

SUMMARY
Metabolic flexibility is a hallmark of many cancers where mitochondrial respiration is critically involved, but the molecular underpinning of mitochondrial control of cancer metabolic reprogramming is poorly understood. Here we show that reverse electron transfer (RET) through respiratory chain complex I (RC-I) is particularly active in brain cancer stem cells (CSC). Although RET generates ROS, NAD+/NADH ratio turns out to be key in mediating RET effect on CSC proliferation, in part through the NAD+-dependent Sirtuin. Mechanistically, Notch acts in an unconventional manner to regulate RET by interacting with specific RC-I proteins containing electron-transporting Fe-S clusters and NAD(H)-binding sites. Genetic and pharmacological interference of Notch-mediated RET inhibited CSC growth in Drosophila brain tumor and mouse glioblastoma multiforme (GBM) models. Our results identify Notch as a regulator of RET and RET-induced NAD+/NADH balance a critical mechanism of metabolic reprogramming and a metabolic vulnerability of cancer that may be exploited for therapeutic purposes.
eTOC Blurb
Metabolic reprogramming is a hallmark of cancer. The role of mitochondria in this process remains enigmatic. Ojha et al. show that cancer cells undergo active reverse electron transfer (RET), and that Notch acts in an unconventional manner to regulate RET. Pharmacological inhibition of RET is beneficial in brain tumor models.
INTRODUCTION
Metabolic reprogramming is one of the hallmarks of cancer (Hanahan and Weinberg, 2011; Vander Heiden et al., 2009). Cancer cells exhibit increased glycolytic metabolism even in the presence of ample oxygen. This phenomenon of aerobic glycolysis (Warburg effect), was initially hypothesized as a consequence of mitochondrial dysfunction in cancer cells (Warburg, 1956). Earlier studies provided evidence of mitochondrial dysfunction in some cancers (Gottlieb and Tomlinson, 2005; Wallace, 2005), and mitochondrial metabolic intermediates acting as oncometabolites (Weinberg and Chandel, 2009; Yang et al., 2013). Recent studies, however, indicate that in many cancers of diverse tissue origins, the capacity for mitochondrial oxidative phosphorylation (OxPhos) is not only maintained, but proved to be essential for oncogenesis (Gui et al., 2016; Janiszewska et al., 2012; Kuntz et al., 2017; Molina et al., 2018; Pasto et al., 2014; Shi et al., 2019; Viale et al., 2014). Warburg effect thus reflects metabolic flexibility of cancer cells rather than permanent switch to glycolysis. Intermediates of the glycolytic pathways are used for the biosynthesis of nucleotides, amino acids, and lipids to support the growth of cancer cells, and presumably all proliferating cells (Vander Heiden et al., 2009). Hence Warburg effect may be interpreted as a normal adaptation to the metabolic requirements of proliferation, both in cancer cells and normal proliferating cells such as tissue stem cells.
Notch signaling is a fundamental signaling pathway operating in almost all multicellular organisms and all cell types in the body (Andersson et al., 2011; Guruharsha et al., 2012). A key function of Notch is to maintain tissue homeostasis by balancing the self-renewal vs. differentiation of stem cells (Liu et al., 2010). In the Drosophila neural stem cell (NSC) setting, loss of Notch signaling causes stem cell depletion and lineage termination, whereas its hyperactivation leads to overproliferation of CSC-like tumorigenic cells (Bowman et al., 2008; Song and Lu, 2011; Weng et al., 2010). Aberrant Notch signaling has been linked to the pathogenesis of brain tumors in mammals (Pierfelice et al., 2008), especially the proneural subtype of GBM (Saito et al., 2014). Deregulated Notch signaling is also frequently observed in other cancer types (Aster et al., 2017). Under certain conditions, Notch can act as tumor suppressor (Nowell and Radtke, 2017). The role of Notch in cancer is thus context-dependent. Beyond its fundamental role in NSC biology and cancer, Notch signaling also critically regulates synaptic plasticity and neuronal survival and its deregulation has been implicated in major brain disorders such as Alzheimer’s disease and stroke (Ho et al., 2020). Thus, Notch signaling is critical for tissue development, function, and regeneration, and of high clinical significance.
Despite intensive studies, the mechanism by which Notch signaling regulates the behavior of CSCs and normal stem cells is not well defined. Previous studies have focused on canonical Notch signaling that involves ligand-receptor interaction, proteolytic cleavage of Notch, release of the intra-cellular domain of Notch (NICD) to the nucleus, and transcription of downstream target genes by NICD through Suppressor of Hairless [Su(H)]-related transcription factors (Kovall et al., 2017). However, Notch also signals in non-canonical fashions in different developmental contexts (Hunter and Giniger, 2020), and recent studies uncovered unconventional Notch signaling mechanisms that involve Notch localization to mitochondria and Notch action in Su(H)-independent fashion (Lee et al., 2013; Li et al., 2020). In the Drosophila brain, the canonical and non-canonical Notch pathways cooperate to regulate NSC homeostasis. The clinical significance of the non-canonical pathway is underscored by the observation that CSC-like tumor-initiating cells in both Drosophila brain tumor models and human GBM are particularly sensitive to its perturbation (Lee et al., 2013). The identification of an unconventional Notch signaling pathway that impinges on RC-I is consistent with the known role of Notch, and an emerging role of OxPhos in NSC homeostasis and brain CSC maintenance (Bonnay et al., 2020; Janiszewska et al., 2012; van den Ameele and Brand, 2019). However, the molecular mechanism by which Notch operates in this pathway is not well understood.
In this study we report an unanticipated role of Notch in directly regulating RET inside mitochondria. RC-I is the first component of the electron transport chain (ETC) that transfers electrons from NADH to ubiquinone through several Fe-S clusters. Under certain conditions, however, electron transfer can reverse direction, moving from ubiquinol to NAD+, producing a significant amount of reactive oxygen species (ROS) in this RET process (Onukwufor et al., 2019; Scialo et al., 2017). Described over six decades ago (Chance and Hollunger, 1961) and initially regarded as some sort of in vitro artifact, RET has been linked to macrophage activation in response to bacterial infection (Mills et al., 2016), adaptation of carotid body to changes in oxygen levels (Fernandez-Aguera et al., 2015), and fly lifespan (Scialo et al., 2016), and RET-generated ROS burst contributes to ischemia-reperfusion injury (Chouchani et al., 2014). Whether RET is involved in cancer has not been tested before, and virtually nothing is known about the key molecular players and the regulation of RET in health and disease.
We found that cancer cells and CSCs undergo active RET, which is important for their growth and survival. We also found that Notch regulates RET through direct interaction with RC-I subunits at the distal arm of the complex involved in electron transfer and NAD(H) metabolism. Surprisingly, we found that NAD+/NADH balance, instead of RET-ROS, critically mediates the physiological effects of RET in the cancer models we studied, at least in part through the NAD+-dependent Sirtuins. Pharmacological inhibition of Notch-regulated RET exhibited anti-cancer effect in mammalian cell culture, in vivo fly model of Notch-induced brain tumor, and orthotopic xenograft model of GBM in the mouse. Our results identify Notch as a non-ETC protein that participates in RET, and RET a mechanism of metabolic reprogramming and a metabolic vulnerability of cancer that can be exploited for therapeutic purposes.
RESULTS
Active RET in GBM and Other Cancer Cells Alters Cancer Cell Metabolism and Promotes Proliferation and Survival
In previous studies on GBM stem cells (Lee et al., 2013), rotenone, a RC-I inhibitor, and Mdivi-1, a presumed mitochondrial fission inhibitor, were identified as potent inhibitors of GBM proliferation and self-renewal. While this result highlighted the importance of mitochondrial function to GBM stem cell maintenance, the exact mechanism involved remained elusive. Intrigued by recent studies implicating Mdivi-1 as a RET inhibitor (Bordt et al., 2017), and the fact that rotenone also acts as a RET inhibitor at the low concentration used in our GBM studies (Cino and Del Maestro, 1989; Liu et al., 2002), we tested if RET is the mitochondrial mechanism regulating GBM stem cells. First, we found that like rotenone and Mdivi-1, metformin and S1QEL1.1, two other RET inhibitors (Batandier et al., 2006; Robb et al., 2018), inhibited tumor sphere formation by pediatric patient-derived GBM-387 cells (Fig. 1A). This was correlated with inhibition of GBM cell viability and proliferation (Fig. 1B; S1A) and promotion of cell death (Fig. 1C). Similar effects were observed in the LN299 GBM cell line (Fig. 1D), the melanoma cell line A375 (Fig. 1E), and the cervical cancer cell line HeLa (Fig. S1B). In contrast, non-cancer cells HEK293 (Fig. S1C, D) and human fibroblasts (Fig. S1E, F) were not sensitive to RET inhibitors. Hence cancer cells are particularly sensitive to RET inhibition. In subsequent studies, we used GBM cells and A375 cells interchangeably in our cancer cell studies.
Figure 1. RET is Active in GBM and Other Cancers.

(A-C) Effect of RET inhibitors on GBM-387 tumor sphere formation (A), viability (B), and cell death (C).
(D, E) Effect of RET inhibitors on cell death of LN299 (D) and A375 (E) cells.
(F-H) Effect of RET inhibitors on H2O2 production as probed with CM-H2DCDA in GBM-387 (F), LN299 (G), and A375 (H) cells.
(I, J) ROS detection by CM-H2FDA and Mito-SOX staining in DES-treated GBM-387 (I) and A375 (J) cells. Scale bar: 100μm (I) and 75μm (J).
(K, L) Effect of DES on tumor sphere formation by GBM-387 (K) and colony formation by A375 cells (L).
(M-O) Effect of RET inhibitors on DES-induced H2O2 production (M) and colony formation (N) by A375 cells, and tumor sphere formation by GBM-387 cells (O).
Data are representative of at least 3 independent experiments. Error bars, ± SEM; *p < 0.05, ** P<0.01,***p < 0.001.
See also Figure S1.
One of the key products of RET is the ROS superoxide, which is quickly converted to H2O2. Compared to non-cancer cells (Fig. S1G, H), GBM cells (Fig. 1F, G) and A375 cells (Fig. 1H) exhibited high basal ROS levels, which were significantly reduced by RET inhibitors, whereas RET inhibitors had no obvious effect on ROS in non-cancer cells (Fig. S1G, H). These results suggest that RET is particularly active in cancer cells. To further demonstrate that RET is driving ROS in cancer cells, we used compounds that disrupt two of the driving forces of RET, mitochondrial membrane potential (MMP) and ubiquinol (CoQH2). CCCP was used to depolarize MMP and dimethyl malonate (DMM), a competitive inhibitor of succinate dehydrogenase (Chouchani et al., 2014; Mills et al., 2016), was used to decrease CoQH2. CCCP and DMM both reduced basal ROS levels in cancer cells (Fig. S1I), supporting that RET is active in cancer cells.
To further test the role of RET in cancer cells, we treated cells with diethylsuccinate (DES), a cell permeable form of the complex II substrate succinate. The accumulation of succinate drives RET and ROS production, as seen in ischemic stroke (Chouchani et al., 2014) and inflammatory immune response (Mills et al., 2016). DES supplementation (DES+) significantly increased ROS in GBM-387 (Fig. 1I) and A375 cells (Fig. 1J). This was correlated with increased tumor sphere formation by GBM-387 cells (Fig 1K) and colony formation on soft agar by A375 cells (Fig. 1L) by DES. DES also increased A375 cell proliferation (Fig. S1J) and cell migration in trans-well migration assay (Fig. S1K). Importantly, the effects of DES on ROS production (Fig. 1M) and colony formation (Fig. 1N) by A375 cells, and on tumor sphere formation by GBM-387 cells (Fig. 1O), were all blocked by RET inhibitors, supporting that DES acts through RET to regulate cancer cell behavior. Furthermore, DES stimulated lactate dehydrogenase (LDH) activity, a key marker of glycolysis, and this effect was blocked by RET inhibition (Fig. S1L), suggesting that RET promotes glycolysis, endowing cancer cells with the Warburg effect. DES had minor effect on ROS production in non-tumor cells, and it did not promote proliferation or tumor sphere formation in these cells (Fig. S1M, N).
A Critical Role of Notch in Regulating RET in Cancer Cells
We probed the effect of DES and RET inhibitors on cancer signaling pathways, with particular focus on unconventional Notch signaling, as previous studies implicated a critical role of mitochondria-localized Notch in regulating RC-I through a transcription-independent mechanism in GBM cells (Lee et al., 2013). DES+ increased the level of Notch2, and stimulated mTORC2 signaling as indicated by increased p-AKT (T473) (Fig. 2A), a marker of non-canonical Notch signaling (Lee et al., 2013). DES also stimulated NF-kB signaling as indicated by increased p-P65 level (Fig. 2A) and increased expression of NLR family pyrin domain containing 3 (NLRP3) inflammasome (Fig. 2A), and the Interleukin 1 beta (IL-1β) and Interleukin 4 (IL-4) cytokines (Fig. 2B). Importantly, RET inhibitors significantly reduced the DES effect on Notch 2, NLRP3, and IL-4/IL-1β expression, whereas the inhibitor effects on p-AKT and p-P65 varied. Consistent with an unconventional role of Notch in RET, DES had no effect on the expression of canonical Notch signaling targets (Fig. S2A). These results implicate unconventional Notch action in RET regulation and the mTORC2/AKT and NF-kB pathways as downstream mediators.
