

Abstract
Since its initial demonstration in 2000, far-field super-resolution light microscopy has undergone tremendous technological developments. In parallel, these developments have opened a new window into visualizing the inner life of cells at unprecedented levels of detail. Here, we review the technical details behind the most commonly used implementations of super-resolution microscopy and highlight some of the recent, promising advances in this field.
eTOC Blurb
Bond et al provide a technologically focused review on common super-resolution light microscopy approaches as well as recent, promising technical developments in this field. The review particularly highlights emerging advanced imaging approaches that hold promise for visualizing the molecular architecture of cells with high spatiotemporal resolution.
Introduction
Over the years, genomic, transcriptomic and proteomic approaches have transformed our understanding of the molecular makeup of tissues under physiological and pathological conditions at the single cell level (Stuart and Satija, 2019). However, cells are not a soup of randomly distributed proteins and nucleic acids. Proteins rarely function in isolation inside the cell but have to form complexes with other proteins at the right place and time (Marsh and Teichmann, 2015). Our genomic DNA is intricately folded to bring together distant genomic sequences in 3D space to regulate gene function (Rowley and Corces, 2018). Myriad molecular mechanisms are at play including phase separation, inter- and intra-molecular interactions to coordinate a temporally and spatially compartmentalized sub-cellular organization. Bulk –omics methods often fail to capture this dynamic spatiotemporal organization of proteins and nucleic acids that helps define a cell’s molecular state. Microscopy, in particular light microscopy, remains an indispensable tool to visualize this dynamic sub-cellular organization. However, until the last decade, far-field light microscopy remained limited in its spatial resolution to ~200–300 nm due to diffraction. To place that number in context, the synaptic cleft between two neuronal processes is ~20 nm and the nucleosome that forms the repeating unit of chromatin is only ~10 nm. The impact of super-resolution microscopy methods that overcome this limitation can therefore not be overstated. The super-resolution revolution started with the demonstration of STED (Stimulated Emission Depletion Microscopy) in 2000 (Klar et al., 2000) and continued with the subsequent development of single molecule localization microscopy (SMLM) techniques STORM (Stochastic Optical Reconstruction Microscopy) (Rust et al., 2006) and PALM/fPALM (Photoactivated Localization Microscopy and Fluorescence Photoactivation Localization Microscopy) (Betzig et al., 2006; Hess et al., 2006) in 2006. Since then, there has been an explosion of technological developments that expanded these methods to 3D imaging, multiplexed/multi-color imaging and multimodal imaging. Here, we overview super-resolution microscopy methods with a particular focus on the most commonly used and widely adopted implementations and highlight some of the exciting recent technological developments that hold high promise for studying biological processes at a level of unprecedented detail. While this review is meant to be a technologically focused review, we provide several examples of biological applications of the various methods. In addition, we discuss the pluses and minuses of each technology to guide the reader in choosing the right method. We also provide guidance in the form of a Table that lists the most common range of spatial and temporal resolutions accessible to each technology as well as other important parameters that should be considered by researchers seeking to apply these methods to their biological question of interest. Finally, we refer the readers to several recent reviews that provide a focus on biological applications of super-resolution microscopy and further guidance on how to choose the best method (Schermelleh et al., 2019; Sigal et al., 2018; Valli et al., 2021).
Super-resolution modalities
STED and RESOLFT
STED (Klar et al., 2000) was the first far-field super-resolution light microscopy modality to be demonstrated. To break the diffraction limit, the sample is scanned not only with an excitation beam, but also a co-aligned STED beam that forces excited fluorophores within a region where the STED beam has high intensity back to a non-fluorescent state through stimulated emission. When the STED beam is shaped like a donut in the lateral dimension (Klar et al., 2001) (Figure 1A(i)), only the molecules at the center of the donut, where the STED beam intensity is minimum, emit observable fluorescence, effectively shrinking the point spread function (PSF) of the illumination. As scanning coordinates are pre-determined with nano-scale precision, the fluorescence intensity at each scanning position is recorded to build a super-resolved image down to ~20 nm in resolution when using common fluorophores (Figure 1).
Figure 1: STED and RESOLFT.
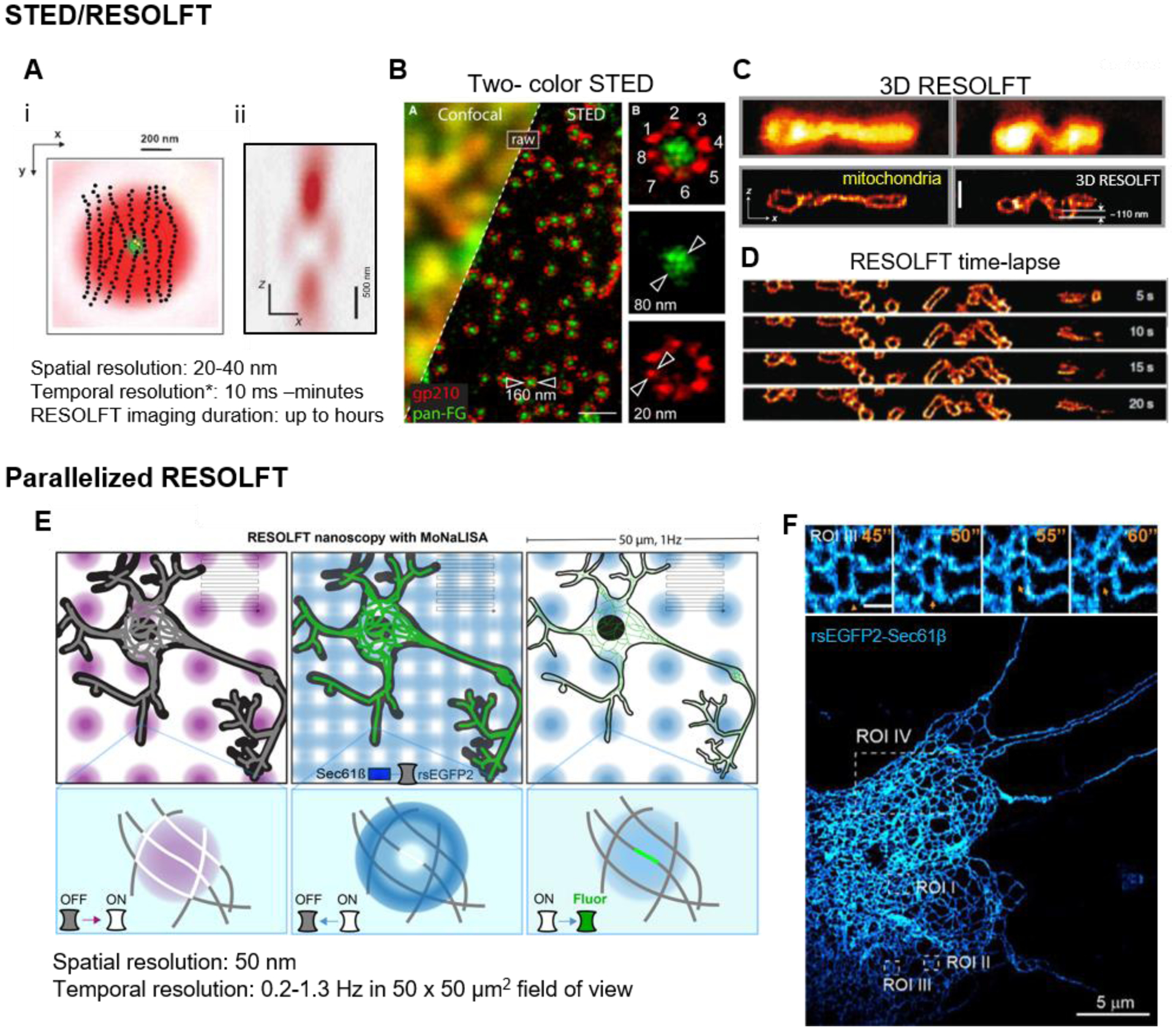
(A) (i) STED depletion beam (red) geometry in lateral (x,y) dimension overlayed with the excitation beam (green), and (ii) cross-section of the z depletion beam to improve axial resolution (Hell, 2015; Willig et al., 2007). The intensity at center of the STED beam is zero, which allows for the few molecules there remain fluorescent (yellow dots) while the surrounding molecules are forced to the dark state (black dots). *temporal resolution scales inversely with the size of the field-of-view. (B) Two-color STED image of Xenopus nuclear pore complex. Scale bar, 500 nm (Göttfert et al., 2013). (C) 3D volumetric RESOLFT image of mitochondria in the axial direction in a U2OS cell expressing rsEGFP2-Omp25. Upper two panels, enhanced confocal images without 3D off-switching. Lower two panels, 3D RESOLFT images. Scale bar, 500 nm (Bodén et al., 2021). (D) Axial mitochondria dynamics (rsEGFP2-Omp25) captured by 3D RESOLFT (Bodén et al., 2021). (E) The scheme of parallelized RESOLFT (MoNaLISA) imaging the neuronal ER labeled with rsEGFP2-Sec61β (Damenti et al., 2021). The imaging setup includes parallelized foci switch-on/off and readout as illustrated. These foci only need to scan a limited fraction of the whole field (indicated by the gray lines) to cover a large 50 μm2 field of view. (F) An extensive ER network (rsEGFP2-Sec61β) in a hippocampal neuron, super-resolved by parallelized RESOLFT. Scale bar, 5 μm. Inset: zoomed in dynamics of ER at 0.2 Hz in the ROI II. Scale bar, 500 nm.
The critical component in STED is to effectively suppress the fluorescence outside of the center of the illumination, which requires high STED beam intensity (MW-GW/cm2) to increase the probability of the STED beam to interact with every electron in the excited state during the lifetime of the excited state. The high intensity and repeated cycles of excitation and depletion cause considerable photobleaching, which degrades image quality, and therefore it is important to choose appropriately bright and photostable fluorophores (Zheng and Lavis, 2017) or replenishable labeling strategies (Nicolai et al., 2011; Spahn et al., 2019). Implementation of STED using continuous wave (cw) lasers (cw-STED) reduces the STED beam illumination intensity compared to the use of STED pulses, hence reducing photodamage albeit with a compromise in spatial resolution (Willig et al., 2007). More recent implementations of STED use a sample adaptive illumination strategy to minimize photobleaching. For example, in RESCue-STED, scan beams are shutdown in regions of the sample that are not occupied by fluorophores (Staudt et al., 2011). MINFIELD is another implementation that restricts the scan field to a region about the same size as the donut minimum “valley” to avoid scanning the high intensity crests over the fluorophores to reduce photobleaching (Gottfert et al., 2017). In a related approach, Dynamic Intensity Minimum (DyMIN) (Heine et al., 2017), STED beam power is gradually increased as a fluorophore is approached and then gradually decreased during the scan away from the fluorophore, once again reducing photobleaching without limiting the field of view to a small region like in MINFIELD.
The shape of the STED beam in the focus can be adjusted to allow resolution improvements in 2D as well as in 3D. To reach an isotropic improvement of resolution, IsoSTED integrates the 4Pi geometry by wedging a sample between two opposing objective lenses and illuminating the sample simultaneously from both sides (Schmidt et al., 2008). This setup requires excellent alignment of the objectives and some adaptability of the sample preparation but yields significant improvement especially in the axial resolution. Recently, adaptive optics have further improved the axial resolution of isoSTED while imaging whole cells and tissues (Hao et al., 2021a; Velasco et al., 2021). With careful choice of fluorescent spectra, dual-color imaging has also been achieved and applied in many biological settings by using one STED beam for simultaneous fluorescence depletion (Carravilla et al., 2019; Damenti et al., 2021; Eggeling et al., 2009; Göttfert et al., 2013; Große et al., 2016; Moors et al., 2021). Remarkably, by combining reversibly switchable fluorescent proteins with organic fluorescent dyes, three-color imaging by STED can be achieved (Willig et al., 2021). Numerous investigators have successfully used STED in fixed and live samples across a wide range of biological contexts, such as observing neuronal actin dynamics or 3D imaging of extracellular space in live mouse brain, mapping of large-scale protein targets in dendritic spines, tracking cellular organelle morphological changes, characterizing lipid molecule movement on the membrane, and evaluating the accessibility of HIV-neutralizing antibodies to single virions. (Berning et al., 2012; Bottanelli et al., 2016; Carravilla et al., 2019; Eggeling et al., 2009; Große et al., 2016; Helm et al., 2021; Schroeder et al., 2018; Tønnesen et al., 2018). However despite recent strategies that reduce photobleaching, photobleaching/phototoxicity remains a concern for the most common implementations of STED microscopy. In addition, in the absence of specialized implementations of STED that use parallelized illumination (Bingen et al., 2011), the acquisition speed of STED is low when imaging large fields of view.
RESOLFT (reversible saturable optical fluorescence transitions) (Hofmann et al., 2005) is another method that uses a similar principle as STED but uses photo-switchable proteins that can change conformation upon exposure to different wavelengths of light to achieve on- and off-switching, instead of stimulated emission. Using the same donut-shape design, the off-switching requires much lower intensity (W-kW/cm2) (Figure 1), which supports continued, gentle imaging in a wide range of biological settings including mammalian cells and Drosophila tissue to give a few examples (Grotjohann et al., 2012; Schnorrenberg et al., 2016; Tiwari et al., 2015). The tradeoff for the gentle laser power of RESOFT is that it takes longer (millisecond pixel dwell time, Table 1) for fluorescent proteins to switch off whereas the depletion by STED is instantaneous (~200 psec, see Table 1 for comparative properties of various super-resolution modalities that should be considered during experimental design).
