

SUMMARY
In diabetes, glucagon secretion from pancreatic α-cells is dysregulated. The underlying mechanisms, and whether dysfunction occurs uniformly amongst cells, remain unclear. We examined α-cells from human donors and mice using electrophysiological, transcriptomic, and computational approaches. Rising glucose suppresses α-cell exocytosis by reducing P/Q-type Ca2+ channel activity, and this is disrupted in type 2 diabetes (T2D). Upon high-fat-feeding of mice, α-cells shift towards a ‘β-cell-like’ electrophysiologic profile in concert with indications of impaired identity. In human α-cells we identify links between cell membrane properties and cell surface signalling receptors, mitochondrial respiratory complex assembly, and cell maturation. Cell type classification using machine learning of electrophysiology data demonstrates a heterogenous loss of ‘electrophysiologic identity’ in α-cells from donors with T2D. Indeed, a sub-set of α-cells with impaired exocytosis is defined by an enrichment in progenitor and lineage markers, and up-regulation of an immature transcriptomic phenotype, suggesting important links between α-cell maturation state and dysfunction.
Keywords: diabetes, glucagon, patch-seq, modelling, islets of Langerhans
Graphical Abstract
eTOC:
In diabetes, glucagon secretion from pancreatic α-cells is dysregulated. Dai et al. examined electrical and transcriptomic α-cell phenotypes and find that dysfunction in type 2 diabetes is linked to cell maturation state and impaired α-cell identity. Notably, a sub-set of α-cells enriched for lineage markers appears uniquely susceptible to dysfunction.
INTRODUCTION
In concert with reduced insulin secretion from pancreatic β-cells in type 2 diabetes (T2D), disrupted glucagon secretion from α-cells contributes to hyperglycemia and impaired hypoglycemia counter-regulation (Girard, 2017). While insulin and glucagon secretion are both dependent upon electrical excitability and Ca2+-dependent exocytosis, the nature of the ion channels involved and their roles, and the impact of glucose-stimulation, differ in α- and β-cells. Insulin granule exocytosis is linked to Ca2+ entry via L-type Ca2+ channels, whereas glucagon secretion is coupled to P/Q-type Ca2+ channels (Marinis et al., 2010). Also, Na+ channels play a more prominent role in glucagon secretion (Barg et al.,2000; Göpel et al., 2000; Ramracheya et al., 2010) and differ in their regulation between these cell types (Zhang et al., 2014). In rodents such differences can distinguish α- from β-cells, either by Na+ current properties (Zhang et al., 2014) or ‘electrophysiological fingerprints’ (Briant et al., 2017).
Similar to β-cells the excitatory and secretory machinery in α-cells, including ion channel activities (Huang et al., 2011a), Ca2+ responses (Marchand and Piston, 2010; Reissaus and Piston, 2017; Shuai et al., 2016), glucagon content (Zadeh et al., 2020) and exocytotic capacity (Huang et al., 2011b) are heterogeneous. Indeed, the intracellular Ca2+ response of α-cells varies, with some suppressed by glucose and others activated (Shuai et al., 2016). This suggests a heterogeneity among these cell types, which is supported by single-cell transcriptomics (Camunas-Soler et al., 2020; Korsunsky et al., 2018). Recent reports suggest a sub-population of α-cells (or ‘α-like-cells’) that are proliferative. In mice these are identified by Slc38a5, which encodes an amino acid transporter (Kim et al., 2017). In humans these may be identified by the presence of ARX and cytosolic Sox9 (Lam et al., 2018). These cells could account for α-cell hyperplasia upon glucagon-receptor antagonism and may be a source of new β-cells (Meulen et al., 2017).
While little evidence so far suggests that α-cells dedifferentiate in diabetes, they can transdifferentiate in rodents following severe β-cell loss (Thorel et al., 2010) or genetic manipulation of transcription factor expression (Chakravarthy et al., 2017; Matsuoka et al., 2017). Interestingly, α-cells may show more plasticity than β-cells (Bramswig et al., 2013) as they appear to exist in distinct states characterized by chromatin accessibility at promotors for GCG, functional genes such as ABCC8, and at sites enriched in motifs for transcription factors of the RFX, GATA and NEUROD families, among others (Chiou et al., 2021). In type 1 diabetes, the expression of α-cell markers is reduced (Brissova et al., 2018) and in T2D α-cells express an immature transcriptomic profile (Avrahami et al., 2020).
We hypothesised that this plasticity influences α-cell membrane function, contributing to dysfunction in T2D. We used correlated electrophysiological and single-cell RNA-seq (patch-seq) of α-cells from human donors and mice to define a cell-autonomous glucose-regulation of α-cell Ca2+ channel activity and exocytosis that is associated with α-cell maturation state, and identify putative regulators of glucagon secretion. These include the mitochondrial respiratory chain complex and numerous cell surface receptors. In mice, high fat-feeding prompts some α-cells to adopt a ‘β-cell-like’ electrical profile and impaired identity. Similarly, in human T2D, α-cells enriched for markers of mitochondrial function and endocrine lineage such as NEUROD1, ISL1, NKX2-2, and ARX exhibit a selective induction of an immature transcriptional profile, impaired electrophysiological phenotype and dysregulated exocytosis. This suggests an important link between α-cell maturation, identity and dysfunction in T2D.
RESULTS
Glucose-mediated suppression of α-cell exocytosis is disrupted in T2D
In β-cells, glucose metabolism amplifies Ca2+-triggered exocytosis and insulin secretion (Ferdaoussi et al., 2015; Gembal et al., 1992; Sato et al., 1992), which is linked to the activation of L-type Ca2+ channels (Barg et al., 2001; Bokvist et al., 1995; Wiser et al., 1999). We examined the impact of glucose on glucagon exocytosis in α-cells from donors with no diabetes (ND) or with T2D (Suppl Table 1) following dispersion to single cells and identification by glucagon immunostaining (Fig 1A). In ND α-cells, exocytosis was highest at low glucose (1 mM) and was suppressed by increasing glucose (to 5–20 mM) (Fig 1B; Suppl Fig 1). In α-cells from donors with T2D, exocytosis was ~75% lower at 1 mM glucose than in α-cells from ND donors, and elevating glucose levels exerted a modest increase (Fig 1B; Suppl Fig 1A). The differences between ND and T2D cells, also seen when grouped by donor (Suppl Fig 1B), are consistent with a recent live-cell imaging study (Omar-Hmeadi et al., 2020). In ND α-cells voltage-dependent Ca2+ channel activity was lower at elevated glucose than in α-cells from donors with T2D, here recorded using Ba2+ as a charge carrier (Fig 1C; Suppl Fig 1C), and the relationship between Ca2+ entry and exocytosis was not altered (Suppl Fig 1D).
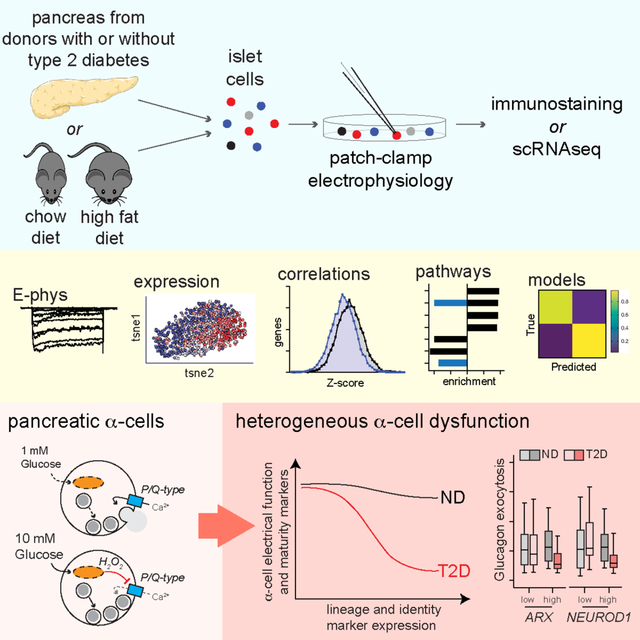
Figure 1. Glucose suppresses human α-cell exocytosis in concert with P/Q-channel activity.
A) Schematic diagram illustrating that human islets were isolated, dispersed and cultured for 1–2 days prior to whole-cell patch-clamp and subsequent α-cell identification by glucagon immunostaining.
B) Total exocytosis upon a series of ten membrane depolarizations with increasing glucose in ND α-cells (grey; n = 42, 28, 24, 20 cells from 9 donors) and T2D α-cells (pink; n = 29, 16, 12, 27 cells from 6 donors).
C) Voltage-dependent Ca2+ channel activity, with Ba2+ as a charge carrier, in ND α-cells with increasing glucose (n = 31, 24, 27 cells from 7 donors).
D) Effect of the P/Q-type Ca2+ channel activator GV-58 (10 μM) on Ca2+ current inactivation (left; n = 24, 28, 22, 21 cells) and charge entry (middle; n = 22, 20, 20, 23 cells) during a 500 ms depolarization from 70 to 0 mV, and total exocytosis (right; n = 23, 22, 28, 34 cells) at 1 and 10 mM glucose (3 donors).
E) Effect of the P/Q-type Ca2+ channel blocker agatoxin (100 nM) and the L-type Ca2+ channel blocker isradipine (10 μM) on ND α-cells exocytosis at 1 mM glucose (n = 34, 30, 21 cells from 5 donors).
F) Voltage-dependent Ca2+ channel activity at 0 mV (Ba2+ as a charge carrier) with increasing glucose in ND α-cells, with agatoxin (100 nM), and in T2D α-cells (n = 31, 16, 24, 15, 27, 18 cells from 7 ND donors, and 34, 31, 27 cells from 5 T2D donors).
G) Effect of agatoxin (100 nM) and the L-type Ca2+ channel blocker isradipine (10 μM) on total exocytosis in T2D α-cells at 1 and 10 mM glucose (n = 30, 31, 29, 32, 31, 29 cells from 5 T2D donors).
H) Putative scheme for glucose-regulation of depolarization-induced exocytosis in α-cells, and the dysfunction seen in T2D.
*P < 0.05; **P < 0.01; and ***P < 0.001 by one-way ANOVA (D,E,G) or two-way ANOVA (B,C,F) and Tukey post test compared with 1 mM glucose control, versus the ND control (C) or as indicated.
The majority of the human α-cell Ca2+ current is mediated by L-type and P/Q-type channels (Ramracheya et al., 2010), and the latter are directly linked to glucagon exocytosis (Dai et al., 2014; Ramracheya et al., 2010, 2018). The low α-cell exocytosis at elevated glucose could be reversed by the P/Q-type Ca2+ channel activator GV-58 (Tarr et al., 2012), which delays Ca2+ current inactivation (Fig 1D). The P/Q-type channel blocker agatoxin inhibited exocytosis from ND α-cells at 1 mM glucose while the L-type channel blocker isradipine did not (Fig 1E). Finally, the greater P/Q-type Ca2+ channel activity seen in ND α-cells at 1 mM glucose was largely absent in T2D (Fig 1F; Suppl Fig 1A), but still contributed to the modest glucose-dependent increase in exocytosis (Fig 1G). Thus, in ND α-cells increasing glucose inhibits P/Q-type Ca2+ channels to limit exocytosis, an effect that requires intact mitochondrial function (Suppl Fig 1E,F). In T2D α-cells, Ca2+ channel activity at low glucose is reduced, and increasing glucose facilitates exocytosis (Fig 1H).
Patch-seq highlights a role for the mitochondrial respiratory chain in α-cell exocytosis, and suggests poor responsiveness in immature α-cells
We performed patch-seq in α-cells, some initially pre-incubated at low glucose for 1 hour. However, exocytosis was low after an extended time at 1 mM glucose, due to depletion of glucagon granules (Suppl Fig 2). These pre-incubated cells were excluded from further analysis, and we performed transcriptome-wide correlations with exocytosis in 400 human α-cells exposed acutely to 1 and 10 mM glucose, from 31 donors (Fig 2A). ND α-cells at 1 mM were enriched for positively-correlated genes while negatively-correlated genes were enriched at 10 mM glucose (Fig 2B; Suppl Table 2). Gene-set enrichment analysis (GSEA) using Z-scores of transcripts found in >20% of cells highlighted pathways associated with elevated α-cell exocytosis at 1 mM glucose and lower exocytosis at 10 mM glucose (Fig 2C). Consistent with a role for metabolism in determining α-cell responsiveness, we found mitochondrial respiratory chain complex assembly as a positive correlate to exocytosis at low glucose and a negative correlate at high glucose (Fig 2C–D). A separate over-representation analysis (ORA) yielded similar results, highlighting the electron transport chain in the glucose-regulation of α-cell exocytosis (Suppl Fig 3A).