Figure 2. Notch Critically Regulates RET in Cancer Cells.

(A) Immunoblots showing the effect of RET inhibitors on Notch2, p-AKT, p-P65, and NLRP3 levels in LN299 cells under basal or DES+ conditions.
(B) qRT-PCR analysis showing induction of IL-1β and IL-4 expression by DES in LN299 cells and effect of rotenone.
(C) CM-H2DFHDA staining of A375 cells showing effect of Notch1, Notch2, and Notch3 knockdown on H2O2 production under basal (control) or DES+ conditions. Scale bar: 25μm.
(D) Effect of Notch1, Notch2, and Notch3 knockdown on H2O2 production in LN299 cells under basal or DES+ conditions.
(E) Effects of Notchi or Notch2 RNAi on the expression of the indicated proteins in A375 cells under basal or DES+ conditions.
(F, G) Effect of Notchi on colony formation by A375 cells (F) or tumor sphere formation by GBM-387 cells (G) under basal or DES+ conditions.
(H) Effect of Notch2 RNAi on LN299 cell proliferation under basal or DES+ conditions.
(I) Effect of Notch2 RNAi on IL-1β and IL-4 expression in LN299 cells under basal or DES+ conditions.
Data are representative of at least 3 independent experiments. Bar graphs show data quantification.
Error bars, ± SEM; *p < 0.05, ** P<0.01,***p < 0.001.
See also Figure S2.
Given the effect of RET on Notch2 protein level, we next tested whether Notch2 participates in RET. Strikingly, Notch2 RNAi completely obliterated the induction of RET-ROS by DES in A375 cells (Fig. 2C) and LN299 cells (Fig. 2D). Notch1 RNAi and Notch3 RNAi also showed some effect, but much weaker than Notch2 RNAi (Fig. 2C, D). Pharmacological inhibition of Notch signaling using a Notch inhibitor (Notchi) FLI-06, which alters the intracellular trafficking of Notch (Kramer et al., 2013) and reduced Notch2 protein level (Fig. S2B), showed similar effect as Notch2 RNAi (Fig. S2C, D). Notch2 RNAi also blocked ROS production driven by oxidation of glycerol-3-phosphate (G3P) (Fig. S2E), a substrate of G3P dehydrogenase that generates the ubiquinol pool, a driving force of RET (Stepanova et al., 2019). It is interesting to note that Notch2 RNAi (Fig. 2C, D, S2E) and Notchi (Fig. S2C, D) both led to increased ROS production under basal, non-DES+ condition. Thus, the effect of Notch inhibition is similar to the action of rotenone, which can block RET-related ROS production driven by elevated succinate level, but under basal conditions can lead to ROS production due to blockage of forward electron transfer (FET) (Liu et al., 2002). This result further suggested that in addition to its critical and unique role in RET, Notch may also regulates FET and OxPhos under normal conditions. Importantly, ROS production under Notch inhibition condition (Fig. S2F) and DES-driven RET condition (Fig. S2G) was blocked by the flavin protein inhibitor diphenyleneiodonium (DPI) (Holland et al., 1973), supporting that both DES-driven RET condition and blockage of FET by Notch inhibition under basal condition generated ROS through the flavin site of RC-I, a major source of ROS production at RC-I (Pryde and Hirst, 2011).
To further support the notion that Notch signals in an unconventional manner to regulate RET-ROS, we examined the effect of pharmacological and genetic manipulation of canonical Notch signaling. Inhibition of γ-secretase activity with a small molecule to prevent NICD generation (Fig. S2H), or RNAi-mediated inhibition of Rbpj (Fig. S2I), a key factor in canonical Notch signaling (Kovall et al., 2017), had no effect on basal ROS or DES-induced RET-ROS. Inhibition of γ-secretase or Rbpj also had no obvious effect on cancer cell viability (Fig. S2J, K).
We next tested the effect of Notch on RET-driven signaling events. DES-induced activation of mTORC2/AKT, NF-kB, and proinflammatory signaling (Fig. 2E) were blocked by Notch2 RNAi or Notchi in A375 cells. Notch inhibition also blocked the stimulating effect of DES on colony formation by A375 cells (Fig. 2F), tumor sphere formation by GBM-387 cells (Fig. 2G), and proliferation (Fig. 2H) and proinflammatory response (Fig. 2I) of LN299 cells. Moreover, Notch2 RNAi attenuated the effect of DES on glycolysis (Fig. S2L). Thus, the effects of RET on cancer cell metabolic reprogramming, proliferation, and survival are mediated by Notch. These results identify Notch as signaling molecule that directly participates in RET.
NAD+/NADH Balance Mediates the Effect of RET in Cancer Cells
Mitochondrial ROS (mito-ROS) can be formed at multiple sites (Goncalves et al., 2015), with RET at RC-I being a major source (Murphy, 2009; Scialo et al., 2017), and ROS can drive tumorigenesis in some cancer types (Sullivan and Chandel, 2014). The fact that Notchi or Notch2 RNAi induced ROS but inhibited cancer proliferation under basal condition suggested that mito-ROS might not be the main mediator of RET effect in the cancer cells studied here. Consistently, treatment with a battery of antioxidants acting in the cytosol or mitochondria (NAC, mito-TEMPO, mito-Q, and Trilox) did not affect tumor sphere formation by GBM-387 (Fig. S3A), or viability of LN299 (Fig. S3B) and A375 (Fig. S3C) cells. Antioxidants also had no significant effect on the viability of A375 (Fig. S3C) or LN299 (Fig. S3D) cells under DES+ condition.
We sought to find the key mediator of the biological effects of RET in cancer cells. The most immediate effect of RET is the reduction of NAD+ to NADH by ubiquinol-derived electrons. Indeed, DES+ significantly decreased NAD+/NADH ratio (Fig. 3A). Moreover, treatment of GBM-387 cells with extra NADH, or lowering cellular NAD+ level with FK866 (Hasmann and Schemainda, 2003), a selective inhibitor of the NAD+ salvage pathway enzyme nicotinamide phosphoribosyltransferase (NAMPT), promoted tumor sphere formation (Fig. 3B), suggesting that NAD+/NADH balance may be an important mediator of the biological effect of RET. Indeed, treatment with the NAD+ precursor nicotinamide (NAM) or nicotinamide mononucleotide (NMN) inhibited tumor sphere formation by GBM-387 (Fig. 3C, S3E), or viability of LN299 cells (Fig. S3F). Treatment with DPI, which prevents conversion of NAD+ to NADH, had similar effects as NAM (Fig. 3D). Both NAM and DPI prevented DES-induced cytokine production (Fig. 3E, F) and cell signaling (Fig. 3G, H).
Figure 3. NAD+/NADH Ratio Mediates RET Effect on Cancer Cell Proliferation and Signaling.

(A) Effect of DES on NAD+/NADH ratio in A375 cells.
(B) Effects of NADH and FK866 on tumor sphere formation by GBM-387 cells.
(C, D) Effects of NAM and DPI on tumor sphere formation by GBM-387 cells under basal or DES+ conditions.
(E, F) Effects of DPI (E) and NAM (F) on DES-induced IL-1β and IL-4 expression in A375 cells.
(G, H) Immunoblots showing effects of DPI (G) and NAM (H) on DES-induced expression of the indicated signaling proteins in A375 cells.
(I-K) Effects of cyto-LbNOX and mito-LbBOX on NAD+/NADH ratio (I), cell viability (J), and signaling (K) in A375 cells. K shows expression of Flag-tagged LbNOX proteins.
(L) Immunoblots showing effects of resveratrol on DES-induced expression of the indicated signaling proteins in LN299 cells.
Data are representative of at least 3 independent experiments. Error bars, ± SEM; *p < 0.05, ** P<0.01,***p < 0.001.
See also Figure S3.
To further test the importance of NAD+/NADH ratio in regulating cancer behavior, we used a NADH oxidase from Lactobacillus brevis (LbNOX) as a genetic tool to alter NAD+/NADH ratio (Martinez-Reyes et al., 2020; Titov et al., 2016). Compared to cytosolically-localized LbNOX (cyto-LbNOX), mitochondrially-localized mito-LbNOX significantly increased NAD+/NADH ratio and reduced cancer cell viability. This was correlated with reduced p-AKT and p-NF-kB signaling (Fig. 3I–K). Colony- or sphere-forming ability of cancer cells was also compromised by mito-LbNOX (Fig. S3G, H). We further altered NAD+/NADH ratio using yeast NADH dehydrogenase (NDI1), a single enzyme that can replace the NAD+ regeneration capability of the mammalian RC-I (Seo et al., 1998). As expected, NDI1 increased NAD+/NADH ratio (Fig. S3I). Since NDI1 transfers electrons from NADH to CoQ to generate CoQH2, a driving force of RET, NDI1 was previously used to induce RET-ROS (Scialo et al., 2016). Indeed, we found that NDI1 increased ROS level, which was abolished by treatment with RET inhibitor or knockdown of RC-I component (Fig. S3J, K), suggesting that NDI1 induces RET-ROS. However, NDI1 expression decreased cancer cell viability and colony-forming ability (Fig. S3L, M), supporting that NAD+/NADH ratio, instead of RET-ROS, is a major mediator of RET effect in these cancer cells. NDI1-induced NAD+/NADH ratio and cell viability changes were rescued by NDUFS2 RNAi (Fig. S3N, O), supporting that these effects were mediated by RET.
NAD+ serves as a central regulator of metabolism and longevity, by acting as a cosubstrate for key enzymes including the Sirtuin family of protein deacetylases (Bonkowski and Sinclair, 2016; Imai and Guarente, 2014). We tested whether Sirtuins might be involved in the regulation of cancer cell behavior by RET. The Sirtuin activator Resveratrol mimicked the effect of DPI or NAM on DES-induced signaling effects (Fig. 3L). Resveratrol also reduced the viability of LN299 and A375 cells under basal condition, and more so under DES+ condition (Fig. S3P, Q). On the other hand, treatment of A375 cells with the SIRT1 inhibitor EX 527 (Solomon et al., 2006), but not SIRT2 inhibitor AGK2 (Outeiro et al., 2007) or SIRT3 inhibitor 3-TYP (Galli et al., 2012), mimicked the effect of DES on cell signaling (Fig. S3R) and tumor sphere formation in GBM-387 cells (Fig. S3S). Thus RET-induced NAD+/NADH ratio change may act through SIRT1 to influence cancer signaling and proliferation.
Notch2 Regulates RET through Interaction with RC-I Proteins Containing Fe-S Clusters and NAD(H)-Binding Site
Identification of Notch2 as a regulator of RET offered a unique opportunity to dissect the molecular events involved. Notch2 exhibited mitochondrial localization (Fig. S4A, B), consistent with previous findings (Lee et al., 2013). This was likely mediated through Notch2 interaction with Tom20, a receptor in the TOM complex involved in import of nuclear-encoded mitochondrial proteins (Fig. S4C). Moreover, the Notch2-Tom20 interaction was enhanced under DES+ condition, suggesting that the mitochondrial import of Notch2 might be regulated by RET. DES enhanced the mitochondrial localization while decreasing the plasma membrane localization of Notch2, an effect reversed by RET inhibitor rotenone (Fig. S4B). The level of Notch2, but not Notch1 or Notch3, was increased in a time-dependent manner by DES (Fig. S4D). Furthermore, Notch2 overexpression (OE) decreased NAD+/NADH ratio, whereas Notch2-RNAi increased NAD+/NADH ratio in DES+ condition (Fig. S4E). These data suggested that Notch2 might act in mitochondria to regulate RET. Indeed, we found Notch2, primarily NICD2, in immunoaffinity-captured RC-I holo-complex (Fig. 4A). Compared to core RC-I proteins, the amount of Notch2 in the immuno-captured complex relative to the input was at a sub-stoichiometric level, consistent with Notch 2 being present in multiple cellular compartments with only a portion inside mitochondria.
Figure 4. Notch2 Regulates RET through Interaction with RC-I Proteins.

(A) Immunoblots showing the presence of NICD2 in immuno-captured RC-I from A375 cells. COX-IV and ATP5A: negatives controls.
(B) co-IP assay of the interaction between Notch2 and the indicated proteins.
(C, D) Effect of C-I30 and NDUFV2 RNAi on H2O2 production under basal and DES+ conditions. Bar graphs show data quantification. Scale bar: 25μm.
(E, F) Effect of NICD2 OE on H2O2 level (E) and signaling (F) in A375 cells under basal and DES+ conditions. Scale bar: 75μm (E).
(G) Effects of NDUFV2 RNAi on NICD2 OE-induced signaling events.
(H) co-IP assay showing effect of NDUFV2 OE on Notch2 interaction with NDUFV1 and C-I30.
(I, J) co-IP assay showing effect of C-I30 OE (I) or NICD2 OE (J) on NDUFV1 interaction with the indicated proteins.
(K) Immunoblots showing the effect of Notchi on the assembly of NDUFV1 into newly formed RC-I in CMP-treated cells after drug wash out.
Data are representative of at least 3 independent experiments. Error bars, ± SEM; *p < 0.05, ** P<0.01,***p < 0.001.