Table 1:
Comparison of super-resolution techniques and parameters that should be considered in choosing a super-resolution modality.
| Technique | Commercial Availability |
Equipment Cost | Lateral Spatial Resolution | Axial Spatial Resolution | Temporal Resolution | Sample penetration depth in standard setup | Specialized fluorophores | Difficulty of Data Analysis |
|---|---|---|---|---|---|---|---|---|
| STORM/PALM | High | Medium | ~20–50 nma | ~40–100 nmb | Seconds-Minutes | Several μm | Yes | High |
| DNA-PAINT | Medium | Medium | ~20–50 nma | ~40–100 nmb | Minutes-Hoursc | Several μm | No | High |
| ROSE/SIMFLUX | Low | Medium/High | ~5–20 nma | N/A (implemented in 2D) | Minutes-Hours | Several μm | Nod | High |
| ROSE-Z | Low | Medium/High | ~2–5 nma | ~2–5 nm | Minutes-Hours | Several μm | Nod | High |
| ModLoc | Low | Medium/High | ~3–5 nma | ~7–10 nm | Minutes-Hours | Several μm | Nod | High |
| Expansion Microscopy | High | Low-Mediume | ~25–70 nmf | ~25–70 nm | Seconds-Minutes | Variableg | No | Low |
| STED | High | High | ~20–50 nm | ~40–100 nmh | 10 ms-Minutesi | Typically 10–15 μm, up to 120 μmj | No | Low |
| RESOLFT | Medium | High | ~20–50 nm | ~40–100 nm | Seconds-Minutesi | Several μm | Yes | Low |
| Parallelized RESOLFT | Low | High | ~45–65 nmk | ~50–80 nmk | Seconds-Minutes | Several μm | Yes | Low-Medium |
| MINFLUX | Medium | High | ~1–3 nm | ~1–3 nm | Down to 80 μsecondsi | Several μm | Yes | High |
| SIM | High | Medium | ~100–150 nm | ~250–350 nm | Sub-seconds | Several μml | No | Medium |
| TIRF-SIM | High | Medium | ~85–120 nm | ~100–200 nm | Sub-seconds | 200 nm | No | Medium |
| ISIM | High | Medium | ~140–200 nm | ~300–400 nm | Down to 10 ms | 20 μm | No | Medium |
| Airyscan | High | Medium | ~120–180 nm | ~350–450 nm | Milliseconds-Secondsi | Several μm | No | Low |
| SOFI | High | Low | ~75–175 nmm | ~200–300 nmm | Sub-seconds-Seconds | Several μm | Yes | High |
| SRRF | High | Low | ~60–150 nm | Diffractionlimited (no improvement) | Seconds | Several μm | no | high |
Values depend on many parameters, importantly the localization precision, which is dependent on the number of photons collected and the fluorophores used. Ranges correspond to most commonly used and bright fluorophores.
Values correspond to the most commonly used PSF engineering implementations (e.g. cylindrical lens) and can further be improved using interferometric approaches (iPALM, 4Pi-SMS and W-4Pi-SMSN)
Values are dependent on number of targets to image; with improved buffers and DNA-sequences this can be decreased to seconds-minutes
These methods can be implemented with STORM fluorophores or DNA-PAINT approaches. When implemented with DNA-PAINT, they do not require specialized fluorophores.
ExM can be done on a simple widefield microscope, but generally a confocal microscope is used due to sample size and thickness
70 nm refers to the most common ~4.5X sample expansion. With improved gel recipes or iterative expansions, larger expansions are possible, bringing resolution down to 25 nm
ExM imaging generally requires many Z-stacks based on increased sample size
Values correspond to most common implementation of 3D STED and can further be improved with the use of 4Pi geometry (isoSTED)
Temporal resolution inversely scales with the size of field of view.
Deep sample penetrance has been achieved with high N.A glycerol immersion lenses (Urban et al., 2011)
Lateral and axial resolution based on numbers reported in (Masullo et al., 2018) and (Bodén et al., 2021), respectively.
Up to 80 um depth has been demonstrated with adaptive optics (Lin et al., 2021).
The resolution here is based on 2nd to 4th order statistical analysis, the most common range for SOFI.
A series of developments to make fluorescent proteins brighter, faster-switching, more photostable, and/or decoupling excitation from photo-switching has made RESOLFT more desirable for live-imaging (Jensen et al., 2020). Imaging throughput has further been improved by using massively parallelized illumination (Bodén et al., 2021; Chmyrov et al., 2013) as well as similar sample adaptive illumination strategies as STED that enable only scanning regions of the sample that are occupied by fluorophores (Dreier et al., 2019). In addition, MoNaLISA (Molecular Nanoscale Live Imaging with Sectioning Ability) used different On- and Off-switching light patterns with optimized shape and periodicity to effectively balance imaging speed, field-of-view and 3D resolution (50 × 50 μm2 at 0.2–1.3 Hz with 45–65 nm resolution) (Masullo et al., 2018) (Figure 1). This setup allowed the investigators to track the ER and mitochondria contact dynamics in different compartments of the neuron simultaneously at seconds and minutes time resolution (Damenti et al., 2021). These recent iterations of RESOLFT that use massively parallel, sample adaptive and optimized light illumination patterns, although not commercialized, hold great promise for live-imaging applications of super-resolution microscopy as they achieve imaging of large fields of view at high spatiotemporal resolution with reduced photobleaching and phototoxicity.
SMLM
SMLM utilizes the principle that a single fluorescent molecule can be localized by pinpointing its center of emission, resulting in a point estimate with a precision on the order of several nanometers. However, this approach requires that ideally only a single fluorophore is imaged in a given diffraction limited volume at a time. SMLM methods overcome this challenge by detecting a “blinking” fluorescent signal (see below). These localizations are then used to reconstruct a higher resolution image after sufficient rounds of blinking have allowed for all labels on the target structure to be localized. This generally necessitates several thousands of frames to be acquired for successful reconstruction of the final image. Different SMLM modalities achieve the required sparseness of the single molecules using different mechanisms of “blinking” as discussed below:
STORM/PALM
STORM and PALM utilize similar principles, and historically the major difference between them has been the type of fluorophore used. Initially, PALM utilized genetically encoded photoactivatable fluorescent proteins (Betzig et al., 2006), while STORM used organic fluorescent dyes (Rust et al., 2006). In both cases, the photophysics of the fluorophores allows for switching between two states (on/off or two different colors) for precise localization of a small number of blinking events at a time, resulting in a resolution reaching ~20 nm in x and y. Both methods have since been extended to 3D imaging with additionally improved z-resolution. One class of 3D methods use various ways of point spread function (PSF) engineering, in which the shape of the PSF is dependent on the z-position of the fluorophore (Hajj et al., 2014; Huang et al., 2008) Alternatively, 3D near-isotropic resolution can be achieved via the use of a dual-objective geometry combined with PSF engineering (Xu et al., 2012) or interferometric methods including iPALM (Shtengel et al., 2009), 4Pi-SMS (Aquino et al., 2011), and W-4PiSMSN with extended depth penetration over a whole cell (Huang et al., 2016).
STORM and PALM have been instrumental for many biological discoveries for over a decade ranging from the discovery of the Membrane-Associated Periodic Skeleton (MPS) in neuronal axons (Xu et al., 2013), the visualization of the folding patterns of DNA in the nucleus (Bintu et al., 2018; Neguembor et al., 2021; Ricci et al., 2015), the determination of the functional clustering of membrane receptors (Rossy et al., 2013) to the characterization of protein aggregates (Gyparaki et al., 2021). For a detailed review on the technical details behind STORM and PALM and their biological applications the reader is directed to a recent primer on this topic (Lelek et al., 2021). STORM and PALM are compatible with traditional widefield microscopes, making them accessible to many researchers, but they are not without limitations. With a few exceptions, imaging is generally limited to 2–3 colors in thin, fixed samples. These techniques have been combined with emerging advanced imaging methods to overcome some of these challenges (see section on Combined and Correlative Approaches).
DNA-PAINT (DNA points accumulation for imaging in nanoscale topography)
DNA-PAINT relies on the same concept of blinking as STORM and PALM, but the method for achieving blinking is unique. In DNA-PAINT, the target is labeled with a short DNA oligonucleotide (the docking strand) rather than a fluorophore (Jungmann et al., 2014). The fluorophore is floating freely in solution, tethered to the complementary, short DNA oligonucleotide (the imager strand). When the imager strand transiently binds to the docking strand and becomes temporarily immobilized, this event is registered as a blink. A major advantage is that the imagers can be washed out after a round of imaging, and hence the number of targets that can be imaged is only limited by the number of unique docking strands. Recently, 124-color DNA-PAINT multiplexing has been shown via a unique process referred to as kinetic barcoding (Wade et al., 2019). This strategy separates targets based on the tightly engineered binding kinetics of the imager oligo leading to distinct “blinking” rates, pushing the limits of super-resolution multiplexing and offering a major advantage over STORM/PALM. Similar to STORM, the DNA-PAINT docking oligos can be conjugated to primary or secondary antibodies, nanobodies as well as Halo-tags (Schlichthaerle et al., 2019), making DNA-PAINT a versatile approach.
While the multiplexing capability of DNA-PAINT is a significant advantage over STORM/PALM, this method has not been without its relative downsides. One downside is the fact that docking and imager oligos can have off-target binding to genomic DNA, leading to high background signal in the nucleus and making it more difficult to image nuclear targets. To overcome this challenge, a method called L-DNA-PAINT has been developed that takes advantage of left-handed DNA (L-DNA) oligomers that do not hybridize with right handed DNA (R-DNA) (Geertsema et al., 2021). Another major downside of traditional DNA-PAINT is imaging speed, which is generally 10–15 times slower than STORM for acquisitions of the same number of frames (Table 1). This is due to the need for long exposure times to blur out the fluorescence from imagers in solution, and to avoid localizing freely floating imagers that transiently move into the field of view. Another limitation that impacts imaging speed is the diffusion limited time that it takes for an imager strand to bind to its target strand. This waiting time is inversely proportional to the imager oligo concentration, however, the imager oligo concentration has to be kept low to avoid excessive background fluorescence. Recently, to increase the number of imager binding events, improved DNA sequence design and buffer conditions were introduced, allowing for imaging times comparable to STORM (Schueder et al., 2019; Strauss and Jungmann, 2020). However, these improvements do not yet address the problem of transient imager diffusion into the field of view without stable binding. This problem can be overcome with the use of fluorogenic dyes, which only fluoresce when bound to their target sequence (Chung et al., 2020). These can dramatically reduce background and off-target signal, enabling faster acquisition times. Combined, these advancements show promise for DNA-PAINT to offer super-resolution imaging of many targets with comparable imaging speeds to other SMLM techniques. A final downside is that DNA-PAINT is not generally compatible with live cell imaging due to both the long acquisition times of traditional DNA-PAINT and also the need for the imager oligo to cross the plasma membrane to visualize intracellular targets.
As DNA-PAINT technology continues to mature, we look forward to improved commercial availability of DNA-conjugated probes, including primary antibodies and Halo-tags, and widespread implementation of fluorogenic probes and oligo sequences to improve speed. We believe these advancements will lead to further adoption of DNA-PAINT as a routinely used and robust SMLM modality. As the method gains popularity it will be exciting to watch further advancements in both the technique and in the biological insights gained from its use.
Image analysis and quantification in SMLM
The tools for analyzing traditional optical microscopy images that consist of pixel-based intensity information have been developed over many years and continue to evolve. Several open-source software packages are available for analyzing these types of images, including ImageJ (Schneider et al., 2012), CellProfiler (Carpenter et al., 2006) and others and these tools are readily applicable to super-resolution images that are intensity based such as STED images. Methods like STED and RESOLFT also require minimal or no pre-processing to obtain a final image whereas SMLM requires specialized pre-processing steps. In addition, SMLM generates pointillist images consisting of a list of fluorophore positions. Therefore, post-processing of SMLM images requires specialized tools that are capable of handling this pointillist information. Consequently, image quantification in SMLM is a challenging bottleneck and large efforts have been devoted to the development of tools that are uniquely capable of analyzing SMLM data.
Pre-processing for SMLM
The first pre-processing task is to identify peaks (i.e. individual fluorophore images or PSFs) and determine their location through the estimation of the PSF’s center position (localization) (Figure 2). The localization step can be achieved through several methods including nonlinear least squares fitting to a Gaussian PSF model or maximum likelihood estimation (MLE) using a Gaussian PSF and Poisson noise model (Smith et al., 2010). However, analytical functions such as a Gaussian may not accurately capture the PSF shape. More recently, other estimation methods have been used, such as fitting to cubic splines (Babcock and Zhuang, 2017) or to experimental PSFs (Li et al., 2018). While the most precise way to localize fluorophores is to obtain sparse fluorophore images, acquisition of high-density fluorophore images can speed up imaging time. Hence, methods have also been developed to handle data containing high emitter density, including multi-emitter fitting algorithms (Holden et al., 2011), compressed sensing (Zhu et al., 2012) and more recently machine learning (Nehme et al., 2018; Speiser et al., 2021) (see below). Several open and closed source SMLM software packages are available for achieving the peak detection and localization steps. Users often face the dilemma of picking the “right” image processing platform. A recent paper evaluated the performance of several open-source software options to assess detection rate, accuracy, software usability and speed (Sage et al., 2019). This comparison serves as a resource to evaluate the tradeoff between SMLM packages in detecting and localizing fluorophores under different experimental conditions. Some of these software packages also include additional pre-processing steps such as linking the same fluorophore across multiple frames, filtering imprecise localizations and drift correction. The reader is directed to a recent primer on SMLM for more in depth reading of SMLM pre-processing (Lelek et al., 2021).
Figure 2: Single Molecule Localization Microscopy and analysis.