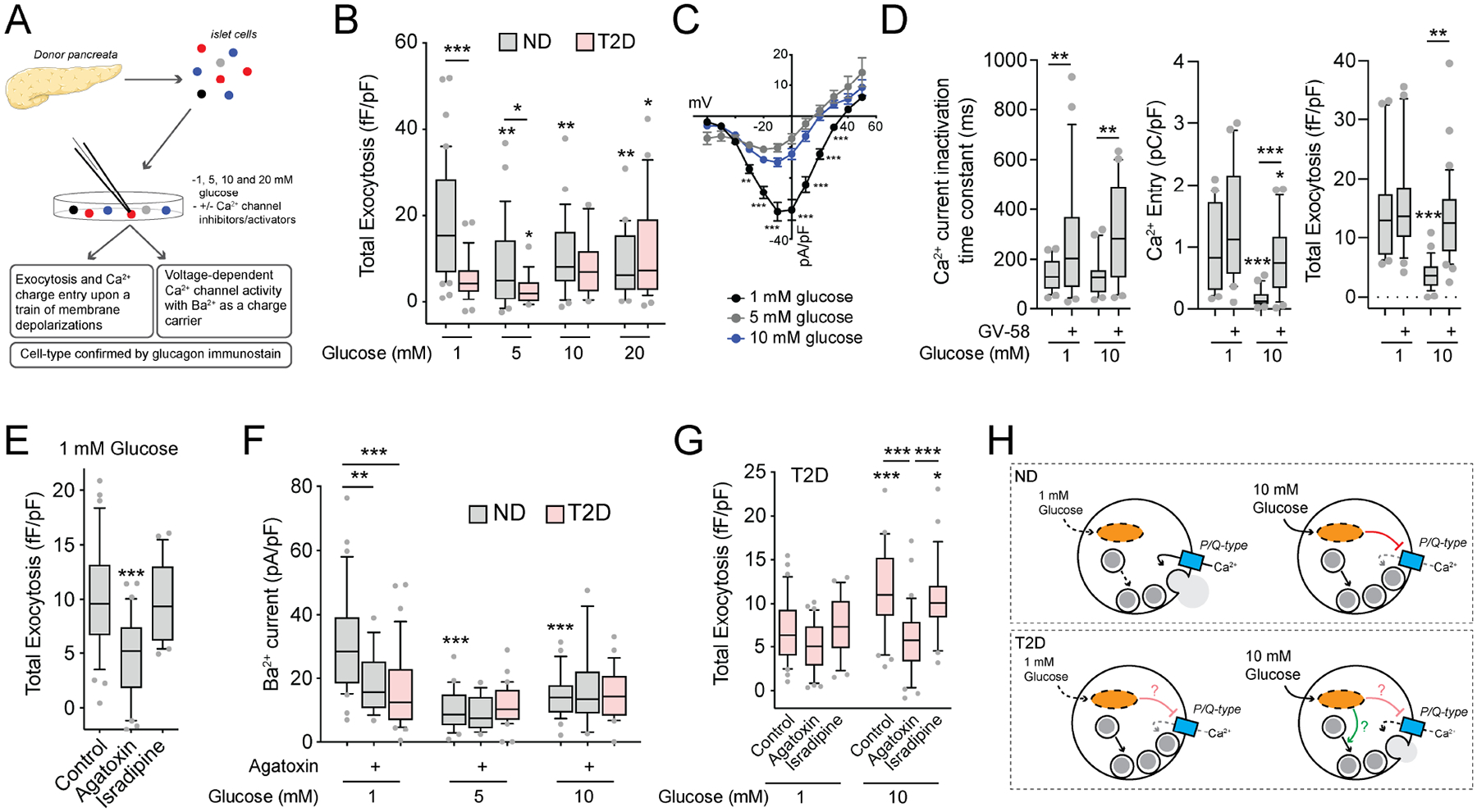
Figure 2. Patch-seq suggests roles for the mitochondrial respiratory complex in α-cell function, and endocrine development and cell fate in α-cell dysfunction.
A) Schematic diagram illustrating the isolation of 400 α-cells from 24 donors without diabetes (ND) and 7 donors with T2D assessed by patch-seq, some of which have been partly published previously (Camunas-Soler et al., 2020).
B) The distribution of exocytosis - transcript correlations from α-cells of ND donors at 1 and 10 mM glucose (see also Suppl Table 2).
C) Gene set enrichment analysis (GSEA) Gene ontology (GO): Biological Pathways using Z-scores as weighting across the transcriptome (using genes expressed in >20% of α-cells), separately at 1 and 10 mM glucose, reveals pathways linked to facilitation (positive values) or suppression (negative values) of exocytosis. FDR < 0.05 unless indicated otherwise, and absence of bars indicates no significant enrichment.
D) Leading-edge transcripts for mitochondrial respiratory chain complex assembly that flip correlation with exocytosis from positive to negative in α-cells of ND donors as glucose increases from 1 to 10 mM.
E) An endocrine system development pathway is enriched in ND α-cells with low exocytotic responses at 1 mM glucose.
F) Leading-edge genes underlying the signal in panel E, which includes transcripts involved in islet cell lineage and α-cell identity.
G) The distribution of exocytosis - transcript correlations α-cells from donors with T2D at 1 and 10 mM glucose (see also Suppl Table 3).
H) GSEA GO: Biological Pathways with Z-scores as weighting (using genes expressed in >20% of α-cells) reveals development and cell fate pathways linked to increased exocytosis in T2D α-cells at 10 mM glucose (FDR < 0.05 unless indicated otherwise).
I) Selected leading-edge transcripts that underlie the cell fate commitment (left) and pancreas development (right) pathways, including islet lineage and α-cell identity markers.
False discovery rate (FDR) for pathways identified by GSEA were < 0.05 unless indicated otherwise.
Ca2+ channel transcripts were not reduced, in α-cells of T2D donors compared with those from ND donors (Suppl Fig 3B–C). Hyperglycemia may induce α-cell mitochondrial dysfunction (Knudsen et al., 2019), and we saw a modest but significantly higher mitochondrial respiratory chain complex transcript expression in α-cells of T2D donors compared with α-cells from ND donors (Suppl Fig 3D,E). Intriguingly, transcripts associated with endocrine development appeared as anti-correlates of exocytosis at 1 mM glucose (Fig 2C,E–F) suggesting a role for cell differentiation state in the responsiveness of α-cells. In α-cells from donors with T2D, the effect of glucose on the distribution of transcriptome-wide correlations was reversed, such that high glucose associated with more positively correlated genes (Fig 2G; Suppl Table 3). GSEA of these correlations highlighted a role for cell development state in the inappropriately high α-cell exocytosis at 10 mM glucose in T2D (Fig 2H). Several leading-edge transcripts enriched in these pathways, including transcription factors important for pancreatic endocrine maturity like GATA6, PAX6, RFX6, and RFX3, overlap with those that correlated with inappropriately low exocytosis in α-cells of ND donors at 1 mM glucose (Fig 2F,I).
Glucose control of mouse α-cell exocytosis, and impaired ‘electrophysiological identity’ following high-fat feeding
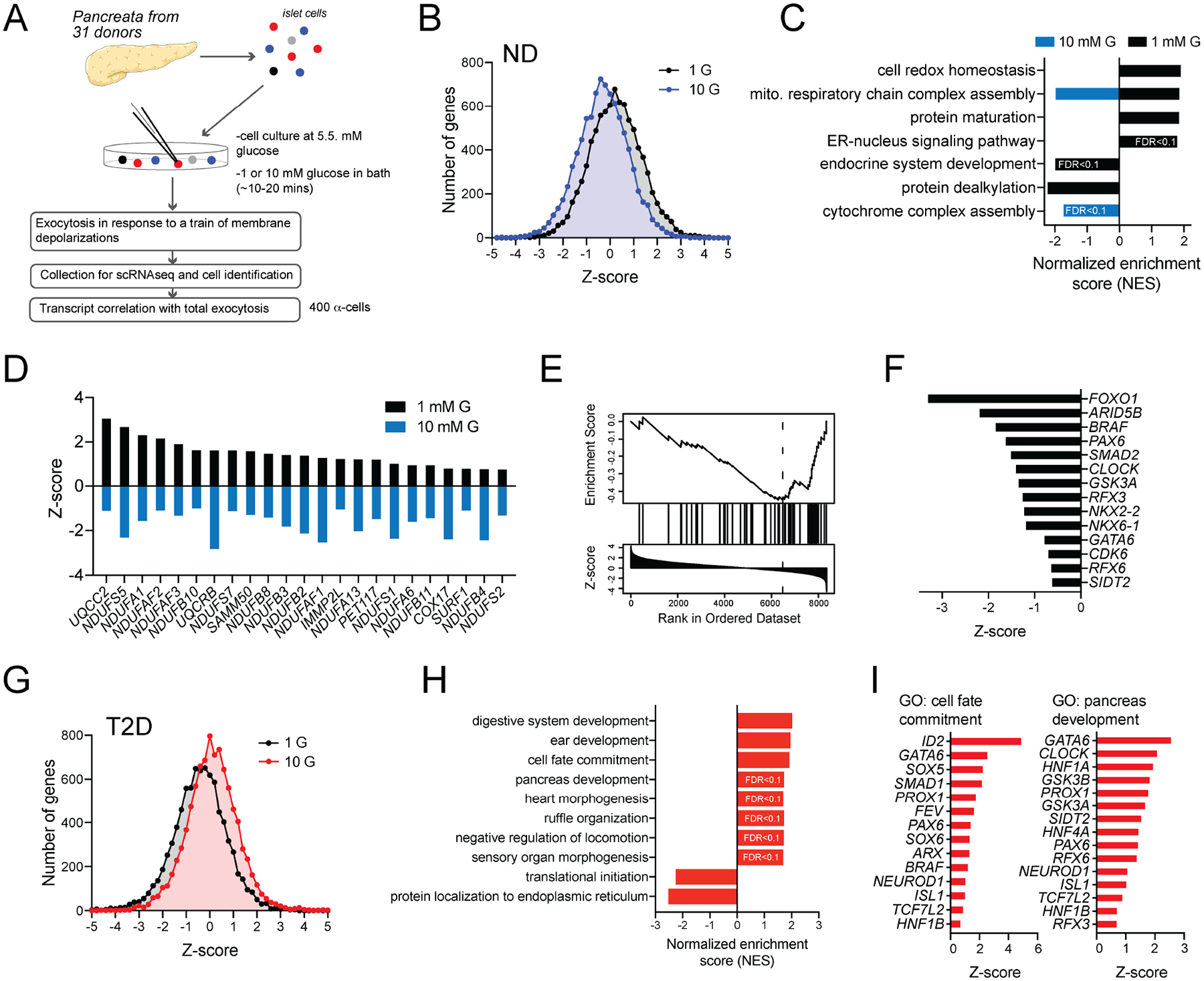
Following an ~10-minute exposure to 1 mM glucose, depolarization of mouse islets with 20 mM KCl elicited a transient stimulation of glucagon release that was blunted in islets kept at 5 mM glucose (Fig 3A), consistent with the glucose-dependent suppression of α-cell exocytosis. These differences could not be explained by the indirect paracrine effects of insulin, as insulin secretion evoked by KCl was slightly higher at 1 mM glucose than at 5 mM glucose (Fig 3B). Similar to human α-cells, increasing glucose suppressed exocytosis in mouse α-cells (Fig 3C). Although the physiological impact of glucose on glucagon secretion involves key paracrine signals (Briant et al., 2016), the suppression of exocytosis by increasing glucose was similar when cells were seeded at 10% of normal density (Fig 3D) and required glucose metabolism since the non-metabolizable analog 2-deoxyglucose (2-DG) did not mimic the effect of glucose (Fig 3E). The glucose-suppression of Ca2+ entry and exocytosis could be prevented by rotenone, but not antimycin A (Fig 3F), suggesting a signal at or between mitochondrial respiratory chain complexes I and III. This could include reactive oxygen species/H2O2 produced by complex I via reverse electron transport (Onukwufor et al., 2019) or by complex III towards the cytosol (Muller et al., 2004). Indeed, direct intracellular dialysis of H2O2 suppressed α-cell Ca2+ influx and exocytosis in mouse (Fig 3F) and human (Suppl Fig 1E–F) α-cells.
Figure 3. Glucose-suppression of mouse α-cell exocytosis requires glucose metabolism and the mitochondrial respiratory chain.
A-B) Glucagon (A) and insulin (B) secretion from islets of 10–12 week old male C57bl6 mice as glucose remains at 5 mM (blue) or drops from 5 to 1 mM (black), and upon subsequent stimulation with 20 mM KCl. A control (grey) was maintained at 5 mM glucose (n = 3, 3, 3 mice).
C-D) Mouse α-cell exocytosis with increasing glucose (n = 32, 30, 14, 27 cells from 4 mice), and at very low (10% of normal) density (D; n = 6, 6, 13, 9 cells from 2 mice).
E) Effect of 2-Deoxy-D-glucose (2-DG) on glucose-regulation of α-cell exocytosis (n = 20, 20, 21, 46 cells from 4 mice).
F) Effect of the complex I inhibitor rotenone (0.5 μM) and the complex III inhibitor antimycin A (0.5 μM) on α-cell Ca2+ charge entry during a 500 ms depolarization from −70 to 0 mV, and exocytosis, at 1 and 10 mM glucose. Also the effect of direct intracellular dialysis of H2O2 (10 μM) via the patch pipette at 1 mM glucose (left n = 19, 15, 18, 17, 11, 19, 16 cells; and right n = 20, 19, 16, 26, 16, 26, 22 cells from 3 mice).
**- P < 0.01; and ***- P < 0.001 by one-way ANOVA (C,D,E) or two-way ANOVA (A,B,F), followed by Tukey post-test to compare points or groups with the 5 mM glucose points (A, B) or with the 1 mM glucose control group.
