

Abstract
Ulcerative colitis (UC) is driven by disruptions in host-microbiota homeostasis, however current treatments exclusively target host inflammatory pathways. To understand how host-microbiota interactions become disrupted in UC, we collected and analyzed six fecal or serum based –omic datasets (metaproteomic, metabolomic, metagenomic, metapeptidomic, and amplicon sequencing profiles of fecal samples and proteomic profiles of serum samples) from 40 UC patients at a single inflammatory bowel disease centre, as well as various clinical, endoscopic, and histologic measures of disease activity. A validation cohort of 210 samples (73 UC, 117 Crohn’s disease (CD), 20 healthy controls) was collected and analyzed separately and independently. Data integration across both cohorts showed that a subset of the clinically active UC patients had an over-abundance of proteases that originated from the bacterium, Bacteroides vulgatus. To test whether B. vulgatus proteases contribute to UC disease activity, we first profiled B. vulgatus proteases found in patients and bacterial cultures. Use of a broad-spectrum protease inhibitor improved B. vulgatus-induced barrier dysfunction in vitro, and prevented colitis in B. vulgatus monocolonized, IL-10 deficient mice. Furthermore, transplantation of feces from UC patients with a high abundance of B. vulgatus proteases into germ-free mice induced colitis dependent on protease activity. These results, stemming from a multi-omics approach, improve understanding of functional microbiota alterations that drive UC and provides a resource for identifying other pathways that could be inhibited as a strategy to treat this disease.
Introduction
Ulcerative colitis (UC), an inflammatory bowel disease (IBD), is characterized by chronic inflammation of the colon, with severity and persistence of mucosal inflammation being associated with morbidity and mortality1. Non-specific immunosuppressive agents targeting the host, such as steroids, thiopurines, and/or biologics, are used to offset the natural history of disease in patients with moderate-severe inflammation. These therapies are, however, associated with significant risks and often ineffective in adequately managing disease2. There are numerous microbiome studies based on genomic techniques that have identified associations with UC, highlighting microbial dysbiosis, and temporal shifts in composition related to UC status3–6. While recent efforts extended profiling of microbiota in UC beyond genomics7,8, it remains poorly understood if these shifts are causal or associative in nature9,10. Our group has previously investigated the integration of metagenomics and metaproteomics 11 to help elucidate host-microbiota interactions through contrasting gene profiles with community-level proteomes through mass spectrometry (MS)12. Adoption of metaproteomics has historically lagged behind other -omic technologies 13, although our efforts have indicated that utilizing developing methods in the field such as sample multiplexing and deep fractionation approaches14, hold potential for uncovering important findings11,15. Here we investigated our metagenomic-metaproteomic approach alongside conventional 16S rRNA gene amplicon sequencing, fecal metabolomics and serum proteomics methods in a large cross-sectional cohort of IBD patients seeking host-microbiota interactions that could be exploited for therapy8,10,16–18.
After broadly analyzing our data, we focus on one finding with multi-omics evidence, that proteases from some Bacteroides spp. could be involved in UC pathogenesis. In particular, our meta-omics data highlighted Bacteroides vulgatus proteases as potential targets for treating UC. B.vulgatus proteases have been previously postulated as therapeutic targets in IBD, although clear understanding of the contribution and identity of the proteases has been less investigated19. Our multi-omics results expand upon and bring more clarity to the recent report that B. vulgatus was correlated with stool protease activity in a small population of patients that were later diagnosed with UC20. Together, our results provide evidence that B. vulgatus protease inhibition may be a therapeutic approach for preventing or treating UC.
Results
Study design
To initiate our study, patient samples from a convenience biobank at a single academic IBD center (University of California, San Diego) who underwent extensive phenotyping with clinical disease activity indices and blinded assessments of endoscopic and histologic severity were collected and analyzed using a multi-omics approach 21–23 (Supplementary Table 1). Our resulting data represents one of the most extensive multi-omic resources on IBD patients to date utilizing patient matched serum and fecal samples for metagenomic and 16S rRNA gene amplicon sequencing, metabolomic, metapeptidomic, serum proteomic, and metaproteomic analyses (Extended Data Fig. 1). An initial discovery group of 40 UC serum and fecal samples was collected and followed-up by a second group of 210 fecal samples which included 73 UC, 117 CD (roughly split by ileal, ileocolonic, and colonic subtypes), and samples from 20 volunteers without IBD. Our previously established integrated metagenomic-metaproteomic approach of shared database assembly and quantification was used for direct comparisons between microbial genes and proteins11. Application of our multiplexing metaproteomic methods provided increased protein identifications and a greater than 10-fold increase in proteins quantified per sample in comparison to conventional label-free metaproteomic methodology. We demonstrate this important technical advantage by comparison with data downloaded from the Human Microbiome Project’s IBD multi-omics database which notably represents a smaller patient population than the cohorts of this study7 (Extended Data Fig. 2).
Meta-omic associations to IBD severity
Despite our cohort representing a diverse group of patients (Supplementary Table 1), many clinical severity metrics showed a high degree of correlation (Fig. 1a). Given the overlap of severity metrics, a representative metric including patient symptoms was chosen, partial Mayo for UC, alongside the patient reported outcomes (stool frequency, abdominal pain, general well-being) from the Crohn’s Disease Activity Index (CDAI) for CD24. Disease severity significantly correlated with both alpha-diversity and the beta-diversity in all meta-omics collected (Fig 1b–c, Supplementary Table 2, Supplementary Fig. 1a–f). CD subtypes and the two separately processed UC cohorts displayed unique microbiota compositions distinct from healthy controls (Fig 2a, Supplementary Fig. 2). We observed stronger correlations between the distributions of data in the fecal based -omics than the serum proteome (Fig. 2b, Supplementary Fig. 1g–h), and that the metaproteome allowed for the strongest prediction of UC activity while closely followed by the combined data and the metabolome (Fig. 2c, Supplementary Table 3). Unlike UC, an influential feature in CD patient microbiomes was the dominance of a member of the Enterobacteriaceae family (Extended Data Fig. 3). In patients with active UC, we also observed an increase in human proteins, and classes of metabolites such as phosphocholines correlated with activity (Fig. 2d–e).
Figure 1. Multi-omic diversity correlates with IBD disease activity.
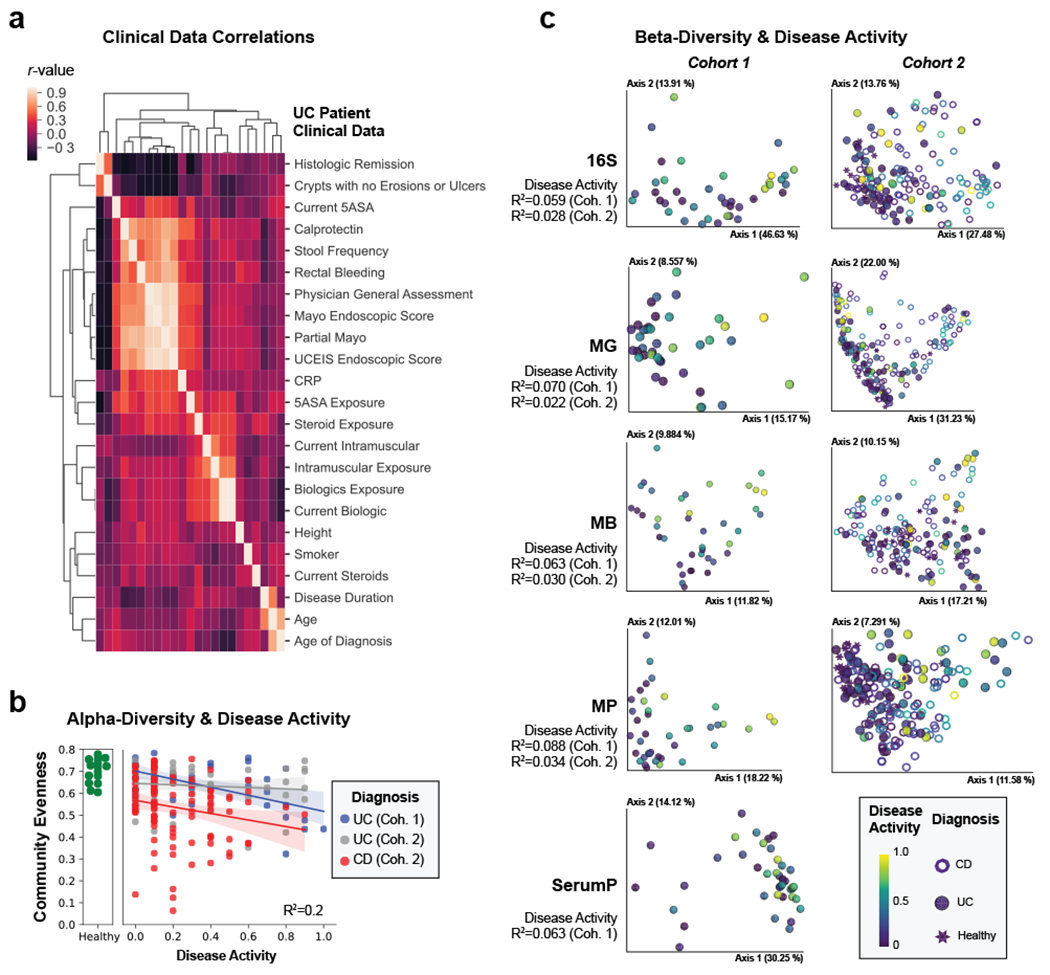
a, Heatmap of the correlation between clinical data. Hierarchical clustering was performed on spearman correlation values between each clinical metric for UC patients identifying groups of closely related clinical measurements. Each metric is represented in the same order on x- and y-axes and only y-axis labels are shown. b, Alpha-diversity decreases with active IBD. Pielou evenness based on 16S data is plotted for each patient with linear regression best-fit lines and 95% confidence intervals per patient group. An R2 value is indicated based on the disease activity, diagnosis and their interaction. c, Beta-diversity correlates with active IBD. Each collected -omic dataset is displayed by a principal coordinate analysis showing the first two axes. Each sample is colored by the disease activity state and has a shape corresponding to diagnosis. Adonis R2 values are shown to demonstrate the effect size of disease activity when accounting for disease activity, diagnosis and their interaction. Distance matrices best separating disease activity are displayed. Distance matrices shown are weighted UniFrac for each dataset other than proteomic datasets, which use the Bray-Curtis distance metric, and the metagenome of UC cohort 1 which uses unweighted UniFrac.
Figure 2. Multi-omic analysis of IBD disease activity.
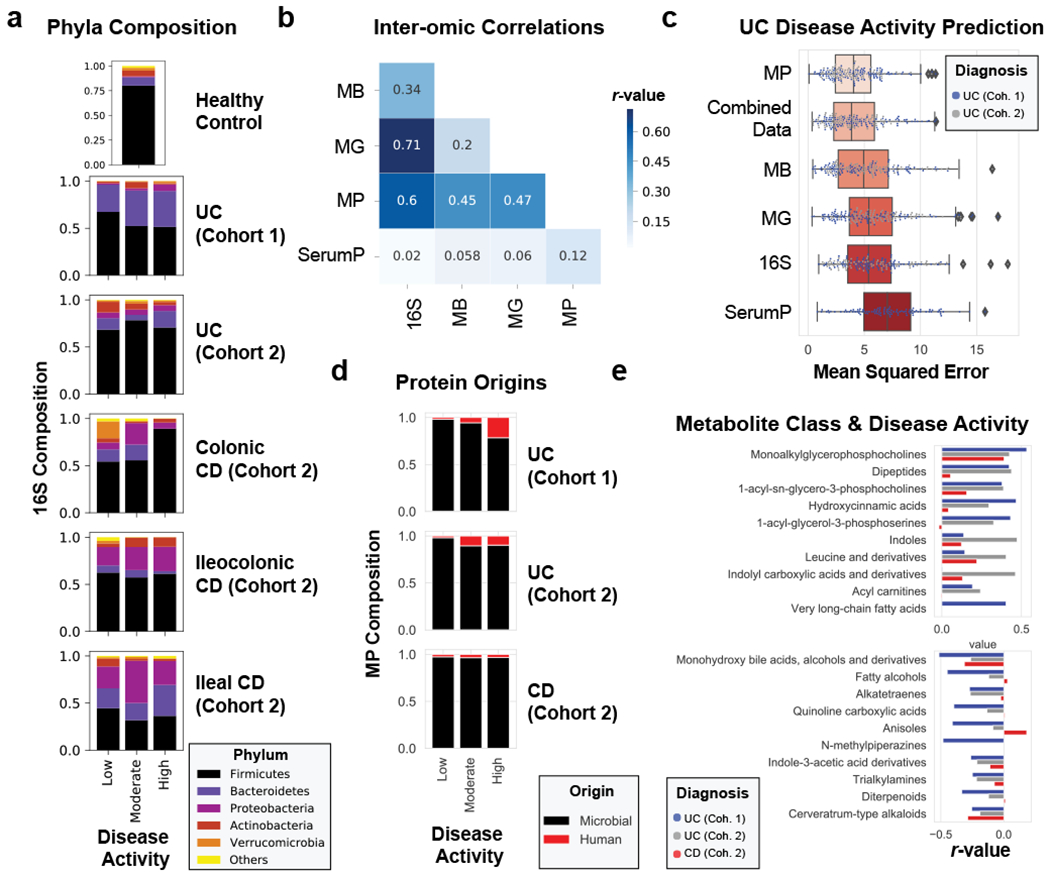
a, 16S phyla composition by disease activity states. The average phyla compositions of groups of patient samples are shown in bar plots. Barplots represent sample sizes of n=18, 12, 10 for UC Cohort 1; n=34, 9, 13 for UC Cohort 2; n=19, 8, 1 for Colonic CD; n=22, 7, 3 for Ileocolonic CD; n=19, 4, 2 for Ileal CD (each ordered low, moderate, high activity respectively); n=15 samples for healthy controls. b, Data type correlations. Pearson correlations between data types are displayed in a heat map. The Bray-Curtis distance metric was used for all data types and correlations were performed on distance matrices through Mantel’s test. c, Evaluating meta –omic performance in predicting UC disease activity. The mean squared error from n=100 iterations of random forest analyses on each UC cohort trained to predict the partial Mayo disease activity (ranging from 0-9) are displayed in boxplots ordered from the strongest predictive capability (metaproteome) to the least predictive capability (serum proteome). d, Metaproteome composition by disease activity states. The relative abundances of human and microbial proteins were averaged by disease activity states and plotted by different patient categories. Barplots represent sample sizes of n=18, 12, 10 for UC Cohort 1; n=38, 11, 14 for UC Cohort 2; n=66, 31, 9 for CD (each ordered low, moderate, high activity respectively). e, Top metabolite classes correlated with UC disease activity. Metabolite abundances summed by chemical class were averaged and linear regressions were performed to disease activity. The r-values of the top 10 positively and negatively correlated classes of chemicals are plotted by diagnosis and cohort, and displayed in order of the summed r-values from UC cohorts. Boxplots in (c) are defined by the median, quartiles and 1.5x inter-quartile range.
Utilizing direct comparison of genes and proteins of the microbiota11, linear regression identified the most correlated features to clinical disease severity (r > 0.3). Comparing genera annotations of positive and negative associations identified that Bacteroides proteins represented 40-60% of proteins positively correlated to UC disease activity (Fig. 3a, Extended Data Fig. 4). This association between disease activity and Bacteroides was confirmed across both UC cohorts, and identified as unique to UC with CD subtypes each presenting unique profiles of disease-correlated proteins (Extended Data Fig. 5). The metagenome largely reflected the direction and magnitude of the genera level bias of the associations identified in the metaproteome, however, Bacteroides genes showed a weaker relationship to high disease severity in UC relative to the metaproteome (Fig. 3b, Extended Data Fig. 4). A functional analysis of proteins associated with disease activity from Bacteroides displayed an increased representation of enzyme families, and more specifically, proteases (Fig. 3c). B. vulgatus and B. dorei, two closely related species prevalent among healthy adults25,26, contributed ~40% of all Bacteroides reads in the metagenome of UC patients (Fig. 3d). We next analyzed the correlation to UC severity of the 119 distinct enzymes and proteases derived from 59 species of Bacteroides. Serine proteases, including six dipeptidases, were among the commonly correlated proteases to UC activity from prevalent Bacteroides species (Fig. 3e). Applying an outlier approach comparing metagenomic and metaproteomic data we identified patient samples with over- or under-production of B. vulgatus and B. dorei proteases, and observed that patients containing increased proteases had significantly higher clinical severity and endoscopic activity in comparison to the decreased proteases group and the typical UC patient sample (Fig. 3f, Extended Data Fig. 6a). From a histological perspective, only 18.8% of patients categorized as “overproducers” were in histological remission, while 38.5% of patients categorized as “underproducers” and 45% of all other patients were in histological remission (Extended Data Fig. 6b). As some of the correlated proteases included serine and metalloproteases, classes of proteases that largely function in the extracellular space19, we hypothesized that these proteins may play roles in extracellular proteolysis and exacerbation of disease activity.
Figure 3. Integrated metagenomic-metaproteomic analyses reveal Bacteroides proteases distinguishing a subset of active UC patients.