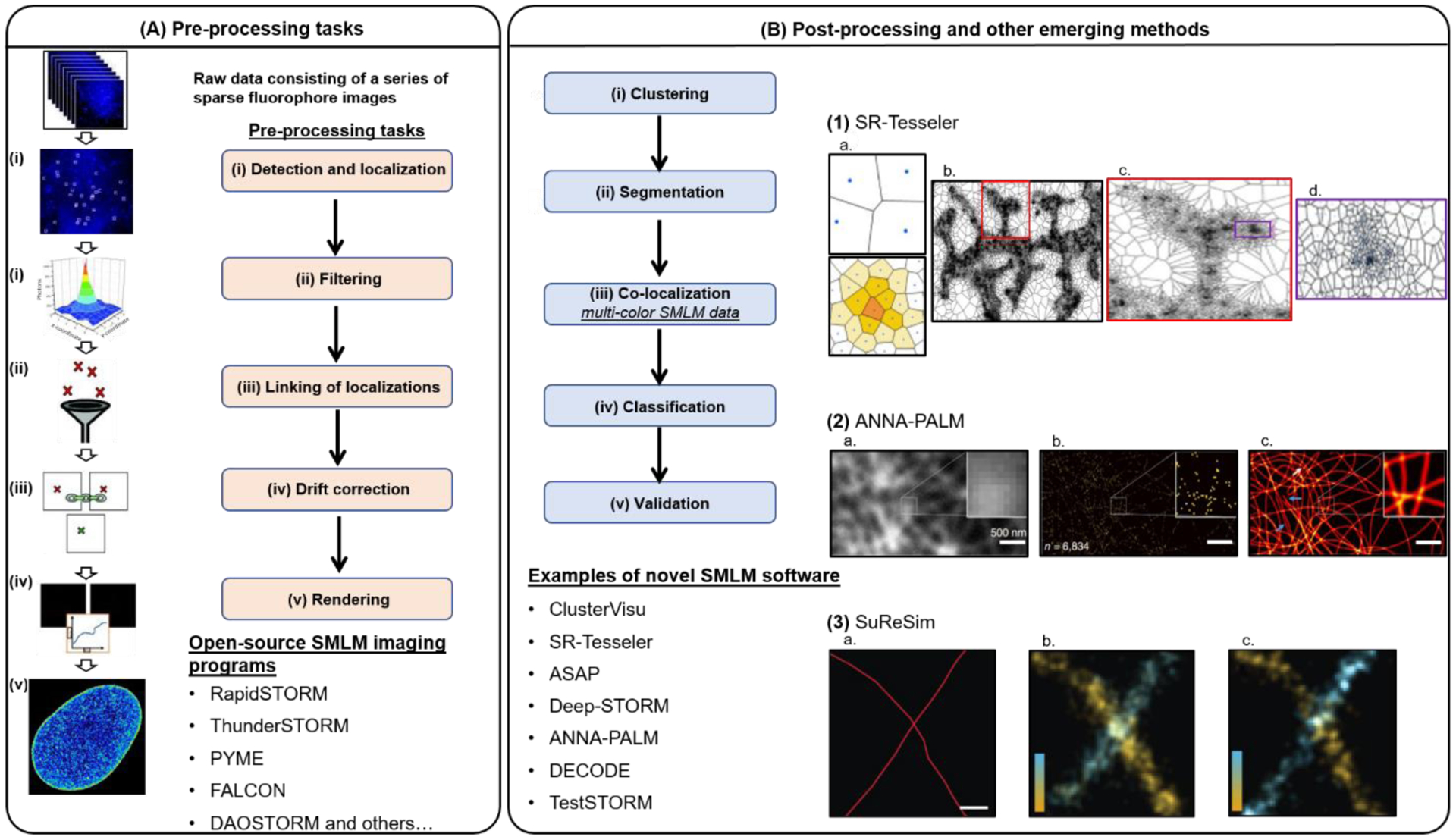
(A) The pre-processing tasks of SMLM data consist of: (i) peak detection and localization, (ii) linking of localizations, (iii) filtering, (iv) drift correction, and (v) rendering. Currently, there are more than 25 open-source software packages that can perform all or some steps of these tasks. Figure was adapted from (Brede and Lakadamyali, 2012) (B) Post-processing of SMLM images include: (i) clustering, (ii) segmentation, (iii) co-localization, (iv) classification, and (v) validation. (1) SR-Tesseler, developed by (Levet et al., 2015), segments SMLM images consisting of a wide-range of shapes and sizes using Voronoi Tessellation (1a), such as entire neuronal processes (1b, c) or nano-clusters (1d). (2) An up-and-coming category of SMLM analysis platforms are those that apply machine- and deep-learning algorithms. For example: ANNA-PALM, developed by (Ouyang et al., 2018), reconstructs super-resolution images (2c) from sparse, rapidly acquired SMLM data (2a,b). (3) Simulation of realistic SMLM data can be used to validate and compare the performance of available SMLM analysis platforms. For example, SuReSim, developed by (Venkataramani et al., 2016), is a software that can simulate biological structures under different experimental conditions; 3a shows the ground truth model, 3b shows the simulated image, and 3c shows the real-experimental data for a pair of crossed-microtubules labeled with antibodies.
Post-processing for SMLM
A striking result of super-resolution studies, particularly using SMLM, has been that many proteins assemble into heterogeneous nano-clusters. Thus, several methods have been developed that can segment and quantify the nature of these nano-clusters (Figure 2).
Initial methods for quantifying clusters in SMLM images took advantage of statistical tools such as Ripley’s K function or Pair Correlation Analysis (PCA) (Khater et al., 2020). These statistical tools work on the principle that randomly distributed points in space generate a known number of localizations within a certain distance of another localization. Deviations from this random distribution can thus be determined statistically. These methods are applicable to 2D and 3D SMLM data as well as multi-color SMLM data for determining whether the distribution of two proteins is correlated in space and have been successfully applied in many biological settings including determination of membrane receptor clustering (Sengupta et al., 2011). However, they only provide statistical averages and hence heterogeneities and properties of individual clusters cannot be obtained. To overcome this challenge, several cluster segmentation methods, often relying on mathematical approaches that have been in use in other fields, have been adapted for use in SMLM, including DBSCAN (Density Based Spatial Clustering of Applications with Noise) (Khater et al., 2020), Voronoi tessellation (Andronov et al., 2016; Levet et al., 2015) (Figure 2) and Bayesian analysis (Rubin-Delanchy et al., 2015). Some of these approaches, in particular Voronoi tessellation, have also been extended to determining co-localization in multi-color SMLM images (Levet et al., 2019).
While each method has its advantages and drawbacks, none of these approaches alone can inform whether the identified clusters are true biological clusters or clustering artifacts. Pseudoclusters are the result of repetitive and stochastic photoblinking of the same fluorophore and the fact that labeling stoichiometry is typically not one to one (Durisic et al., 2014a). As a result, in SMLM it is not possible to assign each localization to a unique protein, leading to clustering artifacts and over- or under-counting of protein copy number. Several approaches have been developed to overcome this problem. A recent example is DDC (Distance Distribution Correction), which relies on obtaining the true pairwise distance distribution of different fluorophores from the imaging sequence at different frame differences (Bohrer et al., 2021). However, methods like DDC that only correct for repeated fluorophore blinking cannot account for repeated localizations arising from amplifications in labeling, for example due to the use of primary and secondary antibodies. A frequently used solution to account for both problems and to determine the underlying molecular stoichiometry incorporates the use of calibration samples (Durisic et al., 2014b; Zanacchi et al., 2017) such as the nuclear pore complexes (NPCs), which have a well-defined subunit stoichiometry (Thevathasan et al., 2019). Another, easily accessible approach for accounting for clustering artifacts involves varying the labeling density, which leads to characteristic changes in the localization maps of each acquisition (Baumgart et al., 2016). DNA-PAINT offers unique advantages over STORM/PALM for quantitative analysis. While the blinking of the fluorophores in STORM/PALM is highly stochastic, the equilibrium dissociation constant (Kd) between the docking and imager strands is precise (Jungmann et al., 2016). At a given concentration of imager oligo and secondary antibody, the number of blinking events visualized per area, thus, scales linearly with the number of labels. Thus, with careful antibody titration controls, quantitative PAINT (qPAINT) (Jungmann et al., 2016) can measure the copy number of proteins more precisely. The precision of this technique can be further improved with the use of DNA-tethered nanobodies, which reliably only have one docking strand, whereas traditional secondary antibodies generally have several.
An exciting, up and coming approach to quantification of super-resolution data is the development of tools that incorporate machine and deep learning. ASAP (Automated Structure Analysis Program) is a machine learning approach that enables rapid and automated detection, segmentation, and classification based on the structural features of SMLM clusters (Danial and Garcia-Saez, 2019). Another recently employed approach is the use of Iterative Hierarchical Clustering, which is an unsupervised classification algorithm (Gyparaki et al., 2021). Machine and deep learning have also been applied to generate high-quality super-resolution images from suboptimal data. One example is ANNA-PALM (Artificial Neuronal Network Accelerated-PALM), which can reconstruct super-resolution images from sparse, rapidly acquired SMLM data and low-resolution images captured with conventional microscopes (Ouyang et al., 2018) (Figure 2). Deep-STORM (Nehme et al., 2018) and DECODE (Deep Context Dependent) (Speiser et al., 2021) are computational tools that employ a trainable deep-convolution network to generate high-resolution images in the presence of challenging signal-to-noise ratio and high emitter density. Finally, data-driven methods have been developed that are capable of converting images acquired from low-resolution systems (widefield and confocal fluorescence microscopes) into super-resolution quality images (Wang et al., 2019). While the advantages of these approaches seem attractive, whether the resulting images are artifact-free is unclear. Hence, tools to validate super-resolution images hold great importance. TestSTORM is a simulator that can be used to optimize critical imaging parameters and understand where imaging artifacts come from (Novák et al., 2017). SuReSim is another simulation software that simulates data sets of arbitrary structures allowing users to test parameters that can potentially affect the imaging outcome (Venkataramani et al., 2016) (Figure 2).
While the caveat of deep learning is the need for robust and large training data sets using synthetic or experimental data, the incorporation of these novel methods holds much promise in pushing forward the toolset available for pre- and post-processing of super-resolution data.
Intermediate resolution modalities
As the SMLM- and STED-based super-resolution microscopy techniques continue to develop towards higher resolution, other alternative intermediate resolution microscopy methods have come into light for their advantage of better temporal resolution and less phototoxicity albeit with a somewhat modest improvement in spatial resolution (typically 1.7 to 3-fold) (Table 1).
SIM (Structured Illumination Microscopy)
The most widely used SIM, or linear SIM, illuminates fluorescent samples with a fine strip pattern so the images appear with a sinusoidal brightness (Gustafsson, 2000; Heintzmann and Cremer, 1999). These images contain information of higher spatial frequencies that are otherwise not captured by the objective lens. Typically, for 2D SIM, collecting 3–5 SIM images that are phase-shifted in illumination pattern at each of the three angles that are rotated laterally is sufficient to gain resolution. In other words, 9–15 raw images are needed to reconstruct an image with enhanced resolution. The higher frequency information that represents the fine details are untangled by a deconvolution step in the Fourier space. The final image is produced through a reverse Fourier transform step, reaching an improvement in resolution by up to two-fold. SIM has gained popularity to study a wide range of biological questions. For example, a recent study found that fission position on a mitochondrion is a strong predictor of mitochondria fate, and the researchers were able to discern the fine morphological and biochemical changes before and after fission via SIM in live cells (Kleele et al., 2021). SIM has also provided new insights into the 3D organization of chromatin and the formation of topologically associating domains (TADs) within the nucleus of single cells (Szabo et al., 2018). Due to its reliance on conventional fluorophores and fluorescent proteins, SIM can be routinely used with multicolor and has also been expanded to 3D imaging (Fiolka et al., 2012; Gustafsson et al., 2008). The signal to noise ratio can be further enhanced by combining SIM with TIRF (total internal reflection) (TIRF-SIM), although the imaging depth is limited to within 200 nm near basal cell membrane (Li et al., 2015). On the other hand, grazing-incidence SIM (GI-SIM), which essentially illuminates the sample with a 1 μm light sheet parallel to the imaging plane gives an excellent penetration for visualizing organelle dynamics at millisecond time scale (Guo et al., 2018b).
Experienced developers have been continuously expanding SIM’s capabilities on both spatial and temporal resolution. For example, non-linear SIM reaches resolution down to 45 nm on biological samples (Gustafsson, 2005; Li et al., 2015; Rego et al., 2012), fastSIM, DMD-SIM and HIT-SIM reach a speed of up to 50–60 Hz (Lu-Walther et al., 2015; Sandmeyer et al., 2021; Van den Eynde et al., 2021), and GI-SIM reaches a remarkable 266 Hz frame rate (Guo et al., 2018b). SIM has also been combined with other modalities, such as lattice light sheet (Chen et al., 2014), two-photon microscopy (Ingaramo et al., 2014; Winter et al., 2014) or adaptive optics for deep sample penetration (Lin et al., 2021). We refer the readers to recent in-depth review articles on SIM for further reading (Prakash et al., 2021; Wu and Shroff, 2018).
Another commercially available alternative, Instant SIM (ISIM), offers favorable temporal resolution by employing rapid, parallelized multifoci scanning (York et al., 2013). Instead of applying a stripe pattern at multiple angles, it combines microlenses and pinhole arrays that locally scale down each excitation and emission foci, and uses optical hardware to scan and sum the raw images for an instantaneous improvement of spatial resolution (~1.4 fold). Typical imaging speed in ISIM can be up to 100 Hz, a two-fold resolution gain can be recovered after a further deconvolution step and this method has been combined with illumination geometries such as TIRF (instant TIRF-SIM) for high contrast imaging near the coverslip (Guo et al., 2018a).
One important consideration when using SIM is that artifacts can arise post-reconstruction due to various experimental and instrumental factors. Some artifacts can be visually spotted as they carry characteristic patterns in the resolved images, which can be reduced or eliminated by adjusting different parameters during reconstruction, improving signal to noise ratio in the biological sample and/or tuning/calibration of the instrument (Demmerle et al., 2017). There are accessible open source algorithms, for example, SIMcheck, an ImageJ plugin suite (Ball et al., 2015), and other advanced methods (Huang et al., 2018; Smith et al., 2021), which are useful for detecting and eliminating artifacts.
Airyscan
Airyscan is a confocal scanning microscopy-based method, and the resolution improvement (~1.7 fold) is largely through improvements in the signal to noise ratio (Huff, 2015; Huff et al., 2017). The signal is recorded by an array of 32 small point detectors (each detector element behaving as a 0.2 airy unit pinhole) that collectively maintain the light collection efficiency of a 1.25 airy unit pinhole, improving the signal to noise without compromising resolution. Through a linear deconvolution step of each of the 32 individual elements to further improve their lateral and axial resolution followed by pixel reassignment of each element back to the center position, Airyscan produces images with enhanced lateral and axial resolution (Table 1). Because Airyscan is commercially available, gentle, easy to operate (see detailed protocol by (Wu and Hammer, 2021)), compatible with multiplexing and capable of acquiring images at millisecond temporal resolution with minimal processing, it has been routinely used in a wide range of biological applications, for example, visualizing the buckling of detyrosinated microtubules in contracting heart muscle cells (Robison et al., 2016), determining how defects in cardiac cytoskeletal organization affect failing cardiomyocytes (Chen et al., 2020), or how actin cables assemble and move at mitochondria during mitosis (Moore et al., 2021).
SOFI (Super-resolution Optical Fluctuation Imaging) and SRRF (Super-resolution Radial Fluctuation)
SOFI, similar to SMLM methods, takes advantage of the blinking nature of fluorescent probes. However, unlike SMLM methods, each image in a SOFI experiment does not require a sparse subset of active fluorophores, which results in a higher emitter density, and thus faster acquisition times. Indeed, stochastically blinking fluorophores produce fluctuations in the fluorescence intensity that are detectable by carrying out higher-order statistical analysis of these temporal fluctuations in the image sequence. This will result in a spatial resolution beyond diffraction (Dedecker et al., 2012; Dertinger et al., 2009). The faster acquisition times make the method compatible with both fixed and live samples in a multicolor setting in 3D. SOFI has shown ~2–4-fold improvement in spatial resolution on structures such as subdomains on the plasma membrane, microtubules, actin, and clathrin-coated pits (Dedecker et al., 2012; Zhang et al., 2015). SOFI can also be used for biosensing experiments. For example, a new class of biosensors termed FLINC (Fluorescence fluctuation increase by contact), in which the fluorescence fluctuation behavior of the readout protein changes when in proximity to a second fluorescent protein, have been combined with SOFI to visualize dynamic biochemical activity within cells with super-resolution (Mo et al., 2017).