Low-glucose stimulation of glucagon secretion was enhanced in mice fed a high fat diet (HFD) for 12–14 weeks compared with age-matched controls (18–20 weeks; Fig 4A), similar to what we have shown previously (Kellard et al., 2020). There was no difference in insulin secretion at 5 and 1 mM glucose but the response to high-K+ was reduced in mice fed a HFD compared with controls (Fig 4B), possibly reflecting the disruption of a direct coupling between Ca2+ channels and insulin granules (Collins et al., 2010). As in humans, exocytosis at 1 mM glucose in α-cells from chow fed mice was inhibited by the P/Q-type Ca2+ channel blocker agatoxin, but not the L-type channel blocker isradipine (Fig 4C). This was reversed in α-cells from HFD mice where exocytosis is resistant to agatoxin but sensitive to isradipine (Fig 4C). Importantly, the P/Q-channel agonist GV-58, which delayed channel inactivation in α-cells from both the chow-fed and HFD mice (Fig 4D), rescued exocytosis following suppression by 5 mM glucose in α-cells from chow-fed, but not HFD-fed, mice, consistent with a loss of P/Q-type channel coupling to exocytosis (Fig 4E). Mouse α-cells are distinguished from β-cells by a characteristic right-shifted Na+ current half-inactivation, and these two cell types primarily express Scn3a and Scn9a, respectively (Zhang et al., 2014). Many α-cells from the HFD-fed mice, identified by glucagon immunostaining, showed a leftward shift in Na+ channel half-inactivation towards a ‘β-cell-like’ phenotype (Fig 4F; Suppl Fig 4A–C).
Figure 4. ‘β-cell like’ properties of α-cells from HFD-fed mice.
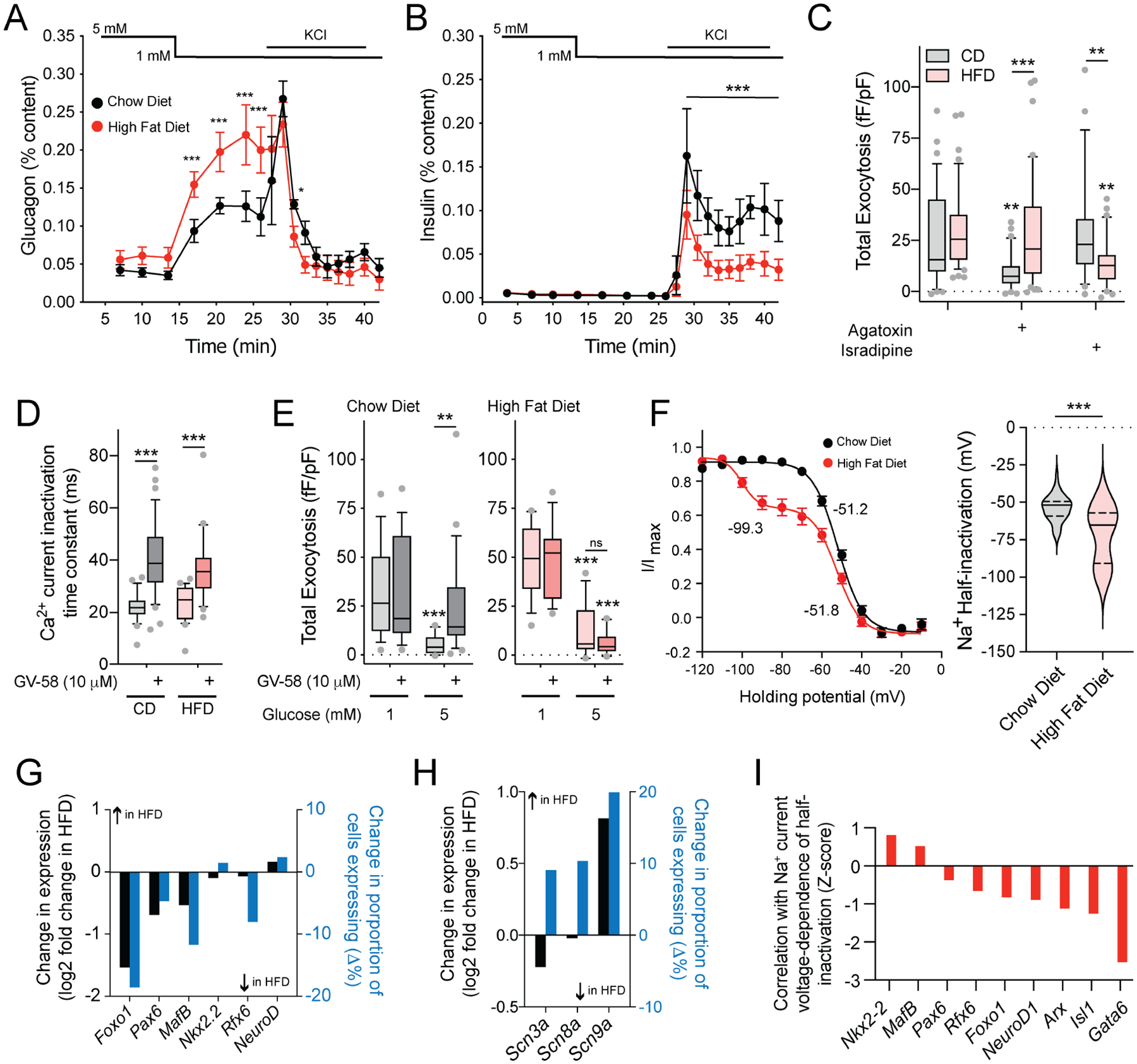
A-B) Glucagon (A) and insulin (B) secretion from islets of male C57/bl6N mice fed HFD for 10–12 weeks starting from 8 weeks of age (red) compared with age matched (18–20 week) chow fed controls (black). (n = 3, 3 mice).
C) Effect of the P/Q-type Ca2+ channel blocker agatoxin (100 nM) and the L-type Ca2+ channel blocker isradipine (10 μM) on total exocytosis from chow diet (CD) and HFD α-cells at 1 mM glucose (n = 34, 42, 34, 40, 29, 39 cells from 8 HFD and 8 CD mice).
D-E) Effect of the P/Q-channel activator GV-58 (10 μM) to delay Ca2+ current inactivation in both CD and HFD α-cells (D, n = 23, 36, 24, 23 cells from 3 CD and 3 HFD-fed mice) and on exocytosis (E) from CD (grey) and HFD (pink) α-cells at 1 and 5 mM glucose (n = 15, 16, 16, 21 cells from 3 CD mice; n = 14, 13, 15, 14 cells from 3 HFD-fed mice).
F) Steady-state voltage-dependent Na+ current inactivation curves (left) and individual half-inactivation voltages (right) from α-cells of chow fed and HFD mice (n = 36, 44 cells from 9 CD and 9 HFD mice, measured at 1 mM glucose). Half-inactivation voltages from fit curves are indicated.
G) From a separate set of CD and HFD α-cells assessed by patch-seq (Suppl Fig 4), mean expression (black) and % of cells expressing (blue) some α-cell identity transcription factors in CD and HFD α-cells.
H) Mean expression (black) and % of cells expressing (blue) the β-cell Na+ channel isoform, Scn9a, in HFD α-cells.
I) Correlation Z-scores of the negative shift in Na+ channel steady-state inactivation (at 1 mM glucose) in HFD α-cells with transcripts involved in α-cell lineage and identity.
*- P < 0.05; **- P < 0.01; and ***- P < 0.001 by the Student’s t-test (F), or by one-way ANOVA (D,E) or two-way ANOVA (A,B,C) followed by Tukey post-test to compare points or groups with the 5 mM glucose points (A,B), with the 1 mM glucose control group, or as indicated.
This switching of both Ca2+-channel-exocytosis coupling and Na+ channel half-inactivation towards ‘β-cell-like’ phenotypes upon HFD feeding is reminiscent of observations from genetically induced α- to β-cell trans-differentiation (Chakravarthy et al., 2017). However, the present changes occurred while glucagon expression was maintained since the α-cells were identified by glucagon immunostaining. We explored these findings in separate experiments where α-cells from chow-fed and HFD-fed mice were collected for patch-seq (Suppl Fig 4A) where we confirmed the negative shift in Na+ current inactivation (Suppl Fig 4B,C) and the switch from P/Q- to L-type Ca2+ channel dependence of exocytosis (Suppl Fig 4D). After HFD feeding some endocrine transcription factors were lower than in α-cells of chow-fed mice (Fig 4G; Suppl Table 4), although there was no clear change in exocytotic or Ca2+ channel transcripts (Suppl Fig 4E). The β-cell Na+ channel transcript Scn9a was higher (Fig 4H) in α-cells from HFD than chow-fed mice, and the HFD-induced shift in Na+ channel inactivation at 1 mM glucose correlated with expression of α-cell identity and islet lineage factors (Fig 4I; Suppl Table 5). Thus, after HFD feeding some α-cells adopted electrophysiological properties typically associated with β-cell function, and a transcriptional profile consistent with altered α-cell identity and/or maturation.
Electrophysiological fingerprint modeling links human α-cell behavior and cell phenotype
We next compiled data from islet cells of 67 donors collected at 1, 5 and 10 mM glucose, and identified either by scRNA-seq or immunostaining (Fig 5A). Unlike in mice, but similar to previous reports (Braun et al., 2008; Ramracheya et al., 2010), Na+ channel properties are similar between human α- and β-cells and could not be reliably used alone to distinguish these cell types (Fig 5B,C; Suppl Fig 4C). In ND α-cells, the peak Na+ current significantly correlated with genes known to impact α-cell activity (Fig 5D). Top positive correlates include genes involved in α-cell or islet function; including Na+ and Ca2+ channels (SCN3A, SCN3B, CACNA1A), α- and islet-cell lineage transcription factors (MAFB, FEV, NKX2.2, NEUROD1), G-protein coupled receptors (GIPR, SSTR1, FFAR1, GPR119), and secreted factors (LOXL4, WNT4, UCN3, IL16). Negative correlates were less abundant but included the vitamin D binding protein (GC) which is known to regulate α-cell Na+ currents (Viloria et al., 2020). GSEA for KEGG Pathways and GO: Biological Process terms revealed additional pathways enriched in ND α-cells with larger Na+ currents (Suppl Fig 5A,B), including the serotonin, GABA and glutamate pathways. Numerous cell surface receptor transcripts also appear as correlates to Na+ channel activity (Fig 5E, Suppl Fig 5C). We validated the ability of agonists for some of these to increase human α-cell Na+ currents (Fig 5F). Transcript correlates of peak Na+ current, and voltage-dependence of inactivation, from α-cells of ND and T2D donors are provided in Suppl Table 6.
Figure 5. Na+ current properties correlate with transcriptomic markers of α-cell function.
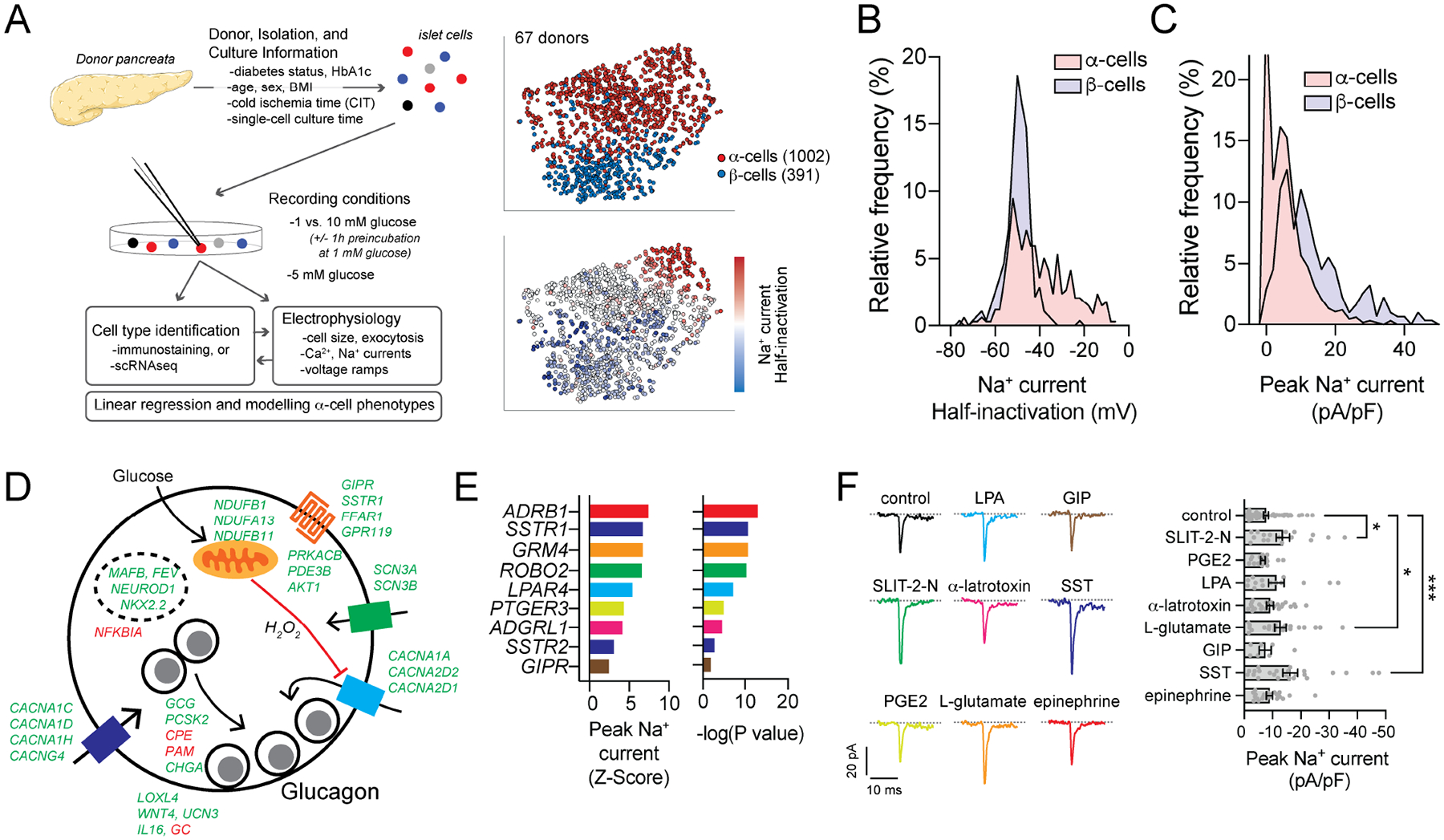
A) Schematic diagram illustrating an expanded dataset of electrically profiled human islet cells, and tSNE representations of α- and β-cells identified by immunostaining or sequencing along with the relative distribution of Na+ current half-inactivation values.