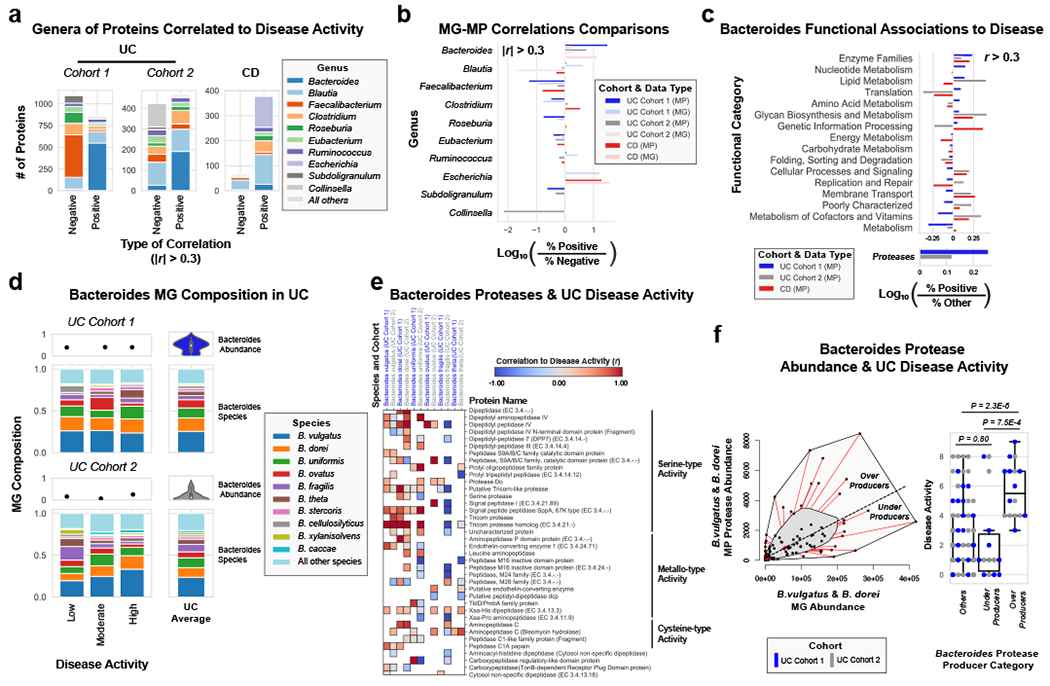
a, Taxonomic biases among proteins correlated to disease activity. Linear regressions against disease activity were performed for each protein quantified and the taxonomic origins of all highly associated proteins (Pearson’s r > 0.3 or r < −0.3) are plotted per patient cohort. b, Comparison of biases in the taxonomic origins of highly associated microbial open-reading frames at the MG or MP level. Linear regressions were performed as in (a), and the percent representation of taxa in positive correlations (r > 0.3) and negative correlations (r < −0.3) are plotted by Log10 transformation. c, Functional shifts in Bacteroides during active IBD. The Bacteroides proteins associated with disease activity (r > 0.3) from (a) were compared to remaining identified Bacteroides proteins to identify putative functional shifts related to UC disease activity. d, Species-level investigation of Bacteroides in MG of UC patients. Bacteroides species composition plots are shown for categories of UC disease activity, as well as the average within each cohort. Above each composition plot are dot plots indicating the average abundance of Bacteroides reads in the MG, or a violin plot showing the kernel density estimate of the general distribution in the UC cohort. Data was compiled from sample sizes of n=18, 12, 10 for UC Cohort 1 and n=38, 13, 13 for UC Cohort 2 (each ordered low, moderate, high activity respectively). e, Correlation of Bacteroides proteases and enzymes to UC disease activity. The species level annotation of proteases identified in different Bacteroides species was compared in a heatmap showing the correlations of each enzyme to UC activity per species. f, Patients with Bacteroides protease overproduction correlates with increased disease activity. An outlier approach comparing B. vulgatus and B. dorei metagenomic abundance to the summed protein abundances from B. vulgatus and B. dorei proteases was taken to identify groups of UC patients with higher or lower than metagenomically expected protease presence. A bagplot is shown with a best-fit line and over or under-producer status was determined by outlier status above or below the best-fit line. The disease activity of overproducers, underproducers, and other UC patients are individually plotted over boxplots. Two-tailed, t-test p-values are displayed above the boxplots. Sample sizes include n=16, 14 and 77 for Over Producers, Under Producers and Others respectively. Boxplots are defined by the median, quartiles and 1.5x inter-quartile range.
Assessing proteolysis in UC patient -omics and Bacteroides supernatant
Metabolomic and metapeptidomic analyses corroborated the importance of proteolysis in UC patients. This was initially observed through the identification of dipeptide abundance being the class of metabolite with the second highest correlation to UC disease activity (Fig. 2h, Fig. 4a). Dipeptides and oligopeptides were the two most common chemical classes among the metabolites positively correlated to disease activity (r > 0.3) accounting for 44% and 5.8% of the total positive correlations. To further analyze oligopeptides, a de novo sequencing approach was taken to analyze the metapeptidome (the peptides from complex multi-species samples). Results identified more peptide fragments within high severity UC fecal samples and patients with overproduction of Bacteroides proteases (Fig. 4b, Extended Data Fig. 7). This data also revealed the identity of peptide fragments from human proteins, including structural proteins from collagens and mucins (Fig. 4c). These human proteins represent potential targets of proteases in UC. The known cleavage patterns of Neutrophil elastase and Proteinase-327 were not strong signals among termini of identified peptides (Extended Data Fig. 7b), indicating that neutrophil proteases were likely not primary contributors to the proteolysis in patients. Network analysis of host proteins correlated to disease activity from the fecal and serum of UC patients and highlighted regulation of proteolysis as a common function (Supplementary Fig. 3).
Figure 4. Assessing proteolysis in UC patients and Bacteroides supernatant.
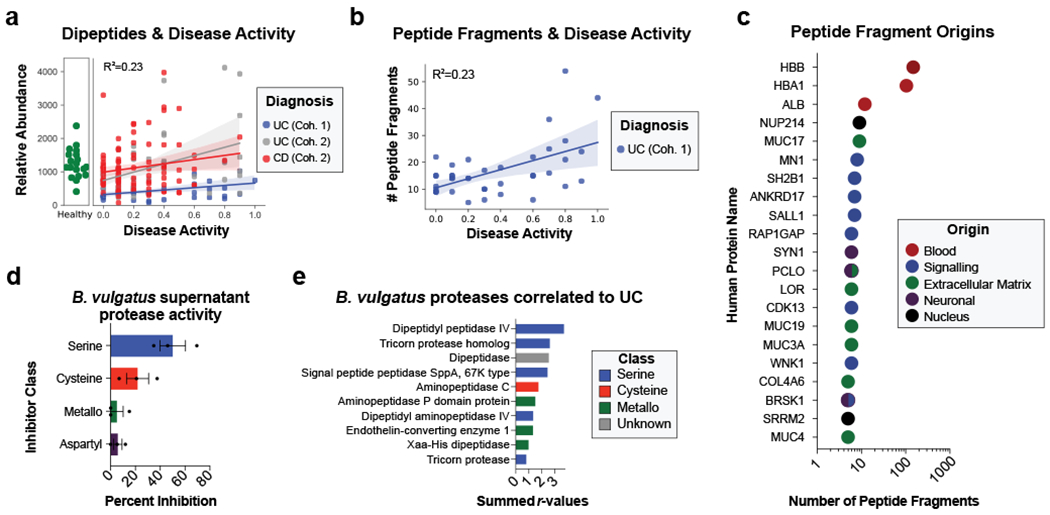
a, Abundances of dipeptides increases with disease activity. The average relative abundance of metabolomic features annotated as dipeptides per sample is plotted according to disease activity with linear regression best-fit lines and 95% confidence intervals shown per patient cohort. b, Peptide fragments are more abundant during active UC. The number of peptides identified through a de-novo peptidomic workflow is plotted alongside UC disease activity. The linear regression best-fit line with a 95% confidence interval is shown for UC cohort 1. c, The number of peptide fragments from human proteins indicates potential targets of UC proteolysis. The gene symbol for the human proteins with the most number of short peptides present are shown on the y-axis and the quantity of peptides is shown on a log10 transformed x-axis. The proteins are colored by common categories of the observed proteins. d, Class of protease activity in B. vulgatus supernatant. Concentrated supernatant from overnight cultures of B. vulgatus was subjected to a protease activity assay in the presence of different classes of protease inhibitors. Vehicle controls were used to determine the percent inhibition from each inhibitor and the mean +/− SEM from n=11 wells per-condition from n=3 independent experiments are displayed. Protease inhibitors included 10 mM AEBSF (Serine), 100 μM E-64 (Cysteine), 2.5 mM GM6001 (Metallo) and 180 μM Pepstatin A (Aspartyl). e, Ranking of B. vulgatus proteases by summed correlations to UC disease activity. The correlation values (r) between UC disease activity and B. vulgatus and B. dorei proteases were summed. The sums from the top-10 ranked proteases are shown with the colors of each bar representing protease class.
To characterize the protease activity present in the Bacteroides species we identified as related to UC disease activity, bacterial cultures were grown and the supernatant was analyzed through proteomics and protease activity assays. Inhibition of serine proteases proved to be the most effective method of disrupting proteolysis from B. vulgatus supernatant (Fig. 4d). Proteomic analysis identified that serine-type activity was the most common class of enzymatic function from proteins in the supernatant of B. vulgatus, B. dorei, and B. thetaiotaomicron (B. theta) (Supplementary Fig. 4a). Identified proteases were next ranked by increased abundance in the supernatant of B. vulgatus compared to B. theta (Supplementary Fig. 4b) and ranked by the summed correlation values in UC cohorts (Fig. 4e). Further, a comparison of the identities of Bacteroides proteases correlated to UC patients and those found in the Bacteroides supernatant has been conducted (Supplementary Fig. 4c, Supplementary Table 4).
Protease inhibition prevents B. vulgatus induced colonic epithelial damage in vitro and in vivo
We next tested the six most abundant Bacteroides species in UC for effects on the intestinal barrier using Caco-2 epithelial monolayers. Our results showed a significant decrease in transepithelial electrical resistance (TEER) after 38 hours of incubation with the two most abundant Bacteroides species, Bacteroides vulgatus and Bacteroides dorei, while the other species increased TEER (Extended Data Fig. 8a). Although both B. vulgatus and B. dorei had similar impacts on TEER and both had numerous proteases correlated to UC activity, we chose to focus our experiments on B. vulgatus as it is a more abundant member of both the UC gut microbiota and the healthy gut microbiota. We next assessed the contribution of protease activity to the disruption of epithelial permeability by adding a protease inhibitor cocktail specific to serine and cysteine proteases. We found that protease inhibition significantly increased TEER at both 22- and 38-hours post-inoculation with B. vulgatus (Adjusted p-value < 0.0001, eta2 = 0.64, Fig. 5a–b). The phenotype was not due to effects on bacterial growth or viability, as colony-forming units (CFUs) were not significantly different between the B. vulgatus wells treated with or without the protease inhibitor cocktail (Adjusted p-value = 0.98, Fig. 5c, Extended Data Fig. 8b). We further tested whether the supernatant from B. vulgatus in log-phase growth had a similar impact on TEER (Extended Data Fig. 8c). No significant effect was found, indicating that either the proteases of interest are membrane-bound or that a stressor (e.g., host-microbe interaction or nutrient deprivation) is necessary for protease secretion.
Figure 5. Protease inhibition protects from Bacteroides vulgatus and fecal transplant induced pathology in vitro and in vivo.
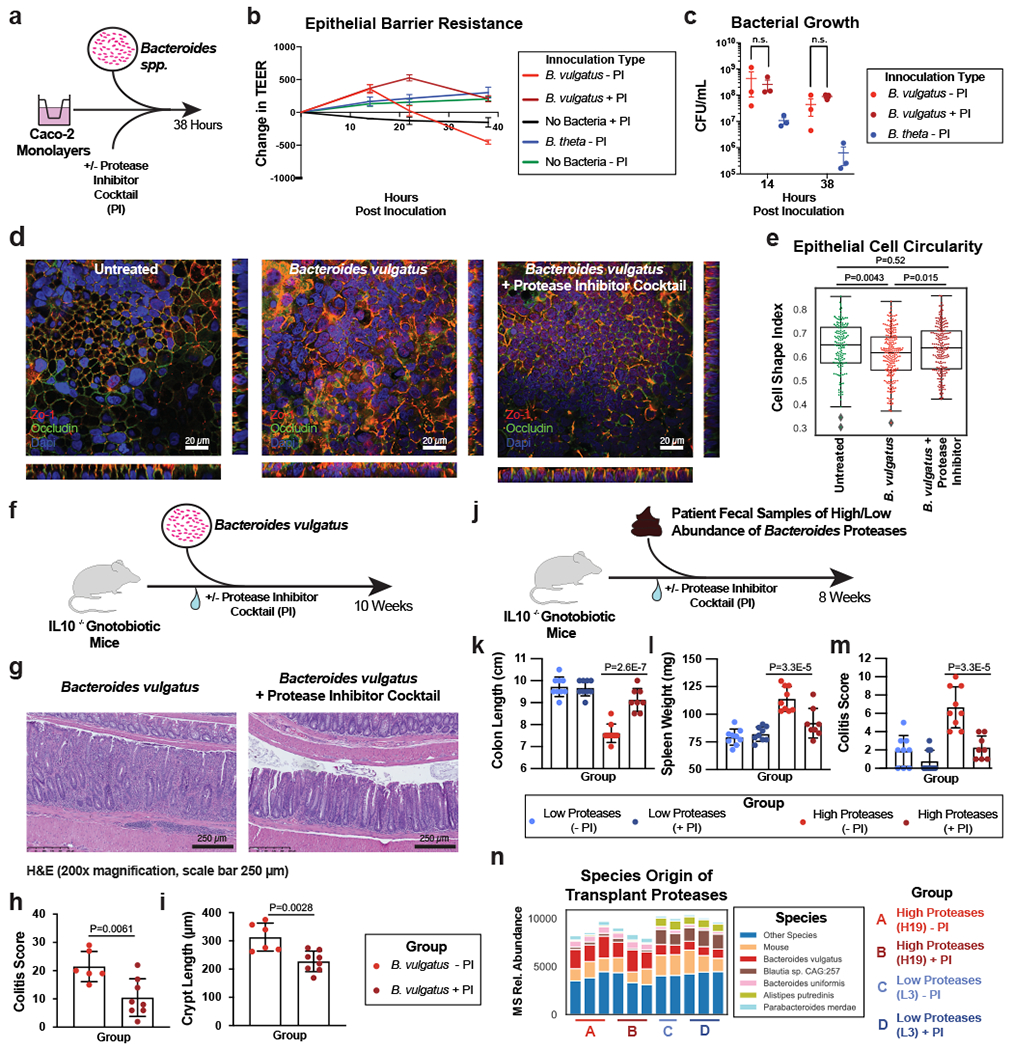
a, Schematic describing the in vitro studies using Caco-2 cell monolayers and Bacteroides spp. b, Protease inhibition significantly restores the Caco-2 epithelial barrier when co-cultured with B. vulgatus. A timeseries of the change in transepithelial electrical resistance (TEER) is plotted with the mean and standard error of the mean (SEM). c, Protease inhibitor cocktail does not significantly influence the number of CFUs during Caco-2 co-culturing with B. vulgatus. Plotted are the mean CFUs +/− SEM from each independent experiment. Two-way ANOVA adjusted for multiple comparisons performed at 14 hours (P=0.69) and 38 hours (P=0.97). Data from (b, c) derived from n=3 independent experiments containing n=3 biological replicates per condition. d, Representative images from confocal microscopy of the transwell experiments. A representative image from untreated, B. vulgatus, and B. vulgatus with a protease inhibitor cocktail are shown. Immunofluorescence of tight junction proteins, Zo-1 and Occludin along with dapi are shown. Below and to the right of each image are the XZ and YZ slices. Scale bars are 20 μm. e, Quantification of cell circularity in the images from panel d. Two-tailed t-test p-values are shown between groups. Statistics were derived from n=151, 221, 198 untreated, B. vulgatus - PI and B. vulgatus + PI cells examined over 1 independent experiment. Boxplots are defined by the median, quartiles and 1.5x inter-quartile range. f, Experimental design of monocolonized IL10−/− mouse study. Mice were inoculated with B. vulgatus. During 10-weeks of colonization, a protease inhibitor cocktail was continuously administered through the drinking water of B. vulgatus mice. g, Representative H&E-stained colon sections of monocolonized mice with a 250 μm scale bar for scale. h, Colitis scores from histological assessment of monocolonized mice. Between group two-tailed t-test P=0.0061. i, Crypt lengths of monocolonized mice. Between group two-tailed t-test P=0.0028. Data from h-j displayed as barplots with mean values +/− SD from n=6 animals for B. vulgatus - PI and n=8 B. vulgatus + PI groups conducted in n=2 independent experiments. j, Experimental design of humanized IL10−/− mouse study. A total of n=9 animals per group (with the exception of n=8 mice for High Proteases + PI group) representing n=3 UC patient samples per group were examined over 2 independent experiments. k-m Protease inhibition improves colitis measurements induced by UC stool. Barplots showing the mean +/− SD are shown with overlaid p-values from one-way ANOVA adjusted for multiple comparisons between groups are for colon length (P=2.6E-7) (k), spleen weight (P=3.3E-5) (l) and histopathological scoring of colonic sections (P=3.3E-5) (m). n, Species representation of proteases in the fecal metaproteome of humanized mice. The fecal samples from one group of humanized mice with abundant Bacteroides proteases and one group without abundant proteases was subjected to LC-MS3 based metaproteomics. The relative abundance from identified proteases is shown based on the species annotation of each protease.
Confocal microscopy of the intestinal monolayers revealed dramatic impact on the B. vulgatus treated epithelial cells, with apparent alteration of tight-junction proteins, Zo-1 and Occludin (Fig. 5d, Supplementary Fig. 5). Imaging studies also demonstrated potential impacts on cell morphology and actin networks of the Caco-2 cells treated with B. vulgatus (Supplementary Fig. 5). Analysis of the cell shape within monolayers showed a significant decrease in the circularity of the cells (P = 0.0043), which could be restored through protease inhibition (Fig. 5e).
To investigate the effect of B. vulgatus proteases in vivo, we performed a monocolonization with B. vulgatus in an IL10−/− germ-free mouse model, supplementing the drinking water of half the mice with our selected protease inhibitor cocktail (Fig. 5f). After 10-weeks of colonization, protease inhibition had a protective effect on the colonic epithelia, decreasing inflammatory cell infiltration of the crypts (Fig. 5g). Histological colitis severity was significantly improved by protease inhibitor treatment (Adjusted p-value = 0.0061, Fig. 5h), along with significantly reduced B. vulgatus-induced crypt hyperplasia (Adjusted p-value = 0.0028, Fig. 5i). Macroscopic features such as the colon length of mice were not significantly different (Supplementary Fig. 6a–h). Further, immune cell profiles of mesenteric lymph nodes revealed no significant differences between the groups for CD4+, Th1, Th17 and Treg cell populations (Supplementary Fig. 6i–l, Supplementary Fig. 7). In this study, we were not able to evaluate the extent to which the protease treatment reflected a state similar to mice colonized with non-proteolytic Bacteroides as this control group was not included.
B. vulgatus proteases present in UC patient’s feces drive colitis severity upon transplant into germ-free mice
Next, we sought to evaluate the extent to which the presence of high levels of B. vulgatus proteases in UC patients impacted development of gut inflammation. To this end, we performed a transplant of feces from UC patients into colitis-prone IL10 deficient germfree mice (Fig. 5j). We selected patient fecal samples with, or without high levels of B. vulgatus proteases (n=3 UC patients per condition) for transplant into groups of colitis-prone IL10-deficient germfree mice. Half of these mice were administered a protease inhibitor cocktail via their drinking water (n=9 mice per condition). Mice administered protease-abundant fecal samples displayed overt colitis based on both gross indicators of disease (colon shortening and splenomegaly- Fig. 5k–l) and histopathologic analysis (Fig. 5m). These phenotypes were not evident in mice receiving feces from UC patients that lacked an abundance of B. vulgatus proteases. Significant differences were not observed on other organs (Extended Data Fig. 9a–f). The protease inhibitor cocktail did not significantly impact these parameters in mice administered the low-protease containing fecal samples but markedly attenuated the colitis exhibited by mice that had received the protease-abundant fecal samples. Assessment of colonic inflammation via measuring fecal lipocalin abundance and splenic bacterial load showed similar trends but did not reach statistical significance (Extended Data Fig. 9g–h). These studies reveal that the microbiomes of UC patients with increased Bacteroides proteases have high colitogenic potential and suggest protease inhibition as a therapeutic intervention in severe UC.