The SOFI image, however, does not linearly correspond to the fluorescent intensity present within each pixel. Therefore, one should be careful when interpreting fluorophore density information. Various factors can degrade the SOFI image quality due to its assumption of independent blinking, such as bleaching and laser instabilities. Artifacts can be evaluated and addressed using open access tools (Hugelier et al., 2021; Moeyaert et al., 2020; Peeters et al., 2017; Vandenberg et al., 2016).
SRRF is an analytical framework that can improve the resolution of images acquired by conventional microscopes, by calculating the radiality of the signals across the entire frame. SRRF is applicable to a wide range of experimental datasets including those acquired for SMLM or confocal microscope images and the resolution improvement depends on the nature of the dataset (Gustafsson et al., 2016). For example, microtubules can be super-resolved in live cells with ~60–120 nm spatial resolution at 1 second temporal resolution using SRRF (Gustafsson et al., 2016).
Both SOFI and SRRF can produce improved resolution in living samples without the need for lengthy acquisition times typically required in SMLM. They provide extra tools when advanced super-resolution microscopy equipment is not accessible, and can be complementary to existing super-resolution methods that have lower temporal resolution (Deschout et al., 2016). The associated analysis algorithms are open source, including parameter guidance and evaluation (Cevoli et al., 2021; Culley et al., 2018; Moeyaert et al., 2020). However, caution is needed in both SOFI and SRRF to avoid artifacts and interpret the resolved images and algorithms have been developed to address some of the pitfalls of these high-density intensity fluctuation methods (Marsh et al., 2018).
Emerging super-resolution modalities
MINFLUX and MINSTED
Despite much advancement in super-resolution, the spatial resolution of most methods does not reach true molecular scale and the temporal resolution has been a limiting factor. MINFLUX (minimal emission fluxes) (Balzarotti et al., 2017), offers a leap forward towards improved spatial and temporal resolution (Figure 3 A–B, Table 1). In MINFLUX, the fluorescent molecule’s position can be precisely determined by exposing the molecule to multiple offsetted excitation donuts. The center of the donut-shaped excitation beam has minimum intensity, but a molecule that is proximal to the center of the donut will emit fluorescence to different degrees depending on its distance from the donut minimum. The number of photons emitted at various coordinates is used to calculate the emitter position.
Figure 3: Emerging super-resolution modalities.
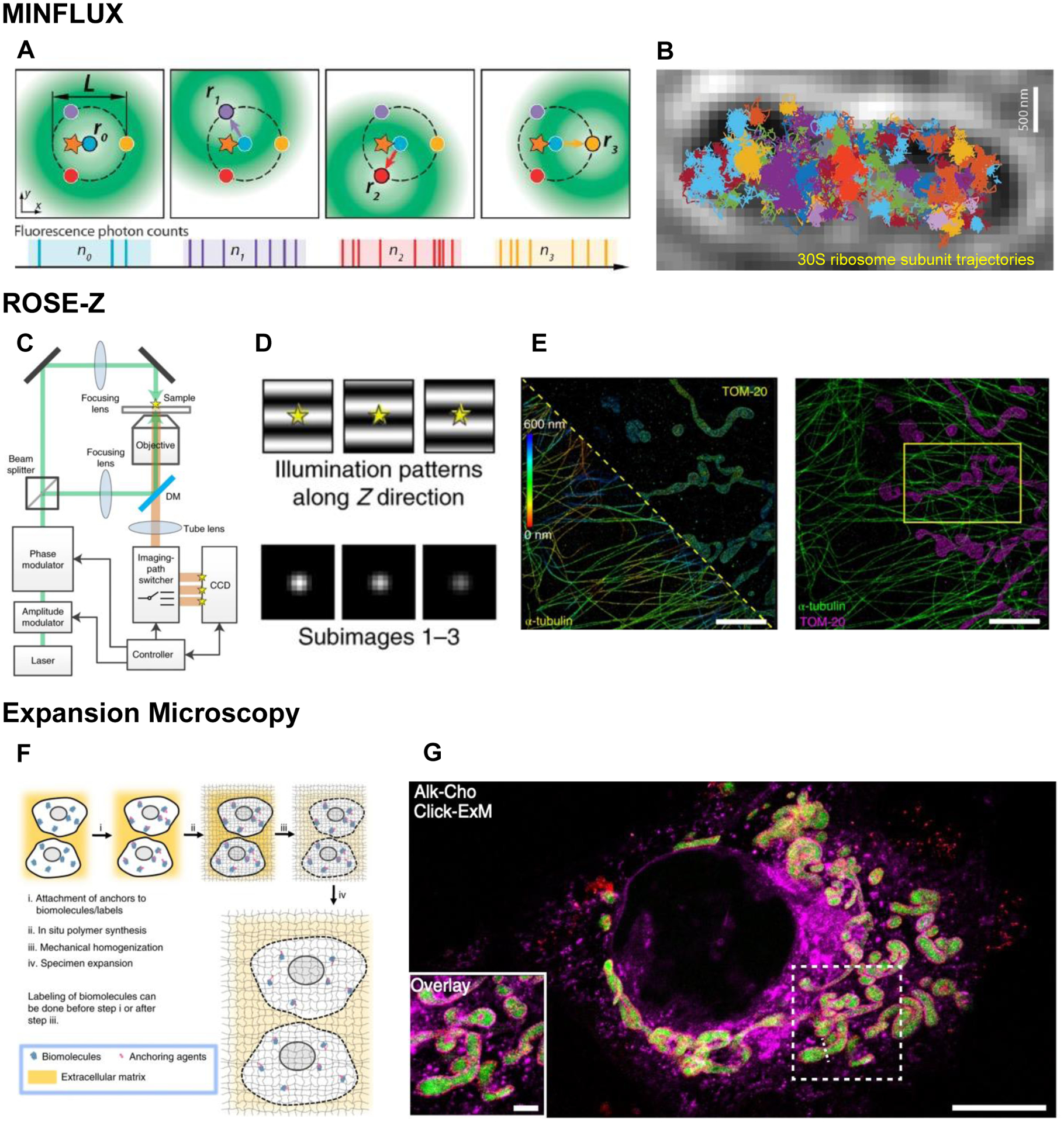
(A) Graphic representation of a fluorescent molecule (star) excited by a moving, donut-shaped excitation beam (green) at 4 different positions (r0, r1, r2, r3) based on the donut minimum in MINFLUX (Balzarotti et al., 2017). Each position is color-coded, and the corresponding photon counts are shown below each panel. (B) Color-coded trajectories of 30S ribosome subunit fused to mEos2 rapidly moving in a E.coli cell (transmission micrograph), captured by MINFLUX at 125 μs temporal resolution and 48 nm localization precision, by detecting on average 9 photons per localization (Balzarotti et al., 2017). Scale bar, 500 nm. (C) Working principle of ROSE-Z. The excitation laser was phase- and amplitude-modulated, split into two paths and interfered at the sample to create an interference fringe pattern. The intensity and phase of the interference fringes are modulated to switch among three different illumination patterns during imaging. In the imaging path, the fluorescence signal is recorded at different areas of the charge-coupled device (CCD) and synchronized with the illumination pattern, resulting in three subimages within a single exposure. DM, dichroic mirror. From (Gu et al., 2021). (D) Illumination patterns along Z direction and corresponding sub-images. (E) ROSE-Z imaging of Tom20 labeled mitochondria and α-tubulin labeled microtubules in a Cos7 cell. Scale bars, 5 μm. (F) General workflow for expansion microscopy. Adapted from (Wassie et al., 2019). (G) Click-ExM image showing mitochondria in a Cos-7 cell labeled with anti-Tom20, Mito-GFP and Alkyne-Choline to label choline-containing lipids. Scale bars, 10 μm in B, 2 μm in inset of B. Adapted from (Sun et al., 2021).
The capability of MINFLUX to capture highly dynamic events was first demonstrated by tracking 30S ribosomal subunit utilizing only 10 photons per localization in live E.coli cells at a localization precision of 48 nm and at time resolution of 125 μs (Balzarotti et al., 2017). To achieve an outstanding spatiotemporal resolution, additional hardware measures such as fast estimate of position were implemented to keep the fluorescent molecules near the donut center (Eilers et al., 2018). Spatial resolution can be further improved to near molecular scale by multiple iterations of scanning and estimation of the emitter location. Additionally, MINFLUX can be implemented in 3D with a precision of a few nanometers on a fixed sample (Gwosch et al., 2020; Pape et al., 2020; Schmidt et al., 2021). Due to the low photon requirement for precise localization, MINFLUX is very powerful when applied to single molecule tracking especially when the signal is dim (Schmidt et al., 2021). MINFLUX microscopes have just begun to be commercialized very recently and may become more widely applied in biology in the future. It is yet unclear if MINFLUX can retain temporal resolution with a large field of view to carry out molecular scale super-resolution imaging at high throughput, which ultimately comes back to the tradeoff between spatial and temporal resolution, a persistent challenge in microscopy.
Using a related concept to MINFLUX, MINSTED employs a donut-shaped STED beam co-aligned with the excitation beam to scan around the fluorophore to localize it (Weber et al., 2021). In contrast to MINFLUX, a molecule at the donut minimum in MINSTED emits the highest number of photons. However, MINSTED also uses the information of photons emitted when the molecule is away from the donut center during the scan, where STED beam intensity is intermediate, and this informs the subsequent scanning iterations to close in on the molecule’s position (providing down to 1~3 nm precision). One advantage of MINSTED over MINFLUX is that it provides better background suppression (Weber et al., 2021).
SIMFLUX, ROSE, and ModLoc
While MINFLUX and MINFIELD were a conceptual leap forward in bringing the resolution even closer to the scale of individual molecules, the scanning nature of these approaches constrains imaging to a small field of view. Several recent techniques have attempted to address this limitation using extended illumination for larger fields of view, while also enhancing resolution. SIMFLUX (Cnossen et al., 2020), SIMPLE (structured illumination microscopy based point localization estimator) (Reymond et al., 2019) and ROSE (repetitive optical selective exposure) (Gu et al., 2019) utilize similar principles in which the sample is illuminated with a shifting pattern of interference fringes. These fringes are shifted three times each in x and y, creating six sub-images of fluorophores with variations in fluorophore intensity. The fluorophore’s position can then be estimated by combing the relative position with respect to the shifting illumination pattern and the estimated center of the fluorescent spot. These methods achieve ~2-fold improvement in localization precision over traditional SMLM techniques in the lateral direction. Related methods like ModLoc (Jouchet et al., 2021) and ROSE-Z (Gu et al., 2021) (Figure 3C–E) also improve the axial resolution. ROSE-Z (Gu et al., 2021) implements interference fringe excitation in the axial direction in addition to inducing an astigmatism via insertion of a cylindrical lens into the imaging path. The result is an axial localization precision of roughly 2 nm, dramatically improved compared to traditional SMLM setups that utilize PSF engineering alone. In a similar approach, ModLoc (Jouchet et al., 2021) uses an illumination pattern composed of tilted fringes and the axial localization is obtained by a combined analysis of the phase of the modulated fluorescent signal produced by the shifting structured illumination and of the lateral localization obtained by centroid fitting of the PSF leading to a ~7 nm axial and ~3 nm lateral localization precision over several microns z-depth. These approaches all utilize a single objective as opposed to the previous interferometric approaches that relied on dual-objective 4Pi geometry (iPALM, 4Pi-SMS, W4PiSMSN) and are hence in principle easier to align and maintain. These relatively new techniques, while requiring specialized setups that are not yet widely or commercially available, hold promise for enhancing resolution in future biological applications.
Expansion Microscopy (ExM)
ExM relies on isotropic expansion of samples by embedding them within a swellable hydrogel, physically increasing the distance between targets to improve imaging resolution. Initially, samples were expanded in all directions by a factor of ~4.5, offering a lateral resolution of about 70 nm (Chen et al., 2015) (Figure 3 F–G). Since then, numerous improvements have increased the method’s accessibility and resolution, broadening its applicability.
In 2016, several groups introduced new protocols for ExM that could be carried out using only commercially available reagents and making ExM a highly cost-effective technique for achieving super-resolution imaging (Chozinski et al., 2016; Ku et al., 2016; Tillberg et al., 2016). Compatible with traditional wide-field or confocal microscopes and conventional fluorophores, ExM is highly accessible compared to other techniques that require more specialized setups, fluorophores, reagents and image processing tools. Recently, for example, ExM was used to investigate the densely packed microtubule networks within neuronal processes, as physical expansion of the sample better allowed for isolated imaging of individual microtubules compared to STED imaging on a non-expanded sample (Katrukha et al., 2021).
Despite its accessibility, ExM has major downsides such as the time cost of imaging large, expanded samples and the fact that this is not a live cell compatible technology. ExM was also initially restricted to labeling proteins only. Several iterations of ExM have addressed this challenge and enabled labeling of other biomolecules, such as RNA (Chen et al., 2016), or lipids (Wen et al., 2020). Click-ExM (Sun et al., 2021), for example, is a new, versatile method that takes advantage of click chemistry to overcome biomolecule labeling limitations. The technique first involves labeling a biomolecule of interest with a clickable functional group such as an azide or alkyne. Next, the corresponding azide or alkyne biotin conjugate is attached via click chemistry. Finally, fluorescently labeled streptavidin binds this biotin and anchors it to the gel matrix before expansion. This technique is compatible with proteins, lipids, glycans, DNA, RNA, and small molecules.