See also Figure S4.
To identify the proteins in RC-I that interact with Notch, we performed Notch2 IP and found specific Notch2 interactions with proteins in the NADH dehydrogenase and hydrogenase modules, including NDUFV1, NDUFV2, NDUFS1, NDUFS2, NDUFS3 (also known as C-I30), many containing electron-transporting Fe-S clusters, and in the case of NDUFV1 also NAD(H)- and FMN-binding sites (Vinothkumar et al., 2014). Weaker interaction with NDUFB6 was also detected. However, no interaction with proteins in the hydrophobic membrane module of RC-I (e.g., ND3 and NDUFA16) or complex III (e.g., COX-IV) was detected (Fig. 4B).
We tested the functional role of the Notch2-interacting RC-I proteins in RET. Like Notch2 RNAi, C-I30 RNAi or NDUFV2 RNAi blocked DES-induced ROS production (Fig. 4C, D). As a control, inhibition of complex V component ATP5A did not affect DES-induced ROS production (Fig. S4F), suggesting that simply inhibiting OxPhos would not affect RET. Notch2 OE boosted ROS production (Fig. 4E) and RET-related cell signaling (Fig. 4F) under basal conditions, an effect blocked by NDUFV2 RNAi (Fig. 4G), suggesting that the Notch2-interacting RC-I proteins are required for Notch2 function in RET. Supporting this notion, NDUFV2 OE enhanced Notch2 interaction with NDUFV1 and C-I30 (Fig. 4H). Thus, although NDUFV2 itself may not be directly involved in electron transfer due to the long distance from its Fe-S cluster to the nearest Fe-S cluster incompatible with electron-transferring (Hinchliffe and Sazanov, 2005), it facilitates the association of Notch2 with RC-I proteins involved in RET. Surprisingly, unlike NDUFV2, C-I30 OE inhibited Notch2 interaction with NDUFV1 (Fig. 4I), reduced Notch2 level, and inhibited RET-induced mTORC2/AKT signaling (Fig. S4G). We hypothesized that C-I30 and Notch2 compete for binding to NDUFV1. Consistently, Notch2 OE reduced C-I30 interaction with NDUFV1 (Fig. 4J). In cells treated with the mitochondrial translational inhibitor chloramphenicol (CMP) to prevent new RC-I assembly, allowing existent complex to turn over and new complex to assemble after CMP washout, Notch inhibition reduced NDUFV1 assembly into RC-I (Fig. 4K). These results reveal unanticipated dynamic interactions between Notch2 and the RC-I proteins involved in RET.
A Small Molecule Inhibits RET by Modulating Notch Interaction with RC-I Proteins
Quinazolinones and their derivatives are building blocks of naturally occurring alkaloids isolated from plants, microorganisms, and animals, which exhibit diverse pharmacological effects. Quinazoline- and quinazolinone-based drugs have made their way to clinics to treat various disease conditions (Hameed et al., 2018). A quinazolinone, 6-chloro-3-(2,4-dichloro-5-methoxyphenyl)-2-mecapto-7-methoxyquinazolin-4(3H)-one (CPT2008 or CPT) (Fig. S5A), was designed by Cerepeut through medicinal chemistry and initially isolated based on its potent anticancer activity in T-ALL cells, which feature hyperactive Notch signaling (Weng et al., 2004). CPT had no effect on canonical Notch signaling (Fig. S5B). We tested whether CPT might interfere with Notch-mediated RET. CPT showed potent anti-cancer activity in LN299 and A375 cells (Fig. 5A), but little toxicity to non-cancer cells (Fig. S5C, D), which were also non-sensitive to other RET inhibitors (Fig. S5E, F). CPT exhibited stronger potency than Mdivi-1 in inhibiting tumor sphere formation by GBM-387 cells (Fig. 5B), and similar activity as Notchi in inhibiting colony formation by A375 cells (Fig. 5C), and its inhibition of DES-induced proliferation correlated with blockage of DES induced MMP increase (Fig. 5D). Like other RET inhibitors, CPT inhibited DES-induced RET-ROS in LN299 cells (Fig. 5E), and RET-ROS production by DES (Fig. 5F), G3P (Fig. 5G), or NDI1 OE (Fig. S5G) in A375 cells. CPT also inhibited DES-induced upregulation of Notch2, p-AKT, p-P65, NLRP3 (Fig. 5H), and proinflammatory cytokines (Fig. 5I). Together, these data support that CPT is a new RET inhibitor.
Figure 5. CPT Inhibits RET by Modulating Notch Interaction with RC-I Proteins.

(A) Effect of CPT and RET inhibitors on viability of A375 and LN299 cells.
(B) Effects of different doses of CPT and Mdivi-1 on tumor sphere formation by GBM-387 cells. Scale bar: 100μm.
(C) Effect of CPT, Mdivi-1, and Notchi on colony formation by A375 cells.
(D) Effects of CPT on DES-induced cell proliferation (TOTO-3) and MMP (JC-1) increase in A375 cells.
(E) Effect of CPT and RET inhibitors on H2O2 level in LN299 cells under basal and DES+ conditions.
(F, G) Effect of CPT on H2O2 level in A375 cells under basal and DES+ (F) or G3P+ (G) conditions.
Scale bar: 50μm.
(H) Effect of CPT on levels of the indicated proteins in A375 cells under basal and DES+ conditions.
(I) Effects of CPT on DES-induced IL-1β and IL-4 expression in A375 cells.
(J) Effect of CPT treatment on C-I30 interaction with the indicated proteins in A375 cells.
(K) Immunoblots detecting proteins pulled down by the CPT-Photolabel (PL) probe.
(L) Effect of NDUFV2 OE on p-AKT level in A375 cells with or without CPT.
(M) Effect of NICD2 OE on cell viability in A375 cells with or without CPT.
Data are representative of at least 3 independent experiments. Bar graphs show data quantification.
Error bars, ± SEM; *p < 0.05, ** P<0.01,***p < 0.001.
See also Figure S5.
We next investigated the mechanism of action of CPT in RET. CPT strongly inhibited the interaction of C-I30 with Notch2 and NDUFV2, but slightly increased C-I30 interaction with NDUFV1 (Fig. 5J). To identify the mitochondrial targets of CPT, we applied a photoaffinity labeling method (Rebecca et al., 2019), using a probe (CPT-PL) consisting of a CPT warhead attached to benzophenone (as a photo label) and linked to desthiobiotin (as an affinity handle) (Fig. S5H). Treatment of A375 cells with CPT-PL reduced Notch2 level (Fig. 5K), suggesting that CPT-PL was cell permeable and retained the activity of CPT. Western blot analysis of protein affinity purified with CPT-PL detected Notch2, C-I30, and NDUFV1 as bound proteins (Fig. 5K). Neither NDUFV2, NDUFS1, NDUFS2 nor other mitochondrial proteins were detected (Fig. 5K), demonstrating specificity of CPT interaction with Notch2, C-I30, and NDUFV1. This data, together with the effect of CPT on the protein-protein interactions within the Notch2-asociated module shown earlier, indicated that CPT inhibits Notch2 interaction, but facilitates C-I30 interaction with NDUFV1. This is likely to have significant impact on RET and NAD+/NADH balance, as NDUFV1 is the only subunit that harbors both Fe-S clusters and NAD(H)- and FMN-binding sites. Supporting the importance of protein-protein interaction dynamics in mediating CPT effect, although NDUFV2 did not directly bind to CPT, its OE antagonized the effect of CPT on mTORC2/AKT signaling (Fig. 5L), presumably by strengthening the complex formation between Notch2 and NDUFV1 (Fig. 4H), and Notch2 OE attenuated the effect of CPT on cancer cell viability (Fig. 5M).
A Key Role of NAD+/NADH Balance in Mediating the Effect of CPT in Cancer Cells
We further tested whether CPT acts through NAD+/NADH balance to regulate cancer cell behavior. CPT treatment increased NAD+/NADH ratio under basal condition, and it blocked DES-induced NAD+/NADH ratio change (Fig. 6A). Importantly, supplementation of cells with NADH blocked the effect of CPT on ROS production (Fig. 6B) and cell viability (Fig. 6C) in A375 cells. NADH supplementation also blocked CPT effect on tumor sphere formation by GBM-387 cells (Fig. 6D). Depletion of NAD+ with FK866 similarly attenuated CPT effect on tumor sphere formation by GBM-387 cells (Fig. 6E), although FK866 alone showed considerable toxicity. FK866 treatment also inhibited CPT effect on cell viability in LN299 cells (Fig. 6F). These data support that NAD+/NADH balance critically mediates the effect of CPT on cancer cell behavior. Further supporting this, NADH blocked the CPT effect on Notch2 and p-AKT and p-P65 levels (Fig. 6G). As a positive control, we showed that the effects of the flavin site RC-I inhibitor DPI on Notch2, p-AKT, p-p65 signaling (Fig. S6A) and on cell viability (Fig. S6B) were also attenuated by added NADH in A375 cells. The effect of CPT on NAD+/NADH ratio change was further validated in the LbNOX system, where we found that CPT-induced NAD+/NADH ratio increase was further enhanced by mito-LbNOX (Fig. S6C), and the CPT effect on cancer cell viability was also enhanced by mito-LbNOX (Fig. S6D). NADH or FK866 treatment blocked CPT effect on proinflammatory cytokine expression (Fig. S6E, F). Moreover, the SIRT1 inhibitor blocked the effect of CPT on A375 cell viability (Fig. 6H) and Notch2, p-AKT, and p-p65 signaling (Fig. 6I). Thus, NAD+/NADH ratio and the ensuing SIRT1 activity change may mediate the effects of RET inhibition by CPT.
Figure 6. NAD+/NADH Ratio Mediates CPT Effect on Cancer Cell Proliferation.

(A) Effect of CPT on NAD+/NADH ratio in A375 cells under basal and DES+ conditions.
(B) Effect of NADH on H2O2 level in A375 cells with or without CPT. Scale bar: 50μm.
(C) Effect of NADH on viability of A375 cells with or without CPT.
(D, E) Effect of NADH (D) or FK866 (E) on tumor sphere formation by GBM-387 cells with or without CPT.
(F) Effect of FK866 on viability of LN299 cells with or without CPT.
(G) Effect of NADH on levels of the indicated proteins in A375 cells with or without CPT.
(H) Effect of SIRT1 or SIRT3 inhibitor on viability of A375 cells with or without CPT.
(I) Effect of SIRT1 or SIRT3 inhibitor on levels of the indicated proteins in A375 cells with or without CPT.
Data are representative of at least 3 independent experiments. Bar graphs show data quantification.
Error bars, ± SEM; *p < 0.05, ** P<0.01,***p < 0.001.
See also Figure S6.
Effect of RET on CSC Metabolism, Proliferation, and Tumorigenesis In Vivo
To test the physiological relevance of Notch-regulated RET on CSC behavior in vivo, we first used a Drosophila brain tumor model induced by Notch OE driven by the 1407-Gal4 driver (Lee et al., 2013; Song and Lu, 2011). We found that Notch-induced brain tumors exhibited elevated ROS production compared to control brains (Fig. 7A). Treatment with CPT significantly reduced the ROS level (Fig. 7A), suggesting that, as in cultured human GBM and other cancer cells, Notch regulated RET and RET-ROS production in vivo in Drosophila NBs. This was further supported by the reduction of ROS level in Notch-induced brain tumors by CCCP, and by the OE of alternative oxidase (AOX) (Fig. S7A), which metabolizes the RET driver CoQH2 (Fernandez-Ayala et al., 2009). OE of mastermind (Mam), a key component of canonical Notch signaling, did not induce ROS production, and inhibition of Mam or the Notch ligand Delta (Dl) did not affect Notch-induced RET-ROS (Fig. S7B), supporting that Notch acts unconventionally to regulate RET-ROS in this brain tumor model. Notch OE in the gut also increased ROS, which was blocked by CPT (Fig. S7C), suggesting that the regulation of RET by Notch occurs in more than one tissues. Moreover, compared to control brains, Notch-induced tumorous brains exhibited lower NAD+/NADH ratio, which was restored by CPT (Fig. 7B) or the flavin site inhibitor DPI (Fig. S7D), but not affected by genetic manipulation of canonical Notch signaling components (Fig. S7E). The NAD+ precursor NAM had similar effect (Fig. 7C). Correlating with their effects on RET-ROS and NAD+/NADH ratio, CPT (Fig. 7D), NAM (Fig. 7E), and DPI (Fig. S7F) inhibited Notch-induced NB overproliferation, which was concomitant with increased expression of the differentiation marker Prospero (Pros). As in mammalian cancer cells, NDI1 OE increased ROS in the fly brain, which was inhibited by CPT or RNAi of NDUFS2, supporting that NDI1 induces RET-ROS (Fig. S7G). However, Notch-induced brain tumor was attenuated by NDI1 OE (Fig. S7H), likely due to the elevation of NAD+/NADH ratio by NDI1 (Fig. S7I). These data support the importance of NAD+/NADH ratio in mediating the effect of RET on CSCs in vivo. Consistent with Sirtuin’s role in mediating the effect of NAD+/NADH, resveratrol also inhibited Notch-induced NB overproliferation (Fig. S7J). Remarkably, as in human cancer cells where C-I30 OE antagonized Notch2 effect on RET signaling (Fig. 4I, S4F), C-I30 OE inhibited Notch-induced NB overproliferation (Fig. 7F) and partially rescued animal viability (Fig. 7G). This was correlated with reduced Notch protein level (Fig. S7K). Consistent with C-I30 RNAi being able to attenuate Notch-induced brain tumor phenotype (Lee et al., 2013), Notch OE induced lethality was also partially rescued by C-I30 RNAi (Fig. S7L). These results indicate that the mechanism of Notch regulation of CSC behavior through RET is conserved across species. C-I30 OE and RNAi may both rescue Notch OE-induced lethality by inhibiting Notch-mediated RET but through different mechanisms: C-I30 OE acts by competing with Notch for NDUFV2 binding and by reducing Notch level (Fig. 4I; S4F), whereas as a core component of the machinery involved in RET (Fig. 4C), C-I30 is required for RET in general and its knockdown will block Notch-mediated RET.
