3065–75. [DOI] [PubMed] [Google Scholar]
- 105.Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity. 2017;47(6):1083–99.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schoenfeld AJ, Hellmann MD. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell. 2020;37(4):443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yum S, Li M, Chen ZJ. Old dogs, new trick: classic cancer therapies activate cGAS. Cell Res. 2020;30(8):639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Griguolo G, Pascual T, Dieci MV, Guarneri V, Prat A. Interaction of host immunity with HER2-targeted treatment and tumor heterogeneity in HER2-positive breast cancer. J Immunother Cancer. 2019;7(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hannesdottir L, Tymoszuk P, Parajuli N, Wasmer MH, Philipp S, Daschil N, et al. Lapatinib and doxorubicin enhance the Stat1-dependent antitumor immune response. Eur J Immunol. 2013;43(10):2718–29. [DOI] [PubMed] [Google Scholar]
- 110.Uzhachenko RV, Bharti V, Ouyang Z, Blevins A, Mont S, Saleh N, et al. Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep. 2021;35(1):108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 112.Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27(1):96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]