Another major limitation of ExM is that initially it did not reach the ~20 nm resolution that SMLM or STED can offer. To get around this challenge, ExM protocols were developed that increase the expansion factor to improve resolution to ~25 nm. The first, termed iterative expansion microscopy (iExM) (Chang et al., 2017), involves two sequential ~4.5X expansions for an expansion factor of ~20X. The second, termed X10 microscopy (Truckenbrodt et al., 2018), expands the sample by a factor of ~10X in all directions in a single step by utilizing a new hydrogel recipe, though this expansion is more labor-intensive than other protocols and may not be suitable for thick tissue samples. Recently, a new protocol termed Ten-fold Robust Expansion Microscopy (TREx) was introduced, allowing for 10X sample expansion with a simple and reproducible protocol (Damstra et al., 2021). Another promising advancement was the introduction of Pan-ExM, which utilizes sequential expansions to achieve a 13–21X expansion factor combined with a global “pan-” proteomic fluorescent marker to achieve electron microscopy like structural contrast (M’Saad and Bewersdorf, 2020). This method offers added molecular context around a target protein of interest detected via normal immunofluorescence labeling. Together, these techniques reach close to the same resolutions as SMLM or STED, but have the advantage that they are more accessible, may offer improved visualization of densely packed proteins, and can achieve high resolution for low cost albeit only in fixed samples.
Combined and Correlative Approaches
ExM combined with super-resolution
Combining ExM with other super-resolution techniques is an emerging field, and the advancements mainly focus on improving the ~70 nm resolution of imaging after ~4–4.5X expansion of the sample. ExM has been combined with SIM, achieving a resolution of ~30 nm (Wang et al., 2018). Combinations of ExM with either SMLM or STED offer the exciting possibility of achieving even higher resolution, but also introduce unique challenges. During the expansion protocol, samples are treated with various harsh agents that either denature or digest proteins, resulting in loss of labeling epitopes. Because of this, most ExM protocols involve antibody labeling before expansion rather than after expansion. This approach presents further challenges, because even the best fluorophores lose a significant amount of fluorescence during the expansion process (Wassie et al., 2019). For SMLM or STED microscopy, labeling efficiency is crucial, and this loss during expansion can result in incomplete staining patterns or undercounting of targets due to signal quenching. This limitation was overcome by labeling targets of interest with multiple primary antibodies recognizing different epitopes to perform STED imaging, but this approach may not be feasible for most target proteins (Gao et al., 2018). Thus, for these combination techniques, post-expansion labeling is generally necessary. Post-labeling ExM has the distinct advantage that the linkage error between target protein and fluorescent label is reduced proportional to the expansion factor. With this advantage in mind, post-expansion labeling ExM was successfully combined with STORM to achieve ~4 nm resolution imaging of microtubules and centrioles, which retained their labeling epitopes after the expansion process (Zwettler et al., 2020).
While achieving this level of resolution is impressive, it was still limited to a subset of proteins that retain labeling epitopes after expansion. In 2021, Label-Retention ExM was introduced, which overcomes this problem using an optimized expansion protocol combined with SNAP and CLIP tags targeting proteins of interest (Shi et al., 2021). These small epitope tags are not degraded during the expansion process and are efficiently labeled with antibodies post-expansion. This technique, combined with STORM, allowed for ~4 nm resolution, and can be used to visualize any target that can be labeled with a SNAP or CLIP tag. This combination of techniques is highly promising, and we look forward to its use for future biological discoveries.
Light-Sheet and Lattice Light-Sheet microscopy combined with super-resolution
Light sheet microscopy addresses two major challenges of fluorescence imaging: imaging samples with greater depth and doing so with minimal photobleaching. Unlike confocal, the sample is illuminated from the side with a sheet of light such that sequential Z-planes can be imaged without illuminating the entire sample, dramatically reducing photobleaching. Lattice light sheet microscopy (Chen et al., 2014) utilizes the principles of light-sheet microscopy, but improves the technique by generating ultra-thin light sheets that illuminate an entire field of view with low peak excitation intensity, thus leading to high axial resolution, reduced photobleaching/phototoxicity and increased imaging speed.
LLS is highly compatible with super-resolution techniques, improving the penetration depth of these methods, which are typically limited to imaging thin specimens. LLS was first combined with both SIM and PALM (Chen et al., 2014). Recently, LLS combined with 3D-PALM was used to image the accessible genome in mouse embryonic stem cells, providing new insights into the organization of accessible regions of chromatin (Xie et al., 2020). LLS has also been combined with STORM to image the movement of plasma membrane receptors on whole live cells, highlighting the promise of combining high-speed 3D imaging with single-molecule precision (Waldchen et al., 2020).
Another exciting combination is LLS-ExM, which has enabled imaging an entire drosophila brain and mouse cortex with sub-cellular detail (Figure 4A). These techniques are theoretically quite compatible, as ExM physically expands samples for greater resolution, and LLS offers high-speed 3D imaging of large samples, but the technical challenges of combining them highlight some of the drawbacks of LLS microscopy. LLS requires highly specialized equipment, and uses a two-objective setup that requires specialized sample prep and sample mounting that generally are not compatible with standard cell culture imaging dishes and microscope slides. Several groups have attempted to create light-sheet microscopy setups that are more compatible with standard biological sample preparation. One particularly promising recent development is a single-objective oblique plane microscope with a high numerical aperture (Sapoznik et al., 2020). This setup offers comparable resolution to LLS microscopy for large, thick samples, while remaining compatible with standard biological sample preparation and mounting.
Figure 4: Combined and correlative approaches.
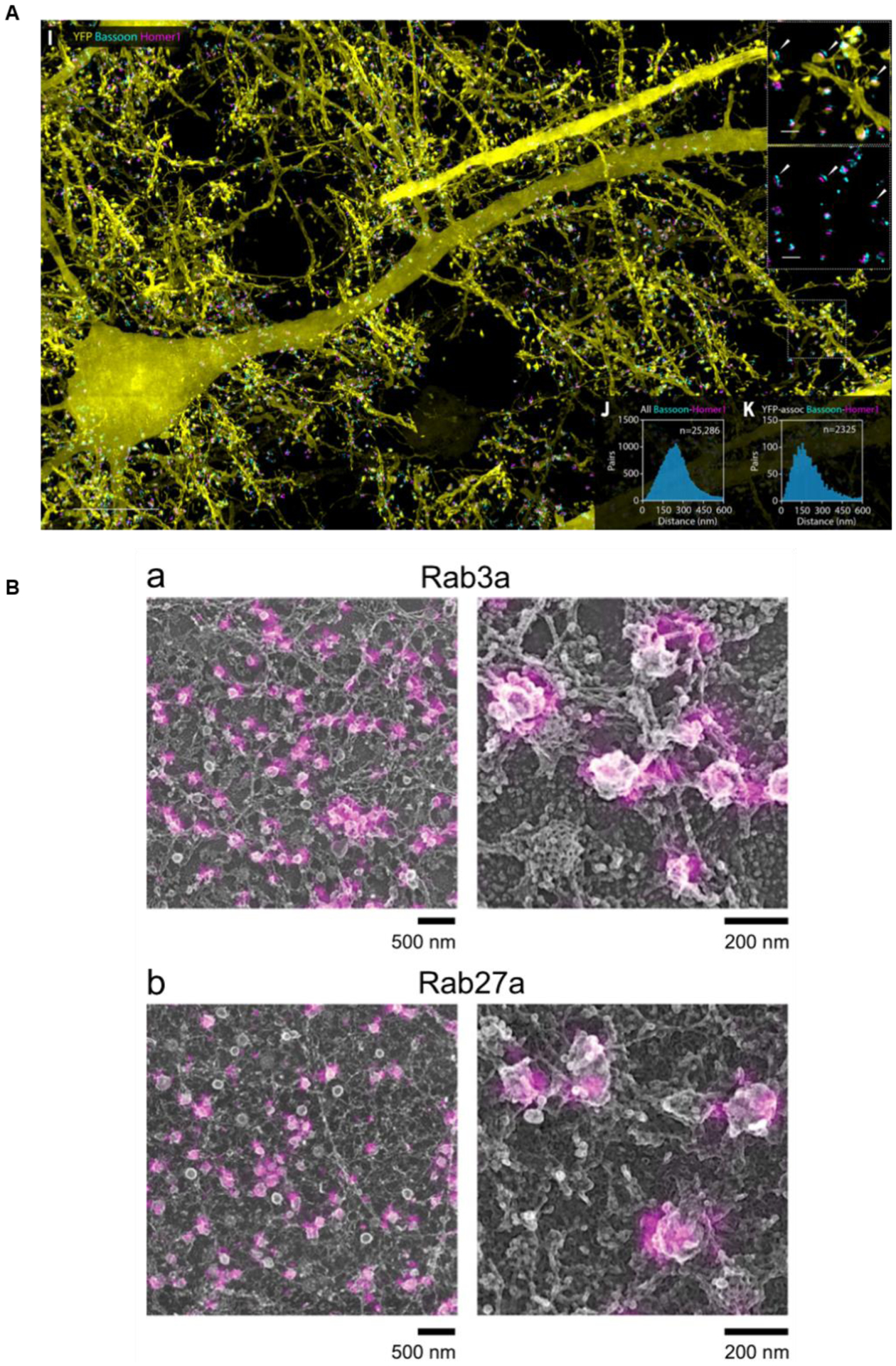
(A) Combined expansion and lattice light sheet microscopy showing individual neurons marked with YFP, as well as two individual synaptic proteins, Bassoon and Homer1, in a mouse cortical brain slice. Adapted from (Gao et al., 2019). (B) Correlative dSTORM and platinum-replica EM of dense core vesicle proteins Rab3a and Rab27a. Adapted from (Prasai et al., 2021)
Electron Microscopy combined with super-resolution:
Correlative light and electron microscopy (CLEM) has emerged as a powerful technique that combines the molecular-scale resolution and ultrastructural context of electron microscopy with the precise target specificity of light microscopy. However, without super-resolution imaging, CLEM is limited to targets that are either relatively large or sparsely distributed within cells. Combining super-resolution (SR) and EM (SR-CLEM), while potentially powerful, is an extremely difficult technical challenge. The EM and SR images must be properly aligned to one another, and since both techniques achieve very high resolution, a misalignment of only a few nanometers can be a major issue. SR-CLEM also requires using sample preparation methods that are compatible with both techniques, and research into how best to prepare samples and set up downstream imaging is ongoing. There have been several promising applications of SR-CLEM in recent years, using different strategies to overcome these technical challenges.
Correlative iPALM and metal replica transmission electron microscopy (TEM) utilizes gold nanorods coated in a thin layer of silica as a fiducial marker for alignment of fluorescence and TEM images. iPALM imaging is done first on targets of interest, and importantly gold nanorods can be visualized in this setup. Next, samples are prepared for TEM, and a metal replica is generated by coating the sample with a thin layer of platinum and carbon. Critically, when this replica is transferred from the sample and onto a TEM grid, the gold nanorods are embedded in the replica and can be visualized in TEM images. Sample warping can occur during this transfer process, adding another layer of technical difficulty. Nonetheless, the two sets of images can be aligned in X and Y using the position of the gold nanorods. One major limitation of this technique is that it relies on iPALM for Z-plane correlation of the fluorescence images to TEM images and is thus only applicable to targets at the plasma membrane of cells. However, it has been used to gain valuable insight into the organization of endocytic proteins at the plasma membrane with unprecedented resolution (Prasai et al., 2021; Sochacki et al., 2017) (Figure 4B).
SR has also been combined with cryogenic-electron tomography (Cryo-ET) in limited cases. The main challenge is achieving high quality SR images after cryo-preservation without disrupting the sample. The sample must remain under cryogenic conditions, while the objective lens for fluorescence imaging typically remains at room temperature. This necessitates the use of air-objectives at long working distances, which have much lower NA (~0.8) than oil immersion objectives (~1.3) used for SR imaging at room temperature. Additionally, while photobleaching is reduced, photoswitching is significantly slower at cryogenic temperatures than at room temperature, increasing the necessary imaging time. Further still, excitation laser intensity must be dramatically reduced, as the high laser intensity typically needed for SMLM can result in an increase in sample temperature, devitrifying ice. This too slows down photoswitching and results in longer acquisition times. These and other technical challenges of combining these techniques have been described in great detail recently (Dahlberg and Moerner, 2021). Despite these limitations, correlative PALM and Cryo-ET has been used to identify type VI secretion systems within Myococcus xanthus bacteria (Chang et al., 2014). Additionally, proof-of-concept of correlative 3D-PALM and cryo-ET has been demonstrated on thin cryosections of HEK293 cells (Liu et al., 2015). Using a custom setup, another group was able to achieve ~9 nm precision with a ~30 nm registration error between SR and cryo-ET images on three specific protein targets in Caulobacter crescentus (Dahlberg et al., 2020). Finally, an exciting application was the combination of focused ion beam scanning electron microscopy (FIB-SEM) with 3D cryogenic SMLM to image entire vitreously frozen cells (Hoffman et al., 2020). This imaging combination offers tremendous insight into the internal organization of human cells. However, it requires custom-built setups, and imaging for a single cell took 1–2 days per color for 3D SMLM, as well as 10–15 days for EM sample preparation and imaging.
Overall, as outlined above, SR-CLEM methods are extremely technically challenging, yet simultaneously extremely powerful. When implemented successfully, the payoff of combining precise target specificity with critical cellular context at molecular-scale resolution can be tremendous. Though this technique will likely not be accessible to the vast majority of researchers in the very near future, the wealth of information generated by SR-CLEM imaging, if shared freely, would allow researchers across different fields to examine their biomolecules of interest in their native context with new molecular-scale insight.
Conclusions and Outlook
Super-resolution microscopy has become an enabling tool for biological discovery in a short time but several challenges remain to be solved. Penetration depth of most commonly used and widely adopted super-resolution methods remains limited. While STED and RESOLFT, being point scanning methods, are more compatible with imaging thicker samples than the wide-field based SMLM methods, imaging very deep into tissues and intact organisms remains challenging. Combining super-resolution modalities with more specialized illumination geometries such as two-photon or light sheet in addition to tissue clearing or adaptive optics based corrections potentially hold promise for using these methods in more challenging biological settings (Bancelin et al., 2021; Chen et al., 2014; Hao et al., 2021b; Mlodzianoski et al., 2018; York et al., 2011). In particular, when combined with the quantitative and multiplexing capabilities of DNA-PAINT, such methods can advance our understanding of the molecular organization of cells, tissues and even intact organisms with high quantitative power. However, implementations of super-resolution microscopy with improved depth penetration are still a specialist’s tool due to being more technically challenging and remain to be widely adopted. Another promising avenue towards super-resolution imaging of thick biological samples is the combination of ExM with light sheet based approaches.