Figure 7. Effect of RET on Cancer Stem Cell Proliferation and Tumorigenesis in vivo.
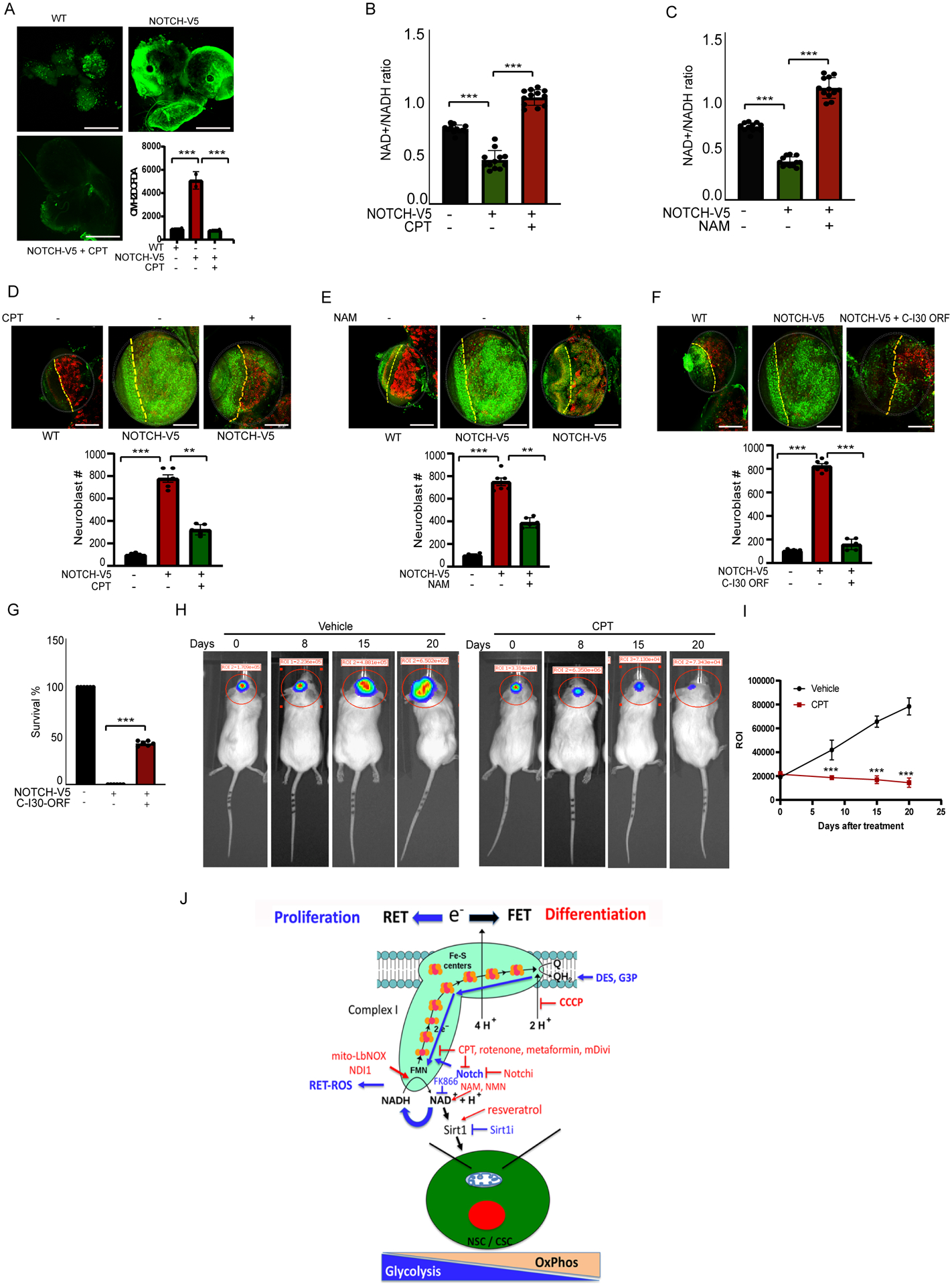
(A) Effect of CPT on H2O2 level in Notch OE larval brain. Scale bar: 50μm.
(B, C) Effects of CPT or NAM on NAD+/NADH ratio in Notch OE larval brain. Scale bar: 20 μm.
(D-F) Effects of CPT (D), NAM (E), or C-I30 OE (F) on NB number in Notch OE larval brain. Brains were immunostained for the NB marker Deadpan (green) and the differentiation marker Prospero (red). White dashed circles mark brain lobes, yellow dashed lines separate optic lobe (left) from central brain (right). Scale bar: 20μm.
(G) Effect of C-I30 OE on the survival of Notch OE flies.
(H, I) Bioluminescence imaging of human GBM-387 growth in mouse brains at different days after transplantation and treated with CPT or vehicle. Graph shows data quantification (I).
(J) Model of metabolic reprogramming dictated by RET that regulates the proliferation vs. differentiation of NSCs or CSCs. The various molecules and drugs used in this study are indicated. Blue lines indicate RET. Promoters of RET or mediators of RET effect are in blue. Inhibitors of RET or inhibitors of the biological effect of RET in red.
Bar graphs show data quantification. Error bars, ± SEM; *p < 0.05, ** P<0.01,***p < 0.001.
See also Figure S7.
To further test the conserved role of RET in cancer, we used an orthotopic transplantation assay to test the in vivo efficacy of RET inhibition on GBM growth in a mouse model. GBM-387 cells expressing a luciferase reporter were stereotaxically injected into the striatum of mice. GBM growth was monitored by tracking bioluminescence from the luciferase reporter. CPT treatment not only significantly arrested the growth of transplanted GBM cells but also caused regression of established tumors in the mouse brain (Fig. 7H, I), supporting the importance of RET to GBM stem cell proliferation and maintenance in vivo in a mammalian system.
DISCUSSION
Mechanisms regulating the metabolic reprogramming of cancer cells are complex and not fully understood. Efforts have been directed toward elucidating roles of oncogenes and tumor suppressors (Schulze and Harris, 2012). Previous studies on Notch function in cancer biology have focused on canonical signaling, implicating transcriptional targets of NICD in cancer metabolism (Slaninova et al., 2016; Xu et al., 2015). It is thus unexpected that we find Notch acts inside mitochondria to directly regulate RET, and that the direction of electron flow in ETC can have profound effect on metabolism and confer growth advantage to cancer cells. RET is sensitive to two key mitochondrial parameters: the redox state of CoQ, which senses the state of electron flow along the ETC, and MMP, which reflects the capacity to produce ATP and drives key mitochondrial events such as protein import and calcium influx (Murphy, 2009; Scialo et al., 2017). RET is well suited to detect a range of physiological changes that require metabolic adaptation.
How is the RET process activated in cancer cells? One possibility is that RC-I is present in a different conformation in cancer cells, being conducive to RET and with or without grossly affected FET. Our data show that Notch is physically associated with RC-I subunits that correspond to the entry point of FET and the end point of RET, and that Notch alters the protein-protein interactions among them. This is likely to change the direction or kinetics of electron flow, as these proteins contain the electron transporting Fe-S clusters. At this point, we cannot exclude the possibility that Notch may interact with a subcomplex of RC-I to effect RET. Future structural studies of RC-I will offer insights into the regulation of RET by Notch or RET inhibitors such as CPT. RET activation in cancer cells may also have to do with events happening downstream of RC-I. For example, succinate accumulation due to blockage of complex II, III or IV, higher levels of ubiquinol due to hyperactivation of enzymes such as G3P dehydrogenase that introduce electrons downstream of RC-I, or higher MMP due to complex V operating in reverse direction.
Mito-ROS is the main source of free radicals in most cells. ROS plays multi-faceted roles, with physiological levels of ROS exerting signaling functions whereas pathological levels causing oxidative damage and contributing to aging and diseases (Schieber and Chandel, 2014). It is thought that RET-ROS is the main contributor of mito-ROS, and RET-ROS has been linked to disease (Murphy, 2009; Scialo et al., 2017). Hence RET-ROS is a reasonable candidate for mediating the effect of RET on cancer cell proliferation and survival. It is thus surprising that in our cancer models, RET-ROS is not as critical as NAD+/NADH ratio in mediating the RET effects. It is still possible that RET-ROS is important in certain cancer types where ROS signaling has been implicated (Sullivan and Chandel, 2014). Interestingly, a recent study implicated CoQH2 oxidation by complex III to drive oxidative TCA cycle and dihydroorotate dehydrogenase activity as an essential event in certain tumor growth (Martinez-Reyes et al., 2020). As RET will also lead to CoQH2 oxidation, future studies will address how RET induced mito-ROS, NAD+/NADH ratio change, and CoQH2 oxidation may be differentially deployed in different cancer contexts.
Our data indicate that the NAD+-dependent Sirtuins at least partially mediate the effect of NAD+/NADH ratio in cancer cells. Sirtuins are NAD-dependent enzymes involved in metabolism, stress response, and longevity (Imai and Guarente, 2014). Mammals express a family of seven Sirtuins (SIRT1–7). Though we have not tested all Sirtuins, our data implicated SIRT1 in mediating the effect of RET. Our results are consistent with the notion that Sirtuins have the ability to suppress the Warburg effect, although Sirtuin action can be context-dependent (Chalkiadaki and Guarente, 2015). Notch acetylation and stability can be regulated by SIRT1 (Guarani et al., 2011), and SIRT1 and Notch functionally interact to regulate NSC behavior (Ma et al., 2014). Our results suggest that through stimulation of RET and regulation of NAD+/NADH ratio, Notch may also reciprocally regulate SIRT1. Future studies will investigate how SIRT1 may mediate the effect of Notch on mTORC2/AKT and NF-kB signaling, whether RET is involved in the cross-regulation between Notch and SIRT1 in the metabolic control of normal NSC behaviors such as proliferation and temporal differentiation, which are known to involve OxPhos and RC-I (Homem et al., 2014; Lee et al., 2013; van den Ameele and Brand, 2019), and the relative contribution of NAD+/NADH and RET-ROS in these processes. Future studies will also test the generality of the intriguing model (Fig. 7J) derived from our studies that the direction of electron flow along RC-I may act as a switch to control the proliferation vs. differentiation decisions of normal NSCs and CSCs through metabolic reprogramming regulated by the signaling molecules studied here.
Our results indicate that RET is a targetable metabolic vulnerability of cancer. Using CPT and other RET inhibitors, we showed that cancer cells are particularly susceptible to RET inhibition. Using Drosophila and mouse models, we showed that RET inhibition with CPT significantly inhibited tumor growth, suggesting that RET-driven CSC proliferation and maintenance is fundamental to brain tumorigenesis. GBM remains one of the deadliest forms of cancer with limited treatment option. Altered protein expression and protein-protein interactions within RC-I (Deighton et al., 2014), and activation of Notch, mTORC2/AKT, NF-kB signaling (Le Rhun et al., 2019) have been observed in GBM, and inflammation plays a critical role in GBM pathogenesis (Salazar-Ramiro et al., 2016), and high Notch2 expression correlates with GBM chemoresistance (Tome et al., 2019). The LN299 GBM cells used in this study, which are sensitive to RET inhibition, are naturally resistant to temozolomide (TMZ), the standard of care chemotherapy for GBM (Lee, 2016). Targeting RET may offer a treatment option for GBM, including TMZ non-responding GBM.
Besides cancer, RET inhibitors like CPT may find application in other clinical indications. Deregulated RET, and RET-ROS in particular, contributes to reperfusion injury in ischemic stroke (Chouchani et al., 2014). Notch activation has long been implicated in the pathophysiology of stroke (Arumugam et al., 2006), although it has not been tested whether RET is involved. Our ongoing studies are testing the effect of CPT in stroke models. Increased RET has been observed in aging human muscle (Capel et al., 2005). Intriguingly, the RC-I proteins shown here to interact with Notch and participate in RET are reported to be downregulated in long lived mammals (Miwa et al., 2014), and their knockdown in worms or flies can extend lifespan (Hur et al., 2014), raising the intriguing possibility that modulation of their function in RET with CPT may extend the lifespan of normal animals. This is supported by the observations that CPT can reduce the effect of RET on inflammation, a hallmark of aging, that CPT restores NAD+/NADH ratio, and that supplementation of NAD+ precursors can counter the age-dependent decline of NAD+, extend lifespan and protect again age-related diseases across species (Rajman et al., 2018). Future studies will test the effect of RET modulation on lifespan and health span in normal and diseased animals.