B-C) The distribution of Na+ current amplitudes (B) and voltage-dependence of half-inactivation values (C) of β-cells (light blue) and α-cells (pink) (see also Suppl Fig 4C).
D) Selected significant positive (green) and negative (red) transcript correlates of peak Na+ current from α-cells of ND donors mapped to a proposed scheme of glucose-regulation of glucagon exocytosis (see also Suppl Table 6).
E) Correlation of α-cell peak Na+ current and selected transmembrane signaling receptor transcripts (see also Suppl Fig 5).
F) In α-cells of 6 donors with no diabetes (ND), Na+ currents measured with receptor agonists (colors matching receptors shown in panel E) upon depolarization from −70 to −10 mV. Peak current is shown at right: control (n = 53 cells); 0.5 μg/ml SLIT-2-N (n = 17 cells); 10 μM prostaglandin E2 (PGE2, n = 17 cells); 0.5 μM lysophosphatidic acid (LPA, n = 14 cells); 0.2 μg/ml α-latratoxin (n = 24 cells); 10 mM L-glutamic acid (n = 21 cells); 100 nM glucose-dependent insulinotropic polypeptide (GIP, n = 7 cells); 200 nM somatostatin (SST, n = 24 cells); 5 μM epinephrine (n = 22 cells).
*- P < 0.05 and ***- P < 0.001 by one-way ANOVA, followed by the Benjamini and Hochburg post-test to method to compare groups controlling for false discovery rate.
Although α-cell lineage markers correlate with Na+ currents, the overlap in Na+ current properties between human α- and β-cells make it impossible to use these alone to interrogate shifts in human α-cell phenotypes. We therefore developed ‘electrophysiological fingerprinting’ classifier models (Fig 6A) that integrate Na+ current, Ca2+ current and exocytosis measures (Fig 6B) to characterize human α-cell ‘functional phenotypes’, similar to the logistic regression (Briant et al., 2017) and random forests (Camunas-Soler et al., 2020) models we used previously. In those models however, cell size was the major predictor of α-cell identity. Here we generated three independent models for cell type classification: Optimizable Ensemble classifiers that include (Model 1) or exclude (Model 2) cell size as an independent variable, and an Extreme Gradient Boosting model that also excludes cell size, but with additional restrictions to donor age, donor BMI and organ cold ischemic time (CIT) applied to the training data (Model 3). These models distinguished ND α- and β-cells well (Fig 6A) and, unlike some underlying parameters, were unaffected by potential confounders, such as donor sex, BMI, CIT, cell culture time and glucose concentration (Fig 6B).
Figure 6. Electrophysiological fingerprints define a loss of ‘functional identity’ in T2D α-cells.
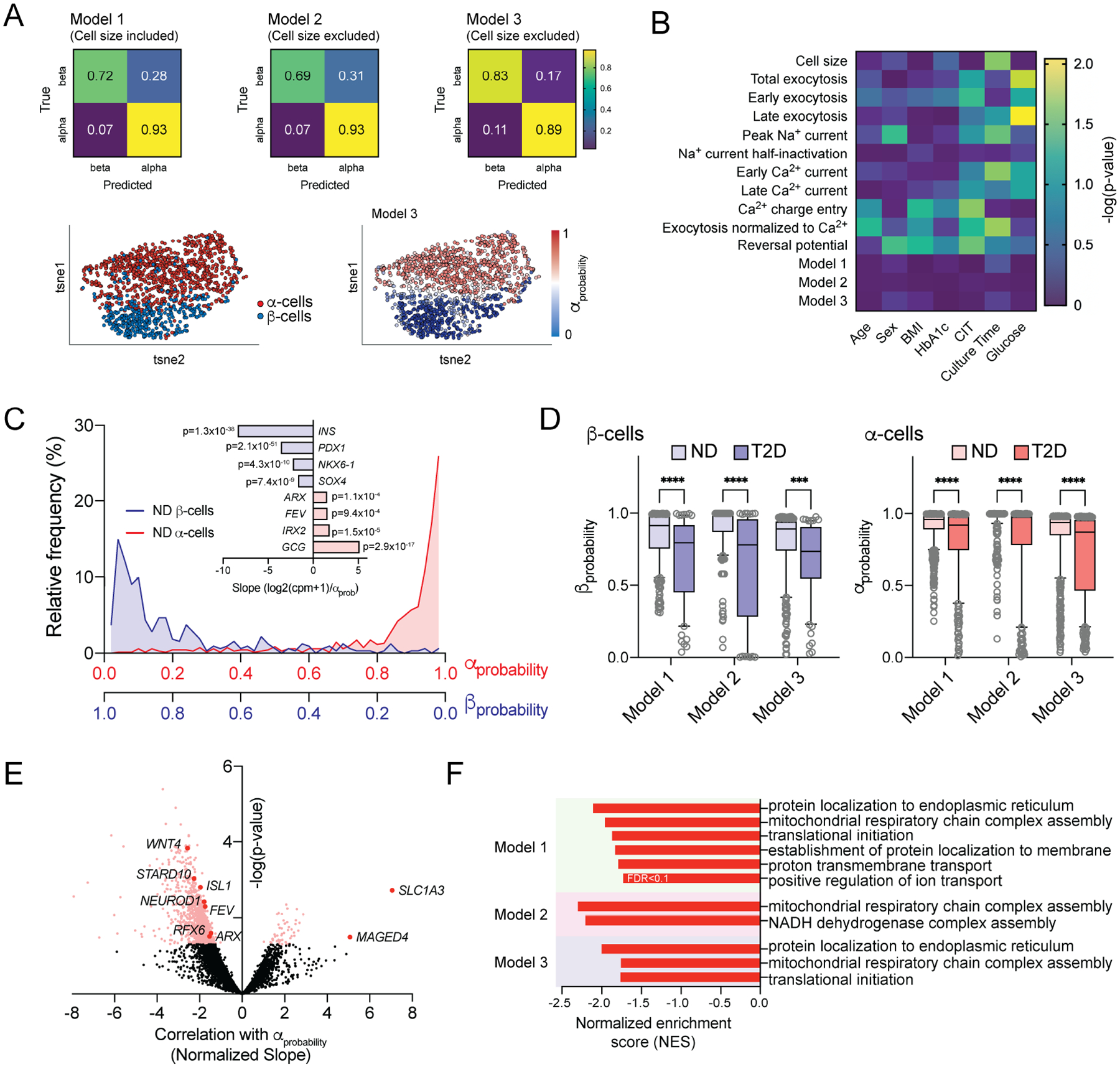
A) Classifier models trained on islet cell electrical properties of α- and β-cells from donors with no diabetes (ND) using Optimizable Ensemble or Extreme Gradient Boosting (XGBoost) approaches, identify cell types with high accuracy regardless of the inclusion or exclusion of cell size from training data. 80% of data was used for training. 20% of data was reserved for validation and generation of confusion matrices. tSNE plots show cell types determined by immunostaining or sequencing (left) and assigned αprobability scores (right).
B) Ordinary least squares multiple regression of electrophysiological properties of α-cells from ND donors, Model scoring, and donor/isolation variables.
C) Calculated αprobability (and βprobability) values from Model 3 applied to cells collected for patch-seq, without a priori knowledge of cell type and correlation with canonical β-cell (light blue) and α-cell (pink) markers.
D) βprobability and αprobability values derived from all three models, of all β- and α-cells from ND or T2D donors.
E) Volcano plot of transcript correlations with Model 3 αprobability values (slope/standard deviation) in α-cells from donors with T2D. A negative correlation indicates transcripts associated with reduced αprobability.
F) Significantly enriched GO: Biological Pathways from Gene Set Enrichment Analysis (GSEA) performed using αprobability - transcript correlation slopes of α-cells from donors with T2D in all three Models as weighting.
***- P < 0.001 and **** P < 0.0001 as indicated within models using non-parametric Kruskal-Wallis test (D) followed by Dunn’s post-test to correct for multiple comparisons. Pink/red points in E indicate significance at P < 0.05. False discovery rate (FDR) for pathways identified by GSEA in F were <0.05 unless indicated otherwise.
Model output between 0 and 1 can be considered the probability an electrophysiological profile matches that of an α-cell, and we called this αprobability (with βprobability = 1-αprobability). Assignment of an αprobability to all ND α- and β-cells for which we had patch-seq data, without a priori knowledge of cell type, showed the effective separation of cell types and correlation with canonical markers (Fig 6C). All three models showed a significantly reduced βprobability and αprobability, in β- and α-cells respectively, from donors with T2D compared with cells from ND donors (Fig 6D). The expression of numerous transcripts correlated with a ‘loss of electrophysiological identity’ in T2D α-cells within one or more of the models, including lineage markers such as ISL1, NEUROD1, FEV, and RFX6 (Fig 6E; Suppl Table 7). GSEA using αprobability correlation slopes as weighting for transcripts expressed in >20% of cells highlighted GO: Biological Process terms related to mitochondrial respiratory chain complex assembly in all three models (Fig 6F), underscoring a link between increased respiratory chain complex expression and α-cell dysfunction.
Impaired functional identity and exocytosis in T2D α-cells enriched in lineage and immaturity markers
Human α-cells exist in states of variable chromatin accessibility at the GCG promoter and sites enriched with transcription factor motifs characterizing endocrine lineage and development (Chiou et al., 2021). Markers of α-cell lineage are heterogenous within our dataset, with evidence for increased expression in T2D (Fig 7A; Suppl Fig 6A,B). Intriguingly, this also included ARX which we confirmed at the protein level, along with MAFB and glucagon itself (Fig 7B). ARX-enriched (ARXhi) cells expressed consistently higher levels of progenitor transcripts like NEUROD1, FEV, GATA6, and others (Suppl Fig 6B). We find no difference in αprobability scores between ND α-cells either enriched or deficient in these (Fig 7C; Suppl Fig 6). In T2D however, an impaired electrical fingerprint occurred selectively in α-cells with higher levels of ARX, NEUROD1, ISL1, PAX6, GATA6, FEV, NKX2–2, RFX6 and MAFB (Fig 7C; Suppl Fig 6). Accordingly, ARX and these other markers did not correlate with exocytosis in α-cells of ND donors but did correlate significantly with impaired exocytosis in T2D (Fig 7D). In separate scRNA-seq data (Avrahami et al., 2020) we confirmed the up-regulation of a juvenile α-cell profile (Arda et al., 2016), tissue development genes and a β-cell-like profile selectively in ARXhi α-cells in T2D compared with α-cells from ND donors (Fig 7E; Suppl Fig 7). Finally, while the α-cells with low ISL1, NKX2–2, NEUROD1 and ARX from donors with T2D had normal exocytosis at 1 mM glucose, α-cells enriched in these transcripts showed impaired exocytotic function in T2D (Fig 7F).
Figure 7. A role for maturation state in α-cell dysfunction in T2D.