Finally, to confirm the presence of B. vulgatus proteases in the fecal transplantation study, metaproteomic analysis of mouse fecal material was performed. Comparing the fecal material of mice transplanted with samples from one patient with overabundant B. vulgatus proteases and one low protease control patient, we were able to detect an increased abundance of Bacteroides proteins and B. vulgatus proteases from the overabundant transplantation irrespective of the presence of the protease inhibitor cocktail (Extended Data Fig. 9i, Fig. 5n). Common functions among the proteases identified from B. vulgatus in these mice included serine-type peptidase activity and dipeptidase activity (Extended Data Fig. 9j). To guide future studies into B. vulgatus proteases, comparisons were performed between the identity of proteases highlighted in UC patients, the in vitro studies, and the in vivo studies (Extended Data Fig. 9k–l, Supplementary Table 4). Of note, several dipeptidyl peptidases (e.g. DPPIV, DPPVII) were consistently identified throughout the study, which have known roles in amino acid metabolism in nutrient limited areas28 and virulence in Porphymonas gingivalis29, a bacterium linked to periodontal disease. Interestingly, human DPPIV is the target of numerous therapeutics for the treatment of diabetes30. DPPIV inhibitors were also shown to have a protective effect in a colitis model (attributed to preventing Glucagon-like peptide-2 degradation)31, therefore we speculate that B. vulgatus DPPIV may be of interest as a potential therapeutic target.
Discussion
Here, we effectively collect and translate an extensive meta-omic profile of IBD patients into a hypothesis of biological and therapeutic value. Through integrating fecal metaproteomics, metabolomics, 16S gene amplicon sequencing, shotgun metagenomic sequencing, metapeptidomics, and serum proteomics, in addition to in vitro and in vivo validation, we demonstrate that certain members of the microbiome, such as B. vulgatus, may contribute to exacerbating UC disease activity through protease activity. Further, given the promise of our in vitro and in vivo experiments, this study sets the stage for further investigation of Bacteroides protease inhibition as a therapeutic approach in UC.
To generate our hypothesis, we utilized several innovative -omic advances that may be of broad interest such as our integrated approach for comparing metagenomic and metaproteomic data11, and the analysis of peptide fragments. Given that previous high-profile IBD data sets that included metaproteomic data7 used methods that generated an order of magnitude more missing values, we had interest in further investigating findings unique to our metaproteome data. One striking observation uniquely highlighted by this data was that ~50% of microbial proteins correlated to UC disease activity were derived from Bacteroides. While metapeptidomic data is rarely collected in microbiome studies, this data type provided an important complementary tool for identifying that proteolysis, potentially derived from Bacteroides proteases, was correlated to UC activity. By integrating metagenomic data, we provided a genomic context to our findings and identified Bacteroides species of interest for in vitro studies. Other -omic profiles (serum proteomics, metabolomics, and 16S) further corroborated and contextualized the core hypothesis of Bacteroides derived proteolysis as a contributing factor to UC severity.
Our study advances what is currently known about B. vulgatus and UC. Bacteroides spp. are among the most abundant species of the gut, residing in the outer mucosal layer of the colon32, with important roles in digesting complex carbohydrates33,34. Early research suggested a potential pathogenic role for B. vulgatus because the bacterium was found to induce experimental colitis in gnotobiotic guinea pigs35. Furthering this connection was the identification of high levels of antibodies directed toward B. vulgatus outermembrane proteins in UC patients36. However, later studies identified mixed roles for B. vulgatus in colitis models37,38, and recent genomic studies only occasionally implicate Bacteroides spp.4,39,40. Therefore, a detailed mechanistic understanding of the role of Bacteroides in IBD has not been well established33. Our in vitro studies found that B. vulgatus and B. dorei (which are close phylogenetic neighbors recently recategorized under the Phocaeicola genus25,41) disrupt colonic epithelial integrity, and further, that this phenotype was related to serine and/or cysteine proteases. The results of our monocolonization studies with B. vulgatus and fecal transplant studies further suggested the importance of serine proteases from B. vulgatus in experimental colitis. In patient samples, cross-referencing the abundance of B. vulgatus proteases to the metagenomic abundance of B. vulgatus allowed us to identify a subset of inflamed patients with unusually high levels of these bacterial proteases. Interestingly, a recent study highlights that the genomic presence of B. vulgatus proteases, alongside increased fecal protease activity is correlated with the onset of UC20. Together, our studies correlate B. vulgatus proteases to both the initiation of disease and to flares in disease activity. Further, our study now shows that inhibiting B. vulgatus proteases may have a therapeutic or preventative effect.
Although extracellular matrix remodeling42 and protease activity19 are known molecular events in IBD, the role of bacterial proteases in IBD has been primarily a source of speculation19,43–46. Work in this area has mostly focused on the contribution of host proteases, such as trypsin, which is decreased in IBD patients47, or matrix metalloproteases which can degrade commonly used therapeutics48,49. Some authors estimate that ~27% of proteolysis in UC patients is from bacterial proteases50. Further evidence of the importance of bacterial protease activity in the gastrointestinal tract was seen when antibiotic exposure in mice reduced the activity of microbiome-derived serine proteases51. Here, we were able to directly identify the species and identities of bacterial proteases that may contribute to IBD. Given that current treatments for UC are focused on targeting host inflammatory pathways, our findings represent an alternative approach for therapeutic development52. Interestingly, the human version of one of our most promising bacterial proteases, DPPIV, has already been considered as a potential target for therapeutic development53. By our estimates, ~40% of UC patients may have overexpression of B. vulgatus serine proteases, which represents a significant subset of patients that might benefit from bacterial protease inhibition.
The role of proteases in Bacteroides remains an underexplored research area. Studies indicated that their proteases may have effects on host digestive enzymes, with B. vulgatus having higher protease activity than other Bacteroides species54. However, studies into the roles of Bacteroides proteases in general are limited beyond the characterization of a metalloprotease enterotoxin from B. fragilis55,56. Increased protease abundance could be related to extracellular membrane vesicles, which in Bacteroides, are abundant in proteases57. Interestingly, extracellular vesicles were linked to IBD from a recent metaproteomic study, and Bacteroides proteins were reported as a major contributor to bacterial extracellular proteins17. Our working hypothesis is that nutrient availability or host-microbe interactions in the UC gut may trigger increased production of B. vulgatus proteases relevant to UC activity (Extended Data Fig. 10). One or a combination of these proteases appear capable of disrupting the colonic epithelium, which may allow for the influx of innate immune cells such as neutrophils which further exacerbate colitis. An alternative hypothesis would be that there are B. vulgatus strains carrying unique proteases, although we do not think this is likely given that our in vitro and in vivo work was performed using a strain of B. vulgatus isolated from healthy stool.
We note several limitations of our study. One limitation of our study is that we utilized non-specific protease inhibitors in our experiments and therefore were not able to distinguish the identities of which protease or proteases were most important to our phenotypes. Second, our monocolonization study did not include an additional control group to compare the extent to which protease inhibition treatment reflected a healthy phenotype. Finally, our experiments using the supernatant of B. vulgatus on the colonic epithelial barrier did not disrupt membrane integrity as observed in co-culture, emphasizing that more work is needed to determine the conditions and mechanisms underlying our observed phenotypes.
The multidimensional meta-omic integration shown here not only represents an important resource for future multi-omic investigation of IBD, but also serves as an example demonstrating the development of hypotheses from multi-omic data integration. Starting with broad-scale analysis of hundreds of IBD patients, and further refining our analyses according to an observation of interest led to compounding evidence of our hypothesis within each dataset. We have further narrowed and validated our primary hypothesis with numerous in vitro and in vivo studies that demonstrate the efficacy of protease inhibition to prevent B. vulgatus induced colitis. In total, our study highlights promising areas of investigation regarding the role of proteolysis in Bacteroides, and demonstrates that proteolysis from B. vulgatus may be relevant to UC pathology and treatment.
METHODS
Monocolonization experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California San Diego. Fecal transplantation experiments were done in accordance with institutional approval from Georgia State University (Atlanta, Georgia, USA) and Cochin Institute (Paris, France) under institutionally approved protocols (IACUC # A18006 and APAFIS#24788-2019102806256593 v8). All studies were conducted in accordance with NIH guidelines for the Care and Use of Laboratory Animals. Human demographics, relevant medical information, disease activity, stool, serum, and mucosal biopsies were collected with informed consent and following regulations at the Biobank at the University of California San Diego.
Patient population and clinical diagnostics
Ulcerative colitis and Crohn’s disease patients were selected from a convenience sampling biobank at the University of California at San Diego (UCSD: PI Dulai). Longitudinal data was collected on patient demographics (age, gender, ethnicity), disease characteristics (prior surgeries, disease-related complications, phenotype classification according to Montreal sub-classifications), current and prior treatments (corticosteroids, immunomodulators, biologics), clinical disease activity (patient reported outcomes using the partial Mayo score and Crohn’s disease activity index), and endoscopic and histologic disease activity. Patients also agreed to stool, serum, and mucosal biopsy collection. When endoscopy was performed as part of routine practice, stool was collected within 24 hours prior to endoscopy and serum was collected the day of endoscopy. A detailed endoscopic disease activity assessment using the Mayo endoscopic sub-score and the Ulcerative Colitis Endoscopic Index of Severity (UCEIS), was conducted by a physician without knowledge of the clinical disease activity score or biomarker data. Routine standard of care biopsies were scored using the Geboes score by a pathologist, who was blinded to clinical, biomarker, and endoscopic data and scores. Further information regarding clinical, endoscopic and histologic activity scoring have been previously discussed23. All serum and stool samples were aliquoted within 24 hours of collection to avoid future freeze-thaw cycles, and samples were stored at −80 °C. For this study, two cohorts of subjects were processed and analyzed separately, years apart, to serve as a discovery and validation cohort.
DNA extraction
Frozen samples were thawed and transferred into 96-well plates containing garnet beads and extracted using Qiagen MagAttract DNA kit adapted for magnetic bead purification as previously described58. DNA was eluted in 100 μl Qiagen elution buffer.
16S gene amplicon sequencing
16S rRNA gene amplicon sequencing was performed according to the Earth Microbiome Project. Briefly, the V4 region of the 16S rRNA gene (515f/806r) was amplified from 1 ul DNA per sample in triplicate59,60. Amplicons were quantified with Quant-iT™ PicoGreen™ dsDNA Assay Kit, and 240 ng, or maximum 15 ul, of each sample was pooled into a final library and cleaned using the QIAquick PCR Purification Kit. Paired-end sequencing was performed on the Illumina MiSeq using MiSeq Reagent Kit v3 (300-cycle).
Shotgun metagenomic sequencing
Extracted DNA was quantified with PicoGreen™ dsDNA Assay Kit, and 1 ng of input, or maximum 3.5 μl, gDNA was used in a 1:10 miniaturized Kapa HyperPlus protocol. Per sample libraries were quantified and pooled at equal nanomolar concentration. The pooled library was cleaned with the QIAquick PCR Purification Kit and size selected for fragments between 300 and 700 bp on the Sage Science PippinHT. The pooled library was sequenced as a paired-end 150-cycle run on an Illumina HiSeq4000 v2 at the UCSD IGM Genomics Center.
Processing of metagenomic reads for a shared reference library
A shared reference database was created from generated metagenomic data for both metagenomic and metaproteomic protein identification11. Individual samples were first trimmed and host-filtered using trimmomatic61 and bowtie262. Reads from each sample were concatenated. MEGAHIT63 was utilized for assembling short reads into contigs. Assembled contigs were searched for possible coding regions through the program Prodigal64. Next, Diamond65 was used for gene alignment to the uniref50 database. Finally, the most likely uniref50 entry, determined through bitScore, was used for the functional annotations. Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology annotations were cross-referenced using GhostKOALA66. Taxonomic assignments were determined by Diamond alignment65 to an extensive database of bacterial and archaeal genomes67. A study-specific databases per cohort with 299,807 and 4,113,467 open reading frames for Cohort 1 and Cohort2 respectively. Scripts used for data processing are available at https://github.com/knightlab-analyses/uc-severity-multiomics.
Unweighted UniFrac analysis of shotgun metagenomic data
Taxonomic profiling of shotgun sequences was performed using Centrifuge 1.0.3 with default parameter settings against the microbial genome database described above. The numbers of reads mapped to individual reference genomes per sample were summarized into a BIOM table. Genomes mapped by less than 0.01% reads per sample were dropped. The beta diversity of samples was assessed using the unweighted UniFrac metric as implemented in QIIME68, with reference to the phylogenetic tree of the microbial genomes (also available at: https://github.com/biocore/wol). The resulting distance matrix was visualized with PCoA, and the hypothesis was tested using PERMANOVA and Adonis as implemented in QIIME68.
Generating copy numbers of metagenomic genes
The program Salmon69 was applied to determine the reads present for each gene from the shared reference library described above. First, an index was created with Salmon inputting the shared reference library’s fasta file. Next, reads were aligned to this index in quasi-mapping mode for each of the metagenomic samples. The results were represented in counts per million sequences, with missing values padded as zeroes.
Serum collection, depletion and analysis
Seppro human depletion kits were used according to manufacturer protocols for depletion of highly abundant proteins. After thawing samples on ice, 14 uL of serum was applied to columns following the depletion protocol, and wash and elution fractions were combined to increase the total protein content. After depletion, protein was processed as described below, with the exception of a TCA precipitation70 being used in place of chloroform methanol extraction. After data collection and processing, large variability was observed dependent on serum coloring, and 7 samples with study identifiers L7, L15, L13, L8, L18, L6 and H17 (which were colored red likely due to the presence of blood in the serum) were removed for PCoA visualization.
Protein preparation
Fecal samples were measured out to ~0.5 g and suspended in 5 mL of ice-cold, sterile TBS. Samples were vortexed until completely suspended. Two 20 μM vacuum, steriflip (Milipore) filters were used per sample to remove particulate. Cells were pelleted through centrifugation at 3220 x g for 10 min at 4 °C. Cells were lysed in 2 mL of buffer containing 75 mM NaCl (Sigma), 3% sodium dodecyl sulfate (SDS, Fisher), 1 mM NaF (Sigma), 1 mM beta-glycerophosphate (Sigma), 1 mM sodium orthovanadate (Sigma), 10 mM sodium pyrophosphate (Sigma), 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma), and 1X Complete Mini EDTA-free protease inhibitors (Roche) in 50 mM HEPES (Sigma), pH 8.571. An equal volume of 8M urea in 50 mM HEPES, pH 8.5 was added to each sample. Cell lysis was achieved through two 15-second intervals of probe sonication at 25% amplitude. Proteins were then reduced with dithiothreitol (DTT, Sigma), alkylated through iodoacetamide (Sigma), and quenched as previously described72. Proteins were next precipitated via chloroform-methanol precipitation and protein pellets were dried73. Pellets were re-suspended in 1M urea in 50 mM HEPES, pH 8.5 and digested overnight at room temperature with LysC (Wako)74. A second 6-hour digestion using trypsin at 37 °C was performed and the reaction was stopped through addition of 10% trifluoroacetic acid (TFA, Pierce). Samples were then desalted through C18 Sep-Paks (Waters) and eluted with a 40% and 80% Acetonitrile solution containing 0.5% acetic acid75. Concentration of desalted peptides was determined, and 50 μg aliquots of each sample were dried in a speed-vac. Bridge channels consisting of 25 μg from each sample were created and 50 μg aliquots of this solution were used in either one or two channels (dependent on the experiment and listed in supplementary files) per Tandem Mass Tag (TMT, Thermo Fisher Scientific) 10 plex MS experiment as previously described76. These bridge channels were used to control for labeling efficiency, inter-run variation, mixing errors and the heterogeneity present in each sample77. Mass defects for each TMT set were accounted for in the database searches according to manufacturer’s report per lot number. The lot numbers for TMT reagents were SF253264 for metaproteomics and SG253268 for serum proteomics of the first cohort of UC patients, TG271363 for the second cohort of IBD patients, and lot number VA296083 for both the bacterial supernatant and metaproteomics of the mouse fecal transplant experiments. Each sample or bridge channel was resuspended in 30% dry acetonitrile in 200 mM HEPES, pH 8.5 for TMT labeling with 7 μL of the appropriate TMT reagent78. Reagents 126 and/or 131 (Thermo Scientific) were used to bridge between mass spec runs. Remaining reagents were used to label samples in random order. Labeling was performed at room temperature for 1 hour, and quenched with 8 μL of 5% hydroxylamine (Sigma). Labeled samples were acidified with adding 50 μL of 1% TFA. After TMT labeling each 10-plex experiment was combined, desalted (C18 Sep-Paks) and dried in a speed-vac.
Generation and processing of LC-LC-MS2/MS3 proteomic data
Basic pH reverse-phase liquid chromatography (LC) followed by data acquisition through LC-MS2/MS3 was performed as previously described76. Briefly, 60-minute linear gradients of acetonitrile were performed on C18 columns using an Ultimate 3000 HPLC (Thermo Scientific). Subsequently, 96 fractions were combined as previously described79, and further separation of fractions was performed with an in-line Easy-nLC 1000 (Thermo Fisher Scientific) and a chilled autosampler. LC-MS2/MS3 data was collected on an Orbitrap Fusion (Thermo Fisher Scientific) mass spectrometer with acquisition and separation settings as previously defined80.
Data was processed using Proteome Discoverer 2.1 (Thermo Fisher Scientific). MS2 data was searched against the shared metagenomic database and Uniprot Human database (uniprot.org, accessed 5/11/2017). The Sequest searching algorithm81 was used to align spectra to database peptides. A precursor mass tolerance of 50 parts per million (ppm)82,83 was specified and 0.6 Da tolerance for MS2 fragments. Included in the search parameters was static modification of TMT 10-plex tags on lysine and peptide n-termini (+229.162932 Da), carbamidomethylation of cysteines (+57.02146 Da), and variable oxidation of methionine (+15.99492 Da). Raw data was searched at a peptide and protein false discovery rate of 1% using a reverse database search strategy84–86.
TMT reporter ion intensities were extracted from MS3 spectra for quantitative analysis and signal-to-noise values were used for quantitation. Additional stringent filtering was used removing any moderate confidence peptide spectral matches (PSMs), or ambiguous PSM assignments. Any peptides with a spectral interference above 25% were removed, as well as any peptides with an average signal to noise ratio less than 10. As metaproteome data represents a complex group of proteins that may contain homologs of similar sequence identity, several steps were taken as previously described15 to reduce false assignments for metaproteome datasets. The standardized methods in Proteome Discoverer (Version 2.1) preferentially assign peptides to proteins that previously had peptides reported. If this does not resolve the assignment, the peptide is assigned to the longest protein. After the first search, all proteins reported in forward or reverse datasets were filtered into a smaller database for a second search87. This method effectively decreased the database search space in cohort 1 from 766 mb to 22 mb and 1 gb to 42 mb in cohort 2. Additionally, a duplicate peptide filter was performed according to the Proteome Discoverer report. All signal from PSMs assigned to the same protein group were summed to represent protein abundance.