Limitations of study
A limitation of the current study is that the involvement of specific Sirtuins in mediating the biological effect of RET-induced NAD+/NADH ratio change was not validated by genetic means in cell culture models or in vivo models of cancer. The other limitation is that it was not tested whether NAD+/NADH ratio or RET-ROS change mediated the anti-cancer effect of CPT in the mouse GBM model, and whether the signaling molecules implicated by the cell culture studies were involved. These issues will be explored in future studies.
STAR★METHODS
Resource Availability
Lead Contact. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bingwei Lu (bingwei@stanford.edu)
Materials Availability. All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
- Data and Code Availability.
- All data reported in this paper will be shared by the lead contact upon request.
- This paper does not report original code.
- Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Experimental Model and Subject Details
Mouse
The mice used in this study were at the age of eight-ten weeks. The animals were maintained under 14 hr light/ 12 hr dark cycle with free access to food and water. Animals were housed in groups of up to 3 per cage. The mouse strain used in the experiment is NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ or NOD-scid gamma (NSG) (#005557 from Jackson Laboratory). As no obvious differences between male and female mice were observed in previous GBM studies, we selected only male mice for the present study. After mice were received from the vendor, they were kept in the animal facility of Stanford University School of Medicine for one week. All animal experiments were performed in accordance with the protocols approved by the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford University and comply with all regulations for ethical conduct of animal research. See Methods Details for model generation and drug treatment studies.
Drosophila
The indicated UAS RNAi and OE fly lines were crossed to 1407-Gal4 driver line for NB expression, and escargot-Gal4 for fly gut expression. To reduce potential toxicity of Notch OE during early development, the Gal80ts system was used to repress transgene expression. Crosses were set up at 25°C, embryos were collected in vials with drug-containing food or control normal food, and after larval hatching, vials were transferred to 29°C for Gal80ts inactivation and transgene activation. Animals were allowed to develop further at 29°C for 120 hrs before dissection. All experiments included in this study were performed on 3rd stage of larva. See Key Resources Table for genotypes. Fly culture and crosses were performed according to standard procedures and raised at indicated temperatures. Adult flies were generally raised at 25°C and with 12/12 hr dark/light cycles. Fly food was prepared with a standard receipt (Water, 17 L; Agar, 93 g; Cornmeal, 1,716 g; Brewer’s yeast extract, 310 g; Sucrose, 517 g; Dextrose, 1033 g). Both males and females were used for the described studies as no phenotypic differences were observed between sexes.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-FLAG | Sigma-Aldrich | F1804 |
| Rabbit anti-FLAG | Sigma-Aldrich | F7425 |
| Mouse anti-TOM20 | Santa Cruz Biotech | sc17764 |
| Rabbit anti-cytochrome C | Abcam | ab90529 |
| Rabbit anti-C-I30 | Abcam | ab14711 |
| Mouse anti-C-IV s.1 | Abcam | ab14705 |
| Mouse anti-Actin | Sigma-Aldrich | A2228 |
| Rabbit anti-NDUFV1 | Proteintech | 11238-1-AP |
| Rabbit anti-NDUFV2 | Proteintech | 15301-1-AP |
| Rabbit anti-HEY1 | Proteintech | 19929-1-AP |
| Rabbit anti-HES5 | Proteintech | 22666-1-AP |
| Rabbit anti-NDUFA10 antibody | Abcam | ab174829 |
| Rabbit anti- NDUFB6 antibody | Abcam | ab241313 |
| Rabbit anti- NDUFA9 Polyclonal antibody | Proteintech | 20312-1-AP |
| Rabbit anti-MT-ND3 Polyclonal Antibody | Thermofisher scientific | PA5-75629 |
| Rabbit Phospho-NF-κB p65 (Ser468) Antibody | Cell signaling Technology | 3039 |
| Rabbit anti Phospho-Akt (Ser473) | Cell signaling Technology | 4060 |
| Rabbit anti NLRP3 (D4D8T) | Cell signaling Technology | 15101 |
| Rabbit anti- Notch1 (D1E11) XP | Cell signaling Technology | 3608 |
| Rabbit anti- Notch2 (D76A6) XP® | Cell signaling Technology | 5732 |
| Rabbit anti- Notch3 (D11B8) | Cell signaling Technology | 5276 |
| Mouse anti-SDHA antibody | Abcam | ab14715 |
| Goat anti-Mouse IgG-HRP | Santa Cruz | sc-2005 |
| Goat anti-Rabbit IgG HRP | Santa Cruz | sc-2004 |
| Goat anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A32723 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | A11036 |
| DAPI | Sigma-Aldrich | D9542 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Diethyl succinate | Sigma-Aldrich | 112402 |
| Diethyl butyl malonate | Sigma-Aldrich | 112038 |
| Dimethyl malonate | Sigma-Aldrich | 136441 |
| G3P | Sigma-Aldrich | P3591-50MG |
| Antimycin A | Sigma-Aldrich | A8674 |
| METFORMIN | Sigma-Aldrich | M0605000 |
| CPT | Cerepeut Inc. | CPT2008 |
| CPT-Photolabel probe | Cerepeut Inc. | CPT-PL |
| S1QEL1.1 | Sigma-Aldrich | SML1948 |
| S3QEL 2 | Sigma-Aldrich | SML1554 |
| Diphenyleneiodonium chloride (DPI) | Sigma-Aldrich | D2926 |
| Resveratrol | Sigma-Aldrich | R5010 |
| Selisistat (EX 527) | Selleckchem | S1541 |
| AGK2 | Selleckchem | S7577 |
| 3-TYP | Selleckchem | S8628 |
| RBPJ Inhibitor-1 (RIN1) | Selleckchem | S3376 |
| Mdivi-1 | Enzo Life Sciences | BML-CM127-0010 |
| Rotenone | Sigma-Aldrich | R8875 |
| Nicotinamide | Sigma-Aldrich | N0636 |
| NADH | Sigma-Aldrich | N8129 |
| Piericidin A | Sigma-Aldrich | P4368 |
| DAPT | Selleckchem | S2215 |
| FLI-06 | Selleckchem | S7399 |
| FK866 | Selleckchem | S2799 |
| MITO TEMPO | Sigma-Aldrich | SML0737 |
| Mitoquinol | Cayman | 89950 |
| Chloramphenicol | Sigma-Aldrich | C0378-25G |
| Lipofectamine 3000 | Invitrogen | L3000015 |
| Lipofectamine RNAi-MAX | Invitrogen | 13778150 |
| Tris base | Sigma-Aldrich | 11814273001 |
| Glycine | Sigma-Aldrich | G8898 |
| SDS | Sigma-Aldrich | L3771 |
| Pierce 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher | 28908 |
| Triton™ X-100 | Sigma Aldrich | T9284 |
| DMEM, high glucose, GlutaMAX Supplement | GIBCO | 10566016 |
| RPMI-1640 | Thermo Fisher | 11875085 |
| EDTA | Sigma-Aldrich | E9884 |
| EGTA | Sigma-Aldrich | E3889 |
| Dimethyl sulfoxide | Sigma-Aldrich | D8418 |
| 4XLaemmli sample buffer | BioRad | 161-0747 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 |
| Critical Commercial Assays | ||
| Complex I Enzyme Activity Microplate Assay Kit | Abcam | ab109721 |
| NAD+/NADH quantification colorimetric kit | BioVision | K337 |
| iScript cDNA synthesis kit | BioRAD | 1708890 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | A25741 |
| Western Lightning Plus-ECL | PerkinElmer Inc. | NEL105001EA |
| HyBlot CL Autoradiography Film | Denville Scientific Inc. | 1159M38 |
| Tetramethylrhodamine (TMRM) | Invitrogen | T668 |
| MitoSox | Invitrogen | M36008 |
| Amplex™ Red Hydrogen Peroxide/Peroxidase Assay Kit | Thermofisher Scientific | A22188 |
| Di(Acetoxymethyl Ester) (6-Carboxy-2’,7’-Dichlorodihydrofluorescein Diacetate) | Thermofisher Scientific | C2938 |
| TOTO™−3 Iodide (642/660) | Thermofisher Scientific | T3604 |
| TO-PRO™−3 Iodide (642/661) | Thermofisher Scientific | T3605 |
| JC-1 | Invitrogen | T3168 |
| NuPAGE® MOPS SDS running buffer | Invitrogen | NP0001 |
| NuPAGE 4–12% Bis-Tris Protein Gels | Invitrogen | NP0321 |
| Oligonucleotides | ||
| qRT-PCR primer: IL-1β Forward: AGCTACGAATCTCCGACCAC | Stanford PAN facility | N/A |
| qRT-PCR primer: IL-1β Reverse: CGTTATCCCATGTGTCGAAG | Stanford PAN facility | N/A |
| qRT-PCR primer: IL-4 Forward: CCAACTGCTTCCCCCTCTG | Stanford PAN facility | N/A |
| qRT-PCR primer: IL-4 Reverse: TCTGTTACGGTCAACTCGGTG | Stanford PAN facility | N/A |
| siRNAs | ||
| Stealth RNAi siRNA Negative Control Hi GC | Invitrogen | 12935-400 |
| Stealth RNAi™ siRNA of ATP5A | Invitrogen | HSS106021 |
| Stealth RNAi™ siRNA of NDUFV2 | Invitrogen | HSS145636 |
| Stealth RNAi™ siRNA of NDUFS3 | Invitrogen | HSS116992 |
| Stealth RNAi™ siRNA of NOTCH1 | Invitrogen | HSS147816 |
| Stealth RNAi™ siRNA of NOTCH2 | Invitrogen | HSS175195 |
| Stealth RNAi™ siRNA of NOTCH3 | Invitrogen | AS029RSI |
| Stealth RNAi™ siRNA of RBPJ | Invitrogen | HSS142633 |
| Stealth RNAi™ siRNA of NDUFS2 | Invitrogen | HSS107038 |
| Recombinant DNA | ||
| pcDNA3.1-Flag-NDUFS3 | Lab reagent | Wu et al., Mol Cell 2019 |
| NDUFV2 (NM_021074) Human Tagged ORF Clone | RC200653 | Origene |
| pcDNA3.1/NDUFV1/myc-His | HG16710-NH | SinoBiologicals |
| 3XFlagNICD2 | 20184 | Addgene |
| pcDNA3.1/myc-His(−)A | 127503 | Addgene |
| pLV-NDI1 | Gift from Navdeep Chandel, Northwestern University, USA | (Martinez-Reyes et al., 2020) |
| pLV-LbNOX-mito | Gift from Navdeep Chandel, Northwestern University, USA | (Martinez-Reyes et al., 2020) |
| pLV-LbNOX-cyto | Gift from Navdeep Chandel, Northwestern University, USA | (Martinez-Reyes et al., 2020) |
| pLV-RFP | Gift from Navdeep Chandel, Northwestern University, USA | (Martinez-Reyes et al., 2020) |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | http://www.graphpad.com/scientific-software/prism/ |
| FlowJo | FlowJo | http://www.flowjo.com/ |
| Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Fiji ImageJ | National Institute of Health | https://imagej.nih.gov/ij/ |
| Experimental Models: Cell Lines | ||
| HEK293 cell line | ATCC | CRC-1573 |
| HeLa cell line | ATCC | CCL-2 |
| A375 cell line | ATCC | CRL-1619 |
| LN299 cell line | ATCC | CRL-2611 |
| GBM-387 | Stanford University | SU-GBM002/387 |
| Normal fibroblast cell line #1 | Coriell Institute | ND29510 |
| Experimental Models: Organisms/Strains | ||
| UAS-NOTCH-V5 | Gift from Mark Fortini (Thomas Jefferson University, USA) | Lab stock |
| 1407-Gal4 | Gift from Liqun Luo (Stanford University, USA) | Lab stock |
| USA-ND30-3XHA(III) | F00300 | FlyORF |
| ND30 RNAi | 44535 | Bloomington Drosophila Stock Center |
| ND51 RNAi | 36701 | Bloomington Drosophila Stock Center |
| ND24 RNAi | 51855 | Bloomington Drosophila Stock Center |
| UAS-NDI1 | Gift from David Walker (UCLA, USA) | Lab stock |
| UAS-Mam | 27743 | Bloomington Drosophila Stock Center |
| UAS-Mam(DN) | 26672 | Bloomington Drosophila Stock Center |
| ND-49 RNAi | 28573 | Bloomington Drosophila Stock Center |
| Serrate RNAi | 28713 | Bloomington Drosophila Stock Center |
| Delta RNAi | 28032 | Bloomington Drosophila Stock Center |
| Su(H) RNAi | 28900 | Bloomington Drosophila Stock Center |
| UAS-AOX | Gift from Howard Jacobs (University of Tampere, Finland) | Lab stock |
| escargot-Gal4 | Gift from David Walker (UCLA, USA) | Lab stock |
| tub-Gal80ts | 7017 | Bloomington Drosophila Stock Center |
Cell Lines
A375, LN299 cells, HeLa cells and HEK293T cells were purchased from ATCC. The control fibroblast line derived from a 55-year old female subject was obtained from the Coriell Institute (Coriell ID ND29510). HEK293T cells and control fibroblasts were cultured under standard tissue culture conditions (1x DMEM medium - GIBCO, 10% FBS, 5% CO2, 37°C). A375 cells and LN299 cells were cultured in RPMI-1640 medium containing 10% FBS. SU-GBM002/387 line was generated from freshly resected human GBM sample acquired under IRB 18672 approved protocol. GBM-387 cells were provided by Dr. Siddhartha Mitra from Sam Cheshier’s Lab in Stem Cell Institute and Department of Neurosurgery at Stanford University.