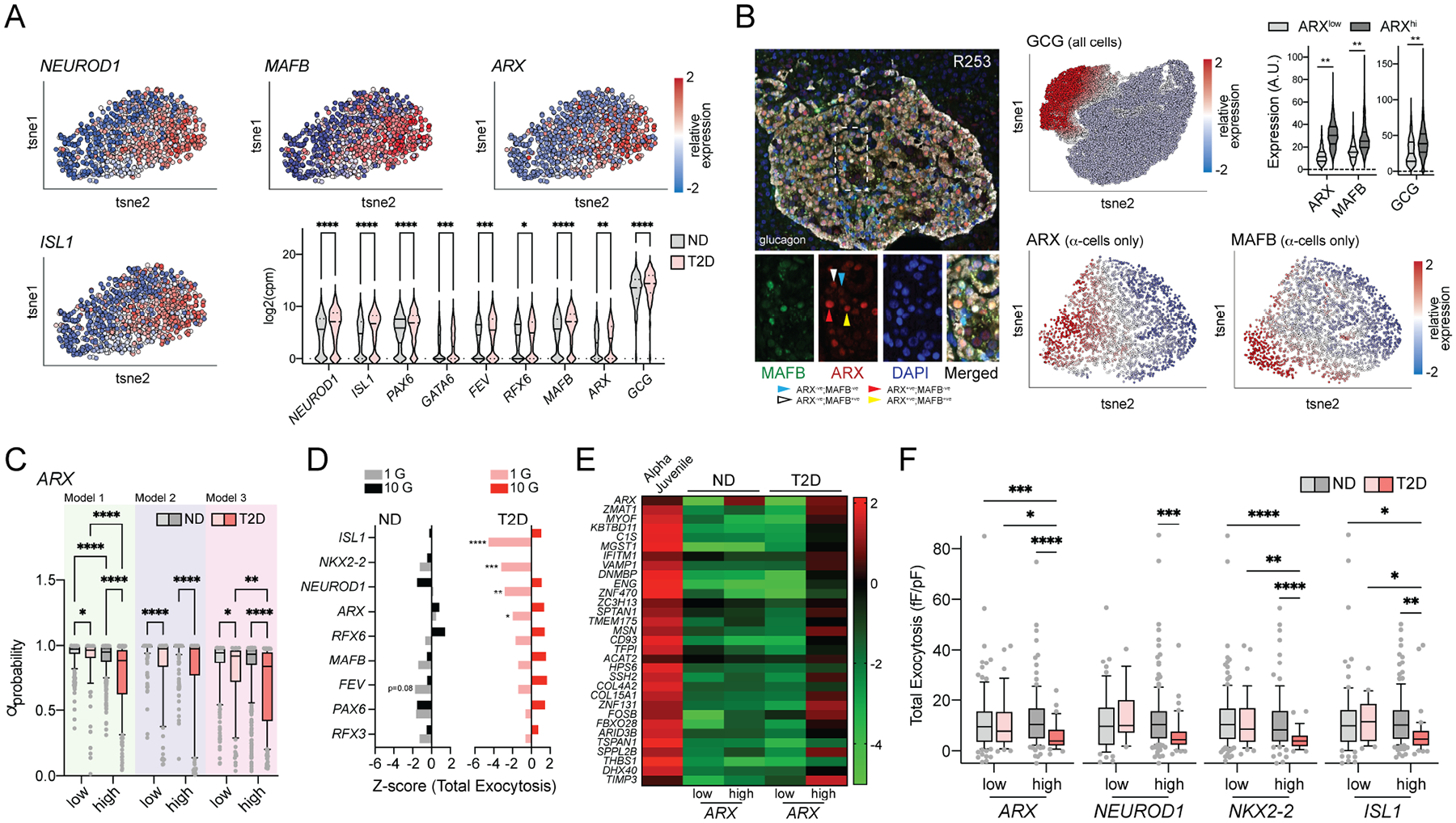
A) Heterogenous expression of transcript makers for islet cell lineage and α-cell maturity in 980 patch-seq α-cells.
B) ARX and MAFB protein expression in α-cells at the protein level in situ by immunostaining. Violin plots show the relative levels of GCG, ARX and MAFB expressed in ARXlow and ARXhi α-cells.
C) αprobability values from the three separate classifier models in ND and T2D α-cells separated by high and low expression of ARX (see also Suppl Fig 6).
D) Correlation of exocytosis in ND and T2D α-cells with α-cell lineage and identity markers. Bars to the left of the centerline indicate correlation with low exocytosis at the given glucose concentration.
E) In a separate human islet single-cell dataset (Avrahami et al., 2020), expression of a juvenile α-cell gene-set in ND and T2D α-cells separated by low and high ARX expression. The heatmap displays relative expression levels as median log2 FPKM values.
F) Total exocytosis in ND and T2D α-cells at 1 mM glucose separated by low and high expression of ISL1, NEUROD1, NKX2-2, and ARX.
*- P < 0.05, **- P < 0.01, ***- P < 0.001 and **** P < 0.0001 two-way ANOVA followed by two-stage step-up method for estimation of FDR (C,D,F) or Tukey post-test (B).
DISCUSSION
Glucagon secretion is under the control of metabolic, paracrine, hormonal and neuronal signals (El et al., 2020). Much debate has centered around the question of whether glucose-suppression of glucagon secretion is mediated via intrinsic, paracrine or autonomic mechanisms although it seems likely that α-cells (like β-cells) adeptly integrate multiple signals for precise physiologic control of glucagon. A role for direct glucose-sensing is supported by the ability of glucose to modulate α-cell ATP-sensitive K+ channels (Zhang et al., 2013), and the impact of α-cell glucokinase manipulation on in vitro cell function and in vivo glucagon secretion and glucose homeostasis (Moede et al., 2020; Bahl et al., 2021; Basco et al., 2018). Here, we show that glucose suppresses human and mouse α-cell exocytosis, consistent with a recent report where exocytosis was measured by live-cell imaging (Omar-Hmeadi et al., 2020). A role for α-cell metabolism is supported by the effect of the non-metabolizable glucose analog 2-DG and the correlation of responses with mitochondrial respiratory complex assembly transcripts, particularly those of complex 1. Indeed, the ability of rotenone to block glucose-suppression of α-cell Ca2+ currents and exocytosis suggests a signaling role for reactive oxygen species and H2O2 generated by complex I. Consistent with this, intracellular application of H2O2 mimics the effect of glucose while reduced glutathione blocks the effect of glucose, and external H2O2 inhibits low glucose-stimulated glucagon secretion (Allister et al., 2013).
The study by Omar-Hmeadi et al. (2020) also show a U-shaped response of isolated α-cells to glucose, suggesting that glucose may increase α-cell exocytosis under some conditions. We find some hints of this in our own data, including in α-cells of donors with T2D. In ND α-cells this may depend on the culture conditions. Following prolonged low-glucose preincubation α-cells are larger, possibly due to continued glucagon granule fusion. Subsequent exocytotic responses are smaller when the α-cells are maintained at 1 mM glucose, while acute glucose-stimulation now increases exocytosis. This is reminiscent of the glucose-dependent increase in glucagon secretion from purified α-cells (Olsen et al., 2005). Thus, glucose acutely suppress P/Q-type Ca2+ channels via a complex I-dependent mechanism to limit depolarization induced α-cell exocytosis, while at the same time facilitating glucagon granule priming or docking to maintain the availability of granules. Altogether these processes will ‘tune’ α-cell responsiveness to myriad paracrine, endocrine, and neuronal inputs.
The P/Q-type Ca2+ current seen at low glucose in α-cells of ND donors appears absent in T2D, but channel expression is not reduced. Therefore, channel regulation appears disrupted. It seems possible that altered mitochondrial function in α-cells in T2D could drive dysregulation of P/Q-type Ca2+ channel activity, and indeed mitochondrial dysfunction has been demonstrated in α-cells from hyperglycemic mice (Knudsen et al., 2019) and mitochondrial morphology is altered in α-cells from western-diet-fed mice (Grubelnik et al., 2020). Genes involved in glucose metabolism appear downregulated in α-cells from donors with type 1 diabetes (Brissova et al., 2018) although these may exhibit a more extreme phenotype than in T2D, with down-regulation of several channels, exocytotic transcripts, and α-cell identity markers. We do find an up-regulation, particularly in ARXhi T2D α-cells, of mitochondrial respiratory chain complex assembly transcripts and a clear correlation of this with the impaired α-cell phenotype by fingerprint modelling, however a demonstration of altered mitochondrial respiratory function and/or reactive oxygen species generation in α-cells from donors with T2D remains to be shown.
The coupling of glucagon exocytosis to P/Q-type Ca2+ channels, which we confirm at low glucose, has been demonstrated previously (Marinis et al., 2010; Ramracheya et al., 2010) and is similar to the direct coupling of insulin granule exocytosis to the activation of L-type Ca2+ channels (Wiser et al., 1999). These and other electrophysiological properties are used to distinguish α- and β-cells from rodents. Most commonly, mouse islet cell types are identified by a combination of cell size (α-cells are smaller) and distinct properties of Na+ current inactivation. In a model of genetically induced α-to-β trans-differentiation in mice, we reported a clear shift in electrophysiological phenotype consistent with the attainment of β-cell properties (Chakravarthy et al., 2017). Intriguingly, following high-fat feeding mouse α-cells undergo a negative-shift in Na+ current inactivation and convert from P/Q- to L-type Ca2+ channel dependence of exocytosis. While we provide some evidence for impaired α-cell identity, this is not associated with a clear trans-differentiation as β-cell markers are not increased and cells maintain positive immunostaining for glucagon. The shift in Na+ channel inactivation towards a ‘β-cell like’ phenotype, which could be related to changes in membrane composition (Godazgar et al., 2018), correlates with the expression of important α-cell lineage and identity transcription factors, suggesting that the change in electrical phenotype occurs more readily in cells with higher levels of these markers.
A subset of human α-cells, even from donors without diabetes, exist in an immature state and may suffer a further loss of mature identity in T2D (Avrahami et al., 2020), perhaps related to their greater epigenetic plasticity (Bramswig et al., 2013) and distinct states of chromatin accessibility (Chiou et al., 2021). We find that α-cells from ND donors with inappropriately low exocytosis are enriched in transcripts and pathways associated with endocrine development (FOXO1, PAX6, RFX6 and others). In T2D we see no obvious loss of α-cell transcription factors, or up-regulation of β-cell-defining transcription factors indicative of trans-differentiation per se. We do however find heterogeneity in many lineage markers, as reported previously by us (Camunas-Soler et al., 2020; Drigo et al., 2019) and others (Li et al., 2016). In our dataset, most of these show a small but significant increase in T2D. To assess a shift in electrophysiological phenotype of these human α-cells we could not use Na+ current inactivation alone as this feature overlaps with measurements from human β-cells. We therefore modified machine learning approaches that we previously used to improve the identification of mouse (Briant et al., 2017) and human (Camunas-Soler et al., 2020) islet cells. We used exocytosis, Na+ current, and Ca2+ properties as training data for three separate models with similar results. Two of these excluded cell size, to solely identify shifts in membrane ‘activity’, and all models accurately assigned probability values for α- and β-cells irrespective of ambient glucose, culture times and other important donor and isolation-related parameters. Correlation of αprobability values with transcriptomic data therefore emphasized pathways linked to a ‘loss of functional phenotype’ in T2D and suggest that α-cells with the most altered electrical phenotypes in T2D are those with higher expression of pancreatic endocrine lineage markers.
In this study we define an impaired α-cell phenotype in T2D that is largely restricted to a subgroup of cells expressing higher levels of markers that define not only α-cell maturity, but pancreatic endocrine lineage. At first look we would have expected that high levels of ARX should be indicative of mature α-cell function, but this is misleading given a strong overlap of ARXhi cells with transcripts more traditionally associated with an endocrine progenitor state such as NEUROD1, ISL1, FEV and others. These cells indeed appear more ‘plastic’ in their electrophysiological responses, which may predispose them to an altered electrical phenotype in T2D. We confirm in a separate dataset the ‘de-repression’ of immature gene-sets associated with a ‘juvenile’ α-cell phenotype and with tissue development restricted to ARXhi α-cells in T2D. Thus, we demonstrate that all α-cells are not equally impacted by disease and that a sub-set of α-cells defined by their maturation state may be key drivers of impaired glucagon responses in T2D.
Limitations of the study
While tempting to link impaired exocytosis in α-cells from donors with T2D to an impaired responsiveness to hypoglycemia in vivo, this must be considered in the context of in situ α-cell function which will be impacted by the local environment and by paracrine or hormonal signals. We studied single isolated α-cells here, and although we demonstrate a similar heterogeneity in ARX/MAFB protein expression in situ, α-cell function may be different within the intact pancreas and would likely be impacted by architectural changes in the disease state. Encouragingly though, we find clear differences between ND and T2D α-cells that persist in vitro, and among the novel findings of this study we also find wellestablished regulators of α-cell function (such as GIPR). Additionally, both α-cell function and transcript expression are likely dynamic and impacted by metabolic status or culture conditions. Indeed, pre-culture at low glucose can alter α-cell exocytotic function. Our approach to ‘electrophysiological fingerprinting’ addresses this in part since it is unaffected by glucose, time in single-cell culture (up to 3 days), or various donor-related parameters. Nonetheless, the relevance of possible dynamic shifts in α-cell phenotype in T2D remains unclear. We do not know, for example, if exocytotic function would be restored if T2D α-cells transition from an ARXhi to ARXlo state, or whether such a transition itself is impaired.
While α-cells enriched in lineage factors appear less mature and maintain some plasticity, whether T2D induces a true reversal of maturation or a more general phenotypic drift could still be questioned. While an enrichment of numerous gene-sets including β-cell genes in the ARXhi cells in T2D could suggest the latter, we note that those are also all enriched in true juvenile α-cells (Avrahami et al., 2020). The exact links between these changes and dysregulated glucagon secretion remains somewhat speculative, particularly since we see no decrease in Ca2+ channel transcripts to explain the reduced Ca2+ currents. Interestingly, mitochondrial respiratory complex assembly transcripts are increased in T2D α-cells, most notably in the α-cells with impaired function and enriched for ARX. While altered mitochondrial function could drive P/Q-type Ca2+ channel inhibition and impaired exocytosis, and we provide some evidence linking the respiratory chain to α-cell dysfunction in T2D, we do not know if mitochondrial respiration is altered in T2D α-cells. This will require assessment of mitochondrial function and oxygen consumption in the ARXhi sub-set of α-cells from donors with T2D as a future priority.