Protein relative abundances were normalized first to the pooled standards for each protein and then to the median signal across the pooled standard. An average of these normalizations was used for the next step. To account for slight differences in amounts of protein labeled, these values were then normalized to the median of the entire dataset and reported as final normalized summed signal-to-noise ratios per protein per sample. Proteomic datasets generated from IBD patient samples resulted in final data tables containing 1,005 proteins for cohort 1 serum samples, 46,398 proteins for cohort 1 fecal samples, and 86,451 proteins for cohort 2 fecal samples.
Metabolite extraction and LC-MS2
Metabolites were extracted by adding a 1:5 weight to volume solution of 70% methanol infused with a 5 μM internal standard sulfamethoxine. The samples were briefly vortexed to mix and stored at 4°C overnight. Extracts were then centrifuged at 1,500 x g for 5 minutes to pellet particulate matter and the supernatant was removed for MS analysis. The extracts were diluted 1:4 in a 96 well plate in pure methanol prior to injection.
LC-MS/MS was performed on a Bruker Daltonics® Maxis qTOF mass spectrometer (Bruker, Billerica, MA USA) with a ThermoScientific UltraMate 3000 Dionex UPLC (Fisher Scientific, Waltham, MA USA). Metabolites were separated using a Kinetex 2.6 μm C18 (30 × 2.10 mm) UPLC column with a guard column for cohort 1, and using a Kinetex C18 1.7 μm C18 (50 × 2.10 mm) column for cohort 2. Mobile phases were A 98:2 and B 2:98 ratio of water and acetonitrile containing 0.1% formic acid and a linear gradient from 0 to 100% for a total run time of 840 s at a flow rate of 0.5 mL min−1 were used. The mass spectrometer was calibrated daily using Tuning Mix ES-TOF (Agilent Technologies) at a 3 mL min−1 flow rate. For accurate mass measurements, lock mass internal calibration used a wick saturated with hexakis (1H,1H,3H-tetrafluoropropoxy) phosphazene ions (Synquest Laboratories, m/z 922.0098) located within the source. Full scan MS spectra (m/z 50 – 2000) were acquired in the qTOF and the top ten most intense ions in a scan were fragmented using collision induced dissociation at 35 eV for +1 ions and 25 eV for +2 ions in the collision cell. Data dependent automatic exclusion protocol was used so that an ion was fragmented when it was first detected, then twice more, but not again unless its intensity was 2.5x the first fragmentation. This exclusion method was cyclical, being restarted after every 30 seconds.
Metabolite annotation
Data was converted to the .mzXML format using the Bruker Data Analysis software and uploaded to GNPS88 through the MassIVE server under ID MSV000082457 for cohort 1 and MSV000084908 for cohort 2. Molecular networking was performed as follows: precursor and fragment ion mass tolerance 0.03 Da, minimum cosine score of 0.65, minimum matched fragment ions of 4, and minimum cluster size of 2. GNPS library searching was performed with the same minimum matched peaks and cosine score. All library hits were inspected for quality with the mirror plot feature in GNPS. Area under the curve feature abundances were calculated to produce a metabolome bucket table with the mzMine software89. Parameters were as follows: Mass Detection (MS1 noise level of 1000, MS2 noise level of 50), ADAP Chromatogram Builder (min group size in # of scans 4, group intensity threshold 3000, min highest intensity 1000, m/z tolerance of 0.005 Da or 10 ppm), Chromatogram deconvolution (Local min search used, chromatographic threshold 0.01%, minimum in RT range 0.50 min, minimum relative height 0.01%, minimum absolute height 3000, min ratio of peak top/edge 2, peak duration 0.05 – 0.50 min, m/z range for MS2 pairing 0.01 Da, RT range for MS2 pairing 0.10 min), Isotopic peaks grouper (m/z tolerance 0.01 m/z or 10 ppm, RT tolerance 0.30 min, maximum charge 5), Join aligner (m/z tolerance 0.01 m/z or 10 ppm, weight for m/z 80, RT tolerance 0.30 min, weight for RT 20) and filtered for at least 2 peaks in a sample and gap filling was performed to produce the final bucket table for statistical analysis. Molecular class annotations were generated through the Qemistree workflow90 on GNPS which utilized the programs Sirius91 and ClassyFire92. Cohort 1 contained 4,267 MS2 features, of which 492 had putative annotations through either GNPS or Qemistree, and the final table from Cohort 2 contained 1,928 MS2 of which 442 had putative annotations.
Generation of metapeptidome data
LC-MS/MS .mzXML formatted files were loaded into PEAKS Studio 8.593 for de novo identification and searching against the Uniprot human protein database as previously described94. De novo error tolerance parameters were used according to PEAKS default qTOF settings, 0.1 Da parent mass error tolerance, 0.1 Da fragment mass error tolerance. The search settings included no added restriction enzymes, variable dehydration, Acetylation (N-Term), Oxidation (M), and Ubiquitination. The max variable post-translational modifications per peptide was set to 3. De novo sequences were filtered to keep only those with an average local confidence above 85% resulting in 651 PSMs for cohort 1 and 369 PSMs for cohort 2.
For human peptides, label-free quantification was run through PEAKS Studio 8.593. A 1% FDR cutoff was used integrating peaks with a 20 ppm mass error tolerance and a 6 min retention time window. Peptides were searched against the human protein database (uniprot.org, accessed 05/11/2017) for identification. Quantification was normalized to the total ion chromatograph.
Comparison of metaproteomic approaches
Raw mass spectra data was downloaded from fecal proteomic data generated from UC patient samples (N=25, n=102) in the IBD multiomics database7. Data was searched using Proteome Discoverer with settings described above using a two-step database approach87 utilizing a generalized human gut metagenome database95. Data from this study was re-searched under identical conditions for direct comparisons between datasets. Datasets were compared for their total protein identifications, proteins identified per sample and the sparsity in the dataset as measured by the percent missing values (number of proteins lacking quantification/total potential quantifications).
Meta -omic data analysis
Data analysis was performed in python (version 3.5), and records of the code are available in corresponding Jupyter Notebooks for this project (https://github.com/knightlab-analyses/uc-severity-multiomics). Clinical data correlations were performed on UC cohort 1 using the package seaborn’s (https://seaborn.pydata.org/) clustermap function. Seaborn’s lmplot function was used to display linear relationships between alpha-diversity measurements and disease activity.
16S fastq were split, demultiplexed, trimmed to 150 base pairs and processed through deblur using QIITA96 (Study ID 11549). A denovo phylogenetic tree was formed for 16S data using the reference hits through QIIME 297 (version 2018.4) commands “qiime alignment mafft”, “qiime alignment mask”, “qiime phylogeny fasttree” and “qiime phylogeny midpoint-root”. 16S alpha-diversity was generated using QIIME 297 (Version 2019.7) through the command “qiime diversity core-metrics-phylogenetic”. Statistical association between disease activity and alpha diversity was performed using the package statsmodel’s ordinary least squares (https://www.statsmodels.org/) accounting for diagnosis and the interaction between patient diagnosis and disease activity. For correlations to each clinical variable to alpha-diversity, Kruskal Wallis tests were performed on categorical variables using the “alpha-group-significance” command in QIIME 297 (Version 2019.7). Quantitative variables were correlated with alpha-diversity measurements using the linregress command from the python package scipy (https://www.scipy.org). All alpha-diversity associations were based on 16S data.
Community diversity analysis was performed using the “qiime diversity core-metrics” command in QIIME 2. Statistical analysis of beta-diversity association to disease activity while accounting for the diagnosis of the patients was performed using ADONIS in QIIME 2. To facilitate faster analysis of the association of beta-diversity and numerous patient variables, QIIME’s compare_categories.py function was used for single variable associations to beta-diversity with ADONIS for quantitative measures and PERMANOVA for categorical measures.
16S taxonomic barplots grouped patients into three categories based on either the partial Mayo activity score for UC patients or the CDAI for CD patients. Patients in the bottom 30% of activity scores were categorized as “Low”, patients in a range between 30% and 50% of the highest activity score were categorized as “Moderate” and patients above 50% were categorized as “High”. All composition plots for –omics data were plotted using the package matplotlib (https://matplotlib.org/). Correlation between –omics data types were performed using scikitbio’s mantel test (http://scikit-bio.org/) and visualized using a seaborn heatmap.
Random forest regressions were performed using QIIME 297 (Version 2018.11) using the sample-classifier regress-sample command. The test size was set to 0.1. Statistics and importance scores for each feature within the 100 independent analyses were compiled. To facilitate comparisons between mass spectrometry datasets, where exact metabolite or protein matches are unfeasible, importance scores were summarized by annotation information about each metabolite or protein. For metaproteomics studies, importance scores were combined when exact protein name and species were found. For metabolomics studies, the importance scores were combined when exact annotations were found by the two annotation methods used and described above (the name of the GNPS spectral library match and the direct parent annotations provided by ClassyFire).
Linear regression of metagenome, metabolome, and metaproteome data to disease activity scores were performed calculating the Pearson correlation coefficient using the linregress function in the python package, scipy (https://www.scipy.org). To identify classes of metabolites correlated with disease activity, the abundance of each metabolite with a direct parent annotation from ClassyFire was averaged, and regression was performed on the average values of metabolite classes. Given the nature of TMT-labeled proteomic data, where protein abundances are frequently normalized to account for differences in the number of peptides identified, missing values in regressions were ignored, and the percentage of missing values in each protein was calculated. When comparing metagenome and metaproteome data, metagenomic data was analyzed accounting for missing values in an identical manner to metaproteomic data where missing values were ignored and the sparsity of each feature was evaluated to prevent spurious correlations. Composition of genes or proteins most correlated with disease activity (Pearson’s r > 0.3) were compared as previously11, where the number of taxonomic or functional annotations related to significantly correlated or anti-correlated genes or proteins were compared. Proteases identified in Bacteroides species were plotted in a heatmap showing the summed r-value of each protein name within a particular species of Bacteroides. The gene ontology (GO) molecular functions of these proteins were then analyzed to group the proteases by activity type.
To identify patient samples containing an over-abundance of Bacteroides vulgatus proteases, an outlier approach was taken using R studio (v. 1.1.383) using the bagplot function from the aplpack package. After applying a BLASTp analysis (https://blast.ncbi.nlm.nih.gov/) to the peptide sequences identified in UC patient metaproteomic studies that were assigned to proteins being correlated to disease activity and derived from B. vulgatus or B. dorei proteases, we determined that we could not specify the origin of these proteases beyond being derived from either B. vulgatus or B. dorei. As a result, outlier analysis and later analyses of B. vulgatus proteases in UC patients were performed using both B. dorei and B. vulgatus proteases. For the outlier approach, summed metaproteomic abundances of all correlated (r > 0.3) proteases from B. vulgatus and B. dorei were compared to the summed abundance of metagenomic reads assigned to B. vulgatus and B. dorei. Outliers identified above the best-fit line were classified as Bacteroides protease “overproducers” while outliers identified below the best-fit line were classified as “underproducers”. All other patient samples were categorized as “others”. Statistical comparisons of patient endoscopic and disease activity scores between these groups of patients were performed using independent t-tests of unequal variance through the package scipy.
Host protein networks were compiled from serum and fecal proteomics data from UC cohort 1. Linregress correlation values (r) between proteins and disease activity (partial Mayo scores) were used to rank associations. Top ranked proteins were uploaded to STRING-db98, with associations between proteins determined through default settings, accounting for textmining, experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence. Networks were next visualized through Cytoscape (version 3.5.1)99.
The program iceLogo’s web application100 was used for consensus sequence analysis of de novo peptides identified in UC patient’s metabolome data. The first and last amino acids from peptides with an average local confidence over 85% were analyzed against a background using the percentage scoring system. For metapeptidome consensus sequences, all residues from peptides with over 85% average local confidence were used as background. For human consensus sequences, the precompiled Homo sapiens Swiss-Prot database was used. Peptide fragment origins analysis was performed from the results of PEAKS studio database search described above, summarizing all PSMs assigned to each protein.
Bacterial supernatant protease activity studies
Bacteroides vulgatus (ATCC 8482) were grown anaerobically in Brain-heart-infusion (BHI, BD) broth supplemented with 5 μg/ml hemin (Sigma) and 0.5 μg/ml vitamin K (Sigma). Overnight supernatant was collected by pelleting cells at 8000 x g. Supernatant was then 8-fold concentrated at 3,300 x g for 15 minutes using 10 kDa Amicon Ultra-15 filters (Millipore). Concentrated supernatant protease activity was tested using the EnzChek protease activity assay (Invitrogen) after incubation for 24 hours at 37 °C measuring fluorescence at 485 nm for excitation and 530 nm for emission. Protease inhibitors were administered at 10% total volume and inhibition was calculated by comparison to vehicle control wells. Protease inhibitors tested included water-solubilized 4(2-Aminoethyl)benzenesulfonyl Fluoride (AEBSF, MP Biomedicals), water-solubilized E-64 (Sigma), DMSO-solubilized GM6001 (EMD Millipore), and DMSO-solubilized Pepstatin A (MP Biomedicals). After analysis of a preliminary dilution series, max inhibition was found for each protease inhibitor at the highest concentration allowed by the solubility of each compound, and these concentrations were used for subsequent studies.
Bacterial supernatant proteomics
Bacteroides vulgatus (ATCC 8482) and Bacteroides thetaiotaomicron (B. theta, ATCC 29148) alongside Human Microbiome Project strain #717 Bacteroides dorei CL02T00C15 were grown in technical triplicate anaerobically in BHI broth supplemented with 5 μg/ml hemin (Sigma) and 0.5 μg/ml vitamin K (Sigma). Supernatant was concentrated using 10 kDa Amicon Ultra-15 filters (Millipore) and prepared for TMT-mediated LC-LC-MS2/MS3 analysis as described above with samples compiled into one TMT-10plex experiment. MS analysis resulted in 219,087 MS/MS spectra that were searched in Proteome Discoverer as described above using uniprot reference proteomes for each strain (www.uniprot.org; proteome identifiers UP000005974, UP000001414 and UP000002861; downloaded 8/24/2020). Data processing resulted in a final table of 2,574 quantified protein groups that were analyzed as described below.
Stacked barplots of proteome enzyme activity type were generated based on the average relative abundance of each species, subsetting proteins annotated with a KEGG functional category annotation of “Enzyme families” and proteins containing the terms “protease” or “peptidase” in their name, and next summing protein abundances by GO molecular functions. Seaborn barplots were used to display the proteases most associated with B. vulgatus as determined by subsetting abundant proteases and comparing the average signal of each protein in samples from B. vulgatus and B. theta. Venn diagrams of the protein names identified in the reference proteomes for each Bacteroides species were generated using the matplotlib_venn function. Code for the normalization and analysis of the bacterial supernatant proteomics data can be found in the github repository for this project (https://github.com/knightlab-analyses/uc-severity-multiomics).
Caco-2 transwell studies
Caco-2 cell transwell studies were performed, as previously described101. Briefly, Caco-2 cells (passage ranging from 14-30; ATCC) were plated into collagen coated 6.5 mm inserts with 0.4 μm pores (Corning). Cells were cultured for 2.5 weeks prior to bacterial inoculation, changing media every 2 days. A day before inoculation, media was changed to media without antibiotics and when indicated, Roche cOmplete EDTA-free protease inhibitor cocktail (Sigma) was dissolved at 1x concentration. TEER was measured prior to inoculation of bacteria, and measurements at each following timepoint referenced the original TEER measurement prior to inoculation. Transwell plates were incubated at 37 °C between measurements and allowed to equilibrate to room temperature for 20 minutes before each TEER measurement. CFU estimates were performed through serial dilution of 10 μLs of media from inside of the transwell insert. Mammalian cell culture media consisted of DMEM with L-Glutamine (Corning) with 10% heat-inactivated fetal bovine serum, 100 μM sodium pyruvate (Corning), 0.75% sodium bicarbonate, 1X Insulin-Transferrin-Selenium (Gibco), 238.3 μM HEPES, and 1x penicillin streptomycin (Thermo). Antibiotic-free media was used during bacterial inoculation containing the same contents with the exception of 2% heat-inactivated fetal bovine serum.
B. vulgatus (ATCC 8482), B. fragilis (ATCC 25285), B. thetaiotaomicron (ATCC 29148), B. uniformis (ATCC 8492), and B.ovatus (ATCC 8483). Bacteroides dorei was derived from the Human Microbiome Project strain #717, Bacteroides dorei CL02T00C15. For inoculation, Bacteroides cultures were grown anaerobically overnight (80% N2, 10% H2, 10% CO2) at 37 °C in BHI. To determine growth conditions with protease inhibitors, B. vulgatus was diluted from overnight cultures into either BHI and grown under anaerobic conditions, or DMEM media (antibiotics-free) grown under mammalian cell culture conditions (37 °C, 5% CO2). Inhibitors tested included PMSF (5 mM, sigma), Ethylenediaminetetraacetic acid (5 mM EDTA, bioPLUS), and 1x concentration of a protease inhibitor cocktail (Roche cOmplete EDTA-free).
Cultures were spun down at 8000 x g, and resuspended in DMEM. Inoculations were performed through normalization by OD600 at an estimated multiplicity of infection of 5. CFUs from above the transwell insert were estimated by serial dilution and plating under anaerobic conditions. Statistical significance between the change in TEER at each timepoint was determined using 2-way ANOVA p-values adjusted for multiple comparisons conducted in GraphPad Prism (Version 7.0b). To calculate the effect size of protease inhibition treatment at the 22- and 38-hour timepoints, the average values from the technical replicates of each condition per biological replicate were compiled and tested for significance using ANOVA in the pingouin package (version 0.3.12). The eta2 effect size for condition was reported from an analysis of the effect between hours incubated and condition on TEER.
The impact of Bacteroides supernatant on TEER was assessed by collecting supernatant from B. vulgatus or B. thetaiotaomicron at mid-log phase growth and concentrating and filtering the supernatant using 10 kDa Amicon Ultra-15 spin filters (Millipore) and a 0.22 μm filter. The supernatants were concentrated by 10-fold and 50 μl of the concentrated supernatant was added to each transwell. Broth not containing any bacteria was used as an additional control. A protease inhibitor cocktail (Roche cOmplete EDTA-free) was added to the media of selected wells a day before the experiment as described above. For these studies Caco-2 cells were cultured in DMEM (ATCC) media with 10% heat-inactivated fetal bovine serum (FBS), 1x MEM Non-Essential Amino Acids (Gibco), and 1x penicillin streptomycin. When cultured with the supernatant, media contained 2% FBS as performed for bacterial co-culture studies. Three biological replicates were performed each containing 3-4 technical replicates per condition, and the average of the technical replicates was plotted alongside the standard error of the mean using GraphPad Prism (Version 7.0b).