Super-resolution modalities, in their most commonly used form also remain limited to imaging fixed samples due to compromise between acquisition speed and spatial resolution, photobleaching and phototoxicity. RESOLFT and derivative approaches with their reduced photobleaching and phototoxicity combined with massively parallelized illumination can push the capability to image large fields of view at high spatiotemporal resolution, providing a promising path forward for both live and fixed cell super-resolution microscopy (Bodén et al., 2021; Chmyrov et al., 2013; Mahecic et al., 2019). Along similar lines, the imaging throughput of most super-resolution microscopy methods is low, making it challenging to acquire sufficient statistics even in fixed samples for robust biological interpretations. Massively parallelized illumination in STED/RESOLFT as well as new developments in SMLM that combine microfluidic sample handling, automated image acquisition and large field-of-view illumination are starting to address this challenge (Almada et al., 2019; Barentine et al., 2019; Beghin et al., 2017; Douglass et al., 2016; Mahecic et al., 2019; Stefko et al., 2018). However, most of these approaches are still in their infancy and remain to be widely adopted and more accessible.
The initial implementations of super-resolution microscopes, while drastically improving spatial resolution, did not reach true molecular scale resolution. While MINFLUX, MINSTED and related approaches are an important leap forward towards imaging at the molecular scale, more effort is needed for the development of bright, photostable, photoswitchable fluorophores as well as labeling approaches that minimize linkage errors and enable endogenous labeling. Development of new super-resolution compatible fluorophores and labeling methods has lagged behind the immense developments we have seen in optical microscopes, limiting the potential of these cutting-edge microscopes to reach true molecular scale resolution. Such improved labeling and sample preparation methods will also enable more robust combination of ExM with SMLM or STED based super-resolution microscopy, further pushing the resolution limit towards molecular scale imaging (Shi et al., 2021).
With advancements in microscopy and biological applications came the need for better quantitative analysis tools, in particular for SMLM methods that heavily rely on pre- and post-processing for image analysis. Several methods have been developed to not only generate/visualize super-resolution images but to also carry out segmentation, co-localization analysis and classification of morphologically diverse structures captured in super-resolution microscopy. While some efforts have been made to develop unified and modular analysis platforms for super-resolution data (Ries, 2020), most methods have remained disperse. There is a need for a concerted effort from method developers to consolidate the wide range of existing algorithms into a user-friendly, modular and robust platform that is easily accessible to new researchers entering the super-resolution field. In particular, incorporation of machine and deep learning approaches hold great promise for extracting novel biological insight from a diverse range of super-resolution images.
Overall, despite its recent history, super-resolution microscopy has been making a big impact in biology and it will be exciting to watch continued advancements in instrumentation, new fluorophores, labeling approaches and quantitative analysis tools, which will enable new biological applications. Commercialization of some of the more advanced modalities will be an important step towards accessibility of these tools to a wider range of researchers. In the future, we envision a multi-modal “smart microscope” that is operated in a fully automated mode, free of user input, and guided by the properties of a specific biological sample. Such a microscope could, for example, acquire lower resolution images with high throughput, analyze these images at high speed using neural networks, and use the analyzed information to perform a guided higher resolution imaging of a specific region of interest or upon a biological trigger. We are already starting to see examples of such “sample adaptive” and “smart” microscopes (Alvelid et al., 2021; Dreier et al., 2019; Gottfert et al., 2017; Heine et al., 2017) and this would be a particularly exciting field to watch.
Acknowledgements:
M.L. acknowledges funding from the National Institute of General Medical Sciences (NIGMS) (Grant number: RO1GM133842 and RM1GM136511), the Center for Engineering and Mechanobiology (CEMB), an NSF Science and Technology Center Pilot Award under grant agreement CMMI 15-48571, and NIH Common Fund 4D Nucleome (4DN) program (Grant number: UO1DA052715). A.N.S.R. is partially funded through the NIH Structural Biology and Molecular Biophysics Training Grant (Grant number: 5T32GM132039-03). We thank Siewert Hugelier for critical reading and comments on the manuscript. We apologize to any researchers whose work we have not been able to cite due to space limitations or our own ignorance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: Authors declare no conflict of interest.
References
- Almada P, Pereira PM, Culley S, Caillol G, Boroni-Rueda F, Dix CL, Charras G, Baum B, Laine RF, Leterrier C, et al. (2019). Automating multimodal microscopy with NanoJ-Fluidics. Nature Communications 10, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvelid J, Damenti M, and Testa I (2021). Event-triggered STED imaging. biooRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronov L, Orlov I, Lutz Y, Vonesch J-L, and Klaholz BP (2016). ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Scientific Reports 6, 24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquino D, Schonle A, Geisler C, Middendorff CV, Wurm CA, Okamura Y, Lang T, Hell SW, and Egner A (2011). Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat Methods 8, 353–359. [DOI] [PubMed] [Google Scholar]
- Babcock HP, and Zhuang X (2017). Analyzing Single Molecule Localization Microscopy Data Using Cubic Splines. Scientific Reports 7, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball G, Demmerle J, Kaufmann R, Davis I, Dobbie IM, and Schermelleh L (2015). SIMcheck: a Toolbox for Successful Super-resolution Structured Illumination Microscopy. Scientific Reports 5, 15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarotti F, Eilers Y, Gwosch KC, Gynnå AH, Westphal V, Stefani FD, Elf J, and Hell SW (2017). Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 355, 606–612. [DOI] [PubMed] [Google Scholar]
- Bancelin S, Mercier L, Murana E, and Nagerl UV (2021). Aberration correction in stimulated emission depletion microscopy to increase imaging depth in living brain tissue. Neurophotonics 8, 035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barentine AES, Lin Y, Liu M, Kidd P, Balduf L, Grace MR, Wang S, Bewersdorf J, and Baddeley D (2019). 3D Multicolor Nanoscopy at 10,000 Cells a Day. bioRxiv. [Google Scholar]
- Baumgart F, Arnold AM, Leskovar K, Staszek K, Fölser M, Weghuber J, Stockinger H, and Schütz GJ (2016). Varying label density allows artifact-free analysis of membrane-protein nanoclusters. Nature Methods 13, 661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beghin A, Kechkar A, Butler C, Levet F, Cabillic M, Rossier O, Giannone G, Galland R, Choquet D, and Sibarita J-B (2017). Localization-based super-resolution imaging meets high-content screening. Nature Methods 14, 1184–1190. [DOI] [PubMed] [Google Scholar]
- Berning S, Willig KI, Steffens H, Dibaj P, and Hell SW (2012). Nanoscopy in a Living Mouse Brain. Science 335, 551–551. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, and Hess HF (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- Bingen P, Reuss M, Engelhardt J, and Hell SW (2011). Parallelized STED fluorescence nanoscopy. Opt Express 19, 23716–23726. [DOI] [PubMed] [Google Scholar]
- Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, and Zhuang X (2018). Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodén A, Pennacchietti F, Coceano G, Damenti M, Ratz M, and Testa I (2021). Volumetric live cell imaging with three-dimensional parallelized RESOLFT microscopy. Nature Biotechnology 39, 609–618. [DOI] [PubMed] [Google Scholar]
- Bohrer CH, Yang X, Thakur S, Weng X, Tenner B, McQuillen R, Ross B, Wooten M, Chen X, Zhang J, et al. (2021). A pairwise distance distribution correction (DDC) algorithm to eliminate blinking-caused artifacts in SMLM. Nature Methods 18, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottanelli F, Kromann EB, Allgeyer ES, Erdmann RS, Wood Baguley S, Sirinakis G, Schepartz A, Baddeley D, Toomre DK, Rothman JE, et al. (2016). Two-colour live-cell nanoscale imaging of intracellular targets. Nature Communications 7, 10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brede N, and Lakadamyali M (2012). GraspJ: an open source, real-time analysis package for super-resolution imaging. Optical Nanoscopy 1, 11. [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carravilla P, Chojnacki J, Rujas E, Insausti S, Largo E, Waithe D, Apellaniz B, Sicard T, Julien J-P, Eggeling C, et al. (2019). Molecular recognition of the native HIV-1 MPER revealed by STED microscopy of single virions. Nature Communications 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevoli D, Vitale R, Vandenberg W, Hugelier S, Van den Eynde R, Dedecker P, and Ruckebusch C (2021). Design of experiments for the optimization of SOFI super-resolution microscopy imaging. Biomed Opt Express 12, 2617–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JB, Chen F, Yoon YG, Jung EE, Babcock H, Kang JS, Asano S, Suk HJ, Pak N, Tillberg PW, et al. (2017). Iterative expansion microscopy. Nat Methods 14, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YW, Chen S, Tocheva EI, Treuner-Lange A, Lobach S, Sogaard-Andersen L, and Jensen GJ (2014). Correlated cryogenic photoactivated localization microscopy and cryo-electron tomography. Nat Methods 11, 737–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B-C, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, Liu Z, et al. (2014). Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346, 1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Salomon AK, Caporizzo MA, Curry S, Kelly NA, Bedi K, Bogush AI, Krämer E, Schlossarek S, Janiak P, et al. (2020). Depletion of Vasohibin 1 Speeds Contraction and Relaxation in Failing Human Cardiomyocytes. Circulation Research 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Tillberg PW, and Boyden ES (2015). Optical imaging. Expansion microscopy. Science 347, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Wassie AT, Cote AJ, Sinha A, Alon S, Asano S, Daugharthy ER, Chang JB, Marblestone A, Church GM, et al. (2016). Nanoscale imaging of RNA with expansion microscopy. Nat Methods 13, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmyrov A, Keller J, Grotjohann T, Ratz M, d’Este E, Jakobs S, Eggeling C, and Hell SW (2013). Nanoscopy with more than 100,000 ‘doughnuts’. Nature Methods 10, 737–740. [DOI] [PubMed] [Google Scholar]
- Chozinski TJ, Halpern AR, Okawa H, Kim HJ, Tremel GJ, Wong RO, and Vaughan JC (2016). Expansion microscopy with conventional antibodies and fluorescent proteins. Nat Methods 13, 485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Zhang Z, Kidd P, Zhang Y, Williams ND, Rollins B, Yang Y, Lin C, Baddeley D, and Bewersdorf J (2020). Fluorogenic probe for fast 3D whole-cell DNA-PAINT. bioRxiv, 2020.2004.2029.066886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnossen J, Hinsdale T, Thorsen RØ, Siemons M, Schueder F, Jungmann R, Smith CS, Rieger B, and Stallinga S (2020). Localization microscopy at doubled precision with patterned illumination. Nature Methods 17, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culley S, Albrecht D, Jacobs C, Pereira PM, Leterrier C, Mercer J, and Henriques R (2018). Quantitative mapping and minimization of super-resolution optical imaging artifacts. Nat Methods 15, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg PD, and Moerner WE (2021). Cryogenic Super-Resolution Fluorescence and Electron Microscopy Correlated at the Nanoscale. Annu Rev Phys Chem 72, 253–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg PD, Saurabh S, Sartor AM, Wang J, Mitchell PG, Chiu W, Shapiro L, and Moerner WE (2020). Cryogenic single-molecule fluorescence annotations for electron tomography reveal in situ organization of key proteins in Caulobacter. Proc Natl Acad Sci U S A 117, 13937–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damenti M, Coceano G, Pennacchietti F, Bodén A, and Testa I (2021). STED and parallelized RESOLFT optical nanoscopy of the tubular endoplasmic reticulum and its mitochondrial contacts in neuronal cells. Neurobiology of Disease 155, 105361. [DOI] [PubMed] [Google Scholar]
- Damstra HGJ, Mohar B, Eddison M, Akhmanova A, Kapitein LC, and Tillberg PW (2021). Visualizing cellular and tissue ultrastructure using Ten-fold Robust Expansion Microscopy (TREx). bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial JSH, and Garcia-Saez AJ (2019). Quantitative analysis of super-resolved structures using ASAP. Nature Methods 16, 711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedecker P, Mo GCH, Dertinger T, and Zhang J (2012). Widely accessible method for superresolution fluorescence imaging of living systems. Proceedings of the National Academy of Sciences 109, 10909–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmerle J, Innocent C, North AJ, Ball G, Müller M, Miron E, Matsuda A, Dobbie IM, Markaki Y, and Schermelleh L (2017). Strategic and practical guidelines for successful structured illumination microscopy. Nature Protocols 12, 988–1010. [DOI] [PubMed] [Google Scholar]
- Dertinger T, Colyer R, Iyer G, Weiss S, and Enderlein J (2009). Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proceedings of the National Academy of Sciences 106, 22287–22292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschout H, Lukes T, Sharipov A, Szlag D, Feletti L, Vandenberg W, Dedecker P, Hofkens J, Leutenegger M, Lasser T, et al. (2016). Complementarity of PALM and SOFI for super-resolution live-cell imaging of focal adhesions. Nat Commun 7, 13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass KM, Sieben C, Archetti A, Lambert A, and Manley S (2016). Super-resolution imaging of multiple cells by optimised flat-field epi-illumination. Nat Photonics 10, 705–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier J, Castello M, Coceano G, Cáceres R, Plastino J, Vicidomini G, and Testa I (2019). Smart scanning for low-illumination and fast RESOLFT nanoscopy in vivo. Nature Communications 10, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durisic N, Cuervo LL, and Lakadamyali M (2014a). Quantitative super-resolution microscopy: pitfalls and strategies for image analysis. Current Opinion in Chemical Biology 20, 22–28. [DOI] [PubMed] [Google Scholar]
- Durisic N, Laparra-Cuervo L, Sandoval-Álvarez Á, Borbely JS, and Lakadamyali M (2014b). Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nature Methods 11, 156–162. [DOI] [PubMed] [Google Scholar]
- Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, Von Middendorff C, Schönle A, et al. (2009). Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 457, 1159–1162. [DOI] [PubMed] [Google Scholar]