METHOD DETAILS
Cell culture and cell transfection conditions
Regular LN299, A375, HeLa cells and HEK293T cells were obtained from American Tissue Culture Collection (ATCC) and were maintained under standard tissue culture conditions (5% CO2, 37°C).
SU-GBM002/387 line was generated from freshly resected human GBM sample acquired under IRB 18672 approved protocol at Stanford University School of Medicine. Tissue sample was dissociated by collagenase IV (1mg/ml), and treated with ACK/RBC lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3 and 0.1 mM Na2-EDTA), and plated at sub clonal density for neurosphere formation in Neurobasal medium (Invitrogen), B27 (-A) (Invitrogen), human-bFGF (20ng/ml) (Shenandoah Biotech), human-EGF (20ng/ml) (Shenandoah Biotech), and heparin (10ng/ml). Neurospheres were maintained in serum-free defined media.
Cell transfections were performed by using Lipofectamine 3000 (cat#: L3000015, Invitrogen), and siRNA knockdown experiments were performed using Lipofectamine RNAiMAX reagent (cat#: 13778150, Invitrogen), according to manufacturer’s instructions. For lentiviral transfection, the Invitrogen™ ViraPower™ Lentiviral Packaging Mix (cat no.# K497500) was used. HEK293T cell transfections were performed by using Lipofectamine 2000. After 72 hrs of transfection, virus particles were collected and concentrated using the Lenti-X™ Concentrator (cat no.# PT4421–2, Takara Bio). After concertation, A375 cells were transfected with virus titrate samples.
Immunofluorescence analysis of cultured cells
For immunofluorescence analysis, cells were washed with 1x PBS three times and fixed with 4% formaldehyde in 1x PBS for 30 min at room temperature, later washed and permeabilized with 1x PBS containing 0.1% Triton X-100 for 5 min. The fixed samples were subsequently blocked with 1x PBS containing 5% normal goat serum and incubated for 1 hour at room temperature followed by incubation with primary antibodies for 3 hrs at room temperature. Thereafter, secondary antibodies were added for 1 hour at room temperature. The primary antibodies used were mouse anti-TOM20 (1:1,000, Santa Cruz) and anti-Notch2 (1:100). The secondary antibodies used were Alexa Fluor® 488 and 594 conjugated antibodies (1:500, Molecular Probes).
Mitochondria isolation
Intact mitochondria from human cells were purified and quality controlled for the absence of contamination by other organelles according to the established procedures. Briefly, samples were homogenized using a Dounce homogenizer. After two steps of centrifugation (1,500 g for 5 min and 13,000 g for 17 minutes), the mitochondria pellet was resuspended and washed twice with HBS buffer (5 mM HEPES, 70 mM sucrose, 210 mM Mannitol, 1 mM EGTA, 1x protease inhibitor cocktail). Mitochondrial pellet was resuspended in appropriate buffers for further analyses. Each experiment was conducted in triplicate and repeated three times.
Mitochondrial respiratory complex I activity assay
Mitochondrial complex-I activities were measured by using a Complex I Enzyme Activity Microplate Assay Kit (Colorimetric) (ab109721, abcam). Briefly, cells were seeded and mitochondrial extracts were prepared and quantified by using Bradford reagent. Mitochondrial extracts were added in coated plates and incubated for 3 hrs. After incubation, wells were washed with wash buffer for three times. Thereafter, wells were blocked with blocking buffer for 1 hour. After blocking, wells were washed with wash buffer and assay buffer was added for 30 min. After incubation, O.D. was observed at 450 nM using a microplate reader (Cytation3, BioTek Inc). Each experiment was conducted in triplicate and repeated three times.
Complex-I immune-capture assay
The Complex I Immuno-capture Kit (ab109711, abcam) was used to perform complex I affinity purification as per manufacturer’s protocol. Briefly, mitochondria were isolated in HBS buffer using dounce homogenizer. Mitochondrial pellet was resuspended in lauryl sulfate buffer and mitochondrial fraction were further purified by cold centrifugation at 20000 g for 30 min. Supernatants were collected and protein concentrations were measured. 250μg protein was used for purification. Thereafter, immunocapture beads were added in 0.1% lauryl sulfate buffer for overnight on shaker. The next day, beads were washed three times for 1.5 hrs at 4°C. After washing, sample buffer was added and boiled for 10 min at 94°C and proceeded for western blotting. Each experiment was conducted in triplicate and repeated three times.
CPT, CPT photolabeling/affinity purification probe, and photoaffinity purification
CPT and CPT photolabeling/affinity purification probe were obtained from Cerepeut Inc. under a Materials Transfer Agreement between Cerepeut Inc. and Stanford University. The compounds were synthesized for Cerepeut Inc. by the Chemistry Branch of WuXi AppTec. Details of chemical synthesis are available upon request.
The cellular targets of CPT were investigated by using the CPT Photoaffinity/Biotinylated Probe. The strategy for chemical modification of CPT to enable photolabeling by UV cross-linking and subsequent Biotin-Streptavidin based affinity purification of target protein(s) was adopted from a previous study (Rebecca et al., 2019). To identify the molecular target of CPT, A375 cells were treated with CPT-PL photolabeling probe (40μM) for 4 hrs. After treatment, cells were irradiated with UV light to conjugate the probe to the bound proteins. Following treatment, mitochondria extracts were prepared and protein concentration determined by Bradford reagent. About 500μg mitochondrial protein was taken for streptavidin affinity precipitation. Streptavidin beads were added for overnight incubation on a shaker at 4°C. The next day, samples were washed and processed for western blot analysis.
Co-immunoprecipitation (co-IP), SDS-PAGE, and western blot analyses
Cells lysates were processed directly for NP40 IP-lysis buffer (5 M NaCl, 10% NP-40, 1 M Tris (pH 8.0), and protease inhibitor cocktail (cat#: 11836170001, Sigma). After centrifugation at 10,000 g for 5 min, the supernatant was subjected to primary antibodies at 4°C overnight with gentle shaking. Subsequently, the magnetic beads were added for 2 hrs and thereafter washed three times (10 minutes each) at 4°C in washing buffer. 1x loading dye was added and samples were boiled at 97°C for 5 min. Samples were loaded onto NuPAGE 4%–12% Bis-Tris Protein Gels (cat#: NP0321BOX, Invitrogen) and ran in MOPS SDS running buffer (cat#: NP0001, Invitrogen) were used for SDS-PAGE and immunoblot analyses according to standard procedures. For data quantification of western blots, signal intensity was measured and calculated using NIH Image J.
Complexosome assay
To study the role of NOTCH in complex-I assembly, we adopted the complexosome assay as described previously (Guerrero-Castillo et al., 2017). In order to detect newly formed complex I, mitochondrial translation was inhibited in A375 cells by treatment with chloramphenicol for 5 days at a final concentration of 50 mg/ml, to allow existing complex I to turn over. After 5 days, cells were washed with PBS twice and new medium with or without CPT (10μM) or NOTCHi (FLI-06, 10μM) was added for 24 hrs. After treatment, cells were scrapped and mitochondria was isolated using Dounce homogenizer in HBS buffer (5 mM HEPES, 70 mM Sucrose, 210 mM Mannitol, 1 mM EGTA, 1x protease inhibitor cocktail). Mitochondrial concentration was determined by using Bradford reagent. About 40 μg protein sample was prepared and run SDS PAGE for western blotting.
Flow cytometric analysis of ROS
Cells were seeded at 0.5×106 cells/ml. Cells were treated for 30 min to 4 hrs and thereafter, cells were trypsinzed and processed for staining of Mitosox (5μM), CM-H2DCFDA (5μM). Cells were incubated with Mitosox and CM-H2DCFDA for 15 min in the dark at 37°C. After incubation, cells were washed and resuspended in PBS. Samples were immediately observed using a LSR II flow cytometer, and data were analyzed using the FlowJo software.
Confocal microscopy analysis of ROS
Cells were plated at 0.3×106 cells/ml on coverslip in six well plate. After treatment, cells were washed and CM-H2DCFDA (5μM) was added in DPBS and incubated for 10 min at 37°C in the dark. Cells were imaged on a Leica SP8 confocal microscope with an excitation laser of 492–495 nm and detection set for 517–527 nm using a 40x oil-objective lens. Multiple images were taken for each treatment.
Mitochondrial ROS were measured by using Mitosox Red following manufacture’s instruction. Briefly, cells were plated at 0.3×106 cells/ml in six well plate. After treatment, cells were washed and Mitosox (5μM) was added in media and incubated for 10 min at 37°C in the dark. Cells were imaged on a Leica SP8 confocal microscope with an excitation laser of 510 nm and detection set for 560–650 nm using a 40x oil-objective lens. Multiple images were taken for each treatment.
For assessing NDI1-mediated ROS production, A375 cells were transfected with NDI1 plasmid or empty vector for 48h and transfection, then metformin, piericidin A, or CPT was given for 30 min and cells were stained with CM-H2DCFDA or MitosoxRed. To study the role of NDUFS2 in NDI1 mediated RET-ROS production, A375 cells were first transfected with NDUFS2 siRNA for 24 hrs, thereafter NDI1 or empty vector was transfected. After 48 hrs, cells were stained with CM-H2DCFDA or MitosoxRed and imaged on a Leica SP8 confocal microscope using a 40x oil-objective lens.
Amplex Red ROS measurement with microplate reader
Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit was used for the measurement of ROS as per manufacturer’s instructions. Briefly, cells were seeded for mitochondria isolation. Mitochondrial fractions were resuspended into mitochondrial resuspension buffer (MRB) (NaCl 135mM, KCl 5mM, Mg2SO4 1mM, K2HPO4 0.4mM, Glucose 5.5mM, HEPES 20mM) and concentration was measured. The master buffer contains the MRB buffer supplemented with 5μM Amplex Red (non-fluorescent form) and 7U/ml horseradish peroxidase (HRP). Mitochondria were added just prior to measurements. Fluorescence spectrum was measured at 571/585 nm for 30 min at 37°C on a Cytation 3 Multi-Mode Reader (BioTek Instruments) with shaking. Freshly prepared H2O2 was used a positive control.
ROS measurement by CM-H2DCFDA with microplate reader
5×104 cells were seeded and treated as indicated in figures. After treatment, cells were trypsinized and stained with CM-H2DCFDA (5μM) for 15 min in the dark at 37°C. After incubation, cells were washed and resuspended in DPBS. Stained cells were added in black-walled, clear-bottom 96-well. Emission was observed at 492–495/517–527 nm for 30 min at 37°C on a Cytation 3 Multi-Mode Reader (BioTek Instruments) with shaking. Each experiment was repeated three times. For measuring NDI1-mediated ROS production, A375 cells were transfected with NDI1 plasmid or empty vector for 48h. After transfection, cells were trypsinzed and stained with CM-H2DCFDA and fluorescence spectrum was observed on a Cytation 3 Multi-Mode Reader (BioTek Instruments). For Basal level of RET-ROS measurement, cells were treated with RET inhibitors for 15–30 min. After treatment, cells were trypsinized and stained with CM-H2DCFDA.
NAD+/NADH measurement
NAD+/NADH was assayed using an NAD+/NADH quantification colorimetric kit according to the manufacturer’s instructions. Briefly, 2×105 cells were seeded and treated with DES (5mM), CPT (20μM), NAM (5mM), and DPI (10μM) for 24h. After treatment, cells were pellet by centrifuging at 2,000 rpm for 5 minutes. Cells were incubated with lysis buffer for 15 min at 4°C and cell lysates were collected after centrifugation at 12,000g for 15 min. 100μl samples were added to 96-well plate. For NADH measurement, NADH reaction mixture was added to the well and incubated at room 37°C for 15 minutes and absorbance was observed at 460 nm. For the measurement of total NAD+/NADH amount, NAD extraction solution into the lysates were added and incubated at 37°C for 15 minutes, thereafter neutralization solution was added to neutralize the NAD extracts. Absorbance was monitored at 460 nm. The ratio of NAD+/NADH was determined by the following equation: ratio = NAD (total) – NADH/NADH.
For the measurement of NAD+/NADH ratio change after expression of NDI1 or LbNOX, cells were transfected with pLV-NDI1, -LbNOX-mito, -LbNOX-cyto, or -RFP control. The constructs were described before (Martinez-Reyes et al., 2020). Briefly, cells were transfected with indicated virus titrates. After 48h of transfection, cells were collected and lysed in lysis buffer and samples were processed for NAD+/NADH measurement as described above.
For fly experiments, larvae brains were isolated and NAD+/NADH was measured using the same protocol as mentioned above. NADH standard, blanks were taken as experimental control. Each experiment was conducted in triplicate and repeated three times.