Confocal microscopy
At the end point of transwell studies (38 hours post bacterial inoculation), cells were fixed and prepared for immunofluorescence. Caco-2 cells were fixed on the transwell membrane at 37 °C for 10 minutes in 1 mL 4% Paraformaldehyde (Thermo) in PHEM (60 mM Piperzine-1,4-bis[2-ethanesulfonic Acid] Monosodium Salt, pH 6.9 [TCI Chemicals], 25 mM HEPES 102, 10 mM EGTA [Oakwood Chemical], 2 mM MgCl2 × 6H2O 102). Cells were permeabilized for 5 minutes in PHEM with 0.5% Triton X-100 (Fisher) at room temperature followed by 3× 5-minute washes were performed in PHEM containing 0.1% Triton X-100 at room temperature. Blocking was performed for 30 minutes in 1 mL AbDil (150 mM NaCl 102, 20 mM Tris-HCl, pH 7.4 [JT Baker], 0.1% Triton X-100 102, 2% Bovine serum albumin [Gemini Bioproducts]) at room temperature. Primary antibodies for Occludin (Thermo, catalog number 33-1500, 0.5 μg/mL) and ZO-1 (Thermo, catalog number 61-7300, 1.5 μg/mL) were added into AbDil and left in a humidified chamber overnight at 4 °C. Cells were washed 4x in PHEM containing 0.1% Triton X-100 for 5 minutes at room temperature. Secondary antibodies, Rhodamine Red Donkey Anti-Rabbit (Jackson ImmunoResearch, Code Number 711-295-152), and Alexa Fluor 488 Donkey Anti-Mouse (Jackson ImmunoResearch, Code Number 715-545-150) were diluted to 3 μg/mL in AbDil containing a 1:1000 dilution of Phalloidin-iFluor 647 (abcam, ab176759) and 1 μg/mL DAPI (Thermo). Secondary antibodies were incubated for 1 hour at room temperature in a humidified chamber followed by 3 washes in PHEM containing 0.1% Triton X-100 for 5 minutes at room temperature. Finally, cells were rinsed in PHEM, removed from transwell insert and fixed onto microscope slides for imaging.
Cells were imaged using a Nikon A1R HD confocal with a four-line (405nm, 488nm, 561nm, and 640nm) LUN-V laser engine and DU4 detector using bandpass and longpass filters for each channel (450/50, 525/50, 595/50 and 700/75), mounted on a Nikon Ti2 using an Apo 60× 1.49 NA objective, or a C2 Plus confocal with a similar four-line LUN-4 laser engine and a DUV-B detector operating in virtual bandpass mode. Images stacks were acquired with the galvo scanning mode on both confocals, and Z-steps of 0.2 μm. To avoid cross-talk between channels, Z-stacks were acquired of the DAPI and Rhodamine Red channels first, and the AlexaFluor 488 and Phalloidin-iFluor 647 channels were acquired subsequently. The laser powers used were 1.5% for the 405 nm laser 2% for the 488 nm laser, 1.5% for the 561 nm laser and 1.5% for the 640 nm laser.
Cell morphology was analyzed in representative images using protocols outlined previously103. Images were processed in ImageJ (https://imagej.nih.gov/ij/) using the MorpholibJ plugin104. In brief, images were converted to binary, image borders were extended, and morphological segmentation was performed. Images were outlined, dilated and analyzed for particles. Circularity values were plotted and significance between groups was assessed using independent t-tests through the package scipy.
Monocolonization studies
Germ-free IL10−/− (7 male, 4 female) mice (B6.129P2-Il10tm1Cgn/J; Jackson Laboratory) were bred and housed in flexible film isolators until 6 - 8 weeks of age, and transferred to micro-isolator cages and maintained with autoclaved food (Lab Diet), bedding and water supplemented with gentamicin at 100 μg/ml. Mice were mono-associated with gentamicin-resistant Bacteroides vulgatus was grown as mentioned above. Bacterial cells were washed twice and resuspended in sterile PBS prior to oral gavage. Both groups of IL10−/− mice were orally gavaged with B. vulgatus. In select experiments, drinking water was supplemented with a 1 X concentration of Roche cOmplete EDTA-free protease inhibitor cocktail (Sigma). Mice were monocolonized for 10 weeks. Statistical significance of measurements in monocolonization studies was determined using unpaired t-tests conducted in GraphPad Prism (Version 7.0B). All procedures were performed in accordance with the approved protocols using IACUC guidelines of the University of California San Diego.
Histological procedure and scoring of monocolonized mouse studies
Colons were removed, flushed with cold PBS, cut longitudinally and prepared as swiss rolls. 5μm formalin–fixed, paraffin–embedded tissue sections were H&E stained and slides were scanned with a NanoZoomer slide scanner (Hamamatsu). Tissue sections were investigated using NDP.2 viewer software (Hamamatsu) in a blinded fashion. Colitis scores were assessed using a semi–quantitative score as previously described105.
Flow Cytometry / Intracellular Cytokine Staining
Mesenteric lymph nodes of monocolonized mice were processed by dissociating tissues through a 100 μm cell strainer (BD Falcon). Single-cell suspensions were stained for 30 min at 4 °C with LIVE/DEAD Aqua (Thermo Fisher, L34957, 1:1000 dilution), eFluor 450-conjugated anti-mouse CD4 (eBioscience, 48-0042-82, clone RM4-5, 1:400 dilution), APC-conjugated anti-mouse CD25 (eBioscience, 17-0251-82, clone PC61.5, 1:400 dilution). BV510-conjugated anti-mouse CD19 (BioLegend, 115545, clone 6D5, 1:400 dilution) was used as a dump channel. Cells were blocked for non-specific binding to Fc receptors with a combination of TruStain FcX anti-mouse CD16/CD32 (BioLegend, 101319, clone 93, 1:400 dilution). For intracellular staining, cells were fixed and permeabilized with the Foxp3/Transcription factor buffer kit (eBioscience). Cells were blocked with 2% normal mouse serum (Jackson Immuno Research Labs) for 15 min. The following antibodies were used for intracellular staining: FITC-conjugated anti-mouse Foxp3 (eBioscience, 11-5773-82, clone FJK-16s, 1:400 dilution), APC-eF780-conjugated anti-mouse IFNγ (eBioscience, 47-7311-82, clone XMG1.2, 1:400 dilution), PE-Cyanine7-conjugated anti-mouse IL-17A (eBioscience, 25-7177-82, clone eBio17B7, 1:400 dilution), and PE-conjugated anti-mouse IL10 as control (eBioscience, 12-7101-82, clone JES5-16E3, 1:400 dilution). Cells were acquired on the Attune NxT Flow Cytometer (Thermo Fisher) and data were analyzed on FlowJo (version 10.6.2).
Fecal microbiota transplantation
Germ-free C57BL/6 IL10−/− male mice (C57BL/6NTac-Il10em8Tac; Taconic model GF-16006) were maintained in isolated ventilated cages Isocages (Techniplast, West Chester, PA, USA) (Hecht et al., 2014). At 5-6 weeks of age, mice were orally administered with 200 μL of fecal suspension from three patients with overabundant B. vulgatus proteases (sample identifiers H5, H7 and H19) and three patients without overabundant B. vulgatus proteases (sample identifiers L3, L15 and L19). Transplanted mice were group-housed (n=3) in isolated ventilated cages, Isocages and fed autoclaved Purina Rodent Chow # 5021. Each sample was administered to two cages of mice (n=2-3 per cage), one with and one without a 1x concentration of Roche cOmplete EDTA-free protease inhibitor cocktail (Sigma) administered in the drinking water of the mice throughout the colonization. Mice were housed at Georgia State University (Atlanta, Georgia, USA) or at Cochin Institute (Paris, France) in a controlled environment (12h day/night cycle, lights off at 7 :00PM, 20. ±2°C, 48±6% of humidity). At the terminal timepoint, mice were weighed, euthanized, and tissue was collected for further analysis. Statistical strength of measurements taken from transplanted mice were assessed by ordinary one-way ANOVA using GraphPad Prism (Version 7.0B), with adjusted P values reported accounting for multiple comparisons.
H&E staining of fecal transplantation colonic tissue and histopathologic analysis
Mouse colons were fixed in Carnoy solution and then embedded in paraffin. Tissues were sectioned at 5-μm thickness and stained with hematoxylin & eosin (H&E) using standard protocols. Images were acquired using a Lamina (Perkin Elmer) at the Hist’IM platform (INSERM U1016, Paris, France). Histological scoring was determined on each colon as previously described106,107. Briefly, each colon was assigned four scores based on the degree of epithelial damage and inflammatory infiltrate in the mucosa, submucosa and muscularis/serosa106. Each of the four scores was multiplied by a coefficient 1 if the change was focal, 2 if it was patchy and 3 if it was diffuse107 and the 4 individual scores per colon were added.
Bacterial load quantification by 16S rRNA qPCR
Bacterial DNA was extracted from the spleen using the DNeasy PowerLyser PowerSoil Kit (Qiagen). Extracted DNAs were amplified by quantitative PCR using the 16S V4 specific primers 515F 5’-GTGYCAGCMGCCGCGGTAA-3’ and 806R 5’-GGACTACNVGGGTWTCTAAT-3’ on a LightCycler 480 II (Roche) using QuantiFast SYBR® Green PCR Kit (Qiagen). Data are expressed as relative values normalized with spleen weight used for DNA extraction.
Quantification of fecal lipocalin-2 (Lcn-2) by ELISA
For quantification of fecal Lcn-2 by ELISA, frozen fecal samples were reconstituted in PBS containing to a final concentration of 100 mg/mL and vortexed for 20 min to get a homogenous fecal suspension107. These samples were then centrifuged for 10 min at 14,000 g and 4°C. Clear supernatants were collected and stored at −20°C until analysis. Lcn-2 levels were estimated in the supernatants using Duoset murine Lcn-2 ELISA kit (R&D Systems, Minneapolis, MN, USA) using the colorimetric peroxidase substrate tetramethylbenzidine, and optical density (OD) was read at 450 nm (SpectraMax ABS Plus microplate reader, Molecular Device).
Metaproteomic analysis of mouse fecal samples in transplant study
At the end of the 8-week colonization, fecal samples were collected and snap-frozen for further analysis. Metaproteomic sample preparation and data acquisition was performed as described above for TMT-mediated LC-LC-MS2/MS3 analysis of fecal samples from mice within cages associated with patient samples H19 and L3. For database search of mass spectra, a custom database was generated using the metagenomic database generation workflow described above on sequencing data from the UC patient donors. Here, to track the origin of protein sequences, H19 and L3 reads were assembled and searched for coding regions separately. Open reading frames from each patient sample were combined and annotated as described above, resulting in a 70 mb fasta file containing 225,056 open reading frames. MS analysis resulted in 500,120 spectra that were subsequently searched in Proteome Discoverer against the custom database and the uniprot mouse reference database (uniprot.org, accessed 09/03/2020). After data processing, a final table of 12,603 quantified protein groups were analyzed as described below.
Stacked barplots were created as described above, plotting the proportion of the metaproteome signal of each sample dedicated to each genus. Data was further subset to contain only enzymes and proteases and stacked barplots were created, stacking each sample by species level annotations. Further subsetting the enzymes and proteases to only those from B. vulgatus, abundance of each sample was plotted in stacked barplots stacked by GO molecular function. Student’s t-tests comparing protein abundances of samples from mice receiving patient samples H19 and L3 were performed in scipy using unequal variance. T-test P values were then combined with the fold-change differences to rank associated proteins via the Pi score statistic108, as previously described109. Top proteins associated with the H19 samples were then presented in a barplot using the seaborn python package.
Extended Data
Extended Data Fig. 1. Study Design and Database Generation.
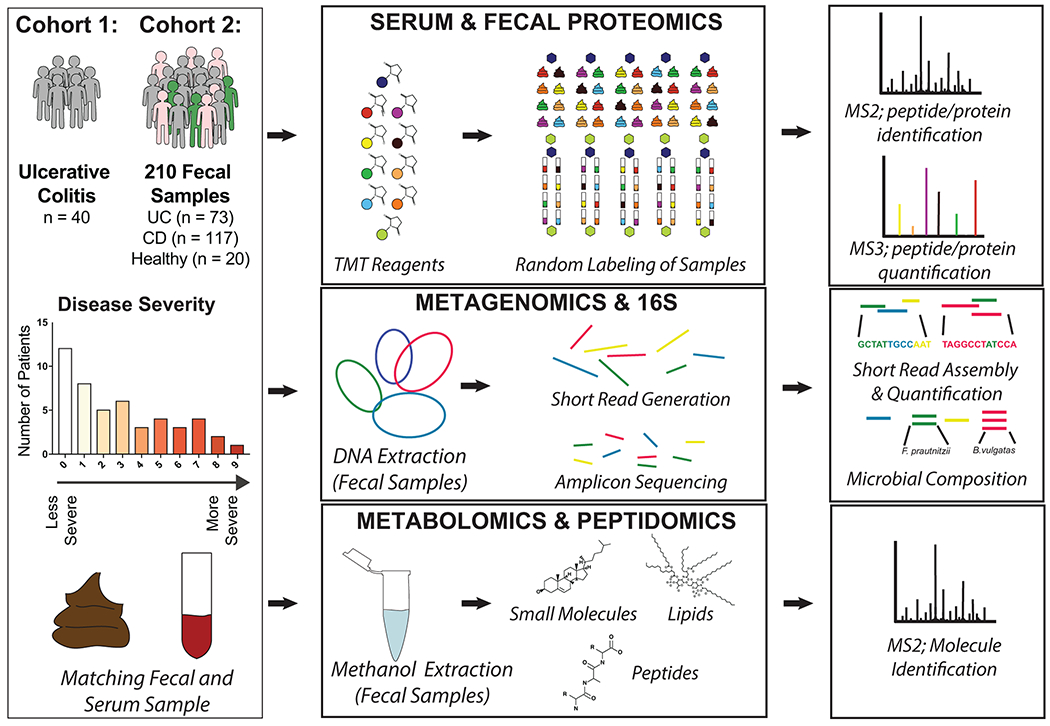
Paired fecal and serum samples were collected from 40 patients with varying severity of Ulcerative Colitis. A separately analyzed cohort of fecal samples was also collected on 210 samples with 73 UC, 117 CD and 20 healthy controls. Samples were processed for proteomics using a Tandem Mass Tag multiplexing workflow. Fecal samples were also subjected to both 16S and shotgun metagenomic analyses for microbial composition and gene quantification respectively. In parallel, a metabolomics workflow was performed on fecal samples where collected MS2 spectra were analyzed for both metabolites and peptides in two separate computational pipelines. A custom database was compiled from the metagenome of fecal samples to mediate a comparative analysis between shotgun metagenomic and metaproteomic data sets. This eliminated database dependent bias and the shared reference was used for estimating copy number.
Extended Data Fig. 2. A multiplexing approach improves the depth and sparsity of metaproteomics data.
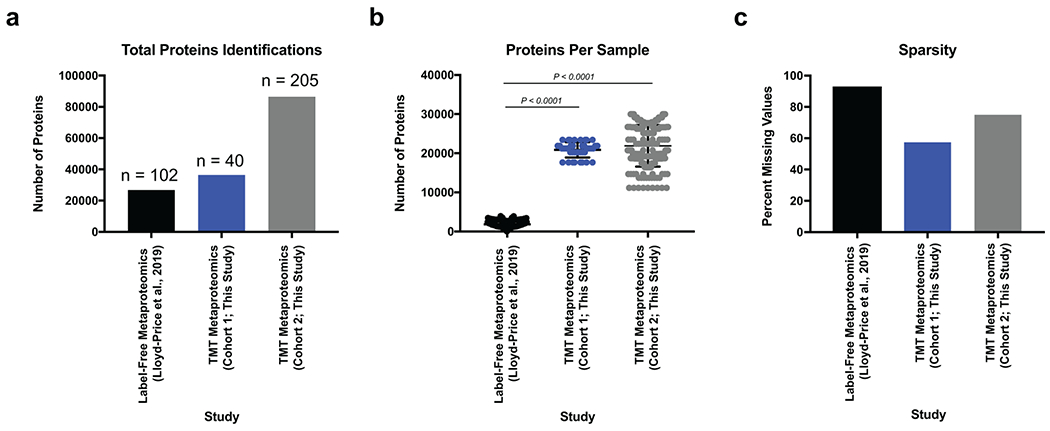
a, Multiplexed metaproteomic methods increase the total number of proteins quantified. Shown is a bar graph showing the total number of proteins identified when using identical database methodology between the 102 UC samples from the IBD multiomics database, the 40 UC samples from cohort 1 of this study, and the 205 samples from cohort 2 of this study. b, Multiplexed metaproteomic methods improve the number of proteins quantified per sample. Displayed are the mean +/− SD of the proteins identified per sample from studies shown in (a). Data derived from n=102, 40, 205 biologically independent samples as described for (a). One-way ANOVA p-values adjusted for multiple comparisons are shown (P < 0.0001). c, Multiplexed metaproteomic methods decrease the sparsity of metaproteomic studies. The percentage of missing quantification values for proteins in each data set is shown.
Extended Data Fig. 3. Characterizing uneven samples.
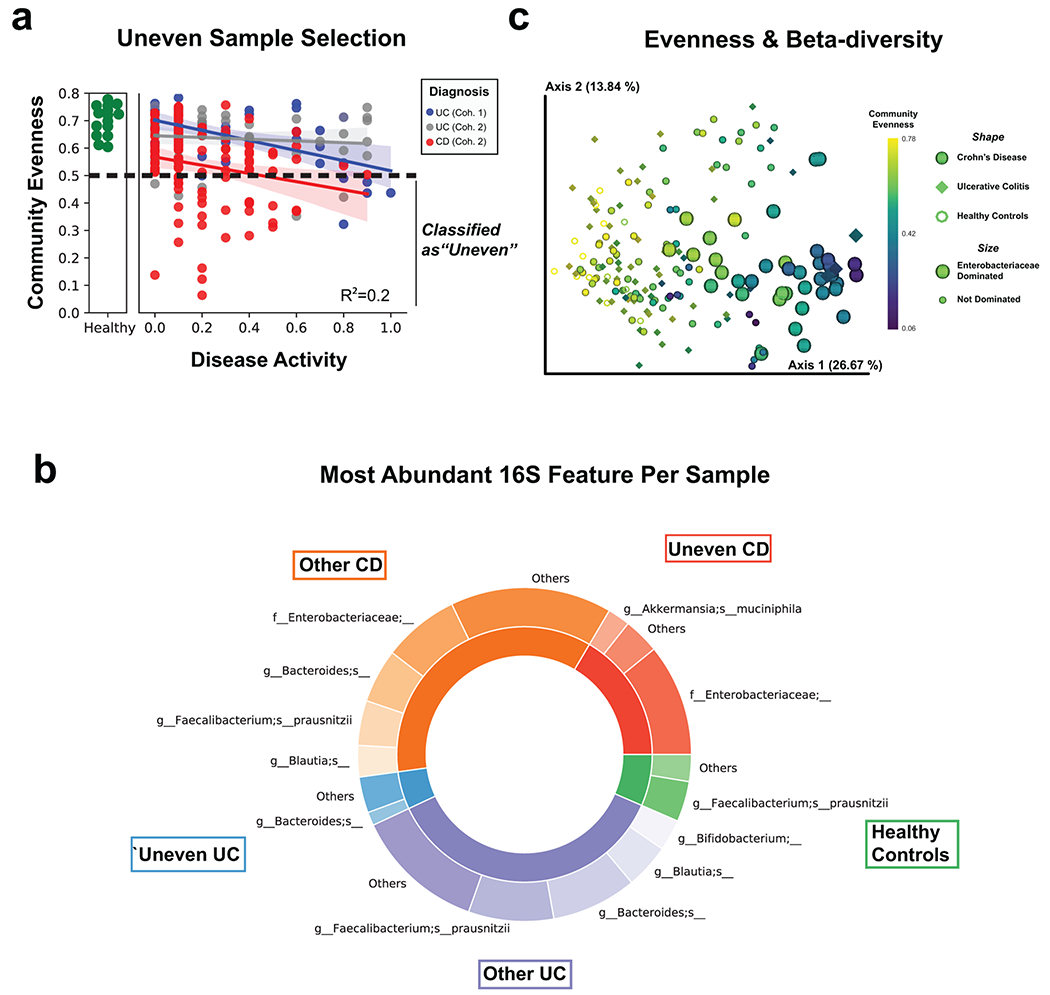
a, Alpha diversity (using Pielou’s evenness metric) by disease activity as shown in Figure 1b, but highlighting classification of samples as uneven when below Pielou Evenness of 0.5. Best-fit linear regression lines with 95% confidence intervals are shown and an R2 statistic is reported from an ordinary least-squares regression using the formula (Disease Activity + Diagnosis + Disease Activity:Diagnosis). b, 16S beta-diversity is strongly influenced by community evenness. The weighted UniFrac distance metric was used and each sample was classified by community evenness, diagnosis and whether the most abundant 16S feature was from the family Enterobacteriaceae. c, Characterizing the most abundant 16S features. Each sample was classified as either “Uneven” (Pielou Evenness < 0.5) or “Other” as shown in (a). Abundances of each amplicon sequence variant were summed by their highest resolution taxonomic annotation and the most abundant feature of samples are represented in a donut plot. The inside ring represents the fractional composition of each patient subgroup and the outside rings represents the number of patients within each subgroup whom share a similar most abundant feature. Less common features for each patient subgroup are counted as “Other”.