Finally, we should be careful when directly comparing the rodent and human studies. One clear difference we find is that human α-cells in T2D show impaired (low) exocytosis at 1 mM glucose and a modest increase with higher glucose, while the mouse HFD α-cells instead show altered Na+ channel inactivation and exocytosis-Ca2+-channel coupling. The exact reasons for these differences are unclear, but perhaps related to obvious differences between humans and the mouse model (disease/HFD duration, degree of dysglycemia, age, etc.). Nonetheless, in both the mice and human cells, we find evidence to suggest that α-cell dysfunction is linked to cell maturation state.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Patrick MacDonald (pmacdonald@ualberta.ca).
Materials availability
This study did not generate new unique reagents.
Raw sequencing reads are available in the NCBI Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA) under accession numbers GSE124742 and GSE164875.
The code and scripts generated during this study, as well as preprocessed datasets, are available at https://github.com/jcamunas/patchseq.
Data S1 represents an Excel file containing the values that were used to create all the graphs in the paper. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human islets
In most cases, human islets were from our in-house human islet isolation and distribution program (www.isletcore.ca) (Lyon et al., 2019). Some human islets were provided by the Clinical Islet Transplant Program at the University of Alberta, by Dr. Rita Bottino at the Alleghany Health Network (Pennsylvania, US), or from the Human Pancreas Analysis Program (Kaestner et al., 2019). Details of donors with no diabetes (ND) or type 2 diabetes (T2D) used in this study are shown in Supplementary Table 1. T2D was determined either by reporting of previous clinical diagnosis at the time of organ procurement, or by assessment of %HbA1c > 6.5 in a few cases that were considered as previously undiagnosed T2D. Human islets and dispersed cells were cultured in DMEM (ThermoFisher, #11885) with 10% FBS (ThermoFisher, #12483020) and 100 U/ml penicillin/streptomycin (ThermoFisher, #15070063) at 37°C and 5% CO2. In our previous study (Camunas-Soler et al., 2020) we saw no obvious effect of sex on patch-seq data, or when analyses were corrected for sex as a co-variate. In the present study we find that electrophysiological fingerprint modelling is not impacted by donor sex (Fig 6B). All donors provided written informed consent for research. Human tissue studies were approved by the Human Research Ethics Board of the University of Alberta (Pro00013094, Pro00001754).
Mouse islets
Mouse islets were isolated from chow-fed (5L0D Picolab Laboratory Rodent Diet, #3005659–220) male C57bl/6N mice (Charles River, #CRL:027; RRID:IMSR_CRL:027) at 10–12 weeks of age or following 1012 weeks of high fat diet (60% of calories from fat; VWR Bio-Serv, #CA89067–471) starting from 8 weeks of age by collagenase digestion and hand-picking (Smith et al., 2020). Mouse islets and dispersed cells were cultured in RPMI (ThermoFisher, #11875) with 10% FBS and 100 U/ml penicillin/streptomycin at 37°C and 5% CO2. Animals were housed at 20–24°C with a 12h:12h light:dark cycle and daily health checks. Studies were performed in accordance with institutional guidelines and were approved by the Animal Care and Use Committee at the University of Alberta (AUP00000291).
METHOD DETAILS
Patch-clamp recordings
Hand-picked islets were dissociated to single cells using StemPro accutase (ThermoFisher, #A1110501). Dispersed islet cells were cultured for 1–3 days, after which media was changed to bath solution containing (in mM): 118 NaCl, 20 TEA, 5.6 KCl, 1.2 MgCl2, 2.6 CaCl2, 5 HEPES, and with glucose as indicated (pH adjusted to 7.4 with NaOH) in a heated chamber (32–35 °C). Modulators of Ca2+ channels (isradipine, Sigma-Aldrich, # I6658; agatoxin, Alomone Labs, #STA-500; GV-58, Alomone Labs, #G-140) or mitochondrial respiratory chain inhibitors (antimycin A, Sigma-Aldrich, #A8674; rotenone, Sigma-Aldrich, #R8875) were added to the bath, and pH adjusted, as indicated in figure legends. For whole-cell patch-clamping, fire polished thin wall borosilicate pipettes coated with Sylgard (3–5 MOhm) contained intracellular solution (in mM): 125 Cs-glutamate, 10 CsCl, 10 NaCl, 1 MgCl2, 0.05 EGTA, 5 HEPES, 0.1 cAMP and 3 MgATP (pH adjusted to 7.15 with CsOH). Electrophysiological measurements were collected using a HEKA EPC10 amplifier and PatchMaster Software (SmartEphys HEKA) within 5 minutes of break-in as described previously (Camunas-Soler et al., 2020). Quality control was assessed stability of the seal (>10 GOhm) and access resistance. Cells were identified by post-hoc immunostaining for insulin with a rabbit anti-insulin primary antibody (Santa Cruz; #SC-9168; RRID:AB_2126540) and goat anti-rabbit Alexa Fluor 488 secondary (ThermoFisher, #A-11076; RRID:AB_141930), and with a guinea pig anti-glucagon primary antibody (Sigma-Aldrich, #G2654; RRID:AB_259852) and goat antiguinea pig Alexa Fluor 594 secondary (ThermoFisher, #A-11076; RRID:AB_141930); or following collection for single-cell RNA sequencing analysis (Camunas-Soler et al., 2020).
For assessment of Ca2+ channel activity using Ba2+ as a charge carrier (Fig 1C,F) bath solution contained (in mM): 100 NaCl, 20 BaCl2, 5 CsCl, 1 MgCl2, 10 HEPES, with 0.2 μM tetrodotoxin and glucose as indicated (pH 7.4 with NaOH); and intracellular solution was (in mM): 140 Cs-glutamate, 1 MgCl2, 20 TEA, 5 EGTA, 20 HEPES, 3 Mg-ATP (pH 7.15 with CsOH). For validation of receptor agonist effects on Na+ currents (Fig 5F), bath solution contained (in mM): 118 NaCl, 20 TEA, 5.6 KCl, 1.2 MgCl2, 5 HEPES, and 5 mM glucose (pH 7.4 with NaOH); and intracellular solution was (in mM): 125 Cs-glutamate, 10 CsCl, 10 NaCl, 1 MgCl2, 0.05 EGTA, 5 HEPES (pH 7.15 with CsOH). Compounds used were: lysophosphatidic acid (LPA; Sigma-Aldrich, #L7260), α-latrotoxin (Enzo Life Sciences, #ALX-630027-C040), Slit guidance ligand 2 (SLIT-2-N; Sigma-Aldrich, #SRP3155), prostaglandin E2 (PGE2; Tocris, #2296), somatostatin (SST; Sigma-Aldrich, #S9129), glucose-dependent insulinotropic polypeptide (GIP;Eurogentec, #AS-65568), L-glutamic acid (Sigma-Aldrich, #G1251), and epinephrine (Sigma-Aldrich, #E4375). These were added in bath solution, at the concentrations indicated in the figure legend, and pH was re-adjusted if needed. Na+ current was activated by a 50 ms depolarization from −70 to −10 mV.
Pancreas patch-seq
Our protocol for pancreas patch-seq is outlined in a recent paper (Camunas-Soler et al., 2020). In brief, following patch-clamp cells were collected using a separate wide-bore collection pipette (0.2–0.5 MOhm) filled with lysis buffer (10% Triton, Sigma-Aldrich, #93443; ribonuclease inhibitor 1:40, Clontech, #2313A), ERCC RNA spike-in mix (1:600000; ThermoFisher, #4456740), 10 mM dNTP, and 100 μM dT (3’-AAGCAGTGGTATCAACGCAGAGTACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN-5’) and then transferred to PCR tubes and stored at −80°C. We generated cDNA and sequencing libraries using an adaption of the SmartSeq-2 protocol for patch-seq plates (Camunas-Soler et al., 2020; Picelli et al.,2014). Libraries were generated from the amplified cDNA by tagmentation with Tn5 and sequenced in the NovaSeq platform (Illumina) using paired-end reads (100 bp) to an average depth of 1 million reads per cell. Sequencing reads were aligned to the human genome (GRCh38 genome with supplementary ERCC sequences) using STAR (Dobin et al., 2013), and gene counts determined using htseq-count (intersection-nonempty) using a GTF annotation with Ensembl 89 release genes (Anders et al., 2015). Gene expression was normalized to counts per million (cpm) after removal of counts corresponding to ERCC spike-ins and transformed to log2 values after addition of a pseudocount. This was followed by QC filtering of the sequenced cells (Camunas-Soler et al., 2020) to remove low-quality cells and potential doublets. Raw sequencing reads are available in the NCBI Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA) under accession numbers GSE124742 and GSE164875. Correlation between transcript expression and electrophysiology was as outlined previously (Camunas-Soler et al., 2020). Visualization as t-distributed stochastic neighbor embedding (tSNE) plots was with the Qlucore Omics Explorer v3.6. Gene-set enrichment analysis (GSEA) was performed with of Z-scores or slopes for transcripts found in >20% of cells using the WEB-based Gene SeT AnaLysis Toolkit (webgestalt.org) and weighted set gene coverage to reduce redundancy in identified terms (Liao et al., 2019).
Hormone secretion measurements
Groups of 150–175 hand-picked mouse islets were perifused using a BioRep perifusion system (BioRep, Miami). Islets were pre-incubated in perfused KRB buffer containing (in mM): 140 NaCl, 3.6 KCl, 2.6 CaCl2, 0.5 NaH2PO4, 0.5 MgSO4, 5 HEPES, 2 NaHCO3 and 0.5 mg/ml Essentially fatty acid free BSA (Sigma A6003) for 30 minutes, and then perifused in the same KRB with changes in glucose and KCl as indicated. Samples were collected at 90 −210 sec intervals and stored for assay of glucagon (U-PLEX Mouse Glucagon Assay, Meso Scale Diagnostics, #K1525YK) and insulin (STELLUX Rodent Insulin Chemiluminescence ELISA, ALPCO, #80-INSMR-CH10) at −20°C.
Immunostaining and single-cell protein analysis
Human paraffin embedded pancreas biopsies were sectioned to 3 μm and immunostained with antibodies against MAFB (Cell Signaling Technologies, #41019; RRID:AB_2799192), ARX (R&D Systems, # AF7068; RRID:AB_10973178) and GCG (Sigma-Aldrich, #G2654; RRID:AB_259852). The secondary antibodies were Cy2, Cy3, or Cy5 conjugated (1:500; Jackson ImmunoResearch, 111–225144, RRID:AB_2338021; 115-165-003; RRID:AB_2338680, 713-175-147, RRID:AB_2340730). Nuclear co-staining was conducted with DAPI Fluoromount G (Southern Biotech, #0100-20). Immunofluorescent mages were acquired on a Zeiss Axio Imager M2 widefield microscope with ApoTome. Images containing islet and exocrine cells were processed for nuclei segmentation, individual cell detection and cytosol border inference using Qupath software v0.2.3 (Bankhead et al., 2017). Parameters for nuclei detection: background radius 20px, median filter 5px, sigma 7px, minimum nuclei area 10px2, maximum nuclei area 1000px2, threshold of 2, nuclei were split by shape, and cell boundaries were determined by an expansion threshold of 12px with smoothing. Next, the relative expression levels of nuclear ARX, nuclear MAFB and cytosolic GCG levels for each detected cell were exported as .csv files and imported into Cytomap v1.4 for spatial analysis (Stoltzfus et al., 2020). In Qlucore Omics Explorer v3.6, we performed dimensionality reduction using tSNE of all imaged islet dataset using default parameters with 30 perplexity and 0.5 theta. Data was normalized for each dataset and the relative GCG, ARX and MAFB levels were displayed in the tSNE plot. Next, we created a gate to select the α-cell population in our dataset and the relative levels of ARX and MAFB were plotted. This information was used to determine the identity of ARXhi and ARXlow cells and the relative expression levels of ARX, MAFB and GCG for each α-cell subpopulation was plotted.
Gene-set and pathway analysis of scRNA-seq
To characterize the expression program of ARXhi and ARXlow α-cells in ND and T2D samples, we conducted gene set enrichment analysis (GSEA) on aggregated ARXhi versus ARXlow α-cell transcriptomes from our recently published data (Avrahami et al., 2020) using a hypergeometric test by Genomica software (http://genomica.weizmann.ac.il/) considering gene sets with a P value <0.01 and an FDR <0.05 to be significantly enriched. Pathway analyses were performed with Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005).
QUANTIFICATION AND STATISTICAL ANALYSIS
Electrophysiological Fingerprint Modelling
Multiple regression was carried out on ND α-cells using Ordinary Least Squared (OLS) Regression with Statsmodel v0.12.2 (Seabold and Perktold, 2010). Independent variables included age, sex, body mass index (BMI), HbA1c, cold ischemia time (CIT), culture time, and glucose concentration. Dependent variables included: cell size (pF), total exocytosis (fF/pF), early exocytosis (fF/pF), late exocytosis (fF/pF), peak Na+ current (pA/pF), Na+ half-Inactivation (mV), early Ca2+ current (pA/pF), late Ca2+ current (pA/pF), Ca2+ charge entry during an initial depolarization (pC/pF), exocytosis normalized to Ca2+ charge entry (fF/pC), reversal potential (mV), and αprobability from Models 1–3 (see below). Cells lacking data for a dependent variable were only dropped from that specific OLS analysis. Classification of cell type was conducted using the above electrophysiological measures as dependent variables from ND donors in an Optimizable Ensemble that either included (Model 1) or excluded (Model 2) cell size in MATLAB, or using Extreme Gradient Boosting (XGBoost v1.0.2) in a Python v3.7.11 framework that excluded cell size and reversal potential and restricted training data to 32 ND donors within an age of 20–70 years, a BMI of 18.5–30.3, and a pancreas CIT of ≤20 hours. Fine tuning was performed with a predetermined minimum accuracy of 80% for both α- and β-cells and training was performed with early stopping set to 50 iterations and utilized AUCPR as the evaluation metric. Models were trained on 80% of the α- and β-cells, with 20% reserved for testing and validation. Confusion matrices were generated using scikit-learn v0.24.2 (Pedregosa et al., 2012). We applied the models across our combined immunostaining and patch-seq database of ND and T2D cells and utilized the classifier’s predicted probability scores to assess fit to α-cell (αprobability = 1.0) and β-cell (αprobability = 0.0) models.