- Eilers Y, Ta H, Gwosch KC, Balzarotti F, and Hell SW (2018). MINFLUX monitors rapid molecular jumps with superior spatiotemporal resolution. Proceedings of the National Academy of Sciences 115, 6117–6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiolka R, Shao L, Rego EH, Davidson MW, and Gustafsson MGL (2012). Time-lapse two-color 3D imaging of live cells with doubled resolution using structured illumination. Proceedings of the National Academy of Sciences 109, 5311–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Maraspini R, Beutel O, Zehtabian A, Eickholt B, Honigmann A, and Ewers H (2018). Expansion Stimulated Emission Depletion Microscopy (ExSTED). ACS Nano 12, 4178–4185. [DOI] [PubMed] [Google Scholar]
- Gao R, Asano SM, Upadhyayula S, Pisarev I, Milkie DE, Liu TL, Singh V, Graves A, Huynh GH, Zhao Y, et al. (2019). Cortical column and whole-brain imaging with molecular contrast and nanoscale resolution. Science 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geertsema HJ, Aimola G, Fabricius V, Fuerste JP, Kaufer BB, and Ewers H (2021). Left-handed DNA-PAINT for improved super-resolution imaging in the nucleus. Nat Biotechnol 39, 551–554. [DOI] [PubMed] [Google Scholar]
- Göttfert F, Christian, Mueller V, Berning S, Volker, Honigmann A, and Stefan(2013). Coaligned Dual-Channel STED Nanoscopy and Molecular Diffusion Analysis at 20 nm Resolution. Biophysical Journal 105, L01–L03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfert F, Pleiner T, Heine J, Westphal V, Gorlich D, Sahl SJ, and Hell SW (2017). Strong signal increase in STED fluorescence microscopy by imaging regions of subdiffraction extent. Proc Natl Acad Sci U S A 114, 2125–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Große L, Wurm CA, Brüser C, Neumann D, Jans DC, and Jakobs S (2016). Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. The EMBO Journal 35, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotjohann T, Testa I, Reuss M, Brakemann T, Eggeling C, Hell SW, and Jakobs S (2012). rsEGFP2 enables fast RESOLFT nanoscopy of living cells. eLife 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Li Y, Zhang S, Xue Y, Li W, Li D, Xu T, and Ji W (2019). Molecular resolution imaging by repetitive optical selective exposure. Nature Methods 16, 1114–1118. [DOI] [PubMed] [Google Scholar]
- Gu L, Li Y, Zhang S, Zhou M, Xue Y, Li W, Xu T, and Ji W (2021). Molecular-scale axial localization by repetitive optical selective exposure. Nature Methods 18, 369–373. [DOI] [PubMed] [Google Scholar]
- Guo M, Chandris P, Giannini JP, Trexler AJ, Fischer R, Chen J, Vishwasrao HD, Rey-Suarez I, Wu Y, Wu X, et al. (2018a). Single-shot super-resolution total internal reflection fluorescence microscopy. Nature Methods 15, 425–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li D, Zhang S, Yang Y, Liu J-J, Wang X, Liu C, Milkie DE, Moore RP, Tulu US, et al. (2018b). Visualizing Intracellular Organelle and Cytoskeletal Interactions at Nanoscale Resolution on Millisecond Timescales. Cell 175, 1430–1442.e1417. [DOI] [PubMed] [Google Scholar]
- Gustafsson MGL (2000). Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. SHORT COMMUNICATION. Journal of Microscopy 198, 82–87. [DOI] [PubMed] [Google Scholar]
- Gustafsson MGL (2005). Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proceedings of the National Academy of Sciences 102, 13081–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MGL, Shao L, Carlton PM, Wang CJR, Golubovskaya IN, Cande WZ, Agard DA, and Sedat JW (2008). Three-Dimensional Resolution Doubling in Wide-Field Fluorescence Microscopy by Structured Illumination. Biophysical Journal 94, 4957–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson N, Culley S, Ashdown G, Owen DM, Pereira PM, and Henriques R (2016). Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nature Communications 7, 12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwosch KC, Pape JK, Balzarotti F, Hoess P, Ellenberg J, Ries J, and Hell SW (2020). MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nature Methods 17, 217–224. [DOI] [PubMed] [Google Scholar]
- Gyparaki MT, Arab A, Sorokina EM, Santiago-Ruiz AN, Bohrer CH, Xiao J, and Lakadamyali M (2021). Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajj B, El Beheiry M, Izeddin I, Darzacq X, and Dahan M (2014). Accessing the third dimension in localization-based super-resolution microscopy. Phys Chem Chem Phys 16, 16340–16348. [DOI] [PubMed] [Google Scholar]
- Hao X, Allgeyer ES, Lee D-R, Antonello J, Watters K, Gerdes JA, Schroeder LK, Bottanelli F, Zhao J, Kidd P, et al. (2021a). Three-dimensional adaptive optical nanoscopy for thick specimen imaging at sub-50-nm resolution. Nature Methods 18, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao X, Allgeyer ES, Lee DR, Antonello J, Watters K, Gerdes JA, Schroeder LK, Bottanelli F, Zhao J, Kidd P, et al. (2021b). Three-dimensional adaptive optical nanoscopy for thick specimen imaging at sub-50-nm resolution. Nat Methods 18, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine J, Reuss M, Harke B, D’Este E, Sahl SJ, and Hell SW (2017). Adaptive-illumination STED nanoscopy. Proc Natl Acad Sci U S A 114, 9797–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzmann R, and Cremer C (1999). Laterally modulated excitation microscopy: improvement of resolution by using a diffraction grating, Vol 3568 (SPIE; ). [Google Scholar]
- Hell SW (2015). Nanoscopy with Focused Light (Nobel Lecture). Angewandte Chemie International Edition 54, 8054–8066. [DOI] [PubMed] [Google Scholar]
- Helm MS, Dankovich TM, Mandad S, Rammner B, Jähne S, Salimi V, Koerbs C, Leibrandt R, Urlaub H, Schikorski T, et al. (2021). A large-scale nanoscopy and biochemistry analysis of postsynaptic dendritic spines. Nature Neuroscience 24, 1151–1162. [DOI] [PubMed] [Google Scholar]
- Hess ST, Girirajan TP, and Mason MD (2006). Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 91, 4258–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DP, Shtengel G, Xu CS, Campbell KR, Freeman M, Wang L, Milkie DE, Pasolli HA, Iyer N, Bogovic JA, et al. (2020). Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M, Eggeling C, Jakobs S, and Hell SW (2005). Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proceedings of the National Academy of Sciences 102, 17565–17569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden SJ, Uphoff S, and Kapanidis AN (2011). DAOSTORM: an algorithm for high-density super-resolution microscopy. Nature Methods 8, 279–280. [DOI] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, and Zhuang X (2008). Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Sirinakis G, Allgeyer ES, Schroeder LK, Duim WC, Kromann EB, Phan T, Rivera-Molina FE, Myers JR, Irnov I, et al. (2016). Ultra-High Resolution 3D Imaging of Whole Cells. Cell 166, 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Fan J, Li L, Liu H, Wu R, Wu Y, Wei L, Mao H, Lal A, Xi P, et al. (2018). Fast, long-term, super-resolution imaging with Hessian structured illumination microscopy. Nature Biotechnology 36, 451–459. [DOI] [PubMed] [Google Scholar]
- Huff J (2015). The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nature Methods 12, i–ii. [Google Scholar]
- Huff J, Bergter A, Birkenbeil J, Kleppe I, Engelmann R, and Krzic U (2017). The new 2D Superresolution mode for ZEISS Airyscan. Nature Methods 14, 1223–1223. [Google Scholar]
- Hugelier S, Vandenberg W, Lukes T, Grussmayer KS, Eilers PHC, Dedecker P, and Ruckebusch C (2021). Smoothness correction for better SOFI imaging. Sci Rep 11, 7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingaramo M, York AG, Wawrzusin P, Milberg O, Hong A, Weigert R, Shroff H, and Patterson GH (2014). Two-photon excitation improves multifocal structured illumination microscopy in thick scattering tissue. Proceedings of the National Academy of Sciences 111, 5254–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen NA, Jansen I, Kamper M, and Jakobs S (2020). Reversibly Switchable Fluorescent Proteins for RESOLFT Nanoscopy. In (Springer International Publishing; ), pp. 241–261. [Google Scholar]
- Jouchet P, Cabriel C, Bourg N, Bardou M, Poüs C, Fort E, and Lévêque-Fort S (2021). Nanometric axial localization of single fluorescent molecules with modulated excitation. Nature Photonics 15, 297–304. [Google Scholar]
- Jungmann R, Avendano MS, Dai M, Woehrstein JB, Agasti SS, Feiger Z, Rodal A, and Yin P (2016). Quantitative super-resolution imaging with qPAINT. Nat Methods 13, 439442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann R, Avendano MS, Woehrstein JB, Dai M, Shih WM, and Yin P (2014). Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat Methods 11, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrukha EA, Jurriens D, Salas Pastene DM, and Kapitein LC (2021). Quantitative mapping of dense microtubule arrays in mammalian neurons. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khater IM, Nabi IR, and Hamarneh G (2020). A Review of Super-Resolution Single-Molecule Localization Microscopy Cluster Analysis and Quantification Methods. Patterns 1, 100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar TA, Engel E, and Hell SW (2001). Breaking Abbe’s diffraction resolution limit in fluorescence microscopy with stimulated emission depletion beams of various shapes. Physical Review E 64. [DOI] [PubMed] [Google Scholar]
- Klar TA, Jakobs S, Dyba M, Egner A, and Hell SW (2000). Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc Natl Acad Sci U S A 97, 8206–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, Ruberto FP, Nemir M, Wai T, Pedrazzini T, et al. (2021). Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439. [DOI] [PubMed] [Google Scholar]
- Ku T, Swaney J, Park JY, Albanese A, Murray E, Cho JH, Park YG, Mangena V, Chen J, and Chung K (2016). Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat Biotechnol 34, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelek M, Gyparaki MT, Beliu G, Schueder F, Griffié J, Manley S, Jungmann R, Sauer M, Lakadamyali M, and Zimmer C (2021). Single-molecule localization microscopy. Nature Reviews Methods Primers 1, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levet F, Hosy E, Kechkar A, Butler C, Beghin A, Choquet D, and Sibarita J-B (2015). SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nature Methods 12, 1065–1071. [DOI] [PubMed] [Google Scholar]
- Levet F, Julien G, Galland R, Butler C, Beghin A, Chazeau A, Hoess P, Ries J, Giannone G, and Sibarita J-B (2019). A tessellation-based colocalization analysis approach for single-molecule localization microscopy. Nature Communications 10, 2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Shao L, Chen BC, Zhang X, Zhang M, Moses B, Milkie DE, Beach JR, Hammer JA, Pasham M, et al. (2015). Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 349, aab3500–aab3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Mund M, Hoess P, Deschamps J, Matti U, Nijmeijer B, Sabinina VJ, Ellenberg J, Schoen I, and Ries J (2018). Real-time 3D single-molecule localization using experimental point spread functions. Nature Methods 15, 367–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Kipreos ET, Zhu J, Khang CH, and Kner P (2021). Subcellular three-dimensional imaging deep through multicellular thick samples by structured illumination microscopy and adaptive optics. Nature Communications 12, 3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Xue Y, Zhao W, Chen Y, Fan C, Gu L, Zhang Y, Zhang X, Sun L, Huang X, et al. (2015). Three-dimensional super-resolution protein localization correlated with vitrified cellular context. Sci Rep 5, 13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu-Walther H-W, Kielhorn M, Förster R, Jost A, Wicker K, and Heintzmann R (2015). fastSIM: a practical implementation of fast structured illumination microscopy. Methods and Applications in Fluorescence 3. [DOI] [PubMed] [Google Scholar]
- M’Saad O, and Bewersdorf J (2020). Light microscopy of proteins in their ultrastructural context. Nat Commun 11, 3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahecic D, Testa I, Griffie J, and Manley S (2019). Strategies for increasing the throughput of super-resolution microscopies. Curr Opin Chem Biol 51, 84–91. [DOI] [PubMed] [Google Scholar]
- Marsh JA, and Teichmann SA (2015). Structure, dynamics, assembly, and evolution of protein complexes. Annu Rev Biochem 84, 551–575. [DOI] [PubMed] [Google Scholar]
- Marsh RJ, Pfisterer K, Bennett P, Hirvonen LM, Gautel M, Jones GE, and Cox S (2018). Artifact-free high-density localization microscopy analysis. Nature Methods 15, 689–692. [DOI] [PubMed] [Google Scholar]
- Masullo LA, Bodén A, Pennacchietti F, Coceano G, Ratz M, and Testa I (2018). Enhanced photon collection enables four dimensional fluorescence nanoscopy of living systems. Nature Communications 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlodzianoski MJ, Cheng-Hathaway PJ, Bemiller SM, McCray TJ, Liu S, Miller DA, Lamb BT, Landreth GE, and Huang F (2018). Active PSF shaping and adaptive optics enable volumetric localization microscopy through brain sections. Nature Methods 15, 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo GCH, Ross B, Hertel F, Manna P, Yang X, Greenwald E, Booth C, Plummer AM, Tenner B, Chen Z, et al. (2017). Genetically encoded biosensors for visualizing live-cell biochemical activity at super-resolution. Nature Methods 14, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeyaert B, Vandenberg W, and Dedecker P (2020). SOFIevaluator: a strategy for the quantitative quality assessment of SOFI data. Biomed Opt Express 11, 636–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Coscia SM, Simpson CL, Ortega FE, Wait EC, Heddleston JM, Nirschl JJ, Obara CJ, Guedes-Dias P, Boecker CA, et al. (2021). Actin cables and comet tails organize mitochondrial networks in mitosis. Nature 591, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moors TE, Maat CA, Niedieker D, Mona D, Petersen D, Timmermans-Huisman E, Kole J, El-Mashtoly SF, Spycher L, Zago W, et al. (2021). The subcellular arrangement of alpha-synuclein proteoforms in the Parkinson’s disease brain as revealed by multicolor STED microscopy. Acta Neuropathologica. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neguembor MV, Martin L, Castells-Garcia A, Gomez-Garcia PA, Vicario C, Carnevali D, AlHaj Abed J, Granados A, Sebastian-Perez R, Sottile F, et al. (2021). Transcription-mediated supercoiling regulates genome folding and loop formation. Mol Cell 81, 3065–3081 e3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehme E, Weiss LE, Michaeli T, and Shechtman Y (2018). Deep-STORM: super-resolution single-molecule microscopy by deep learning. Optica 5, 458–464. [Google Scholar]
- Nicolai Katrin, Stefan, and U (2011). STED Nanoscopy of Actin Dynamics in Synapses Deep Inside Living Brain Slices. Biophysical Journal 101, 1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák T, Gajdos T, Sinkó J, Szabó G, and Erdélyi M (2017). TestSTORM: Versatile simulator software for multimodal super-resolution localization fluorescence microscopy. Scientific Reports 7, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Aristov A, Lelek M, Hao X, and Zimmer C (2018). Deep learning massively accelerates super-resolution localization microscopy. Nature Biotechnology 36, 460–468. [DOI] [PubMed] [Google Scholar]