Extended Data Fig. 4. Comparison of genera annotations from genes and proteins correlated to disease severity.
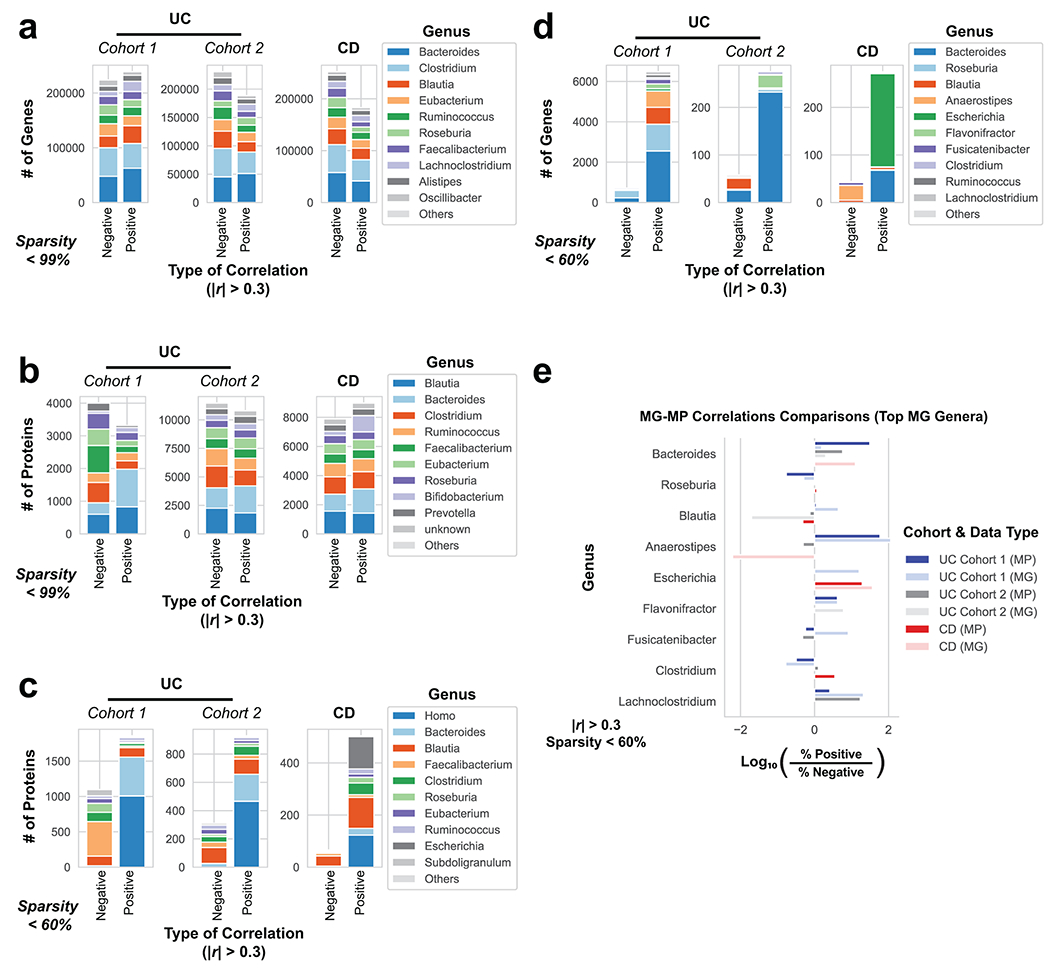
The genus composition of genes and proteins correlated to disease activity were compared with different levels of sparsity as a requirement for being deemed “correlated”. Stacked bar charts summarize the number of genes or proteins from the 10 most common genus assignments when correlated to either partial Mayo severity in UC cohorts or CDAI in CD patients. Only genes or proteins with |r| > 0.3 from linear regression were included. a, Genus composition of significant positively and negatively correlated genes from the MG with no sparsity requirement. b, Genus composition of significantly positively and negatively correlated proteins from the MP with no sparsity requirement. c, Genus composition of associated proteins as in Fig. 3a, but without removing host proteins (genus Homo). d, Genes correlated to disease activity from the MG when filtering out genes appearing in less than 40% of patients within each category. e, Summary of comparing the portions of positively and negatively correlated genes and proteins from each patient cohort when examining the top 10 genera identified in the MG. This analysis is analogous to Fig. 3b, but displaying the top MG genera.
Extended Data Fig. 5. Comparison of genera and functional annotations from genes and proteins correlated to disease severity in CD subtypes.
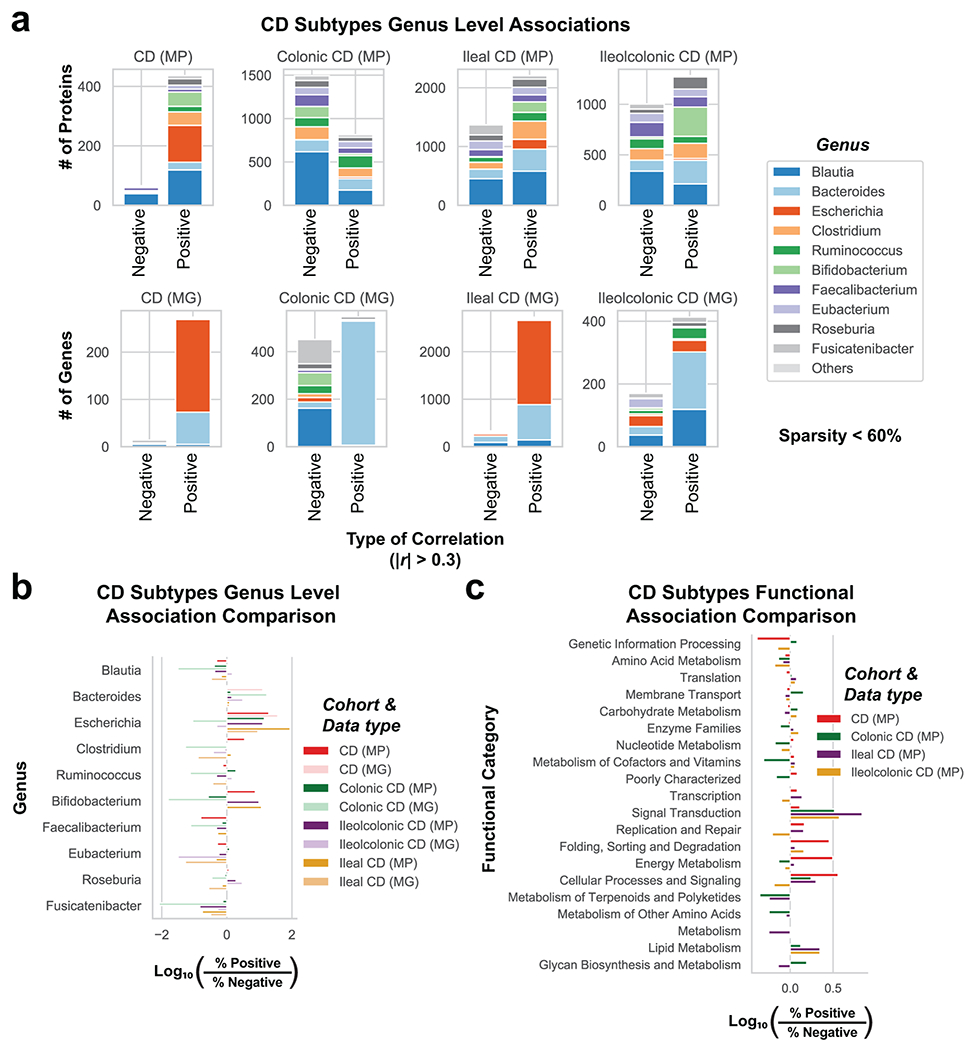
a, Genus level barcharts of significantly correlated genes or proteins stratified by CD subtype. The genus composition of genes and proteins from either the MG or MP were correlated to CDAI and shown in stacked bar charts. Only genes or proteins with |r| > 0.3 from linear regression were included, and the top 10 genera are displayed with other genera compiled into an “Others” category. b, CD subtypes genus level association comparison. The portion of genes or proteins correlated with disease activity from (a) are plotted by a Log10 comparison between the proportion of positive to negative correlations. Genes correlated to disease activity from the MG when filtering out genes appearing in less than 40% of patients within each category. c, CD subtypes functional association comparison. This analysis is analogous to (b) but summarizing the associations to KEGG functional category annotations in the MP.
Extended Data Fig. 6. Patients with overproduction of Bacteroides vulgatus proteases have increased endoscopic and histological severity.
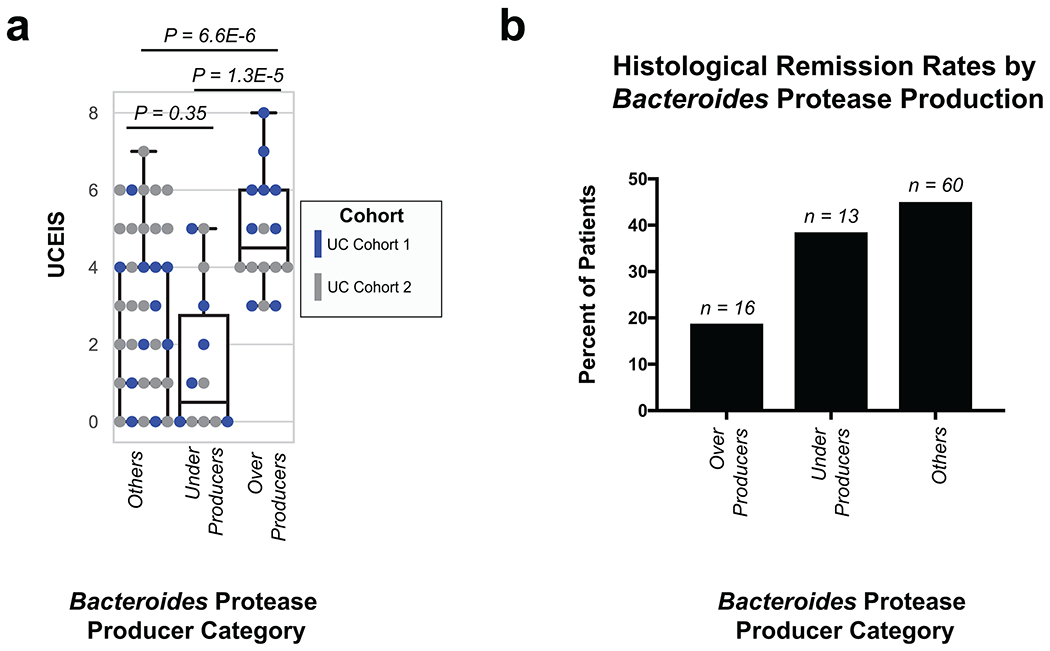
a, Bacteroides protease production corresponds to increased endoscopic severity. The disease activity of overproducers, underproducers, and other patients are individually plotted over boxplots. Two-tailed, t-test p-values are displayed above the boxplots. Sample sizes include n=16, 14 and 71 for overproducers, underproducers and others respectively. Boxplots are defined by the median, quartiles and 1.5x inter-quartile range. b, Bacteroides protease production corresponds to a patient population with a decreased proportion of patients in histological remission. Each UC patient sample was categorized by Bacteroides vulgatus protease production category and the percent of patients in histological remission is shown in a bargraph with the number of samples in each category displayed above each bar. Histological remission is defined here as Geboes Grade 3 = 0.
Extended Data Fig. 7. Peptide fragments are increased in active UC patients and Bacteroides protease enriched patients.
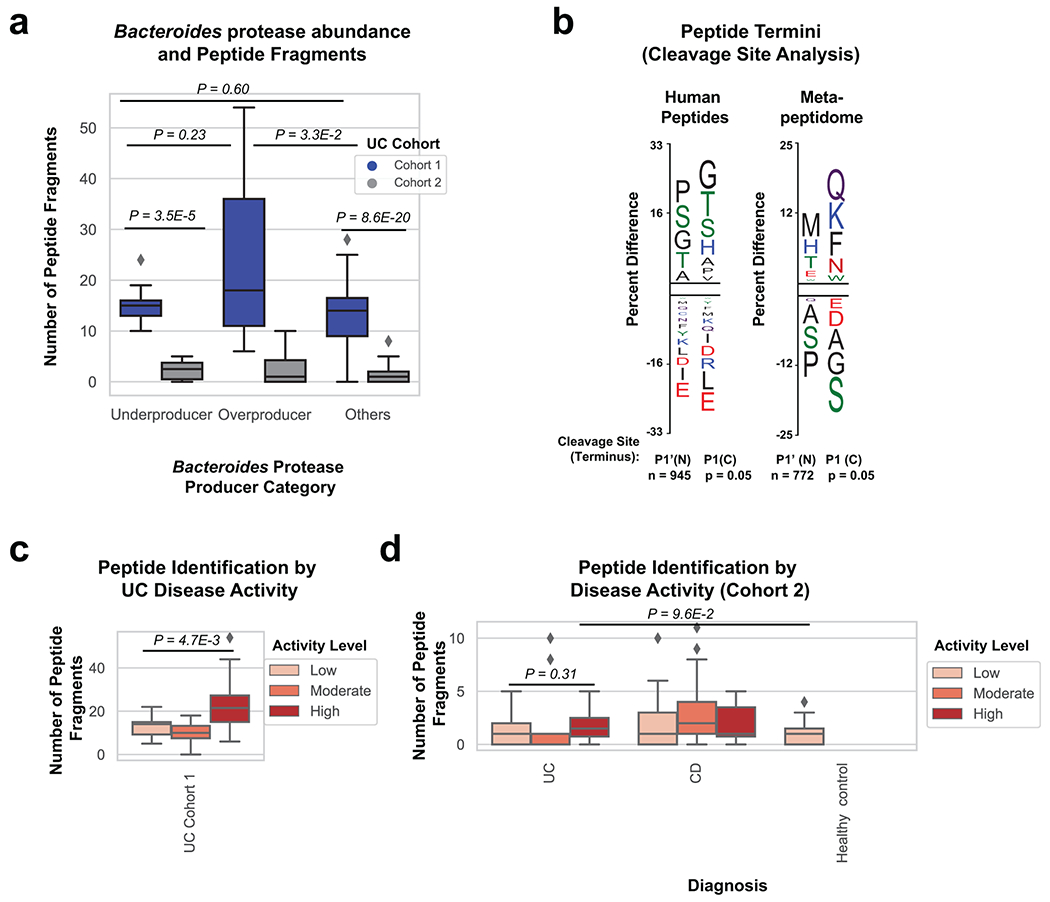
a, Comparison of peptide fragments identified in patients with varying abundance of Bacteroides proteases. Overproducers from UC cohort 1 had increased peptide fragments in comparison to other patients (Two-tailed t-test P=3.5E-2). Data was derived from n=8, 9, 23 UC cohort 1 samples and n=6, 6, 49 UC cohort 2 samples from patients classified as underproducer, overproducer and other respectively. b, Peptide termini indicate unique proteolysis of human and microbial proteins. The frequency of each amino acid within the N and C terminus of human and de-novo peptides was compared to either the human proteome or the total amino acid content of de novo peptides. The Y-axis represents the percent difference of each residue and the letter indicates the amino acid associated with the difference. The N and C terminus are shown separately and each residue is colored by chemical property (Green = polar, Black = Hydrophobic, Red = Acidic, Blue = Basic, Purple = Neutral). c, Peptide fragment identification comparison by disease activity in UC cohort 1. Boxplots with a two-tailed t-test p-value is shown (P=4.7E-3). Data was derived from n=18, 12, 10 patient samples with low moderate or high disease activity respectively. d, Peptide fragment identification comparison by disease and disease activity state for cohort 2 samples. Boxplots are shown with overlaid two-tailed t-test p-values. Data was derived from n=19 healthy controls, n=39, 30, 12 UC samples, and n=64, 30, 8 CD samples from patients of low, moderate and high activity respectively. Boxplots in (a,c,d) are defined by the median, quartiles and 1.5x inter-quartile range.
Extended Data Fig. 8. Determining the impact of Bacteroides species on TEER using co-culture, supernatants and protease inhibitors.