LDH activity assay
2×105 cells were seeded and treated with DES (5mM), CPT (10μM), Mdivi-1 (10μM), S1QEL1.1 (10 μM), or rotenone (250nM) for 24h. After treatment, medium was collected and centrifuged at 2000 rpm for 5 min. 200μl medium was added into clear, flat bottom 96-well plate. 20μl assay buffer was added into the wells and incubated at room temperature for one hour. Thereafter 50μl of LDH reaction buffer was added for 30 min and O.D. was measured at 490nm. For siRNA experiments, cells were transfected with control and NOTCH2 siRNA for 24h. After 24h transfection, cells were treated with DES (5mM) for 24h. After treatment, medium was collected and processed as mentioned above. Each experiment was conducted in duplicate and repeated three times.
Cytotoxicity assay
TOTO™-3 Iodide and TO-PRO™-3 Iodide dyes were used for cell proliferation and cell death respectively. Protocols were followed as per manufacture’s instructions. Briefly, 3×106 cells were seeded and treated with DES (5mM), CPT (10 μM), Mdivi-1 (10μM), S1QEL1.1 (10μM), metformin (10μM), rotenone (250nM), Notch inhibitor FL-06 (10μM), DAPT (10 μM), NADH (500μM), FK866 (50nM), NAM (5mM), DPI (10μM), NMN (5mM), SIRT1i (10μM), SIRT2i (10μM), SIRT3i (10μM), for 24–48 hrs. After treatment, cells were trypsinized and washed with PBS. Cell pellets were stained with To-To3 (5μM) or To-Pro 3 (5μM) for 15 min in the dark at 37°C. After incubation, cells were immediately observed using flow cytometer (LSR II) and data was analyzed by using the FlowJo software. Each experiment was conducted in duplicated and repeated three times.
MTT assay
For MTT assays, cells were plated in 96-well plates (2,000 cells/well), allowed to adhere overnight, Next day, cells were treated with DES (5mM), CPT (10–40μM), Mdivi-1 (10μM), S1QEL1.1 (10μM), metformin (10μM), rotenone (250nM), piericidin A (10 μM), Notch inhibitor FL-06 (10μM), DAPT (10 μM), RBPJ inhibitor (RIN1; 10 μM), NADH (500μM), FK866 (50nM), NAM (5mM), DPI (10μM), NMN (5mM), SIRT1i (10μM), SIRT2i (10μM), SIRT3i (10μM), or DMSO vehicle control for 72 hrs. After treatment, MTT (5mg/ml) was added and incubated for 4 hrs. Thereafter, MTT solubilizing buffer was added and O.D. was observed after 30 min at 570 nm. Data was analyzed using Microsoft excel and statistical significance was calculated by using Graphpad. For siRNA experiments, cells were transfected with control and NOTCH2 siRNA, RBPJ-siRNA, or NDUFS2-siRNA for 24 hrs. After transfection, cells were treated with indicated inhibitors for 72 hrs. After treatment, MTT was added for 4 hrs and MTT solubilizing buffer was added. O.D. was measured at 570 nm. Each experiment was repeated three times. A375 and GBM-387 cells were transfected with NDI1, cyto-LbNOX, mito-LbNOX or RFP control using Lipofectamine 2000. After 48 hrs of transfection, MTT was added for 4 hrs and thereafter MTT solubilizing buffer was added and O.D. was measured at 570 nm.
Clonogenicity assay
For colony formation assay, A375 cells were plated in 96-well plates (500 cells/well), allowed to adhere overnight, Next day, cells were treated with DES (5mM), CPT (10–40μM), Mdivi (10 μM), S1QEL1.1 (10μM), metformin (10μM), rotenone (250nM), piericidin A (10μM), Notch inhibitor FL-06 (10μM), DAPT (10 μM), RBPJ inhibitor (RIN1; 10 μM), NADH (500μM), FK866 (50nM), NAM (5mM), DPI (10μM), NMN (5mM), SIRT1i (10μM), SIRT2i (10μM), SIRT3i (10μM), or DMSO vehicle control for two weeks. After treatment, cells were washed with PBS and crystal violet solution (0.5% crystal violet solution in 25% methanol) was added for 15 min at room temperature. After incubation, plates were washed with water. After drying the plates, colonies were scanned and quantified in Adobe Photoshop (Adobe Photoshop CC2017). Each experiment was repeated three times. A375 cells were transfected with virus titrates of pLV-NDI1, -mito-LbNOX, or -RFP control. Colonies were continuously observed for 10 days. Thereafter, colonies were stained with crystal violet solution. After staining, colonies were scanned and quantified using Adobe Photoshop.
Tumor sphere formation assay
To test for the effect of RET inhibitor on GBM-387 CSCs, 200 cells/well were plated in a 96 well plate and treated with DES (6.5μM), CPT (2.5μM), Mdivi-1 (5μM), S1QEL1.1 (2μM), metformin (2μM), rotenone (10nM), Notch inhibitor FL-06 (2μM), NADH (100μM), FK866 (5nM), NAM (1mM), DPI (1μM), NMN (1mM), SIRT1i (5μM), SIRT2i (2μM), SIRT3i (5μM), or DMSO vehicle control. Spheroids were observed for one to two weeks. Number of spheroids formed was counted under an inverted microscope and graphs were plotted using GraphPad. Each experiment was repeated three times. Approximately 1×103 GBM-387 cells were seeded in 96 well plate and transfected with virus titrates of pLV-NDI1, -mito-LbNOX, or -RFP control. After 10 days of transfection, tumor spheroids were counted under inverted microscope and statistical significance was calculated by Graph-Pad.
Transwell migration assay
A375 cells were re-suspended at the concentration of 1 × 106 cells/ml in the migration buffer consisting of DMEM medium and 0.1% BSA without serum. In the transwell cell migration assay, 24-well inserts from Corning were used. Conditioned media from NIH3T3 fibroblast cells grown in DMEM medium containing 10% fetal bovine serum was used as the chemo-attractant. 100 μl of A375 cells was added on top of the transwell membrane in the upper chamber and 600 μl of chemo-attractant was added to the lower chamber or the same volume of migration buffer added as a negative control. The number of migrated cells could be quantified by counting underneath a microscope or in the pictures taken. Each experiment was repeated three times.
qRT-PCR
RNA was isolated using Trizol from cells treated under various conditions, and qPCR analysis was performed for IL-1β and IL-4 mRNA expression. Briefly, 2×106 cells were seeded and treated with DES (5mM), CPT (10μM) and NADH (500μM) for 24h. After incubation, cells were scrapped and washed with PBS. Cell pellet were lysed with 1 ml Trizol and incubated at room temperature for 15 min. Thereafter, 250μl chloroform was added and mixed properly by vortexing followed by centrifugation at 12000g for 15min. Top layer (clear solution) was collected and 500μl isopropanol was added and incubated at −20°C for overnight. The next day, samples were centrifuged at 20000g for 30 min. Pellets were washed with 75% ethanol and centrifuged at 7000g for 5min. After that, pellets were air dried and resuspended in DEPC-treated water and RNA concentration was measured. cDNA was immediately synthesized using iScript cDNA synthesis kits (Bio-Rad). Thereafter, powerUp SYBR green master mix (A25741, Applied Biosystems) was used for determination of IL-1β and IL-4 expression. Primer sequences were described in oligonucleotide section. Fold change was measured using Microsoft Excel and statistical significance was performed using GraphPad. Each experiment was conducted in triplicate and repeated three times.
Protease protection assay
3×106 A375 cells were seeded for overnight. Next day, cells were scrapped and mitochondrial extracts were prepared using a Dounce homogenizer in HBS buffer (5 mM HEPES, 70 mM sucrose, 210 mM Mannitol, 1 mM EGTA, 1x protease inhibitor cocktail). 50 μg mitochondrial extracts were treated with digitonin (2%) and dose dependent trypsin (0–2.5μg) for 30 min on ice. Thereafter, centrifugation was done at 10,000×g for 15min. The mitochondrial pellets were used for western blot analysis. Tom20 and Cyto-C was used as markers for mitochondrial outer membrane and intermembrane space, respectively. C-I30 was used as inner membrane marker.
Pharmacological treatment of fly larvae
CPT (10–20μM), CCCP (10μM), S1QEL1.1 (10μM) rotenone (250nM), Mdivi-1 (10μM), NAM (5mM), resveratrol (100μM) and DPI (5μM) were mixed in instant Drosophila media. 0.5% DMSO alone was used as vehicle control. Embryos were collected on drug-containing food or control food for 6 hrs at 25°C, shifted to 29°C to activate transgene expression and allowed to develop further at 29°C to 120h after larval hatching before larval brain dissection and immunostaining.
Immunohistochemistry and ROS imaging on fly tissues
For larval brain immunostaining, larvae were dissected in Schneider’s medium (Invitrogen) and fixed with 4% formaldehyde in PEM buffer (100 mM PIPES at pH 6.9, 1 mM EGTA, 1 mM MgCl2) for 23 min at room temperature. Immunohistochemistry with anti-Pros and anti-Dpn in Drosophila brain was performed as described (Song and Lu, 2011). The primary antibodies used were mouse anti-Pros (1:200) and guinea pig anti-Dpn (1:1000; J. Skeath). Corresponding secondary antibodies were from Molecular Probe (all at 1:200 dilution). Samples were imaged on a Leica SP8 confocal microscope and neuroblast numbers were counted and quantified.
For ROS staining in fly tissues, samples were dissected in 1xPBS and were incubated, protected from light, in 10μM CM-H2DCFDA (Thermofisher) in 1xPBS for 30 min at room temperature. Dye was freshly reconstituted each time in anhydrous DMSO right before use. The samples were briefly washed 3 times using 1xPBS. Midguts and whole brains were imaged immediately after staining procedure under confocal microscope (Leica SP8). For monitoring total ROS production from the midgut, Z stacks of region 200–500 μm anterior to the pylorus region were evaluated for mean signal intensity acquired at 488nm and quantified in NIH Image J. For brain samples, Z stacks of images from whole-mount brain samples were acquired at 488nm and signal intensity quantified in NIH Image J. For ROS quantification in larval brain, ROS staining intensity was normalized to brain area. Around 10 tissue samples were used for quantification in each condition.
Mouse xenograft GBM model
Patient derived BFP-GBM-387 cells stably expressing a luciferase reporter were sorted by flowcytometry. After sorting, 0.5×105 cells were prepared for supratentorial injection at the coordinates of 2 millimeters posterior to the bregma, 1 millimeter laterally, and 2 millimeters deep to the dura. After injection, surgical site was closed and anesthetized mice were observed until recovery. Tumor growth was observed after 8 days of injection by bioluminescence imaging following intraperitoneal injection of D-luciferin. The in vivo imaging was done at Stanford Center for innovation in in vivo imaging (Sci3) on IVIS spectrum (Parkin Elmer), to monitor effect of CPT on tumor growth. Animals exhibiting tumor growth were randomly separated into vehicle and drug-treatment groups. Thereafter, we started giving CPT (50mg/kg body weight) or vehicle only (DMSO:PEG300:cyclodextran:H2O=5:45:10:40) via IP route. All animal experiments were performed in accordance with the protocols approved by the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford University.
Quantification and statistical analysis
Comparisons between two groups were calculated using one- or two-tailed Student’s t tests, using GraphPad Prism software. Data are reported as mean ± SEM. Statistical values, including number of replicates (n), can be found in the figure legends. *p < 0.05, **p < 0.01, ***p < 0.001. For comparing multiple groups, we used one-way ANOVA test followed by Student–Newman–Keuls test (SNK test) plus Bonferroni correction (multiple hypotheses correction).
Supplementary Material
Highlights.
Cancer cells undergo active reverse electron transfer (RET) along complex I (C-I)
NAD+/NADH ratio change critically mediates RET effect on cancer cell behavior
Notch acts unconventionally to promote RET by interacting with C-I proteins
Pharmacological inhibition of RET retards brain tumor growth in flies and mice
ACKNOWLEDGEMENTS
We are grateful to Drs. D. Walker, H. Jacobs, M. Fortini, L. Luo, the VDRC, FlyORF, and Bloomington Stock Centers for fly stocks; Dr. N. Chandel for plasmids; Stanford Flowcytometry Core facility, Sci3 Imaging Core, Stanford PAN facility for services; Dr. M. Bogyo’s lab in the Department of Pathology for equipment use. Special thanks go to J. Gaunce for maintaining fly stocks and providing technical supports and members of the Lu lab for discussions. Supported by the NIH (NS084412 to B.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
B.L. is a scientific founder of Cerepeut, a member of Cerepeut’s scientific advisory board, and an inventor on patents related to CPT2008.