- Pape JK, Stephan T, Balzarotti F, Büchner R, Lange F, Riedel D, Jakobs S, and Hell SW (2020). Multicolor 3D MINFLUX nanoscopy of mitochondrial MICOS proteins. Proceedings of the National Academy of Sciences 117, 20607–20614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters Y, Vandenberg W, Duwe S, Bouwens A, Lukes T, Ruckebusch C, Lasser T, and Dedecker P (2017). Correcting for photodestruction in super-resolution optical fluctuation imaging. Sci Rep 7, 10470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash K, Diederich B, Reichelt S, Heintzmann R, and Schermelleh L (2021). Super-resolution structured illumination microscopy: past, present and future. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasai B, Haber GJ, Strub M-P, Ahn R, Ciemniecki JA, Sochacki KA, and Taraska JW (2021). The nanoscale molecular morphology of docked exocytic dense-core vesicles in neuroendocrine cells. Nature Communications 12, 3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego EH, Shao L, Macklin JJ, Winoto L, Johansson GA, Kamps-Hughes N, Davidson MW, and Gustafsson MGL (2012). Nonlinear structured-illumination microscopy with a photoswitchable protein reveals cellular structures at 50-nm resolution. Proceedings of the National Academy of Sciences 109, E135–E143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond L, Ziegler J, Knapp C, Wang FC, Huser T, Ruprecht V, and Wieser S (2019). SIMPLE: Structured illumination based point localization estimator with enhanced precision. Opt Express 27, 24578–24590. [DOI] [PubMed] [Google Scholar]
- Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, and Cosma MP (2015). Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160, 1145–1158. [DOI] [PubMed] [Google Scholar]
- Ries J (2020). SMAP: a modular super-resolution microscopy analysis platform for SMLM data. Nature Methods 17, 870–872. [DOI] [PubMed] [Google Scholar]
- Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, and Prosser BL (2016). Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossy J, Owen DM, Williamson DJ, Yang Z, and Gaus K (2013). Conformational states of the kinase Lck regulate clustering in early T cell signaling. Nat Immunol 14, 82–89. [DOI] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nature Reviews Genetics 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin-Delanchy P, Burn GL, Griffié J, Williamson DJ, Heard NA, Cope AP, and Owen DM (2015). Bayesian cluster identification in single-molecule localization microscopy data. Nature Methods 12, 1072–1076. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, and Zhuang X (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage D, Pham T-A, Babcock H, Lukes T, Pengo T, Chao J, Velmurugan R, Herbert A, Agrawal A, Colabrese S, et al. (2019). Super-resolution fight club: assessment of 2D and 3D single-molecule localization microscopy software. Nature Methods 16, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmeyer A, Lachetta M, Sandmeyer H, Hübner W, Huser T, and Müller M (2021). Cost-Effective Live Cell Structured Illumination Microscopy with Video-Rate Imaging. ACS Photonics 8, 1639–1648. [Google Scholar]
- Sapoznik E, Chang BJ, Huh J, Ju RJ, Azarova EV, Pohlkamp T, Welf ES, Broadbent D, Carisey AF, Stehbens SJ, et al. (2020). A versatile oblique plane microscope for large-scale and high-resolution imaging of subcellular dynamics. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L, Ferrand A, Huser T, Eggeling C, Sauer M, Biehlmaier O, and Drummen GPC (2019). Super-resolution microscopy demystified. Nat Cell Biol 21, 72–84. [DOI] [PubMed] [Google Scholar]
- Schlichthaerle T, Strauss MT, Schueder F, Auer A, Nijmeijer B, Kueblbeck M, Jimenez Sabinina V, Thevathasan JV, Ries J, Ellenberg J, et al. (2019). Direct Visualization of Single Nuclear Pore Complex Proteins Using Genetically-Encoded Probes for DNA-PAINT. Angew Chem Int Ed Engl 58, 13004–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Weihs T, Wurm CA, Jansen I, Rehman J, Sahl SJ, and Hell SW (2021). MINFLUX nanometer-scale 3D imaging and microsecond-range tracking on a common fluorescence microscope. Nature Communications 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Wurm CA, Jakobs S, Engelhardt J, Egner A, and Hell SW (2008). Spherical nanosized focal spot unravels the interior of cells. Nature Methods 5, 539–544. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnorrenberg S, Grotjohann T, Vorbrüggen G, Herzig A, Hell SW, and Jakobs S (2016). In vivo super-resolution RESOLFT microscopy of Drosophila melanogaster. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder LK, Barentine AES, Merta H, Schweighofer S, Zhang Y, Baddeley D, Bewersdorf J, and Bahmanyar S (2018). Dynamic nanoscale morphology of the ER surveyed by STED microscopy. Journal of Cell Biology 218, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueder F, Stein J, Stehr F, Auer A, Sperl B, Strauss MT, Schwille P, and Jungmann R (2019). An order of magnitude faster DNA-PAINT imaging by optimized sequence design and buffer conditions. Nat Methods 16, 1101–1104. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, and Lippincott-Schwartz J (2011). Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nature Methods 8, 969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Li Q, Dai Z, Tran AA, Feng S, Ramirez AD, Lin Z, Wang X, Chow TT, Chen J, et al. (2021). Label-retention expansion microscopy. J Cell Biol 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtengel G, Galbraith JA, Galbraith CG, Lippincott-Schwartz J, Gillette JM, Manley S, Sougrat R, Waterman CM, Kanchanawong P, Davidson MW, et al. (2009). Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci U S A 106, 3125–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal YM, Zhou R, and Zhuang X (2018). Visualizing and discovering cellular structures with super-resolution microscopy. Science 361, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CS, Joseph N, Rieger B, and Lidke KA (2010). Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nature Methods 7, 373–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CS, Slotman JA, Schermelleh L, Chakrova N, Hari S, Vos Y, Hagen CW, Müller M, Van Cappellen W, Houtsmuller AB, et al. (2021). Structured illumination microscopy with noise-controlled image reconstructions. Nature Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochacki KA, Dickey AM, Strub M-P, and Taraska JW (2017). Endocytic proteins are partitioned at the edge of the clathrin lattice in mammalian cells. Nature Cell Biology 19, 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn C, Grimm JB, Lavis LD, Lampe M, and Heilemann M (2019). Whole-Cell, 3D, and Multicolor STED Imaging with Exchangeable Fluorophores. Nano Letters 19, 500–505. [DOI] [PubMed] [Google Scholar]
- Speiser A, Müller L-R, Hoess P, Matti U, Obara CJ, Legant WR, Kreshuk A, Macke JH, Ries J, and Turaga SC (2021). Deep learning enables fast and dense single-molecule localization with high accuracy. Nature Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt T, Engler A, Rittweger E, Harke B, Engelhardt J, and Hell SW (2011). Far-field optical nanoscopy with reduced number of state transition cycles. Opt Express 19, 5644–5657. [DOI] [PubMed] [Google Scholar]
- Stefko M, Ottino B, Douglass KM, and Manley S (2018). Autonomous illumination control for localization microscopy. Opt Express 26, 30882–30900. [DOI] [PubMed] [Google Scholar]
- Strauss S, and Jungmann R (2020). Up to 100-fold speed-up and multiplexing in optimized DNA-PAINT. Nat Methods 17, 789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, and Satija R (2019). Integrative single-cell analysis. Nature Reviews Genetics 20, 257–272. [DOI] [PubMed] [Google Scholar]
- Sun DE, Fan X, Shi Y, Zhang H, Huang Z, Cheng B, Tang Q, Li W, Zhu Y, Bai J, et al. (2021). Click-ExM enables expansion microscopy for all biomolecules. Nat Methods 18, 107–113. [DOI] [PubMed] [Google Scholar]
- Szabo Q, Jost D, Chang JM, Cattoni DI, Papadopoulos GL, Bonev B, Sexton T, Gurgo J, Jacquier C, Nollmann M, et al. (2018). TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci Adv 4, eaar8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevathasan JV, Kahnwald M, Cieśliński K, Hoess P, Peneti SK, Reitberger M, Heid D, Kasuba KC, Hoerner SJ, Li Y, et al. (2019). Nuclear pores as versatile reference standards for quantitative superresolution microscopy. Nature Methods 16, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillberg PW, Chen F, Piatkevich KD, Zhao Y, Yu CC, English BP, Gao L, Martorell A, Suk HJ, Yoshida F, et al. (2016). Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat Biotechnol 34, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari DK, Arai Y, Yamanaka M, Matsuda T, Agetsuma M, Nakano M, Fujita K, and Nagai T (2015). A fast- and positively photoswitchable fluorescent protein for ultralow-laser-power RESOLFT nanoscopy. Nature Methods 12, 515–518. [DOI] [PubMed] [Google Scholar]
- Tønnesen J, Inavalli VVGK, and Nägerl UV (2018). Super-Resolution Imaging of the Extracellular Space in Living Brain Tissue. Cell 172, 1108–1121.e1115. [DOI] [PubMed] [Google Scholar]
- Truckenbrodt S, Maidorn M, Crzan D, Wildhagen H, Kabatas S, and Rizzoli SO (2018). X10 expansion microscopy enables 25-nm resolution on conventional microscopes. EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban NT, Willig KI, Hell SW, and Nagerl UV (2011). STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys J 101, 1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli J, Garcia-Burgos A, Rooney LM, Vale de Melo EOB, Duncan RR, and Rickman C (2021). Seeing beyond the limit: A guide to choosing the right super-resolution microscopy technique. J Biol Chem 297, 100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynde R, Vandenberg W, Hugelier S, Bouwens A, Hofkens J, Müller M, and Dedecker P (2021). Self-contained and modular structured illumination microscope. Biomed Opt Express 12, 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg W, Duwe S, Leutenegger M, Moeyaert B, Krajnik B, Lasser T, and Dedecker P (2016). Model-free uncertainty estimation in stochastical optical fluctuation imaging (SOFI) leads to a doubled temporal resolution. Biomed Opt Express 7, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco MGM, Zhang M, Antonello J, Yuan P, Allgeyer ES, May D, M’Saad O, Kidd P, Barentine AES, Greco V, et al. (2021). 3D super-resolution deep-tissue imaging in living mice. Optica 8, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataramani V, Herrmannsdörfer F, Heilemann M, and Kuner T (2016). SuReSim: simulating localization microscopy experiments from ground truth models. Nature Methods 13, 319–321. [DOI] [PubMed] [Google Scholar]
- Wade OK, Woehrstein JB, Nickels PC, Strauss S, Stehr F, Stein J, Schueder F, Strauss MT, Ganji M, Schnitzbauer J, et al. (2019). 124-Color Super-resolution Imaging by Engineering DNA-PAINT Blinking Kinetics. Nano Lett 19, 2641–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldchen F, Schlegel J, Gotz R, Luciano M, Schnermann M, Doose S, and Sauer M (2020). Whole-cell imaging of plasma membrane receptors by 3D lattice light-sheet dSTORM. Nat Commun 11, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Rivenson Y, Jin Y, Wei Z, Gao R, Günaydın H, Bentolila LA, Kural C, and Ozcan A (2019). Deep learning enables cross-modality super-resolution in fluorescence microscopy. Nature Methods 16, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yu Z, Cahoon CK, Parmely T, Thomas N, Unruh JR, Slaughter BD, and Hawley RS (2018). Combined expansion microscopy with structured illumination microscopy for analyzing protein complexes. Nat Protoc 13, 1869–1895. [DOI] [PubMed] [Google Scholar]
- Wassie AT, Zhao Y, and Boyden ES (2019). Expansion microscopy: principles and uses in biological research. Nat Methods 16, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Leutenegger M, Stoldt S, Jakobs S, Mihaila TS, Butkevich AN, and Hell SW (2021). MINSTED fluorescence localization and nanoscopy. Nature Photonics 15, 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen G, Vanheusden M, Acke A, Valli D, Neely RK, Leen V, and Hofkens J (2020). Evaluation of Direct Grafting Strategies via Trivalent Anchoring for Enabling Lipid Membrane and Cytoskeleton Staining in Expansion Microscopy. ACS Nano 14, 7860–7867. [DOI] [PubMed] [Google Scholar]
- Willig KI, Harke B, Medda R, and Hell SW (2007). STED microscopy with continuous wave beams. Nature Methods 4, 915–918. [DOI] [PubMed] [Google Scholar]
- Willig KI, Wegner W, Müller A, Clavet-Fournier V, and Steffens H (2021). Multi-label in vivo STED microscopy by parallelized switching of reversibly switchable fluorescent proteins. Cell Reports 35, 109192. [DOI] [PubMed] [Google Scholar]
- Winter PW, York AG, Nogare DD, Ingaramo M, Christensen R, Chitnis A, Patterson GH, and Shroff H (2014). Two-photon instant structured illumination microscopy improves the depth penetration of super-resolution imaging in thick scattering samples. Optica 1, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Hammer JA (2021). ZEISS Airyscan: Optimizing Usage for Fast, Gentle, Super-Resolution Imaging. In (Springer; US: ), pp. 111–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, and Shroff H (2018). Faster, sharper, and deeper: structured illumination microscopy for biological imaging. Nature Methods 15, 1011–1019. [DOI] [PubMed] [Google Scholar]
- Xie L, Dong P, Chen X, Hsieh TS, Banala S, De Marzio M, English BP, Qi Y, Jung SK, Kieffer-Kwon KR, et al. (2020). 3D ATAC-PALM: super-resolution imaging of the accessible genome. Nat Methods 17, 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Babcock HP, and Zhuang X (2012). Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods 9, 185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G, and Zhuang X (2013). Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York AG, Chandris P, Nogare DD, Head J, Wawrzusin P, Fischer RS, Chitnis A, and Shroff H (2013). Instant super-resolution imaging in live cells and embryos via analog image processing. Nature Methods 10, 1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York AG, Ghitani A, Vaziri A, Davidson MW, and Shroff H (2011). Confined activation and subdiffractive localization enables whole-cell PALM with genetically expressed probes. Nature Methods 8, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanacchi FC, Manzo C, Alvarez AS, Derr ND, Garcia-Parajo MF, and Lakadamyali M (2017). A DNA origami platform for quantifying protein copy number in super-resolution. Nature Methods 14, 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen X, Zeng Z, Zhang M, Sun Y, Xi P, Peng J, and Xu P (2015). Development of a reversibly switchable fluorescent protein for super-resolution optical fluctuation imaging (SOFI). ACS Nano 9, 2659–2667. [DOI] [PubMed] [Google Scholar]
- Zheng Q, and Lavis LD (2017). Development of photostable fluorophores for molecular imaging. Current Opinion in Chemical Biology 39, 32–38. [DOI] [PubMed] [Google Scholar]
- Zhu L, Zhang W, Elnatan D, and Huang B (2012). Faster STORM using compressed sensing. Nature Methods 9, 721–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwettler FU, Reinhard S, Gambarotto D, Bell TDM, Hamel V, Guichard P, and Sauer M (2020). Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM). Nat Commun 11, 3388. [DOI] [PMC free article] [PubMed] [Google Scholar]