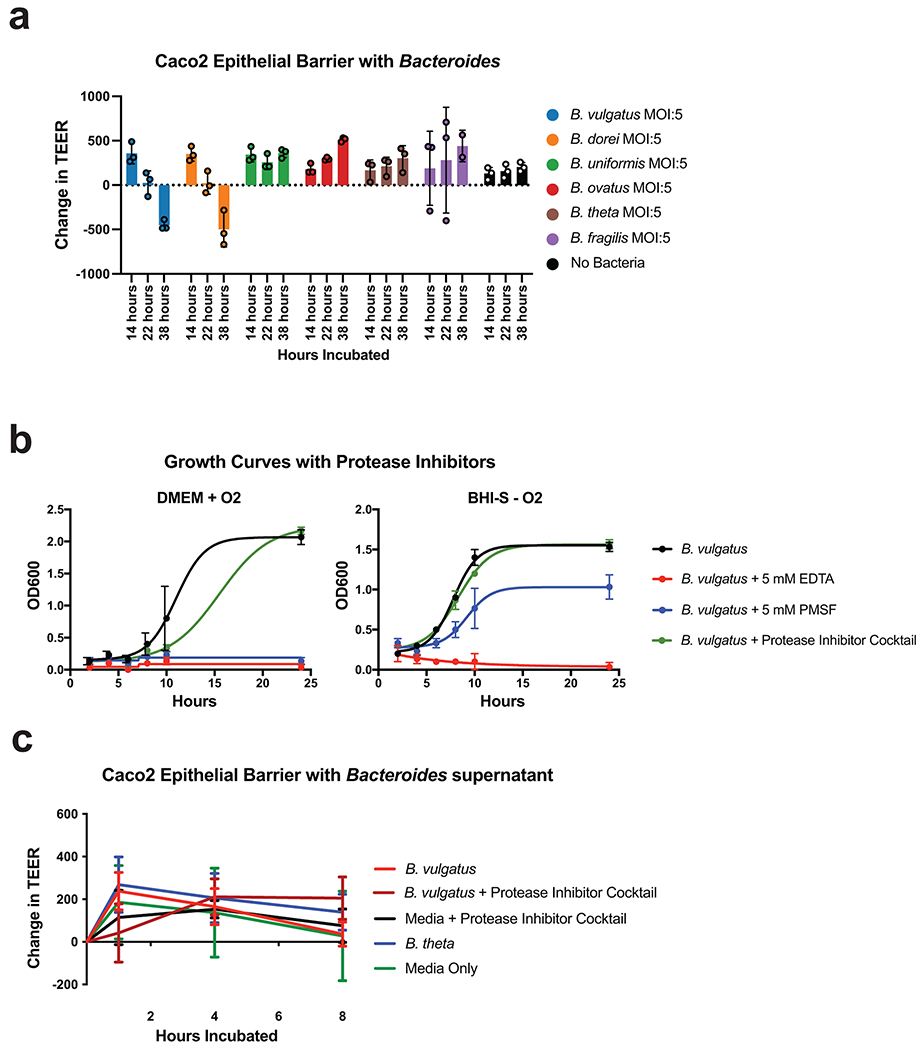
a, Bacteroides vulgatus and Bacteroides dorei, but not other Bacteroides species disrupt Caco-2 epithelial barriers. Barplots are showing the mean and standard deviation of the change in TEER at different time points. Data was derived from n=3 independent cultures collected over n=2 independent experiments. b, Growth curves of Bacteroides vulgatus with protease inhibitors under different growth conditions. OD600 was measured at indicated time points and a non-linear fit is shown. Data was derived from n=3 independent cultures collected over n=1 independent experiments. c, Supernatants from Bacteroides in mid-log phase growth do not significantly impact TEER. B. vulgatus and B. theta were grown to mid-log phase, and their supernatants were concentrated and added to Caco-2 monolayers. TEER was measured at the initial time-point and compared to TEER measured after 1, 4, and 8 hours of incubation. Plotted are the mean and SEM from n=3 independent experiments each representing the mean of n=3 independent wells/experiment (n=4 wells/experiment for B. vulgatus group). No significant differences were found at any timepoint.
Extended Data Fig. 9. Additional measurements from fecal transplant experiments.
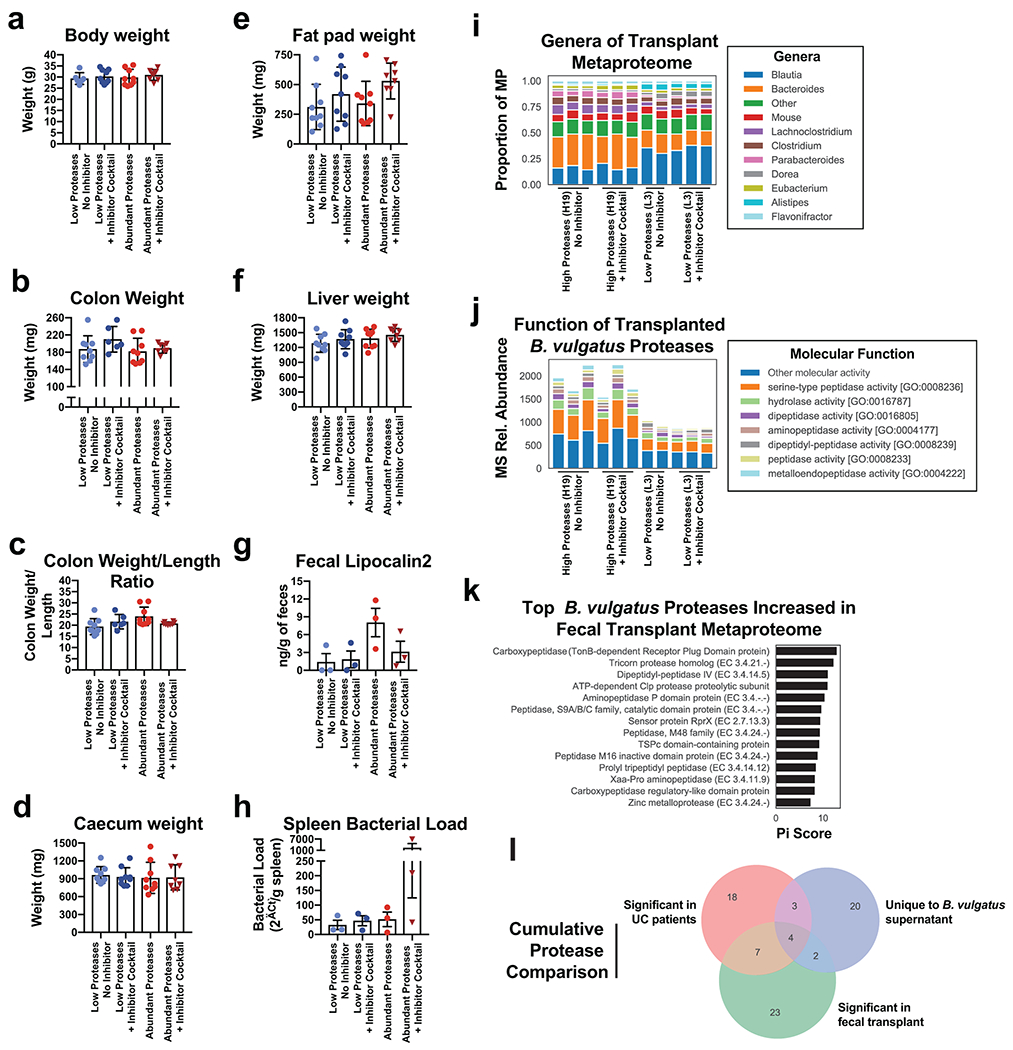
a-f Barplots showing the mean +/− SD of macroscopic organ measurements from fecal transplant of UC patients samples in IL10−/− mice with or without administration of a protease inhibitor. Dots represent one mouse, with each group representing results from 3 UC patient fecal samples with each sample given to 3 co-housed mice. Measurements include final weight of the mice (a), colon weight (b), ratios of the colon weight to length (c), caecum weight (d), fat pad weight (e), liver weight (f). g-h Barplots showing the mean +/− SEM for the concentration of an intestinal inflammatory marker, fecal lipocalin2 (g), and amount of 16S rRNA in the spleen of mice for an estimate of the splenic bacterial load (h). Each dot in g-h represents the mean of n=3 mice transplanted with the same UC fecal sample (with the exception of a mean from n=2 mice for one patient sample in the Abundant Proteases + Inhibitor Cocktail group) from n=2 independent experiments. i, Metaproteome genera composition of mice transplanted with UC fecal samples. Fecal samples taken at 8-weeks from mice transplanted with one high protease containing sample (H19) and one control patient sample (L3) were analyzed by mass spectrometry based metaproteomics. Stacked barplots are shown for each mouse displaying the proportion of protein signal derived from the most common genera. j, Molecular function of B. vulgatus proteases identified in mice receiving UC fecal samples. The relative abundance of each B. vulgatus protease is shown in stacked barplots grouped by the Gene Ontology molecular function associated with each protein. k, Top B. vulgatus or B. dorei proteases associated with the fecal samples of mice receiving the H19 sample. Each protein is ranked by pi-score, which combines two-sided t-test p-values and the fold-change difference between all H19 and L3 samples. l, Cumulative protease comparisons. A venndiagram is shown comparing the protein names of B. vulgatus or B. dorei proteases from four independent proteomics experiments performed in this study. A full list of the Bacteroides proteases identified in this analysis can be found in Supplementary Table 4.
Extended Data Fig. 10. Working hypothesis.
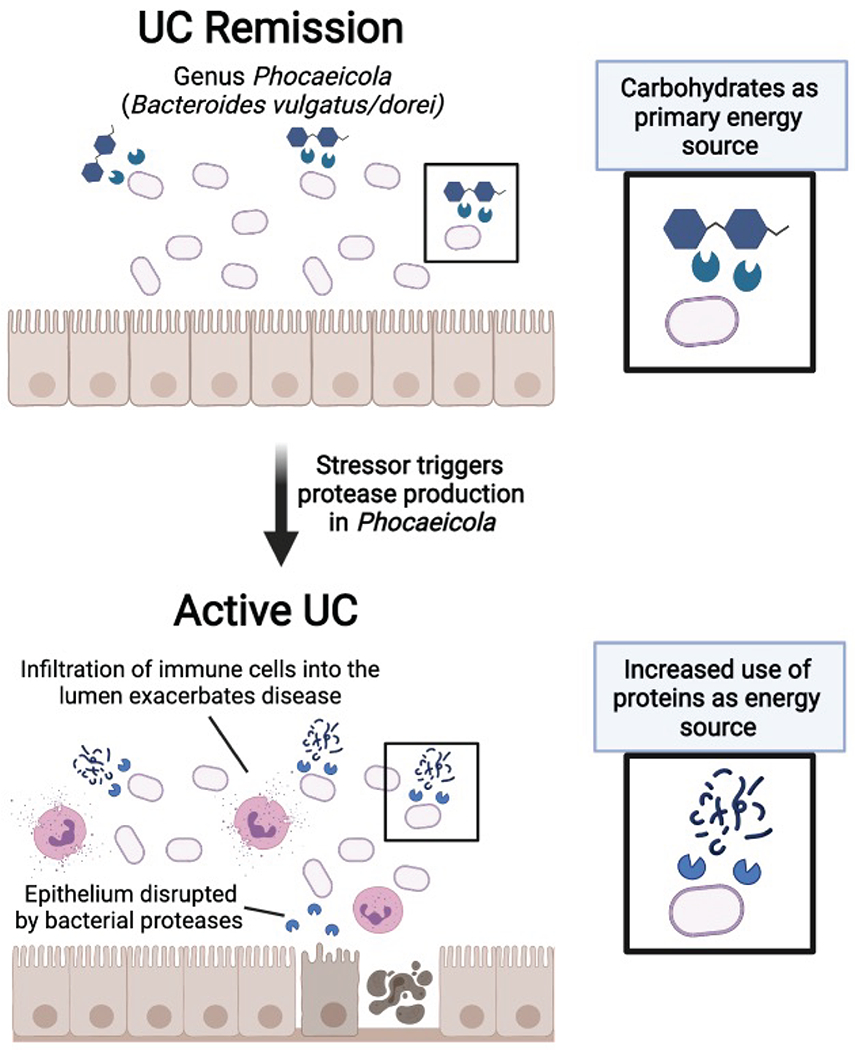
The results of our study may indicate that certain species from the genus Bacteroides, particularly those recently reclassified under the genus Phocaeicola (e.g. Bacteroides vulgatus & Bacteroides dorei), may be implicated in the transition from remission to active disease in UC. We hypothesize that a stressor in the UC gut such as nutrient deprivation or cell-to-cell competition may increase protease production, and a switch in the utilization of carbohydrates to proteins as a nutrient source. Some of these proteases may be involved in the disruption of the epithelial barrier, allowing an influx of innate immune cells which further exacerbate disease.
Supplementary Material
Bacteroides proteases identified in UC patients, bacterial supernatants, and fecal material from humanized mice. Protein names are listed of peptidases or proteases identified throughout the multiple proteomic experiments from this study. Lists are provided for any proteases from B. vulgatus or B. dorei that were positively associated (r > 0.3) with UC patient disease activity from either cohort. Additionally, proteases or peptidases identified in the supernatant from different species of Bacteroides are listed. Finally, from a metaproteomic analysis of the fecal material from mice humanized by UC patient fecal samples, B. vulgatus or B. dorei proteases increased (π > 1) in mice transplanted with a sample overabundant in proteases in comparison to mice transplanted without overabundant proteases.
Association of UC clinical variables to alpha- and beta-diversity. For alpha-diversity, Kruskal-Wallis tests were performed on each of the listed categorical variables, and linear regression was applied for quantitative variable. P-values are reported for all tests, and r-values are reported for quantitative variables. When more than two categories were present in categorical variables, the p-value between the two largest categories was reported. For beta-diversity, categorical variables were tested through PERMANOVA, continuous variables were tested through Adonis. P-values followed by Pseudo-F values for each category are reported. For continuous variables, R2 values are reported after Pseudo-F values. Testing was based on Bray-Curtis distance matrices unless otherwise specified. Significance level is indicated according to p-value (* < 0.05, ** < 0.01, *** < 0.001).
Features of importance to predicting UC disease activity. Feature importance values from the 100 random forest iterations predicting UC disease activity are reported for each -omic data type from both UC cohorts. The top 100 features from each data type are provided ranked by the summed importance scores from both cohorts. From the combined datasets, the top 100 features and annotation information related to each feature’s data type is also provided. This data corresponds to the random forest results shown in Fig. 2c.
Tables containing the underlying data from Extended Data Figure 2.
Tables containing the underlying data from Extended Data Figure 8.
Multiple files related to in vitro and in vivo studies displayed in Figure 5. This includes the raw data from Caco-2 co-cultures with Bacteroides vulgatus and protease inhibitors (Figure 5b–c), the original image files from Figure 5d, the quantification of cell morphology (Figure 5e), original data from the monocolonization experiments (Figure 5f–i), and the original data from fecal transplant studies (Figure 5j–n).
Tables containing denovo peptide identifications and data from experiments of B. vulgatus supernatant protease activity with different inhibitors.
ACKNOWLEDGMENTS
P.S.D and R.H.M. were supported through a UCSD training grant from the NIH/NIDDK Gastroenterology Training Program (T32 DK007202, R.H.M., P.S.D). P.S.D was also supported by an American Gastroenterology Association Research Scholar Award. We would like to acknowledge Eric Griffis, Daphne Bindels and the Nikon Imaging Center at UCSD for help with confocal microscopy. We also acknowledge the UCSD Neuroscience Microscopy Shared Facility (NS047101). This study was supported in part by NIDDK-funded San Diego Digestive Diseases Research Center (P30 DK120515, D.J.G., P.S.D). This study was also funded in part by the UCSD Collaborative Center of Multiplexed Proteomics.
Footnotes
Code Availability
The code used in analysis and visualization of data is available at https://github.com/knightlab-analyses/uc-severity-multiomics.
Statistics & Reproducibility
Multi -omic data was collected and analyzed in two independent experiments to increase the likelihood of reproducibility. Sample sizes from each cohort were largely driven by technical and financial constraints as opposed to power analysis, but our sample sizes are similar to those reported in previous publications7,8,17. Several samples were removed after the identification of bacterial blooms or red blood cell contamination as indicated in the metadata. Experiments were randomized during sample processing.
COMPETING INTERESTS
R.H.M., P.S.D and D.J.G. have jointly filed for a patent based on this work (International Application No. PCT/US2020/057784). Over the course of the publication process, R.H.M. started employment at Precidiag Inc., a company which has licensed the patent based on this work. All other authors declare no competing interests.
Data Availability
Metabolomic data, proteomic data and additional supplementary files for reanalyzing the data collected here are available online at https://massive.ucsd.edu. (Cohort 1 proteomics & metabolomics study ID MSV000082094, Cohort 2 study ID MSV000086509, Cohort 2 metabolomics study ID MSV000084908). Proteomic data and supplementary files for reanalyzing data collected from the fecal transplant study and Bacteroides supernatant are under MassIVE identifiers MSV000086510 and MSV000086511 respectively. Genomic data has been uploaded through EBI https://www.ebi.ac.uk/ena under the study identifiers PRJEB42151 for Cohort 1 and PRJEB42155 for Cohort 2. Comparisons with data generated from this study were also made with proteomics data downloaded from the IBD multi-omics database (https://ibdmdb.org/tunnel/public/HMP2/Proteomics/1633/rawfiles). Databases used in this study include UniRef50 (https://www.uniprot.org/downloads), the human proteome (https://www.uniprot.org/proteomes/UP000005640), mouse proteome (https://www.uniprot.org/proteomes/UP000000589), B. vulgatus proteome (https://www.uniprot.org/proteomes/UP000002861), B. theta proteome (https://www.uniprot.org/proteomes/UP000001414), B. dorei proteome (https://www.uniprot.org/proteomes/UP000005974), a microbial genome database (https://biocore.github.io/wol/), and a human gut microbiome database (https://db.cngb.org/microbiome/genecatalog/genecatalog_human/). Source data is available for in vitro and in vivo experiments.