REFERENCES
- Andersson ER, Sandberg R, and Lendahl U (2011). Notch signaling: simplicity in design, versatility in function. Development 138, 3593–3612. [DOI] [PubMed] [Google Scholar]
- Arumugam TV, Chan SL, Jo DG, Yilmaz G, Tang SC, Cheng A, Gleichmann M, Okun E, Dixit VD, Chigurupati S, et al. (2006). Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat Med 12, 621–623. [DOI] [PubMed] [Google Scholar]
- Aster JC, Pear WS, and Blacklow SC (2017). The Varied Roles of Notch in Cancer. Annu Rev Pathol 12, 245–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batandier C, Guigas B, Detaille D, El-Mir MY, Fontaine E, Rigoulet M, and Leverve XM (2006). The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr 38, 33–42. [DOI] [PubMed] [Google Scholar]
- Bonkowski MS, and Sinclair DA (2016). Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnay F, Veloso A, Steinmann V, Kocher T, Abdusselamoglu MD, Bajaj S, Rivelles E, Landskron L, Esterbauer H, Zinzen RP, et al. (2020). Oxidative Metabolism Drives Immortalization of Neural Stem Cells during Tumorigenesis. Cell 182, 1490–1507 e1419. [DOI] [PubMed] [Google Scholar]
- Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, et al. (2017). The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell 40, 583–594 e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman SK, Rolland V, Betschinger J, Kinsey KA, Emery G, and Knoblich JA (2008). The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev Cell 14, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel F, Rimbert V, Lioger D, Diot A, Rousset P, Mirand PP, Boirie Y, Morio B, and Mosoni L (2005). Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech Ageing Dev 126, 505–511. [DOI] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Developmental cell 14, 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, and Guarente L (2015). The multifaceted functions of sirtuins in cancer. Nat Rev Cancer 15, 608–624. [DOI] [PubMed] [Google Scholar]
- Chance B, and Hollunger G (1961). The interaction of energy and electron transfer reactions in mitochondria. I. General properties and nature of the products of succinate-linked reduction of pyridine nucleotide. J Biol Chem 236, 1534–1543. [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cino M, and Del Maestro RF (1989). Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch Biochem Biophys 269, 623–638. [DOI] [PubMed] [Google Scholar]
- Deighton RF, Le Bihan T, Martin SF, Gerth AMJ, McCulloch M, Edgar JM, Kerr LE, Whittle IR, and McCulloch J (2014). Interactions among mitochondrial proteins altered in glioblastoma. J Neurooncol 118, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieteren CE, Willems PH, Swarts HG, Fransen J, Smeitink JA, Koopman WJ, and Nijtmans LG (2011). Defective mitochondrial translation differently affects the live cell dynamics of complex I subunits. Biochim Biophys Acta 1807, 1624–1633. [DOI] [PubMed] [Google Scholar]
- Dongre A, Surampudi L, Lawlor RG, Fauq AH, Miele L, Golde TE, Minter LM, and Osborne BA (2014). Non-Canonical Notch Signaling Drives Activation and Differentiation of Peripheral CD4(+) T Cells. Front Immunol 5, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Aguera MC, Gao L, Gonzalez-Rodriguez P, Pintado CO, Arias-Mayenco I, Garcia-Flores P, Garcia-Perganeda A, Pascual A, Ortega-Saenz P, and Lopez-Barneo J (2015). Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab 22, 825–837. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ayala DJ, Sanz A, Vartiainen S, Kemppainen KK, Babusiak M, Mustalahti E, Costa R, Tuomela T, Zeviani M, Chung J, et al. (2009). Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab 9, 449–460. [DOI] [PubMed] [Google Scholar]
- Galli U, Mesenzani O, Coppo C, Sorba G, Canonico PL, Tron GC, and Genazzani AA (2012). Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2. Eur J Med Chem 55, 58–66. [DOI] [PubMed] [Google Scholar]
- Goncalves RL, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, and Brand MD (2015). Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J Biol Chem 290, 209–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, and Tomlinson IP (2005). Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer 5, 857–866. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, and Levy DE (2009). Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324, 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarani V, Deflorian G, Franco CA, Kruger M, Phng LK, Bentley K, Toussaint L, Dequiedt F, Mostoslavsky R, Schmidt MHH, et al. (2011). Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 473, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, and Nijtmans L (2017). The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab 25, 128–139. [DOI] [PubMed] [Google Scholar]
- Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ, and Vander Heiden MG (2016). Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 24, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha KG, Kankel MW, and Artavanis-Tsakonas S (2012). The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet 13, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameed A, Al-Rashida M, Uroos M, Ali SA, Arshia, Ishtiaq M, and Khan KM (2018). Quinazoline and quinazolinone as important medicinal scaffolds: a comparative patent review (2011–2016). Expert Opin Ther Pat 28, 281–297. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hasmann M, and Schemainda I (2003). FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res 63, 7436–7442. [PubMed] [Google Scholar]
- Hinchliffe P, and Sazanov LA (2005). Organization of iron-sulfur clusters in respiratory complex I. Science 309, 771–774. [DOI] [PubMed] [Google Scholar]
- Ho DM, Artavanis-Tsakonas S, and Louvi A (2020). The Notch pathway in CNS homeostasis and neurodegeneration. Wiley Interdiscip Rev Dev Biol 9, e358. [DOI] [PubMed] [Google Scholar]
- Holland PC, Clark MG, Bloxham DP, and Lardy HA (1973). Mechanism of action of the hypoglycemic agent diphenyleneiodonium. J Biol Chem 248, 6050–6056. [PubMed] [Google Scholar]
- Homem CCF, Steinmann V, Burkard TR, Jais A, Esterbauer H, and Knoblich JA (2014). Ecdysone and mediator change energy metabolism to terminate proliferation in Drosophila neural stem cells. Cell 158, 874–888. [DOI] [PubMed] [Google Scholar]
- Hunter GL, and Giniger E (2020). Phosphorylation and Proteolytic Cleavage of Notch in Canonical and Noncanonical Notch Signaling. Adv Exp Med Biol 1227, 51–68. [DOI] [PubMed] [Google Scholar]
- Hur JH, Stork DA, and Walker DW (2014). Complex-I-ty in aging. J Bioenerg Biomembr 46, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, and Guarente L (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol 24, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, and Stamenkovic I (2012). Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev 26, 1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsyuba E, Romani M, Hofer D, and Auwerx J (2020). NAD(+) homeostasis in health and disease. Nat Metab 2, 9–31. [DOI] [PubMed] [Google Scholar]
- Kovall RA, Gebelein B, Sprinzak D, and Kopan R (2017). The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev Cell 41, 228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Mentrup T, Kleizen B, Rivera-Milla E, Reichenbach D, Enzensperger C, Nohl R, Tauscher E, Gorls H, Ploubidou A, et al. (2013). Small molecules intercept Notch signaling and the early secretory pathway. Nat Chem Biol 9, 731–738. [DOI] [PubMed] [Google Scholar]
- Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, Helgason GV, and Gottlieb E (2017). Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med 23, 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landor SK, Mutvei AP, Mamaeva V, Jin S, Busk M, Borra R, Gronroos TJ, Kronqvist P, Lendahl U, and Sahlgren CM (2011). Hypo- and hyperactivated Notch signaling induce a glycolytic switch through distinct mechanisms. Proc Natl Acad Sci U S A 108, 18814–18819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P, Reifenberger G, and Weller M (2019). Molecular targeted therapy of glioblastoma. Cancer Treat Rev 80, 101896. [DOI] [PubMed] [Google Scholar]
- Lee KS, Wu Z, Song Y, Mitra SS, Feroze AH, Cheshier SH, and Lu B (2013). Roles of PINK1, mTORC2, and mitochondria in preserving brain tumor-forming stem cells in a noncanonical Notch signaling pathway. Genes Dev 27, 2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY (2016). Temozolomide resistance in glioblastoma multiforme. Genes Dis 3, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wu Z, Tantray I, Li Y, Chen S, Dong J, Glynn S, Vogel H, Snyder M, and Lu B (2020). Quality-control mechanisms targeting translationally stalled and C-terminally extended poly(GR) associated with ALS/FTD. Proc Natl Acad Sci U S A 117, 25104–25115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sato C, Cerletti M, and Wagers A (2010). Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol 92, 367–409. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, and Schubert D (2002). Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 80, 780–787. [DOI] [PubMed] [Google Scholar]
- Ma CY, Yao MJ, Zhai QW, Jiao JW, Yuan XB, and Poo MM (2014). SIRT1 suppresses self-renewal of adult hippocampal neural stem cells. Development 141, 4697–4709. [DOI] [PubMed] [Google Scholar]
- Majander A, Finel M, and Wikstrom M (1994). Diphenyleneiodonium inhibits reduction of iron-sulfur clusters in the mitochondrial NADH-ubiquinone oxidoreductase (Complex I). J Biol Chem 269, 21037–21042. [PubMed] [Google Scholar]
- Martinez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, Kihshen H, Reczek CR, Weinberg SE, Gao P, et al. (2020). Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, et al. (2016). Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S, Jow H, Baty K, Johnson A, Czapiewski R, Saretzki G, Treumann A, and von Zglinicki T (2014). Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun 5, 3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell CS, and Radtke F (2017). Notch as a tumour suppressor. Nat Rev Cancer 17, 145–159. [DOI] [PubMed] [Google Scholar]
- Onukwufor JO, Berry BJ, and Wojtovich AP (2019). Physiologic Implications of Reactive Oxygen Species Production by Mitochondrial Complex I Reverse Electron Transport. Antioxidants (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, et al. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519. [DOI] [PubMed] [Google Scholar]
- Pasto A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, Rasola A, Frasson C, Nardo G, Zulato E, et al. (2014). Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 5, 4305–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierfelice TJ, Schreck KC, Eberhart CG, and Gaiano N (2008). Notch, neural stem cells, and brain tumors. Cold Spring Harb Symp Quant Biol 73, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde KR, and Hirst J (2011). Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J Biol Chem 286, 18056–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajman L, Chwalek K, and Sinclair DA (2018). Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab 27, 529–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecca VW, Nicastri MC, Fennelly C, Chude CI, Barber-Rotenberg JS, Ronghe A, McAfee Q, McLaughlin NP, Zhang G, Goldman AR, et al. (2019). PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov 9, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, and Murphy MP (2018). Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 293, 9869–9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito N, Fu J, Zheng S, Yao J, Wang S, Liu DD, Yuan Y, Sulman EP, Lang FF, Colman H, et al. (2014). A high Notch pathway activation predicts response to gamma secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 32, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Ramiro A, Ramirez-Ortega D, Perez de la Cruz V, Hernandez-Pedro NY, Gonzalez-Esquivel DF, Sotelo J, and Pineda B (2016). Role of Redox Status in Development of Glioblastoma. Front Immunol 7, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M, and Chandel NS (2014). ROS function in redox signaling and oxidative stress. Curr Biol 24, R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A, and Harris AL (2012). How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491, 364–373. [DOI] [PubMed] [Google Scholar]
- Scialo F, Fernandez-Ayala DJ, and Sanz A (2017). Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front Physiol 8, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialo F, Sriram A, Fernandez-Ayala D, Gubina N, Lohmus M, Nelson G, Logan A, Cooper HM, Navas P, Enriquez JA, et al. (2016). Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab 23, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BB, Kitajima-Ihara T, Chan EK, Scheffler IE, Matsuno-Yagi A, and Yagi T (1998). Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc Natl Acad Sci U S A 95, 9167–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lim SK, Liang Q, Iyer SV, Wang HY, Wang Z, Xie X, Sun D, Chen YJ, Tabar V, et al. (2019). Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 567, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, and Guarente L (2014). Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol 54, 363–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing A, Tsatskis Y, Fabian L, Hester I, Rosenfeld R, Serricchio M, Yau N, Bietenhader M, Shanbhag R, Jurisicova A, et al. (2014). The atypical cadherin fat directly regulates mitochondrial function and metabolic state. Cell 158, 1293–1308. [DOI] [PubMed] [Google Scholar]
- Slaninova V, Krafcikova M, Perez-Gomez R, Steffal P, Trantirek L, Bray SJ, and Krejci A (2016). Notch stimulates growth by direct regulation of genes involved in the control of glycolysis and the tricarboxylic acid cycle. Open Biol 6, 150155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, and Huber LJ (2006). Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol 26, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, and Lu B (2011). Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes & development 25, 2644–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova A, Konrad C, Manfredi G, Springett R, Ten V, and Galkin A (2019). The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A. J Neurochem 148, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, and Chandel NS (2014). Mitochondrial reactive oxygen species and cancer. Cancer Metab 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, and Mootha VK (2016). Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tome M, Tchorz J, Gassmann M, and Bettler B (2019). Constitutive activation of Notch2 signalling confers chemoresistance to neural stem cells via transactivation of fibroblast growth factor receptor-1. Stem Cell Res 35, 101390. [DOI] [PubMed] [Google Scholar]
- van den Ameele J, and Brand AH (2019). Neural stem cell temporal patterning and brain tumour growth rely on oxidative phosphorylation. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR, Zhu J, and Hirst J (2014). Architecture of mammalian respiratory complex I. Nature 515, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Warburg O, Wind F, and Negelein E (1927). The Metabolism of Tumors in the Body. J Gen Physiol 8, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, and Chandel NS (2009). Mitochondrial metabolism and cancer. Ann N Y Acad Sci 1177, 66–73. [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris J.P.t., Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, and Aster JC (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271. [DOI] [PubMed] [Google Scholar]
- Weng M, Golden KL, and Lee CY (2010). dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila. Dev Cell 18, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chi F, Guo T, Punj V, Lee WN, French SW, and Tsukamoto H (2015). NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest 125, 1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Soga T, and Pollard PJ (2013). Oncometabolites: linking altered metabolism with cancer. J Clin Invest 123, 3652–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.