Statistical Analysis
Data are expressed as mean and standard error (line plots), mean and 10–90 percentile range (box and whisker plots), or as violin plots with median and quartiles indicated. When comparing two groups we used Student’s t-test or the non-parametric Mann-Whitney test to compare ranks. When comparing more than two groups we used one-way or two-way ANOVA followed by either the Tukey post-test or two-stage step-up method for estimation of false discovery rate (FDR); or alternatively the non-parametric Kruskal-Wallis test followed by Dunn’s post-test to correct for multiple comparisons. Statistical tests used are indicated in the figure legends. p-values less than 0.05 were considered as significant. For correlation of electrophysiology with transcript expression, Spearman tie-corrected correlations were computed for each gene and significance was tested by bootstrapping (1,000 iterations) as described in our previous work (Camunas-Soler et al., 2020). For ORA and GSEA, false discovery rate (FDR) of reported pathways was <0.05 or <0.1 as indicated in the figures using Bengamini-Hochberg (BH) correction within the WEB-based GEne SeT AnaLysis Toolkit (webgestalt.org). We did not perform power calculations prior to experiments, and experimenters were not blinded. Animals were assigned randomly to CD and HFD groups, and all human donors were accepted for islet isolations if they met the requirement for negative serology reports. No cells that passed standard electrophysiology and sequencing quality control (described above) were excluded from analysis.
Supplementary Material
Supplementary Table 2. Transcript correlations with total exocytosis from alpha-cells of donors with no diabetes (ND) at 1 mM and 10 mM glucose, related to Figure 2B
Supplementary Table 3. Transcript correlations with total exocytosis from alpha-cells of donors with type 2 diabetes (T2D) at 1 mM and 10 mM glucose, related to Figure 2G
Supplementary Table 4. Differential expression analysis of transcripts in mouse alpha-cells from chow-fed versus high-fat-fed mice, related to Figure 4
Supplementary Table 5. Transcript correlations with Na+ current voltage-dependence of half-inactivation of alpha-cells from Chow-fed and HFD-fed mice, related to Figure 4
Supplementary Table 6. Correlation of transcripts with peak Na+ current and Na+ current half-inactivation in human ND and T2D alpha-cells, related to Figure 5
Supplemental Table 7. Correlation of transcript expression with αprobability values from three different classifier models applied to ND and T2D alpha-cells, related to Figure 6
Data Supplement S1. Source data used to generate all graphs, related to Figures 1, 3–7 and Supplemental Figures 1, 2, 4 and 6.
Supplementary Figures. Supplementary Figure legends and Supplementary Figures 1–7
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Insulin Antibody (H-86) | Santa Cruz | SC-9168; RRID:AB_2126540 |
| Anti-Glucagon Antibody, clone 13D11.33 | EMD Millipore | MABN238; RRID:AB_433707 |
| Goat anti-Guinea Pig IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | ThermoFisher Scientific | A-11076; RRID:AB_141930 |
| Monoclonal Anti-Glucagon antibody produced in mouse | Sigma-Aldrich | G2654; RRID:AB_259852 |
| Human ARX Affinity Purified Polyclonal Ab antibody | R&D Systems | AF7068; RRID:AB_10973178 |
| MAFB Antibody | Cell Signaling Technology | 41019; RRID:AB_2799192 |
| Cy3-AffiniPure Goat Anti-Mouse IgG | Jackson ImmunoResear ch | 115-165-003; RRID:AB_2338680 |
| Cy2-AffiniPure Goat Anti-Rabbit IgG | Jackson ImmunoResear ch | 111-225-144, RRID:AB_2338021 |
| Cy5-AffiniPure Donkey Anti-Sheep IgG | Jackson ImmunoResear ch | 713-175-147, RRID:AB_2340730 |
| Biological Samples | ||
| Human pancreatic islets | Alberta Diabetes Institute IsletCore | See Suppl Table 1 |
| Human pancreatic islets | University of Alberta Clinical Islet Laboratory | See Suppl Table 1 |
| Human pancreatic islets | Allegheny Health Network | See Suppl Table 1 |
| Human pancreatic islets | Human Pancreas Analysis Program | See Suppl Table 1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI 1640 media | ThermoFisher Scientific | 11875 |
| low glucose DMEM media | ThermoFisher Scientific | 11885 |
| Fetal Bovine Serum - Canadian origin | ThermoFisher Scientific | 12483020 |
| Bovine Serum Albumin | Sigma-Aldrich | A6003 |
| Penicillin-Streptomycin | ThermoFisher Scientific | 15070063 |
| StemPro Accutase Cell Dissociation Reagent | ThermoFisher Scientific | A1110501 |
| chemicals/salts for patch-clamp and KRB buffers | Sigma-Aldrich | n/a |
| isradipine | Sigma-Aldrich | I6658 |
| agatoxin | Alomone Labs | STA-500 |
| GV-58 | Alomone Labs | G-140 |
| rotenone | Sigma-Aldrich | R8875 |
| antimycin A | Sigma-Aldrich | A8674 |
| lysophosphatidic acid | Sigma-Aldrich | L7260 |
| alpha-latrotoxin | Enzo Life Sciences | ALX-630–027-C040 |
| SLIT-2-N human | Sigma-Aldrich | SRP3155 |
| prostaglandin E2 | Tocris | 2296 |
| somatostatin | Sigma-Aldrich | S9129 |
| GIP, rat | Eurogentec | AS-65568 |
| L-glutamic acid | Sigma-Aldrich | G1251 |
| epinephrine | Sigma-Aldrich | E4375 |
| LIVE/DEAD Fixable near-IR dead cell dye | Life Technologies | L10119 |
| ERCC spike-in control | ThermoFisher Scientific | 4456740 |
| 5X All-In-One RT Master Mix | Applied Biological Materials | G486 |
| 10% Triton | Sigma-Aldrich | 93443 |
| Recombination RNase inhibitor | Clontech | 2313A |
| SUPERase-In RNase Inhibitor | ThermoFisher Scientific | AM2696 |
| 10 mM dNTP | ThermoFisher Scientific | 18427088 |
| DAPI Fluoromount-G | SouthernBiotech | 0100–20 |
| Critical Commercial Assays | ||
| U-PLEX Mouse Glucagon Assay | Meso Scale Diagnostics | K1525YK |
| STELLUX Rodent Insulin Chemiluminescence ELISA | ALPCO | 80-INSMR-CH10 |
| KAPA HiFi HotStart ReadyMix | KAPA Biosystems | KK2601 |
| Nextera XT | Illumina | FC-131–1096 |
| Deposited Data | ||
| Single cell mRNA-seq data | this paper | GEO: GSE164875 |
| Single cell mRNA-seq data | Camunas-Soler et al., Cell Metab, 2020 | GEO: GSE124742 |
| Single cell mRNA-seq data | Avrahami et al., 2020 | GEO: GSE154126 |
| Processed patch-seq datasets | Camunas-Soler et al., 2020 | https://github.com/jcamunas/patchseq |
| Experimental Models: Organisms/Strains | ||
| mouse C57BL/6NCrl Inbred | Charles River Laboratories | CRL:027; RRID:IMSR_CRL:027 |
| 5L0D Picolab Laboratory Rodent Diet | LabDiet | 3005659–220 |
| Mouse Diet, High Fat Calories | VWR / Bio-Serv | CA89067–471 |
| Oligonucleotides | ||
| SmartSeq2 OligodT: 5’–AAGCAGTGGTATCAACGCAGAGT ACT30VN-3’ | Picilli et al., 2014 | n/a |
| SmartSeq2 TSO: 5’-AAGCAGTGGTATCAACGCAGAGTA CATrGrG-3’ | Picilli et al., 2014 | n/a |
| SmartSeq2 ISPCR: 5’-AAGCAGTGGTATCAACGCAGAGT-3’ | Picilli et al., 2014 | n/a |
| Software and Algorithms | ||
| Qlucore Omics Explorer v3.6 | Qlucore | www.qlucore.com |
| WEB-based Gene SeT AnaLysis Toolkit | Liao et al., 2019 | webgestalt.org |
| Graphpad Prism v9.0.0 | GraphPad | https://www.graphpad.com |
| Custom analysis software | Camunas-Soler et al., 2020 | https://github.com/jcamunas/patchseq |
| PatchMaster 2×90.1 | Smart Ephys HEKA | https://www.heka.com |
| FitMaster 2×90.1 | Smart Ephys HEKA | https://www.heka.com |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq | Anders et al., 2015 | https://github.com/simon-anders/htseq |
| MATLAB R2020a | Mathworks | https://uk.mathworks.com/products/matlab.html |
| Qupath v0.2.3 | Bankhead et al., 2013 | https://qupath.github.io |
| Cytomap v1.4 | Stoltzfus et al., 2020 | https://gitlab.com/gernerlab/cytomap |
| Genomica | Segal Lab of Computational Biology | http://genomica.weizmann.ac.il/ |
| GSEA 4.1.0 | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| Python v3.7.11 | Python Software Foundation | http://www.python.org |
| XGBoost v1.0.2 | XGBoost | https://xgboost.readthedocs.io/en/latest/python/index.html |
| Scikit-learn v0.24.2 | Pedregosa et al., 2012 | https://scikit-learn.org/stable/ |
| Statsmodel v0.12.2 | Seabold et al., 2010 | https://www.statsmodels.org/stable/index.html |
Highlights:
Glucose suppresses α-cell exocytosis by inhibiting P/Q-type Ca2+ currents
Patch-seq links maturation, respiration, and receptor expression to α-cell function
Dysfunction of α-cells associates with a ‘β-cell-like’ electrophysiologic signature
Impaired exocytosis occurs in α-cells enriched for lineage and immaturity markers
ACKNOWLEDGEMENTS
The University of Alberta is situated on Treaty 6 territory, traditional lands of First Nations and Métis people. We thank Dr. Jesper Grud Skat Madsen (University of Southern Denmark), Dr. Jakob Knudsen (University of Copenhagen) and Dr. Lori Sussel (University of Colorado) for helpful discussion, and Dr. Francis Lynn (University of British Columbia) for critical reading of the draft manuscript. We thank the Human Organ Procurement and Exchange (HOPE) program and Trillium Gift of Life Network (TGLN) for their work in procuring human donor pancreas for research. We also thank Dr. Rita Bottino (Allegheny Health Network) and Drs. James Shapiro and Tatsuya Kin (University of Alberta Clinical Islet Program) for contributing some islet preparations for this study. Finally, we especially thank the organ donors and their families for their kind gift in support of diabetes research.
TdS was supported by the Alberta-Helmholtz Diabetes Research School, the Alberta Innovates Scholarship in Data-Enabled Innovation, and the Sir Fredrik Banting and Dr. Charles Best Canada Graduate Scholarship. This work was funded by grants to PEM from the Canadian Institutes of Health Research (CIHR: 148451), from the JDRF to PEM, LB and PR (SRA-2019-698-S-B), and from the National Institutes of Health to EMW (F32 DK109577), to RS, PEM, SK (R01 DK126482) and to PEM, RAD, SK (U01 DK120447). Some islet samples were from the Human Pancreas Analysis Program (HPAP-RRID:SCR_016202), a Human Islet Research Network (RRID:SCR_014393) consortium (UC4DK-112217 and UC4-DK-112232). PEM holds the Canada Research Chair in Islet Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no conflict of interest relating to this study.