REFERENCES
- 1.Fumery M et al. Natural History of Adult Ulcerative Colitis in Population-based Cohorts: A Systematic Review. Clin Gastroenterol Hepatol 16, 343–356 e343, doi: 10.1016/j.cgh.2017.06.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dulai PS, Siegel CA, Colombel JF, Sandborn WJ & Peyrin-Biroulet L Systematic review: Monotherapy with antitumour necrosis factor alpha agents versus combination therapy with an immunosuppressive for IBD. Gut 63, 1843–1853, doi: 10.1136/gutjnl-2014-307126 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Sartor RB & Wu GD Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 152, 327–339 e324, doi: 10.1053/j.gastro.2016.10.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schirmer M et al. Compositional and Temporal Changes in the Gut Microbiome of Pediatric Ulcerative Colitis Patients Are Linked to Disease Course. Cell Host Microbe 24, 600–610 e604, doi: 10.1016/j.chom.2018.09.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen ZH et al. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J Gastroentero 24, 5–14, doi: 10.3748/wjg.v24.i1.5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halfvarson J et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol 2, 17004, doi: 10.1038/nmicrobiol.2017.4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662, doi: 10.1038/s41586-019-1237-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franzosa EA et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol, doi: 10.1038/s41564-018-0306-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campieri M & Gionchetti P Bacteria as the cause of ulcerative colitis. Gut 48, 132–135, doi: 10.1136/gut.48.1.132 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan I et al. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 8, doi: 10.3390/pathogens8030126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mills RH et al. Evaluating Metagenomic Prediction of the Metaproteome in a 4.5-Year Study of a Patient with Crohn’s Disease. mSystems 4, e00337–00318, doi: 10.1128/mSystems.00337-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verberkmoes NC et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J 3, 179–189, doi: 10.1038/ismej.2008.108 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Li L, Butcher J, Stintzi A & Figeys D Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 7, 154, doi: 10.1186/s40168-019-0767-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CW et al. Isobaric Labeling Quantitative Metaproteomics for the Study of Gut Microbiome Response to Arsenic. J Proteome Res 18, 970–981, doi: 10.1021/acs.jproteome.8b00666 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Tran HQ et al. Associations of the fecal microbial proteome composition and proneness to diet-induced obesity. Mol Cell Proteomics, doi: 10.1074/mcp.RA119.001623 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jansson JK & Baker ES A multi-omic future for microbiome studies. Nat Microbiol 1, 16049, doi: 10.1038/nmicrobiol.2016.49 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Zhang X et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nature Communications 9, doi:ARTN 2873 10.1038/s41467-018-05357-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erickson AR et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One 7, e49138, doi: 10.1371/journal.pone.0049138 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergnolle N Protease inhibition as new therapeutic strategy for GI diseases. Gut 65, 1215–1224, doi: 10.1136/gutjnl-2015-309147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galipeau HJ et al. Novel Fecal Biomarkers That Precede Clinical Diagnosis of Ulcerative Colitis. Gastroenterology 160, 1532–1545, doi: 10.1053/j.gastro.2020.12.004 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Lewis JD et al. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis 14, 1660–1666, doi: 10.1002/ibd.20520 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narula N, Alshahrani AA, Yuan Y, Reinisch W & Colombel JF Patient-Reported Outcomes and Endoscopic Appearance of Ulcerative Colitis: A Systematic Review and Meta-Analysis. Clin Gastroenterol Hepatol, doi: 10.1016/j.cgh.2018.06.015 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Dulai PS, Levesque BG, Feagan BG, D’Haens G & Sandborn WJ Assessment of mucosal healing in inflammatory bowel disease: review. Gastrointest Endosc 82, 246–255, doi: 10.1016/j.gie.2015.03.1974 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh AJ, Bryant RV & Travis SP Current best practice for disease activity assessment in IBD. Nat Rev Gastroenterol Hepatol 13, 567–579, doi: 10.1038/nrgastro.2016.128 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Bakir MA, Sakamoto M, Kitahara M, Matsumoto M & Benno Y Bacteroides dorei sp. nov., isolated from human faeces. Int J Syst Evol Microbiol 56, 1639–1643, doi: 10.1099/ijs.0.64257-0 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Kulagina EV et al. Species Composition of Bacteroidales Order Bacteria in the Feces of Healthy People of Various Ages. Biosci Biotech Bioch 76, 169–171, doi: 10.1271/bbb.110434 (2012). [DOI] [PubMed] [Google Scholar]
- 27.O’Donoghue AJ et al. Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS One 8, e75141, doi: 10.1371/journal.pone.0075141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nemoto TK & Ohara-Nemoto Y Exopeptidases and gingipains in Porphyromonas gingivalis as prerequisites for its amino acid metabolism. The Japanese dental science review 52, 22–29, doi: 10.1016/j.jdsr.2015.08.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumagai Y et al. Enzymatic properties of dipeptidyl aminopeptidase IV produced by the periodontal pathogen Porphyromonas gingivalis and its participation in virulence. Infect Immun 68, 716–724 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deacon CF & Lebovitz HE Comparative review of dipeptidyl peptidase-4 inhibitors and sulphonylureas. Diabetes Obes Metab 18, 333–347, doi: 10.1111/dom.12610 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Mimura S et al. Dipeptidyl peptidase-4 inhibitor anagliptin facilitates restoration of dextran sulfate sodium-induced colitis. Scand J Gastroenterol 48, 1152–1159, doi: 10.3109/00365521.2013.832366 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Donaldson GP, Lee SM & Mazmanian SK Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14, 20–32, doi: 10.1038/nrmicro3552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wexler HM Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20, 593–621, doi: 10.1128/CMR.00008-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foley MH, Cockburn DW & Koropatkin NM The Sus operon: a model system for starch uptake by the human gut Bacteroidetes. Cell Mol Life Sci 73, 2603–2617, doi: 10.1007/s00018-016-2242-x (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onderdonk AB, Franklin ML & Cisneros RL Production of experimental ulcerative colitis in gnotobiotic guinea pigs with simplified microflora. Infect Immun 32, 225–231, doi: 10.1128/iai.32.1.225-231.1981 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bamba T, Matsuda H, Endo M & Fujiyama Y The pathogenic role of Bacteroides vulgatus in patients with ulcerative colitis. Journal of gastroenterology 30 Suppl 8, 45–47 (1995). [PubMed] [Google Scholar]
- 37.Waidmann M et al. Bacteroides vulgatus protects against Escherichia coli-induced colitis in gnotobiotic interleukin-2-deficient mice. Gastroenterology 125, 162–177, doi: 10.1016/s0016-5085(03)00672-3 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Sellon RK et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 66, 5224–5231, doi: 10.1128/iai.66.11.5224-5231.1998 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vich Vila A et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci Transl Med 10, doi: 10.1126/scitranslmed.aap8914 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Zhou Y & Zhi F Lower Level of Bacteroides in the Gut Microbiota Is Associated with Inflammatory Bowel Disease: A Meta-Analysis. Biomed Res Int 2016, 5828959, doi: 10.1155/2016/5828959 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.García-López M et al. Analysis of 1,000 Type-Strain Genomes Improves Taxonomic Classification of Bacteroidetes. Front Microbiol 10, 2083, doi: 10.3389/fmicb.2019.02083 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimshoni E, Yablecovitch D, Baram L, Dotan I & Sagi I ECM remodelling in IBD: innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation. Gut 64, 367–372, doi: 10.1136/gutjnl-2014-308048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Spaendonk H et al. Regulation of intestinal permeability: The role of proteases. World J Gastroenterol 23, 2106–2123, doi: 10.3748/wjg.v23.i12.2106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steck N, Mueller K, Schemann M & Haller D Bacterial proteases in IBD and IBS. Gut 61, 1610–1618, doi: 10.1136/gutjnl-2011-300775 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Carroll IM & Maharshak N Enteric bacterial proteases in inflammatory bowel disease-pathophysiology and clinical implications. World J Gastroenterol 19, 7531–7543, doi: 10.3748/wjg.v19.i43.7531 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kriaa A et al. Serine proteases at the cutting edge of IBD: Focus on gastrointestinal inflammation. Faseb j 34, 7270–7282, doi: 10.1096/fj.202000031RR (2020). [DOI] [PubMed] [Google Scholar]
- 47.Denadai-Souza A et al. Functional Proteomic Profiling of Secreted Serine Proteases in Health and Inflammatory Bowel Disease. Sci Rep 8, 7834, doi: 10.1038/s41598-018-26282-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Sullivan S, Gilmer JF & Medina C Matrix metalloproteinases in inflammatory bowel disease: an update. Mediators Inflamm 2015, 964131, doi: 10.1155/2015/964131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biancheri P et al. Proteolytic cleavage and loss of function of biologic agents that neutralize tumor necrosis factor in the mucosa of patients with inflammatory bowel disease. Gastroenterology 149, 1564–1574 e1563, doi: 10.1053/j.gastro.2015.07.002 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Gordon MH et al. N-Terminomics/TAILS Profiling of Proteases and Their Substrates in Ulcerative Colitis. Acs Chem Biol 14, 2471–2483, doi: 10.1021/acschembio.9b00608 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Roka R et al. Colonic luminal proteases activate colonocyte proteinase-activated receptor-2 and regulate paracellular permeability in mice. Neurogastroenterol Motil 19, 57–65, doi: 10.1111/j.1365-2982.2006.00851.x (2007). [DOI] [PubMed] [Google Scholar]
- 52.Ordas I, Eckmann L, Talamini M, Baumgart DC & Sandborn WJ Ulcerative colitis. Lancet 380, 1606–1619, doi: 10.1016/S0140-6736(12)60150-0 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Sałaga M, Sobczak M & Fichna J Inhibition of proteases as a novel therapeutic strategy in the treatment of metabolic, inflammatory and functional diseases of the gastrointestinal tract. Drug Discov Today 18, 708–715, doi: 10.1016/j.drudis.2013.03.004 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Riepe SP, Goldstein J & Alpers DH Effect of secreted Bacteroides proteases on human intestinal brush border hydrolases. J Clin Invest 66, 314–322, doi: 10.1172/JCI109859 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Obiso RJ Jr., Lyerly DM, Van Tassell RL & Wilkins TD Proteolytic activity of the Bacteroides fragilis enterotoxin causes fluid secretion and intestinal damage in vivo. Infect Immun 63, 3820–3826 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valguarnera E & Wardenburg JB Good Gone Bad: One Toxin Away From Disease for Bacteroides fragilis. J Mol Biol 432, 765–785, doi: 10.1016/j.jmb.2019.12.003 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Elhenawy W, Debelyy MO & Feldman MF Preferential packing of acidic glycosidases and proteases into Bacteroides outer membrane vesicles. MBio 5, e00909–00914, doi: 10.1128/mBio.00909-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marotz C et al. DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques 62, 290–293, doi: 10.2144/000114559 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Caporaso JG et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624, doi: 10.1038/ismej.2012.8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson LR et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463, doi: 10.1038/nature24621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, doi: 10.1093/bioinformatics/btu170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li DH, Liu CM, Luo RB, Sadakane K & Lam TW MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676, doi: 10.1093/bioinformatics/btv033 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119, doi: 10.1186/1471-2105-11-119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60, doi: 10.1038/nmeth.3176 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Kanehisa M, Sato Y & Morishima K BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol 428, 726–731, doi: 10.1016/j.jmb.2015.11.006 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Zhu Q et al. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat Commun 10, 5477, doi: 10.1038/s41467-019-13443-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caporaso JG et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336, doi: 10.1038/nmeth.f.303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419, doi: 10.1038/nmeth.4197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koontz L TCA precipitation. Methods Enzymol 541, 3–10, doi: 10.1016/B978-0-12-420119-4.00001-X (2014). [DOI] [PubMed] [Google Scholar]
- 71.Villen J & Gygi SP The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc 3, 1630–1638, doi: 10.1038/nprot.2008.150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haas W et al. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics 5, 1326–1337, doi: 10.1074/mcp.M500339-MCP200 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Wessel D & Flugge UI A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138, 141–143 (1984). [DOI] [PubMed] [Google Scholar]
- 74.Van Rechem C et al. Lysine Demethylase KDM4A Associates with Translation Machinery and Regulates Protein Synthesis. Cancer Discov 5, 255–263, doi: 10.1158/2159-8290.Cd-14-1326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tolonen AC & Haas W Quantitative proteomics using reductive dimethylation for stable isotope labeling. J Vis Exp, doi: 10.3791/51416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lapek JD Jr. et al. Defining Host Responses during Systemic Bacterial Infection through Construction of a Murine Organ Proteome Atlas. Cell Syst, doi: 10.1016/j.cels.2018.04.010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tolonen AC et al. Proteome-wide systems analysis of a cellulosic biofuel-producing microbe. Mol Syst Biol 7, 461, doi: 10.1038/msb.2010.116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thompson A et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75, 1895–1904 (2003). [DOI] [PubMed] [Google Scholar]
- 79.Wang Y et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026, doi: 10.1002/pmic.201000722 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lapek JD Jr., Lewinski MK, Wozniak JM, Guatelli J & Gonzalez DJ Quantitative Temporal Viromics of an Inducible HIV-1 Model Yields Insight to Global Host Targets and Phospho-Dynamics Associated with Vpr. Mol Cell Proteomics, doi: 10.1074/mcp.M116.066019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989, doi: 10.1016/1044-0305(94)80016-2 (1994). [DOI] [PubMed] [Google Scholar]
- 82.Beausoleil SA, Villen J, Gerber SA, Rush J & Gygi SP A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24, 1285–1292, doi: 10.1038/nbt1240 (2006). [DOI] [PubMed] [Google Scholar]
- 83.Huttlin EL et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189, doi: 10.1016/j.cell.2010.12.001 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods 4, 207–214, doi: 10.1038/nmeth1019 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Elias JE, Haas W, Faherty BK & Gygi SP Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods 2, 667–675, doi: 10.1038/nmeth785 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Peng J, Elias JE, Thoreen CC, Licklider LJ & Gygi SP Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2, 43–50 (2003). [DOI] [PubMed] [Google Scholar]
- 87.Jagtap P et al. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics 13, 1352–1357, doi: 10.1002/pmic.201200352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang M et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol 34, 828–837, doi: 10.1038/nbt.3597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pluskal T, Castillo S, Villar-Briones A & Oresic M MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395, doi: 10.1186/1471-2105-11-395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tripathi A et al. Chemically informed analyses of metabolomics mass spectrometry data with Qemistree. Nat Chem Biol, doi: 10.1038/s41589-020-00677-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duhrkop K, Shen H, Meusel M, Rousu J & Bocker S Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc Natl Acad Sci U S A 112, 12580–12585, doi: 10.1073/pnas.1509788112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Djoumbou Feunang Y et al. ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J Cheminform 8, 61, doi: 10.1186/s13321-016-0174-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang J et al. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics 11, M111 010587, doi: 10.1074/mcp.M111.010587 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Quinn RA et al. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 7, 23, doi: 10.1186/s40168-019-0636-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li J et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32, 834–841, doi: 10.1038/nbt.2942 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Gonzalez A et al. Qiita: rapid, web-enabled microbiome meta-analysis. Nat Methods 15, 796–798, doi: 10.1038/s41592-018-0141-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bolyen E et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37, 852–857, doi: 10.1038/s41587-019-0209-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Szklarczyk D et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452, doi: 10.1093/nar/gku1003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504, doi: 10.1101/gr.1239303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Colaert N, Helsens K, Martens L, Vandekerckhove J & Gevaert K Improved visualization of protein consensus sequences by iceLogo. Nat Methods 6, 786–787, doi: 10.1038/nmeth1109-786 (2009). [DOI] [PubMed] [Google Scholar]
- 101.Wang F et al. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol 166, 409–419, doi: 10.1016/s0002-9440(10)62264-x (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tremelling M et al. IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology 132, 1657–1664, doi: 10.1053/j.gastro.2007.02.051 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wakula M et al. Quantification of Cell-Substrate Adhesion Area and Cell Shape Distributions in MCF7 Cell Monolayers. J Vis Exp, doi: 10.3791/61461 (2020). [DOI] [PubMed] [Google Scholar]
- 104.Legland D, Arganda-Carreras I & Andrey P MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32, 3532–3534, doi: 10.1093/bioinformatics/btw413 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Moschen AR et al. Lipocalin 2 Protects from Inflammation and Tumorigenesis Associated with Gut Microbiota Alterations. Cell Host Microbe 19, 455–469, doi: 10.1016/j.chom.2016.03.007 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Katakura K et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest 115, 695–702, doi: 10.1172/jci22996 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chassaing B et al. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 7, e44328, doi: 10.1371/journal.pone.0044328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiao Y et al. A novel significance score for gene selection and ranking. Bioinformatics 30, 801–807, doi: 10.1093/bioinformatics/btr671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mills RH et al. Organ-level protein networks as a reference for the host effects of the microbiome. Genome Res 30, 276–286, doi: 10.1101/gr.256875.119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacteroides proteases identified in UC patients, bacterial supernatants, and fecal material from humanized mice. Protein names are listed of peptidases or proteases identified throughout the multiple proteomic experiments from this study. Lists are provided for any proteases from B. vulgatus or B. dorei that were positively associated (r > 0.3) with UC patient disease activity from either cohort. Additionally, proteases or peptidases identified in the supernatant from different species of Bacteroides are listed. Finally, from a metaproteomic analysis of the fecal material from mice humanized by UC patient fecal samples, B. vulgatus or B. dorei proteases increased (π > 1) in mice transplanted with a sample overabundant in proteases in comparison to mice transplanted without overabundant proteases.
Association of UC clinical variables to alpha- and beta-diversity. For alpha-diversity, Kruskal-Wallis tests were performed on each of the listed categorical variables, and linear regression was applied for quantitative variable. P-values are reported for all tests, and r-values are reported for quantitative variables. When more than two categories were present in categorical variables, the p-value between the two largest categories was reported. For beta-diversity, categorical variables were tested through PERMANOVA, continuous variables were tested through Adonis. P-values followed by Pseudo-F values for each category are reported. For continuous variables, R2 values are reported after Pseudo-F values. Testing was based on Bray-Curtis distance matrices unless otherwise specified. Significance level is indicated according to p-value (* < 0.05, ** < 0.01, *** < 0.001).
Features of importance to predicting UC disease activity. Feature importance values from the 100 random forest iterations predicting UC disease activity are reported for each -omic data type from both UC cohorts. The top 100 features from each data type are provided ranked by the summed importance scores from both cohorts. From the combined datasets, the top 100 features and annotation information related to each feature’s data type is also provided. This data corresponds to the random forest results shown in Fig. 2c.
Tables containing the underlying data from Extended Data Figure 2.
Tables containing the underlying data from Extended Data Figure 8.
Multiple files related to in vitro and in vivo studies displayed in Figure 5. This includes the raw data from Caco-2 co-cultures with Bacteroides vulgatus and protease inhibitors (Figure 5b–c), the original image files from Figure 5d, the quantification of cell morphology (Figure 5e), original data from the monocolonization experiments (Figure 5f–i), and the original data from fecal transplant studies (Figure 5j–n).
Tables containing denovo peptide identifications and data from experiments of B. vulgatus supernatant protease activity with different inhibitors.
Data Availability Statement
Metabolomic data, proteomic data and additional supplementary files for reanalyzing the data collected here are available online at https://massive.ucsd.edu. (Cohort 1 proteomics & metabolomics study ID MSV000082094, Cohort 2 study ID MSV000086509, Cohort 2 metabolomics study ID MSV000084908). Proteomic data and supplementary files for reanalyzing data collected from the fecal transplant study and Bacteroides supernatant are under MassIVE identifiers MSV000086510 and MSV000086511 respectively. Genomic data has been uploaded through EBI https://www.ebi.ac.uk/ena under the study identifiers PRJEB42151 for Cohort 1 and PRJEB42155 for Cohort 2. Comparisons with data generated from this study were also made with proteomics data downloaded from the IBD multi-omics database (https://ibdmdb.org/tunnel/public/HMP2/Proteomics/1633/rawfiles). Databases used in this study include UniRef50 (https://www.uniprot.org/downloads), the human proteome (https://www.uniprot.org/proteomes/UP000005640), mouse proteome (https://www.uniprot.org/proteomes/UP000000589), B. vulgatus proteome (https://www.uniprot.org/proteomes/UP000002861), B. theta proteome (https://www.uniprot.org/proteomes/UP000001414), B. dorei proteome (https://www.uniprot.org/proteomes/UP000005974), a microbial genome database (https://biocore.github.io/wol/), and a human gut microbiome database (https://db.cngb.org/microbiome/genecatalog/genecatalog_human/). Source data is available for in vitro and in vivo experiments.