References
- Allister EM, Robson-Doucette CA, Prentice KJ, Hardy AB, Sultan S, Gaisano HY, Kong D, Gilon P, Herrera PL, Lowell BB, et al. (2013). UCP2 regulates the glucagon response to fasting and starvation. Diabetes 62, 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, et al. (2016). Age-dependent pancreatic gene regulation reveals mechanisms governing human β cell function. Cell Metabolism 23, 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrahami D, Wang YJ, Schug J, Feleke E, Gao L, Liu C, HPAP Consortium, Naji A, Glaser B, and Kaestner KH (2020). Single cell transcriptomics of human islet ontogeny defines the molecular basis of β cell dedifferentiation in T2D. Mol Metab 42, 101057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl V, May CL, Perez A, Glaser B, and Kaestner KH (2021). Genetic activation of α-cell glucokinase in mice causes enhanced glucose-suppression of glucagon secretion during normal and diabetic states. Mol Metab 49, 101193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci Rep-Uk 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg S, Galvanovskis J, Gopel SO, Rorsman P, and Eliasson L (2000). Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting alpha-cells. Diabetes 49, 1500–1510. [DOI] [PubMed] [Google Scholar]
- Barg S, Ma X, Eliasson L, Galvanovskis J, Göpel SO, Obermüller S, Platzer J, Renström E, Trus M, Atlas D, et al. (2001). Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic B cells. Biophysical Journal 81, 3308–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basco D, Zhang Q, Salehi A, Tarasov A, Dolci W, Herrera P, Spiliotis I, Berney X, Tarussio D, Rorsman P, et al. (2018). α-cell glucokinase suppresses glucose-regulated glucagon secretion. Nat Commun 9, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokvist K, Eliasson L, Ammälä C, Renström E, and Rorsman P (1995). Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. The EMBO Journal 14, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, Streeter PR, Naji A, Grompe M, and Kaestner KH (2013). Epigenomic plasticity enables human pancreatic α to β cell reprogramming. The Journal of Clinical Investigation 123, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, and Rorsman P (2008). Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628. [DOI] [PubMed] [Google Scholar]
- Briant L, Salehi A, Vergari E, Zhang Q, and Rorsman P (2016). Glucagon secretion from pancreatic α-cells. Upsala Journal of Medical Sciences 121, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briant LJB, Zhang Q, Vergari E, Kellard JA, Rodriguez B, Ashcroft FM, and Rorsman P (2017). Functional identification of islet cell types by electrophysiological fingerprinting. Journal of the Royal Society, Interface 14, 20160999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, Bottino R, CampbellThompson M, Aramandla R, Poffenberger G, et al. (2018). α cell function and gene expression are compromised in type 1 diabetes. Cell Reports 22, 2667–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camunas-Soler J, Dai X-Q, Hang Y, Bautista A, Lyon J, Suzuki K, Kim SK, Quake SR, and MacDonald PE (2020). Patch-seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metabolism 31, 1017–1031.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, Downie C, Liu K, Wang J, Xing Y, et al. (2017). Converting adult pancreatic islet α cells into β cells by targeting both Dnmt1 and Arx. Cell Metabolism 25, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou J, Zeng C, Cheng Z, Han JY, Schlichting M, Miller M, Mendez R, Huang S, Wang J, Sui Y, et al. (2021). Single-cell chromatin accessibility identifies pancreatic islet cell type– and statespecific regulatory programs of diabetes risk. Nature Genetics 52, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Hoppa MB, Walker JN, Amisten S, Abdulkader F, Bengtsson M, Fearnside J, Ramracheya R, Toye AA, Zhang Q, et al. (2010). Progression of diet-induced diabetes in C57BL6J mice involves functional dissociation of Ca2+ channels from secretory vesicles. Diabetes 59, 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X-Q, Spigelman AF, Khan S, Braun M, Fox JEM, and MacDonald PE (2014). SUMO1 enhances cAMP-dependent exocytosis and glucagon secretion from pancreatic α-cells. The Journal of Physiology 592, 3715–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drigo R.A. e, Erikson G, Tyagi S, Capitanio J, Lyon J, Spigelman AF, Bautista A, Fox JEM, Shokhirev M, MacDonald PE, et al. (2019). Aging of human endocrine pancreatic cell types is heterogeneous and sex-specific. Biorxiv 10.1101/729541. [DOI] [Google Scholar]
- El K, Capozzi ME, and Campbell JE (2020). Repositioning the alpha cell in postprandial metabolism. Endocrinology 161, bqaa169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, Ilkayeva O, Smith N, Miller N, Hajmrle C, et al. (2015). Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. The Journal of Clinical Investigation 125, 3847–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gembal M, Gilon P, and Henquin JC (1992). Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. The Journal of Clinical Investigation 89, 1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard J (2017). Glucagon, a key factor in the pathophysiology of type 2 diabetes. Biochimie 143, 33–36. [DOI] [PubMed] [Google Scholar]
- Godazgar M, Zhang Q, Chibalina MV, and Rorsman P (2018). Biphasic voltage‐dependent inactivation of human NaV1.3, 1.6 and 1.7 Na+ channels expressed in rodent insulin‐secreting cells. J Physiology 596, 1601–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpel SO, Kanno T, Barg S, Weng XG, Gromada J, and Rorsman P (2000). Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. The Journal of Physiology 528, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubelnik V, Markovič R, Lipovšek S, Leitinger G, Gosak M, Dolenšek J, Valladolid-Acebes I, Berggren P-O, Stožer A, Perc M, et al. (2020). Modelling of dysregulated glucagon secretion in type 2 diabetes by considering mitochondrial alterations in pancreatic α-cells. Roy Soc Open Sci 7, 191171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylfe E (2016). Glucose control of glucagon secretion-’There’s a brand-new gimmick every year’. Upsala Journal of Medical Sciences 121, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-C, Gaisano HY, and Leung Y-M (2011a). Electrophysiological identification of mouse islet α-cells: from isolated single α-cells to in situ assessment within pancreas slices. Islets 3, 139–143. [DOI] [PubMed] [Google Scholar]
- Huang Y-C, Rupnik M, and Gaisano HY (2011b). Unperturbed islet α-cell function examined in mouse pancreas tissue slices. The Journal of Physiology 589, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Powers AC, Naji A, HPAP Consortium, and Atkinson MA (2019). NIH initiative to improve understanding of the pancreas, islet, and autoimmunity in type 1 diabetes: The Human Pancreas Analysis Program (HPAP). Diabetes 68(7), 1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellard JA, Rorsman NJG, Hill TG, Armour SL, van de Bunt M, Rorsman P, Knudsen JG, and Briant LJB (2020). Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high-fat diet. Mol Metab 40, 101021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Okamoto H, Huang Z, Anguiano G, Chen S, Liu Q, Cavino K, Xin Y, Na E, Hamid R, et al. (2017). Amino acid transporter Slc38a5 controls glucagon receptor inhibition-induced pancreatic α cell hyperplasia in mice. Cell Metabolism 25, 1348–1361.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen JG, Hamilton A, Ramracheya R, Tarasov AI, Brereton M, Haythorne E, Chibalina MV, Spégel P, Mulder H, Zhang Q, et al. (2019). Dysregulation of glucagon secretion by hyperglycemiainduced sodium-dependent reduction of ATP production. Cell Metabolism 29, 430–442.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, and Raychaudhuri S (2019). Fast, sensitive, and accurate integration of single cell data with Harmony. Nature Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam CJ, Cox AR, Jacobson DR, Rankin MM, and Kushner JA (2018). Highly proliferative α-cellrelated islet endocrine cells in human pancreata. Diabetes 67, 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Klughammer J, Farlik M, Penz T, Spittler A, Barbieux C, Berishvili E, Bock C, and Kubicek S (2016). Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Reports 17, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon J, Kin T, Bautista A, Smith N, O D, MacDonald P, and Manning J (2019). Isolation of human pancreatic islets of Langerhans for research v3. Protocols.Io doi: 10.17504/protocols.io.bt55nq86. [DOI] [Google Scholar]
- Marchand SJL, and Piston DW (2010). Glucose suppression of glucagon secretion: metabolic and calcium responses from alpha-cells in intact mouse pancreatic islets. The Journal of Biological Chemistry 285, 14389–14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinis YZD, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, Braha O, Braun M, Ramracheya R, Amisten S, et al. (2010). GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metabolism 11, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka T-A, Kawashima S, Miyatsuka T, Sasaki S, Shimo N, Katakami N, Kawamori D, Takebe S, Herrera PL, Kaneto H, et al. (2017). Mafa enables Pdx1 to effectively convert pancreatic islet progenitors and committed islet α-cells into β-cells in vivo. Diabetes 66, 1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dólleman S, Liu S, Ackermann AM, Cáceres E, Hunter AE, et al. (2017). Virgin beta cells persist throughout life at a neogenic niche within pancreatic islets. Cell Metabolism 25, 911–926.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moede T, Leibiger B, Sanchez PV, Daré E, Köhler M, Muhandiramlage TP, Leibiger IB, and Berggren P-O (2020). Glucokinase intrinsically regulates glucose sensing and glucagon secretion in pancreatic alpha cells. Sci Rep-Uk 10, 20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Liu Y, and Remmen HV (2004). Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279, 49064–49073. [DOI] [PubMed] [Google Scholar]
- Olsen HL, Theander S, Bokvist K, Buschard K, Wollheim CB, and Gromada J (2005). Glucose stimulates glucagon release in single rat α-cells by mechanisms that mirror the stimulus-secretion coupling in β-cells. Endocrinology 146, 4861–4870. [DOI] [PubMed] [Google Scholar]
- Omar-Hmeadi M, Lund P-E, Gandasi NR, Tengholm A, and Barg S (2020). Paracrine control of α-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat Commun 11, 1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onukwufor JO, Berry BJ, and Wojtovich AP (2019). Physiologic implications of reactive oxygen species production by mitochondrial complex I reverse electron transport. Antioxidants Basel Switz 8, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Müller A, Nothman J, Louppe G, et al. (2012). Scikit-learn: Machine Learning in Python. Arxiv. 1201.0490v4. [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Fulllength RNA-seq from single cells using Smart-seq2. Nature Protocols 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, Johnson PR, Rorsman P, and Braun M (2010). Membrane potential-dependent inactivation of voltage-gated ion channels in alpha-cells inhibits glucagon secretion from human islets. Diabetes 59, 2198–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramracheya R, Chapman C, Chibalina M, Dou H, Miranda C, Gonzalez A, Moritoh Y, Shigeto M, Zhang Q, Braun M, et al. (2018). GLP-1 suppresses glucagon secretion in human pancreatic alpha-cells by inhibition of P/Q-type Ca2+ channels. Physiological Reports 6, e13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissaus CA, and Piston DW (2017). Reestablishment of glucose inhibition of glucagon secretion in small pseudoislets. Diabetes 66, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Aizawa T, Komatsu M, Okada N, and Yamada T (1992). Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic B-cell. Diabetes 41, 438–443. [DOI] [PubMed] [Google Scholar]
- Seabold S, and Perktold J (2010). Statsmodels: Econometric and statistical modeling with Python. Proc 9th Python Sci Conf 92–96. [Google Scholar]
- Shuai H, Xu Y, Yu Q, Gylfe E, and Tengholm A (2016). Fluorescent protein vectors for pancreatic islet cell identification in live-cell imaging. Pflugers Archiv : European Journal of Physiology 468, 1765–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N, Spigelman A, Lin H, and MacDonald PE (2020). Mouse pancreatic islet isolation. Protocols.Io doi: 10.17504/protocols.io.sqaedse. [DOI] [Google Scholar]
- Stoltzfus CR, Filipek J, Gern BH, Olin BE, Leal JM, Wu Y, Lyons-Cohen MR, Huang JY, Paz-Stoltzfus CL, Plumlee CR, et al. (2020). CytoMAP: A spatial analysis toolbox reveals features of myeloid cell organization in lymphoid tissues. Cell Reports 31, 107523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: A knowledgebased approach for interpreting genome-wide expression profiles. Proc National Acad Sci 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr TB, Valdomir G, Liang M, Wipf P, and Meriney SD (2012). New calcium channel agonists as potential therapeutics in Lambert–Eaton myasthenic syndrome and other neuromuscular diseases. Ann Ny Acad Sci 1275, 85–91. [DOI] [PubMed] [Google Scholar]
- Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, and Herrera PL (2010). Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464, 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viloria K, Nasteska D, Briant LJB, Heising S, Larner DP, Fine NHF, Ashford FB, Xavier G. da S., Ramos MJ, Hasib A, et al. (2020). Vitamin-D-binding protein contributes to the maintenance of α cell function and glucagon secretion. Cell Reports 31, 107761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiser O, Trus M, Hernández A, Renström E, Barg S, Rorsman P, and Atlas D (1999). The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc National Acad Sci 96, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh EHG, Huang Z, Xia J, Li D, Davidson HW, and Li W-H (2020). ZIGIR, a granule-specific Zn2+ indicator, reveals human islet α cell heterogeneity. Cell Reports 32, 107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Ramracheya R, Lahmann C, Tarasov A, Bengtsson M, Braha O, Braun M, Brereton M, Collins S, Galvanovskis J, et al. (2013). Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metabolism 18, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJG, Leiss V, Nassar MA, Welling A, Gribble FM, et al. (2014). Na+ current properties in islet α- and β-cells reflect cell-specific Scn3a and Scn9a expression. The Journal of Physiology 592, 4677–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 2. Transcript correlations with total exocytosis from alpha-cells of donors with no diabetes (ND) at 1 mM and 10 mM glucose, related to Figure 2B
Supplementary Table 3. Transcript correlations with total exocytosis from alpha-cells of donors with type 2 diabetes (T2D) at 1 mM and 10 mM glucose, related to Figure 2G
Supplementary Table 4. Differential expression analysis of transcripts in mouse alpha-cells from chow-fed versus high-fat-fed mice, related to Figure 4
Supplementary Table 5. Transcript correlations with Na+ current voltage-dependence of half-inactivation of alpha-cells from Chow-fed and HFD-fed mice, related to Figure 4
Supplementary Table 6. Correlation of transcripts with peak Na+ current and Na+ current half-inactivation in human ND and T2D alpha-cells, related to Figure 5
Supplemental Table 7. Correlation of transcript expression with αprobability values from three different classifier models applied to ND and T2D alpha-cells, related to Figure 6
Data Supplement S1. Source data used to generate all graphs, related to Figures 1, 3–7 and Supplemental Figures 1, 2, 4 and 6.
Supplementary Figures. Supplementary Figure legends and Supplementary Figures 1–7